Introduction
The protein kinase C (PKC) family of kinases is
comprised of 11 members. Based on their molecular structures and
modes of activation, they are divided into the following three
major categories: Conventional PKCs; novel PKCs; and atypical PKCs
(aPKC, ζ and ι/λ) (1,2). aPKCs serve key roles during
embryonic development. aPKCs form polarity complexes with other
components and translocate between the apical or basolateral
membranes to regulate the direction of epithelial cell division
(3,4). Furthermore, aPKCs can interact with
partitioning-defective (Par)-6 in the subapical epithelial region
alongside the aPKC substrate Par-3 (bazooka in Drosophila)
(3). This aPKC/Par-6/Par-3
complex is key for the establishment of apical-basal polarity and
for the maturation of epithelial junctions in both
Drosophila and mammals (4). In turn, this complex regulates the
direction of asymmetric cell division during development (5), which is a key step in determining
the fate of the majority of tissues during the embryonic growth
stage.
Within the aPKC family, there are two isozymes, PKCζ
and PKCι/λ. Although their regulatory regions differ from those of
other members in the PKC family, they do share 84% sequence
homology (6). aPKC isozymes are
co-translationally phosphorylated by mTORC2 on the turn motif,
followed by phosphorylation by phosphoinositide-dependent protein
kinase-1 on the activation loop (7). Similar to other PKC family members,
aPKCs maintain their phosphorylated status after maturation, which
keeps it in an auto-inhibited conformation. Thereafter, activation
of aPKCs requires the phosphorylation of the activation loop
(8).
Unlike other PKC family members, aPKCs do not have
established activators since their activation is not dependent on
phospholipid hydrolysis. Instead, they are activated by binding to
protein scaffolds (9). After
binding to scaffold proteins, aPKCs are harbored near the plasma
membrane in proximity to their substrates. In addition, this type
of binding can relieve auto-inhibitory constraints by moving the
pseudo-substrate domain away from the substrate-binding cavity. The
interaction of PKCζ with the scaffolding protein p62 results in the
tethering of the basic PKCζ pseudo-substrate to the acidic surface
of the Phox-Bem1 domain of p62, which maintain PKCζ in an open and
active conformation (10).
Similarly, PKCζ can also be maintained in an open conformation when
bound to the cell polarity-associated protein Par6 (11). There are several known substrates
that can form complexes with aPKCs to transduce signals, including
Par3, Par6, LLGL scribble cell polarity complex component 2,
Rho-associated coiled-coil containing protein kinase 1, microtubule
affinity regulating kinase 2 and the Hippo pathway component KIBRA
(8,12–16).
Although aPKCs are better known for their function
in regulating cell polarity during embryonic development, previous
studies also revealed their potential role during adulthood. The
N-terminal truncated PKCζ was found to be necessary for maintaining
synaptic potentiation in hippocampal slices (17,18). Another previous study found that
the intraventricular injection of PKCζ can activate synapses, but
inhibiting aPKCs could suppress late-stage long-term potentiation
(LTP) in vivo (19). Using
the ζ inhibitory peptide, a non-selective aPKC inhibitory
pseudo-substrate peptide, blocking aPKC function in the brain is
found to reverse LTP and impair spatial memory in the rat
hippocampus (20). Furthermore,
it was previously demonstrated that PKCζ is involved in Alzheimer's
disease (AD), such that PKCζ could regulate β-secretase 1 (BACE1)
trafficking and distribution in the hippocampus neuron and
therefore reduce Aβ accumulation (21).
The crystal structure of PKCι has been resolved and
widely available for a number of years. However, there remains to
be a limited number of small molecules, if any, that can regulate
aPKC activity. The only known aPKC inhibitor to date is the
pseudo-substrate peptide (22),
which has limited translational development potential due to its
large molecular size.
To discover novel aPKC agonists that may have
therapeutic effects on AD, the present study first performed an
in silica docking screening from a kinase hit library
(K66-X4436) (23). Following the
virtual docking, hit compounds were synthesized and affinity
selection mass spectrometry (ASMS) and in vitro kinase
activity assay performed as secondary binding confirming assays.
Cell based assay was also performed to evaluate compound hits
protective effects in amyloid-β (Aβ) toxicity cell model and to
investigate potential mechanisms such as reactive oxygen species
(ROS) generation, mitochondria function, amyloid protein precursor
(APP) processing and Aβ accumulation.
Materials and methods
Cell line and reagents
The Neuro-2a or N2a cell line (a mouse neuroblastoma
cell line) was purchased from American Type Culture Collection
(cat. no. CCL-131). The WT7 cell line, which is N2a cells stably
expressing both the human APP695 Swedish mutant and wild-type human
presenilin-1 (PS1), was a generously provided by Professor Sangram
Sisodia, Department of Neurobiology of University of Chicago
(Chicago, USA).
The Aβ 25–35 fragment was purchased from Dalian
Meilun Biology Technology Co., Ltd. The CCK8 cell viability kit and
RIPA cell lysis buffer (strong; cat. no. P0013B) was purchased from
Beyotime Institute of Biotechnology. The mitochondrial membrane
potential assay kit (cat. no. JC-1) and ROS staining dye
dichloro-dihydro-fluorescein diacetate (DCFH-DA) were purchased
from Shanghai Yeasen Biotechnology Co., Ltd. The PKCζ (cat. no.
v9731) and PKCι (cat. no. v3751) Kinase Enzyme Systems were
purchased from Promega Corporation. Human Aβ (1–40)
ELISA kit (cat. no. 298-64601) was purchased from FUJIFILM Wako
Pure Chemical Corporation. Z640 was synthesized by WuXi AppTec.
Computational docking
Molecular docking aims to calculate the binding
orientation of small molecules to their targets, to search for
small molecules that can interact with target proteins with high
affinity and selectivity. Computational docking was performed using
Discovery studio 2016 (DS 2016; http://www.3ds.com/products-services/biovia/) in the
present study. Briefly, the small molecule database (ID K66-X4436
KINASet), which contained 11,021 molecules, was obtained from
J&K Scientific, Ltd. and used as the screening library. The
small molecules in this database were prepared using DS 2016. The
crystal structure of PKCι [protein databank (PDB) code: 3A8W] was
downloaded from the PDB for the present study. The PKCι structure
was prepared for molecular docking analysis using DS 2016. The
active sites of PKC-ι were defined using the PDB site records,
where the radius of the active site is 10.3 Å. The PKCι and the
small molecule database were docked using the ‘libdock’ function in
DS 2016. According to the libdock score, 5,000 small molecules were
chosen for docking with PKC-ι using the ‘CDOCKER’ tool in DS 2016.
The strength of the interaction was evaluated based on CDOCKER
energy and CDOCKER interaction energy. Finally, hit compounds from
this virtual screening yielding positive interactions were
synthesized.
ASMS
ASMS was performed as described previously (24–26). Briefly, an automated ligand
identification system was used to detect signal. This is a
dual-chromatography liquid chromatography (LC)/mass spectrometry
(MS) system that can separate the unbound compounds from
protein-bound compounds at the first step before using the
reversed-phase of LC/MS to identify any binding compounds. A
positive ionization method was used and nitrogen was used as
nebulizing gas and drying gas. MS detection was accomplished using
a high-resolution Exactive Orbitrap mass spectrometer (Thermo
Fisher Scientific, Inc.) scanning from 150 to 800 m/z at 100,000
resolutions with a mass accuracy of <5 ppm and a scan rate of 1
Hz. The nebulizer was set at 40 psi, drying gas temperature at
350°C with a flow rate of 1.2 l/min.
Preparation of Amyloid-β oligomer
aggregates
The Aβ 25–35 fragment was dissolved in distilled
water to a concentration of 500 µM (stock solution). To obtain the
neurotoxic form of Aβ 25-35, the peptide solution was placed in an
incubator at 37°C for 7 days and stored at −80°C until further use.
