Introduction
Hereditary multiple exostoses (HME) is an autosomal
dominant skeletal disorder that is characterized by multiple
cartilage-capped tumors growing outward from the metaphyseal region
of the long tubular bones and other skeletal structures (1,2).
HME can lead to physical and psychological problems in patients,
causing huge burdens to both families and society, although it is a
rare disease (1,3,4).
The incidence of HME was reported to be 0.4-1 per 50,000 in the
Western countries (1,5).
The appearance of a palpable mass near the knees,
shoulders, ankles or wrists is the most common clinical
presentation in patients with HME (1). Hennekam (6) suggest that a patient with HME is
generally burdened with an average of six exostoses. In addition to
patient appearance D'Ambrosi et al (3) reported that children affected by HME
report lower sports activity, particularly among female patients.
In addition, neurological syndromes, pains, functional disorders
and deformities are also common clinical presentations of HME
(7–9). According to previous studies,
although most of the masses were benign, ≤2% of cases develop into
malignant chondrosarcoma or osteosarcoma (1,10).
These data and results were summarized and reported in western
countries; however, few reports of clinical features of HME have
been studied in Chinese populations. Therefore, it is essential to
study the clinical course of this disease in Chinese populations
and compare the similarities and differences in natural history,
clinical presentation and characteristics with other ethnicities to
facilitate a further understanding of HME. This is one of the
objectives of the present study.
Study of pathogenesis is useful for further
understanding of diseases and provides guidance for treatments. As
to the pathogenesis of HME, a number of risk factors have been
identified and pathogenic mechanisms have been speculated (10–15), such as genes and genetic factors,
abnormal embryonic development, and growth and development
disfunction of epiphysis at long bones. For example, Inubushi et
al (16) reported a
connection between increased BMP signaling and
osteochondromagenesis, which serves an essential role in skeletal
development by regulating chondrocyte proliferation and
differentiation; they also suggest that treatment with a BMP
inhibitor may be effective in patients with HME, which showed
promising results in their study conducted on mouse models.
Bukowska-Olech et al (12)
hypothesized that heparanase, an enzyme that cleaves the heparan
sulfate (HS) chains and stimulates chondrogenesis, is
physiologically found only in the hypertrophic zone and
perichondrium, and its wider distribution and increased activity
possibly served a role in the development of osteochondromas.
Although several risk factors and pathogenic
mechanisms have been hypothesized, it is most frequently suggested
that mutations in exostosin glycosyltransferase 1 (EXT-1)
and EXT-2, genes with autosomal dominant inheritance, are
the most likely responsible factors for HME (2,12),
having been detected in 28–65% and in 21–61% of the affected
patients, respectively (1).
EXT-1 is located on chromosome 8q24,11-q24.13 and
EXT-2 on chromosome 11p11-12 (11); both encode glycosyltransferases
and serve vital roles in the synthesis of HS. HS is a
polysaccharide material that binds to core proteins to produce HS
proteoglycans, which are present to a large extent in the cell
membrane and extracellular matrix and interact with BMPs
participating in the regulation of bone and cartilage formation
(11). Therefore, heterozygous
mutations of the EXT-1 or EXT-2 gene lead to an HS
deficiency of ~50%, which is inadequate for giving rise to
osteochondroma formation in patients with HME (11). An increasing number of mutations
of EXT-1 and EXT-2 have been observed (12,17). In Guo et al (18), a novel heterozygous splice
mutation (c.1284+2del) in EXT-1 was identified in a
three-generation family with HME. Medek et al (10) identified different EXT-1
gene mutations in five probands that resulted in premature
stop-codons (p.Gly124Argfs*65, p.Leu191*, p.Trp364Lysfs*11,
p.Val371Glyfs*10, p.Leu490Profs*31). In two of the probands,
nonsense mutations were found in EXT-2 gene
(p.Val187Profs*115, p.Cys319fs*46). In total, five mutations were
novel and two mutations have occurred de novo in probands.
Xian et al (19) also
found a novel heterozygous splice site mutation (c.1173+2T>A) in
the EXT-2 gene. Chen et al (20) identified an original nonsense
mutation in the EXT-2 gene (9c.526C>T; p.Gln176*) in a
Chinese family with HME from Hangzhou. Indeed, >70% of HME cases
arise from monoallelic mutations in either EXT-1 or
EXT-2 (10,11,21), and in 5–34% of the patients with
HME, mutations EXT-1 or EXT-2 are not detected
(22), suggesting that there
might be other mutations of EXT genes in patients with HME
which may serve important roles in pathogenesis.
Therefore, the object of the present study was to
report the clinical presentations and characteristics of HME in a
large Chinese pedigree and to analyze the molecular background
using DNA sequencing in this pedigree.
Materials and methods
Patients and a large Chinese
pedigree
The proband was a 24-year-old female patient from
Anhui, Jiangsu (China), with multiple exostoses involving the
scapula, bilateral distal femurs and bilateral proximal tibiae and
bilateral proximal fibula, who received osteochondroma surgery for
the scapula at Shanghai Changhai Hospital (Shanghai, China) in 2012
and osteochondroma surgery for the bilateral proximal tibiae and
fibula in 2016 due to pain, functional limitation and deformity.
Post-biopsy analysis indicated that the masses were multiple
exostoses. Several generations of family members from the proband
were investigated by two surgeons (WW and YS). A total of 20 family
members were recruited in the present study, including 10 males and
10 females (age, 6–83 years). The dates patients were recruited
started at 9 March 2017 and ended at 28 March 2017. Diagnostic
criterium was X-ray examination revealing that the long bone
metaphysis had at least two or more osteochondromas. In addition,
three patients (two males and one female, age, 45 to 55 years old)
with arthritis and without HME who received surgery in Shanghai
Changhai Hospital in June–December 2017 were recruited and their
cartilage specimens were resected and used as controls. The present
study was approved by the Institutional Review Board of Shanghai
Changhai Hospital (approval no. CHEC20180184), and all patients
provided written informed consent for the study. In addition, since
minor patients were not able to understand the study design,
purpose and cautions of this study, the parents of the patients
provided informed consent.
