Introduction
Ischemic stroke is a problematic health issue
worldwide. The major pathophysiological interference of ischemic
stroke is the sudden obstruction of cerebral arteries, which
immediately causes neurons to suffer from hypoxia (1,2).
As a highly energy-dependent organ, the brain is vulnerable to
hypoxia, which is also the reason for the high mortality associated
with ischemic stroke (3). It is
of great importance to understand the mechanisms of neuron injury
under ischemic stroke conditions to provide a more precise
interference to manage this condition.
Different mechanisms have been reported to be
involved in the pathological process of ischemic stroke. Neuron
apoptosis (4), necrosis (5) and even ferroptosis (6) have important roles. Of note,
disturbed energy supply is reported to be critical to
hypoxia-induced neuron death (7).
This is understandable, since neurons are highly energy-dependent
cells and provides hints that maintaining energy supply may be
useful in treating ischemic stroke. In fact, Li et al
(8) reported that maintaining
mitochondrial function prevented nicotine-induced exacerbation of
ischemic brain damage, which indicated that mitochondrial function
is involved in the pathological processes of ischemic stroke.
As the power factory of the cell, mitochondrial
normal function is of importance for neuron survival (9). Mitochondrial dysfunction is involved
in several ischemic diseases, including ischemic stroke (10), myocardial infarction (11) and kidney infarction (12). The oxidative phosphorylation
processes inside the matrix of mitochondria provides the majority
of energy to the cell. However, mitochondria are also vulnerable to
stresses and this may result in mitochondrial dysfunction. The
present study provided evidence that mitochondrial dysfunction is
critically involved in ischemic stroke-induced neuron injury via
mitochondrial calcium overload. 75 kDa glucose-regulated protein
(GRP75) is a member of the heat shock protein (HSP) family and
mediates endoplasmic reticulum (ER)-mitochondrial calcium transfer,
maintaining mitochondrial calcium homeostasis (13). However, the role of GRP75 in
hypoxia-induced neuron damage has so far remained elusive. The
present study examined whether inhibiting GRP75 may ameliorate
excessive mitochondrial calcium overload and whether it may be a
promising target in preventing hypoxia-induced neuron injury.
Materials and methods
Agents
MKT077 (cat. no. HY-15096) was purchased from
MedChemExpress. Penicillin-streptomycin mixed solution (cat. no.
G4003), pancreatic enzyme (cat. no. G4001), primary antibody
diluent (cat. no. G2025), phenylmethylsulphonyl fluoride (PMSF;
cat. no. G2008), protein phosphatase inhibitors (cat. no. G2007),
DMSO, minimal essential medium (MEM; cat. no. G4553) and 5X loading
buffer (cat. no. G2013) were purchased from Wuhan Servicebio
Technology Co., Ltd. High-glucose DMEM (cat. no. 10569010),
glucose-free DMEM (cat. no. 10966025), Opti-MEM (cat. no. 31985062)
and fetal bovine serum (FBS; cat. no. 10091148) were obtained from
Gibco (Thermo Fisher Scientific, Inc.). The enhanced
chemiluminescence (ECL) kit (cat. no. P0018FS) and bicinchoninic
acid (BCA) assay kit (cat. no. P0010S) were purchased from Beyotime
Institute of Biotechnology. Primary antibodies against β-actin
(cat. no. AC026), GRP75 (cat. no. A112560), mitochondrial fission
factor (MFF; cat. no. A8700), mitofusin (MFN)1 (cat. no. A9880),
MFN2 (cat. no. A19678), dynamin-1-like protein (DRP1; cat. no.
A17069), phosphorylated (p)-DRP1 (cat. no. AP0812) and
hypoxia-inducible factor 1-alpha (HIF1α; cat. no. A11945) were
purchased from ABclonal Biotech Co., Ltd. Horseradish peroxidase
(HRP)-conjugated goat anti-rabbit IgG (cat. no. AS014) and
HRP-conjugated goat anti-mouse (cat. no. AS003) were purchased from
ABclonal Biotech Co., Ltd. Lipofectamine® 3000
transfection reagent (cat. no. L3000015) and
MitoTracker® Deep Red FM kits (cat. no. M7531) were from
Thermo Fisher Scientific, Inc.
Animals
Male C57BL/6 mice (age, 10 weeks; weight, 25–30 g)
were purchased from the Animal Experiment Center of Tongji Medical
College, Huazhong University of Science and Technology (Wuhan,
China). The mice were housed in a specific pathogen-free room with
free access to rodent chow and water. The experimental protocol was
reviewed and approved by the Animal Research and Care Committee of
Tongji Medical College, Huazhong University of Science and
Technology [Wuhan, China; no. SYXK(E)2016-0057].
Mouse middle cerebral artery
obstruction (MCAO) model
A mouse model of MCAO was generated as reported
previously with minor modifications (14,15). In brief, 14 mice were randomized
into the control and MCAO groups (n=7 per group). Individual mice
were intraperitoneally injected with 10% chloral hydrate (350
mg/kg) and no peritonitis was present. When the mice were
anesthetized, an incision was made in the neck of the mice. After
careful exposure of the right external carotid artery (ECA), common
carotid artery (CCA) and internal carotid artery (ICA), the CCA was
clamped and the ECA was tied, followed by insertion of a 6–0
monofilament nylon suture with a rounded tip from the CCA to the
ICA to occlude the left MCA at its origin (~10 mm) and the skin was
sutured. The mice were kept on a heating pad to maintain their
temperature at 37°C. After recovery from anesthesia, the mice were
returned into their cages and they were euthanized at 8 h
post-ischemia. The mice were decapitated without anesthesia and
four brains were used for 2,3,5-triphenyltetrazolium chloride (TTC)
staining and three were analyzed using transmission electron
microscopy (TEM).
TTC staining
The mice were euthanized by decapitation at 8 h of
ischemia and their brains were rapidly removed and frozen. The
frozen brains were cut into 2-mm sections. The brain slices were
stained with 2% TTC solution with incubation at 37°C for 15 min.
Subsequently, the brain slices were fixed with 10% paraformaldehyde
for 10 min. The percentage of infarct volume of the total brain
volume in individual mice was measured using Image J software
(v1.51j8) [National Institutes of Health (NIH)] in a blinded
manner.
TEM
The mouse brain tissues were immersed in 2.5%
glutaraldehyde (4°C) for 24 h and fixed with 1% osmotic acid-1.5%
potassium ferrocyanide for 1.5 h at room temperature. An alcohol
gradient was used for dehydration: 75% ethanol for 4 h, 85% ethanol
for 2 h, 90% ethanol for 1 h, 100% ethanol for 1 h and another 100%
ethanol for 1 h. Next, samples were immersed in 100% xylene for 30
min and 100% xylene for 1 h. Finally, samples were
paraffin-embedded for 90 min. Ultrathin tissue sections were
stained with 3% uranyl acetate and lead citrate (50 nm) and the
mitochondrial morphology was observed by TEM. The length/diameter
ratio of the mitochondria was calculated and those with a ratio of
>2 were considered as long mitochondria (long-mito) (16).
Cell culture
HT22 mouse hippocampus neuronal cells (Procell Life
Science & Technology Co., Ltd.) and N2a neuroblastoma cells
(Wuhan Servicebio Technology Co., Ltd) were cultured in 10% FBS
high-glucose DMEM and MEM, respectively, with 1% penicillin and
streptomycin (complete medium) in a 5% CO2 incubator at
37°C.