Each batch of Aβ oligomers was examined by Thioflavin (THT)
staining before use.
Cell viability assay
The proliferation of the cells was detected via
CCK-8 assay (Beyotime Institute of Biotechnology). Briefly, prior
to the treatment, N2a or WT7 cells were plated in 96-well plates at
a density of 1×104 cells/well in DMEM media with 10% FBS
for 24 h. Growth medium was replaced with fresh culture medium
without FBS then and cells were treated with 50 µM Aβ-oligomers
with or without different concentrations of Z640 (1, 3, 10, 30 or
100 µM) at 37°C for 24 h. After incubation, cell viability assay
was conducted by treating cells with CCK-8 reagents (10 µl/well) at
37°C for 2 h. Plates were read at 450 nm using a the BioTek Synergy
HT Multi Mode Microplate Reader. The difference in optical density
(OD) relative to untreated control group or Aβ-oligomers treatment
alone group were measured to draw dose-response curve. Half maximal
effective concentration (EC50) was calculated by
GraphPad Prism Software Version 6. All groups were performed in 5
replicates.
Measurement of ROS generation
ROS generation was measured using the DCFH-DA dye.
Cells were incubated at 37°C for 1 h in the dark in HEPES
containing DCFH-DA (200 µM). Intracellular fluorescence was
measured using a spectrofluorometer (BioTek Synergy HT Multi-Mode
Microplate Reader; Agilent Technologies, Inc.) at an emission
wavelength of 525 nm and an excitation wavelength of 488 nm. The
images were also captured using a confocal microscope (Leica TCS
SP8, with 4× objective lens; Leica Microsystems GmbH). The
intensity of fluorescence staining was analyzed using ImageJ
software (1.50i; National Institutes of Health).
Measurement of mitochondrial membrane
potential (MMP)
MMP was measured by using the fluorescent probe
JC-1. Cells were incubated with the JC-1 staining solution (5
mg/ml) for 20 min at 37°C. The fluorescence intensity of both
mitochondrial JC-1 monomers (excitation, 514 nm; emission, 529 nm)
and aggregates (excitation, 585 nm; emission, 590 nm) were detected
using a microplate reader (BioTek Synergy HT Multi-Mode Microplate
Reader; Agilent Technologies, Inc.) and the images were also
captured using a confocal microscope (Leica TCS SP8, with 4×
objective lens; Leica Microsystems GmbH). The collected
Fluorescence Unit from microplate reader was shown as fluorescence
ratio of red to green.
Aβ40 ELISA
The WT7 cells were plated into 96-well plates at a
density of 3×104 cells/ml and incubated with the
Aβ-oligomers and indicated concentrations of Z640 (0, 0.03, 0.1,
0.3, 1, 3, or 10 µM) at 37°C for 24 h. After incubation, the
culture medium was collected, treated with the protease inhibitor
cocktail and stored at −70°C until use. Aβ40 levels in the culture
medium was assessed using the Human β Amyloid (1–40)
ELISA kit according to manufacturer's protocol. The obtained value
for each sample was normalized to that of the total amount of
protein in the sample.
In vitro kinase activity assay
PKCι and PKCζ kinase activity was performed
according to manufacturer's protocol. Briefly, 25 ng purified PKCζ
or 35 ng purified PKCι was used to incubate together with 10 µM
ATP, 0.2 µg/µl substrate and Z640 at the indicated concentrations
(0, 0.1, 0.3, 1, 3, 10 or 30 µM) for 60 min in room temperature.
After incubation, ADP-Glo was added to the system and incubated at
room temperature for another 40 min. Luminescence signals (LU) were
detected using the BioTek Synergy HT Multi-Mode Microplate Reader
(Agilent Technologies, Inc.).
Western blot analysis
Cells after various treatments were lysed using
protein lysis buffer (Beyotime Institute of Biotechnology) on ice
for 15 min. Following centrifugation (10,000 × g, 7 min at 4°C),
supernatant was collected and protein concentration was determined
by BCA protein assay (Thermo Fisher Scientific, Inc.), reading
absorbance at 560 nm by using plate reader (BioTek Synergy HT
Multi-Mode Microplate; BioTek Instruments, Inc.). Following protein
denaturing, 50 µg total protein was loaded onto 10% SDS PAGE gel
and then transferred onto PVDF membranes. Membranes were then
blocked with 5% BSA (Beyotime Institute of Biotechnology) for 1 h
at room temperature. Following blocking, membranes were incubated
with primary antibody overnight at 4°C and then the secondary
antibody 1 h at room temperature. The primary antibodies used in
the present study included mouse-anti-APP (1:2,000; cat. no. 6E10;
Biolegend, Inc.), rabbit anti-BACE1 (1:1,000; cat. no. 5606T, Cell
Signaling Technology, Inc.), rabbit anti-pBACE1-496 (1:1,000; cat.
no. PA5-105747; Invitrogen; Thermo Fisher Scientific, Inc.), rabbit
anti-PS1 (1:5,000; cat. no. MAT5232; MilliporeSigma), rabbit
anti-PKCζ (1:500; cat. no. 9368T; Cell Signaling Technology, Inc.),
rabbit anti-APP C-terminal fragment (1:10,000; cat. no. 18717;
CTFβ; MilliporeSigma) and β-actin (1:5,000; cat. no. sc-47778,
Santa Cruz Biotechnology, Inc.). The secondary antibody used were
HRP-labelled anti-Rabbit IgG (1:5,000; cat. no. 7074, Cell
Signaling Technology, Inc) and HRP-labelled anti-mouse IgG
(1:5,000; cat. no. 7076, Cell Signaling Technology, Inc). Bands
were visualized by using enhanced ECL reagent (cat. no. WBULS0500,
MilliporeSigma) and developed in dark room. The bands were
semi-quantified by using ImageJ software (1.50i; National
Institutes of Health).
Statistical analysis
Results were analyzed using GraphPad Prism Software
Version 6.0 (GraphPad Software, Inc.). All data were presented as
the mean ± standard deviation. The significance of difference was
determined using unpaired Student's t-test or one-way analysis of
variance followed by Tukey's test. The difference of treatments
among cell types were determined by using Two-way ANOVA followed
with Bonferroni and Sidak's test. The EC50 was
calculated automatically by using non-linearized curve regression.
P<0.05 was considered to indicate a statistically significant
difference.
