Introduction
Bronchial asthma (asthma) is the most common chronic
respiratory disease in children. There are ~300 million individuals
with asthma worldwide and the number of patients is increasing year
by year (1). In China ~30 million
individuals suffer from asthma and the prevalence rate is 1–4%
(2). Asthma is a chronic
inflammatory disease characterized by airway hyperresponsiveness,
inflammatory cell infiltration, excessive mucus production and
airway remodeling and it can be induced by respiratory tract
infection, allergen exposure, strenuous exercise, climate change
and other factors (3,4). Although the specific pathogenesis of
asthma remains to be elucidated, airway inflammation is recognized
as the most fundamental pathological change in its pathogenesis.
Therefore, the inhibition of inflammation is an important strategy
for asthma therapy. Glucocorticoids, among the most effective drugs
for controlling airway inflammation, are widely used to treat
asthma (3,4). However, long-term use of
glucocorticoid treatment can cause a number of side effects, such
as increased blood pressure, hyperglycemia, edema, osteoporosis,
ulcer formation and weight gain (5). Therefore, it is essential to study the
molecular mechanism of airway inflammation in asthma.
Airway epithelial cells are mainly composed of
secretory and ciliated cells and they serve a critical role in the
body's resistance to pathogenic microorganisms and the removal of
foreign substances, such as dust particles (6,7). In
asthmatic patients, the presence of airway inflammation leads to
airway epithelial damage, including airway epithelial apoptosis,
shedding and airway structure remodeling and these changes can
weaken or cause the loss of the original function of the airway
epithelium (8,9). Airway epithelium damage leads to the
weakening of its physical barrier function, thereby increasing the
risk of infection, forming a vicious circle that feeds on itself
(8,9). Studying the apoptosis and inflammatory
mechanisms of airway epithelial cells is therefore crucial for the
treatment of asthma.
Sirtuins are a protein family that regulate cell
health and serve a key role in regulating cell homeostasis
(10). They are considered a new
target for the treatment of bronchial inflammation (11). Sirtuin (SIRT)3 is a member of the
sirtuin family that promotes longevity in a number of organisms,
regulates inflammation through the modulation of mitochondrial
function (12) and improves the
antioxidant defense mechanisms (13,14).
Although no studies of SIRT3 in asthma are reported, the sirtuin
protein family serves an essential role in chronic obstructive
pulmonary disease (15); SIRT3 can
not only resist lung injury by resisting inflammation (16) and oxidation (17) but also protects bronchial epithelial
cells by inhibiting oxidative stress (18) and autophagy (19). In addition, SIRT1, an upstream
molecule that regulates the expression of SIRT3, is related to the
severity of asthma (20,21). A study by Colley et al
(22) also found that defective
SIRT1 increases IL-4 expression through acetylation of GATA-3 in
patients with severe asthma. Combined with the evidence by Liu
et al (12) that SIRT1
suppresses inflammation by promoting SIRT3 expression, it was
hypothesized that SIRT3 also served an essential role in
asthma.
SIRT3 is also an apoptosis-related protein (23) and aberrant apoptosis of airway
epithelial cells is also a contributing factor in development of
asthma (24,25). Previous studies have found that
downregulation of SIRT3 promotes the apoptosis of alveolar
epithelial cells (26) and EC9706
cells (27). Studies have also
shown that SIRT3 protects tumor cells against apoptosis under
unfavorable environments, such as hypoxia (28), oxidized low-density lipoprotein
(29) and high glucose levels
(30). However, the expression of
SIRT3 and its effect on apoptosis and bronchial inflammation in
asthma remains to be elucidated. The present study attempted to
investigate the role of SIRT3 in epithelial cells apoptosis induced
by oxidative stress (H2O2) and the effect of
SIRT3 overexpression on inflammation and airway epithelial
structural integrity in asthmatic mice.
Materials and methods
Animals and experimental asthma
model
The animal experiment was performed according to the
protocols approved by the Beijing Luhe Hospital, Capital Medical
University (approval no. 2021-LHKY-055-02). A total of 28 female
C57BL/6 mice (age, 5–6 weeks; weight, 18–22 g) were used in the
present study. Mice were purchased from Shanghai Lingchang
Biological Technology Co., Ltd. and were kept at ~20-26°C, 40%
relative humidity with 12 h light/dark cycles. After being housed
under conventional conditions for one week prior to any
experiments, 21 mice were used in the establishment of the asthma
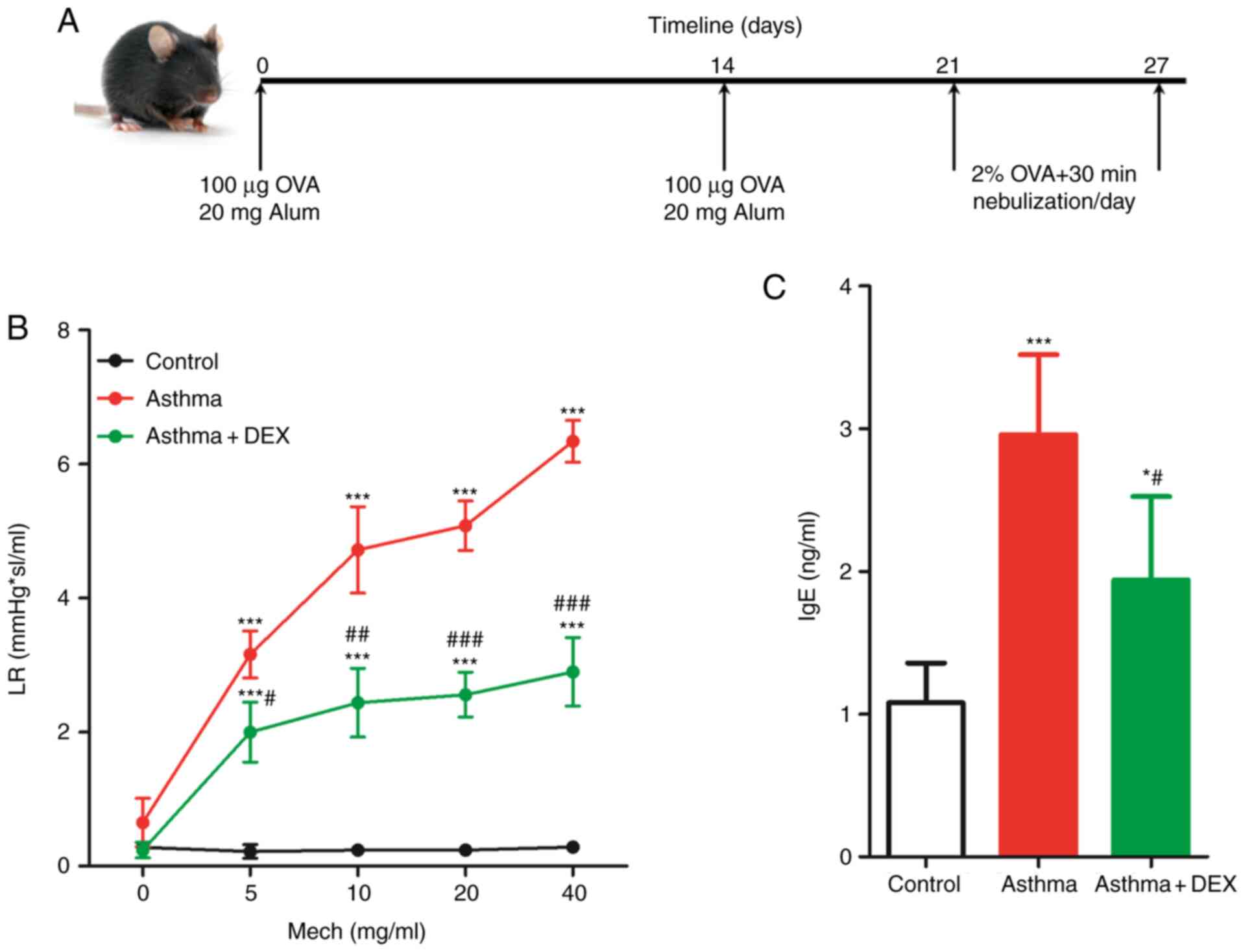
model, as illustrated in Fig. 1A
(31). In brief, 200 µl of
sensitizing fluid [containing 100 µg of ovalbumin (OVA) and 20 mg
of Alum] was administered by intraperitoneal (i.p.) injection on
day 0 and 14. From day 21, mice were given 40 ml of 2% OVA atomized
solution through spray inhalation for 30 min/day for seven days.
Control mice were given the same amount of saline solution at the
same time. All mice were randomly distributed into 4 groups (7
mice/group): i) Control group; healthy mice; ii) Asthma group;
experimental asthma mice model without any treatment; iii) Asthma +
DEX group: experimental asthma mice model treated with 1 mg/Kg
dexamethasone (DEX; cat. no. D4902; MilliporeSigma), i.p., for 7
days during the last week of OVA inhalation (32) and iv) Asthma + Ad-m-SIRT3 group;
experimental asthma mice model infected with mice SIRT3 adenovirus
(cat. no. AAV-272009; Vector BioLabs).
