Introduction
Tuberculosis (TB) imposes an important health burden
worldwide. The 2020 Global World Health Organization TB report
stated that an estimated 10 million people fell ill with TB in 2019
globally (1). Tracheobronchial TB
(TBTB) is defined as Mycobacterium tuberculosis (Mtb)
infection of the tracheobronchial tree. Although TB affects all age
groups, the highest burden is in adult men. Indeed, adult men
accounted for 56% of all cases in 2019, compared with 32% in adult
women and 12% in children (1).
However, previous studies have reported that TBTB is most common in
young and middle-aged females (2–4).
However, the reasons for this are unclear. Estradiol levels are
significantly augmented in patients with TB compared with healthy
controls (5,6). In addition to its effects on sexual
differentiation and reproduction, estrogen may regulate autophagy
by affecting the production of reactive oxygen species (ROS)
(7,8). In certain cases, estrogen serves a
role in promoting autophagy (9),
but on other occasions, when cellular autophagy is stimulated by
hypoxia or lipopolysaccharides, estrogen shows a restrictive effect
on the expression of genes associated with autophagy (10,11). Previous studies have reported that
ER modulators suppress intracellular Mtb growth by enhancing
autophagy in infected macrophages (7). Therefore, we hypothesized that
estrogen may serve an important role in the occurrence and
development of TBTB.
Epithelial cells are known to contribute to the
immune responses in lungs and can sense intra- and extracellular
pathogens (12). In vitro
studies have demonstrated that primary human airway epithelial
cells and the type-II alveolar cell line A549 are capable of
internalizing Mtb, although at a slower rate than macrophages
(13,14) Bacteria have been shown to
replicate extensively inside A549 cells. The replicated bacteria
may then escape from the cell after bacteria-induced apoptosis or
necrosis, which facilitates systemic dissemination (15,16). Autophagy also protects type-II
alveolar epithelial cells, which are non-phagocytic cells, from Mtb
infection and is not conducive to the spread of Mtb inside the
cells (17). However, a previous
study found that blocking autophagy using 3-methyladenine is
advantageous to the infected A549 cells and inhibits bacterial
replication and results in a significant decrease in bacterial
viability compared with the untreated infected A549 cells (18). Therefore, it may be hypothesized
that estradiol could serve a key role in the development of TBTB by
affecting the intracellular Mtb proliferation and the autophagy of
Mtb-infected bronchial epithelial cells.
To address this question, granulomatous tissue from
patients with TBTB was collected to determine the expression of
estrogen receptor (ER). Mtb is already known to be capable of
infecting alveolar epithelial cells. However, whether Mtb could
infect bronchial epithelial cells, including ciliated cells, goblet
cells, basal cells and secretory cells, remains to be elucidated.
Therefore, whether Mtb could infect above bronchial epithelial
cells was determined. Additionally, a model that mimics the
bronchial epithelial cell-Mtb interaction was employed using the
human bronchial epithelial cell line 16HBE infected with Mtb and
treating the 16HBE-infected cells with estradiol at different
concentrations in vitro.
Materials and methods
Patients and tissue collection
Three lobar bronchial granulomatous tissues of 1
male and 2 female patients (age, 21–31 years) with lobar bronchial
TBTB (the etiology test of these tissues was positive for Mtb) were
obtained between March and June 2021 from the First Affiliated
Hospital of Chongqing Medical University (Chongqing, China). For
inclusion in the study patients needed to be confirmed to have TBTB
and have positive etiology test of biopsy tissue for Mtb. Patients
who was unable to tolerate bronchoscopy were excluded. Specimens
for immunofluorescence analysis were fixed with 4% formaldehyde at
4°C for 24 h and embedded in paraffin wax. The present study was
approved by the Ethics Committee of The First Affiliated Hospital
of Chongqing Medical University (approval no. 20188502; Chongqing,
China). Written informed consent was obtained from all
patients.
Bacterial culture
The Mtb H37Rv and Bacillus Calmette-Guerin
(BCG) strains were donated by Beijing Chest Hospital, Capital
Medical University. For in vitro cell infection, frozen Mtb
(strain H37Rv) or BCG was inoculated on Löwenstein-Jensen culture
medium (Baso Diagnostic, Inc.) at 37°C until colony formation.
Subsequently the cultures were transferred to bottles containing 60
ml Middlebrook 7H9 (BD Biosciences) culture medium supplemented
with 10% Middlebrook albumin dextrose catalase (BD Biosciences) and
0.1 mM calcium chloride (Beijing Solarbio Science & Technology
Co., Ltd.). Cultures were incubated in an orbital shaker at 5 × g
at 37°C for 7–12 days until they reached McFarland turbidity
standard no. 5 (Shenzhen Kangtai Biological Products). Bacterial
concentrations were then estimated according to the McFarland
turbidity standard (19).
Cell preparation
The human bronchial epithelial cell line 16HBE
(Shanghai Fuheng Biotechnology, Co., Ltd.) was grown in complete
RPMI-1640 medium (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% heat-inactivated fetal bovine serum (SORFA
Life Science; Beijing Shuohua Biotech Co., Ltd.) at 37°C in 5%
CO2.
Infection with Mtb and stimulation of
cells with estradiol
Estradiol (Sigma-Aldrich; Merck KGaA) was diluted in
DMSO (concentration 10−2 M) and stored in the
refrigerator at −80°C. Before the experiment, estradiol was diluted
to 10−4 M and 10−6 M using PBS. Then 20 µl
estradiol at 10−4 M and 10−6 M were added to
cells containing 2ml medium to ensure final concentrations of
estradiol were 10−6 M (E2) and 10−8 M (E1).
Bacteria growing on the 7H9 broth were centrifuged at 400 × g at
room temperature for 10 min and resuspended in PBS. Bacterial
concentrations were then estimated according to the McFarland
turbidity standard (19). To
evaluate whether different concentrations of estradiol affect the
growth of intracellular Mtb, 16HBE cells were infected with Mtb at
a multiplicity of infection (MOI) of 10:1 at 37°C in 5%
CO2 for 24 h in the absence of estradiol and cells were
then washed four times with PBS to remove extracellular bacilli.
Infected 16HBE cells were incubated at 37°C in 5% CO2
for 1 h (day 0), 24 h (day 1) and 72 h (day 3) in the presence or
absence of estradiol. Mtb-infected cells were lysed with 1% SDS
followed by 20% BSA (Sigma-Aldrich; Merck KGaA), serially diluted
10–104 times in Middlebrook 7H9 medium, and plated in
triplicate on Löwenstein-Jensen culture medium at 37°C for 4–6
weeks. The number of colony-forming units (CFUs) were manually
quantified.