Collection of data, post-operation
biopsy and peripheral blood
The authors (WW and YS) went to Jiangsu (China) to
collect data on 9 March 2017 and 28 March 2017, respectively. Each
participant in this pedigree underwent careful physical and/or
radiographic examinations by two surgeons (MY and DH). Multiple
exostoses were diagnosed as at least two osteochondromas, arising
from the lateral ends of the humerus, ulna, femur, tibia, fibula,
knee joint or scapula. Those patients with multiple exostoses were
classified using a the multiple osteochondromas classification
system through a Switching Neural Networks approach (23). Clinical data were collected from
this family pedigree, including the number of patients with
multiple exostoses and healthy subjects, age, height, the age when
the first exostosis was recognized, deformity or functional
disabled and locations of exostoses.
Resection surgery was performed in the scapula and
bilateral knees of the proband (III-9) and multiple exostoses were
resected in bilateral knees of the patient III-8 for pains and
deformity. These surgeries were performed by the same surgeon (DH)
and the postoperative biopsy was conducted by the Department of
Pathology at Shanghai Changhai Hospital. These two patients were
followed up at two years after the operation.
In addition, EDTA-anticoagulated peripheral blood (5
ml) was drawn from each subject of this pedigree for DNA
sequencing.
Genetic analysis
Preliminary whole-genome sequencing in three HME
patients (II-3, III-8 and III-9) showed that the deletion mutation
at c.824-826delGCA in the EXT-2 gene (in exon 5 of the
EXT-2) was the only mutation site in these three HME patients by
comparing with the published EXT-1 and EXT-2
reference sequences from Leiden Open Variation Database (LOVD;
databases.lovd.nl/shared/genes), which might have disastrous
effects on the etiology of HME. According to preliminary results
(data not shown) the deletion mutation at c.824–826delGCA in the
EXT-2 gene may affect production of glycosyltransferases,
which are vital for the synthesis of heparin sulfate, and
interaction with bone morphogenetic protein that participates in
the regulation of bone and cartilage formation. However, the exact
mechanism by which this deletion mutation influences the occurrence
and development of HME needs further study. Therefore, to explore
whether this mutation at c.824–826delGCA, in exon 5 of the EXT-2
was the only mutation in this pedigree, genomic DNA was extracted
from peripheral blood leukocytes using the GeneJET Whole Blood Mini
kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. The exon 5 of the EXT-2 gene
including the mutation site was amplified by PCR using the upstream
primer 5′-CTGGGAAGTAAGGAAAGGG-3′ and the downstream primer
5′-GAGGTGGGAAAGATGGGTG-3′ (amplicon length: 497; annealing
temperature: 54°C). The amplifications were performed to a final
volume of 20 µl, containing 1 µl genomic DNA, 10 µl 2X Premix Taq
(Vazyme Biotech Co., Ltd.), 1 µl of each primer and 7 µl sterilized
distilled water. The thermocycling conditions were as follows:
Initial denaturation of 5 min at 95°C; followed by 35 cycles of 30
sec at 95°C, 45 sec at annealing temperature and 30 sec at 72°C;
