Introduction
Glioblastoma is the most frequent type of
adult-onset malignant brain cancer, and the progression of the
disease is rapid and aggressive. Multidisciplinary treatments
combined with surgical therapy, chemotherapy, and radiation therapy
are used for the treatment of glioblastoma; however, the median
survival time of patients with glioblastoma is about 15 months. In
addition, the 5 years survival rate is less than 10%. Temozolomide
is used as standard chemotherapy for glioblastoma treatment, but
the prognosis of glioblastoma remains very poor (1,2). A
combination of bevacizumab and multidisciplinary treatment is also
used, but bevacizumab treatment does not extend overall survival.
Therefore, novel therapeutic strategies are needed to improve
outcomes in glioblastoma (3).
GSCs are involved in mechanisms of therapy
resistance. A cell population in glioblastoma tissue retains
characteristics similar to neural stem cells, showing high
resistance to treatment (4). This
is called the cancer stem-cell hypothesis, and is based on the
notion that cell populations that have higher tumorigenicity than
surrounding cells are the cause of treatment resistance and the
starting point of recurrence (5).
Specific inhibition of GSC proliferation is expected to improve the
therapeutic outcome of glioblastoma (6).
In recent years, the importance of mitochondria in
the pathogenesis of cancer has been realized (7). Metformin, which is a mitochondrial
complex I inhibitor, reduces the resistance of glioblastoma to
temozolomide (8). In addition,
enhanced mitochondrial functions are involved in the resistance of
glioblastoma to radiation therapy (9). Therefore, inhibiting mitochondrial
functions may be a new therapeutic strategy for glioblastoma
treatment.
The synthesis of JCI-20679 was based on the
structure of solamin (10), which
is a naturally occurring acetogenin analogue contained in plants of
the Annonaceae family. JCI-20679 inhibits the proliferation
of various types of cancer cells (11–14). Upon treatment, JCI-20679 localizes
in the mitochondria and shows anti-proliferative effects on the
human lung cancer cell line NCI-H23. JCI-20679 also shows
anticancer effects without evidence of serious side effects in a
mouse model that uses transplantation of NCI-H23 cells (15).
In this study, we hypothesized that JCI-20679 would
be an effective therapeutic strategy for glioblastoma. We evaluated
the effects of JCI-20679 in primary GSCs models and investigated
its mechanisms of action.
Materials and methods
Reagents
JCI-20679 was synthesized as described previously
(11) and dissolved in dimethyl
sulfoxide (DMSO; 13445-45, Nacalai Tesque, Kyoto, Japan). Compound
C (044-33751, Wako Pure Chemical Industries, Osaka, Japan), inosine
(I4125, Sigma-Aldrich, St. Louis, MO, USA), CCCP (M34152, Thermo
Fisher Scientific, Waltham, MN, USA), rotenone (R8875,
Sigma-Aldrich) and cyclosporin A (031-24931, Wako Pure Chemical
Industries) were dissolved in DMSO.
Animals
Wild-type C57BL/6 and BALB/c mice were obtained from
Oriental BioService (Kyoto, Japan). The mice were handled in the
animal facility of the Bioscience Research Center at Kyoto
Pharmaceutical University according to a protocol approved by the
Institution Animal Care and Use Committee.
Glioblastoma induction
The procedure was performed as described previously
(16,17). Briefly, to generate
Sleeping-Beauty transposon-mediated de novo
glioblastoma, neonatal mice under hypothermia anesthesia were
placed in a stereotaxic instrument (51730D, Stoelting Co., Wood
Dale, IL, USA). DNA complexed with polyethyleneimine was injected
into the lateral cerebral ventricle at 1 µl/min for 2 min using a
10 µl Hamilton syringe with a 30 gauges needle and an automated
infusion system (Legato 130, KD Scientific, Holliston, MA, USA).
The coordinates for DNA injection were +1.5 AP, 0.7 ML, and −1.5 DV
from λ. In vivo-JetPEI (101000040, Polyplus Transfection, New York,
NY, USA), which is an in vivo-compatible DNA transfection
reagent, was used. The following DNA plasmids were used for
glioblastoma induction: PT2/C-Luc//PGK-SB13 (0.2 µg, 20207,
Addgene, Watertown, MA, USA), PT/Caggs-NRASV12 (0.4 µg, 20205,
Addgene), PT3.5/CMV-EGFRvIII (0.4 µg, 20280, Addgene), and
PT2-shP53 (0.4 µg, 124261, Addgene). Tumor formation was confirmed
10 min after injection of D-luciferin (126-05116, Wako) into the
abdominal cavity of the 1–3 months old mice using an IVIS Lumina XR
imaging system (Summit Pharmaceuticals International, Tokyo,
Japan). Mice were sacrificed by using cervical dislocation for
glioblastoma tissue collection.
Cell culture
The procedure used to establish primary GSCs was
performed as described previously (17). Briefly, tumor tissues were diced
with scalpels and digested with accutase (AT-104, Innovative Cell
Technologies, San Diego, CA, USA) at 37°C for 20–30 min, and then
washed and incubated with serum-free neural stem-cell culture
medium consisting of neurobasal medium supplemented with B27, N2
(17504044, 17502001, Gibco/Thermo Fisher Scientific), and 10 ng/ml
EGF and bFGF (236-EG-200, 233-FB-025, R&D Systems, Minneapolis,
MN, USA) to maintain GSCs as neurospheres. The neurospheres were
split into single cells using accutase and maintained in neural
stem-cell culture medium. The procedure to establish differentiated
glioblastoma cell lines in an adherent culture system was performed
as described previously (18).
A172 and U251 human adherent glioblastoma cells obtained from Riken
BRC (Tsukuba, Japan) were maintained in DMEM supplemented with 10%
FBS (175012, HyClone, South Logan, UT, USA) and
penicillin-streptomycin (15140-148, Gibco/Thermo Fisher
Scientific). All cells were incubated at 37°C in a 5%
CO2 atmosphere.
BrdU incorporation assay
GSCs were treated with JCI-20679 at the indicated
concentrations for 72 h. The cells were harvested, and the
proportion of cells in the DNA synthesis phase was detected by
using an APC BrdU flow kit (552598, BD Biosciences, San Diego, CA,
USA) following the manufacturer's instructions. At least 3,000
cells per specimen were measured and assessed by flow cytometry
using a BD LSRFortessa X-20 cell analyzer (BD Biosciences).
Measurement of mitochondrial membrane
potential
Mitochondrial membrane potentials were detected
using JC-1 MitoMP Detection kit (MT09, Dojindo Laboratories,
Kumamoto, Japan) according to the manufacturer's instructions and
assessed by flow cytometry using a BD LSRFortessa X-20 cell
analyzer (BD Biosciences). For monitoring the mitochondrial
membrane potential, we used the PE and FITC fluorescence as the
JC-1 red and JC-1 green fluorescence. The mitochondrial membrane
potential was analyzed by using the ratio of the mean of the JC-1
red/green fluorescence signal intensities.
Measurement of oxygen consumption
rate
The oxygen consumption rate was measured using an
extracellular oxygen consumption assay (ab197243, Abcam, Cambridge,
UK) according to the manufacturer's instructions. At least 700,000
cells per specimen were analyzed every minute for 1 h. These data
were normalized by cell numbers.