Primary neuron extraction
Primary mouse neuron extraction was performed to
investigate mitochondrial function. In brief, one pregnant (E15-17)
C57 mouse which was purchased from the Animal Experiment Center of
Tongji Medical College, Huazhong University of Science and
Technology, was anesthetized using 10% chloral hydrate (350 mg/kg)
and the abdominal cavity was quickly opened and fetal mice (six
mice) were removed. The pregnant and fetal mice were subsequently
decapitated and the cortical regions were isolated and digested
with 0.25% trypsin (cat. no. G4001; Wuhan Servicebio Technology
Co., Ltd.), followed by termination of digestion with medium (5%
FBS neurobasal medium; cat. no. H430717; Shanghai Basalmedia
Technologies Co., Ltd.). After centrifugation, the cells were
rapidly resuspended in medium. The prepared single cells were
cultured in petri dishes coated with polylysine (Gibco; Thermo
Fisher Scientific, Inc.). After 12 h of culture, the cells were
placed in 1% B27 (FBS-free) medium for subsequent experiments. The
experimental protocol was reviewed and approved by the Animal
Research and Care Committee of Tongji Medical College, Huazhong
University of Science and Technology [approval no.
SYXK(E)2017-0012].
Transfections
The cells were transfected when they had grown to
~70% confluency. For this, 2 µl transfection reagent, 2 µg of probe
plasmid or 5 pM of specific small inhibitory (si)RNA were added to
100 µl of Opti-MEM and incubated for 5 min. Opti-MEM with
transfection reagents was then added to Opti-MEM with plasmid or
siRNA. After resting for 15 min, the mixed suspension was added to
the cell culture medium. In addition, a negative control group
remained untransfected. The medium was replaced 6 h after
transfection, the cells were cultured for another 24 h and then
used in the following experiments. The sequences of negative
control siRNA (si-NC) and GRP75-specific siGRP75 were as follows:
si-NC forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′; siGRP75 forward,
5-GCGUCUUUACCAAACUUAUTT-3′ and reverse,
5′-AUAAGUUUGGUAAAGACGCTT-3′.
Mitotracker staining
For mitotracker staining, MitoTracker®
Deep Red FM kits were used. In brief, cells were stained in the
dark using mitotracker (500 nM) for 15 min and then observed with a
fluorescence confocal microscope. ImageJ software v.1.51j8 (NIH)
was used for the quantification of fluorescence.
Cellular oxygen and glucose
deprivation (OGD) model
The cells were cultured in 10% FBS glucose-free DMEM
at 37°C in an incubator with 91% N2, 5% CO2
and 4% O2 as the OGD conditions for varying time
periods. Control cells were cultured under normal cell culture
conditions in 95% air and 5% CO2; N2a cells were
cultured in MEM with 10% FBS and 1% penicillin-streptomycin, while
HT22 cells were cultured in DMEM with 10% FBS and 1%
penicillin-streptomycin. MKT077 was added to the medium at a final
concentration of 5 µM with incubation for 8 h in a 37°C cell
incubator.
Western blot analysis
The different groups of cells were lysed in RIPA
buffer containing 1 mM PMSF and 1 mM phosphatase inhibitor cocktail
and centrifuged at 14,000 × g for 12 min at 4°C. After determining
the protein concentration of each lysate sample using the BCA kit,
the lysates (30 µg/lane) were separated by SDS-PAGE on 10–12% gels
and transferred onto PVDF membranes (cat. no. WGPVDF45; Wuhan
Servicebio Technology Co., Ltd.). The membranes were incubated with
5% dry skimmed milk in 0.1% Tween-20 in Tris-buffered saline for 1
h at room temperature and probed at 4°C overnight with primary
antibodies (1:1,000 dilution for all). Subsequently, the membranes
were incubated with HRP-conjugated goat anti-rabbit IgG and
(1:10,000 dilution) and HRP-goat anti-mouse IgG and visualized with
the ECL kit using a Bio-Rad exposure system (Bio-Rad Gel Doc XR;
Bio-Rad Laboratories, Inc.) and the Image Lab 5.1 software (Bio-Rad
Laboratories, Inc.). Quantitative results were obtained by
densitometric scanning using ImageJ v.1.51j8 software (NIH).
Fluorescent analysis of mitochondrial
Ca2+
The levels of mitochondrial Ca2+ in N2a
cells were determined after transfection with mitochondrial
Ca2+ probe plasmids which had been used previously
(17). In each confocal dish,
5×104 N2a cells were cultured for 24 h. The calcium
plasmid was transfected into N2a cells for 24 h using the cell
transfection method described above. After washing the cells with
pre-warmed PBS solution, they were examined under a laser confocal
microscope (A1R; Nikon Corporation) using Nikon Imaging Software
Elements Viewer 4.20 (Nikon Corporation). The mitochondrial
Ca2+ levels were determined by measuring average
intensity levels using ImageJ software 1.51j8 (NIH).
Measurement of ATP
The levels of intracellular ATP in individual groups
of cells were measured using a specific kit (cat. no. S0026B;
Beyotime Institute of Biotechnology) following the manufacturer's
protocol. In brief, the same numbers of cells in each group were
lysed on ice and centrifuged. The supernatants were collected for
the measurement of ATP levels using a luminometer (Promega
Corporation) and the levels of ATP were calculated using the
standard curve.
Measurement of the mitochondrial
membrane potential (MMP)
The MMP of each group of cells was measured using
the specific kit (cat. no. C2006; Beyotime Institute of
Biotechnology) following the manufacturer's protocol. In brief,
after treatment, the different groups of cells were stained with
JC-1 solution at 37°C for 20 min. After being washed, the cells
were imaged under a Nikon confocal microscope (A1R; Nikon
Corporation) and the red and green fluorescent intensity of 100
cells from 6 randomly selected fields were analyzed using ImageJ
software v.1.51j8 (NIH).
Measurement of reactive oxygen species
(ROS) levels
The levels of intracellular ROS were measured using
a specific kit (cat. no. S0033; Beyotime Institute of
Biotechnology) following the manufacturer's protocol. In brief, the
dichloro-dihydro-fluorescein diacetate (DCFH-DA) probe was diluted
to 10 mM with FBS-free DMEM. The different groups of cells were
stained with 10 mM DCFH-DA in FBS-free DMEM. The fluorescent
signals in each group of cells were imaged under a Nikon confocal
microscope (A1R; Nikon Corporation) and the green, fluorescent
intensity of ~100 cells from 10 randomly selected fields were
measured using ImageJ software v.1.51j8 (NIH).