and a final extension for 10 min at 72°C. Direct sequencing of the
PCR products was performed using BigDye Terminator v3.1 (Applied
Biosystems) with the ABI 3730 XL automated sequencer (Applied
Biosystems).
Results
Clinical characteristics of the
pedigree
A total of 20 family members were recruited in the
present study with seven members (five male and two female)
diagnosed as HME with the incidence of 35%; these include II-3,
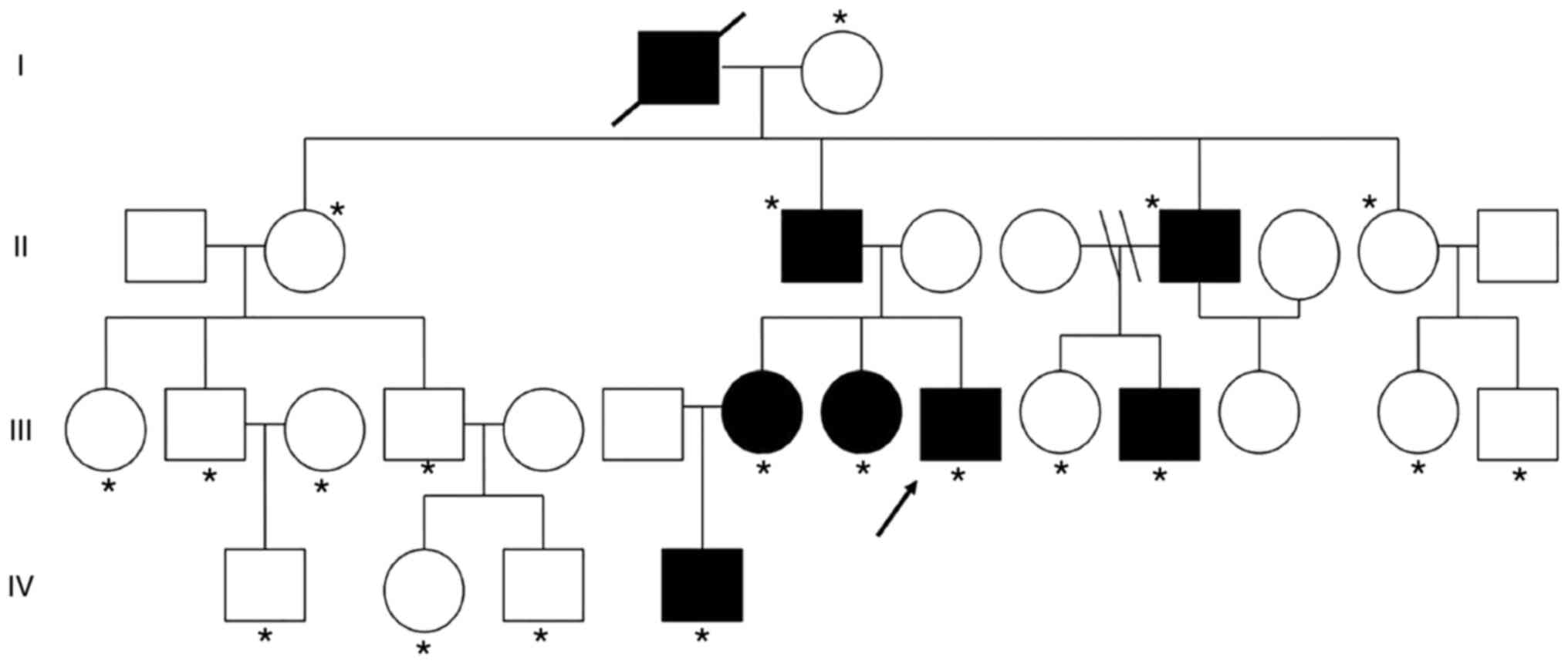
II-6, III-7, III-8, III-9, III-11 and IV-4 (Fig. 1). The grandfather (I-1) of the
proband also had HME according to medical records; however, he died
in 2016 so was not recruited. The pedigree of this family with HME
was shown in Fig. 1. The proband
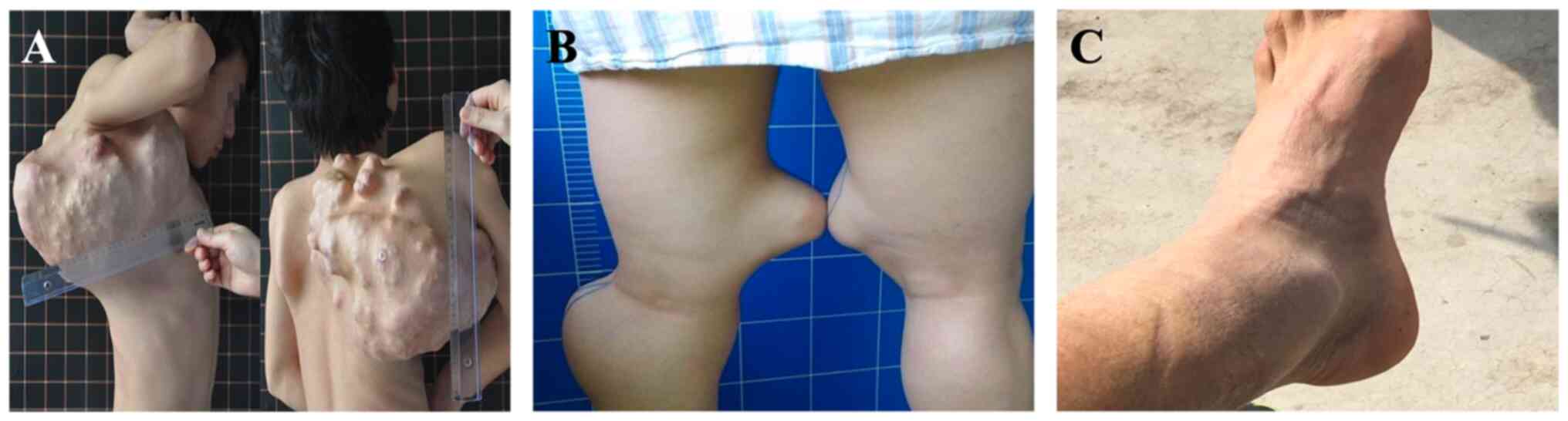
(III-9) was a 24-year-old male patient with multiple exostoses
involving the scapula, bilateral distal femurs, and bilateral
proximal tibiae and fibula (Fig.
2) and other family members was subject without HME.
In this pedigree, the number of male patients with
HME was larger than that of female HME patients. All the patients
with HME in second generation were males and all the male
descendants of II generation with HME (II-3 and II-6), named as
III-9 and III-11 were diagnosed as HME. These findings suggested
that males were more susceptible to HME compared with females.
The age of all patients when the first exostosis was
recognized was <10 years old (Table I). The average age of second
generation, third generation and fourth generation when the first
exostosis was recognized was 8.5 years old, 5.25 years old and 4
years old, respectively, indicating that the age of onset of HME
became earlier in each generation.
 | Table I.Characteristics of HME patients in
the pedigree. |
Table I.
Characteristics of HME patients in
the pedigree.
|
|
|
|
| Affected bone and
joint |
|
|---|
|
|
|
|
|
|
|
|---|
| Subject | Age, year | Age of onset,
year | Height, cm | Scapula | Tibiae | Radius | Rib | Femur | Fibula | Humerus | HME
Classificationa |
|---|
| II-3 | 50 | 9 | 173 | No | Yes | No | No | No | No | No | IA |
| II-6 | 39 | 8 | 174 | No | No | Yes | No | No | No | No | IA |
| III-7 | 30 | 6 | 170 | No | Yes | Yes | Yes | No | No | No | IB |
| III-8 | 25 | 6 | 163 | Yes | Yes | No | Yes | Yes | Yes | Yes | IIB |
| III-9b | 24 | 4 | 156 | Yes | Yes | No | No | Yes | Yes | No | IIIA |
| III-11 | 12 | 5 | 140 | No | Yes | No | Yes | Yes | No | No | IB |
| IV-4 | 10 | 4 | 129 | No | Yes | No | Yes | No | No | No | IA |
The height of II-3, II-6, III-7, III-8 and III-9 was
173, 174, 170, 163 and 156 cm, respectively (Table I). Patients III-11 and IV-4 were
juveniles; based on the data of other five adult patients with HME,
it was found that the height of each affected subject might have a
decreasing trend with generations. Whether the height of patients
III-11 and IV-4 will be shorter than that of second generation
needs further follow-up.
In this pedigree, exostoses were most frequently in
the tibia with occurrence in six patients (85.7%), followed by rib
(4/7; 57.1%), femur (3/7; 42.9%), radius (2/7; 28.6%), fibula (2/7;
28.6%) and scapula (2/7; 28.6%; Table
I). Exostoses was also found in humerus in III-8.