Measurement of mitochondrial ROS
Mitochondrial reactive oxygen species (ROS) were
measured using MitoRos 580 dye optimized for detecting ROS in
mitochondria (16052, AAT Bioquest, Sunnyvale, CA, USA) according to
the manufacturer's instructions, and were assessed by flow
cytometry using a BD LSRFortessa X-20 cell analyzer (BD
Biosciences). At least 10,000 cells per specimen were measured.
Measurement of the AMP/ATP ratio
The procedure was carried out as described
previously (19). Briefly,
cellular ATP was measured using a CellTiter-Glo luminescent cell
viability assay kit, and cellular AMP was measured using an AMP-Glo
assay kit (G7570, V5011, Promega, Madison, WI, USA), and then the
AMP/ATP ratio was calculated.
Knockdown of AMPKβ
The procedure was performed as described previously
(17). Briefly, RNAi clones
targeting AMPKβ were obtained from Sigma-Aldrich. Transduction
using lentiviruses with non-targeting shRNA (SHC016, Sigma-Aldrich,
Sequence:
CCGGGCGCGATAGCGCTAATAATTTCTCGAGAAATTATTAGCGCTATCGCGCTTTTT) or shRNA
targeting AMPKβ was performed at a multiplicity of infection of 10.
The following shRNAs were used: sh-AMPKβ #1 (TRCN0000025105,
Sequence:
CCGGCCCTCCTCTACAAGCCGATATCTCGAGATATCGGCTTGTAGAGGAGGGTTTTT),
sh-AMPKβ #2 (TRCN0000274638, Sequence:
CCGGCCATGATCCTTCTGAGCCAATCTCGAGATTGGCTCAGAAGGATCATGGTTTTT). The
cells were maintained with 0.75 µg/ml puromycin (164-23154, Wako
Pure Chemical Industries).
Western blot analysis
To extract proteins, cells were lysed by 1% SDS
buffer supplemented with a protease inhibitor cocktail for use with
Mammalian Cell and Tissue Extracts (25955-11, Nacalai Tesque) and
PhosSTOP EASYpack (04 906 845 001, Roche Diagnostics, Indianapolis,
IN, USA). Total proteins were separated by SDS-PAGE and transferred
to PVDF membranes (IPVH00010, Millipore, Billerica, MA, USA). The
membranes were blocked with 3–5% fat-free dried milk or 5% BSA in
Tris-buffered saline (TBS; 50 mM Tris, 2.68 mM KCl, 137 mM NaCl, PH
7.4) with 0.05% Tween20 (TBST) or Blocking One-P (05999-84, Nacalai
Tesque), and then incubated with primary and secondary antibodies.
The chemiluminescence of proteins was detected using a ChemiDoc XRS
Plus system (Bio-Rad) through the reaction with Clarity Western ECL
substrate (170-5060, Bio-Rad Laboratories, Hercules, CA, USA) or
ChemiLumi One Super substrate (02230-30, Nacalai Tesque). The
following antibodies were used; p-AMPKα (1:1,000; 2535, Cell
Signaling Technology, Danvers, MA, USA), AMPKα (1:1,000; 5832, Cell
Signaling Technology), GAPDH (1:1,000; 016-25523, Wako Pure
Chemical Industries), p-CAMKII (1:1,000; 12716, Cell Signaling
Technology), CAMKII (1:1,000; 4436, Cell Signaling Technology),
NFATc2 (1:1,000; 5861, Cell Signaling Technology), Vinculin
(1:2,000; 66305-1-Ig, Proteintech), Lamin A/C (1:1,000; 2032, Cell
Signaling Technology), horseradish peroxidase-conjugated horse
anti-mouse IgG (1:2,000; PI-2000, Vector Laboratories, Burlingame,
CA, USA), and HRP-conjugated goat anti-rabbit IgG (1:2,000; 7074,
Cell Signaling Technology).
Fractionation of nuclear and
cytoplasmic proteins
GSCs were treated with JCI-20679 at the indicated
concentrations for the indicated durations, and then cellular
proteins were fractionated into nuclear and cytoplasmic fractions
using a LysoPure nuclear and cytoplasmic extractor kit (295-73901,
Wako) according to the manufacturer's protocol.
Calcineurin activity assay
The differentiated glioblastoma cells were treated
with JCI-20679 at the indicated concentrations for 72 h. The cells
were harvested, and calcineurin phosphatase activity in the same
number of the cells was measured using a calcineurin cellular
activity assay kit (BML-AK816, Enzo Life Sciences, Farmingdale, NY,
USA) following the manufacturer's instructions. At least 10,000
cells per specimen were measured.
Reverse transcription-quantitative
PCR
Cells were lysed with Trizol (15596026, Thermo
Fisher Scientific) to extract total RNA. The RNAs were purified
with an RNeasy mini kit (74106, Qiagen, Hilden, Germany).
Quantitative PCR analysis was performed with THUNDERBIRD SYBR qPCR
mix (QPS-201, TOYOBO, Osaka, Japan) using cDNA synthesized from the
extracted RNAs using ReverTra Ace qPCR RT master mix with gDNA
(genome DNA) remover (FSQ-301, TOYOBO) and a Light Cycler 96 system
(Roche). A standard ΔCt method which had been reported previously
(20) was used for data analysis.
Gene expression levels were normalized to mGAPDH. Primers were
obtained from Eurofins Genomics (Tokyo, Japan). The following
primers were used: mNFATc2 primer #1 (F: GTGCAGCTCCACGGCTACAT, R:
GCGGCTTAAGGATCCTCTCA), mNFATc2 primer #2 (F: GAGAAGACTACAGATGGGCAG,
R: ACTGGGTGGTAGGTAAAGTG), mGAPDH (F: GGTGTGAACGGATTTGGCCGTATTG, R:
CCGTTGAATTTGCCGTGAGTGGAGT).
NFATc2 overexpression
NFATc2 overexpression was performed using a mouse
neural stem-cell nucleofector kit (VPG-1004; Lonza, Tokyo, Japan)
and a nucleofector 2b device (AAB-1001, Lonza) with an A-033
program optimized for mouse GSCs. The NFATc2 gene (MR209524,
Origene Technology, Rockville, USA) or empty vector (PS100001,
Origene) was introduced into mouse GSCs, and cells containing the
NFATc2 gene or empty vector were selected using 50–250 µg/ml G418
(09380-86, Nacalai Tesque) for 2 weeks. The procedure was performed
following the manufacturer's instructions. The nucleofection
efficacy was confirmed by western blot analysis.
Transplantation assay
GSCs were injected into 20 sex-matched mice (6 weeks
old; n=10 in each treatment group) for the analyzing the event-free
survival rate, and GSCs of a deferent line were injected into 14
sex-matched mice (6 weeks old; n=7 in each treatment group) for the
assessing the glioblastoma size. Cells (1×103) were
suspended in 2 µl PBS (166-23555, Wako Pure Chemical Industries).
