Sepsis is a severe life-threatening form of organ
dysfunction caused by dysregulated host response to infection
(1) and it has high morbidity and
mortality rates, Fleischmann et al (2) searched 15 international citation
databases to estimate population-level sepsis morbidity and
mortality in adult populations, the global estimator of
hospital-treated sepsis morbidity from 2003 to 2015 was 437 sepsis
cases per 100,000 person-years, hospital mortality was 17%. The
heart is essential for maintenance of adequate organ perfusion and
is one of the major organs affected during sepsis. Therefore,
cardiac dysfunction is a common complication of sepsis that has a
poor prognostic outcome (2). The
pathological mechanism of sepsis-induced cardiac dysfunction is
complex and multifactorial. Numerous factors, such as hemodynamic
and myocardial energy metabolism disorder, oxygen free radicals,
myocardial inhibitors and cardiomyocyte death, are involved in
cardiac dysfunction, of which cardiomyocyte death is one of the
primary elements that cause myocardial dysfunction. To the best of
our knowledge, however, its mechanism is not yet fully
understood.
A certain number of cardiomyocytes is needed for
maintenance of normal heart function. Cell death in the heart is
detrimental because the majority of adult cardiomyocytes are
terminally differentiated and non-regenerative cells with a limited
capacity to perform key functions (3). According to the Nomenclature
Committee on Cell Death, there are multiple modes of cell death
(4). Cardiomyocyte death modality
exhibits tissue specificity and key cardiomyocyte death modalities
include apoptosis, necroptosis, mitochondrial-mediated necrosis,
pyroptosis, ferroptosis and autophagic cell death (5). Cell membrane remains intact when
cells die via apoptosis, ferroptosis and autophagy. On the other
hand, death by necroptosis, mitochondrial-mediated necrosis and
pyroptosis leads to disruption of the cell membrane (5). Multiple types of cell death can
occur simultaneously or in succession during disease progression
(4).
Sepsis is characterized by acute release of multiple
inflammatory mediators (such as TNF-α, IL-6 and IL-1β); excessive
release of inflammatory mediators damage tissue and organs. There
is increasing evidence that inflammation is associated with cell
death (6–8); cells with a certain number and
proper function are important for maintaining normal organ
function, which play a crucial role in fighting against microbial
infection. Inflammation and cell death can occur simultaneously
(9) or in sequence. Different
types of cell death do not act independently but interact with each
other (10). Mechanisms of
cellular death are associated with organ function, therefore,
targeting the mechanism of cardiomyocyte death in sepsis may
identify potential options for treatment of sepsis-induced
myocardial dysfunction. The present review aimed to summarize the
effects of inflammatory activation on cardiomyocyte death and the
association between modes of cell death modes to identify potential
targets for novel therapeutic strategies based on pathogenesis.
Apoptosis is a regulated cell death program and is
the most common type of cell death (11). It is morphologically characterized
by cellular shrinkage, chromatin condensation, nuclear
fragmentation and formation of apoptotic bodies (12). Apoptosis is highly regulated in
normal healthy tissue but is activated under certain pathological
conditions, such as when cells are damaged by disease or a toxic
agent (13). Intracellular
cysteine-dependent aspartate-specific proteases (caspases) are
effector proteins associated with activation of apoptotic signaling
(14,15). Numerous studies have shown that
apoptosis typically exerts a beneficial effect in anti-inflammatory
and immunosuppressive processes (16,17). However, this programmed cell death
may have different roles in different tissues, moreover,
insufficient or excessive apoptosis promotes organ dysfunction
(18).
Apoptosis serves a crucial role in cardiovascular
disease, apoptosis of myocytes is among those processes that have
been extensively studied in vitro (19). Sepsis notably increases
cardiomyocyte apoptosis (20);
this negatively impacts cardiac function, as apoptosis serves a
major role in the loss of cardiomyocytes (21). Adult cardiomyocytes are terminally
differentiated and loss of cardiomyocytes through apoptosis has
been recognized as the underlying mechanism in the development of
cardiac dysfunction following sepsis (22). In vitro and in vivo
experiments have shown that a high levels of cardiomyocyte
apoptosis result in decreased cardiac function (23,24). As shown by numerous studies, the
pathological changes of the myocardium in sepsis are associated
with inflammation and cardiomyocyte apoptosis, which may impair
myocardial function directly (25,26). Wencker et al (27) demonstrated that very low levels of
myocyte apoptosis (23 compared with 1.5 myocytes per 105 nuclei in
controls) cause life-threatening dilated cardiomyopathy.
Cardiomyocyte apoptosis induced by lipopolysaccharide (LPS) is
completely prevented by treatment with broad-spectrum caspase
inhibitor z-Val-Ala-Asp-fluoromethylketone (28). MicroRNAs (miRNAs or miRs) are a
class of small non-coding RNA involved in numerous types of
disease, including cardiovascular disease. It is reported that
>30 miRNAs are involved in sepsis-induced cardiac dysfunction;
among these, >10 miRNAs have been implicated in regulating
sepsis-induced cardiac apoptosis (29–31). Certain miRNAs (such as miR-155,
−24 and miR-192-5p) activate cell apoptosis but others (such as
miR-214, −25, −93-3p, −23b, −146a, −98 and −150-5p) inhibit cell
apoptosis (29).
Necroptosis is an important form of cell death that
leads to the disruption of cellular membranes and leakage of
cellular substances, which causes inflammation (32). Necroptosis, also named programmed
necrosis (33), is a
pro-inflammatory form of cell death with features of both necrosis
and apoptosis (34). Necroptosis
is similar to necrosis in morphological features, but, like
apoptosis, is strictly regulated by multiple signalling pathways.
Necroptosis is a caspase-independent mode of programmed cell death
and is negatively regulated by caspases (33,35). Necroptosis is regulated by
signalling molecules receptor-interacting protein kinase-1 (RIPK1)
and RIPK3 in a kinase-dependent manner (36–38), resulting in activation of mixed
lineage kinase domain-like (MLKL) and rapid loss of plasma membrane
integrity (39). The necroptosis
and apoptosis pathways affect each other (40). In the absence of apoptotic caspase
activation, cells undergo necroptosis via alternative routes
(38).
Numerous studies have shown that necroptosis serves
an important role in the regulation of inflammatory conditions and
the course of infectious disease (33,41). Sepsis is characterized by
inflammatory response imbalance and excessive production of
pro-inflammatory cytokines, such as TNF-α and IL-1β, in a cytokine
storm (42). TNF activation can
trigger two signalling pathways associated with cell death, namely
apoptosis and necroptosis (43,44). RIPK1, RIPK3 and MLKL are three key
proteins involved in TNF-induced necroptosis (45,46). Necroptosis is regulated by the
RIPK1/RIPK3/MLKL pathway and is associated with organ injury
(47,48). Necrostatin-1 (Nec-1) prevents
necroptosis by blocking RIPK1 kinase activity in various injury
models (49,50). Schenck et al (51) reported that RIPK3, a marker of
necroptosis, is positively correlated with mortality and organ
dysfunction in sepsis. However, in vivo, necroptosis
promotes Staphylococcus aureus clearance by limiting
excessive inflammation to improve prognosis in a mouse model of
sepsis (52). These findings
indicate that necroptosis may be associated with different
pathological effects and mechanisms in sepsis caused by different
etiologies and different stages of sepsis development.