Lung resistance (LR) assessment and
BALF collection
After 48 h from the last atomization with OVA, an
AniRes2005 pulmonary function meter (Beijing Beilanbo Technology
Co., Ltd.) was used to measure the lung resistance of mice to
evaluate the experimental asthma model: Briefly, following
intratracheal intubation, normal saline nebulization was used to
determine basic lung resistance. After 2 min of rest, acetylcholine
solutions of 50, 20, 10 and 5 mg/ml were nebulized for 2 min and
lung resistance was subsequently determined. After assessing the
lung function, the mice were euthanized by exposure to
CO2 (20–30% of chamber volume per min) after being
anesthetized with sodium pentobarbital (50 mg/kg i.p.) and the
trachea was exposed. A 5 ml sterile syringe was then used to inject
2 ml of sterile PBS into the trachea and then the PBS solution was
recovered. This process was repeated twice. The resultant collected
cells in the PBS were bronchoalveolar lavage fluid (BALF). For cell
count, cells were centrifuged at 500 × g for 5 min at room
temperature for cell collection, resuspended in PBS and the cell
suspension was smeared on a glass slide. Wright's Giemsa staining
solution (cat. no. G3990; Beijing Solarbio Science & Technology
Co., Ltd.) was used for cell staining for 1 min at room temperature
and the cells were counted under an optical light microscope.
Infection of adenovirus in mice and
16HBE cells
After being anesthetized with sodium pentobarbital
(50 mg/kg i.p.), 4×105 PFU/40 µl mice SIRT3 adenovirus
(cat. no. AAV-272009, Vector Biolabs) was administered by nasal
spray inhalation and the mice in the control group were infected
with equivalent empty adenovirus, as previously described (33,34).
After being infected with the adenovirus for 48 h, the mice were
sacrificed to collect the bronchial tissues.
For adenovirus infection of 16HBE cells to knockdown
or overexpress SIRT3, 1.0×106 16HBE cells were first
seeded in a 6-well cell culture plate and cultured for 12 h at
37°C. To infect 16HBE cells 1×1010 PFU/100 µl SIRT3-AAV
(cat. no. AAV-223017; Vector Biolabs) or SIRT3-short hairpin
(sh)RNA adenovirus (cat. no. shAAV-272009; Vector Biolabs) were
used to infect 16HBE cells and 16HBE cells were also infected with
equivalent empty adenovirus [NC-shRNA (non-targeting sequence)
(cat. no. shAAV-272009; Vector Biolabs) or NC-AAV (cat. no.
shAAV-223017; Vector Biolabs)] as a negative control. After being
infected with the adenovirus for 48 h at 37°C, the cells were
collected for analysis.
Reverse transcription-quantitative
(RT-q) PCR
An RNA extraction kit (cat. no. RC101-01, Vazyme
Biotech Co., Ltd.) was used to extract the total RNA from cells
(2×106) and tissues according to the manufacturer's
protocol. Total RNA was reverse transcribed into cDNA using the
PrimeScript RT reagent (cat. no. RR047A; Takara Bio, Inc.)
according to the manufacturer's protocol. Next, 20 µl of RT-qPCR
system was prepared, as described in the qPCR master mix kit
instructions (cat. no. A6001; Promega Corporation) and the
following thermocycling conditions were used: 95°C for 2 min; and
40 cycles at 95°C for 5 sec and 65°C for 15 sec. The relative
expression of the genes was calculated using the 2−DDCq
method (35). β-actin was used as a
loading control. The primers used for qPCR analysis were: SIRT3-F:
5′-GGCTCTATACACAGAACATCGAC-3′, SIRT3-R:
5′-TAGCTGTTACAAAGGTCCCGT-3′; TNF-α-F: 5′-GACGTGGAACTGGCAGAAGAG-3′,
TNF-α-R: 5′-TTGGTGGTTTGTGAGTGTGAG-3′; IL-4-F:
5′-ATCATCGGCATTTTGAACGAGG-3′, IL-4-R: 5′-TGCAGCTCCATGAGAACACTA-3′;
IL-5-F: 5′-TCAGGGGCTAGACATACTGAAG-3′, IL-5-R:
5′-CCAAGGAACTCTTGCAGGTAAT-3′; IL-13-F: 5′-CAGCCTCCCCGATACCAAAAT-3′,
IL-13-R: 5′-GCGAAACAGTTGCTTTGTGTAG-3′; β-actin-F:
5′-AGGGAAATCGTGCGTGACAT-3′, β-actin-F: 5′-GCCTCAGGAGTTTTGTCACCT-3′.
These experiments were repeated three times.
Western blot analysis
Total protein was extracted from tissues using a
Tissue Protein Extraction Reagent (cat. no. 78510; Thermo Fisher
Scientific, Inc.) and a BCA kit (cat. no. 23227; Thermo Fisher
Scientific, Inc.) was used to determine the protein concentration.
Then, 50 µg total protein was analyzed using a 10% SDS-PAGE. After
transferring to PVDF membranes (cat. no. LC2002; Thermo Fisher
Scientific, Inc.) blocking was performed using 5% skimmed milk
powder for 1 h at room temperature. The membranes were subsequently
probed with primary antibodies against SIRT3 (1:1,000; cat. no.
2627; Cell Signaling Technology, Inc.), Bax (1:2,000; cat. no.
ab32503; Abcam), Bcl2 (1:2,000; cat. no. ab196495; Abcam) and
β-actin (1:4,000; cat. no. AF5001; Beyotime Institute of
Biotechnology) at 4°C overnight. Subsequently, PVDF membranes were
probed with goat anti-rabbit IgG heavy chain & light chain
(H&L) HRP (1:2,000; cat. no. ab6721; Abcam) or goat anti-mouse
IgG H&L HRP (1:2,000; cat. no. ab6789; Abcam) secondary
antibodies for 1 h at room temperature. The proteins were
visualized with an ECL solution (cat. no. WBKLS0100; Beijing
Xinjingke Biotechnologies Co., Ltd.), followed by densitometry
analysis using ImageJ v3.0 (National Institutes of Health). β-actin
was used as control.
Immunohistochemistry staining of
SIRT3
The expression of SIRT3 was detected in bronchial
tissues using immunohistochemistry staining. Briefly, the bronchial
tissues were harvested and fixed using 4% paraformaldehyde (cat.
no. P1110; Beijing Solarbio Science & Technology Co., Ltd.) at
4°C overnight. Then histological paraffin sections (5 µm) were made
after being embedded in paraffin and procedurally dehydrated
(tissues were placed in 30, 50, 70, 80, 95 and 100% ethanol for 1 h
at room temperature). There were seven mice in each group and ≥10
histological sections were prepared for each animal. The bronchial
sections were incubated overnight at 4°C with the anti-SIRT3
antibody (1:500; cat. no. 2627; Cell Signaling Technology, Inc.).
After a mild wash with PBS buffer, the bronchial sections were
incubated with HRP-conjugated goat anti-rabbit antibody (1:2,000;
cat. no. ab6721; Abcam) for 1 h at room temperature. The peroxidase
activity was assessed via a 2 min reaction at room temperature with
diaminobenzidine (DAB). The bronchial tissue sections were further
counterstained with hematoxylin for 2 min at room temperature and
images captured under a bright-field light microscope (Olympus
BX51; Olympus Corporation).
TUNEL staining
To determine the apoptosis in the bronchial tissues,
TUNEL staining was used as described previously (36). In brief, the mice bronchial tissue
sections were fixed with 4% paraformaldehyde for 24 h at 4°C and
were embedded in paraffin. Subsequently the samples were
procedurally dehydrated (tissues were placed in 30, 50, 70, 80, 95
and 100% ethanol for 1 h at room temperature) and sectioned at 5
µm. After dewaxing and hydrating, tissue sections were incubated
with proteinase K to permeate the cells for 30 min at 37°C. Next,
the sections were incubated with the TUNEL reaction solution for 1
h at 37°C and then color development was achieved with DAB solution
for 30 min at room temperature. The TUNEL Cell Apoptosis Detection
kit (cat. no. TA201-02; TransGen Biotech Co., Ltd.) was used.
Caspase-3 activity measurement
Apoptosis was quantified by the caspase-3 activity
measurement using the Ac-DEVD-7-Amino-4-trifluoromethylcoumarin
(AFC) caspase-3 Fluorogenic Substrate (cat. no. 556574; BD
Pharmingen; BD Biosciences) according to the manufacturer's
instructions. Briefly, bronchial tissues from the control or asthma
mice were lysed on ice. After centrifugation (12,000 × g for 10 min
at 4°C), the supernatants were collected and the protein
concentration was quantified using the BCA method. A total of 50 µg
of proteins was pipetted and mixed with the assay buffer
supplemented with 10 mM dithiothreitol (DTT). The fluorescence
emission of the AFC (400 nm) was measured via the Spectra Max-Plus
Microplate Spectrophotometer (Molecular Devices, LLC). The
caspase-3 activity was expressed as nmol AFC/h/mg protein.
Detection of cytokines and serum
immunoglobulin (Ig)E
The concentration of TNF-α, IL-4, IL-5 and IL-13 in
bronchoalveolar lavage fluid (BALF) and serum IgE from mice were
evaluated by ELISA kits as described in the manufacturer's
instructions. The ELISA kits were: TNFα (cat. no. ab236712; Abcam),
IL-4 (cat. no. ab100710; Abcam), IL-5 (cat. no. ab204523; Abcam),
IL-13 (cat. no. ab100700; Abcam) and mice IgE (cat. no. ab157718;
Abcam).