Subjects were classified according to Switching
Neural Networks approach for classification (23) (Table
I). The results showed that the clinical presentation of each
of the four patients in the third generation was classified as IB,
IIB, IIIA and IB, which was more serious compared with that of
second generation, both IA. Taken together, the data suggest that
the younger patients exhibited more severe conditions compared with
the others. The clinical presentation of III-9 (proband, 24 years
old) and III-8 (26 years old) was classified as IIIA and IIB,
respectively, which was more severe than that of the older
relatives, III-7, II-6 and II-3. The clinical presentation of
proband was the most serious because she had multiple exostoses
involving the scapula, bilateral distal femurs and bilateral
proximal tibiae and fibula.
Biopsy and two years' follow-up
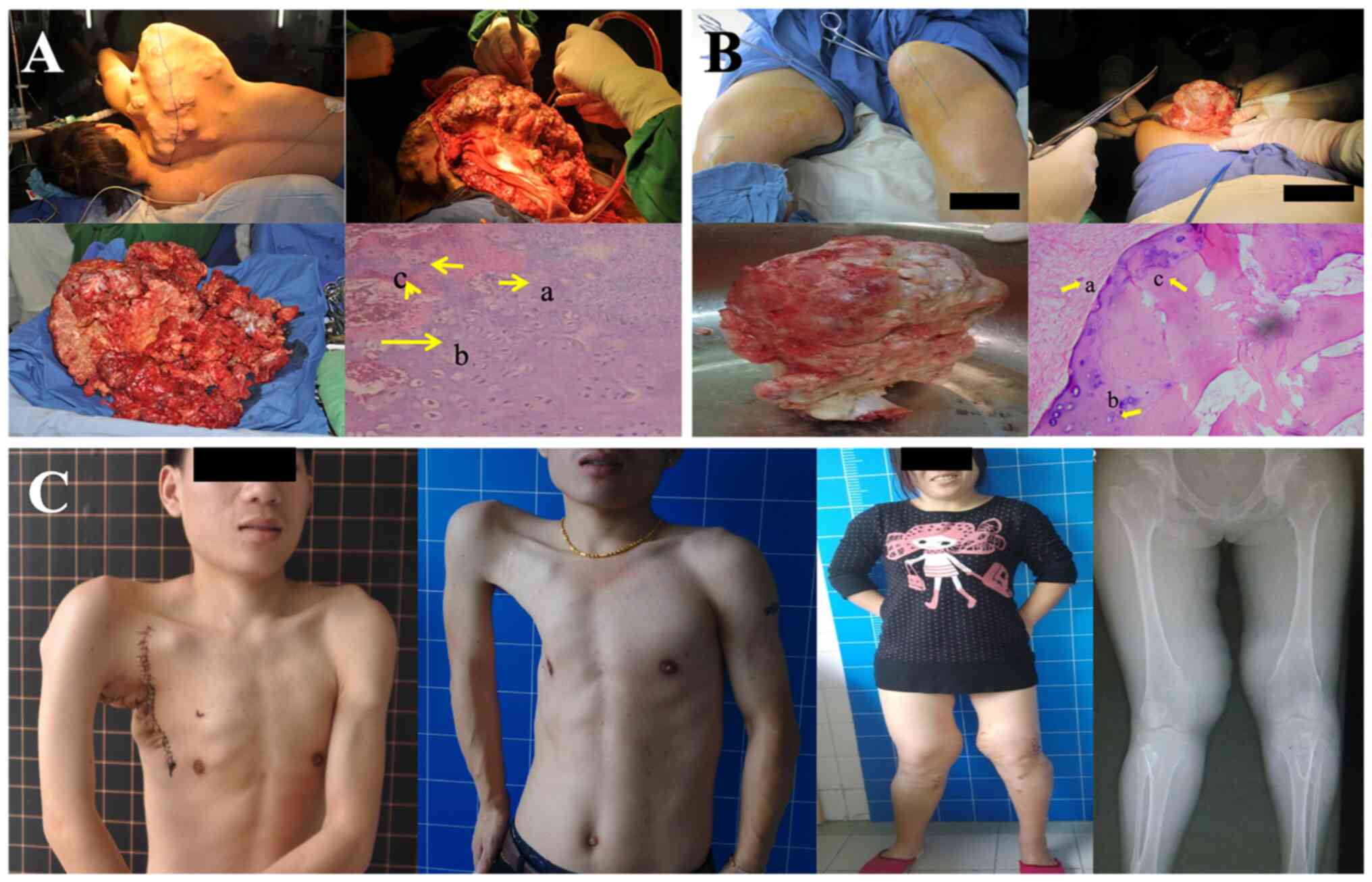
Patients III-8 and the III-9 (proband) received
resection surgery in Shanghai Changhai Hospital for the pains and
skeletal deformations caused by multiple exostoses in the scapula
and the bilateral proximal tibiae and fibula of the proband, and in
the bilateral proximal tibiae and fibula of the patient III-8. The
masses resected from the scapula and bilateral proximal tibiae and
fibula of the proband were verified as malignant cartilage-capped
tumor and benign exostoses, respectively. The post-operation biopsy
of the bilateral proximal tibiae and fibula in patient III-8
indicated that the resected masses were benign exostoses. The
intraoperative pictures and biopsy are shown in Fig. 3A and B. As shown in Fig. 3A, patient III-9 (proband) was
placed in lateral position, and huge masses could be seen in his
scapula. The masses from proband's scapula were resected, and the
postoperative biopsy showed that it was malignant cartilage-capped
tumor. The tumor cells were arranged in small clusters with
occasional binuclear nuclei, some lobular margins contained tumor
cells with obvious heteromorphism, with nuclear hypertrophy, deep
staining, occasional multinucleated and binucleated giant cells.