Similar to the pathology of apoptosis in
cardiomyocytes, cardiomyocyte loss through necroptosis serves a key
role in the pathogenesis of cardiac dysfunction (53). To investigate the importance of
necroptosis in cardiomyocyte death and heart injury in sepsis, a
number of studies both in vivo and in vitro have been
performed (53,54). Beno et al (55) reported that cardiomyocyte death
and heart damage are necroptosis-dependent in a Streptococcus
pneumoniae mouse model in which necroptosis inhibition
attenuated myocardial injury. The promotive effects of necroptosis
on sepsis-associated myocardial damage have been confirmed by cell
experiments (56,57). In vivo and vitro
study reported that necroptosis caused by doxorubicin in
cardiomyocytes is inhibited by potent necroptosis inhibitor Nec-1
(58).
Experimental studies have shown that peroxisome
proliferator-activated receptor γ, a protein receptor with
cardioprotective effects, decreases cardiac inflammation and
alleviates sepsis-associated cardiomyopathy by inhibiting apoptosis
and necroptosis (54,59). In vitro experiments have
confirmed that heparan sulfate fragments (a class of
danger/damage-associated molecular patterns) induce apoptosis in
cardiomyocytes and RIP3-mediated necroptosis occurs over time,
indicating that necroptosis is associated with sepsis-associated
cardiomyopathy (56,60). Another study by Fu et al
(61) suggested that necroptosis
is activated by LPS in cardiomyocytes via the RIPK3/PGam5
signalling pathway.
Pyroptosis is a caspase-dependent inflammatory form
of programmed cell death in response to diverse pathogen- and
host-derived danger signals (75,76). Its morphological features differ
from apoptosis in that they involve cell swelling and lysis
(77). Pyroptosis is a key host
innate immune defense mechanism against pathogens (78). However, excess pyroptosis results
in excessive inflammation and multiple organ dysfunction.
Pyroptosis occurs via two pathways: Classical caspase-1
(caspase-1-mediated) and non-classical caspase-4/5/11 pathways
(caspase-4/5/11-mediated, human homologs caspase-4- and
caspase-5-mediated and murine caspase-11-mediated) (79,80). In the classical caspase-1 pathway,
NLRP3 inflammasome activation occurs via caspase-1 (81,82). Studies (4,80)
have shown, non-classical pathways, intracellular LPS directly
binds with caspase-4, −5, and −11 with high affinity, resulting in
caspase-4, −5, and −11 self-assembly and triggering cell
pyroptosis. Activating caspase-4, −5, and −11 indirectly promotes
cleavage of pro-inflammatory factor precursors (pro-IL-1β and
pro-IL-18) by activating NLRP3 inflammasome and caspase-1 (83).
Pyroptosis has been described as inflammatory death
and is directly associated with inflammatory response (84,85). Therefore, studying the molecular
mechanism of pyroptosis is key for elucidation of the pathological
mechanism of sepsis. Kang et al (86) established a sepsis model using
glutathione peroxidase 4 (GPX4) Mye−/− mice and showed
that caspase-11-dependent pyroptosis mediates septic death. NLRP3
inflammasome is activated in sepsis, while NLRP3
inflammasome-mediated caspase-1 activation induces cell death via
pyroptosis (81,87). Extracellular LPS activates
toll-like receptor (TLR)4 on the cell surface, thereby indirectly
activating caspase-11, which is also activated by directly binding
to intracellular LPS (88). Cheng
et al (89) studied
caspase-1−/− and −11−/− gene knockout mice;
double inflammatory caspase gene-deficient mice exhibited a 90%
survival rate, while mice lacking caspase-1 but expressing
caspase-11 exhibited 0% survival within 72 h, indicating that
caspase-11 serves a greater role in the mechanism of
endotoxemia-induced death in mice. Sphingosine-1-phosphate is a
biomarker of sepsis severity (90); sphingosine-1-phosphate receptor
increases macrophage caspase-11 activity and promotes macrophage
pyroptosis during sepsis (91).
Pyroptosis promotes cell swelling, membrane pore
formation and plasma membrane rupture in sepsis, resulting in
leakage of inflammatory factors from the cell and inducing cell
death (92–94). Studies have modulated NLRP3
inflammasome activation to affect pyroptosis (95,96). Chu et al (97) inhibited activation of atypical
macrophage inflammasomes using oxidized phospholipids, which
decreased the inflammatory response in septic mice. Lee et
al (98) demonstrated that
phospholipase D1 inhibitor VU0155069 has antibacterial activity and
inhibits formation of inflammasomes, thus exerting an
anti-pyroptosis effect. Li et al (99) found that normal saline containing
methane decreases release of inflammatory mediators TNF-α and IL-β,
ROS production and NLRP3-mediated pyroptosis in sepsis.
Iron is one of the trace elements necessary for the
human body. It participates in the mitochondrial respiratory chain,
nucleic acid replication and repair and metabolism (105). Iron is also a key biological
element in microbial life (106). It has been shown that iron
promotes bacterial growth and enhance the virulence of bacteria
(107–109). In order to improve
anti-infection ability, it is necessary to enhance uptake of iron
by the host, but prevent uptake of iron by bacteria (110). However, intracellular iron
overload induces ferroptosis and causes organ dysfunction (111). Therefore, how to maintain this
balance needs more research.
Ferroptosis is a ROS- and iron-dependent form of
non-autophagic and non-apoptotic programmed cell death (112). Ferroptosis is activated by iron
oxidation, which differs from other modes of cell death on
morphological, biochemical and genetic levels (113). The mechanism of ferroptosis
primarily involves two pathways: Consumption of glutathione (GSH)
and reduction of GPX4 activity (GSH/GPX4) pathway (3,112) and reduction of ferroptosis
suppressor protein 1 (FSP1) activity and consumption of co-enzyme
Q10 (FSP1/CoQ/NADPH) pathway (3).
Ferroptosis causes notable iron accumulation and lipid peroxidation
during cell death. It is distinct from apoptosis or necroptosis
because it is inhibited by iron chelators and lipophilic
antioxidants but is not inhibited by caspase or RIPK1 inhibitors
(114). Genes and pathways
involved in iron, lipid and amino-acid metabolism have been found
to modulate ferroptosis (115–120).
It is hypothesized that ferroptosis is associated
with cancer suppression and neurodegenerative disease (121–126). Sepsis is often accompanied by
increased ROS generation, which induces ferroptosis in cells
(127). GPX4 decreases ROS
production, thereby inhibiting ferroptosis (118). Previous reports have suggested
that ferroptosis modulated by GPX4 may be a novel
pathophysiological mechanism (112,128) that leads to organ dysfunction in
sepsis.
The morphological hallmarks of ferroptosis include
mitochondrial shrinkage and condensation with a decreased number of
mitochondrial ridges (122,129). As myocardial tissue containing
abundant mitochondria, ferroptosis studies have primarily focused
on myocardial injury in sepsis compared with kidney, brain and
other organs (130,131). Fang et al (132) found that high-iron diet in mice
lacking ferritin H in cardiomyocytes caused severe cardiac injury
and hypertrophic cardiomyopathy with morphological features of
ferroptosis, decreased GSH levels and increased lipid peroxidation;
ferrostatin-1 (Fer-1), a specific inhibitor of ferroptosis,
reversed these effects (132).
This suggested that inhibition of ferroptotic cell death via iron
metabolism interference improves cardiac function. By affecting
lipid composition, Acyl-CoA thioesterase 1 prevents
doxorubicin-induced ferroptosis in cardiomyocytes and offers a
potential therapeutic approach to the treatment of myocardial
injury and prevention of heart failure (133). Additional studies have
demonstrated that inhibition of ferroptosis-induced cardiomyocyte
death protects against myocardial ischemia-reperfusion injury
(134,135). Moreover, an experiment in
cardiomyocytes confirmed that GPX4 overexpression protects against
palmitic acid-induced ferroptosis, whereas GPX4 knockdown reverses
the anti-ferroptotic effect (136). The aforementioned studies
demonstrated that ferroptosis plays a pathophysiological role in
the heart. Li et al (137) studied sepsis models in
vivo and in vitro and demonstrated that iron-dependent
ferroptosis serves a crucial role in sepsis-induced cardiomyopathy.