Mice were anchored to the stereotaxic instrument. Cells were
injected at a speed of 1 µl/min with an infusion system (Legato
130). The syringe was withdrawn from the brain 2 min after the
injection. JCI-20679 was injected intraperitoneally at 20 mg/kg
three times a week. JCI-20679 was dissolved at 10 mg/ml in DMSO
with Kolliphor EL (C5135, Sigma-Aldrich) (final 10%) and then
diluted in saline (total 200 µl/20 g body weight) to the required
concentration. DMSO was used as a control. The occurrence of death,
obvious neurological symptoms, or a decrease in body weight of more
than 2 g was used to generate event-free survival curves. The
JCI-20679 treatment was started after 3 days from the GSCs
transplantation to analyze the event-free survival rate. Tumor
formation was analyzed every week 10 min after injection of
D-luciferin (126-05116, Wako) into the abdominal cavity of mice
using an IVIS Lumina XR imaging system. For comparison of
bioluminescence intensity, tumor initiation
(1×104−5×105 photons/sec) was confirmed and
then randomized before starting treatment. Mice with neurological
symptoms by the tumor formation, such as slow moving or abnormal
posture, were sacrificed by using cervical dislocation.
Statistical analysis
Two-tailed unpaired Student's t-test, two-tailed
Welch's t-test, one-way ANOVA with Dunnett's multiple comparison
test or 2-way ANOVA with Bonferroni's multiple comparison test were
used to determine the statistical significance for in vitro
studies. Data are displayed as the mean ± SD unless otherwise
indicated. All analyzed data were obtained from at least 3
independent experiments. For the in vivo study, log-rank
tests were used to determine significant differences in the
survival curves by the Kaplan-Meier method. Bioluminescence
intensities were compared by Wilcoxon rank sum test. P-values were
calculated using Microsoft Excel software. P<0.05 was considered
statistically significant.
Results
JCI-20679 suppresses the proliferation
of glioblastoma stem cells
First, we analyzed the effects of JCI-20679 on the
proliferation of GSCs maintained as neurospheres and
differentiation-induced cells in adherent culture; both cell types
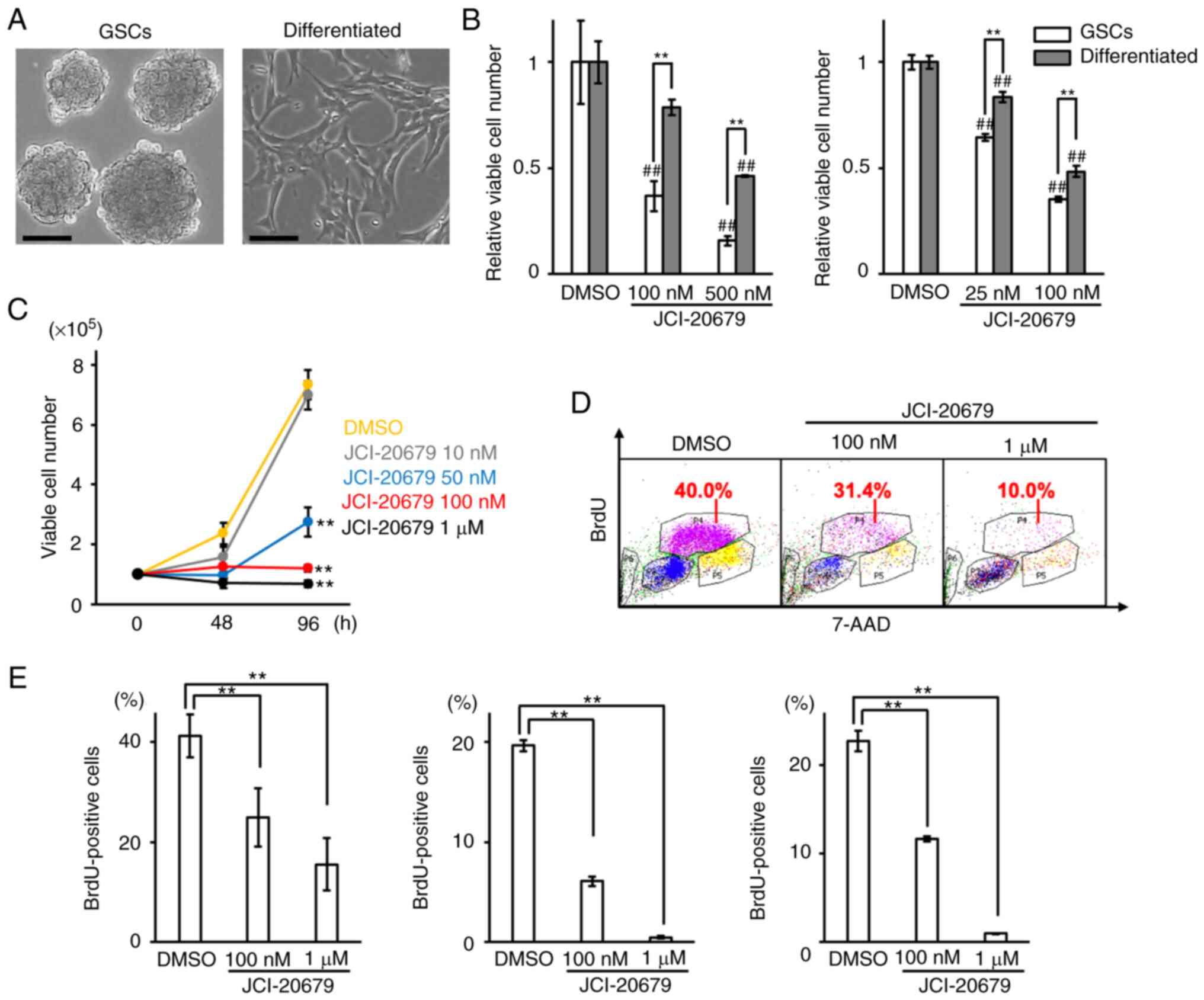
were derived from identical murine glioblastoma tissues (Fig. 1A). The properties of GSCs and
differentiation-induced cells have been established and described
previously (17). We found that
the proliferation of GSCs was more effectively suppressed by
JCI-20679 than the proliferation of differentiated cells (Fig. 1B). JCI-20679 suppressed the
proliferation of GSCs in a concentration-dependent manner (Fig. 1C). JCI-20679 treatment did not
increase the number of trypan blue-positive dead cells (data not
shown). In addition, the BrdU incorporation assay showed that
JCI-20679 significantly reduced the number of cells entering the
DNA synthesis phase of the cell cycle (Fig. 1D, E). However, we observed no
significant induction of apoptosis by the Annexin-V assay and
sub-G1 population by the cell cycle analysis (data not shown).
These results suggest that JCI-20679 suppresses GSCs proliferation
by inducing not apoptosis but cell cycle arrest.