Cortical destruction and surrounding soft tissue infiltration were
seen. As shown in Fig. 3B,
patient III-8 suffered from huge masses in the bilateral proximal
tibiae and fibula, and the masses from the bilateral proximal
tibiae and fibula were resected and the postoperative biopsy showed
that the tumor tissue was lobulated and composed of chondrocytes
and cartilage matrix. At the 2-year follow-up, no recurrence of
multiple exostoses was observed in either patient with the
significant improvements of quality of life (no pain and improved
activity ability) in these patients according to the follow-ups of
these patients, as shown in Fig.
3C.
Genetic analysis of EXT-2
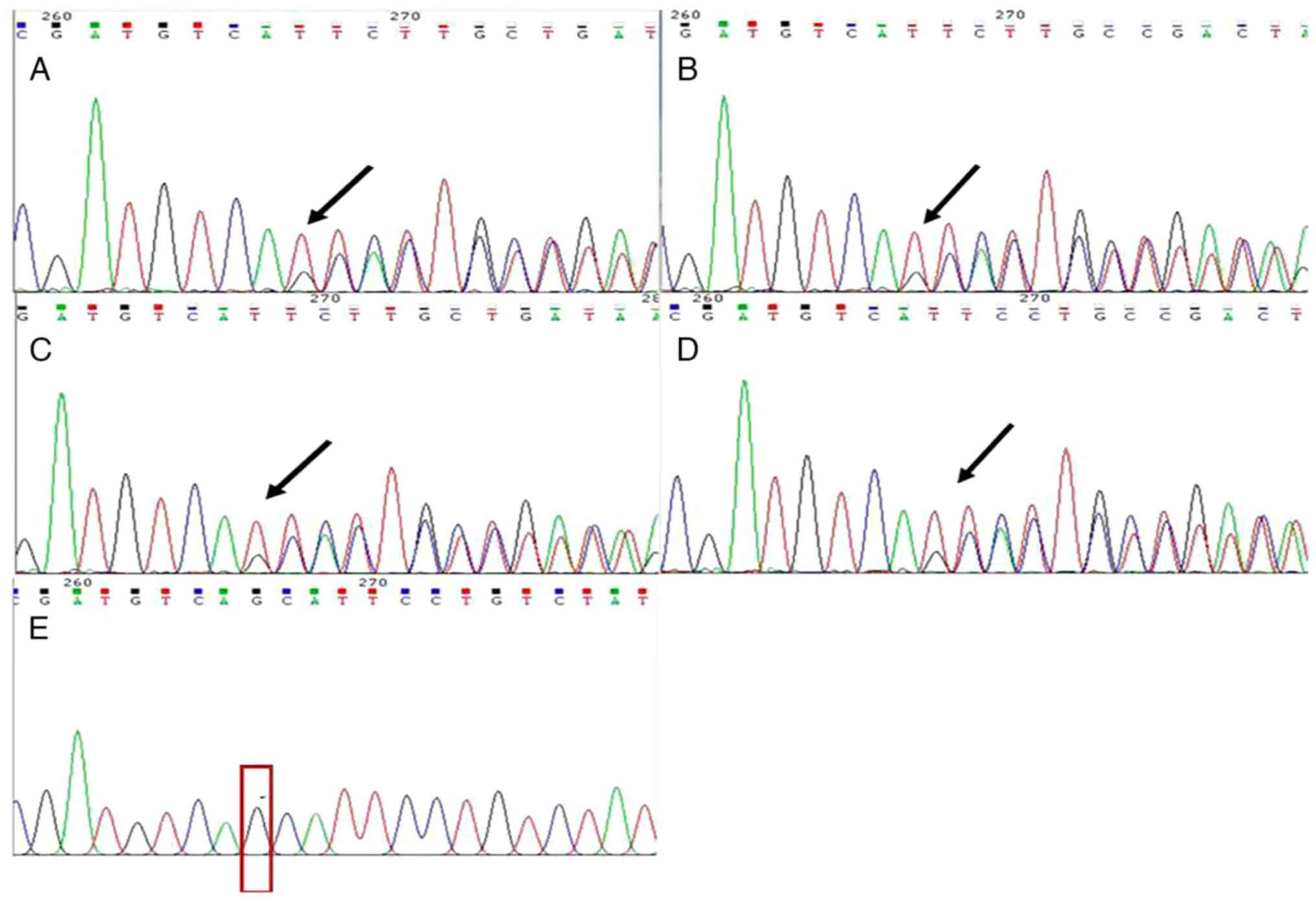
A 3-bp heterozygous deletion (GCA) at the 267–269 of
EXT-2 was found in HME patients (Fig. 4A-D). Beyond nucleotide 267, two
overlapping sequences were observed: ATGTCAGCATTCCT and
ATGTCATTCCT, indicating that the deletion was a heterozygous event;
however, no overlapping sequences was observed in the unaffected
subject (II-8, Fig. 4E). After
genetic analysis of c.824-826delGCA at EXT-2 in all family
members in this pedigree except for III-8, III-9 and II-3 who
received Whole-Genome Sequencing, The results showed that deletion
mutation at c.824-826delGCA in the exon 5 of EXT-2 gene was
detected in four of the affected individuals (II-6, III-7, III-11
and IV-4), whereas none of the unaffected family members examined
carried this mutation (I-2, II-2, II-8, III-1, III-2, III-3, III-4,
III-10, III-13, III-14, IV-1, IV-2 and IV-3). Thus, this finding
was consistent with preliminary results (III-8, III-9 and II-3).
This mutation was found in all patients (II-3, II-6, III-7, III-8,
III-9, III-11 and IV-4) rather than in unaffected family members,
suggesting that the mutation site at c.824-826delGCA in the
EXT-2 gene was the causative mutation for HME in this
family. No other mutations were reported in any of the analyzed
samples.
Discussion
The present study surveyed 20 family members of four
generations in a large pedigree with seven subjects diagnosed with
HME and the relatives without HME used as controls. The study not
only reported clinical characteristics of HME, providing valuable
baseline assessments of the nature and characteristics of HME, but
also found a mutation of EXT-2 gene in a large Chinese
pedigree, extending the known mutational spectrum of EXT-2
gene for improved understanding of the genetic basis of patients
with HME.