Detection of reactive oxygen species
(ROS), glutathione (GSH), superoxide dismutase (SOD), glutathione
peroxidase (Gpx) and malondialdehyde (MDA)
Homogenates were prepared with 50 mg of fresh
bronchial tissue and the supernatant collected following bronchial
tissue homogenate centrifugation (12,000 × g, 4°C, 10 min). Then, a
ROS Detection kit (cat. no. S0058; Beyotime Institute of
Biotechnology) was used to quantify the ROS levels. A GSH detection
kit (cat. no. S0058; Beyotime Institute of Biotechnology) was used
to detect the GSH levels. In addition, a SOD assay kit (cat. no.
S0101S; Beyotime Institute of Biotechnology) was used to detect the
SOD levels and an MDA assay kit (cat. no. S0101S; Beyotime
Institute of Biotechnology) was used to detect the MDA levels.
Finally, a Gpx assay kit (cat. no. S0058; Beyotime Institute of
Biotechnology) was used to detect the levels of Gpx. All these
assays were performed as according to the manufacturer's
protocols.
Cell treatment and apoptosis
assay
16HBE cells (1.5×106) were seeded in the
6-well cell culture plate and cultured for 12 h. After stimulating
them with 0, 100, 200, or 400 µmol/l H2O2 for
12 h, the cells were collected and washed twice with cold-PBS
buffer, then an Annexin V-PE/7-AAD Apoptosis Detection kit (cat.
no. A213-01; Vazyme) was used to evaluate the apoptosis by flow
cytometry (CytoFLEX S; Beckman Coulter, Inc.). In brief, cells were
stained for 20 min at 4°C in the dark. Data were analyzed using
FlowJo software (version 10.7.1; BD Biosciences) and the apoptotic
rate was calculated as the percentage of early + late apoptotic
cells.
Statistical analysis
Data were analyzed using SPSS v20.0 (IBM Corp.) and
shown as mean ± SD. Student's t-test was used to compare the
difference between two groups and one-way ANOVA followed by the
Tukey's post hoc test was used to compare the difference between
multiple groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
SIRT3 expression decreases in the
bronchial tissues of the asthmatic mice
First, an asthma model was established through OVA
sensitization induction, as represented in Fig. 1A. Symptoms such as sneezing,
irritability, shortness of breath and scratching of the ears were
observed in the asthma model mice after aerosolization. At the same
time, DEX treatment could relieve the above symptoms, which
indicated that the performance of the C57BL/6 as an asthma mice
model after the challenge was consistent with the acute attack of
asthma. Airway hyperresponsiveness (AHR) is a critical indicator of
asthma and LR was used, which can reflect AHR to a certain extent,
to represent AHR (37). As shown in
Fig. 1B, acetylcholine increased
mice LR in a dose-dependent manner and DEX reduced it. Similarly,
the total content of IgE in the asthmatic mice serum was
significantly higher when compared with the normal mice and DEX
also significantly reduced it (Fig.
1C). Therefore, these results indicated that a mice asthma
model had been successfully established.
Sirtuins, a group of class III deacetylases, have
gained considerable attention for their positive effects on
age-related diseases, such as cancer, cardiovascular disease,
neurodegenerative diseases, osteoporosis and chronic obstructive
pulmonary disease (15). SIRT3
serves an essential role in the sirtuin protein family and is also
involved in mediating inflammation (14,38).
As a disease with chronic airway inflammation as the primary
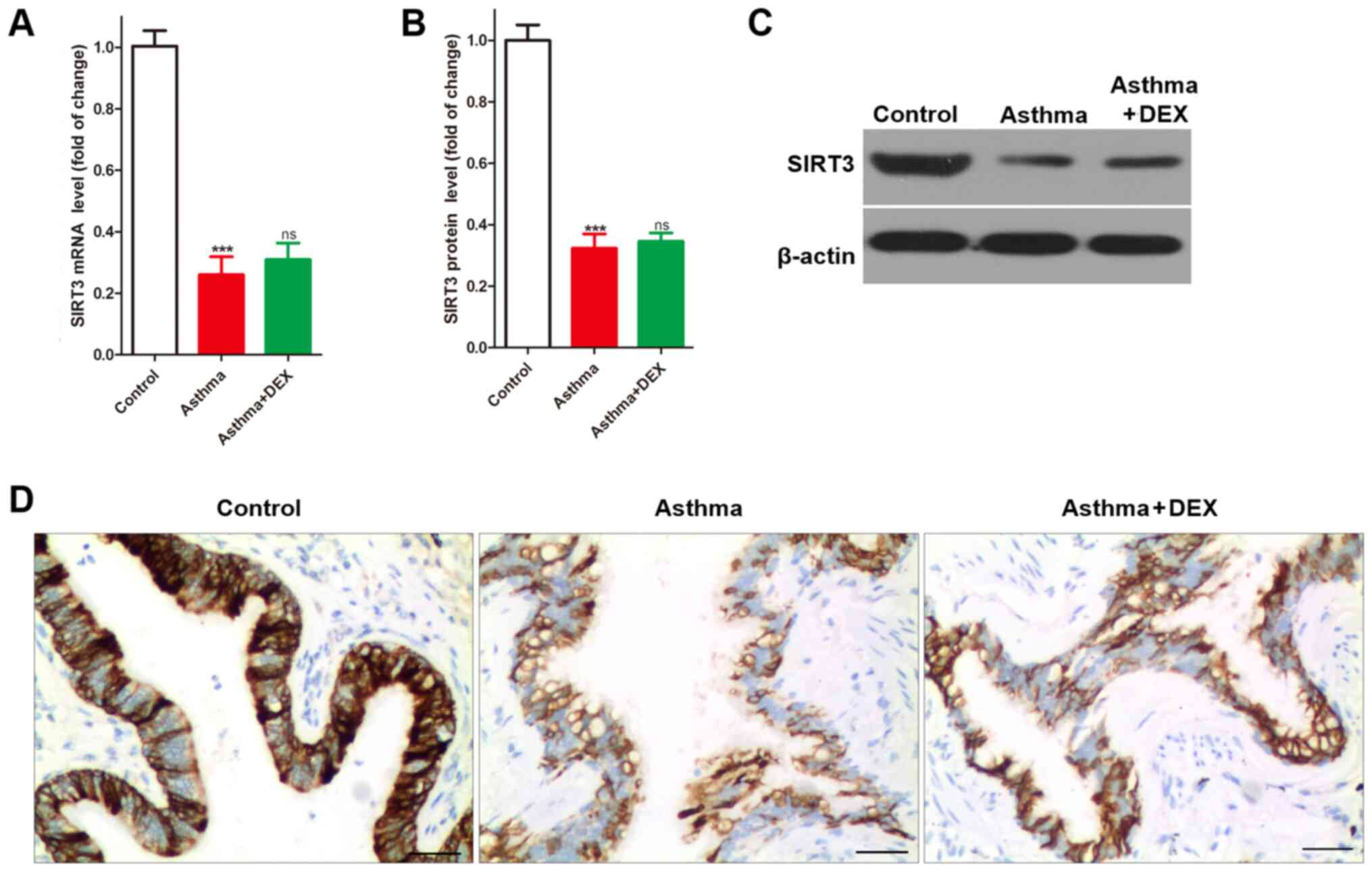
clinical symptom, the present study found that the expression of
SIRT3 mRNA was significantly lower in the bronchial tissues of
asthmatic mice compared with the control (Fig. 2A). Similarly, the results of SIRT3
protein detection using western blotting (Fig. 2B and C) and immunochemistry
(Fig. 2D) indicated that SIRT3
expression was significantly lower in bronchial tissues of
asthmatic mice compared with the control. However, it was also
found that DEX treatment could not change the decreased SIRT3
expression in the bronchial tissues of asthmatic mice (Fig. 2). Taken together, SIRT3 expression
decreased in the bronchial tissues of asthmatic mice.