Fer-1 and deferoxamine decrease levels of ferroptosis in
cardiomyocytes and improve cardiac function and survival rate in
septic mice (137). GSH release
and expression of GPX4 are significantly decreased in
sepsis-induced myocardial injury in mice and dexmedetomidine
decreases ferroptosis by decreasing iron concentration and
hemeoxygenase-1 protein expression, as well as increasing
expression of GPX4 pathway molecules to exert cardioprotective
effects (138,139). The aforementioned results
confirm the cardioprotective effect of dexmedetomidine, supporting
the hypothesis that ferroptosis serves a key role in the
pathogenesis of myocardial injury induced by sepsis (139).
The precise role of autophagic cell death in sepsis
is controversial. Certain studies have suggested that the
activation of autophagy alleviates multiple organ dysfunction
caused by sepsis (145–148); the primary mechanism may be
associated with suppression of inflammation by regulating
activation of macrophages and inhibiting release of inflammatory
factors (149). By eliminating
damaged organelles, autophagy can maintain cellular homeostasis and
cell viability. Another study confirmed that the viability of T
cells in cellular immunity is decreased following autophagy
inhibition (150). One study
revealed that autophagy activation aggravates lung injury and
respiratory muscle dysfunction in sepsis, increases aggregation of
granulocytes and other inflammatory cells and decreases the ability
of macrophages and granulocytes to phagocytose pathogenic bacteria.
Conversely, inhibition of mitochondrial autophagy of macrophages
promotes macrophage activation and enhances host antibacterial
activity (151). Autophagy
activation decreases the viability of immunosuppressive T cells
(CD4+, CD25+ and T regulatory cells) and
increases the immune response in septic patients (152–154). However, a prior clinical study
has shown that autophagy level of neutrophils is positively
correlated with survival rate in septic patients (155). The aforementioned studies
demonstrate that inhibiting autophagy is typically harmful.
Although increasing autophagy has shown certain beneficial results
in experimental research, the potential use of autophagy activators
in the clinic requires further investigation.
The pathogenesis of sepsis is characterized by
excessive inflammatory response and secondary immune dysfunction
(156). The role of autophagy in
response to sepsis is a dynamic process and autophagy serves
different roles in different stages of disease (157). Therefore, effects of autophagy
in different stages of sepsis are key for treatment and protection
of vital organs during sepsis.
Autophagy has become a focus of research and a
potential therapeutic target to protect cardiomyocytes from damage.
Studies confirmed that autophagy plays a protective role in
sepsis-induced cardiac dysfunction by inhibiting the mTOR pathway
associated with autophagy activation (173,174). Similarly, Hsieh et al
(175) used rapamycin to induce
autophagy in myocardial cells of septic mice to improve cardiac
function. Numerous studies have revealed that activation of cardiac
autophagy attenuates myocardial damage induced by sepsis (176–178). The effect of miRNAs on autophagy
was studied by inducing or suppressing autophagy, certain miRNAs
(such as miR-1, miR-22, miR-145 and −144) activate cell apoptosis
but others (such as miR-20b-5p, miR-21, miR-34a, −101, miR-30a and
−122) inhibit apoptosis (179).
Up to date, the current research on cardiac autophagy in sepsis is
still only at the basic research level, further basic and clinical
studies of how autophagy affects myocardial function in sepsis are
required.
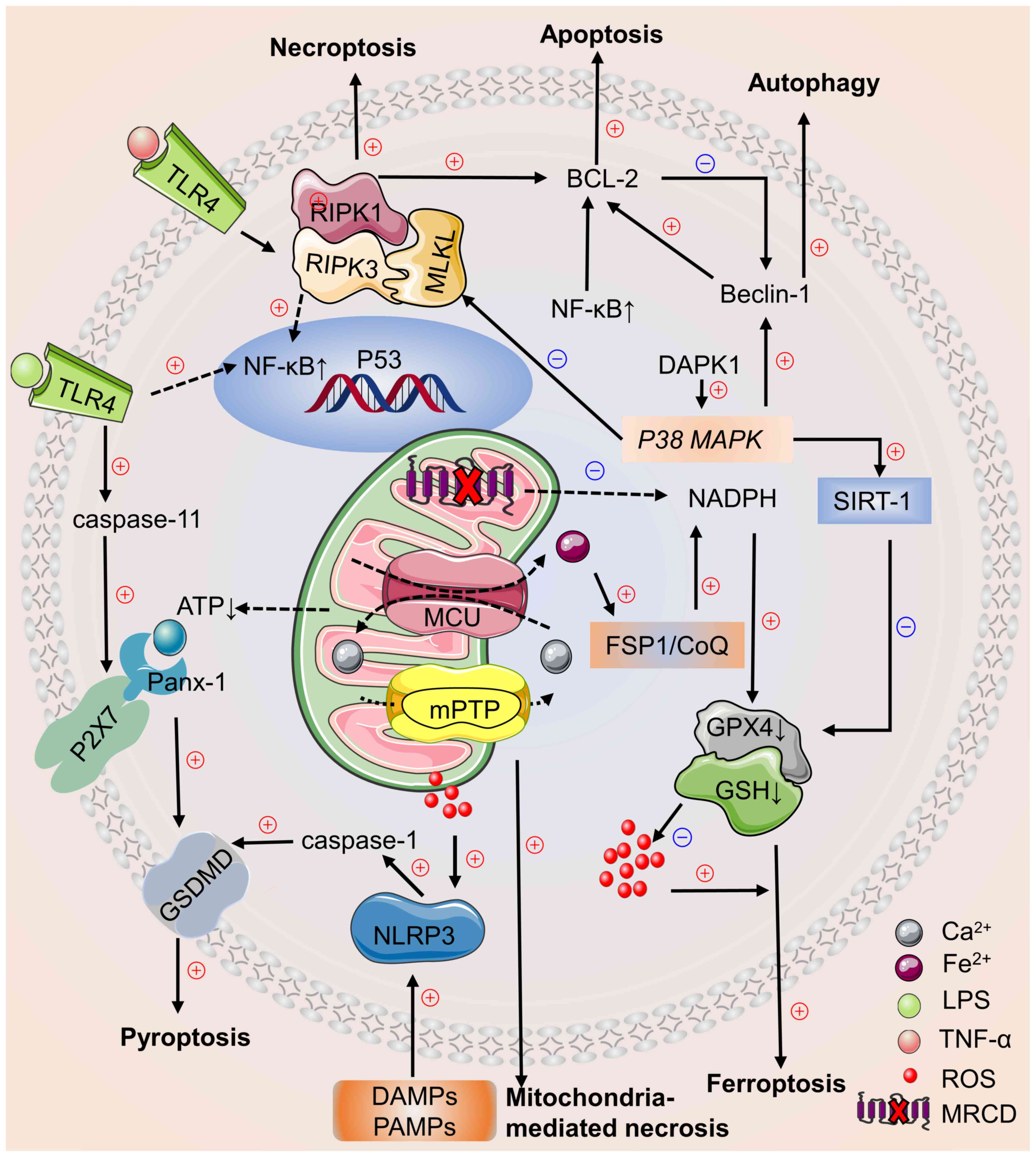
Cell death pathways involve complex interactions in
cardiomyocyte death signalling during sepsis (180,181). In different stages, activation
of multiple cell death pathways may co-occur and affect each other
during the development of sepsis-induced myocardial injury and
crosstalk between signalling cascades has been observed in
cardiomyocyte death pathways (Fig.
1) (180–182).