JCI-20679 inhibits mitochondrial
function
A previous study showed that JCI-20679 inhibits the
function of the bovine mitochondria complex I in vitro
(15). We next investigated the
effects of JCI-20679 on mitochondrial function in cancer cells. It
was difficult to detect the mitochondrial membrane potential, which
directly reflects mitochondrial function, in GSCs so we used the
differentiated glioblastoma cells and A172 human glioblastoma cells
as models. JCI-20679 significantly reduced the mitochondrial
membrane potential, as did the established mitochondrial inhibitor
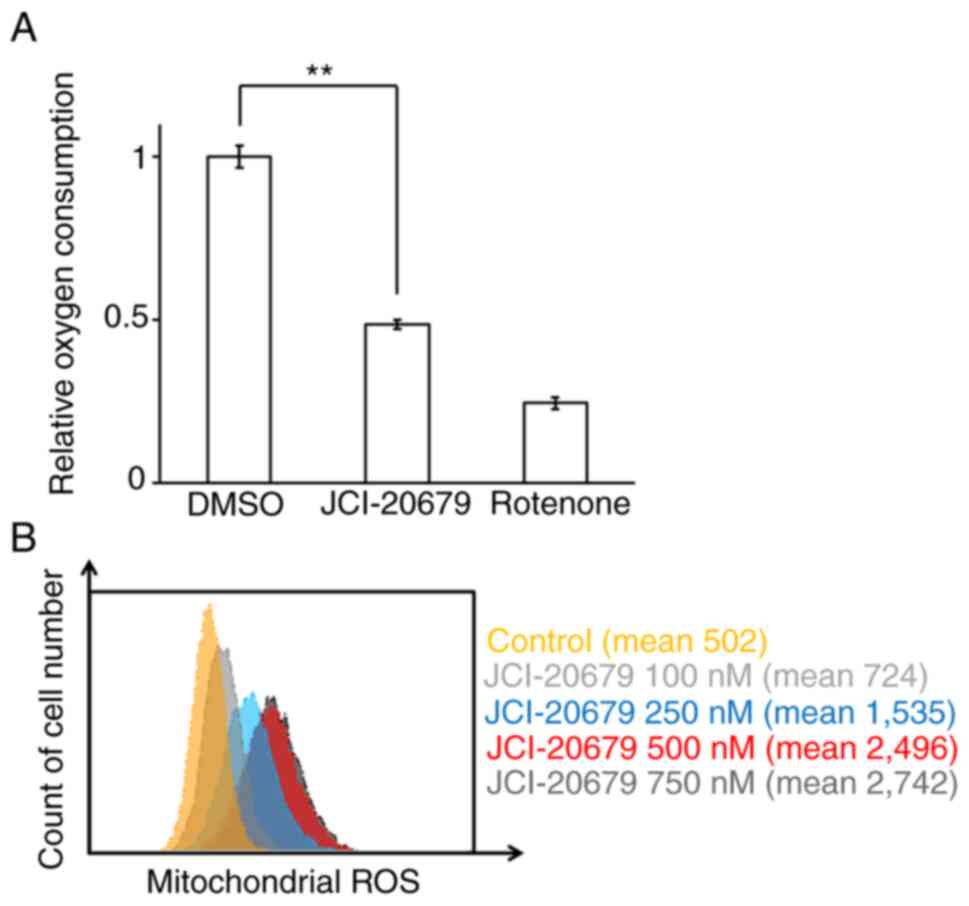
CCCP, which was used as a positive control (Fig. S1A, B). We next measured the
oxygen consumption rate of GSCs. JCI-20679 inhibited the oxygen
consumption rate, which is an index of oxidative phosphorylation.
Rotenone, an established mitochondrial complex I inhibitor, was
used as a positive control (Fig.
2A). Moreover, we found that JCI-20679 increased mitochondrial
ROS generation in the differentiated glioblastoma cells in a
concentration-dependent manner (Fig.
2B). These results indicate that JCI-20679 inhibits
mitochondrial function in cancer cells.
JCI-20679 activates AMPK
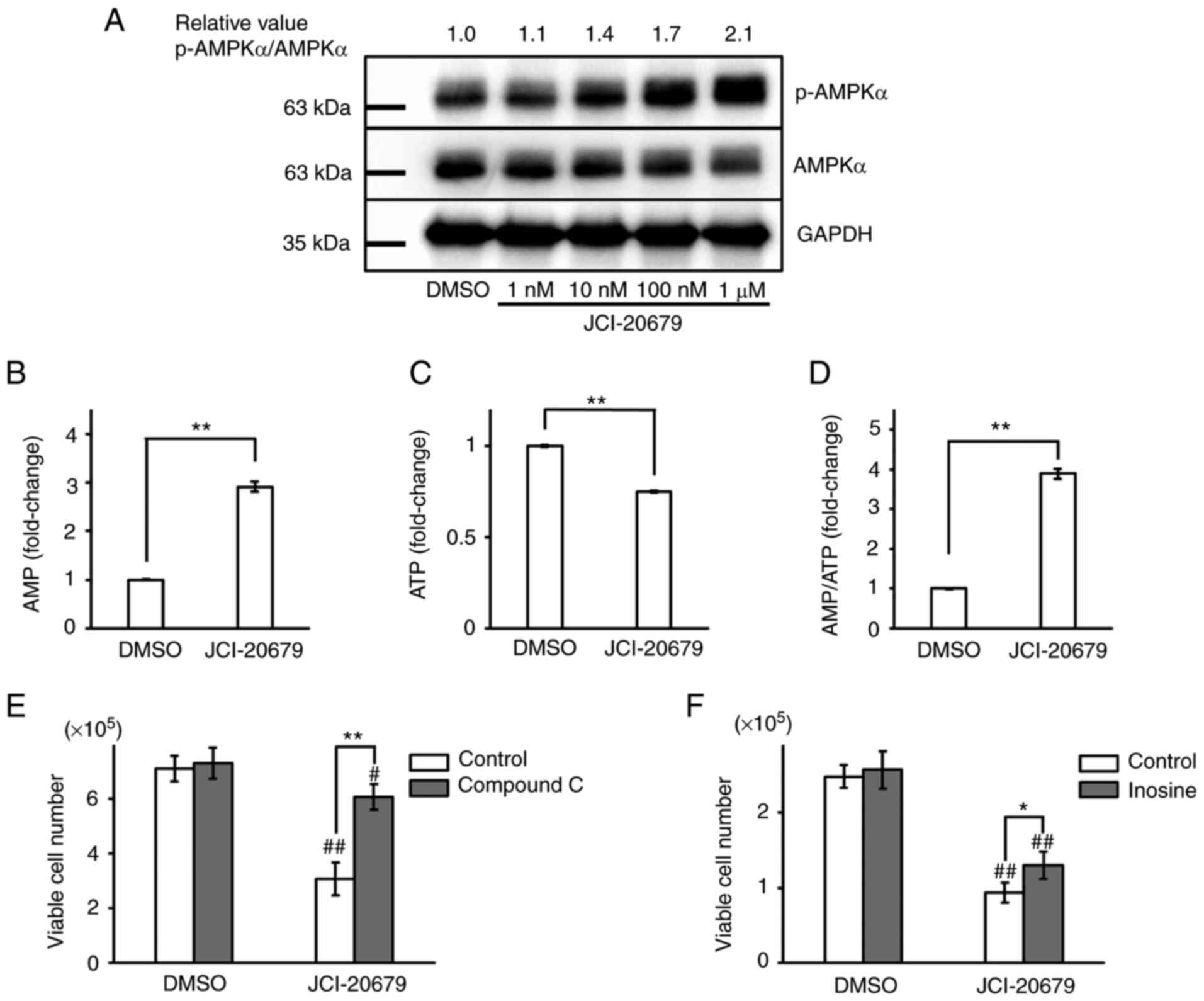
Inhibition of mitochondrial function activates AMPK
through its phosphorylation (21), so we evaluated p-AMPKα protein
levels in GSCs treated with JCI-20679. Our results showed that
JCI-20679 concentration-dependently increased p-AMPKα protein
levels (Fig. 3A). Consistent with
these results, treatment of GSCs with JCI-20679 decreased
intracellular ATP levels and increased intracellular AMP levels,
resulting in a significant increase in the AMP/ATP ratio (Fig. 3B-D). Co-treatment with compound C
or inosine, which are known AMPK inhibitors, significantly reversed
the JCI-20679-mediated suppression of cell proliferation (Fig. 3E, F). These results suggest that
JCI-20679 activates AMPK to suppress GSC proliferation.