Upregulation of SIRT3 reduces the
bronchial epithelial cell apoptosis in the asthmatic mice
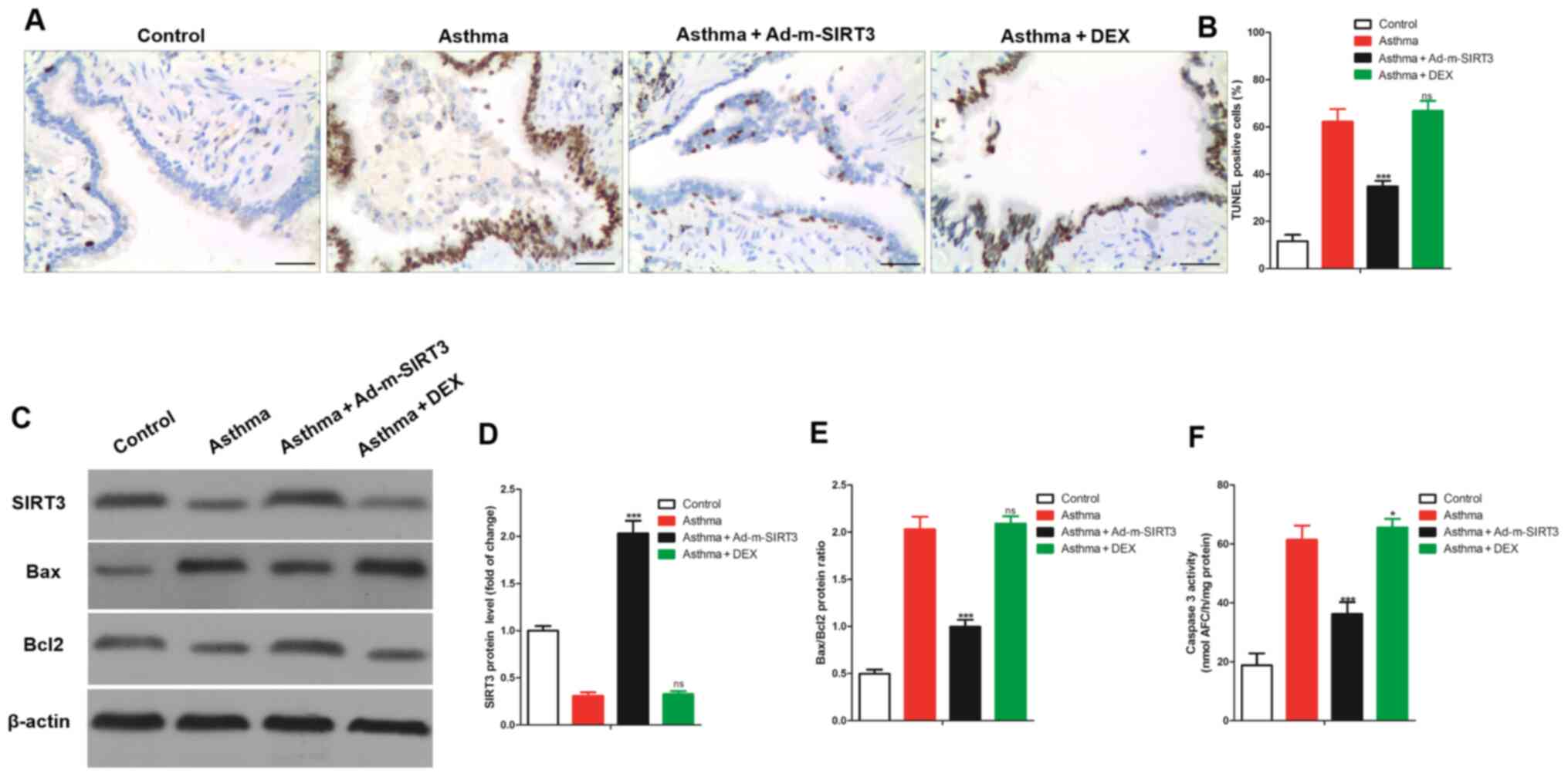
As a mitochondrial protein, SIRT3-mediated mitophagy
protects tumor cells against apoptosis under hypoxia (28) and the loss of SIRT3 promotes the
apoptosis of EC9706 cells (27).
This indicates that the loss of SIRT3 promotes apoptosis. It was
hypothesized that the loss of SIRT3 was also involved in regulating
the apoptosis of bronchial epithelial cells in asthmatic mice. To
test this hypothesis, bronchial tissues were collected from the
mice and apoptosis detected using TUNEL staining. It was found that
the apoptotic (TUNEL positive) cells in bronchial tissue of
asthmatic mice were significantly higher compared with the control
mice (Fig. 3A and B). The results
of the western blotting (Fig. 3C)
demonstrated that Bax (a pro-apoptotic protein) protein expression
levels in the asthma group were markedly higher compared with the
control group, whereas Bcl2 (an anti-apoptotic protein) protein
expression levels in the asthma group were markedly lower compared
with the control group. SIRT3 protein expression levels were also
markedly increased in the asthma group compared with the control
group (Fig. 3D). The ratio of
Bax/Bcl2 was significantly increased (Fig. 3E) in the bronchial tissues of
asthmatic mice. Simultaneously, the caspase 3 activity in the
bronchial tissues of asthmatic mice was significantly higher than
in normal mice (Fig. 3F). To study
the effect of SIRT3 expression on the apoptosis of bronchial
epithelial cells in asthmatic mice, the expression of SIRT3 was
upregulated using adenovirus overexpressing SIRT3 (Ad-m-SIRT3). It
was found that Ad-m-SIRT3 successfully increased the expression of
SIRT3 protein in bronchial tissues of asthmatic mice. Notably,
overexpression of SIRT3 significantly reduced the increased TUNEL
positive cells in bronchia from asthmatic mice and significantly
reduced the increased Bax/Bcl2 ratio and caspase 3 activity in the
bronchia from asthmatic mice (Fig.
3). In conclusion, upregulation of SIRT3 reduced bronchial
epithelial cell apoptosis in asthmatic mice.
Inflammation in BALF of the asthmatic
mice is suppressed by SIRT3 overexpression
SIRT3 is an essential molecule in inflammation
regulation and was previously found to attenuate palmitate-induced
ROS production and inflammation in proximal tubular cells (14). In the present study, the number of
immune cells (macrophages, eosinophils, lymphocytes and
neutrophils) in BALF from asthmatic mice increased significantly
(Fig. 4A). The cytokines expression
in BALF, such as TNF-α, IL-4, IL-5 and IL-13, was analyzed and it
was found that the mRNAs of all these cytokines were also increased
in BALF from asthmatic mice (Fig.
4B). Similarly, the ELISA results revealed that the content of
TNF-α, IL-4, IL-5 and IL-13 in BALF from asthmatic mice were
significantly higher compared with the BALF from normal mice
(Fig. 4C). To study the effect of
SIRT3 expression on BALA inflammation in asthmatic mice, the
expression of SIRT3 was upregulated using adenovirus overexpressing
SIRT3 (Ad-m-SIRT3). The results showed that overexpression of SIRT3
not only significantly reduced the increased number of immune cells
(macrophages, eosinophils, lymphocytes and neutrophils) in BALF
from asthmatic mice but also reduced the increased expression and
content of cytokines from cells in BALF of asthmatic mice (Fig. 4). Together, these results suggested
that the overexpression of SIRT3 reduced airway inflammation in
asthmatic mice, which has the same effect as the specific drug DEX,
which was used as a positive control, for the treatment of allergic
rhinitis.
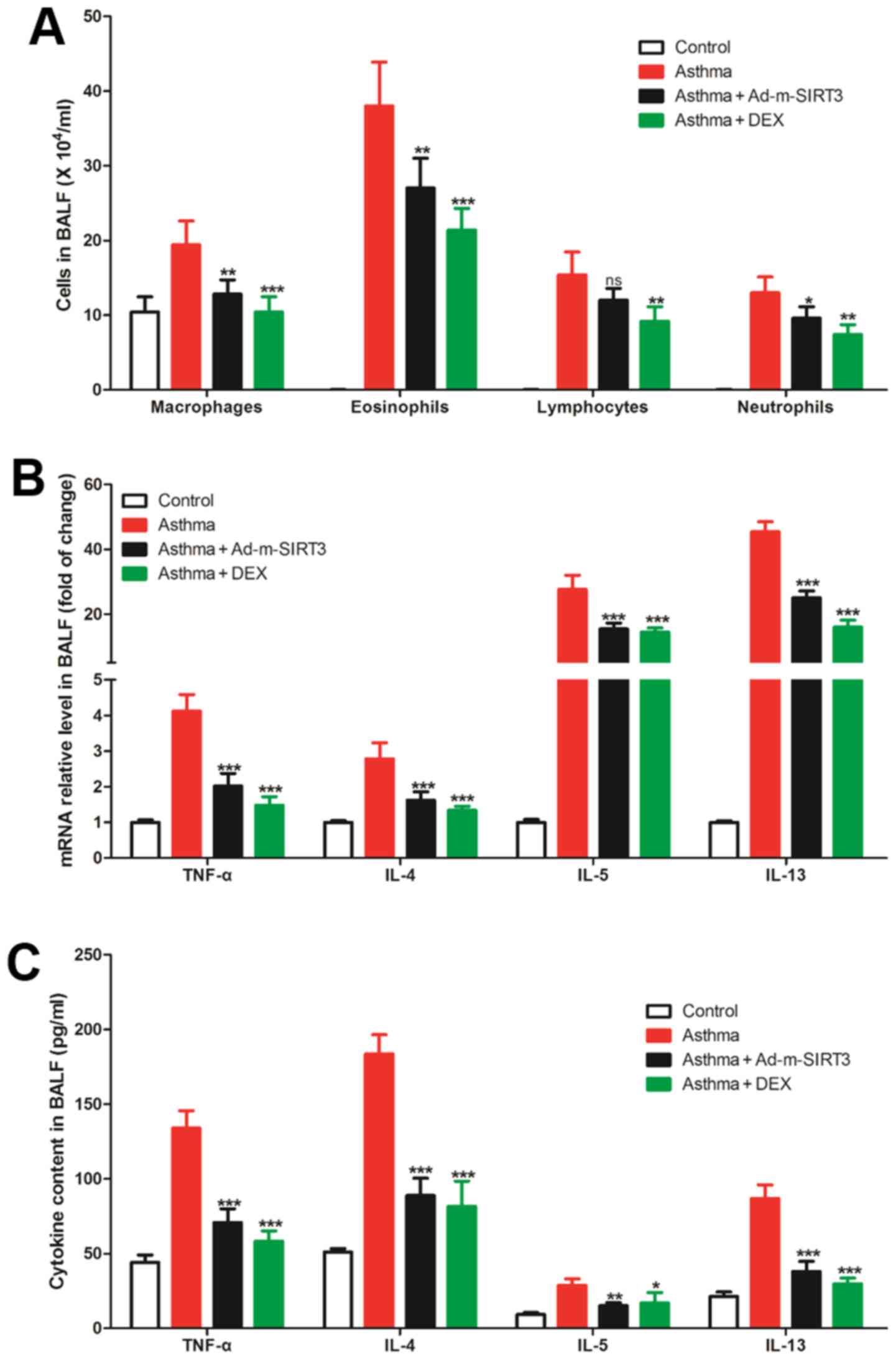 | Figure 4.Upregulation of SIRT3 reduced
increased inflammation of BALF in asthmatic mice. (A) The number of
cells in BALF as determined by Wright's Giemsa staining. (B) The
expression of cytokines (TNF-α, IL-4, IL-5 and IL-13) mRNA in cells
of BALF determined by reverse transcription-quantitative PCR. (C)
The secretion levels of cytokines (TNF-α, IL-4, IL-5 and IL-13) in
BALF determined with ELISA assay. DEX treatment was used as the
positive control. There were seven mice in each group, the data
were shown as mean ± SD and the P-value was calculated by
calculated by post-hoc comparisons. ns, P>0.05, *P>0.05,
**P<0.01 and ***P<0.001 vs. Asthma group. SIRT3, sirtuin 3;
BALF, bronchoalveolar lavage fluid; DEX, dexamethasone. |
Upregulation of SIRT3 reduces the
bronchial oxidative stress in the asthmatic mice
SIRT3 protects cells and inhibits inflammation by
preventing oxidative stress in a number of diseases, such as
diabetes (39), oral cancer
(40) and myocardial
ischemia-reperfusion (41).