In experimental myocardial injury models, inhibition
of Beclin-1 haplotype inhibits ferroptosis and mitochondrial
damage, as well as myocardial remodeling and systolic dysfunction
(196,197). Experiments have confirmed that
there is an overlap between the substrates of caspase-1 and −3, key
effectors of cell apoptosis (198,199). Therefore, there may be a
connection between pyroptosis and apoptosis. Dysfunctional
autophagy promotes NLRP3 inflammasome assembly and leads to
pyroptotic cell death (94).
Pyroptosis and apoptosis share common reaction substrates (198), caspase-8, which promotes
apoptosis, and caspase-11, which mediates pyroptosis, that are not
dependent on RIPK1 and RIPK3 and cause inflammation by activating
TNF (200). GPX4 is a key factor
that inhibits lipid peroxidation, ferroptosis and pyroptosis
(201). During sepsis,
mitochondrial damage and release large amounts of ROS to trigger
apoptosis, pyroptosis, autophagy and ferroptosis. Following cell
death, release of inflammatory mediators and excessive production
of ROS induce other types of cell death, which interact with each
other (202,203). Inflammatory mediators released
during apoptosis and necroptosis induce pyroptosis via NLRP3 and
caspase-1 activation, which activate marker of pyroptosis gasdermin
D (61). Death-related protein
kinase 1 inhibits necroptosis by activating P38 MAPK pathway
and regulates mitochondrial autophagy by promoting expression of
NAD-dependent protein deacetylase Sirtuin 1, which simultaneously
decreases ferroptosis via solute carrier family 7 member 11
(132,134).
The present review discusses the mechanisms of
sepsis-induced cardiomyocyte death and interaction of pathways, as
well as current experimental treatment strategies in sepsis-induced
myocardial injury and prospects for the future. It is unclear which
mode of cell death occurs first and which mode of cell death is
most important during sepsis-induced cardiac injury. It remains to
be determined whether cardiomyocytes exhibit different
characteristics from other types of cell during sepsis-induced cell
death. Determining the role of cell death in sepsis-induced cardiac
dysfunction requires further studies to identify the underlying
mechanisms. Increased knowledge of cardiomyocyte death in sepsis
and the molecular mechanisms may facilitate development targeted
therapy options for sepsis-induced myocardial injury in future.
Not applicable.
The present study was supported by Natural Science Foundation of
Liaoning Province (grant no. 20180551029) and Scientific Research
Project of Liaoning Provincial Department of Education (grant no.
LZ2020036).
Not applicable.
GZ and YZ conceived and designed the review. YZ
drafted and edited the manuscript. GZ, DD, XW and YZ reviewed the
manuscript and contributed to the discussion. All authors have read
and approved the final manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleischmann C, Scherag A, Adhikari NKJ,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K;
International Forum of Acute Care Trialists, : Assessment of global
incidence and mortality of hospital-treated sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bergmann O, Bhardwaj RD, Bernard S, Zdunek
S, Barnabé-Heider F, Walsh S, Zupicich J, Alkass K, Buchholz BA,
Druid H, et al: Evidence for cardiomyocyte renewal in humans.
Science. 324:98–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mishra PK, Adameova A, Hill JA, Baines CP,
Kang PM, Downey JM, Narula J, Takahashi M, Abbate A, Piristine HC,
et al: Guidelines for evaluating myocardial cell death. Am J
Physiol Heart Circ Physiol. 317:H891–H922. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nedeva C: Inflammation and cell death of
the innate and adaptive immune system during sepsis. Biomolecules.
11:10112021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Picca A, Calvani R, Coelho-Junior HJ and
Marzetti E: Cell death and inflammation: The role of mitochondria
in health and disease. Cells. 10:5372021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yu Y, Yan Y, Niu F, Wang Y, Chen X, Su G,
Liu Y, Zhao X, Qian L, Liu P and Xiong Y: Ferroptosis: A cell death
connecting oxidative stress, inflammation and cardiovascular
diseases. Cell Death Discov. 7:1932021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pinheiro Da Silva F and Nizet V: Cell
death during sepsis: Integration of disintegration in the
inflammatory response to overwhelming infection. Apoptosis.
14:509–521. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lengeler JW: Metabolic networks: A
signal-oriented approach to cellular models. Biol Chem.
381:911–920. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hotchkiss RS, Strasser A, McDunn JE and
Swanson PE: Cell death. N Engl J Med. 361:1570–1583. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Raff M: Cell suicide for beginners.
Nature. 396:119–122. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Norbury CJ and Hickson ID: Cellular
responses to DNA damage. Annu Rev Pharmacol Toxicol. 41:367–401.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi Y: Mechanisms of caspase activation
and inhibition during apoptosis. Mol Cell. 9:459–470. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Guo R and Li G: Tanshinone modulates the
expression of Bcl-2 and Bax in cardiomyocytes and has a protective
effect in a rat model of myocardial ischemia-reperfusion. Hellenic
J Cardiol. 59:323–328. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Savill J and Fadok V: Corpse clearance
defines the meaning of cell death. Nature. 407:784–788. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Haslett C: Granulocyte apoptosis and
inflammatory disease. Br Med Bull. 53:669–683. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zorc-Pleskovic R, Alibegović A, Zorc M,
Milutinović A, Radovanović N and Petrović D: Apoptosis of
cardiomyocytes in myocarditis. Folia Biol (Praha). 52:6–9.
2006.PubMed/NCBI
|
|
19
|
Fajardo G, Zhao M, Powers J and Bernstein
D: Differential cardiotoxic/cardioprotective effects of
beta-adrenergic receptor subtypes in myocytes and fibroblasts in
doxorubicin cardiomyopathy. J Mol Cell Cardiol. l40:375–383. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zechendorf E, O'riordan CE, Stiehler L,
Wischmeyer N, Chiazza F, Collotta D, Denecke B, Ernst S,
Müller-Newen G, Coldewey SM, et al: Ribonuclease 1 attenuates
septic cardiomyopathy and cardiac apoptosis in a murine model of
polymicrobial sepsis. JCI Insight. 5:e1315712020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Díez J: Apoptosis in cardiovascular
diseases. Rev Esp Cardiol. 53:267–274. 2000.(In Spanish).
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chao J, Yin H, Yao YY, Shen B, Smith RS Jr
and Chao L: Novel role of kallistatin in protection against
myocardial ischemia-reperfusion injury by preventing apoptosis and
inflammation. Hum Gene Ther. 17:1201–1213. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J,
Guo Y, Bolli R and Rokosh G: Stromal cell derived factor-1 alpha
confers protection against myocardial ischemia/reperfusion injury:
Role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis.
Circulation. 116:654–663. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Saxena A, Fish JE, White MD, Yu S, Smyth
JW, Shaw RM, Dimaio JM and Srivastava D: Stromal cell-derived
factor-1alpha is cardioprotective after myocardial infarction.