JCI-20679 reduces the expression of
NFATc2
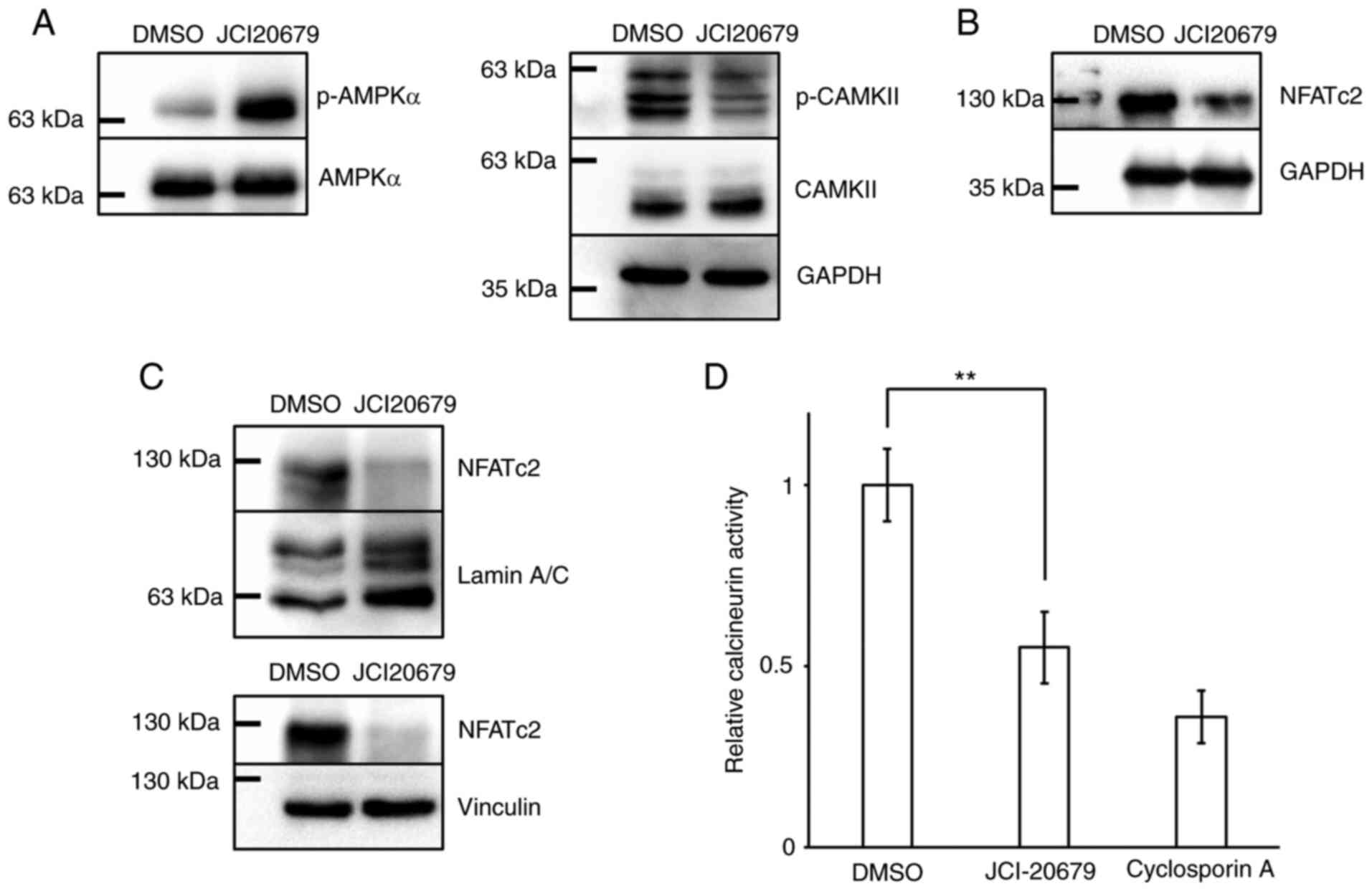
Inhibition of mitochondrial function modulates the
calcium signaling pathway (22),
so we investigated the effect of JCI-20679 on factors involved in
the calcium signaling pathway. Our results showed that JCI-20679
significantly reduced the protein levels of p-CaMKII and NFATc2 in
GSCs (Fig. 4A, B). NFATc2 protein
levels decreased in both nuclear and cytoplasmic protein fractions
(Fig. 4C). Adequate quality of
cellular fractionation was confirmed by western blot analysis
(Fig. S1C). Moreover, JCI-20679
reduced calcineurin phosphatase activity (Fig. 4D). These results suggest that
JCI-20679 suppresses the calcium signaling pathway.