Results
In silico library docking and
screening
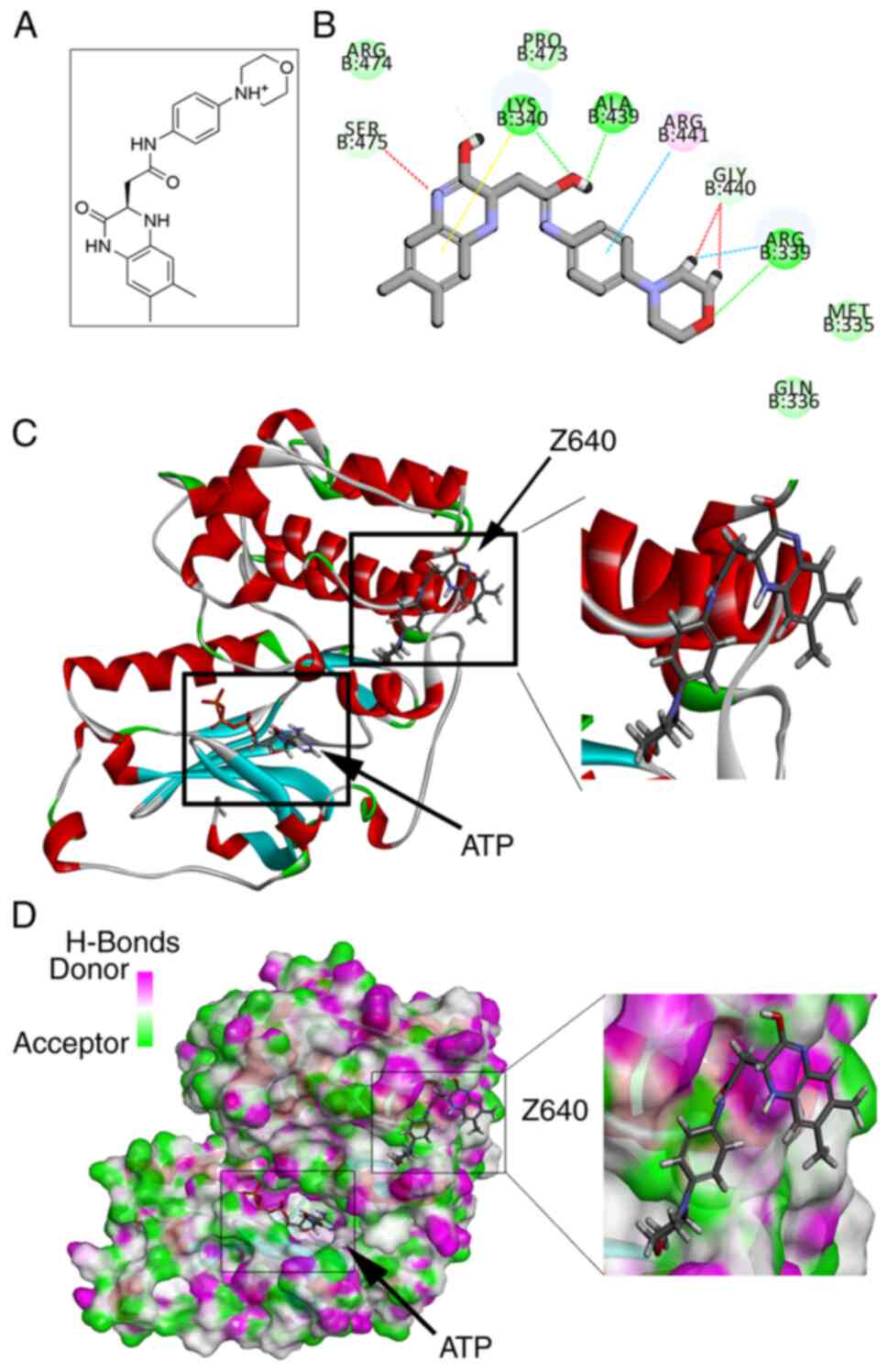
After in silico screening, Z640 was selected
(Fig. 1A). As shown in Fig. 1B and C, Z640 was able to bind PKCι
in an allosteric binding position. The CDOCKER energy was
calculated to be-24.075 kcal/mol whereas the CDOCKER interaction
energy was calculated to be-38.4802 kcal/mol. Molecular studies
revealed that Z640 may form conventional hydrogen bonds with the
Arg339, Lys340 and Ala439 residues of PKCι, in addition to forming
carbon hydrogen bonds with Ser475, Gly440 and Arg339. Furthermore,
Z640 may form pi-cation interactions with Lys340 and pi-alkyl
interactions with Arg441 (Fig.
1D).
Z640 can activate aPKC activity in
vitro
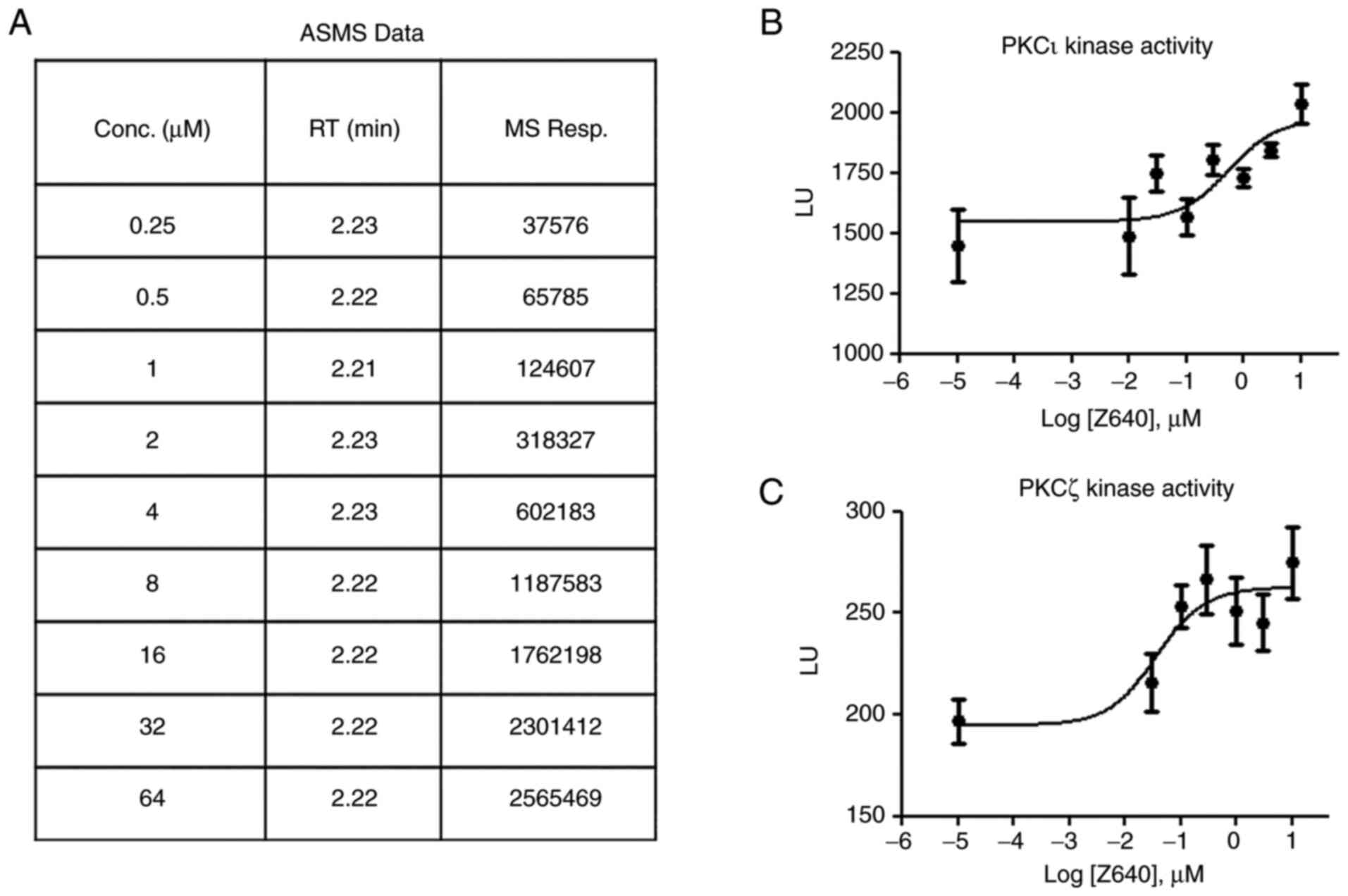
The ASMS results indicated that Z640 can bind to
PKCι with an affinity value of 1.37 µM (Fig. 2A), which is comparable to the
binding affinity of ATP to the majority of the kinases. The present
study then tested if such binding could regulate aPKC kinase
activity. By using in vitro kinase activity assays, both
recombinant PKCζ and PKCι were treated with Z640 in ascending
doses. Z640 was found to increase PKCζ and PKCι kinase activity in
a dose-dependent manner, with an in vitro EC50 of
1.09 and 3.47 µM, respectively (Fig.