Statistical analysis
Data were analyzed by GraphPad Prism 5 software
(GraphPad Software, Inc.) and are expressed as the mean ± standard
error of the mean. Statistical significance was assessed by one-way
ANOVA followed by Tukey's post-hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
Mitochondrial fragmentation and
dysfunction induced by ischemia in vivo
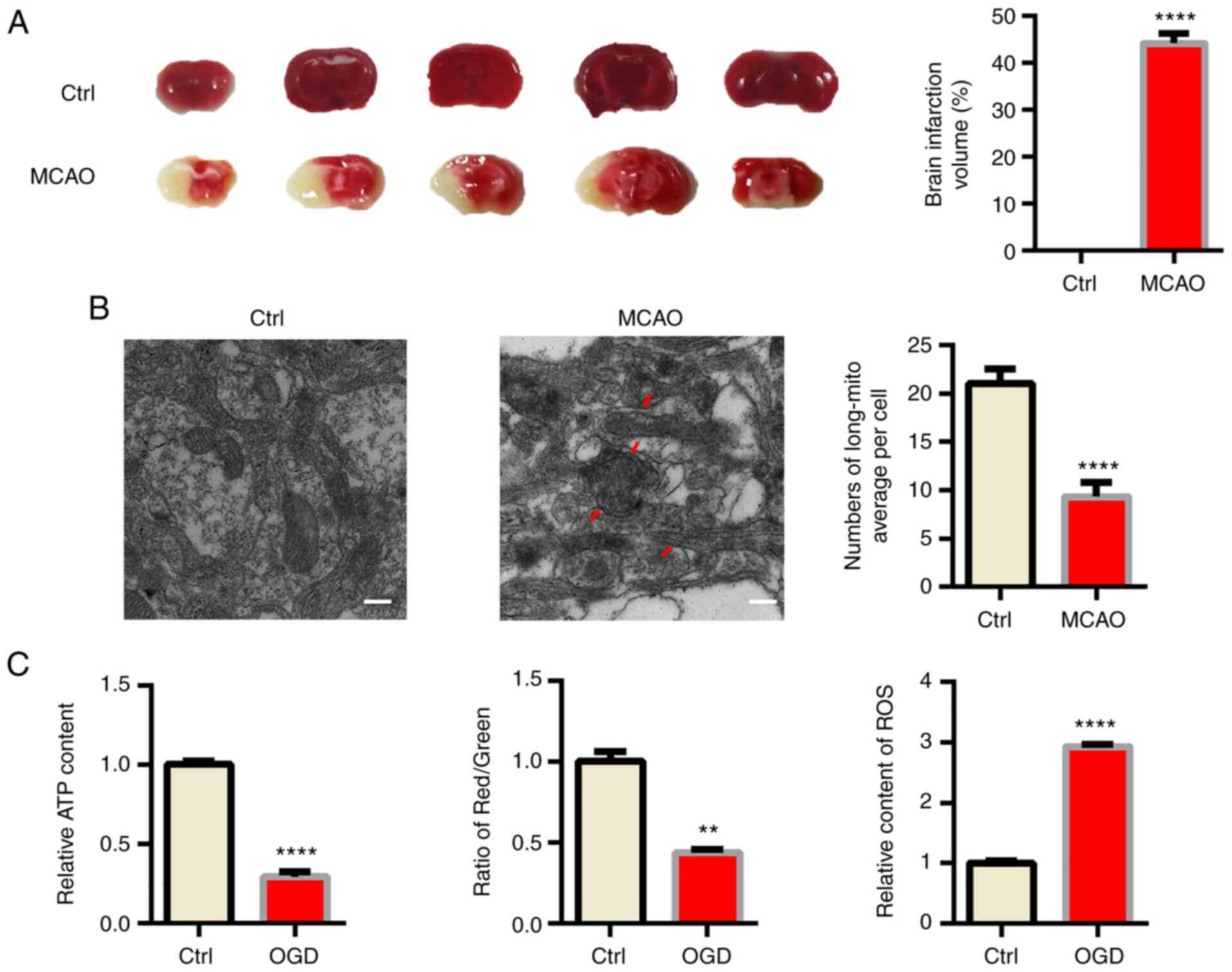
First, male C57BL/6 mice were subjected to MCAO as a
classical in vivo model of ischemic stroke (18). TTC staining indicated obvious
infraction in the brain of MCAO model mice (Fig. 1A). Acute hypoxia significantly
influenced mitochondrial function; hence, TEM was used to confirm
the morphology changes of mitochondria in ischemic mouse brains. As
indicated in Fig. 1B, swollen and
fragmented mitochondria were apparent in the MCAO group and the
cristae of mitochondria were broken and fractured. However, the
morphology of mitochondria was normal in the sham mice. Since
mitochondrial function is tightly associated with the morphology of
mitochondria (19), several
indexes reflecting mitochondrial function, including ATP content,
ROS production and the MMP were also determined and it was
indicated that mitochondrial function was significantly impaired by
OGD (Figs. 1C, S1 and S2).
Mitochondrial fragmentation and
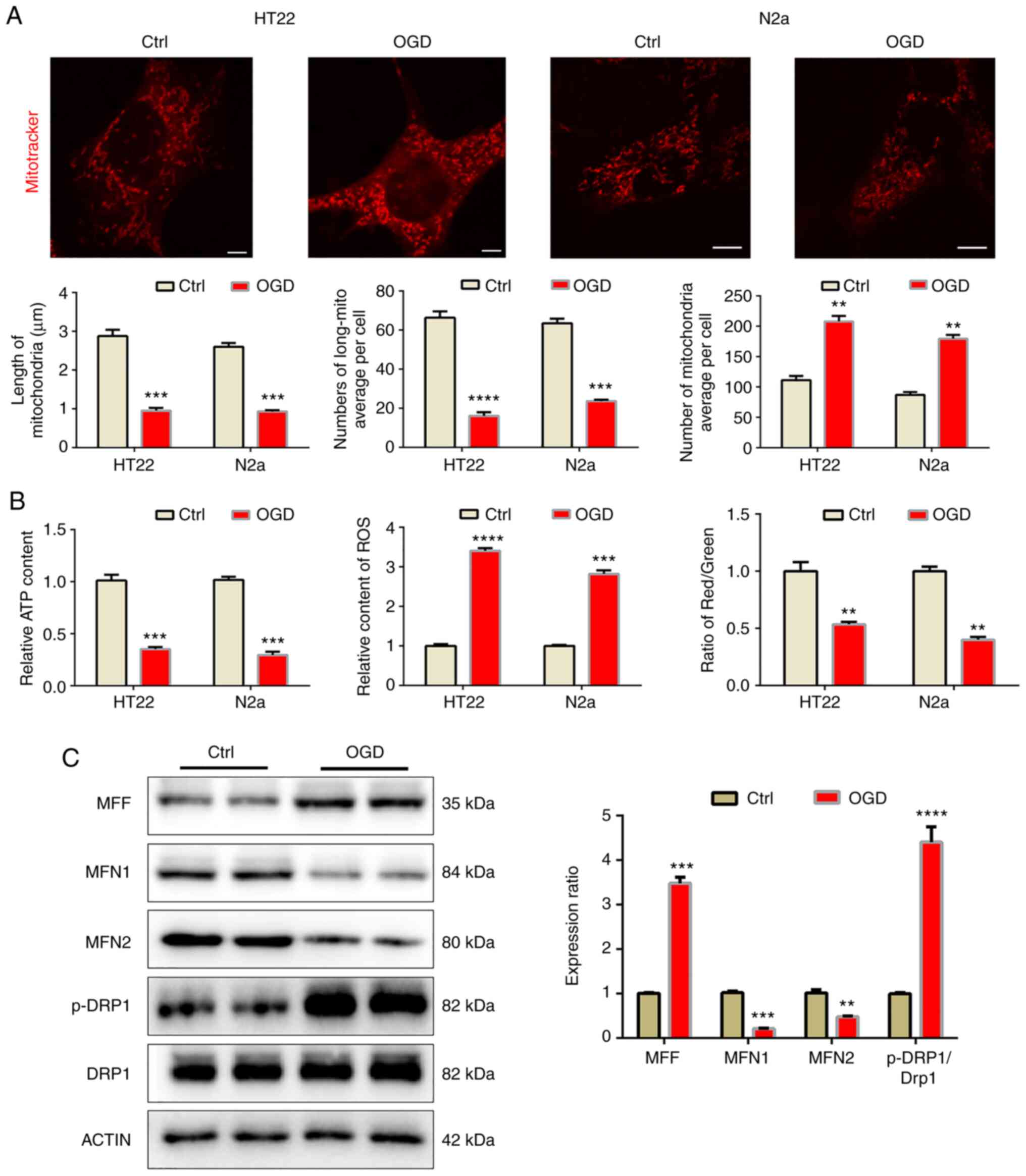
dysfunction induced by OGD in vitro
In order to further confirm the above results in an
in vitro model, OGD was implemented to resemble the effects
of MCAO in the HT22 and N2a cell lines. After 8 h of OGD, cellular
mitochondria were observed by mitotracker staining (Fig. 2A). The mitochondria became
fragmented after OGD, as the length of mitochondria decreased while
the number of mitochondria per cell increased after OGD, which led
to a decreased mitochondrial ratio. Similarly, OGD also impeded
mitochondrial function (Fig. 2B)
and decreased the MMP and ATP levels but increased ROS levels. In
N2a cells, western blot analysis was used to detect mitochondrial
dynamic-related proteins. The results indicated that under OGD
conditions, mitochondrial fusion proteins MFN1 and MFN2 were
downregulated, while mitochondrial fission proteins MFF and p-DRP1
increased significantly, indicating that the mitochondria became
fragmented (Fig. 2C).