Compared with other studies in which unrelated
healthy subjects, matched for geographical ancestry, were included
as controls (21,24), the present study recruited family
members without HME, which decreased the selection bias and the
results were more reliable. Moreover, seven out of the twenty
family members examined were found to have multiple exostoses with
the incidence of 35% in this pedigree. The incidence of HME in
total populations was reported to be 0.4-1 per 50,000 in Western
populations and 40 out of 50,000 in the Chamorro people (1). Guo et al (25) investigated the epidemiology of HME
in mainland China by searching databases. In their study, a total
of 1,051 cases with more detailed documentations were reported in
mainland China since 1990, among which 4% were diagnosed as
sporadic vs. 96% as familial cases and the estimated penetrance was
39%. Whatever the incidence in pedigree or in total populations, it
varies in different studies since these data were taken from
hospital reports and health databases of the affected patients and
families, with a focus on people who reported the symptoms and were
classified as affected. Therefore, studies with large sample size
in multicenter and various ethnicities are necessary.
A total of five out of seven (71.4%) patients with
HME in this pedigree were males, indicating that males might be
more susceptible to HME compared with females, which was consistent
with previous studies (1,21,24,26). Guo et al (25) performed a systematic review by
searching several databases and the prevalence was higher in males
(male:female ratio 637:414; 1.5:1), which might provide more
reliable information for the susceptibility of HME in different
sexes. However, these findings were proved wrong by other authors
(27,28). For example, Wicklund et al
(27) showed that the male to
female ratio was unskewed in nuclear families (probands, affected
sibs, and parents). The present study recommended that males should
be paid more attention by physical and/or radiographic
examinations, especially in HME pedigrees, for early diagnosis and
treatment.
The median age of patients with HME in this pedigree
was 26 years-old, with the oldest age at 50 years-old (II-3) and
youngest age of 10 years-old (IV-4). Xia et al (21) reported a median age of 31, with
the oldest and youngest in their pedigree at 62 and 25 years old,
respectively. In Ruan et al (24), the youngest patient was 6 years
old and the oldest was 75 years old. This difference between the
present study and theirs might be due to the time point of the
survey and the health problems of the subjects. It is hypothesized
that, in general, most patients are diagnosed before the age of 12
and the median age of diagnosis is 3 years old (1,29).
Guo et al (25) also
indicates that most of patients with HME (83%) were diagnosed by
the age of ≤10 years (six cases detected at birth) and 14% of
patients were diagnosed by the age of 10–20 years. The results of
the present study showed that the age of onset varied from 4–9
years-old, which was consistent with the general consensus,
including Guo et al (25).
According to these findings, early diagnosis and early treatment
should be paid attentions in hereditary susceptible subjects.
The present study also reported the height of
patients with HME and the results of adult affected patients
suggested that the height of each adult subject might have a
decreased trend in subsequent generations, consistent with Clement
et al (30) and Ruan et
al (24). Since few studies
have examined the relationship between patients' height throughout
generations, it is hypothesized that the present study might
provide valuable baseline assessments of the nature and
characteristics of HME.
Patients are often diagnosed with HME by a physician
following the appearance of a palpable mass near the joints where
is the lateral side of the most active growth plate of a long bone
(31). Knees, shoulders, ankles
and wrists are the most commonly affected joints (1,25,26). The present study found that
tibiae, femurs, radii and fibulae were the primary locations of
exostoses. These findings suggested that if patients come to
clinics with masses in these locations and with a family history,
HME should be considered. However, ribs are also a common location
of exostoses in this study, which was inconsistent with Guo et
al (25). That study showed
that ribs were less often involved with the incidence of 17% in
studied populations. Selection of patients might be the primary
contributor. In Beltrami et al (31), osteochondromas were often first
discovered on the ribs and the proximal tibia, where they can be
clearly visible and palpable, which might be an another reason. In
addition, other locations including pelvis, vertebrae and
carpal/tarsal bones were reported in previous studies (26,32), whereas these locations were not
observed in the pedigree of the present study. Recently, surgeons
have also reported osteochondroma at rare locations, such as C1 and
C2 (33,34). It has been reported that a patient
with HME is generally burdened with an average of six exostoses
(1), and the results of the
present study were consistent with this finding. Facial bones are
largely unaffected, which may be due to intramembranous
ossification at these locations (35), and no exostoses were found in
these bones in the present study. All these findings indicated that
clinical characteristics of HME often varied among individuals, and
exostoses could occur at several bones and joints, which could be a
great challenge for surgeons.
Although HME may be asymptomatic, a wide spectrum of
severity of clinical manifestations are found in patients with this
disorder (31,36). The present study showed that the
severity of the disease varied in pedigree by using the
classification system of Mordenti et al (23). In the present study, the severity
of clinical presentations in patients with HME was associated with
the generation, as it was found that the patients whose conditions
were more severe were third or fourth generation. The results were
consistent with Shen et al (26), who hypothesized that the patients
whose conditions were more severe were younger than the others.
Therefore, the younger patients with HME in third or fourth
generations should be paid more attention since their clinical
symptoms might be more severe and treatments should be performed
earlier.
Malignant transformation was only found in one case,
the proband (III-9), though it is relatively rare. Beltrami et
al (31) showed that the
places of malignant transformation more frequently involved were
the pelvis, the scapula, the proximal part of the femur and the
humerus. In addition, the authors of that study hypothesized that
malignant transformation in HME is diagnosed at a younger age than
chondrosarcomas are in patients without HME, most often in the
early 20s, which was also verified in the present study in the
24-year-old proband with chondrosarcomas in the scapula. In
addition, no recurrence of chondrosarcomas and exostoses was
observed at the two-years follow-up in the proband and in the
patient III-8, indicating that surgical resection may be a useful
method for exostoses and may greatly affect the quality of life of
patients with HME; this was consistent with Liang et al
(32).