Immunofluorescence (IF)
According to the methods of Hui et al
(20), 5-mm-thick sections were
deparaffinized in xylene followed by rehydration using an ethanol
gradient. Sections were subjected to antigen retrieval using 1X
citric acid antigen repair solution (Wuhan Boster Biological
Technology, Ltd.), which was heated with high fire for 8 min and
medium fire for 8 min. Membrane disruption was performed using
0.03% Triton X-100 for 30 min at room temperature. The sections
were then blocked with 5% BSA for 20 min at room temperature. Mouse
anti-human p63 (1:200; cat. no. NBP3-07430; Novus Biologicals,
Ltd.), mouse anti-human secretoglobin 1A member 1 (SCGB1A1; 1:500;
cat. no. H00007356-M01; Novus Biologicals, Ltd.), mouse anti-human
mucin 5AC (MUC5AC; 1:200; cat. no. ab3649; Abcam), mouse anti-human
forkhead box J1 (FOXJ1; 1:500; cat. no. 14-9965-82; Thermo Fisher
Scientific, Inc.) or rabbit anti-Mtb antibody (1:100; cat. no.
ab905; Abcam) was added overnight at 4°C. FITC-labeled goat
anti-mouse IgG secondary antibody (1:200; cat. no. E031210;
EarthOx; Beijing Canlife Technology Co.) was added and smears were
incubated at 37°C for 50 min. Cy3-labeled goat anti-rabbit IgG
secondary antibody (1:200; cat. no. E031620; EarthOx; Beijing
Canlife Technology Co.) was simultaneously added and smears were
incubated at 37°C for 50 min. Sections and 16HBE cells on
coverslips were fixed, permeabilized and blocked with 5% BSA at
37°C for 20 min. Mouse anti-human ERα (1:500; cat. no. NBP2-61764;
Novus Biologicals, Ltd.) and mouse anti-human ERβ (1:500; cat. no.
NBP2-44366; Novus Biologicals, Ltd.) monoclonal antibodies were
added and incubated overnight at 4°C. Cy3-labeled goat anti-mouse
IgG secondary antibody (1:200; cat. no. E031610-02; EarthOx;
Beijing Canlife Technology Co.) was added and incubated at 37°C for
50 min. DAPI solution was added and incubated at room temperature
for 5 min. An appropriate amount of anti-fluorescence quenching
medium was dropped on the tissue, which was then observed under a
fluorescence microscope (Olympus Corporation).
Transmission electron microscopy
(TEM)
According to the methods of Hui et al
(20), for transmission electron
microscopy examination, 16HBE cells were infected with Mtb at a MOI
of 10:1 for 24 h in the absence of estradiol and cells were then
washed four times with PBS to remove extracellular bacilli.
Infected 16HBE cells were then treated with different
concentrations of estradiol at 37°C for 24 h and were centrifuged
at 410 × g at room temperature for 5 min. Cells were fixed in 1%
glutaraldehyde dissolved in 0.1 M cacodylate buffer (pH 7.0) at 4°C
for 24 h, postfixed in 2% osmium tetroxide at room temperature for
24 h, dehydrated with increasing concentrations of ethanol and
gradually infiltrated with Epon resin (Ted Pella, Inc.) at room
temperature for 2 h. Thin sections (60 nm) were contrasted with 90%
uranyl acetate at 37°C for 30 min and 90% lead citrate at 45°C for
10 min. Finally, thin sections were observed using a transmission
electron microscope JEOL JEM-1400PLUS (JEOL, Ltd.), which was
equipped with DigitalMicrograph 3.9 (Gatan, Inc.) image management
software.
Necrosis assays
16HBE cells at 80–90% confluence in 24-well dishes
were infected with Mtb at MOI=10:1. The infected cells were
designated as Mtb cells and divided into the following five groups:
i) Untreated infection control group (Mtb group); and infected
cells treated with ii) 3-Methyladenine (3-MA; 10 mM; Sigma-Aldrich;
Merck KGaA); iii) rapamycin (RAPA; 100 nM; Sigma-Aldrich; Merck
KGaA); or estradiol at iv) E1; and v) E2. Uninfected and untreated
cells were used as negative controls (PBS group). Uninfected cells
treated with 3-MA, RAPA or estradiol at E1 and E2 were used as the
treatment control. After 24 h, the cells were assessed for necrosis
by incubation with 0.5 ml PBS and propidium iodide (PI) (Shanghai
Biyuntian Biotechnology, Co., Ltd.) at 37°C for 20 min, followed by
observation them under a fluorescence microscope (Olympus
Corporation) according to the manufacturer's instructions. Cells
were harvested at 6, 24 and 72 h to measure the level of released
lactate dehydrogenase (LDH) in the medium using the
LDH-Cytotoxicity Assay Kit II (cat. no. C0017; Shanghai Biyuntian
Biotechnology, Co., Ltd.). The LDH reaction results were determined
by reading the absorbance at 490 nm using a microplate reader
(iMark; Bio-Rad Laboratories, Inc.).
Western blot analysis
Infected 16HBE cells (2.75×106 cells)
were incubated with ERα-specific agonist [4, 4, 4,
4(4-propyl-[1H]-pyrazole-1, 3, 5-triyl) trisphenol (PPT);
10−6 M] or ERβ-specific agonist [diarylpropionitrile
(DPN); 10−6 M] at 37°C in 5% CO2 for 24 h. Infected
16HBE cells were pre-treated with ERα-specific antagonist (AZD9496;
10–6 M) or nonspecific ER modulator [Bazedoxifene (BAZ); 0.5 µM] at
37°C in 5% CO2 for 1 h and co-incubated with E2 at 37°C
in 5% CO2 for 24 h. ER agonists and antagonists were purchased from
Tocris Bioscience and Sigma-Aldrich; Merck KGaA. Stock solutions of
PPT, DPN, AZD9496 and BAZ were prepared using absolute ethyl
alcohol and serially diluted using PBS to the aforementioned
concentrations. Additionally, 3-MA (10 mM) and RAPA (100 nM) were
used as negative and positive controls. After 24 h, 16HBE cells
were washed with PBS and cells were lysed in RIPA lysis buffer
(Cell Signaling Technology, Inc.). The protein concentration in the
lysates was measured with a BCA protein kit (Beyotime Institute of
Biotechnology). The protein samples were boiled for 10 min. Total
protein (25 µg total protein/lane) was separated via SDS-PAGE on
4–20% gels (Nanjing ACE Biological Technology Co., Ltd.), then
transferred onto a PVDF membrane (MilliporeSigma). The membrane was
blocked with protein-free rapid blocking buffer (cat. no. PS108P;
Epizyme, Inc.) for 1 h at room temperature, then incubated with
antibodies against β-actin (1:1,000; cat. no. ab8227; Abcam), LC3B
(1:1,000; cat. no. ab192890; Abcam), beclin1 (1:1,000; cat. no.
ab207612; Abcam), P62 (1:500; cat. no. ab207305; Abcam), p-AKT
(1:500; cat. no. AF0016; Affinity Biosciences), AKT (1:500; cat.
no. AF6263; Affinity Biosciences), p-mTOR (1:500; cat. no. AF3308;
Affinity Biosciences) and mTOR (1:500; cat. no. AF6308; Affinity
Biosciences) at 4°C for 12 h. After washing for 30 min using TBS
with 0.1% Tween-20, the membrane was incubated for 2 h at room
temperature with either goat anti-mouse, HRP-linked antibody
(1:3,000; cat, no. 7076; Cell Signaling Technology, Inc.) or goat
anti-rabbit biotinylated antibody (1:3,000; cat. no. 14708; Cell
Signaling Technology, Inc.) secondary antibodies. The protein band
was visualized using an ECL detection solution (Thermo Fisher
Scientific, Inc.). The digital images of protein bands were
acquired using an ImageQuant LAS 4000 system (Cytiva).
Measurement of ROS
ROS can be detected using a fluorescence microplate
reader, laser scanning confocal microscope and flow cytometry. Due
to the infectivity of Mtb, the biosafety level of the laboratory
where the above three detection methods were located did not meet
the requirements. Only the biosafety level of the laboratory where
flow cytometry was located met the requirements of BCG experimental
biosafety. Therefore, ROS levels were determined using flow
cytometry using BCG instead of Mtb infected 16HBE. BCG-infected
16HBE cells (2.75×106) were incubated with PPT or DPN in
the absence of estradiol at 37°C in 5% CO2 for 24 h.