Mitochondrial calcium overload induced
by OGD
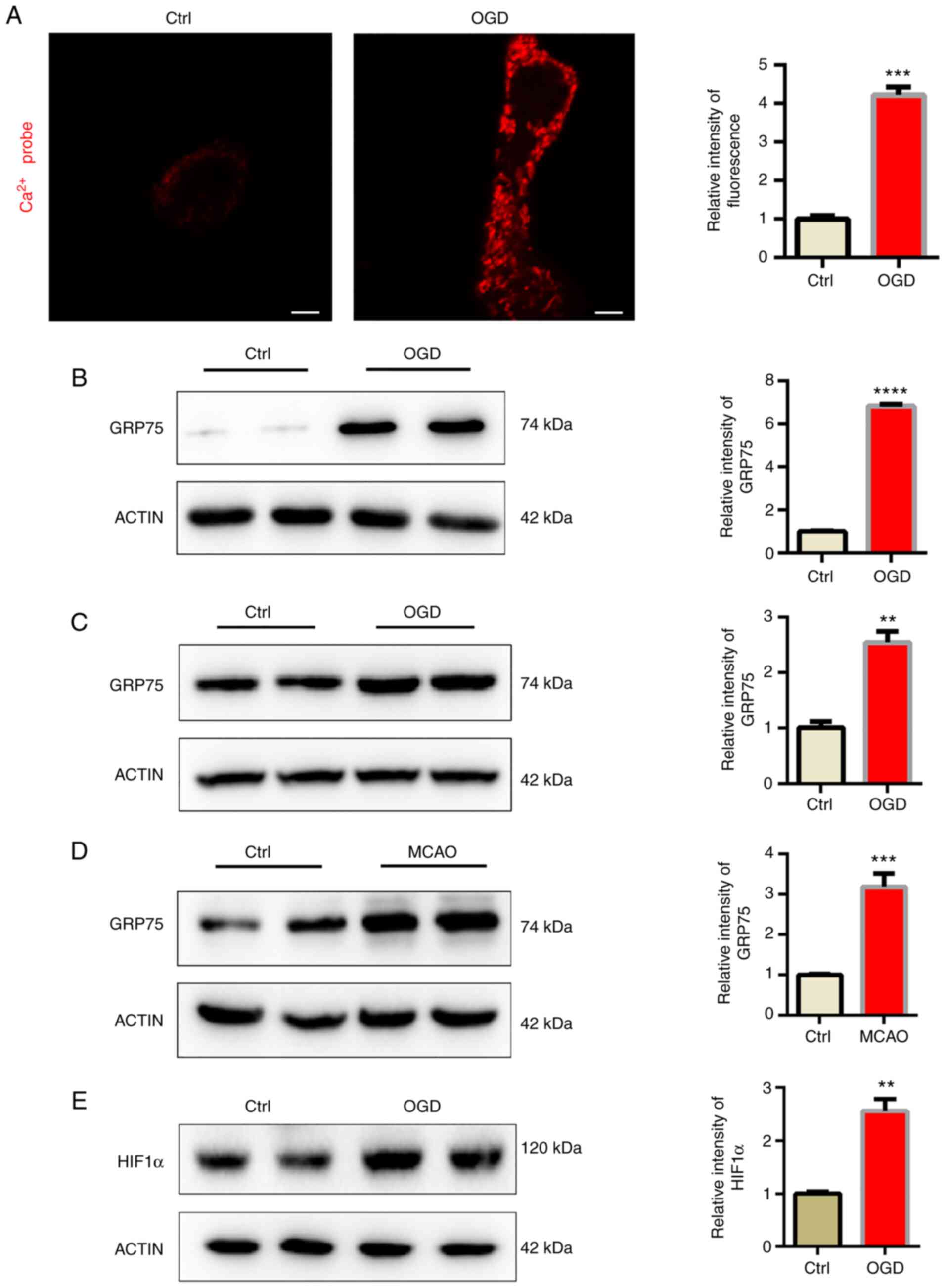
GRP75 acts as a bridge, linking the ER-anchored
inositol 1,4,5-trisphosphate receptor type 1 (IP3R) and
mitochondria-located voltage-dependent anion-selective channel
protein (VDAC) to form a protein complex and induce calcium
transfer between ER and mitochondria (13). In the present study, a
mitochondrial tagged calcium sensor vector was used to monitor
calcium levels in mitochondria and it was indicated that
mitochondrial calcium levels were significantly elevated after OGD
(Fig. 3A). It was then examined
whether mitochondrial overload was related to abnormal GRP75
expression. Consistent with the present hypothesis, GRP75 was
significantly elevated after OGD (Fig. 3B and C). This indicated that GRP75
may be related to OGD-induced mitochondrial calcium overload. The
expression of GRP75 in the infarction area of the MCAO model was
also examined, suggesting that GRP75 was also elevated in
vivo (Fig. 3D). HIF1α is one
of the most important modulators of hypoxia and the expression of
HIF1α was thus determined; the results indicated that HIF1α was
upregulated (Fig. 3E). Therefore,
it may be hypothesized that the increase of HIF1α induced the
upregulation of GRP75.