Circulation. 117:2224–2231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sun Z, Shen L, Sun X, Tong G, Sun D, Han
T, Yang G, Zhang J, Cao F, Yao L and Wang H: Variation of NDRG2 and
c-Myc expression in rat heart during the acute stage of
ischemia/reperfusion injury. Histochem Cell Biol. 135:27–35. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oberholzer C, Oberholzer A, Clare-Salzler
M and Moldawer LL: Apoptosis in sepsis: A new target for
therapeutic exploration. FASEB J. 15:879–892. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wencker D, Chandra M, Nguyen K, Miao W,
Garantziotis S, Factor SM, Shirani J, Armstrong RC and Kitsis RN: A
mechanistic role for cardiac myocyte apoptosis in heart failure. J
Clin Invest. 111:1497–1504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nevière R, Fauvel H, Chopin C, Formstecher
P and Marchetti P: Caspase inhibition prevents cardiac dysfunction
and heart apoptosis in a rat model of sepsis. Am J Respir Crit Care
Med. 163:218–225. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Manetti AC, Maiese A, Paolo MD, Matteis
AD, La Russa R, Turillazzi E, Frati P and Fineschi V: MicroRNAs and
sepsis-induced cardiac dysfunction: A systematic review. Int J Mol
Sci. 22:3212020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lv H, Tian M, Hu P, Wang B and Yang L:
Overexpression of miR-365a-3p relieves sepsis-induced acute
myocardial injury by targeting MyD88/NF-κB pathway. Can J Physiol
Pharmacol. 99:1007–1015. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mirna M, Paar V, Rezar R, Topf A, Eber M,
Hoppe UC, Lichtenauer M and Jung C: MicroRNAs in inflammatory heart
diseases and sepsis-induced cardiac dysfunction: A potential scope
for the future? Cells. 8:13522019. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pasparakis M and Vandenabeele P:
Necroptosis and its role in inflammation. Nature. 517:311–320.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Han J, Zhong CQ and Zhang DW: Programmed
necrosis: Backup to and competitor with apoptosis in the immune
system. Nat Immunol. 12:1143–1149. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Newton K, Dugger DL, Maltzman A, Greve JM,
Hedehus M, Martin-Mcnulty B, Carano RaD, Cao TC, Van Bruggen N,
Bernstein L, et al: RIPK3 deficiency or catalytically inactive
RIPK1 provides greater benefit than MLKL deficiency in mouse models
of inflammation and tissue injury. Cell Death Differ. 23:1565–1576.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Christofferson DE and Yuan J: Necroptosis
as an alternative form of programmed cell death. Curr Opin Cell
Biol. 22:263–268. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cho YS, Challa S, Moquin D, Genga R, Ray
TD, Guildford M and Chan FK: Phosphorylation-driven assembly of the
RIP1-RIP3 complex regulates programmed necrosis and virus-induced
inflammation. Cell. 137:1112–1123. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
He S, Wang L, Miao L, Wang T, Du F, Zhao L
and Wang X: Receptor interacting protein kinase-3 determines
cellular necrotic response to TNF-alpha. Cell. 137:1100–1111. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang DW, Shao J, Lin J, Zhang N, Lu BJ,
Lin SC, Dong MQ and Han J: RIP3, an energy metabolism regulator
that switches TNF-induced cell death from apoptosis to necrosis.
Science. 325:332–336. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun L, Wang H, Wang Z, He S, Chen S, Liao
D, Wang L, Yan J, Liu W, Lei X and Wang X: Mixed lineage kinase
domain-like protein mediates necrosis signalling downstream of RIP3
kinase. Cell. 148:213–227. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhu H and Sun A: Programmed necrosis in
heart disease: Molecular mechanisms and clinical implications. J
Mol Cell Cardiol. 116:125–134. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Moreno-Gonzalez G, Vandenabeele P and
Krysko DV: Necroptosis: A novel cell death modality and its
potential relevance for critical care medicine. Am J Respir Crit
Care Med. 194:415–428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Van Der Poll T, Van De Veerdonk FL,
Scicluna BP and Netea MG: The immunopathology of sepsis and
potential therapeutic targets. Nat Rev Immunol. 17:407–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Vanden Berghe T, Kaiser WJ, Bertrand MJ
and Vandenabeele P: Molecular crosstalk between apoptosis,
necroptosis, and survival signaling. Mol Cell Oncol. 2:e9750932015.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lafont E, Hartwig T and Walczak H: Paving
trail's path with ubiquitin. Trends Biochem Sci. 43:44–60. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Weinlich R, Oberst A, Beere HM and Green
DR: Necroptosis in development, inflammation and disease. Nat Rev
Mol Cell Biol. 18:127–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Oeckinghaus A, Hayden MS and Ghosh S:
Crosstalk in NF-κB signalling pathways. Nat Immunol. 12:695–708.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Newton K and Manning G: Necroptosis and
inflammation. Annu Rev Biochem. 85:743–763. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wu J, Huang Z, Ren J, Zhang Z, He P, Li Y,
Ma J, Chen W, Zhang Y, Zhou X, et al: Mlkl knockout mice
demonstrate the indispensable role of Mlkl in necroptosis. Cell
Res. 23:994–1006. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rosenbaum DM, Degterev A, David J,
Rosenbaum PS, Roth S, Grotta JC, Cuny GD, Yuan J and Savitz SI:
Necroptosis, a novel form of caspase-independent cell death,
contributes to neuronal damage in a retinal ischemia-reperfusion
injury model. J Neurosci Res. 88:1569–1576. 2010.PubMed/NCBI
|
|
50
|
Kaczmarek A, Vandenabeele P and Krysko DV:
Necroptosis: The release of damage-associated molecular patterns
and its physiological relevance. Immunity. 38:209–223. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Schenck EJ, Ma KC, Price DR, Nicholson T,
Oromendia C, Gentzler ER, Sanchez E, Baron RM, Fredenburgh LE, Huh
JW, et al: Circulating cell death biomarker trail is associated
with increased organ dysfunction in sepsis. JCI Insight.
4:e1271432019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kitur K, Wachtel S, Brown A, Wickersham M,
Paulino F, Peñaloza HF, Soong G, Bueno S, Parker D and Prince A:
Necroptosis promotes Staphylococcus aureus clearance by
inhibiting excessive inflammatory signalling. Cell Rep.
16:2219–2230. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Vucur M, Roderburg C, Kaiser L, Schneider
AT, Roy S, Loosen SH, Luedde M, Trautwein C, Koch A, Tacke F and
Luedde T: Elevated serum levels of mixed lineage kinase domain-like
protein predict survival of patients during intensive care unit
treatment. Dis Markers. 2018:19834212018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Peng S, Xu J, Ruan W, Li S and Xiao F:
PPAR-γ activation prevents septic cardiac dysfunction via
inhibition of apoptosis and necroptosis. Oxid Med Cell Longev.
2017:83267492017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Beno SM, Riegler AN, Gilley RP, Brissac T,
Wang Y, Kruckow KL, Jadapalli JK, Wright GM, Shenoy AT, Stoner SN,
et al: Inhibition of necroptosis to prevent long-term cardiac
damage during pneumococcal pneumonia and invasive disease. J Infect
Dis. 222:1882–1893. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zechendorf E, Vaßen P, Zhang J, Hallawa A,
Martincuks A, Krenkel O, Müller-Newen G, Schuerholz T, Simon TP,
Marx G, et al: Heparan sulfate induces necroptosis in murine
cardiomyocytes: A medical-in silico approach combining in vitro
experiments and machine learning. Front Immunol. 9:3932018.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yu S, Yang H, Guo X and Sun Y: Klotho
attenuates angiotensin II-induced cardiotoxicity through
suppression of necroptosis and oxidative stress. Mol Med Rep.
23:662021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yu X, Ruan Y, Huang X, Dou L, Lan M, Cui
J, Chen B, Gong H, Wang Q, Yan M, et al: Dexrazoxane ameliorates
doxorubicin-induced cardiotoxicity by inhibiting both apoptosis and
necroptosis in cardiomyocytes. Biochem Biophys Res Commun.
523:140–146. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Drosatos K, Khan RS, Trent CM, Jiang H,
Son NH, Blaner WS, Homma S, Schulze PC and Goldberg IJ: Peroxisome
proliferator-activated receptor-γ activation prevents
sepsis-related cardiac dysfunction and mortality in mice. Circ
Heart Fail. 6:550–562. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yang X, Lu H, Xie H, Zhang B, Nie T, Fan
C, Yang T, Xu Y, Su H, Tang W and Zhou B: Potent and selective
RIPK1 inhibitors targeting dual-pockets for the treatment of
systemic inflammatory response syndrome and sepsis. Angew Chem Int
Ed Engl. 61:e2021149222022.PubMed/NCBI
|
|
61
|
Fu G, Wang B, He B, Feng M and Yu Y: LPS
induces cardiomyocyte necroptosis through the Ripk3/Pgam5 signaling
pathway. J Recept Signal Transduct Res. 41:32–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Kroemer G, Galluzzi L, Vandenabeele P,
Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS,
Golstein P, Green DR, et al: Classification of cell death:
Recommendations of the nomenclature committee on cell death 2009.