Infected 16HBE cells (2.75×106) were pre-treated with
AZD9496 or BAZ at 37°C in 5% CO2 for 1 h and
co-incubated with estradiol at 37°C in 5% CO2 for 24 h.
The cells were incubated with 2,7-dichlorodihydrofluorescein
diacetate (10 µM; Shanghai Biyuntian Biotechnology, Co., Ltd.) for
15 min at 37°C in the dark. Fluorescence intensity was measured
using flow cytometer FACS Aria II (BD Biosciences) and analyzed
using FlowJo software (version 6.1; Tree Star, Inc.) according to
the manufacturer's protocol.
Statistical analysis
With the exception of the TEM experiment, each in
vitro experiment was performed independently in triplicate.
After the emergence of COVID-19, due to the infectivity of Mtb, TEM
laboratory (Biosafety Level II Laboratory) was not allowed to
conduct Mtb related tests, but we repeated the test twice with BCG
instead of Mtb, the results were consistent with those of Mtb. The
data are presented as the mean ± SEM. Statistical comparisons were
performed using GraphPad Prism 8 (GraphPad Software, Inc.). One-way
ANOVA (followed by Dunnett's post hoc test) was used to analyze the
differences in ROS levels. Two-way ANOVA (followed by Tukey's post
hoc test) was used to analyze the difference in LDH release and CFU
between the groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
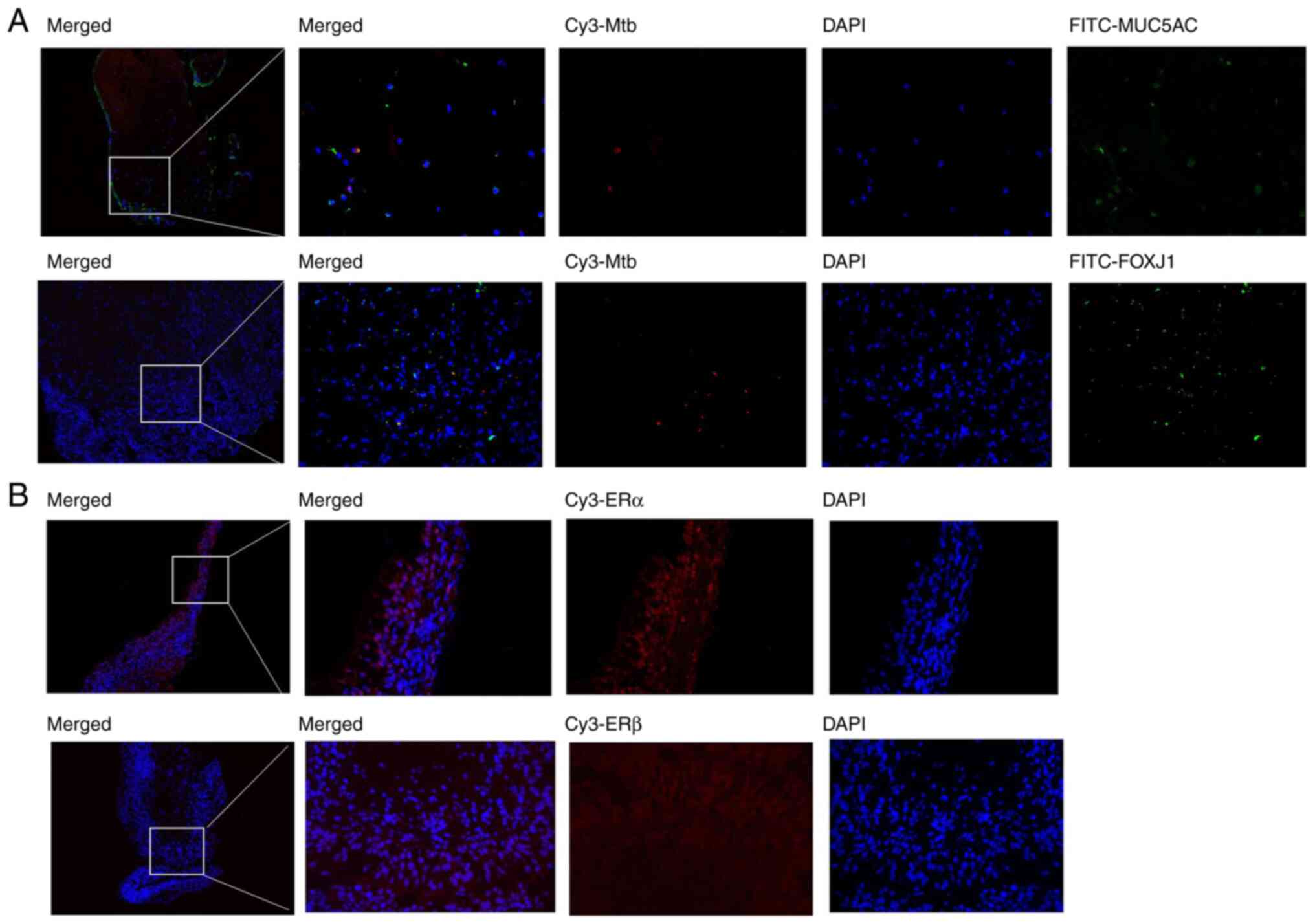
ERα is strongly expressed in
granulomatous tissue of patients with TBTB
It was known from previous studies that Mtb could
enter alveolar type II epithelial cells (21,22). To gain a better understanding in
whether Mtb could enter human bronchial epithelial cells, a
double-label IF analysis was conducted using an antibody against
Mtb combined with antibodies against each of the following markers:
p63 (data not shown), SCGB1A1 (data not shown), MUC5AC and FOXJ1,
commonly used to identify basal cells, secretory club cells, goblet
cells and ciliated cells, respectively. The IF analysis revealed
that Mtb could enter human lobar bronchial goblet cells and
ciliated cells in patients with TBTB (Fig. 1A). The IF analysis also revealed
that there were no Mtb infected basal cells (p63-marked) or Mtb
infected secretory club cells (SCGB1A1-marked) in granulomatous
tissue (data not shown). Clipping normal bronchial epithelial cells
from TBTB patients is likely to spread Mtb, which was not approved
by the ethics committee. Therefore, only collected granulomatous
tissue was used, which is mainly Langerhans cells. Consequently the
number of epithelial cells was very few and it was hard to find
Mtb-infected epithelial cells. Therefore, only a small number of
infected bronchial cells were observed. As aforementioned, estrogen
acts mainly by binding to ER. Therefore, the present study aimed to
elucidate whether ER was expressed in TBTB tissues and found that
only ERα was expressed in granulomatous tissues in patients with
TBTB. More specifically, ERα was expressed in lobar bronchial
epithelial cells (Fig. 1B).