The present study performed Sanger sequencing) on 20
family members in a large HME pedigree to screen for potential gene
mutations and to expand our knowledge of the mutation hotspots of
EXT-2; for example, c.824-826delGCA, in exon 5 of the
EXT-2, according to the findings of preliminary experiments
(data not shown). The present study showed that the c.824-826delGCA
EXT-2 deletion mutation was found in all patients with HME
in this pedigree, but not in the unaffected family members, and was
also the only mutation site in the patients with HME. This mutation
site of EXT-2 was first found and reported by us, indicating
that EXT-2 may serve a key important role in the
pathogenesis of HME, which were consistent with Xia et al
(21) who found a novel
heterozygous frameshift mutation, c.119_120delCT (p.Thr40ArgfsX15),
in exon 2 of the EXT2 gene in HME patients with HME and
hypothesized that EXT-2 mutation might be responsible for
more cases of HME in the Chinese population. In addition, it is
hypothesized (1,2) that mutations of either EXT-1
or EXT-2 will critically disrupt the HS chain elongation
process and subsequently the function of numerous proteins, such as
fibroblast growth factor, vascular endothelial growth factor and
transforming growth factor-β (1,2).
These aberrant proteins include a number of important signaling
receptors and paracrine growth, proliferation and differentiation
factors involved in various normal and pathological processes such
as morphogenesis, angiogenesis, inflammation and tumor metastasis.
Therefore, the findings of the present study may extend the
EXT-2 mutational spectrum in HME pathogenesis. However, how
this novel deletion mutation of EXT-2 influences the
pathogenesis of HME and what signaling pathways requires further
study.
The severity of HME varies, even within the same
family whose affected members all share the same mutation (36). The present study was consistent
with Porter et al (36),
as c.824-826delGCA of EXT-2 gene was observed in all
patients with HME however severe their clinical symptoms. One
previous study (37) reported
that the malignant transformation of preexisting osteochondromas
relied on wild-type cells with functional EXT; that is,
secondary peripheral chondrosarcomas occur through
EXT-independent mechanisms. This may be the reason that the
exostoses in the scapula of the proband (III-9) was a malignant
cartilage-capped tumor, which is a severe complication of HME,
whereas other family members in this pedigree exhibited less severe
symptoms.
Although some new findings have been explored in the
present study, the sample size was relatively small and further
studies at a larger scale are required.
In conclusion, the present study reported clinical
characteristics of patients with HME in a large Chinese pedigree
and identified a novel c.824–826delGCA of EXT-2 gene in this
family. Results from the present study increased our understanding
of the nature of HME, extended the known mutational spectrum of
EXT-2 and suggested further application of mutation
screening in genetic counseling and subsequent prenatal diagnosis
of HME.
Acknowledgements
Not applicable.
Funding
The present study was supported by The National Natural Science
Foundation of China (grant nos. 81572636 and 82102525).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
WW, MY, YS and KC performed the screening and data
collection procedure. DW and JB performed statistical analysis. WW,
MY and CY performed data interpretation and drafted the manuscript.
WW, MY, YS, KC, DW, JB, DW, JG and CY confirm the authenticity of
all the raw data. DH and JG designed the study and supervised the
performance of the study. DH and JG confirm the authenticity of all
the raw data. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Review Board of Shanghai Changhai hospital (approval number
CHEC20180184).
Patient consent for publication
All patients or their parents/guardians in the
present study provided written informed consent for the study and
publication of patient information and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
D'Arienzo A, Andreani L, Sacchetti F,
Colangeli S and Capanna R: Hereditary multiple exostoses: Current
insights. Orthop Res Rev. 11:199–211. 2019.PubMed/NCBI
|
|
2
|
Komura S, Matsumoto K, Hirakawa A and
Akiyama H: Natural history and characteristics of hand exostoses in
multiple hereditary exostoses. J Hand Surg Am. 46:815.e1–815.e12.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
D'Ambrosi R, Caldarini C, Ragone V and
Facchini RM: Effect of multiple hereditary exostoses on sports
activity in children. J Orthop. 15:927–930. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
D'Ambrosi R, Ragone V, Caldarini C, Serra
N, Usuelli FG and Facchini RM: The impact of hereditary multiple
exostoses on quality of life, satisfaction, global health status,
and pain. Arch Orthop Trauma Surg. 137:209–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ryckx A, Somers JF and Allaert L:
Hereditary multiple exostosis. Acta Orthop Belg. 79:597–607.
2013.PubMed/NCBI
|
|
6
|
Hennekam RC: Hereditary multiple
exostoses. J Med Genet. 28:262–266. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Clement ND and Porter DE: Can deformity of
the knee and longitudinal growth of the leg be predicted in
patients with hereditary multiple exostoses? A cross-sectional
study. Knee. 21:299–303. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Noonan KJ, Feinberg JR, Levenda A, Snead J
and Wurtz LD: Natural history of multiple hereditary
osteochondromatosis of the lower extremity and ankle. J Pediatr
Orthop. 22:120–124. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matsumoto Y, Matsumoto K, Harimaya K,
Okada S, Doi T and Iwamoto Y: Scoliosis in patients with multiple
hereditary exostoses. Eur Spine J. 24:1568–1573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Medek K, Zeman J, Honzík T, Hansíková H,
Švecová Š, Beránková K, Kučerová Vidrová V, Kuklík M, Chomiak J and
Tesařová M: Hereditary multiple exostoses: Clinical, molecular and
radiologic survey in 9 families. Prague Med Rep. 118:87–94. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tepelenis K, Papathanakos G, Kitsouli A,
Troupis T, Barbouti A, Vlachos K, Kanavaros P and Kitsoulis P:
Osteochondromas: An updated review of epidemiology, pathogenesis,
clinical presentation, radiological features and treatment options.
In Vivo. 35:681–691. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bukowska-Olech E, Trzebiatowska W, Czech
W, Drzymała O, Frąk P, Klarowski F, Kłusek P, Szwajkowska A and
Jamsheer A: Hereditary multiple exostoses-a review of the molecular
background, diagnostics, and potential therapeutic strategies.