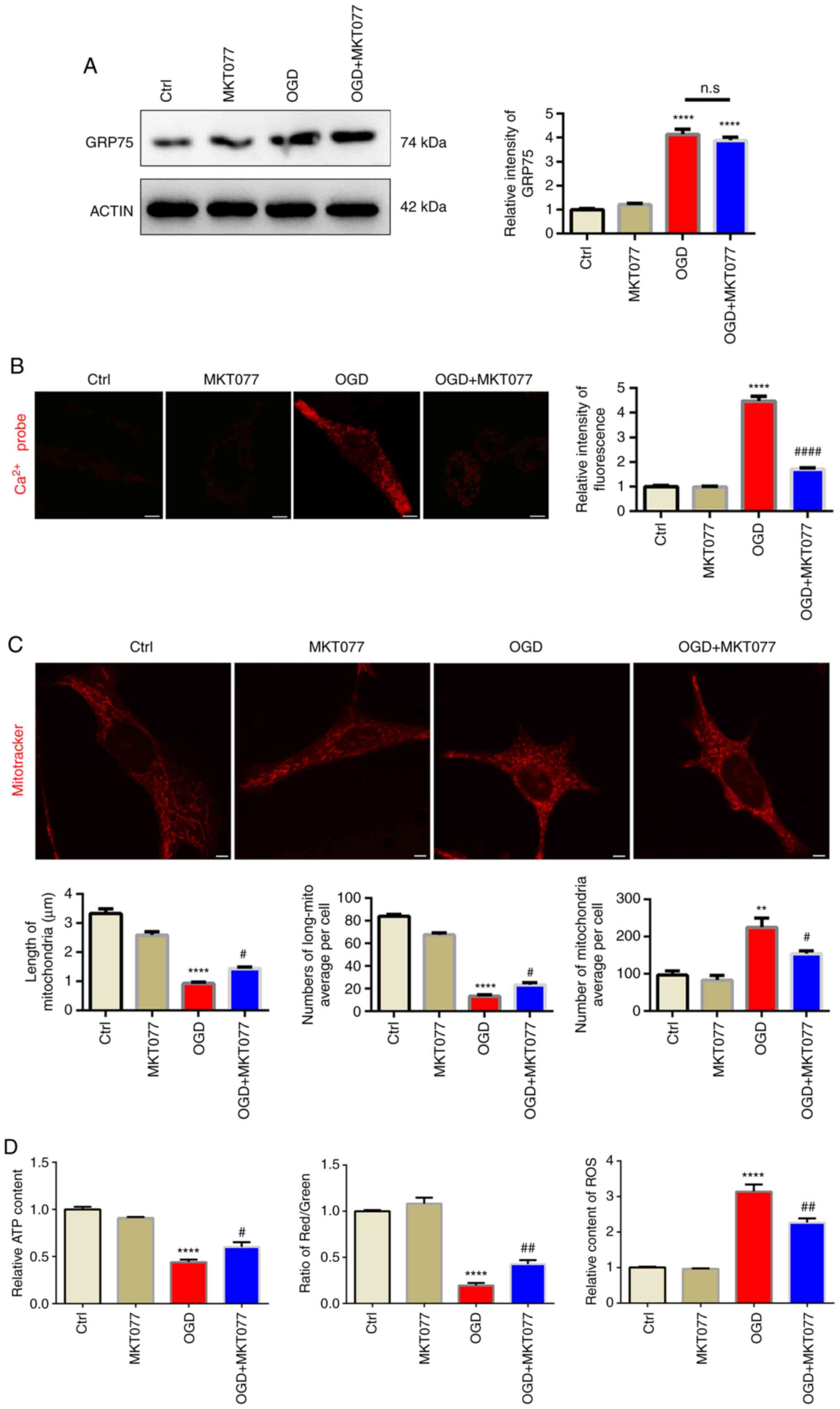
Inhibition of GRP75 by pharmacological
agent preserves mitochondrial morphology and function under
OGD
After confirming abnormal elevation of GRP75 after
OGD, it was investigated whether interfering with GRP75 is an
effective strategy to protect mitochondrial function after OGD.
MKT077, a GRP75-specific inhibitor (20), which is able to bind to GRP75 and
abrogate its activity (21), was
applied and it was observed that, although MKT077 did not
downregulate GRP75 expression after OGD (Fig. 4A), mitochondrial calcium overload
was obviously inhibited after MKT077 treatment (Fig. 4B), which indicated that inhibiting
GRP75 function prevented mitochondrial calcium overload. As
expected, mitochondrial morphology was well-preserved by MKT077
even after OGD (Fig. 4C). This
further supported the notion that mitochondrial calcium overload
was able to trigger mitochondrial fragmentation and induce
mitochondrial dysfunction (Figs.
4D, S1 and S2).
Knockdown of GRP75 preserves
mitochondrial morphology and function under OGD
In order to exclude any possible off-target effects
of the small molecule antagonist, siRNA targeting at GRP75 was used
to further confirm its effect on mitochondrial overload. siGRP75
efficiently reduced GRP75 expression levels after OGD, while si-NC
did not influence the expression of GRP75 (Figs. 5A and S3). Similar to the effect of MKT077,
siGRP75 was also able to reduce mitochondrial calcium overload
(Fig. 5B) and preserve
mitochondrial morphology and function after OGD (Figs. 5C and D, S1 and S2), which further confirmed that
inhibiting GRP75 preserved mitochondrial morphology and
function.