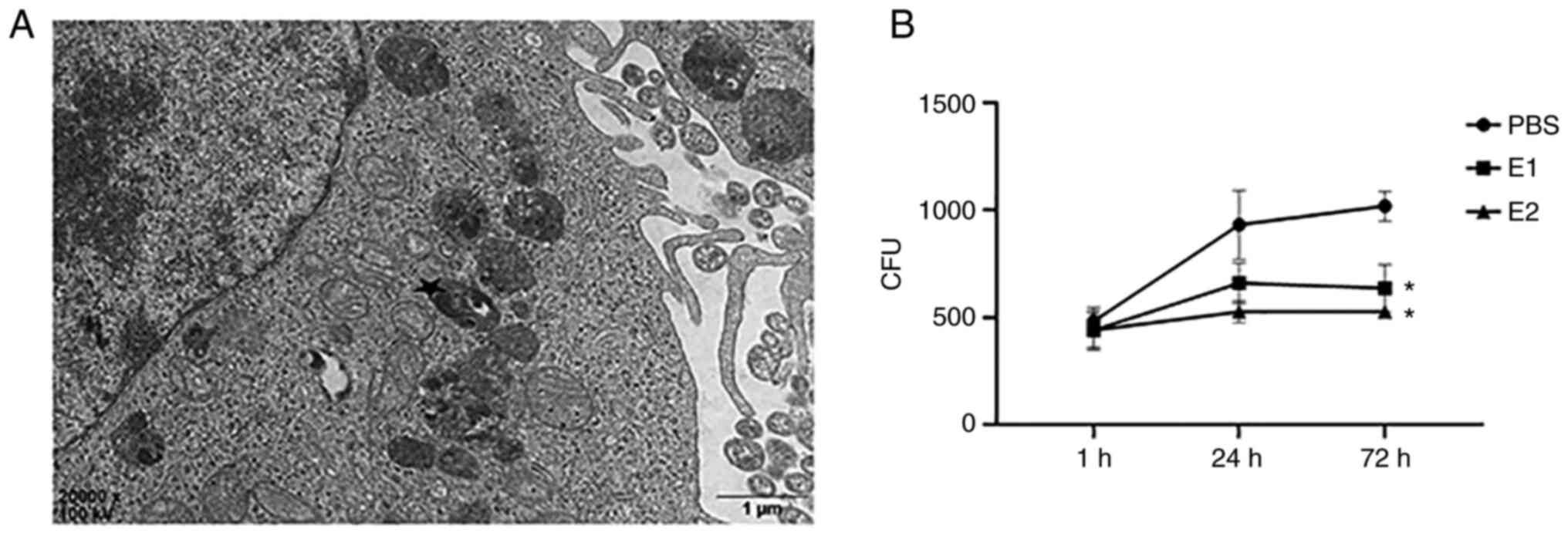
Estradiol decreases CFUs in 16HBE
cells as the time elapses
In order to study the role of estradiol in
Mtb-infected bronchial epithelial cells in vitro, a model
that mimics the bronchial epithelial cell-Mtb interaction was
established by treating with estradiol the 16HBE cells infected
with Mtb. Mtb was internalized into 16HBE cells, as indicated by
TEM (Fig. 2A). To further confirm
whether estradiol affects the intracellular Mtb growth, infected
16HBE cells were treated with estradiol at E1 and E2. The number of
CFUs did not differ between the groups at day 0. 16HBE cells
exposed to Mtb alone (PBS group) showed an increase in the number
of CFUs from day 0 to day 3. However, the differences were not
significant as the time elapsed in estradiol treated 16HBE cells
from day 0 to day 3 (Fig. 2B).
Compared with the PBS group, the number of CFUs in E1 and E2 groups
was significantly decreased at 72 h.
Estradiol impacts the proliferation of
intracellular Mtb and the survival of the infected host cells by
inhibiting autophagy
Fine et al (18) showed that the level of infected
cell necrosis and autophagy determines the proliferation of
intracellular Mtb. Therefore, the effect of estradiol on necrosis
and autophagy of infected cells was examined. LDH release
experiments and PI staining were conducted using the same infected
monolayers to determine whether bacterial viability was associated
with cell survival (Fig. 3A).
Cells treated with estradiol and infected with Mtb released less
LDH at 72 h and exhibited decreased necrosis at 24 h compared with
the untreated cells. Compared with the PBS group, Mtb-infected
16HBE cells in the E1 and E2 groups had markedly fewer
intracellular autophagolysosomes. Furthermore, compared with the
PBS group the protein expression levels of LC3B II/I and beclin1
significantly decreased, whereas P62 protein expression were
significantly increased in the E1 and E2 groups. Autophagy of 16HBE
cells infected with Mtb was inhibited following treatment with
estradiol (Fig. 3B).
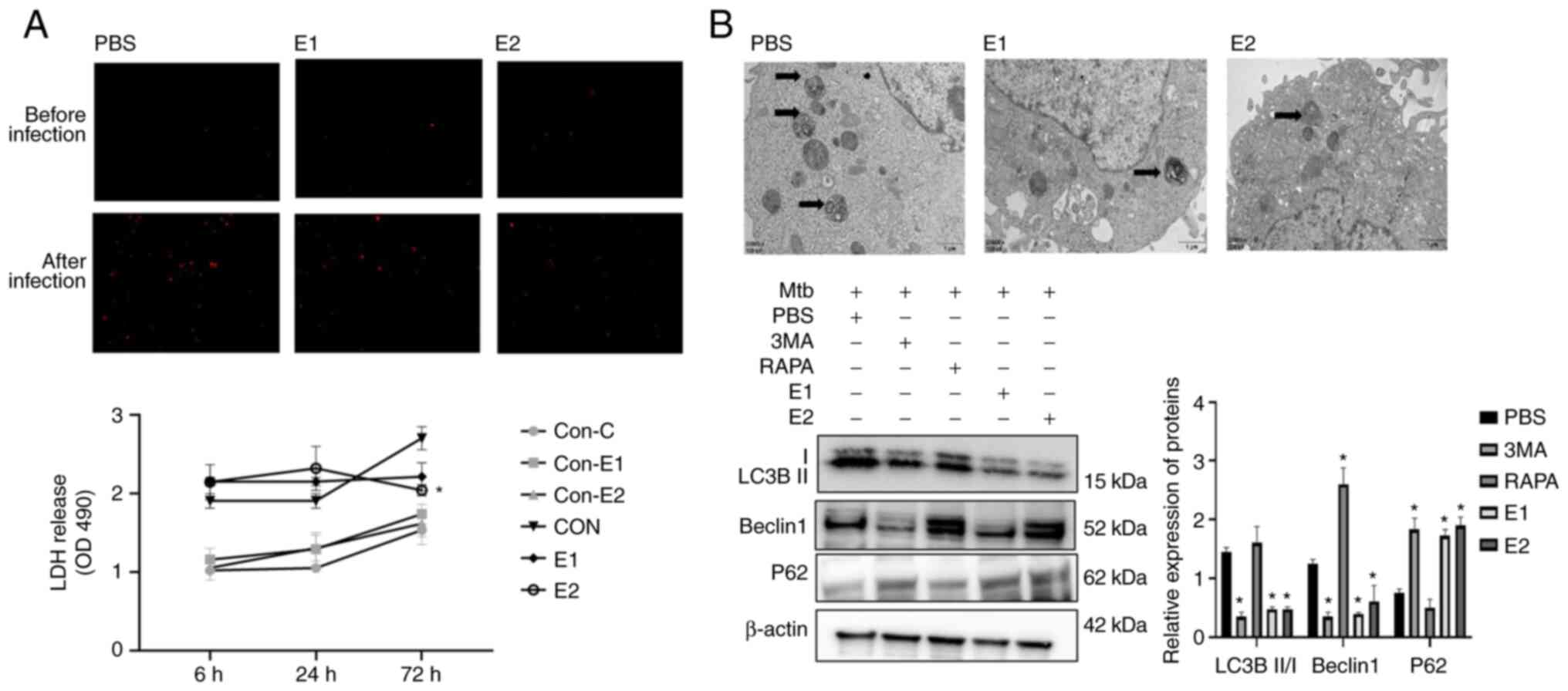 | Figure 3.Estradiol inhibits the growth of
intracellular Mtb and necrosis of the infected host cells by
inhibiting autophagy. (A) Cells treated with estradiol and infected
with Mtb released less LDH at 72 h and displayed decreased necrosis
levels at 24 h compared with the untreated cells. Magnification,
×100. Compared with the con-PBS group, estradiol treatment of
uninfected 16HBE for 72 h did not affect LDH release. However,
compared with the PBS group, estradiol treatment of infected 16HBE
cells for 72 h significantly reduced LDH release, especially at
10−6 M. *P<0.05 vs. con-PBS. (B) Compared with PBS
group, Mtb-infected 16HBE cells in the E1 and E2 groups had