Oxidative stress serves an important role in the establishment of
the asthma model by OVA sensitization induction (42) and oxidative stress can cause
inflammation and apoptosis (43,44).
Therefore, it was hypothesized that SIRT3 was involved in
regulating airway inflammation and bronchial epithelial cell
apoptosis by the modulation of bronchial oxidative stress in
asthmatic mice. To test this hypothesis, the level of ROS was
detected in the bronchial tissue by a ROS probe and it was found
that the increased levels of ROS in the bronchial tissues of
asthmatic mice were reduced by overexpression of SIRT3 (Fig. 5A). ELISA was used to observe the
change in the endogenous antioxidants, GSH, SOD and Gpx, which are
the antioxidants that help to reduce the content of ROS (45). The results of ELISA showed that the
levels of GSH, SOD and Gpx in the bronchial tissues of asthmatic
mice were all significantly lower compared with the control mice,
while the decreased levels of GSH (Fig.
5B), SOD (Fig. 5C) and Gpx
(Fig. 5D) in the bronchial tissues
of asthmatic mice were reduced by overexpression of SIRT3. The
levels of MDA, an indicator of the end products of the cellular
membrane under oxidative injury, were detected and it was found
that MDA levels increased in the bronchial tissues of asthmatic
mice and overexpression of SIRT3 was able to reverse this (Fig. 5E). Therefore, these data suggested
that the upregulation of SIRT3 reduced bronchial oxidative stress
in the asthmatic mice.
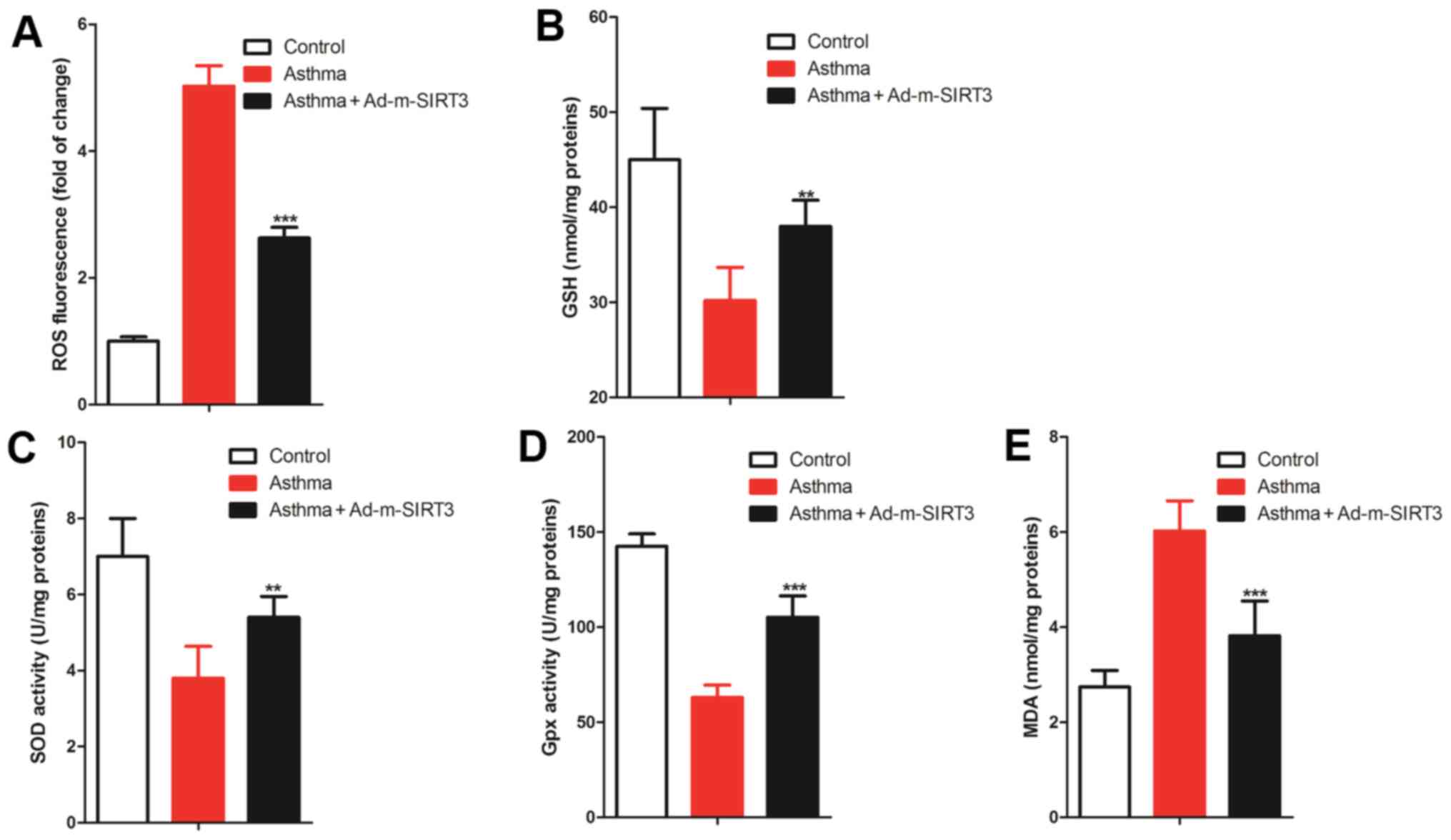 | Figure 5.SIRT3 regulated redox balance in
bronchial tissues of asthmatic mice. (A) ROS probe was used to
observe the change in ROS in bronchial and the relative
fluorescence intensity was used to quantify ROS levels. (B-D) ELISA
was used to observe the change in cellular antioxidants [(B) GSH,
(C) SOD and (D) Gpx]. (E) MDA, the end product of the cellular
membrane under oxidative injury, was detected by ELISA. There were
seven mice in each group, the data were shown as mean ± SD and the
P-value was calculated by Student's t-test. **P<0.01 and
***P<0.001 vs. Asthma group. SIRT3, sirtuin 3; ROS, reactive
oxygen species; GSH, glutathione; SOD, superoxide dismutase; Gpx,
glutathione peroxidase; MDA, malondialdehyde. |
SIRT3 regulated
H2O2-induced apoptosis and inflammation in
16HBE cells
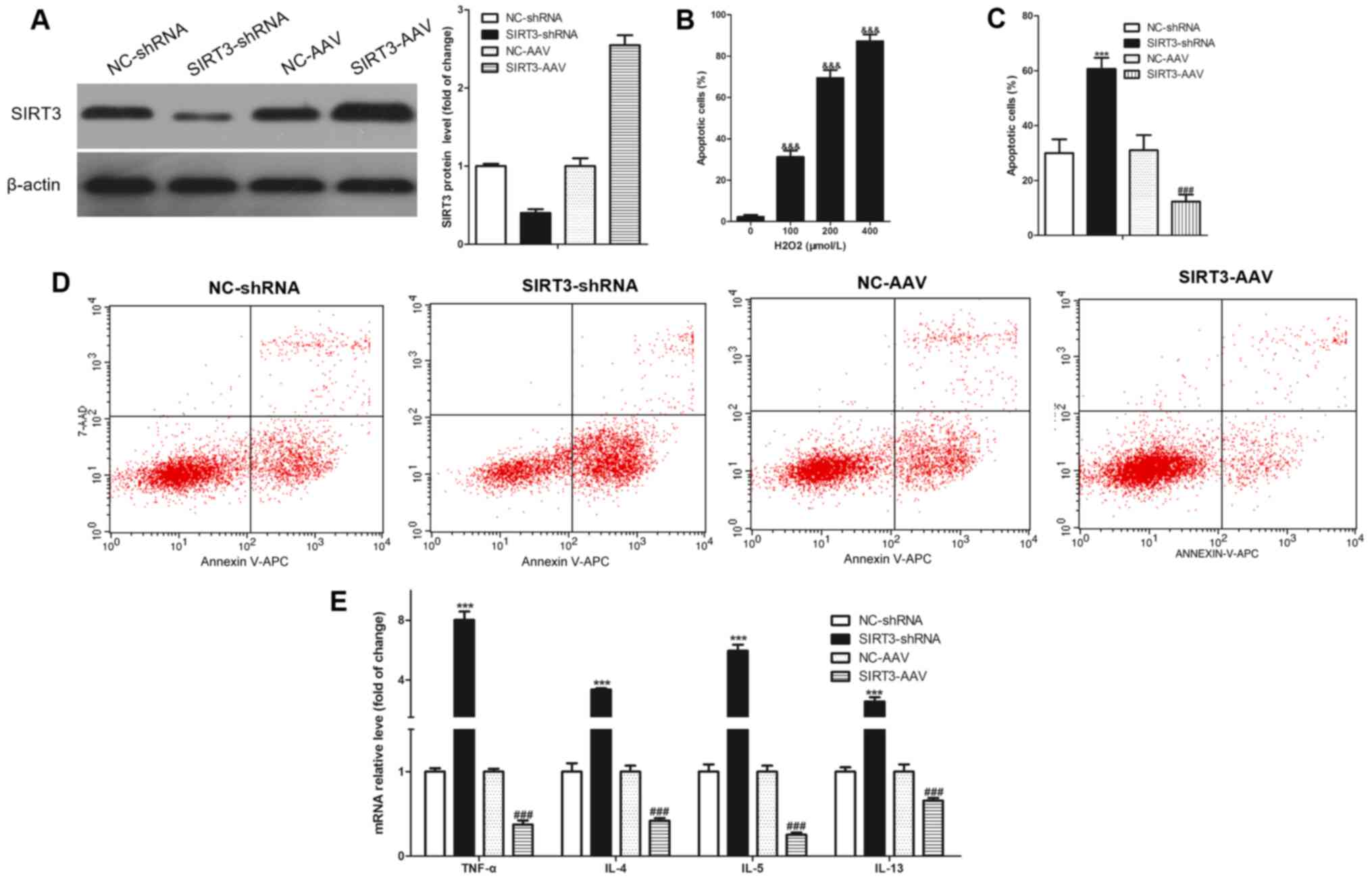
To study the relationship between SIRT3-regulated
bronchial epithelial inflammation/apoptosis and oxidative stress,
an oxidative stress cell model of bronchial epithelial cells
(16HBE) was established, induced by H2O2
in vitro. Simultaneously, SIRT3 was knocked down through
SIRT3-shRNA adenovirus infection and NC-shRNA adenovirus used as a
negative control; SIRT3 was overexpressed through SIRT3-AAV
adenovirus infection and the NC-AAV adenovirus used as a negative
control. SIRT3-shRNA successfully knocked down SIRT3 expression and
SIRT3-AAV successfully overexpressed SIRT3 in 16HBE cells (Fig. 6A). Subsequently, the induction
concentration of H2O2 was analyzed and it was
found that it induced the 16HBE cell apoptosis in a dose-dependent
manner (Fig. 6B). The 100 µmol/l
H2O2 dosage was chosen as the induction
concentration since this concentration could induce a cell
apoptosis rate of ~30%. After stimulating with 100 µmol/l
H2O2 for 12 h, 16HBE cells were collected to
analyze the apoptosis using flow cytometry. The results revealed
that the knockdown of SIRT3 significantly increased
H2O2-induced apoptosis and overexpression of
SIRT3 significantly decreased H2O2-induced
apoptosis in the 16HBE cells (Fig. 6C
and D). Knockdown of SIRT3 significantly increased the
H2O2-induced expression of cytokines (TNF-α,
IL-4, IL-5 and IL-13) and the overexpression of SIRT3 significantly
decreased the expression of these cytokines in 16HBE cells
(Fig. 6E).
Discussion
The characteristics of an ideal asthma animal model
include sudden onset of spasm, the occurrence of immediate and
delayed allergies, airway hyperresponsiveness, chronic airway
remodeling and lung function degradation (46,47).
Previous studies have shown that OVA sensitization induction can
establish this ideal asthma animal model (46,47).
The present study established an asthma model through OVA
sensitization and determined the expression of the SIRT3 gene by
RT-qPCR, western blotting and immunochemistry. All the results
suggested that SIRT3 expression was significantly decreased in the
bronchia of asthma mice and was significantly associated with the
pathologic features of asthma. Notably, upregulation of SIRT3 in