2B and C).
Z640 can protect neuronal cell lines
against Aβ oligomer-induced toxicity
Considering that aPKC expression has been previously
reported to be downregulated in AD and may therefore be involved in
AD pathogenesis (27,28), the present study next considered
the hypothesis that upregulating aPKC activity may be therapeutic
for AD. A cell-based assay mimicking AD toxicity was first
established by treating N2a or WT7 cells with Aβ oligomers. WT7
cell was generated by stably overexpress human APP Swedish-mutation
and human presenilin-1 (PS1) into N2a cell. Generally, APP will get
through sequential cleavages by several different enzymes and Aβ40
or 42 will be produced after the final cleavage conducted through
PS1 (γ secretase complex). The human APP-Swedish mutation has long
been proved having much stronger affinity to γ secretase compare
with wildtype APP and thus will produce a higher level of Aβ40 or
42. In this regard, WT7 cell is an ideal cell model to mimic in
vivo AD pathology due to its high Aβ generation and
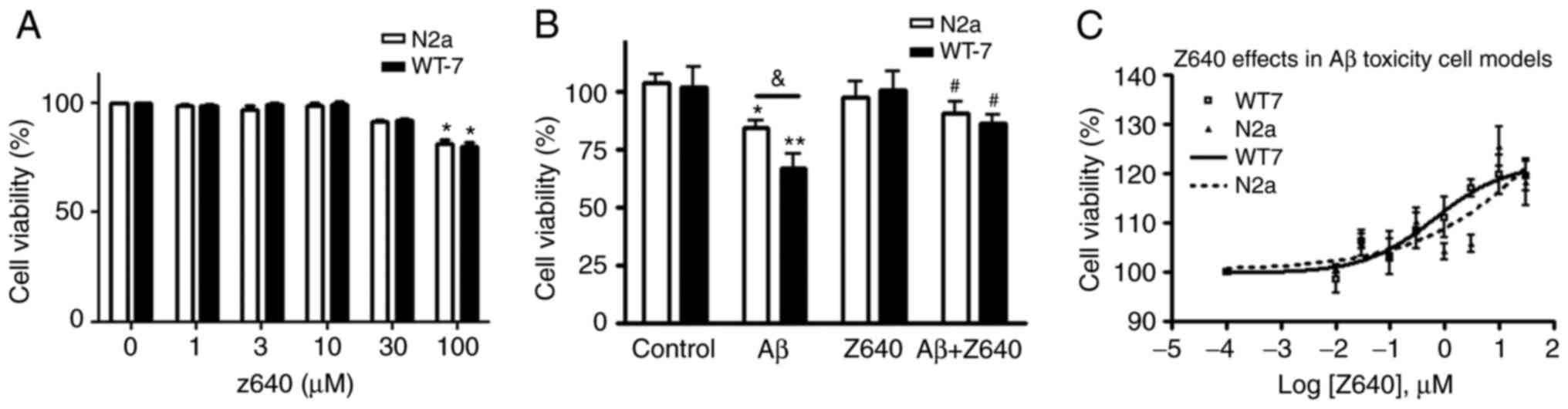
accumulation (29,30). Aβ oligomers were found to induce
significant cell toxicity at concentrations of 50 µM and higher. In
particular, at 50 µM concentration, Aβ oligomers induced higher
levels of toxicity in WT7 cells compared with N2a cells (data not
shown). The cells were then treated with different concentrations
of Z640 alone. Z640 could not induce cell death at concentrations
<100 µM (Fig. 3A). Since 10 µM
is the most frequently applied concentration for testing hit
compound function in cell-based assays without inducing significant
off-target effects (31), 10 µM
was deemed to be a safe concentration for the use of Z640 for
subsequent cell experiments thereafter.
Z640 was next applied in the cell-based assays to
examine the effects of Z640 against Aβ oligomer induced toxicity.
Z640 was found to significantly reduce Aβ oligomer-caused cell
death at 10 µM, with more potent protective effects observed in WT7
cells compared with N2a cells (Fig.
3B). This finding suggests that the protective effects of Z640
may be specifically associated with APP processing and Aβ
generation, since the only difference between these two cell lines
is the level of Aβ expression. Furthermore, the EC50 of
Z640 in both Aβ oligomer toxicity models was also evaluated and
results indicated that Z640 has a EC50 of 4.57 µM in WT7
cells while 419.8 µM in N2a cells (Fig. 3C). This data also showed a much
stronger protective effect of Z640 in WT7 compare with N2a
cell.
Z640 can protect cells by correcting
Aβ oligomer-induced ROS elevation and mitochondria damage
Mitochondria damage is one of the most extensively
reported mechanisms underlying Aβ oligomer-induced cell death. As
Z640 was found to protect Aβ oligomer-mediated cytotoxicity, the
present study subsequently tested if Z640 treatment can protect
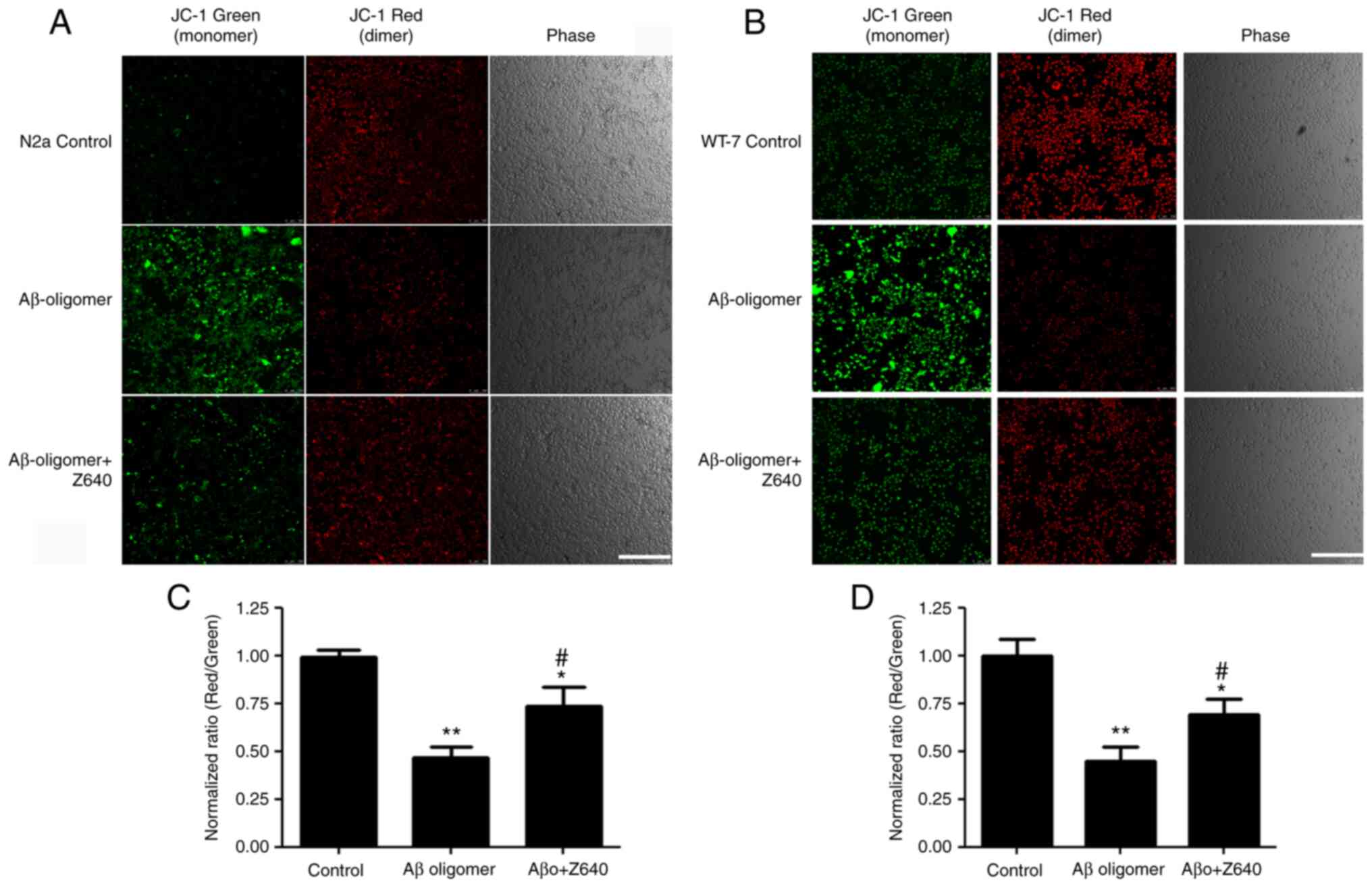
against cell death by reducing mitochondria damage. JC-1 staining
was used to test the effects of Z640 on MMP. In healthy cells, JC-1
exists as a monomer in the cytoplasm (green) but forms aggregates
(red) in the mitochondria due to the higher MMP. However, in
apoptotic and necrotic cells, JC-1 only exists in its monomeric
form and is therefore only stained in the cytoplasm (green). Using
JC-1 live cell staining followed by both confocal microscopy and
fluorescence intensity reading, the degree of mitochondria
impairment was next examined after Z640 and/or Aβ oligomer
treatments. The results showed that the Aβ oligomers could damage
mitochondria membrane integrity, whereas 10 µM Z640 could largely
restore this impairment (Fig.