markedly fewer intracellular autophagolysosomes and the protein
expression levels of LC3B II/I and beclin1 significantly decreased,
whereas the protein expression levels of P62 were significantly
increased. Arrows indicate autophagic lysosomes. Magnification,
×20,000. Mtb, Mycobacterium tuberculosis; RAPA, 100 nM
rapamycin; 3-MA, 10 mM 3-methyladenine; E1, 10−8 M
estradiol, E2, 10−6 M estradiol; Con, uninfected 16HBE
cells; LDH, lactate dehydrogenase; OD, optical density. |
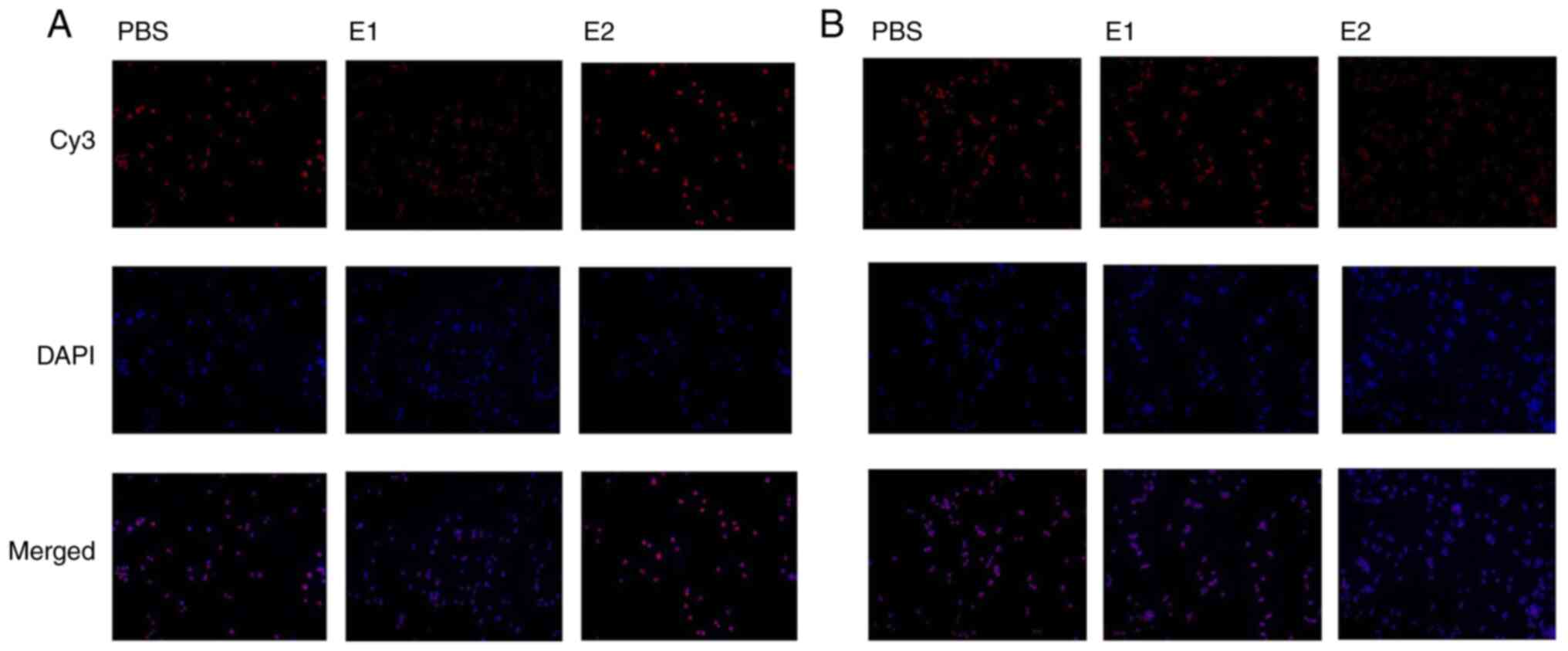
Estradiol-inhibited autophagy is
associated with ROS production and phosphorylation of AKT/mTOR by
binding to ERα
The aforementioned results showed that autophagy of
Mtb-infected 16HBE cells was inhibited by estradiol. To examine
whether estradiol inhibited autophagy by binding to ERα and/or ERβ,
further experiments were conducted to determine whether 16HBE cells
expressed ER. Both ERα and ERβ were strongly expressed in 16HBE
cells. Compared with the PBS group, the fluorescence intensity of
ERα in E1 group was markedly decreased, whereas it was
significantly changed in the E2 group (Fig. 4A). Compared with the PBS group,
fluorescence intensity of ERβ had no significant change in the E1
group. However, the fluorescence intensity of ERβ was markedly
deceased in the 1 E2 group (Fig.
4B). Subsequently, Mtb-infected 16HBE cells were treated with
ER agonists or antagonists. Compared with the PBS group, the
protein expression levels of LC3B II/I and beclin1 significantly
increased and the protein expression levels of p62 significantly
decreased in the DPN group. The protein expression levels of LC3B
II/I and beclin1 significantly decreased, whereas the protein
expression levels of P62 significantly increased in the E2 and PPT
group compared with the PBS group. Furthermore, these results
showed that autophagy of Mtb-infected 16HBE cells was inhibited by
PPT (a specific ERα agonist), which was similar to the effect of
E2. Moreover, increased expression levels of LC3B II/I and beclin1
and decreased protein expression level of P62 by estradiol was
ameliorated when Mtb-infected 16HBE cells were treated with both
estradiol and AZD9496 (a special ERα antagonist) (Fig. 5A). The protein expression levels
of LC3B II/I, beclin1 and p62 in the BAZ + E2 group were similar to
those observed in the PBS group. ROS levels in Mtb-infected 16HBE
cells treated with estradiol and PPT were similar and significantly
lower than that in Mtb-infected 16HBE cells treated with PBS. When
Mtb-infected 16HBE cells were treated with both E2 and AZD9496, the
ROS levels were similar compared with that in the Mtb-infected
16HBE cells treated with PBS. The ROS levels in Mtb-infected 16HBE
cells treated with DPN or BAZ + E2 were not significantly different
compared with the PBS group (Fig.
5B).