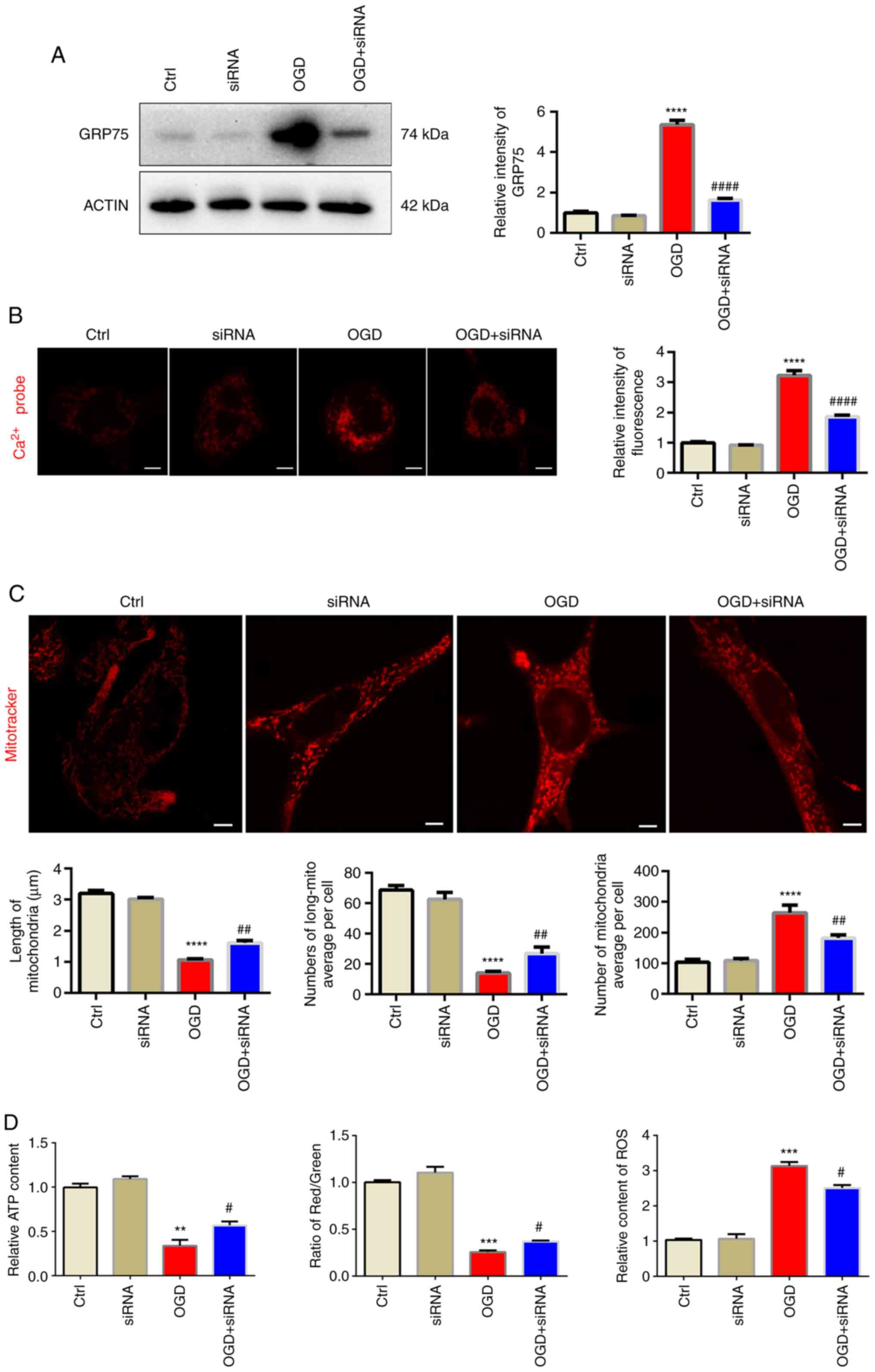 | Figure 5.Knockdown of GRP75 preserves
mitochondrial morphology and function under OGD. (A) Western blot
analysis confirmed siRNA-mediated knockdown of GRP75 expression in
N2a cells. (B) Treatment with siRNA targeting GRP75 to inhibit
GRP75 mitigated the OGD-stimulated calcium overload in the
mitochondria of N2a cells. (C) Fluorescent imaging analysis of
mitochondrial morphology of N2a cells (scale bars, 5 µm). The
average length of mitochondria, number of long mitochondria per
cell and average number of mitochondria per cell were determined.
(D) Mitochondrial function in N2a cells from the different groups;
the relative ATP content (left panel), mitochondrial membrane
potential (relative to red/green fluorescence, middle panel) and
relative content of ROS (right panel) were determined.
Representative images are provided and quantitative data are
expressed as the mean ± standard error of the mean of each group
from three separate experiments. **P<0.01, ***P<0.001,
****P<0.0001 vs. Ctrl; #P<0.05,
##P<0.01, ####P<0.0001 vs. OGD group.
Ctrl, control; OGD, oxygen-glucose deprivation; GRP75, 75 kDa
glucose-regulated protein; siRNA, small interfering RNA; long-mito,
long mitochondria with a length/diameter ratio of >2; ROS,
reactive oxygen species. |
Discussion
As mitochondria are the major energy-producing
organelles, their normal function is vital to cell survival,
particularly to cells with high energy demand, such as neurons or
cardiomyocytes (22). In the
present study, it was indicated that mitochondrial dysfunction took
place both in vivo and in vitro under hypoxia stress.
GRP75 was significantly upregulated by MCAO in vivo or OGD
in vitro, and by inhibiting GRP75 via antagonist or siRNA,
it was further confirmed that GRP75 was key to mitochondrial
overload triggered by hypoxia. The present study supports the idea
that GRP75 may be a promising target in treating ischemic stroke in
future research.
Ischemic stroke affects the quality of life of
patients. Neurons are vulnerable to lack of oxygen (23), which may cause dysfunction of
mitochondria, the power factory of the cell, leading to various
neuronal disturbances. Qadri et al (24) reported that mitochondrial
dysfunction was one of the important causes of Parkinson's disease.
Zhao et al (25) indicated
that after MCAO treatment, the expression of mitochondrial fission
protein increased while the expression of mitochondrial fusion
protein decreased, indicating that mitochondrial fragmentation
occurred in the MCAO model, resulting in mitochondrial dysfunction
and affecting the activity of neurons. Therefore, it is essential
to maintain normal function of mitochondria to protect neurons.
GRP75 belongs to the mammalian HSP70 family, which
is highly conserved and has various cellular functions, including
cell survival (26), the cell
cycle and apoptosis (27). It can
help maintain intracellular homeostasis under various stresses,
including hypoxia (28). However,
GRP75 has another important cellular function as a ‘molecular
bridge’ between IP3R and VDAC to form a calcium channel between the
ER and mitochondrion. Although calcium is required by normal
mitochondrial oxidative phosphorylation, excessive calcium disturbs
this process and trigger more ROS production (29). Calcium overload is thought to be
an important pathological process during several ischemic diseases
such as stroke and myocardial infarction, under which circumstances
excessive calcium floods into the mitochondrion through different
calcium channels, leading to mitochondrial dysfunction (30). In the present study, it was
indicated that under the conditions of OGD, the expression of GRP75
in neurons increased significantly and the mitochondria appeared to
have calcium overload. This indicates that calcium flooded into the
mitochondria. GRP75 is an important component in the OGD model,
resulting in mitochondrial calcium overload and mitochondrial
damage. Furthermore, evidence was provided that inhibiting GRP75
via MKT077 or direct knockdown of GRP75 alleviated OGD induced
mitochondrial calcium overload and preserved cell viability.
However, as a member of the HSP family, GRP75 also has various
physiological functions, such as iron-sulfur cluster assembly,
protein refolding (31) and
erythrocyte differentiation (32). Considering such important
physiological functions of GRP75, how to titer optimized dosage of
GRP75 inhibition under different stress conditions may be a
challenge and this is the obvious weakness of inhibiting GRP75 and
requires further investigation. In conclusion, GRP75 may be a
promising target but requires further investigation.
MKT-077 is a rhodacyanine dye and also an HSP70
inhibitor, which exhibits significant antitumor activity (33). A previous study by our group
indicated that MKT077 did not reduce the expression of GRP75 but
inhibited the GRP75-mediated ER-mitochondrial calcium exchange
(16). Miyata et al
(34) reported that a derivative
of MKT077 had blood-brain-barrier permeability properties and
reduced Tau levels in the brain. Thus, further studies by our group
will use YM-08 (a derivative of MKT077) for intragastric
administration of mice to observe its therapeutic effect. This
noteworthy pharmacological evolution of MKT077 provided evidence
that GRP75 was able to regulate cellular processes more than
maintaining calcium homeostasis. However, concerns about off-target
effects of small-molecule antagonists led us to use siRNA to more
precisely interfere with GRP75 and the results further confirmed
the function of GRP75 in the pathophysiological process of ischemic
stroke.