4).
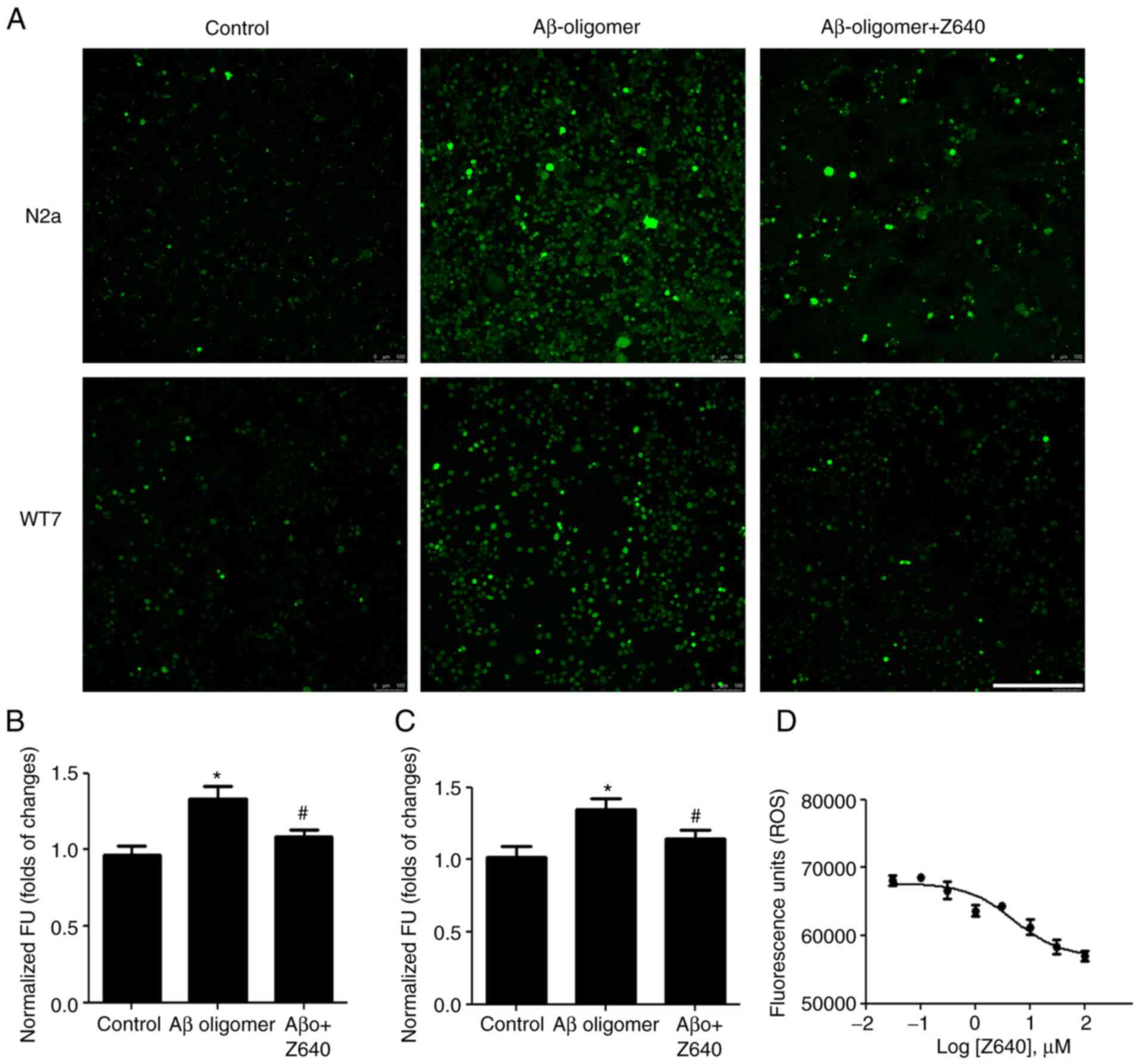
Since elevated intracellular ROS is frequently
accompanied with mitochondria impairments and is used as a
biomarker for measuring mitochondrial stress, the effects of Z640
on intracellular ROS levels were next tested. Aβ oligomers were
found to significantly increase intracellular ROS levels but 10 µM
Z640 could markedly reverse this (Fig. 5A-C). Furthermore, an experiment
was performed to assess any potential dose-dependent effects. Z640
was able to reduce Aβ oligomer-induced ROS elevations in a
dose-dependent manner, with an EC50 of 5.11 µM (Fig. 5D).