Cell Death Differ. 16:3–11. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Kayar SR and Banchero N: Volume density
and distribution of mitochondria in myocardial growth and
hypertrophy. Respir Physiol. 70:275–286. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beretta M, Santos CX, Molenaar C, Hafstad
AD, Miller CC, Revazian A, Betteridge K, Schröder K,
Streckfuß-Bömeke K, Doroshow JH, et al: Nox4 regulates
InsP3 receptor-dependent Ca2+ release into
mitochondria to promote cell survival. EMBO J. 39:e1035302020.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kim J, Kwon J, Kim M, Do J, Lee D and Han
H: Low-dielectric-constant polyimide aerogel composite films with
low water uptake. Polym J. 48:829–834. 2016. View Article : Google Scholar
|
|
66
|
Weiss JN, Korge P, Honda HM and Ping P:
Role of the mitochondrial permeability transition in myocardial
disease. Circ Res. 93:292–301. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kroemer G, Galluzzi L and Brenner C:
Mitochondrial membrane permeabilization in cell death. Physiol Rev.
87:99–163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Izzo V, Bravo-San Pedro JM, Sica V,
Kroemer G and Galluzzi L: Mitochondrial permeability transition:
New findings and persisting uncertainties. Trends Cell Biol.
26:655–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Isoyama S and Nitta-Komatsubara Y: Acute
and chronic adaptation to hemodynamic overload and ischemia in the
aged heart. Heart Fail Rev. 7:63–69. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Larche J, Lancel S, Hassoun SM, Favory R,
Decoster B, Marchetti P, Chopin C and Neviere R: Inhibition of
mitochondrial permeability transition prevents sepsis-induced
myocardial dysfunction and mortality. J Am Coll Cardiol.
48:377–385. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nesci S: The mitochondrial permeability
transition pore in cell death: A promising drug binding
bioarchitecture. Med Res Rev. 40:811–817. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bauer TM and Murphy E: Role of
mitochondrial calcium and the permeability transition pore in
regulating cell death. Circ Res. 126:280–293. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Azzolin L, Antolini N, Calderan A, Ruzza
P, Sciacovelli M, Marin O, Mammi S, Bernardi P and Rasola A:
Antamanide, a derivative of amanita phalloides, is a novel
inhibitor of the mitochondrial permeability transition pore. PLoS
One. 6:e162802011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhou Q, Xie M, Zhu J, Yi Q, Tan B, Li Y,
Ye L, Zhang X, Zhang Y, Tian J and Xu H: PINK1 contained in
huMSC-derived exosomes prevents cardiomyocyte mitochondrial calcium
overload in sepsis via recovery of mitochondrial Ca2+
efflux. Stem Cell Res Ther. 12:2692021. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Fernandes-Alnemri T, Wu J, Yu JW, Datta P,
Miller B, Jankowski W, Rosenberg S, Zhang J and Alnemri ES: The
pyroptosome: A supramolecular assembly of ASC dimers mediating
inflammatory cell death via caspase-1 activation. Cell Death
Differ. 14:1590–1604. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Robinson N, Ganesan R, Hegedűs C, Kovács
K, Kufer TA and Virág L: Programmed necrotic cell death of
macrophages: Focus on pyroptosis, necroptosis, and parthanatos.
Redox Biol. 26:1012392019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Jorgensen I and Miao EA: Pyroptotic cell
death defends against intracellular pathogens. Immunol Rev.
265:130–142. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Frank D and Vince JE: Pyroptosis versus
necroptosis: Similarities, differences, and crosstalk. Cell Death
Differ. 26:99–114. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Shi J, Gao W and Shao F: Pyroptosis:
Gasdermin-mediated programmed necrotic cell death. Trends Biochem
Sci. 42:245–254. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Fink SL and Cookson BT:
Caspase-1-dependent pore formation during pyroptosis leads to
osmotic lysis of infected host macrophages. Cell Microbiol.
8:1812–1825. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Kayagaki N, Stowe IB, Lee BL, O'Rourke K,
Anderson K, Warming S, Cuellar T, Haley B, Roose-Girma M, Phung QT,
et al: Caspase-11 cleaves gasdermin D for non-canonical
inflammasome signalling. Nature. 526:666–671. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Espinosa-Oliva AM, García-Revilla J,
Alonso-Bellido IM and Burguillos MA: Brainiac caspases: Beyond the
wall of apoptosis. Front Cell Neurosci. 13:5002019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhang Y, Liu X, Bai X, Lin Y, Li Z, Fu J,
Li M, Zhao T, Yang H, Xu R, et al: Melatonin prevents endothelial
cell pyroptosis via regulation of long noncoding RNA
MEG3/miR-223/NLRP3 axis. J Pineal Res. 64:e124492018. View Article : Google Scholar
|
|
85
|
Vande Walle L and Lamkanfi M: Pyroptosis.
Curr Biol. 26:R568–R572. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Kang R, Zeng L, Zhu S, Xie Y, Liu J, Wen
Q, Cao L, Xie M, Ran Q, Kroemer G, et al: Lipid peroxidation drives
gasdermin D-mediated pyroptosis in lethal polymicrobial sepsis.
Cell Host Microbe. 24:97–108.e4. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Xue Z, Xi Q, Liu H, Guo X, Zhang J, Zhang
Z, Li Y, Yang G, Zhou D, Yang H, et al: miR-21 promotes NLRP3
inflammasome activation to mediate pyroptosis and endotoxic shock.
Cell Death Dis. 10:4612019. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hagar JA, Powell DA, Aachoui Y, Ernst RK
and Miao EA: Cytoplasmic LPS activates caspase-11: Implications in
TLR4-independent endotoxic shock. Science. 341:1250–1253. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Cheng KT, Xiong S, Ye Z, Hong Z, Di A,
Tsang KM, Gao X, An S, Mittal M, Vogel SM, et al:
Caspase-11-mediated endothelial pyroptosis underlies
endotoxemia-induced lung injury. J Clin Invest. 127:4124–4135.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Nierhaus A, Winkler MS, Holzmann M,
Mudersbach E, Bauer A, Robbe L, Zahrte C, Schwedhelm E, Daum G,
Kluge S and Zoellner C: Sphingosine-1-phosphate is a novel
biomarker in sepsis severity. Intensive Care Med Exp. 3 (Suppl
1):A7892015. View Article : Google Scholar
|
|
91
|
Song F, Hou J, Chen Z, Cheng B, Lei R, Cui
P, Sun Y, Wang H and Fang X: Sphingosine-1-phosphate receptor 2
signalling promotes caspase-11-dependent macrophage pyroptosis and
worsens scherichia coli sepsis outcome. Anesthesiology.