Front Genet. 12:7591292021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sinha S, Mundy C, Bechtold T, Sgariglia F,
Ibrahim MM, Billings PC, Carroll K, Koyama E, Jones KB and Pacifici
M: Unsuspected osteochondroma-like outgrowths in the cranial base
of hereditary multiple exostoses patients and modeling and
treatment with a BMP antagonist in mice. PLoS Genet.
13:e10067422017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Piombo V, Jochmann K, Hoffmann D, Wuelling
M and Vortkamp A: Signaling systems affecting the severity of
multiple osteochondromas. Bone. 111:71–81. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mundy C, Chung J, Koyama E, Bunting S,
Mahimkar R and Pacifici M: Osteochondroma formation is independent
of heparanase expression as revealed in a mouse model of hereditary
multiple exostoses. J Orthop Res. Jan 7–2022.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Inubushi T, Lemire I, Irie F and Yamaguchi
Y: Palovarotene inhibits osteochondroma formation in a mouse model
of multiple hereditary exostoses. J Bone Miner Res. 33:658–666.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Al-Zayed Z, Al-Rijjal RA, Al-Ghofaili L,
BinEssa HA, Pant R, Alrabiah A, Al-Hussainan T, Zou M, Meyer BF and
Shi Y: Mutation spectrum of EXT1 and EXT2 in the Saudi patients
with hereditary multiple exostoses. Orphanet J Rare Dis.
16:1002021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo X, Lin M, Yan W, Chen W and Hong G: A
novel splice mutation induces exon skipping of the EXT1 gene in
patients with hereditary multiple exostoses. Int J Oncol.
54:859–868. 2019.PubMed/NCBI
|
|
19
|
Xian C, Zhu M, Nong T, Li Y, Xie X, Li X,
Li J, Li J, Wu J, Shi W, et al: A novel mutation in ext2 caused
hereditary multiple exostoses through reducing the synthesis of
heparan sulfate. Genet Mol Biol. 44:e202003342021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen Z, Ruan W, Li M, Cao L, Lu J, Zhong F
and Bi Q: A novel nonsense mutation in the EXT2 gene identified in
a family with hereditary multiple osteochondromas. Genet Test Mol
Biomarkers. 24:478–483. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xia P, Xu H, Shi Q and Li D:
Identification of a novel frameshift mutation of the EXT2 gene in a
family with multiple osteochondroma. Oncol Lett. 11:105–110. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ishimaru D, Gotoh M, Takayama S, Kosaki R,
Matsumoto Y, Narimatsu H, Sato T, Kimata K, Akiyama H, Shimizu K
and Matsumoto K: Large-scale mutational analysis in the EXT1 and
EXT2 genes for Japanese patients with multiple osteochondromas. BMC
Genet. 17:522016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mordenti M, Ferrari E, Pedrini E, Fabbri
N, Campanacci L, Muselli M and Sangiorgi L: Validation of a new
multiple osteochondromas classification through switching neural
networks. Am J Med Genet A. 161A:556–560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ruan W, Cao L, Chen Z, Kong M and Bi Q:
Novel exostosin-2 mutation identified in a Chinese family with
hereditary multiple osteochondroma. Oncol Lett. 15:4383–4389.
2018.PubMed/NCBI
|
|
25
|
Guo XL, Deng Y and Liu HG: Clinical
characteristics of hereditary multiple exostoses: A retrospective
study of mainland chinese cases in recent 23 years. J Huazhong Univ
Sci Technolog Med Sci. 34:42–50. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shen Y, Zhang L, Chen B, Dong L, Wang Y
and Wang S: Novel deletion and 2397 G>T mutations of the EXT1
gene identified in two Chinese pedigrees with hereditary multiple
exostoses using exon sequencing. Transl Pediatr. 9:619–628. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wicklund CL, Pauli RM, Johnston D and
Hecht JT: Natural history study of hereditary multiple exostoses.
Am J Med Genet. 55:43–46. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kumar A, Jain VK, Bharadwaj M and Arya RK:
Ollier disease: Pathogenesis, diagnosis, and management.
Orthopedics. 38:e497–e506. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schmale GA, Conrad EU III and Raskind WH:
The natural history of hereditary multiple exostoses. J Bone Joint
Surg Am. 76:986–992. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Clement ND, Duckworth AD, Baker AD and
Porter DE: Skeletal growth patterns in hereditary multiple
exostoses: A natural history. J Pediatr Orthop B. 21:150–154. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Beltrami G, Ristori G, Scoccianti G,
Tamburini A and Capanna R: Hereditary multiple exostoses: A review
of clinical appearance and metabolic pattern. Clin Cases Miner Bone
Metab. 13:110–118. 2016.PubMed/NCBI
|
|
32
|
Liang C, Wang YJ, Wei YX, Dong Y and Zhang
ZC: Identification of novel EXT mutations in patients with
hereditary multiple exostoses using whole-exome sequencing. Orthop
Surg. 12:990–996. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tbini M, Lahiani R, Kharrat O, Riahi I and
Bensalah M: Anterior C1 osteochondroma. Joint Bone Spine.
89:1053052021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jemel N, Gader G, Bedioui A, Zammel I and
Badri M: C1 C2 spinal cord compression in hereditary multiple
exostoses: Case report and review of the literature. Int J Surg
Case Rep. 89:1065762021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stieber JR and Dormans JP: Manifestations
of hereditary multiple exostoses. J Am Acad Orthop Surg.
13:110–120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Porter DE, Lonie L, Fraser M, Dobson-Stone
C, Porter JR, Monaco AP and Simpson AH: Severity of disease and
risk of malignant change in hereditary multiple exostoses. A
genotype-phenotype study. J Bone Joint Surg Br. 86:1041–1046. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
de Andrea CE, Reijnders CM, Kroon HM, de
Jong D, Hogendoorn PC, Szuhai K and Bovée JV: Secondary peripheral
chondrosarcoma evolving from osteochondroma as a result of
outgrowth of cells with functional EXT. Oncogene. 31:1095–1104.
2012. View Article : Google Scholar : PubMed/NCBI
|