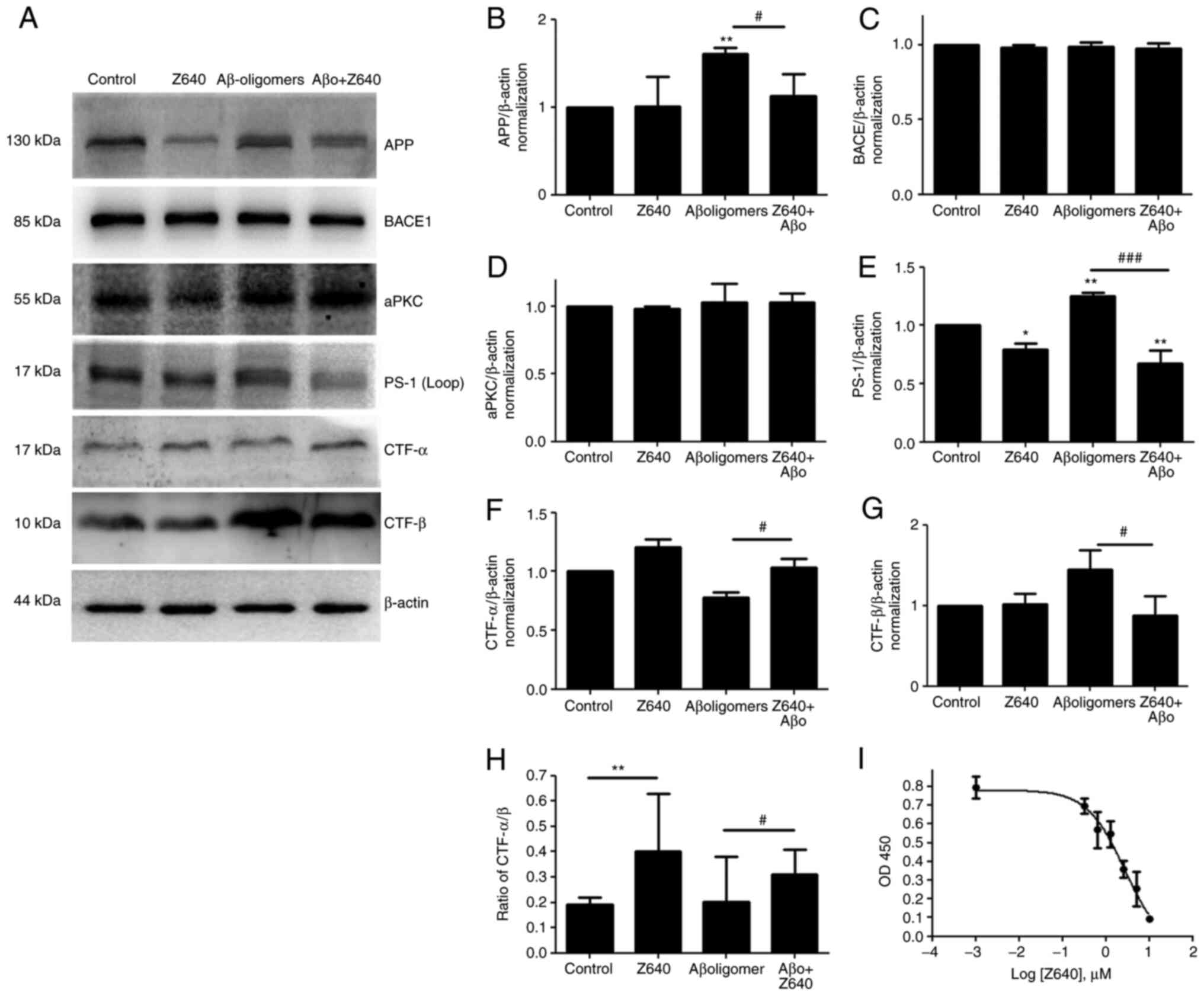
Z640 can regulate APP processing and
reduce Aβ generation
A previous study demonstrated that PKCζ knockdown or
inhibition can promote the retrograde trafficking of BACE1, thereby
increasing Aβ generation and intracellular accumulation (32). However, there is no evidence
demonstrating how the overexpression of PKCζ or elevating its
kinase activity can regulate Aβ generation. By using the WT7 cell
line, the effects of Z640 on APP processing and Aβ generation was
assessed. Consistent with previous reports (33,34), Aβ oligomers treatment promoted APP
processing towards the β-cleavage pathway and therefore increased
both CTF-β and Aβ generation. Following treatment with 10 µM Z640
it was found that although Z640 alone could not induce significant
changes in full-length APP and APP-CTFβ, it could significantly
increase the expression of APP-CTFα and the ratio of
APP-CTFα/APP-CTFβ (Fig. 6). This
observation suggested that Z640 could regulate the direction of APP
processing. Following co-treatment together with 50 µM
Aβ-oligomers, 10 µM Z640 could almost completely reverse the
effects of Aβ-oligomers on APP processing to the level similar to
that in Z640 alone. This finding strongly suggested that both Aβ
oligomers and Z640 are competitors in the regulation of APP
processing through the same signaling pathway. However, the present
study also showed that although Z640 could not alter BACE1
expression levels, it could reduce PS1 expression. To the best of
the authors' knowledge, there have been no reports indicating that
aPKCs can regulate PS1 expression. Therefore, this finding may shed
light on the possibly novel functions of aPKCs on regulating PS1
expression. The effects of Z640 on soluble Aβ generation was next
investigated by measuring Aβ40 levels in the culture medium. The
results showed that Z640 could effectively reduce Aβ40 generation
in a dose-dependent manner with a EC50 of 2.73 µM
(Fig. 6I).
Discussion
The present study first performed the virtual
docking screen of aPKCs with K66-X4436 KINASet database. The reason
virtual docking was chosen as a starting point was because this is
a much more convenient way to select a hit compound than other
ways, such as screening from a real compound library by ASMS or
surface plasmon resonance. K66-X4436 is a diverse kinase library,
which means the chance to obtain positive hits from this library
are higher than other non-organized libraries. After docking, a
novel aPKCs agonist structure was screened and named as Z640.
Furthermore, By using ASMS, its binding affinity to PKCι was
determined to be 1.37 µM in vitro. Although aPKCs have
distinct structures compared with other families of PKCs, they do
have an active catalytic domain that can utilize ATP to
phosphorylate their substrates. In the present study, computational
docking was performed through the entire PKCι structure instead of
only in the ATP adapting pocket, which is typically performed
instead for searching for aPKC inhibitors. Based on the binding
position, the orientation of Z640 with the lowest energy was found
to be at a site far away from the ATP binding site (35,36). Therefore, it was predicted that
the binding of Z640 to PKCι may cause allosteric structural
changes, which increases the in vitro kinase activity of
aPKCs without altering the catalytic site. However, the detailed
structure-activity relationship must be determined by comparing apo
and co-crystallization structures collected using X-ray
crystallography or nuclear magnetic resonance in future.
Another discovery of the present study was that aPKC
activation could reduce Aβ generation by shifting APP processing
according to the cell-based assay. So far, there have been several
hypotheses for the occurrence of AD, the Aβ hypothesis is one of
the most studied among them. People have known that Aβ oligomers
could damage neuron from multiple aspects, such as oxidative
stress, calcium flux hyperactivation, hyper-active microglia,
suppress synaptic morphology and function and may even induce
Tauopathy (37,38). After the AD pathology is
initiated, Aβ oligomers will continuously accumulate in the
cortex/hippocampus microenvironment and keep attacking synapse and
thus worsen the disease situation (39). In a previous study, in patients
with AD, the activity of the Par3/aPKC complex was found to be
reduced (40). In addition, the
Par3/aPKC complex was previously found to regulate BACE1
trafficking in cultured primary hippocampal neurons (21,32,41). These previous studies demonstrated
that after inhibiting aPKC function in the primary neurons, BACE1
would be translocated in a largely retrograde manner from the cell
membrane into the trans-Golgi network to increase Aβ generation.
However, due to the lack of effective agonists, it was not possible
to test if activating the Par3/aPKC complex could reduce Aβ
generation, which is a key concept for the translational
development of aPKC agonists for potential AD therapeutics. In the
present study, by using Z640 it was demonstrated that following
aPKC activation, APP processing can be shifted towards the
α-cleavage pathway to generate CTF-α instead of CTF-β, which
decreases the levels of Aβ. To the best of the authors' knowledge,
these results demonstrated for the first time that aPKC agonists
have potential benefits in an AD model, which strongly supported
the feasibility of developing aPKC agonists for AD
therapeutics.
Aβ monomers have been observed to interact with
higher order oligomers and fibrils to directly form aggregation
nuclei and pathogenic dimers (42). Furthermore, Aβ oligomers have been
confirmed to upregulate Aβ generation through several pathways,
including activating the glutamate synapse or enhancing calcium
influx (31). These previous
findings suggest that Aβ monomers and Aβ oligomers may function
synergistically to induce neuronal cell death. Based on this Aβ
oligomer theory, the hypothesis that inhibiting Aβ monomer
generation by activating aPKC may reduce Aβ oligomer-induced
neuronal cell toxicity was tested in the present study. The
comparative data collected from N2a and WT7 cells suggested that
Z640 can exert more potent protective effects against Aβ oligomers
in WT7 cells, consistent with this hypothesis. However, it was also
noted that Z640 could exert protective effects against Aβ
oligomer-induced N2a cells toxicity, which does not express
endogenous Aβ. This result indicated activated aPKC might also
protect cells from pathways other than by regulating Aβ generation.