However, there are several limitations of the
present study. Mitochondrial function may only be roughly evaluated
via the MMP, as well as ROS and ATP production. Furthermore,
mitochondrial calcium was only determined in N2a cells, while it
was not possible to measure calcium ions in the mitochondria of
HT22 cells due to issues with the transfection of the calcium probe
plasmid. In addition, the mechanism by which ischemic stress
increases GRP75 expression remains elusive. Under hypoxia
circumstances, HIF1α is rapidly upregulated and is able to regulate
the expression of various genes. It was reported that HIF1α was
able to regulate the expression of GRP75 (35); hence, it may be the mechanism of
GRP75 upregulation by MCAO or OGD in the present study.
In conclusion, the present study provided evidence
that GRP75 is an important factor in hypoxia-induced neuron injury
both in vivo and in vitro. GRP75 was able to augment
mitochondria-associated endoplasmic reticulum membranes formation
and lead to mitochondrial calcium overload, ultimately leading to
mitochondrial dysfunction and disruption of energy supply to
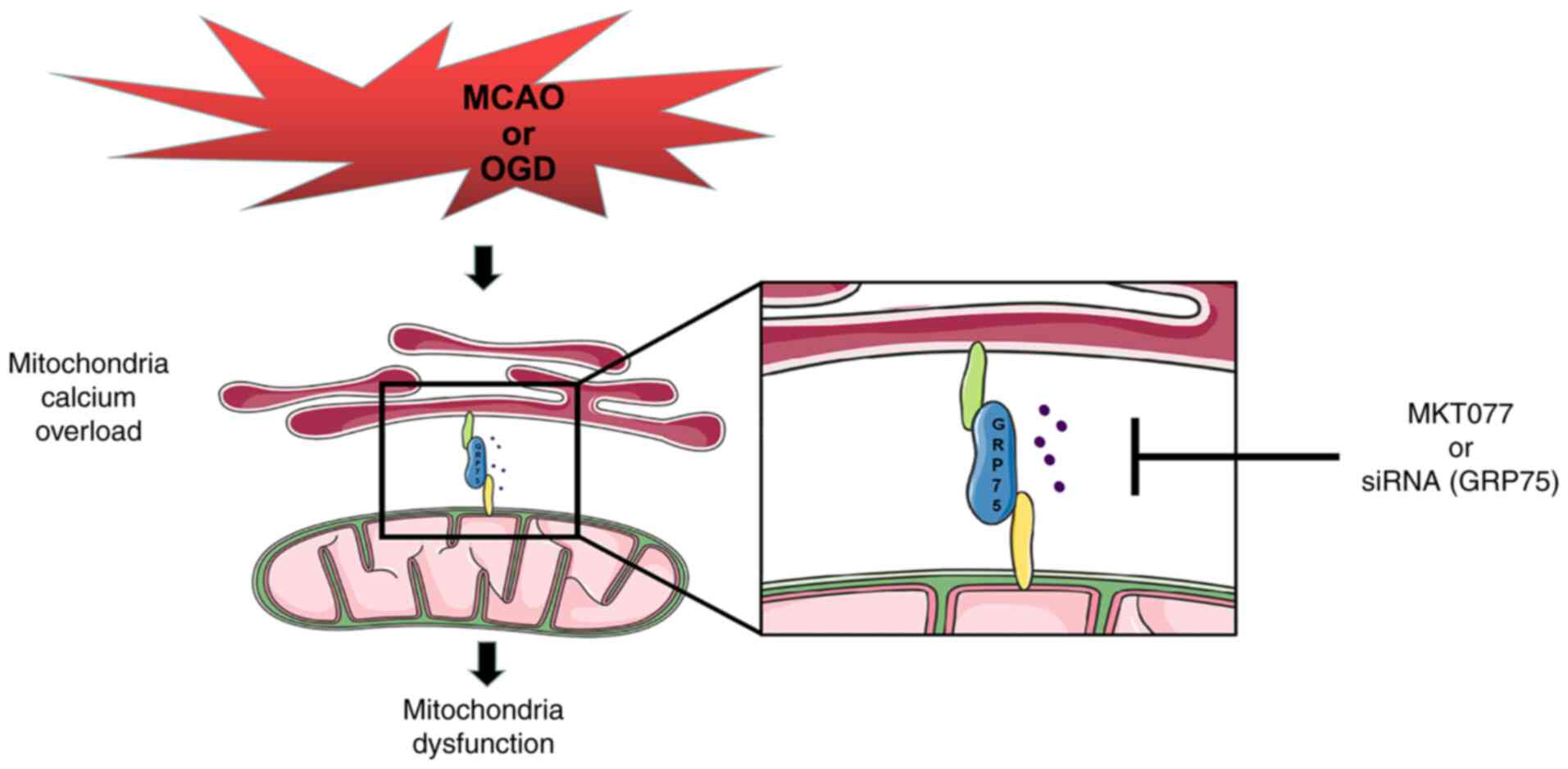
neurons, making neurons vulnerable to hypoxia (Fig. 6). Inhibiting GRP75 may be a
promising strategy to ameliorate injury following ischemic stroke
in future research.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science
Foundation of China (grant no. 81873725 to JC) and Hubei Province's
Outstanding, Medical Academic Leader program (to BH).
Availability of data and materials
The raw data and materials generated and used during
this study are available from the corresponding author upon
reasonable request.
Author's contributions
BW drafted the manuscript. BW and KX established the
cell culture and animal model. JiC and RH performed the western
blotting analysis and confocal microscopy. TJ and JW performed
biochemical measurements. BH and JuC participated in the design and
preparation of the manuscript, as well as conceived the study and
participated in the coordination and preparation of the manuscript.
All authors read and approved the final version of the manuscript
and confirmed the authenticity of the raw data.
Ethics approval and consent to
participate
The animal experiments were approved by the Animal
Care and Use Committee at the Huazhong University of Science and
Technology (Wuhan, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Paul S and Candelario-Jalil E: Emerging
neuroprotective strategies for the treatment of ischemic stroke: An
overview of clinical and preclinical studies. Exp Neurol.
335:1135182021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Su XT, Wang L, Ma SM, Cao Y, Yang NN, Lin
LL, Fisher M, Yang JW and Liu CZ: Mechanisms of acupuncture in the
regulation of oxidative stress in treating ischemic stroke. Oxid
Med Cell Longev. Oct 24–2020.(Epub ahead of print). View Article : Google Scholar
|
|
3
|
Ranjbar Taklimie F, Gasterich N, Scheld M,
Weiskirchen R, Beyer C, Clarner T and Zendedel A: Hypoxia induces
astrocyte-derived Lipocalin-2 in ischemic stroke. Int J Mol Sci.
20:12712019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lv J, Guan W, You Q, Deng L, Zhu Y, Guo K,
Gao X, Kong J and Yang C: RIPC provides neuroprotection against
ischemic stroke by suppressing apoptosis via the mitochondrial
pathway. Sci Rep. 10:53612020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gao CL, Hou GG, Liu J, Ru T, Xu YZ, Zhao
SY, Ye H, Zhang LY, Chen KX, Guo YW, et al: Synthesis and target
identification of benzoxepane derivatives as potential
anti-neuroinflammatory agents for ischemic stroke. Angew Chem Int
Ed Engl. 59:2429–2439. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Alim I, Caulfield JT, Chen Y, Swarup V,
Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie
EJ, et al: Selenium drives a transcriptional adaptive program to
block ferroptosis and treat stroke. Cell. 177:1262–1279.e1225.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guadagno JV, Jones PS, Fryer TD, Barret O,
Aigbirhio FI, Carpenter TA, Price CJ, Gillard JH, Warburton EA and
Baron JC: Local relationships between restricted water diffusion
and oxygen consumption in the ischemic human brain. Stroke.