129:311–320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Bordon Y: Mucosal immunology:
Inflammasomes induce sepsis following community breakdown. Nat Rev
Immunol. 12:400–401. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Guo H, Callaway JB and Ting JP:
Inflammasomes: Mechanism of action, role in disease, and
therapeutics. Nat Med. 21:677–687. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Pu Q, Gan C, Li R, Li Y, Tan S, Li X, Wei
Y, Lan L, Deng X, Liang H, et al: Atg7 deficiency intensifies
inflammasome activation and pyroptosis in pseudomonas sepsis. J
Immunol. 198:3205–3213. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Man SM and Kanneganti TD: Regulation of
inflammasome activation. Immunol Rev. 265:6–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Lamkanfi M and Dixit VM: In retrospect:
The inflammasome turns 15. Nature. 548:534–535. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chu LH, Indramohan M, Ratsimandresy RA,
Gangopadhyay A, Morris EP, Monack DM, Dorfleutner A and Stehlik C:
The oxidized phospholipid oxPAPC protects from septic shock by
targeting the non-canonical inflammasome in macrophages. Nat
Commun. 9:9962018. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Lee SK, Kim YS, Bae GH, Lee HY and Bae YS:
VU0155069 inhibits inflammasome activation independent of
phospholipase D1 activity. Sci Rep. 9:143492019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Li Z, Jia Y, Feng Y, Cui R, Miao R, Zhang
X, Qu K, Liu C and Zhang J: Methane alleviates sepsis-induced
injury by inhibiting pyroptosis and apoptosis: In vivo and in vitro
experiments. Aging (Albany NY). 11:1226–1239. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24:1012152019. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu J, Zhao N, Shi G and Wang H:
Geniposide ameliorated sepsis-induced acute kidney injury by
activating PPARγ. Aging (Albany NY). 12:22744–22758.
2020.PubMed/NCBI
|
|
102
|
Wong WT, Li LH, Rao YK, Yang SP, Cheng SM,
Lin WY, Cheng CC, Chen A and Hua KF: Repositioning of the β-blocker
carvedilol as a novel autophagy inducer that inhibits the NLRP3
inflammasome. Front Immunol. 9:19202018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Tong R, Jia T, Shi R and Yan F: Inhibition
of microRNA-15 protects H9c2 cells against CVB3-induced myocardial
injury by targeting NLRX1 to regulate the NLRP3 inflammasome. Cell
Mol Biol Lett. 25:62020. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen J, Wang B, Lai J, Braunstein Z, He M,
Ruan G, Yin Z, Wang J, Cianflone K, Ning Q, et al: Trimetazidine
attenuates cardiac dysfunction in endotoxemia and sepsis by
promoting neutrophil migration. Front Immunol. 9:20152018.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Dev S and Babitt JL: Overview of iron
metabolism in health and disease. Hemodial Int. 21 (Suppl
1):S6–S20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Drakesmith H and Prentice AM: Hepcidin and
the iron-infection axis. Science. 338:768–772. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Flo TH, Smith KD, Sato S, Rodriguez DJ,
Holmes MA, Strong RK, Akira S and Aderem A: Lipocalin 2 mediates an
innate immune response to bacterial infection by sequestrating
iron. Nature. 432:917–921. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sheldon JR, Laakso HA and Heinrichs DE:
Iron acquisition strategies of bacterial pathogens. Virulence Mech
Bact Pathog. 4:43–85. 2016. View Article : Google Scholar
|
|
109
|
Liu Q, Wu J, Zhang X, Wu X, Zhao Y and Ren
J: Iron homeostasis and disorders revisited in the sepsis. Free
Radic Biol Med. 165:1–13. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Ganz T: Iron in innate immunity: Starve
the invaders. Curr Opin Immunol. 21:63–67. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Hentze MW, Muckenthaler MU and Andrews NC:
Balancing acts: Molecular control of mammalian iron metabolism.
Cell. 117:285–297. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Gaschler MM, Andia AA, Liu H, Csuka JM,
Hurlocker B, Vaiana CA, Heindel DW, Zuckerman DS, Bos PH, Reznik E,
et al: FINO2 initiates ferroptosis through GPX4 inactivation and
iron oxidation. Nat Chem Biol. 14:507–515. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Lei P, Bai T and Sun Y: Mechanisms of
ferroptosis and relations with regulated cell death: A review.
Front Physiol. 10:1392019. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Zhu S, Zhang Q, Sun X, Zeh HJ III, Lotze
MT, Kang R and Tang D: HSPA5 regulates ferroptotic cell death in
cancer cells. Cancer Res. 77:2064–2077. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Yuan H, Li X, Zhang X, Kang R and Tang D:
CISD1 inhibits ferroptosis by protection against mitochondrial
lipid peroxidation. Biochem Biophys Res Commun. 478:838–844. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Gao M, Monian P, Quadri N, Ramasamy R and
Jiang X: Glutaminolysis and transferrin regulate ferroptosis. Mol
Cell. 59:298–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Yu H, Guo P, Xie X, Wang Y and Chen G:
Ferroptosis, a new form of cell death, and its relationships with
tumourous diseases. J Cell Mol Med. 21:648–657. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Cao JY and Dixon SJ: Mechanisms of
ferroptosis. Cell Mol Life Sci. 73:2195–2209. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Fang X, Wang H, Han D, Xie E, Yang X, Wei
J, Gu S, Gao F, Zhu N, Yin X, et al: Ferroptosis as a target for
protection against cardiomyopathy. Proc Natl Acad Sci USA.
116:2672–2680. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Park SJ, Cho SS, Kim KM, Yang JH, Kim JH,
Jeong EH, Yang JW, Han CY, Ku SK, Cho IJ and Ki SH: Protective
effect of sestrin2 against iron overload and ferroptosis-induced
liver injury. Toxicol Appl Pharmacol. 379:1146652019. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Weiland A, Wang Y, Wu W, Lan X, Han X, Li
Q and Wang J: Ferroptosis and its role in diverse brain diseases.
Mol Neurobiol. 56:4880–4893. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bogdan AR, Miyazawa M, Hashimoto K and
Tsuji Y: Regulators of iron homeostasis: New players in metabolism,
cell death, and disease. Trends Biochem Sci. 41:274–286. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Zhu H, Santo A, Jia Z and Li YR: GPx4 in
bacterial infection and polymicrobial sepsis: Involvement of
ferroptosis and pyroptosis. React Oxyg Species (Apex). 7:154–160.
2019.PubMed/NCBI
|
|
129
|
Beatty A, Singh T, Tyurina YY, Tyurin VA,
Samovich S, Nicolas E, Maslar K, Zhou Y, Cai KQ, Tan Y, et al:
Ferroptotic cell death triggered by conjugated linolenic acids is
mediated by ACSL1. Nat Commun. 12:22442021. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Friedmann Angeli JP, Schneider M, Proneth
B, Tyurina YY, Tyurin VA, Hammond VJ, Herbach N, Aichler M, Walch
A, Eggenhofer E, et al: Inactivation of the ferroptosis regulator
Gpx4 triggers acute renal failure in mice. Nat Cell Biol.
16:1180–1191. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Gao G and Chang YZ: Mitochondrial ferritin
in the regulation of brain iron homeostasis and neurodegenerative
diseases. Front Pharmacol. 5:192014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Fang X, Cai Z, Wang H, Han D, Cheng Q,
Zhang P, Gao F, Yu Y, Song Z, Wu Q, et al: Loss of cardiac ferritin
H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ
Res. 127:486–501. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liu Y, Zeng L, Yang Y, Chen C, Wang D and
Wang H: Acyl-CoA thioesterase 1 prevents cardiomyocytes from
doxorubicin-induced ferroptosis via shaping the lipid composition.
Cell Death Dis. 11:7562020. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ma S, Sun L, Wu W, Wu J, Sun Z and Ren Z:
USP22 protects against myocardial ischemia-reperfusion injury via
the SIRT1-P53/SLC7A11-dependent inhibition of ferroptosis-induced
cardiomyocyte death. Front Physiol. 11:5513182020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Lillo-Moya J, Rojas-Solé C,
Muñoz-Salamanca D, Panieri E, Saso L and Rodrigo R: Targeting
ferroptosis against ischemia/reperfusion cardiac injury.