For example, some studies indicated that aPKC activation might
promote cell survival and proliferation by activating β-Catenin/Wnt
signal pathway (43–45). The Wnt/β-Catenin pathway is a very
conserved signal pathway and exists extensively in the majority of
types of tissue and cells. The activation of β-Catenin/Wnt signal
pathway might increase cell resistance to undesired or toxic
microenvironments and thus show protective effects in N2a cells.
However, potential off-target effects of Z640 cannot be excluded in
the present study. By using the DS 2016 program, other potential
bio-targets of Z640 were also explored. The results indicated Z640
may bind to several enzymes, including lyases or hydrolases (data
not shown). The unspecific binding may also lead to this extra
protection. A comprehensive kinase crosstalk panel screening should
be performed in the future for clarification.
Another key finding in the present study is that
Z640 yielded similar EC50 values in all assays tested.
Z640 was able to act against Aβ induced toxicity with a
EC50 of 4.57 µM, reduce Aβ 40 generation with a
EC50 of 2.73 µM and could reduce Aβ oligomer-mediated
ROS production with a EC50 of 5.11 µM. Furthermore, Z640
could activate aPKCs kinase activity with a EC50 of 3.46
and 1.08 µM. Considering the similar activation performance of Z640
on above bio-events, it was hypothesized that these bio-events
might occur in a cascade way and the common upstream modulator is
Z640. Z640 could activate aPKCs in the cell and the activated aPKC
will then reduce Aβ generation and thus reduce Aβ oligomer-mediated
cell toxicity.
The present study had several limitations that
should be addressed. The optimization of the dose of Z640 is
required. In the present study, Z640 showed an EC50
value ≤5 µM for the majority of the experiments, which would serve
as a good starting point for translational development in the
future. In addition, future studies should be focused on modifying
Z640 to optimize its binding affinity. A co-crystallization or
soaking structure will assist in this type of investigation.
Another limitation is that the selectivity was not optimized. A
panel kinase activity screening for Z640 is required to determine
its selectivity. Finally, more hits libraries should be screened in
future work to find better starting structures.
In summary, by using a computational
docking/modeling program, an in silico screening of a
commercial hits library was performed, which revealed the
non-selective aPKC agonist Z640. Its ability to bind to aPKCs was
verified in vitro using ASMS and kinase activity assays.
Z640 was next applied in a cell-based Aβ toxicity model and it was
found that Z640 could alleviate Aβ oligomer-induced cell apoptosis
by reducing ROS generation to preserve mitochondria function.
Finally, Z640 could regulate APP processing to reduce Aβ
production, which may be one of the mechanisms underlying the
protective function of Z640. To the best of the authors' knowledge,
the present study was the first to identify a non-selective aPKC
agonist, which has therapeutic potential for AD by reducing Aβ
oligomer-induced neuronal toxicity.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81801080 and 82071174 to MS) and
The Science and Technology Project Fund of Nantong City (grant no.
JC2019097) to XB.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
MS and XB designed the whole study. DZ and MS
performed general experiments. QL performed LC/MS experiments. DZ,
WP and PC performed analysis of the data. MS and XB confirm the
authenticity of all the raw data. WP and PC drafted the manuscript.
MS and XB finalized the manuscript and supplied the funding to
conduct this study. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Breitkreutz D, Braiman-Wiksman L, Daum N,
Denning MF and Tennenbaum T: Protein kinase C family: On the
crossroads of cell signaling in skin and tumor epithelium. J Cancer
Res Clin Oncol. 133:793–808. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moscat J, Diaz-Meco MT and Wooten MW: Of
the atypical PKCs, Par-4 and p62: Recent understandings of the
biology and pathology of a PB1-dominated complex. Cell Death
Differ. 16:1426–1437. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Gunaratne A and Di Guglielmo GM: Par6 is
phosphorylated by aPKC to facilitate EMT. Cell Adh Migr. 7:357–361.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Horikoshi Y, Suzuki A, Yamanaka T, Sasaki
K, Mizuno K, Sawada H, Yonemura S and Ohno S: Interaction between
PAR-3 and the aPKC-PAR-6 complex is indispensable for apical domain
development of epithelial cells. J Cell Sci. 122:1595–1606. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Golub O, Wee B, Newman RA, Paterson NM and
Prehoda KE: Activation of Discs large by aPKC aligns the mitotic
spindle to the polarity axis during asymmetric cell division.
Elife. 6:e321372017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Selbie LA, Schmitz-Peiffer C, Sheng Y and
Biden TJ: Molecular cloning and characterization of PKC iota, an
atypical isoform of protein kinase C derived from insulin-secreting
cells. J Biol Chem. 268:24296–24302. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tobias IS, Kaulich M, Kim PK, Simon N,
Jacinto E, Dowdy SF, King CC and Newton AC: Protein kinase Cζ
exhibits constitutive phosphorylation and
phosphatidylinositol-3,4,5-triphosphate-independent regulation.
Biochem J. 473:509–523. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
McCaffrey LM and Macara IG: The Par3/aPKC
interaction is essential for end bud remodeling and progenitor
differentiation during mammary gland morphogenesis. Genes Dev.
23:1450–1460. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kajimoto T, Caliman AD, Tobias IS, Okada
T, Pilo CA, Van AN, Andrew McCammon J, Nakamura SI and Newton AC:
Activation of atypical protein kinase C by sphingosine 1-phosphate
revealed by an aPKC-specific activity reporter. Sci Signal.
12:eaat66622019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Tsai LC, Xie L, Dore K, Xie L, Del Rio JC,
King CC, Martinez-Ariza G, Hulme C, Malinow R, Bourne PE and Newton
AC: Zeta inhibitory peptide disrupts electrostatic interactions
that maintain atypical protein kinase C in its active conformation
on the scaffold p62. J Biol Chem. 290:21845–21856. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ozdamar B, Bose R, Barrios-Rodiles M, Wang
HR, Zhang Y and Wrana JL: Regulation of the polarity protein Par6
by TGFbeta receptors controls epithelial cell plasticity. Science.
307:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Aranda V, Haire T, Nolan ME, Calarco JP,
Rosenberg AZ, Fawcett JP, Pawson T and Muthuswamy SK: Par6-aPKC
uncouples ErbB2 induced disruption of polarized epithelial
organization from proliferation control. Nat Cell Biol.
8:1235–1245. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chalmers AD, Pambos M, Mason J, Lang S,
Wylie C and Papalopulu N: aPKC, Crumbs3 and Lgl2 control apicobasal
polarity in early vertebrate development. Development. 132:977–986.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hartmann S, Ridley AJ and Lutz S: The
function of Rho-associated kinases ROCK1 and ROCK2 in the
pathogenesis of cardiovascular disease. Front Pharmacol. 6:2762015.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chen YM, Wang QJ, Hu HS, Yu PC, Zhu J,
Drewes G, Piwnica-Worms H and Luo ZG: Microtubule
affinity-regulating kinase 2 functions downstream of the
PAR-3/PAR-6/atypical PKC complex in regulating hippocampal neuronal
polarity. Proc Natl Acad Sci USA. 103:8534–8539. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yoshihama Y, Chida K and Ohno S: The
KIBRA-aPKC connection: A potential regulator of membrane
trafficking and cell polarity. Commun Integr Biol. 5:146–151. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Amini N, Azad RR, Motamedi F,
Mirzapour-Delavar H, Ghasemi S, Aliakbari S and Pourbadie HG:
Overexpression of protein kinase Mζ in the hippocampus mitigates
Alzheimer's disease-related cognitive deficit in rats. Brain Res
Bull. 166:64–72. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Crary JF, Shao CY, Mirra SS, Hernandez AI
and Sacktor TC: Atypical protein kinase C in neurodegenerative
disease I: PKMzeta aggregates with limbic neurofibrillary tangles
and AMPA receptors in Alzheimer disease. J Neuropathol Exp Neurol.