the bronchial tissues could reduce the increased apoptosis of
bronchial epithelial cells and significantly decrease the elevated
airway inflammation in asthmatic mice. In addition, upregulation of
SIRT3 reduced bronchial oxidative stress in asthmatic mice. In
vitro, SIRT3 was found to regulate
H2O2-induced apoptosis and inflammation in
16HBE cells. Asthma is the result of the interaction between
genetic and environmental factors and it is a complex pathological
process involving multiple gene regulations (9). However, chronic airway inflammation,
abnormal apoptosis of airway epithelial cells, airway epithelial
damage and repair disorders are the essential pathological basis of
asthma (48,49). Therefore, studies on the
pathogenesis of asthma, such as chronic airway inflammation and
airway epithelial cell apoptosis, might find new targets for asthma
treatment.
The SIRT3 gene is located on human chromosome 11
(11p15.5) and consists of 21,902 bases. SIRT3 protein is an
evolutionarily highly conserved deacetylase whose activity depends
on nicotinamide adenine dinucleotide and belongs to one of the
proteins in the sirtuin family. In addition to regulating the
body's energy metabolism, cell aging and tumor formation, SIRT3
also serves a pivotal role in regulating cell apoptosis through its
enzymatic catalytic activity (23).
However, to the best of the authors' knowledge, no data exist
concerning the influence of SIRT3 on airway epithelial cell
apoptosis. The present study found that the upregulation of SIRT3
in bronchial tissues could reduce the increased apoptosis of
bronchial epithelial cells; however, the corresponding mechanism
remains to be elucidated. Based on the antioxidant function of
SIRT3 (50), it was hypothesized
that SIRT3 might resist apoptosis by inhibiting oxidation. It was
later found that the upregulation of SIRT3 reduced bronchial
oxidative stress in asthmatic mice and reduced the
H2O2-induced apoptosis in the 16HBE cells
in vitro, while the loss of SIRT3 promoted
H2O2-induced apoptosis in the 16HBE
cells.
The body produces a large number of free radicals in
metabolism and ROS are the main components. Under normal
conditions, the level of ROS in the body maintains a dynamic
balance, but this balance is broken by endogenous or exogenous
harmful stimuli, resulting in a large amount of ROS accumulating in
the body, destroying the balance between the oxidative system and
the antioxidant system, developing oxidative stress and causing
cell oxidative damage or apoptosis (51,52).
It has been confirmed that oxidative stress is closely related to
cell apoptosis and ROS serves a vital role in this process
(51,52). A previous study has shown that ROS
can activate nuclear transcription factor NF-κB leading to cell
apoptosis by activating proteinase C (53). ROS can also activate the P53-induced
apoptosis pathway by damaging DNA (54) and can also lead to cell apoptosis by
damaging mitochondria and initiating the mitochondrial-mediated
apoptosis pathways (39). In brief,
inhibiting the generation of ROS and/or increasing the body's
ability to eliminate it will inhibit cell apoptosis to some extent.
Notably, SIRT3 is closely related to oxidative stress: Fu et
al (50) and Zhang et al
(55) confirmed that the activity
of Mn-SOD in the liver of SIRT3 knockout mice is reduced and SIRT3
can significantly enhance the activity of Mn-SOD by reducing the
acetylation levels of K53 and K89 or by deacetylating the 122
lysine group of Mn-SOD, thereby reducing the ROS levels in the
cells.
It is hypothesized that in the pathogenesis of
asthma, the ratio or functional imbalance of T helper (Th)1 and Th2
cells, especially the increase of Th2 cells hyperfunction, are
important immunological abnormal factors (56). Th2 cells mainly secrete IL-4, IL-5,
IL-13 and other cytokines and mediate the occurrence of asthma
through multiple pathways, such as efficient recruitment and
activation of eosinophils, stimulation of Ig conversion to produce
IgE and others (57). TNF-α is a
pro-inflammatory cytokine that participates in the immune and
inflammatory responses and can mediate the elevation of several
inflammatory cell numbers such as eosinophils and neutrophils,
generate a variety of inflammatory mediators and aggravate the
inflammatory response (58,59). TNF-α can also stimulate endothelial
cells and macrophages to release IL-6 and other inflammatory
mediators, causing airway inflammation, destroying collagen
networks and participating in airway remodeling (60,61).