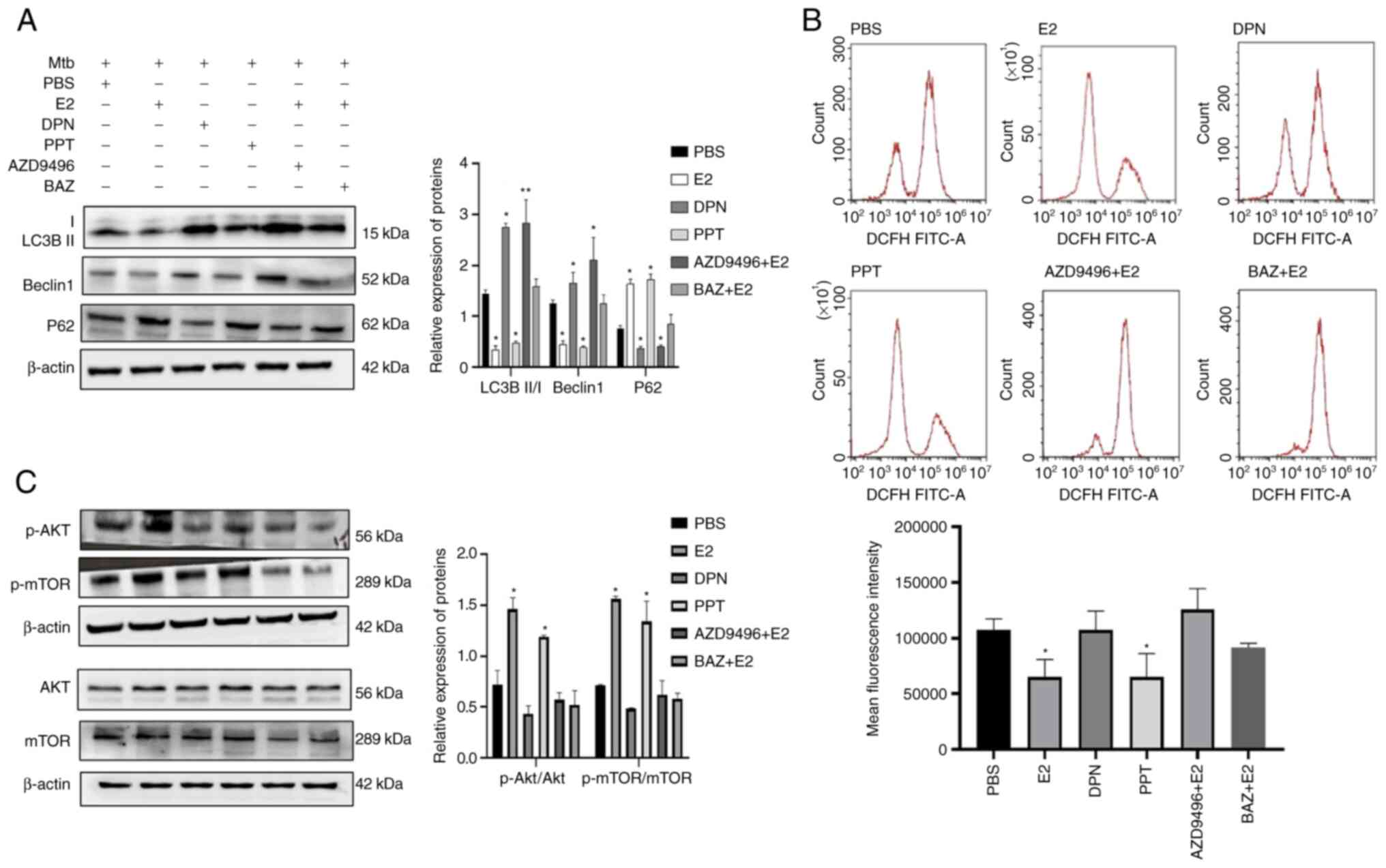 | Figure 5.Inhibition of autophagy by estradiol
promotes ROS production and phosphorylation of AKT/mTOR by binding
to ERα. (A) E2 inhibited autophagy by binding to ERα. Infected
16HBE cells were incubated with PPT or DPN for 24 h in the absence
of estradiol. Moreover, infected 16HBE cells were pre-treated with
AZD9496 or BAZ for 1 h and co-incubated with estradiol for 24 h.
(B) ROS levels in infected 16HBE cells treated with ER agonist or
antagonist for 24 h were detected using the DCFH-diacetate probe
(10 µM). Mean fluorescence intensity of ROS was analyzed by flow
cytometry. (C) Protein levels of p-AKT, total AKT, p-AMPK, total
AMPK, p-mTOR and total mTOR in Mtb-infected 16HBE cells were
analyzed using western blotting. *P<0.05 and **P<0.01 vs.
PBS. E2, 10−6 M estradiol; PPT, 4, 4, 4,
4(4-propyl-[1H]-pyrazole-1, 3, 5-triyl) trisphenol or ERα-specific
agonist (10−6 M); DPN, diarylpropionitrile or
ERβ-specific agonist (10−6 M); AZD9496, ERα-specific
antagonist (10−6 M); BZA, Bazedoxifene or nonspecific ER
modulator (0.5 µM); Mtb, Mycobacterium tuberculosis; p,
phosphorylated; DCFH, 2,7-dichlorodihydrofluorescein. |
Phosphorylation of AKT/mTOR is a key signaling step
for estradiol-associated autophagy (23,24). To understand the signaling pathway
involved in the estradiol-mediated inhibition of autophagy, the
phosphorylation levels of mTOR and AKT were measured. Similar total
protein amounts of AKT and mTOR among all groups were observed. By
contrast, the levels of p-mTOR and p-AKT were notably increased in
E2 and PPT-treated Mtb-infected 16HBE cells compared with that in
the other groups (Fig. 5C).
Discussion
It is well known that women have stronger immunity
to infection than men (25).
People in all age groups can be affected by TB, although the
highest burden is in adult men (1). However, previous studies have
demonstrated that TBTB is most common in young and middle-aged
female (2–4). Therefore, it may be hypothesized
that estrogen is involved in the pathogenesis of TBTB.
Previous studies have demonstrated that Mtb could
enter alveolar and interstitial macrophages, type-II pneumocytes,
endothelial cells and fibroblasts (17,18,21,22). In the present study, it was
demonstrated that Mtb could enter goblet cells and ciliated cells.
There have been numerous studies on the expression of ER in the
lung and gonad (26,27), few studies on the airway (28,29) and none in patients with pulmonary
TB and TBTB. Previous studies have demonstrated that ERs were
expressed in human airway mast cells (28) and human airway smooth muscle cells
(29). In the present study, it
was found that only ERα was expressed in TBTB tissues and that ERβ
was not expressed in TBTB tissues. Therefore, it was hypothesized
that estradiol, through binding to ERα, may serve a role in the
pathogenesis of TBTB.
Several studies have shown that autophagy of
non-phagocytic cells infected with Mtb is closely related to cell
necrosis and dissemination of intracellular Mtb and autophagy
protects type II alveolar epithelial cells from Mtb infection
(14,17,18). Behar et al (30) found that Mtb infection caused
different levels of death in human macrophages and alveolar
epithelial cells. With respect to non-phagocytic cells, Mtb
infection mainly causes necrosis in Mtb-infected cells (31). Inhibition of the autophagy pathway
using 3-MA has been indicated to improve host cell viability and
decreased numbers of viable intracellular Mtb (18). The present study also suggested
that estradiol controlled the growth of intracellular Mtb by
inhibiting the autophagy of infected cells. The regulation of
autophagy in Mtb-infected macrophages is a complex process. ROS
have been reported to induce autophagy (32) through inhibition of the AKT and
mTOR signaling pathway (33). Jin
et al (34) suggested that
estradiol alleviates intervertebral disc degeneration through
modulating the antioxidant enzymes and inhibiting autophagy in a
menopausal rat model. Cook et al (35) showed that knockdown of ERα induces
autophagy and promotes ROS-induced breast cancer cell death.