65:319–326. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Patel H and Zamani R: The role of PKMζ in
the maintenance of long-term memory: A review. Rev Neurosci.
32:481–494. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yao Y, Kelly MT, Sajikumar S, Serrano P,
Tian D, Bergold PJ, Frey JU and Sacktor TC: PKM zeta maintains late
long-term potentiation by N-ethylmaleimide-sensitive
factor/GluR2-dependent trafficking of postsynaptic AMPA receptors.
J Neurosci. 28:7820–7827. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun M and Zhang H: Par3 and aPKC regulate
BACE1 endosome-to-TGN trafficking through PACS1. Neurobiol Aging.
60:129–140. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bogard AS and Tavalin SJ: Protein kinase C
(PKC)ζ pseudosubstrate inhibitor peptide promiscuously binds PKC
family isoforms and disrupts conventional PKC targeting and
translocation. Mol Pharmacol. 88:728–735. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen C, Lu Y, Siu HM, Guan J, Zhu L, Zhang
S, Yue J and Zhang L: Identification of novel vacuolin-1 analogues
as autophagy inhibitors by virtual drug screening and chemical
synthesis. Molecules. 22:8912017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Andrews CL, Ziebell MR, Nickbarg E and
Yang X: Mass Spectrometry-Based Screening and Characterization of
Protein-Ligand Complexes in Drug Discovery. Protein and Peptide
Mass Spectrometry in Drug Discovery. 253–286. 2011. View Article : Google Scholar
|
|
25
|
Annis DA, Athanasopoulos J, Curran PJ,
Felsch JS, Kalghatgi K, Lee WH, Nash HM, Orminati JPA, Rosner KE,
Shipps GW Jr, et al: An affinity selection-mass spectrometry method
for the identification of small molecule ligands from self-encoded
combinatorial libraries: Discovery of a novel antagonist of E.
coli dihydrofolate reductase. Int J Mass Spectrom. 238:77–83.
2004. View Article : Google Scholar
|
|
26
|
Annis A, Chuang CC and Nazef N: ALIS: An
Affinity Selection-Mass Spectrometry System for the Discovery and
Characterization of Protein-Ligand Interactions. Mass Spectrometry
in Medicinal Chemistry: Applications in Drug Discovery. 121–156.
2007. View Article : Google Scholar
|
|
27
|
Sajan MP, Hansen BC, Higgs MG, Kahn CR,
Braun U, Leitges M, Park CR, Diamond DM and Farese RV: Atypical
PKC, PKCλ/ι, activates β-secretase and increases
Aβ1−40/42 and phospho-tau in mouse brain and isolated
neuronal cells, and may link hyperinsulinemia and other aPKC
activators to development of pathological and memory abnormalities
in Alzheimer's disease. Neurobiol Aging. 61:225–237. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Etcheberrigaray R, Tan M, Dewachter I,
Kuipéri C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ,
Kozikowski AP, et al: Therapeutic effects of PKC activators in
Alzheimer's disease transgenic mice. Proc Natl Acad Sci USA.
101:11141–11146. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sun M, Zhou T, Zhou L, Chen Q, Yu Y, Yang
H, Zhong K, Zhang X, Xu F, Cai S, et al: Formononetin protects
neurons against hypoxia-induced cytotoxicity through upregulation
of ADAM10 and sAβPPα. J Alzheimers Dis. 28:795–808. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu J, Sun M, Chen Z, Lu J, Liu Y, Zhou L,
Xu X, Fan D and Chui D: Magnesium modulates amyloid-beta protein
precursor trafficking and processing. J Alzheimers Dis.
20:1091–1106. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cline EN, Bicca MA, Viola KL and Klein WL:
The amyloid-β oligomer hypothesis: Beginning of the third decade. J
Alzheimers Dis. 64 (Suppl 1):S567–S610. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sun M, Huang CY, Wang H and Zhang HY: Par3
regulates polarized convergence between APP and BACE1 in
hippocampal neurons. Neurobiol Aging. 77:87–93. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rolland M, Powell R, Jacquier-Sarlin M,
Boisseau S, Reynaud-Dulaurier R, Martinez-Hernandez J, André L,
Borel E, Buisson A and Lanté F: Effect of Aβ oligomers on neuronal
APP triggers a vicious cycle leading to the propagation of synaptic
plasticity alterations to healthy neurons. J Neurosci.
40:5161–5176. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schützmann MP, Hasecke F, Bachmann S,
Zielinski M, Hänsch S, Schröder GF, Zempel H and Hoyer W:
Endo-lysosomal Aβ concentration and pH trigger formation of Aβ
oligomers that potently induce tau missorting. Nat Commun.
12:46342021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang H, Neimanis S, Lopez-Garcia LA,
Arencibia JM, Amon S, Stroba A, Zeuzem S, Proschak E, Stark H,
Bauer AF, et al: Molecular mechanism of regulation of the atypical
protein kinase C by N-terminal domains and an allosteric small
compound. Chem Biol. 21:754–765. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Dong W, Lu J, Zhang X, Wu Y, Lettieri K,
Hammond GR and Hong Y: A polybasic domain in aPKC mediates
Par6-dependent control of membrane targeting and kinase activity. J
Cell Biol. 219:e2019030312020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
De Felice FG, Velasco PT, Lambert MP,
Viola K, Fernandez SJ, Ferreira ST and Klein WL: Abeta oligomers
induce neuronal oxidative stress through an N-methyl-D-aspartate
receptor-dependent mechanism that is blocked by the Alzheimer drug
memantine. J Biol Chem. 282:11590–11601. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nunomura A, Perry G, Pappolla MA,
Friedland RP, Hirai K, Chiba S and Smith MA: Neuronal oxidative
stress precedes amyloid-beta deposition in down syndrome. J
Neuropathol Exp Neurol. 59:1011–1017. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bayer TA and Wirths O: Intracellular
accumulation of amyloid-beta-a predictor for synaptic dysfunction
and neuron loss in Alzheimer's disease. Front Aging Neurosci.
2:82010.PubMed/NCBI
|
|
40
|
Farese RV and Sajan MP: Atypical PKC:
Therapeutic target for Alzheimer's? Aging (Albany NY). 11:13–14.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sun M, Asghar SZ and Zhang HY: The
polarity protein Par3 regulates APP trafficking and processing
through the endocytic adaptor protein Numb. Neurobiol Dis. 93:1–11.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rosenman DJ, Connors CR, Chen W, Wang C
and Garcia AE: Aβ monomers transiently sample oligomer and
fibril-like configurations: Ensemble characterization using a
combined MD/NMR approach. J Mol Biol. 425:3338–3359. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sabherwal N, Tsutsui A, Hodge S, Wei J,
Chalmers AD and Papalopulu NJD: The apicobasal polarity kinase aPKC
functions as a nuclear determinant and regulates cell proliferation
and fate during Xenopus primary neurogenesis. Development.
136:2767–2777. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song G, Ouyang G and Bao S: The activation
of Akt/PKB signaling pathway and cell survival. J Cell Mol Med.
9:59–71. 2010. View Article : Google Scholar
|
|
45
|
Islam SMA, Patel R and Acevedo-Duncan M:
Protein kinase C-ζ stimulates colorectal cancer cell carcinogenesis
via PKC-ζ/Rac1/Pak1/β-catenin signaling cascade. Biochim Biophys
Acta Mol Cell Res. 1865:650–664. 2018. View Article : Google Scholar : PubMed/NCBI
|