In asthma, TNF-α expression in the airways and lungs is
significantly increased, promoting the occurrence and development
of inflammatory reactions (60,61).
Inflammation and oxidative stress are not independent of each other
(62,63): Inflammation can cause oxidative
stress and oxidative stress can also cause inflammation, including
oxidative stress-induced airway inflammation (64). In a number of previous studies, a
cell oxidative stress induced by H2O2 model
was used to study inflammation, such as Cao et al (65) and de Oliveira-Marques et al
(66), including
H2O2-induce 16HBE cells (67). The results of the present study
suggested that the upregulation of SIRT3 could decrease the
elevated airway inflammation in asthmatic mice and reduce the
H2O2-induced inflammation in the 16HBE cells
in vitro, while the loss of SIRT3 improved the
H2O2-induced inflammation in the 16HBE
cells.
To the best of the authors' knowledge, no relation
has been established between SIRT3 and inflammation in the
bronchial tissue in asthma. However, the study of SIRT3 inhibiting
inflammation has been widely reported: Koyama et al
(14) found that SIRT3 attenuates
palmitate-induced ROS production and inflammation in proximal
tubular cells and Boniakowski et al (38) describes that SIRT3 suppresses
macrophage-mediated inflammation in diabetic wound repair. The
findings of the present study shared broad similarities with an
earlier study by Kim et al (68) who report that KIF3A deficiency in
mice results in increased inflammation. Thus, it was hypothesized
that decreased expression of SIRT3 in the bronchial epithelium of
asthmatic animals may modulate the airway inflammation, which needs
further validation and support. In conclusion, the results of the
present study indicated that the SIRT3 gene may be a key molecule
for targeted therapeutics for asthma. However, the lack of data on
SIRT3 expression in clinical studies is limited and therefore needs
to be explored in future work.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JS conceived the study, wrote the manuscript,
performed the data analysis, participated in the experiments and
data collection. JW designed and supervised the study and edited
the manuscript. JS and JW confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Beijing Luhe
Hospital, Capital Medical University (Tongzhou, China; approval no.
2021-LHKY-055-02).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Boulet LP, Reddel HK, Bateman E, Pedersen
S, FitzGerald JM and O'Byrne PM: The global initiative for asthma
(GINA): 25 years later. Eur Respir J. 54:19005982019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin J, Wang W, Chen P, Zhou X, Wan H, Yin
K, Ma L, Wu C, Li J, Liu C, et al: Prevalence and risk factors of
asthma in mainland China: The CARE study. Respir Med. 137:48–54.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lambrecht BN, Hammad H and Fahy JV: The
cytokines of asthma. Immunity. 50:975–991. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pavord ID, Beasley R, Agusti A, Anderson
GP, Bel E, Brusselle G, Cullinan P, Custovic A, Ducharme FM, Fahy
JV, et al: After asthma: Redefining airways diseases. Lancet.
391:350–400. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heffler E, Madeira LNG, Ferrando M,
Puggioni F, Racca F, Malvezzi L, Passalacqua G and Canonica GW:
Inhaled corticosteroids safety and adverse effects in patients with
asthma. J Allergy Clin Immunol Pract. 6:776–781. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kato A and Schleimer RP: Beyond
inflammation: Airway epithelial cells are at the interface of
innate and adaptive immunity. Curr Opin Immunol. 19:711–720. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hiemstra PS, McCray PB Jr and Bals R: The
innate immune function of airway epithelial cells in inflammatory
lung disease. Eur Respir J. 45:1150–1162. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lopez-Guisa JM, Powers C, File D, Cochrane
E, Jimenez N and Debley JS: Airway epithelial cells from asthmatic
children differentially express proremodeling factors. J Allergy
Clin Immunol. 129:990–997.e6. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Haddad A, Gaudet M, Plesa M, Allakhverdi
Z, Mogas AK, Audusseau S, Baglole CJ, Eidelman DH, Olivenstein R,
Ludwig MS and Hamid Q: Neutrophils from severe asthmatic patients
induce epithelial to mesenchymal transition in healthy bronchial
epithelial cells. Respir Res. 20:2342019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wątroba M, Dudek I, Skoda M, Stangret A,
Rzodkiewicz P and Szukiewicz D: Sirtuins, epigenetics and
longevity. Ageing Res Rev. 40:11–19. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma K, Lu N, Zou F and Meng FZ: Sirtuins as
novel targets in the pathogenesis of airway inflammation in
bronchial asthma. Eur J Pharmacol. 865:1726702019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu TF, Vachharajani V, Millet P,
Bharadwaj MS, Molina AJ and Mccall CE: Sequential actions of
SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication
during immunometabolic adaptation to acute inflammation and sepsis.
J Biol Chem. 290:396–408. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ren JH, Xiang C, Li Z, Tao NN, Zhou HZ,
Liu B, Li WY, Huang AL and Chen J: Protective role of sirtuin3
(SIRT3) in Oxidative stress mediated by hepatitis B virus X protein
expression. PLoS One. 11:e01509612016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Koyama T, Kume S, Koya D, Araki S, Isshiki
K, Chin-Kanasaki M, Sugimoto T, Haneda M, Sugaya T, Kashiwagi A, et
al: SIRT3 attenuates palmitate-induced ROS production and
inflammation in proximal tubular cells. Free Radic Biol Med.
51:1258–1267. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chun P: Role of sirtuins in chronic
obstructive pulmonary disease. Arch Pharm Res. 38:1–10. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kurundkar D, Kurundkar AR, Bone NB, Becker
EJ Jr, Liu W, Chacko B, Darley-Usmar V, Zmijewski JW and Thannickal
VJ: SIRT3 diminishes inflammation and mitigates endotoxin-induced
acute lung injury. JCI Insight. 4:e1207222019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tian YG and Zhang J: Protective effect of
SIRT3 on acute lung injury by increasing manganese superoxide
dismutase-mediated antioxidation. Mol Med Rep. 17:5557–5565.
2018.PubMed/NCBI
|
|
18
|
Zhang M, Zhang Y, Roth M, Zhang L, Shi R,
Yang X, Li Y and Zhang J: Sirtuin 3 inhibits airway epithelial
mitochondrial oxidative stress in cigarette smoke-induced COPD.
Oxid Med Cell Longev. 2020:75829802020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen IC, Huang HH, Chen PF and Chiang HC:
Sirtuin 3 protects against urban particulate matter-induced
autophagy in human bronchial epithelial cells. Toxicol Sci.
152:113–127. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang Y, Li D, Ma G, Li W, Wu J, Lai T,
Huang D, Zhao X, Lv Q, Chen M and Wu B: Increases in peripheral
SIRT 1: A new biological characteristic of asthma. Respirology.
20:1066–1072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsilogianni Z, Baker JR, Papaporfyriou A,
Papaioannou AI, Papathanasiou E, Koulouris NG, Daly L, Ito K,
Hillas G, Papiris S, et al: Sirtuin 1: Endocan and sestrin 2 in
different biological samples in patients with asthma. Does severity
make the difference? J Clin Med. 9:4732020.PubMed/NCBI
|
|
22
|
Colley T, Mercado N, Kunori Y, Brightling
C, Bhavsar PK, Barnes PJ and Ito K: Defective sirtuin-1 increases
IL-4 expression through acetylation of GATA-3 in patients with
severe asthma. J Allergy Clin Immunol. 137:1595–1597. –e7. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Giralt A and Villarroya F: SIRT3, a
pivotal actor in mitochondrial functions: Metabolism, cell death
and aging. Biochem J. 444:1–10. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Roscioli E, Hamon R, Ruffin RE, Lester S
and Zalewski P: Cellular inhibitor of apoptosis-2 is a critical
regulator of apoptosis in airway epithelial cells treated with
asthma-related inflammatory cytokines. Physiol Rep. 1:e001232013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Trautmann A, Schmid-Grendelmeier P, Krüger
K, Crameri R, Akdis M, Akkaya A, Bröcker EB, Blaser K and Akdis CA:
T cells and eosinophils cooperate in the induction of bronchial
epithelial cell apoptosis in asthma. J Allergy Clin Immunol.
109:329–337. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jablonski RP, Kim SJ, Cheresh P, Williams
DB, Morales-Nebreda L, Cheng Y, Yeldandi A, Bhorade S, Pardo A,
Selman M, et al: SIRT3 deficiency promotes lung fibrosis by
augmenting alveolar epithelial cell mitochondrial DNA damage and
apoptosis. FASEB J. 31:2520–2532. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang M, Yang C and Pei Y: Effects of
downregulation of SIRT3 expression on proliferation and apoptosis
in esophageal squamous cell carcinoma EC9706 cells and its
molecular mechanisms. Biomed Mater Eng. 24:3883–3890.
2014.PubMed/NCBI
|
|
28
|
Qiao A, Wang K, Yuan Y, Guan Y, Ren X, Li
L, Chen X, Li F, Chen AF, Zhou J, et al: Sirt3-mediated mitophagy
protects tumor cells against apoptosis under hypoxia. Oncotarget.