Another study also found that BAZ inhibits the intracellular Mtb
growth in macrophages by increasing autophagy, associated with ROS
production and phosphorylation of mTOR and AKT signaling (7). A previous study on osteoarthritis
suggested that estradiol can protect chondrocytes against mitophagy
by activating the PI3K/AKT signaling pathway (36). Lambert et al (37) showed that when ERα-deficient
peritoneal macrophages were exposed to Mycobacterium avium in
vitro, the bacterial load decreased significantly compared with
wild-type mouse macrophages. ERβ-deficient peritoneal macrophages,
on the other hand, did not differ from wild-type peritoneal
macrophages in the bacterial load. The results of these studies are
consistent with the findings of the present study in that estradiol
inhibits autophagy primarily by binding to ERα rather than ERβ,
which resulted in reduced production of ROS and affected the AKT
pathway. However, the current data have not addressed the exact
signaling pathway from ROS to autophagy during estradiol treatment.
BCG is known as Mycobacterium bovis, which is a subtype of
Mtb. BCG and Mtb not only have several similar antigens, but also
can induce macrophage necrosis by enhancing the accumulation of ROS
(7,17,38,39). In addition, BCG could induce
upregulation of ROS in A549 cells (40). Thus, the effect of estrogen and
its receptor modulator on ROS was measured using BCG-infected 16HBE
cells. The number of necrotic cells among different groups was not
further compared by flow cytometry and only fluorescence
observation after PI staining was carried out.
In summary, the present study demonstrated that
estradiol may serve a key role in the development of TBTB through
binding to ERα and affect the growth of Mtb in bronchial epithelial
cells. Previous studies have shown that ER modulators (tamoxifen
and BAZ) have anti-TB effects in vitro (7,41–43). Rey et al showed (44) that endocrine changes, including an
increase in estradiol, in patients with TB would favor a reduction
in protective cell-mediated immunity and an exacerbation of
inflammation, leading to perpetuation of the lung injury. A
cross-sectional study found that low serum estradiol levels are
related to Mycobacterium avium complex lung disease
(45). Exogenous estradiol
supplementation was found to be beneficial in controlling lung
disease in a Mycobacterium avium lung infection animal model
(46). In another animal study,
exogenous administration of 2-methoxyestradiol (an endogenous
metabolite of estradiol) accelerated disease progression in the
early stage of Mtb infection by inhibiting hypoxia-inducible factor
1α. In the late stage, Mtb load was reduced by inducing apoptosis
of Mtb-infected macrophages in BALB/c mice (47). Therefore, estrogen or ER
modulators may be promising for the treatment of TBTB, but further
animal experiments are needed to explore the value of estrogen and
its receptor modulators in the treatment of TBTB.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Science and
Technology Major Project of China (grant no.
2018ZX10302302003).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
SLG contributed to the conception and design of the
study and manuscript preparation. YLG conducted experiments,
analyzed the data and prepared the manuscript. QFH assisted in the
experiments, analyzed the experimental data and drafted parts of
the manuscript. AML and LG contributed to the design of the study
and drafted parts of the manuscript. AML and LG confirm the
authenticity of all raw data. All authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of The First Affiliated Hospital of Chongqing Medical
University (approval no. 20188502; Chongqing, China). Written
informed consent was obtained from all the patients whose tissues
were used in the present study.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
World Health Organisation, . Global
tuberculosis report 2020. https://www.who.int/publications/i/item/9789240013131July
3–2021
|
|
2
|
Lee JH, Park SS, Lee DH, Shin DH, Yang SC
and Yoo BM: Endobronchial Tuberculosis: Clinical and bronchoscopic
features in 121 cases. Chest. 102:990–994. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jung SS, Park HS, Kim JO and Kim SY:
Incidence and clinical predictors of endobronchial tuberculosis in
patients with pulmonary tuberculosis. Respirology. 20:488–495.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xue Q, Wang N, Xue X and Wang J:
Endobronchial tuberculosis: An overview. Eur J Clin Microbiol
Infect Dis. 30:1039–1044. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bottasso O, Bay ML, Besedovsky H and del
Rey A: The immuno-endocrine component in the pathogenesis of
tuberculosis. Scand J Immunol. 66:166–175. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fernández R, Díaz A, D'Attilio L,
Bongiovanni B, Santucci N, Bertola D, Besedovsky H, Del Rey A, Bay
ML and Bottasso O: An adverse immune-endocrine profile in patients
with tuberculosis and type 2 diabetes. Tuberculosis (Edinb).
101:95–101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ouyang Q, Zhang K, Lin D, Feng CG, Cai Y
and Chen X: Bazedoxifene Suppresses Intracellular Mycobacterium
tuberculosis growth by enhancing autophagy. mSphere. 5:e00124–20.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xiang J, Liu X, Ren J, Chen K, Wang HL,
Miao YY and Qi MM: How does estrogen work on autophagy? Autophagy.
15:197–211. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Totta P, Busonero C, Leone S, Marino M and
Acconcia F: Dynamin II is required for 17β-estradiol signaling and
autophagy-based ERα degradation. Sci Rep. 6:237272016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lin CW, Chen B, Huang KL, Dai YS and Teng
HL: Inhibition of autophagy by estradiol promotes locomotor
recovery after spinal cord injury in rats. Neurosci Bull.
32:137–144. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang F, Xiao J, Shen Y, Yao F and Chen Y:
Estrogen protects cardiomyocytes against lipopolysaccharide by
inhibiting autophagy. Mol Med Rep. 10:1509–1512. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Invernizzi R, Lloyd CM and Molyneaux PL:
Respiratory microbiome and epithelial interactions shape immunity
in the lungs. Immunology. 160:171–182. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reuschl AK, Edwards MR, Parker R, Connell
DW, Hoang L, Halliday A, Jarvis H, Siddiqui N, Wright C, Bremang S,
et al: Innate activation of human primary epithelial cells broadens
the host response to Mycobacterium tuberculosis in the airways.
PLoS Pathog. 13:e10065772017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Scordo JM, Knoell DL and Torrelles JB:
Alveolar epithelial cells in Mycobacterium tuberculosis infection:
Active players or innocent bystanders? J Innate Immun. 8:3–14.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Birkness KA, Deslauriers M, Bartlett JH,
White EH, King CH and Quinn FD: An in vitro tissue culture bilayer
model to examine early events in Mycobacterium tuberculosis
infection. Infect Immun. 67:653–658. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mehta PK, King CH, White EH, Murtagh JJ Jr
and Quinn FD: Comparison of in vitro models for the study of
Mycobacterium tuberculosis invasion and intracellular replication.
Infect Immun. 64:2673–2679. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Guo XG, Ji TX, Xia Y and Ma YY: Autophagy
protects type II alveolar epithelial cells from Mycobacterium
tuberculosis infection. Biochem Biophys Res Commun. 432:308–313.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fine KL, Metcalfe MG, White E, Virji M,
Karls RK and Quinn FD: Involvement of the autophagy pathway in
trafficking of Mycobacterium tuberculosis bacilli through cultured
human type II epithelial cells. Cell Microbiol. 14:1402–1414. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Walker SV, Wolke M, Plum G, Weber RE,
Werner G and Hamprecht A: Failure of Vitek2 to reliably detect
vanB-mediated vancomycin resistance in Enterococcus faecium. J
Antimicrob Chemother. 76:1698–1702. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hui KPY, Ching RHH, Chan SKH, Nicholls JM,
Sachs N, Clevers H, Peiris JSM and Chan MCW: Tropism, replication
competence, and innate immune responses of influenza virus: An
analysis of human airway organoids and ex-vivo bronchus cultures.