JCI-20679 suppresses the proliferation
of glioblastoma stem cells by activating AMPK and reducing NFATc2
expression
We next investigated the implication of the
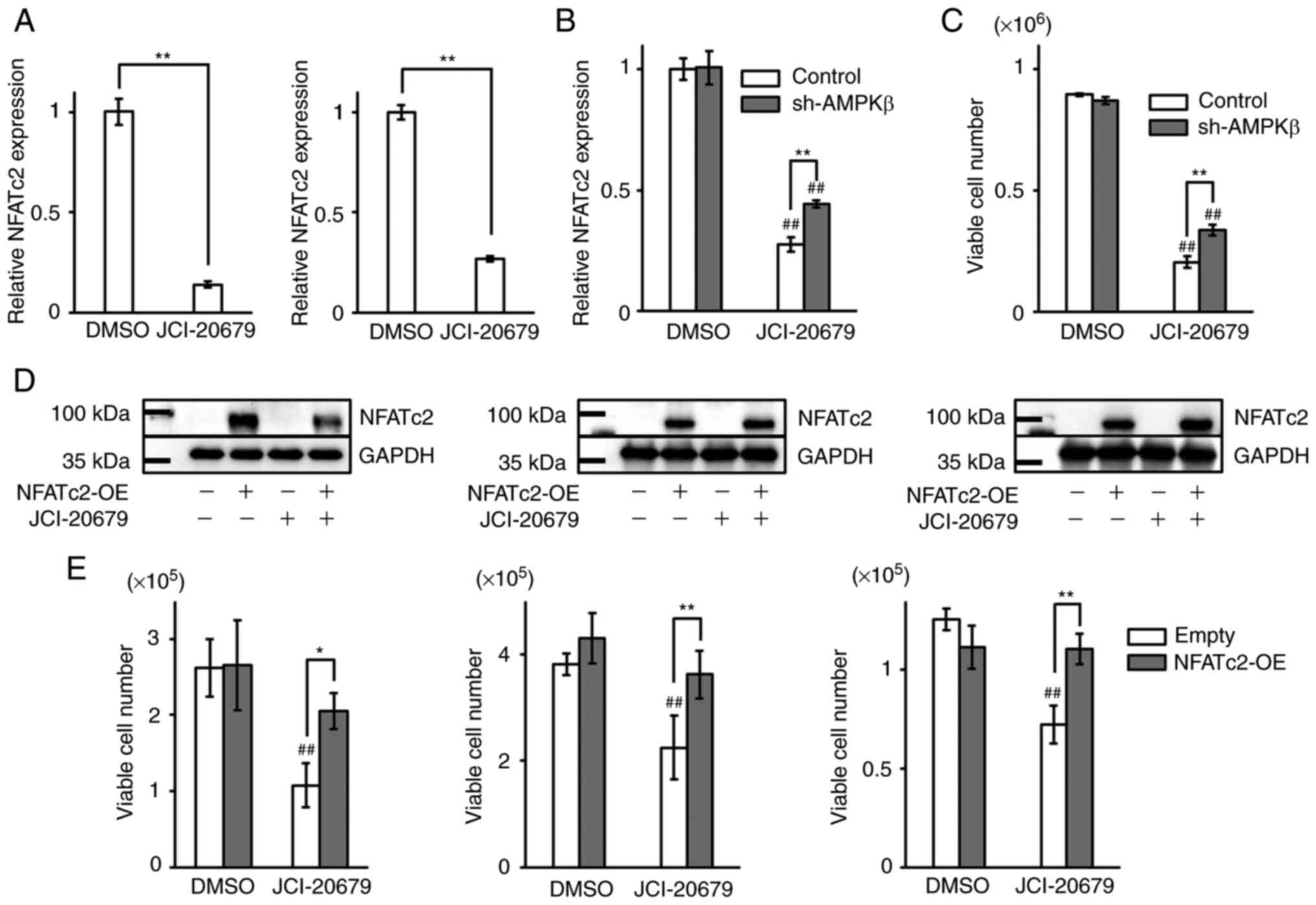
JCI-20679-mediated reduction in NFATc2 expression. JCI-20679
decreased NFATc2 mRNA levels in GSCs (Fig. 5A). It has been reported that AMPKα
phosphorylation is suppressed by knockdown of AMPKβ, which is
crucial for AMPKα phosphorylation (21). Thus, we used AMPKβ shRNA for
suppressing AMPK activation. We confirmed suppressing AMPK
activation successfully by western blot analysis. In addition,
depleted AMPKβ has no effect on expression levels of NFATc2 protein
(Fig. S1D). The decrease of
NFATc2 mRNA levels mediated by JCI-20679 treatment was recovered
when phosphorylation of AMPKα was suppressed (Fig. 5B). Furthermore, the experiment
used another shRNA sequence and another NFATc2 primer gave similar
result (Fig. S1E). In addition,
cell viability of GSCs treated by JCI-20679 were recovered by
knockdown of AMPKβ (Fig. 5C).
Moreover, we tested the effects of NFATc2 overexpression in GSCs
and differentiated glioblastoma cells. We confirmed NFATc2
overexpression by western blot analysis (Fig. 5D). NFATc2 overexpression
significantly reversed the JCI-2067-mediated suppression of
proliferation (Fig. 5E). These
results indicate that the anti-proliferative effects of JCI-20679
are mediated by decreased NFATc2 expression levels that are caused
by AMPK activation.
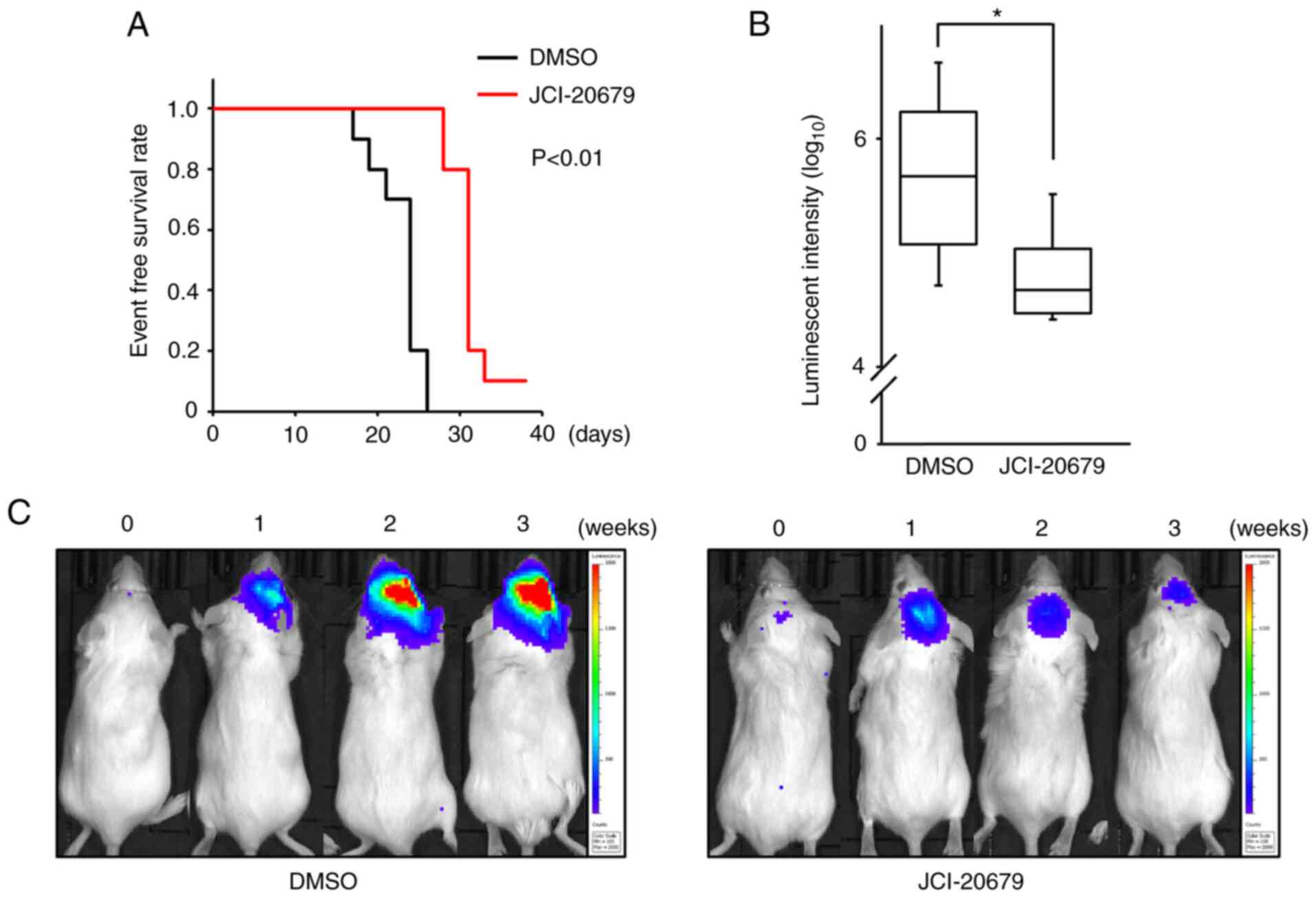
JCI-20679 suppresses tumorigenicity in
vivo
To examine the antitumor effects of JCI-20679 in
vivo, JCI-20679 was administered intraperitoneally (20 mg/kg
three times a week) to a mouse model orthotopically transplanted
with GSCs. JCI-20679 significantly improved event-free survival
(Fig. 6A). We confirmed the
efficacy of JCI-20679 using orthotopically transplanted with a
different GSC line. JCI-20679 significantly decreased the
luminescent signal, which reflects glioblastoma size, 3 weeks after
JCI-20679 treatment was initiated (Fig. 6B, C). These results indicate that
JCI-20679 suppresses glioblastoma proliferation in vivo.