37:1741–1748. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li C, Sun H, Xu G, McCarter KD, Li J and
Mayhan WG: Mito-tempo prevents nicotine-induced exacerbation of
ischemic brain damage. J Appl Physiol (1985). 125:49–57. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rose J, Brian C, Woods J, Pappa A,
Panayiotidis MI, Powers R and Franco R: Mitochondrial dysfunction
in glial cells: Implications for neuronal homeostasis and survival.
Toxicology. 391:109–115. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mondal NK, Behera J, Kelly KE, George AK,
Tyagi PK and Tyagi N: Tetrahydrocurcumin epigenetically mitigates
mitochondrial dysfunction in brain vasculature during ischemic
stroke. Neurochem Int. 122:120–138. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao D, Sun Y, Tan Y, Zhang Z, Hou Z, Gao
C, Feng P, Zhang X, Yi W and Gao F: Short-duration swimming
exercise after myocardial infarction attenuates cardiac dysfunction
and regulates mitochondrial quality control in aged mice. Oxid Med
Cell Longev. 2018:40790412018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Galvan DL, Green NH and Danesh FR: The
hallmarks of mitochondrial dysfunction in chronic kidney disease.
Kidney Int. 92:1051–1057. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thoudam T, Ha CM, Leem J, Chanda D, Park
JS, Kim HJ, Jeon JH, Choi YK, Liangpunsakul S, Huh YH, et al: PDK4
augments ER-mitochondria contact to dampen skeletal muscle insulin
signaling during obesity. Diabetes. 68:571–586. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jiang J, Dai J and Cui H: Vitexin reverses
the autophagy dysfunction to attenuate MCAO-induced cerebral
ischemic stroke via mTOR/Ulk1 pathway. Biomed Pharmacother.
99:583–590. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Smith HK, Russell JM, Granger DN and
Gavins FN: Critical differences between two classical surgical
approaches for middle cerebral artery occlusion-induced stroke in
mice. J Neurosci Methods. 249:99–105. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Y, Liu X, Bai J, Tian X, Zhao X, Liu
W, Duan X, Shang W, Fan HY and Tong C: Mitoguardin regulates
mitochondrial fusion through MitoPLD and is required for neuronal
homeostasis. Mol Cell. 61:111–124. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liang T, Hang W and Chen J, Wu Y, Wen B,
Xu K, Ding B and Chen J: ApoE4 (Δ272-299) induces
mitochondrial-associated membrane formation and mitochondrial
impairment by enhancing GRP75-modulated mitochondrial calcium
overload in neuron. Cell Biosci. 11:502021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Knauss S, Albrecht C, Dirnagl U, Mueller
S, Harms C, Hoffmann CJ, Koch SP, Endres M and Boehm-Sturm P: A
semiquantitative non-invasive measurement of PcomA patency in
C57BL/6 mice explains variance in ischemic brain damage in filament
MCAO. Front Neurosci. 14:5767412020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seo SJ, Yoon SH and Do JT: Mitochondrial
dynamics in stem cells and differentiation. Int J Mol Sci.
19:38932018. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu PK, Hong SK, Starenki D, Oshima K, Shao
H, Gestwicki JE, Tsai S and Park JI: Mortalin/HSPA9 targeting
selectively induces KRAS tumor cell death by perturbing
mitochondrial membrane permeability. Oncogene. 39:4257–4270. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rousaki A, Miyata Y, Jinwal UK, Dickey CA,
Gestwicki JE and Zuiderweg ER: Allosteric drugs: The interaction of
antitumor compound MKT-077 with human Hsp70 chaperones. J Mol Biol.
411:614–632. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Escobar-Henriques M and Joaquim M:
Mitofusins: Disease gatekeepers and hubs in mitochondrial quality
control by E3 Ligases. Front Physiol. 10:5172019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Head E, Rofina J and Zicker S: Oxidative
stress, aging, and central nervous system disease in the canine
model of human brain aging. Vet Clin North Am Small Anim Pract.
38:167–178. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Qadri R, Goyal V, Behari M, Subramanian A,
Datta SK and Mukhopadhyay AK: Alteration of mitochondrial function
in oxidative stress in parkinsonian neurodegeneration: A
cross-sectional study. Ann Indian Acad Neurol. 24:506–512.
2021.PubMed/NCBI
|
|
25
|
Zhao J, Dong L, Huo T, Cheng J, Li X,
Huangfu X, Sun S, Wang H and Li L: O-GlcNAc Transferase (OGT)
protects cerebral neurons from death during Ischemia/Reperfusion
(I/R) injury by modulating Drp1 in mice. Neuromolecular Med. Oct
27–2021.(Epub ahead of print). View Article : Google Scholar
|
|
26
|
Starenki D, Sosonkina N, Hong SK, Lloyd RV
and Park JI: Mortalin (GRP75/HSPA9) promotes survival and
proliferation of thyroid carcinoma cells. Int J Mol Sci.
20:20692019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang G, Han M, Wang X and Xiao A: GRP75
involves in retinal ganglion cell apoptosis after rat optic nerve
crush. J Mol Neurosci. 56:422–430. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
D'Eletto M, Rossin F, Occhigrossi L,
Farrace MG, Faccenda D, Desai R, Marchi S, Refolo G, Falasca L,
Antonioli M, et al: Piacentini, transglutaminase type 2 regulates
ER-mitochondria contact sites by interacting with GRP75. Cell Rep.
25:3573–3581. e35742018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–C833. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Verma M, Wills Z and Chu CT: Excitatory
dendritic mitochondrial calcium toxicity: Implications for
Parkinson's and other neurodegenerative diseases. Front Neurosci.
12:5232018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Shan Y and Cortopassi G: Mitochondrial
Hspa9/Mortalin regulates erythroid differentiation via iron-sulfur
cluster assembly. Mitochondrion. 26:94–103. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen TH, Kambal A, Krysiak K, Walshauser
MA, Raju G, Tibbitts JF and Walter MJ: Knockdown of Hspa9, a
del(5q31.2) gene, results in a decrease in hematopoietic
progenitors in mice. Blood. 117:1530–1539. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Srinivasan SR, Connarn J, Ahmad A,
Young ZT, Kabza AM, Zuiderweg ER, Sun D and Gestwicki JE: Analogs
of the allosteric heat shock protein 70 (Hsp70) inhibitor, MKT-077,
as anti-cancer agents. ACS Med Chem Lett. 4:1042–1047. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miyata Y, Li X, Lee HF, Jinwal UK,
Srinivasan SR, Seguin SP, Young ZT, Brodsky JL, Dickey CA, Sun D
and Gestwicki JE: Synthesis and initial evaluation of YM-08, a
blood-brain barrier permeable derivative of the heat shock protein
70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem
Neurosci. 4:930–939. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Mylonis I, Kourti M, Samiotaki M,
Panayotou G and Simos G: Mortalin-mediated and ERK-controlled
targeting of HIF-1α to mitochondria confers resistance to apoptosis
under hypoxia. J Cell Sci. 130:466–479. 2017.PubMed/NCBI
|