Lancet Respir Med. 6:846–854. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hernández-Pando R, Jeyanathan M, Mengistu
G, Aguilar D, Orozco H, Harboe M, Rook GA and Bjune G: Persistence
of DNA from Mycobacterium tuberculosis in superficially normal lung
tissue during latent infection. Lancet. 356:2133–2138. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rodrigues TS, Alvarez ARP, Gembre AF,
Forni MFPAD, de Melo BMS, Alves Filho JCF, Câmara NOS and Bonato
VLD: Mycobacterium tuberculosis-infected alveolar epithelial cells
modulate dendritic cell function through the HIF-1α-NOS2 axis. J
Leukoc Biol. 108:1225–1238. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang L, Zhao Y and Guo L: 17β-estradiol
protects INS-1 insulinoma cells from mitophagy via G
protein-coupled estrogen receptors and the PI3K/Akt signaling
pathway. Int J Mol Med. 41:2839–2846. 2018.PubMed/NCBI
|
|
24
|
Xue JF, Shi ZM, Zou J and Li XL:
Inhibition of PI3K/AKT/mTOR signaling pathway promotes autophagy of
articular chondrocytes and attenuates inflammatory response in rats
with osteoarthritis. Biomed Pharmacother. 89:1252–1261. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moulton VR: Sex hormones in acquired
immunity and autoimmune disease. Front Immunol. 9:22792018.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carey MA, Card JW, Voltz JW, Germolec DR,
Korach KS and Zeldin DC: The impact of sex and sex hormones on lung
physiology and disease: Lessons from animal studies. Am J Physiol
Lung Cell Mol Physiol. 293:L272–L278. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tang ZR, Zhang R, Lian ZX, Deng SL and Yu
K: Estrogen-Receptor expression and function in female reproductive
disease. Cells. 8:11232019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhao XJ, McKerr G, Dong Z, Higgins CA,
Carson J, Yang ZQ and Hannigan BM: Expression of oestrogen and
progesterone receptors by mast cells alone, but not lymphocytes,
macrophages or other immune cells in human upper airways. Thorax.
56:205–211. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhallamudi S, Connell J, Pabelick CM,
Prakash YS and Sathish V: Estrogen receptors differentially
regulate intracellular calcium handling in human nonasthmatic and
asthmatic airway smooth muscle cells. Am J Physiol Lung Cell Mol
Physiol. 318:L112–L124. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Behar SM, Martin CJ, Booty MG, Nishimura
T, Zhao X, Gan HX, Divangahi M and Remold HG: Apoptosis is an
innate defense function of macrophages against Mycobacterium
tuberculosis. Mucosal Immunol. 4:279–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lam A, Prabhu R, Gross CM, Riesenberg LA,
Singh V and Aggarwal S: Role of apoptosis and autophagy in
tuberculosis. Am J Physiol Lung Cell Mol Physiol. 313:L218–L229.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shin DM, Jeon BY, Lee HM, Jin HS, Yuk JM,
Song CH, Lee SH, Lee ZW, Cho SN, Kim JM, et al: Mycobacterium
tuberculosis ers regulates autophagy, inflammation, and cell death
through redox-dependent signaling. PLoS Pathog. 6:e10012302010.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bolisetty S and Jaimes EA: Mitochondria
and reactive oxygen species: Physiology and pathophysiology. Int J
Mol Sci. 14:6306–6344. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jin LY, Lv ZD, Wang K, Qian L, Song XX, Li
XF and Shen HX: Estradiol alleviates intervertebral disc
degeneration through modulating the antioxidant enzymes and
inhibiting autophagy in the model of menopause rats. Oxid Med Cell
Longev. 2018:78902912018. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cook KL, Clarke PA, Parmar J, Hu R,
Schwartz-Roberts JL, Abu-Asab M, Wärri A, Baumann WT and Clarke R:
Knockdown of estrogen receptor-α induces autophagy and inhibits
antiestrogen-mediated unfolded protein response activation,
promoting ROS-induced breast cancer cell death. FASEB J.
28:3891–3905. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Fan DX, Yang XH, Li YN and Guo L:
17β-Estradiol on the expression of G-Protein coupled estrogen
receptor (GPER/GPR30) mitophagy, and the PI3K/Akt signaling pathway
in ATDC5 chondrocytes in vitro. Med Sci Monit. 24:1936–1947. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lambert KC, Curran EM, Judy BM, Lubahn DB
and Estes DM: Estrogen receptor-alpha deficiency promotes increased
TNF-alpha secretion and bacterial killing by murine macrophages in
response to microbial stimuli in vitro. J Leukoc Biol.
75:1166–1172. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wu X, Deng G, Li M, Li Y, Ma C, Wang Y and
Liu X: Wnt/β-Catenin signaling reduces Bacillus
Calmette-Guerin-induced macrophage necrosis through a ROS-mediated
PARP/AIF-dependent pathway. BMC Immunol. 16:162015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shah G, Zielonka J, Chen F, Zhang G, Cao
Y, Kalyanaraman B and See W: H2O2 generation by bacillus
Calmette-Guérin induces the cellular oxidative stress response
required for bacillus Calmette-Guérin direct effects on urothelial
carcinoma biology. J Urol. 192:1238–1248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Méndez-Samperio P, Pérez A and Torres L:
Role of reactive oxygen species (ROS) in Mycobacterium bovis
Bacillus Calmette-Guerin mediated up-regulation of the human
cathelicidin LL-37 in A549 cells. Microb Pathog. 47:252–257. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen FC, Liao YC, Huang JM, Lin CH, Chen
YY, Dou HY and Hsiung CA: Pros and cons of the tuberculosis drugome
approach-an empirical analysis. PLoS One. 9:e1008292014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Abdmouleh F, El Arbi M, Saad HB, Jellali
K, Ketata E, Amara IB, Pigeon P, Hassen HB, Top S, Jaouen G, et al:
Antimicrobial, antitumor and side effects assessment of a newly
synthesized tamoxifen analog. Curr Top Med Chem. 20:2281–2288.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jang WS, Kim S, Podder B, Jyoti MA, Nam
KW, Lee BE and Song HY: Anti-Mycobacterial activity of tamoxifen
against drug-resistant and intra-macrophage Mycobacterium
tuberculosis. J Microbiol Biotechno. 25:946–950. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rey AD, Mahuad CV, Bozza VV, Bogue C,
Farroni MA, Bay ML, Bottasso OA and Besedovsky HO: Endocrine and
cytokine responses in humans with pulmonary tuberculosis. Brain
Behav Immun. 21:171–179. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Uwamino Y, Nishimura T, Sato Y, Tamizu E,
Asakura T, Uno S, Mori M, Fujiwara H, Ishii M, Kawabe H, et al: Low
serum estradiol levels are related to Mycobacterium avium complex
lung disease: A cross-sectional study. BMC Infect Dis. 19:10552019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Tsuyuguchi K, Suzuki K, Matsumoto H,
Tanaka E, Amitani R and Kuze F: Effect of oestrogen on
Mycobacterium avium complex pulmonary infection in mice. Clin Exp
Immunol. 123:428–434. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Baay-Guzman GJ, Duran-Padilla MA,
Rangel-Santiago J, Tirado-Rodriguez B, Antonio-Andres G,
Barrios-Payan J, Mata-Espinosa D, Klunder-Klunder M, Vega MI,
Hernandez-Pando R and Huerta-Yepez S: Dual role of
hypoxia-inducible factor 1 alpha in experimental pulmonary
tuberculosis: Its implication as a new therapeutic target. Future
Microbiol. 13:785–798. 2018. View Article : Google Scholar : PubMed/NCBI
|