Discussion
In this study, we showed that JCI-20679 suppresses
GSC proliferation by causing cell-cycle arrest. JCI-20679 decreased
the mitochondrial membrane potential and suppressed the oxygen
consumption rate, and increased the generation of mitochondrial
ROS. These results are consistent with previous findings showing
that JCI-20679 has inhibitory effects on mitochondrial function.
AMPK is the molecule that senses the metabolic stress caused by
inhibition of oxidative phosphorylation in mitochondria; an
increase in the AMP/ATP ratio causes phosphorylation of AMPK
(23). We found that JCI-20679
increased the AMP/ATP ratio and phosphorylated AMPK protein levels.
AMPK inhibitors prevented JCI-206790-mediated suppression of GSC
proliferation, suggesting that JCI-20679 suppresses GSCs
proliferation by inhibiting mitochondrial functions, which activate
AMPK.
We also found that JCI-20679 suppresses NFATc2
expression at the mRNA and protein levels; NFATc2 expression is
regulated by calmodulin and calcineurin (24). Consistent with this finding,
inhibition of mitochondrial complex I by knockdown of the GRIM-19
subunit suppresses NFATc2 expression and impairs heart development
(25). Importantly, NFATc2 is
highly expressed in glioblastoma tissue and is involved in cancer
metastasis and invasion, and the calcineurin inhibitor cyclosporin
A suppresses the growth of human glioblastoma cell lines (26). NFATc2 is more highly expressed in
human GSCs than in differentiated glioblastoma cells (27). Moreover, an inhibitor of the
calcineurin-NFAT pathway has anticancer effects in mice
transplanted with glioblastoma cells (28). These findings suggest that NFATc2
is a promising molecular target for glioblastoma treatment. We
found that JCI-20679 suppressed calcineurin activity, resulting in
decreased NFATc2 expression levels. NFATc2 overexpression reversed
the anti-proliferative effects of JCI-20679. These results indicate
that the mechanism of action for JCI-20679 involves a decrease in
NFATc2 expression.
Furthermore, we investigated the mechanisms
underlying the JCI-20679-mediated suppression of NFATc2 expression.
Inhibition of AMPKα recovered JCI-20679-mediated decreases in
NFATc2 expression levels. It has been reported that activation of
AMPK suppresses NFAT transcription by resveratrol treatment in a
cardiac hypertrophy model (29).
These results suggest that JCI-20679 inhibits mitochondrial
function, which activates AMPK, leading to reduced NFATc2
expression levels. AMPK is also activated by the generation of
mitochondrial ROS (30). Given
that JCI-20679 induced mitochondrial ROS activity, AMPK may be
activated by the generation of mitochondrial ROS in GSCs.
We showed that JCI-20679 inhibited the progression
of glioblastoma in vivo. Mitochondrial inhibition is a
promising novel therapeutic strategy for the treatment of cancer
(31), including glioblastoma
(32). Metformin, which inhibits
oxidative phosphorylation in mitochondria, is being tested
clinically in combination with temozolomide (33). In addition, AMPK activation is
involved in the anticancer mechanisms of metformin (34). These findings support the notion
that JCI-20679 may be an effective glioblastoma treatment in
vivo.
Taken together, our results show that JCI-20679
inhibits mitochondrial function in cancer cells and exhibits
anti-proliferative effects in GSCs. We also show that JCI-20679
inhibits proliferation by activating AMPK and suppressing NFATc2
expression levels. Moreover, our in vivo studies show that
JCI-20679 has potential as a new therapeutic agent against
glioblastoma.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms. Satomi Toma, Ms.
Shoko Inoie, Ms. Seiko Okamoto, Ms. Natsuko Tanaka and Mr. Hitoshi
Iwasaki (Kyoto Pharmaceutical University, Kyoto, Japan) for
experimental support.
Funding
This work was supported by the Japan Society for the Promotion
of Science (grant nos. 16K08330, 20K07623 and 21K09167); the
Ministry of Education, Culture, Sports, Science and
Technology-Supported Program for the Strategic Research Foundation
at Private Universities (grant no. S1511024L; between 2015 and
2019).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Author's contributions
SA, NK, CM, KO, HI and SN conducted experiments. SA,
CM, HI and SN performed data analysis. NK, MF and SN participated
in research design and supervised the study. SA and SN wrote the
manuscript. SA, HI and SN confirmed the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were carried out under the
approval of the Institutional Ethics Committee for Animal
Experiments of Kyoto Pharmaceutical University (approval no.
A21-001).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
GSCs
|
glioblastoma stem cells
|
|
DMSO
|
dimethyl sulfoxide
|
|
NFATc2
|
nuclear factor of activated T-cells
2
|
|
ROS
|
reactive oxygen species
|
|
CAMKII
|
Ca2+/calmodulin-dependent
protein kinase II
|
|
CCCP
|
carbonyl cyanide
m-chlorophenylhydrazone
|
References
|
1
|
Stupp R, Mason WP, van den Bent MJ, Weller
M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn
U, et al: Radiotherapy plus concomitant and adjuvant temozolomide
for glioblastoma. N Engl J Med. 352:987–996. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Marenco-Hillembrand L, Wijesekera O,
Suarez-Meade P, Mampre D, Jackson C, Peterson J, Trifiletti D,
Hammack J, Ortiz K, Lesser E, et al: Trends in glioblastoma:
Outcomes over time and type of intervention: A systematic evidence
based analysis. J Neurooncol. 147:297–307. 2020. View Article : Google Scholar
|
|
3
|
Chinot OL, Wick W, Mason W, Henriksson R,
Saran F, Nishikawa R, Carpentier AF, Hoang-Xuan K, Kavan P, Cernea
D, et al: Bevacizumab plus radiotherapy-temozolomide for newly
diagnosed glioblastoma. N Engl J Med. 370:709–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sancho P, Barneda D and Heeschen C:
Hallmarks of cancer stem cell metabolism. Br J Cancer.
114:1305–1312. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Singh SK, Hawkins C, Clarke ID, Squire JA,
Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB:
Identification of human brain tumour initiating cells. Nature.