7:43390–43400. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Luo X, Yang Z, Zheng S, Cao Y and Wu Y:
Retracted: Sirt3 activation attenuated oxidized low-density
lipoprotein-induced human umbilical vein endothelial cells'
apoptosis by sustaining autophagy. Cell Biol Int. 41:9322017.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jiao X, Li Y, Zhang T, Liu M and Chi Y:
Role of Sirtuin3 in high glucose-induced apoptosis in renal tubular
epithelial cells. Biochem Biophys Res Commun. 480:387–393. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Geng G, Du Y, Dai J, Tian D, Xia Y and Fu
Z: KIF3A knockdown sensitizes bronchial epithelia to apoptosis and
aggravates airway inflammation in asthma. Biomed Pharmacother.
97:1349–1355. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yilmaz O, Karaman M, Bagriyanik HA,
Firinci F, Kiray M, Turkeli A, Karaman O and Yuksel H: Comparison
of TNF antagonism by etanercept and dexamethasone on airway
epithelium and remodeling in an experimental model of asthma. Int
Immunopharmacol. 17:768–773. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang C, Li J, Lin H, Zhao K and Zheng C:
Nasal mucosa derived-mesenchymal stem cells from mice reduce
inflammation via modulating immune responses. PLoS One.
10:e01188492015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Karra L, Haworth O, Priluck R, Levy BD and
Levi-Schaffer F: Lipoxin B4 promotes the resolution of
allergic inflammation in the upper and lower airways of mice.
Mucosal Immunol. 8:852–862. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hu J, Zhang G and Selzer ME: Activated
caspase detection in living tissue combined with subsequent
retrograde labeling, immunohistochemistry or in situ hybridization
in whole-mounted lamprey brains. J Neurosci Methods. 220:92–98.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wang H, Liu Y, Shi J and Cheng Z: ORMDL3
knockdown in the lungs alleviates airway inflammation and airway
remodeling in asthmatic mice via JNK1/2-MMP-9 pathway. Biochem
Biophys Res Commun. 516:739–746. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Boniakowski AE, denDekker AD, Davis FM,
Joshi A, Kimball AS, Schaller M, Allen R, Bermick J, Nycz D,
Skinner ME, et al: SIRT3 regulates macrophage-mediated inflammation
in diabetic wound repair. J Invest Dermatol. 139:2528–2537.e2.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng J, Shi L, Feng L, Xu W, Li T, Gao L,
Sun Z, Yu J and Zhang J: Sirt3 ameliorates oxidative stress and
mitochondrial dysfunction after intracerebral hemorrhage in
diabetic rats. Front Neurosci. 12:4142018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kao YY, Chou CH, Yeh LY, Chen YF, Chang
KW, Liu CJ, Fan Chiang CY and Lin SC: MicroRNA miR-31 targets SIRT3
to disrupt mitochondrial activity and increase oxidative stress in
oral carcinoma. Cancer Lett. 456:40–48. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chang G, Chen Y, Zhang H and Zhou W: Trans
sodium crocetinate alleviates ischemia/reperfusion-induced
myocardial oxidative stress and apoptosis via the SIRT3/FOXO3a/SOD2
signaling pathway. Int Immunopharmacol. 71:361–371. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sahiner UM, Birben E, Erzurum S, Sackesen
C and Kalayci Ö: Oxidative stress in asthma: Part of the puzzle.
Pediatr Allergy Immunol. 29:789–800. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mishra V, Banga J and Silveyra P:
Oxidative stress and cellular pathways of asthma and inflammation:
Therapeutic strategies and pharmacological targets. Pharmacol Ther.
181:169–182. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chandra J, Samali A and Orrenius S:
Triggering and modulation of apoptosis by oxidative stress. Free
Radic Biol Med. 29:323–333. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pisoschi AM and Pop A: The role of
antioxidants in the chemistry of oxidative stress: A review. Eur J
Med Chem. 97:55–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yuk JE, Lee MY, Kwon OK, Cai XF, Jang HY,
Oh SR, Lee HK and Ahn KS: Effects of astilbic acid on airway
hyperresponsiveness and inflammation in a mouse model of allergic
asthma. Int Immunopharmacol. 11:266–273. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ji W, Chen X, Zhengrong C, Yumin H, Huang
L and Qiu Y: Therapeutic effects of anti-B7-1 antibody in an
ovalbumin-induced mouse asthma model. Int Immunopharmacol.
8:1190–1195. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xie CH, Cao YM, Huang Y, Shi QW, Guo JH,
Fan ZW, Li JG, Chen BW and Wu BY: Long non-coding RNA TUG1
contributes to tumorigenesis of human osteosarcoma by sponging
miR-9-5p and regulating POU2F1 expression. Tumour Biol.
37:15031–15041. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Marszalek JR, Liu X, Roberts EA, Chui D,
Marth JD, Williams DS and Goldstein LS: Genetic evidence for
selective transport of opsin and arrestin by kinesin-II in
mammalian photoreceptors. Cell. 102:175–187. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Fu Y, Kinter M, Hudson J, Humphries KM,
Lane RS, White JR, Hakim M, Pan Y, Verdin E and Griffin TM: Aging
promotes sirtuin 3-dependent cartilage superoxide dismutase 2
acetylation and osteoarthritis. Arthritis Rheumatol. 68:1887–1898.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Loh KP, Huang SH, De Silva R, Tan BK and
Zhu YZ: Oxidative stress: Apoptosis in neuronal injury. Curr
Alzheimer Res. 3:327–337. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lushchak VI: Free radicals, reactive
oxygen species, oxidative stress and its classification. Chem Biol
Interact. 224:164–175. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Yijing GF and Tangxue-Ming SY: Activation
of Transcription Factor NF-kB and Accumulation of Reactive Oxygen
Species Are Involved in Arsenic Trioxide-induced Apoptosis of
Esophageal. Carcinoma Cells. 5:140. 2000.(In Chinese).
|
|
54
|
Ye J, Wang S, Leonard SS, Sun Y,
Butterworth L, Antonini J, Ding M, Rojanasakul Y, Vallyathan V,
Castranova V and Shi X: Role of reactive oxygen species and p53 in
chromium(VI)-induced apoptosis. J Biol Chem. 274:34974–34980. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhang J, Song X, Cao W, Lu J, Wang X, Wang
G, Wang Z and Chen X: Autophagy and mitochondrial dysfunction in
adjuvant-arthritis rats treatment with resveratrol. Sci Rep.
6:329282016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Riiser A: The human microbiome, asthma and
allergy. Allergy Asthma Clin Immunol. 11:352015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fahy JV: Type 2 inflammation in
asthma-present in most, absent in many. Nat Rev Immunol. 15:57–65.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Almishri W, Santodomingo-Garzon T, Le T,
Stack D, Mody CH and Swain MG: TNFα augments cytokine-induced NK
cell IFNγ production through TNFR2. J Innate Immun. 8:617–629.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Lampropoulou IT, Stangou Μ, Sarafidis P,
Gouliovaki A, Giamalis P, Tsouchnikas I, Didangelos T and
Papagianni Α: TNF-α pathway and T-cell immunity are activated early
during the development of diabetic nephropathy in type II diabetes
mellitus. Clin Immunol. 215:1084232020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Brightling C, Berry M and Amrani Y:
Targeting TNF-alpha: A novel therapeutic approach for asthma. J
Allergy Clin Immunol. 121:5–10; quiz 11–2. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Berry M, Brightling C, Pavord I and
Wardlaw A: TNF-alpha in asthma. Curr Opin Pharmacol. 7:279–282.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Tabet F and Rye KA: High-density
lipoproteins, inflammation and oxidative stress. Clin Sci (Lond).
116:87–98. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Yoshikawa T: Inflammation and oxidative
stress. Nihon Naika Gakkai Zasshi. 95:1870–1875. 2006.(In
Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Chen Q, Zhou Y, Zhou L, Fu Z, Yang C, Zhao
L, Li S, Chen Y, Wu Y, Ling Z, et al: TRPC6-dependent
Ca2+ signaling mediates airway inflammation in response
to oxidative stress via ERK pathway. Cell Death Dis. 11:1702020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cao YJ, Zhang YM, Qi JP, Liu R, Zhang H
and He LC: Ferulic acid inhibits H2O2-induced oxidative stress and
inflammation in rat vascular smooth muscle cells via inhibition of
the NADPH oxidase and NF-κB pathway. Int Immunopharmacol.
28:1018–1025. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
de Oliveira-Marques V, Cyrne L, Marinho HS
and Antunes F: A quantitative study of NF-kappaB activation by
H2O2: Relevance in inflammation and synergy with TNF-alpha. J
Immunol. 178:3893–3902. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Suwara MI, Borthwick LA, Mann J, Fisher AJ
and Mann DA: Inflammation: An important Regulator of the Fibrotic
Response: S120 IL-1 is a Key Epithelial Alarmin Which Oromotes
Fibroblast Activation. Newcastle University; Newcastle: 2010
|
|
68
|
Kim D, Park W, Lee S, Kim W, Park SK and
Kang KP: Absence of Sirt3 aggravates cisplatin nephrotoxicity via
enhanced renal tubular apoptosis and inflammation. Mol Med Rep.
18:3665–3672. 2018.PubMed/NCBI
|