432:396–401. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Schonberg DL, Lubelski D, Miller TE and
Rich JN: Brain tumor stem cells: Molecular characteristics and
their impact on therapy. Mol Aspects Med. 39:82–101. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bueno MJ, Ruiz-Sepulveda JL and
Quintela-Fandino M: Mitochondrial inhibition: A treatment strategy
in cancer? Curr Oncol Rep. 23:492021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang SH, Li S, Lu G, Xue H, Kim DH, Zhu JJ
and Liu Y: Metformin treatment reduces temozolomide resistance of
glioblastoma cells. Oncotarget. 7:78787–78803. 2016. View Article : Google Scholar
|
|
9
|
Gao X, Yang Y, Wang J, Zhang L, Sun C,
Wang Y, Zhang J, Dong H, Zhang H, Gao C, et al: Inhibition of
mitochondria NADH-Ubiquinone oxidoreductase (complex I) sensitizes
the radioresistant glioma U87MG cells to radiation. Biomed
Pharmacother. 129:1104602020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Myint SH, Cortes D, Laurens A,
Hocquemiller R, Lebȩuf M, Cavé A, Cotte J and Quéro AM: Solamin, a
cytotoxic mono-tetrahydrofuranic γ-lactone acetogenin from Annona
muricata seeds. Phytochemistry. 30:3335–3338. 1991. View Article : Google Scholar
|
|
11
|
Kojima N, Fushimi T, Tatsukawa T, Tanaka
T, Okamura M, Akatsuka A, Yamori T, Dan S, Iwasaki H and Yamashita
M: Thiophene-3-carboxamide analogue of annonaceous acetogenins as
antitumor drug lead. Eur J Med Chem. 86:684–689. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ohta K, Akatsuka A, Dan S, Iwasaki H,
Yamashita M and Kojima N: Structure-activity relationships of
thiophene carboxamide annonaceous acetogenin analogs: Shortening
the alkyl chain in the tail part significantly affects their growth
inhibitory activity against human cancer cell lines. Chem Pharm
Bull (Tokyo). 69:1029–1033. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Matsumoto T, Akatsuka A, Dan S, Iwasaki H,
Yamashita M and Kojima N: Synthesis and cancer cell growth
inhibition effects of acetogenin analogs bearing ethylene glycol
units for enhancing the water solubility. Tetrahedron.
76:1310582020. View Article : Google Scholar
|
|
14
|
Matsumoto T, Kojima N, Akatsuka A, Yamori
T, Dan S, Iwasaki H and Yamashita M: Convergent synthesis of
stereoisomers of THF ring moiety of acetogenin thiophene analogue
and their antiproliferative activities against human cancer cell
lines. Tetrahedron. 73:2359–2366. 2017. View Article : Google Scholar
|
|
15
|
Akatsuka A, Kojima N, Okamura M, Dan S and
Yamori T: A novel thiophene-3-carboxamide analog of annonaceous
acetogenin exhibits antitumor activity via inhibition of
mitochondrial complex I. Pharmacol Res Perspect. 4:e002462016.
View Article : Google Scholar
|
|
16
|
Wiesner SM, Decker SA, Larson JD, Ericson
K, Forster C, Gallardo JL, Long C, Demorest ZL, Zamora EA, Low WC,
et al: De novo induction of genetically engineered brain tumors in
mice using plasmid DNA. Cancer Res. 69:431–439. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tanigawa S, Fujita M, Moyama C, Ando S, Ii
H, Kojima Y, Fujishita T, Aoki M, Takeuchi H, Yamanaka T, et al:
Inhibition of Gli2 suppresses tumorigenicity in glioblastoma stem
cells derived from a de novo murine brain cancer model. Cancer Gene
Ther. 28:1339–1352. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fujita M, Scheurer ME, Decker SA, McDonald
HA, Kohanbash G, Kastenhuber ER, Kato H, Bondy ML, Ohlfest JR and
Okada H: Role of type 1 IFNs in antiglioma immunosurveillance-using
mouse studies to guide examination of novel prognostic markers in
humans. Clin Cancer Res. 16:3409–3419. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Taniguchi K, Kageyama S, Moyama C, Ando S,
Ii H, Ashihara E, Horinaka M, Sakai T, Kubota S, Kawauchi A and
Nakata S: γ-Glutamylcyclotransferase, a novel regulator of HIF-1α
expression, triggers aerobic glycolysis. Cancer Gene Ther.
29:37–48. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Garcia D and Shaw RJ: AMPK: Mechanisms of
cellular energy sensing and restoration of metabolic balance. Mol
Cell. 66:789–800. 2017. View Article : Google Scholar
|
|
22
|
Pathak T and Trebak M: Mitochondrial Ca2+
signaling. Pharmacol Ther. 192:112–123. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar
|
|
24
|
Mognol GP, Carneiro FR, Robbs BK, Faget DV
and Viola JP: Cell cycle and apoptosis regulation by NFAT
transcription factors: New roles for an old player. Cell Death Dis.
7:e21992016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen Y, Yuen WH, Fu J, Huang G, Melendez
AJ, Ibrahim FB, Lu H and Cao X: The mitochondrial respiratory chain
controls intracellular calcium signaling and NFAT activity
essential for heart formation in Xenopus laevis. Mol Cell Biol.
27:6420–6432. 2007. View Article : Google Scholar
|
|
26
|
Tie X, Han S, Meng L, Wang Y and Wu A:
NFAT1 is highly expressed in, and regulates the invasion of,
glioblastoma multiforme cells. PLoS One. 8:e660082013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang Y, Song Y, Wang R, Hu T, Zhang D,
Wang Z, Tie X, Wang M and Han S: NFAT1-mediated regulation of NDEL1
promotes growth and invasion of glioma stem-like cells. Cancer Res.
79:2593–2603. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Z, Li H, He L, Xiang Y, Tian C, Li C,
Tan P, Jing J, Tian Y, Du L, et al: Discovery of small-molecule
inhibitors of the HSP90-calcineurin-NFAT pathway against
glioblastoma. cell Chem Biol. 26:352–365.e7. 2019. View Article : Google Scholar
|
|
29
|
Chan AY, Dolinsky VW, Soltys CL, Viollet
B, Baksh S, Light PE and Dyck JR: Resveratrol inhibits cardiac
hypertrophy via AMP-activated protein kinase and Akt. J Biol Chem.
283:24194–201. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Rabinovitch RC, Samborska B, Faubert B, Ma
EH, Gravel SP, Andrzejewski S, Raissi TC, Pause A, St-Pierre J and
Jones RG: AMPK maintains cellular metabolic homeostasis through
regulation of mitochondrial reactive oxygen species. Cell Rep.
21:1–9. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ghosh P, Vidal C, Dey S and Zhang L:
Mitochondria targeting as an effective strategy for cancer therapy.
Int J Mol Sci. 21:33632020. View Article : Google Scholar
|
|
32
|
Guntuku L, Naidu VG and Yerra VG:
Mitochondrial dysfunction in gliomas: pharmacotherapeutic potential
of natural compounds. Curr Neuropharmacol. 14:567–583. 2016.
View Article : Google Scholar
|
|
33
|
Maraka S, Groves MD, Mammoser AG,
Melguizo-Gavilanes I, Conrad CA, Tremont-Lukats IW, Loghin ME,
O'Brien BJ, Puduvalli VK, Sulman EP, et al: Phase 1 lead-in to a
phase 2 factorial study of temozolomide plus memantine, mefloquine,
and metformin as postradiation adjuvant therapy for newly diagnosed
glioblastoma. Cancer. 125:424–433. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao B, Luo J, Yu T, Zhou L, Lv H and
Shang P: Anticancer mechanisms of metformin: A review of the
current evidence. Life Sci. 254:1177172020. View Article : Google Scholar
|