Antioxidants (Basel). 10:6672021. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Wang N, Ma H, Li J, Meng C, Zou J, Wang H,
Liu K, Liu M, Xiao X, Zhang H and Wang K: HSF1 functions as a key
defender against palmitic acid-induced ferroptosis in
cardiomyocytes. J Mol Cell Cardiol. 150:65–76. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Hu H, Chen Y, Jing L, Zhai C and Shen L:
The link between ferroptosis and cardiovascular diseases: A novel
target for treatment. Front Cardiovasc Med. 8:7109632021.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang
CF, Li Y and Zhang Z: Dexmedetomidine alleviated sepsis-induced
myocardial ferroptosis and septic heart injury. Mol Med Rep.
22:175–184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Antonioli M, Di Rienzo M, Piacentini M and
Fimia GM: Emerging mechanisms in initiating and terminating
autophagy. Trends Biochem Sci. 42:28–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Zheng L, Terman A, Hallbeck M, Dehvari N,
Cowburn RF, Benedikz E, Kågedal K, Cedazo-Minguez A and Marcusson
J: Macroautophagy-generated increase of lysosomal amyloid β-protein
mediates oxidant-induced apoptosis of cultured neuroblastoma cells.
Autophagy. 7:1528–1545. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Anand SK, Sharma A, Singh N and Kakkar P:
Entrenching role of cell cycle checkpoints and autophagy for
maintenance of genomic integrity. DNA Repair (Amst). 86:1027482020.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Denton D and Kumar S: Autophagy-dependent
cell death. Cell Death Differ. 26:605–616. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Jiang Y, Gao M, Wang W, Lang Y, Tong Z,
Wang K, Zhang H, Chen G, Liu M, Yao Y and Xiao X: Sinomenine
hydrochloride protects against polymicrobial sepsis via autophagy.
Int J Mol Sci. 16:2559–2573. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Chung MT, Lee YM, Shen HH, Cheng PY, Huang
YC, Lin YJ, Huang YY and Lam KK: Activation of autophagy is
involved in the protective effect of 17β-oestradiol on
endotoxaemia-induced multiple organ dysfunction in ovariectomized
rats. J Cell Mol Med. 21:3705–3717. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Jia J, Gong X, Zhao Y, Yang Z, Ji K, Luan
T, Zang B and Li G: Autophagy enhancing contributes to the organ
protective effect of alpha-lipoic acid in septic rats. Front
Immunol. 10:14912019. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Lu LH, Chao CH and Yeh TM: Inhibition of
autophagy protects against sepsis by concurrently attenuating the
cytokine storm and vascular leakage. J Infect. 78:178–186. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Cui SN, Chen ZY, Yang XB, Chen L, Yang YY,
Pan SW, Wang YX, Xu JQ, Zhou T, Xiao HR, et al: Trichostatin A
modulates the macrophage phenotype by enhancing autophagy to reduce
inflammation during polymicrobial sepsis. Int Immunopharmacol.
77:1059732019. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Oami T, Watanabe E, Hatano M, Sunahara S,
Fujimura L, Sakamoto A, Ito C, Toshimori K and Oda S: Suppression
of t cell autophagy results in decreased viability and function of
T cells through accelerated apoptosis in a murine sepsis model.
Crit Care Med. 45:e77–e85. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Patoli D, Mignotte F, Deckert V, Dusuel A,
Dumont A, Rieu A, Jalil A, Van Dongen K, Bourgeois T, Gautier T, et
al: Inhibition of mitophagy drives macrophage activation and
antibacterial defense during sepsis. J Clin Invest. 130:5858–5874.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Dong G, Si C, Zhang Q, Yan F, Li C, Zhang
H, Ma Q, Dai J, Li Z, Shi H, et al: Autophagy regulates
accumulation and functional activity of granulocytic
myeloid-derived suppressor cells via STAT3 signaling in endotoxin
shock. Biochim Biophys Acta Mol Basis Dis. 1863:2796–2807. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Jin L, Batra S and Jeyaseelan S: Deletion
of NLRP3 augments survival during polymicrobial sepsis by
decreasing autophagy and enhancing phagocytosis. J Immunol.
198:1253–1262. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Ge Y, Huang M, Dong N and Yao YM: Effect
of interleukin-36β on activating autophagy of CD4+CD25+ regulatory
T cells and its immune regulation in sepsis. J Infect Dis.
222:1517–1530. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Park SY, Shrestha S, Youn YJ, Kim JK, Kim
SY, Kim HJ, Park SH, Ahn WG, Kim S, Lee MG, et al: Autophagy primes
neutrophils for neutrophil extracellular trap formation during
sepsis. Am J Respir Crit Care Med. 196:577–589. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Napolitano LM: Sepsis 2018: Definitions
and guideline changes. Surg Infect (Larchmt). 19:117–125. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Jiang P and Mizushima N: Autophagy and
human diseases. Cell Res. 24:69–79. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Lemasters JJ: Selective mitochondrial
autophagy, or mitophagy, as a targeted defense against oxidative
stress, mitochondrial dysfunction, and aging. Rejuvenation Res.
8:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Carchman EH, Whelan S, Loughran P, Mollen
K, Stratamirovic S, Shiva S, Rosengart MR and Zuckerbraun BS:
Experimental sepsis-induced mitochondrial biogenesis is dependent
on autophagy, TLR4, and TLR9 signaling in liver. FASEB J.
27:4703–4711. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Kubli DA, Quinsay MN, Huang C, Lee Y and
Gustafsson AB: Bnip3 functions as a mitochondrial sensor of
oxidative stress during myocardial ischemia and reperfusion. Am J
Physiol Heart Circ Physiol. 295:H2025–H2031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Pohl C and Dikic I: Cellular quality
control by the ubiquitin-proteasome system and autophagy. Science.
366:818–822. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Ma J, Wang Y, Zheng D, Wei M, Xu H and
Peng T: Rac1 signalling mediates doxorubicin-induced cardiotoxicity
through both reactive oxygen species-dependent and -independent
pathways. Cardiovasc Res. 97:77–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Wu C, Zhou XX, Li JZ, Qiang HF, Wang Y and
Li G: Pretreatment of cardiac progenitor cells with bradykinin
attenuates H2O2-induced cell apoptosis and
improves cardiac function in rats by regulating autophagy. Stem
Cell Res Ther. 12:4372021. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Jiang YJ, Sun SJ, Cao WX, Lan XT, Ni M, Fu
H, Li DJ, Wang P and Shen FM: Excessive ROS production and enhanced
autophagy contribute to myocardial injury induced by branched-chain
amino acids: Roles for the AMPK-ULK1 signaling pathway and α7nAChR.
Biochim Biophys Acta Mol Basis Dis. 1867:1659802021. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Huang J, Lam GY and Brumell JH: Autophagy
signalling through reactive oxygen species. Antioxid Redox Signal.
14:2215–2231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Takahashi W, Watanabe E, Fujimura L,
Watanabe-Takano H, Yoshidome H, Swanson PE, Tokuhisa T, Oda S and
Hatano M: Kinetics and protective role of autophagy in a mouse
cecal ligation and puncture-induced sepsis. Crit Care. 17:R1602013.
View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Yen YT, Yang HR, Lo HC, Hsieh YC, Tsai SC,
Hong CW and Hsieh CH: Enhancing autophagy with activated protein C
and rapamycin protects against sepsis-induced acute lung injury.
Surgery. 153:689–698. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci
B, Xie Y, Carlson D, Rothermel BA, Sun Y, et al: Beclin-1-dependent
autophagy protects the heart during sepsis. Circulation.
138:2247–2262. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Liu JJ, Li Y, Yang MS, Chen R and Cen CQ:
SP1-induced ZFAS1 aggravates sepsis-induced cardiac dysfunction via
miR-590-3p/NLRP3-mediated autophagy and pyroptosis. Arch Biochem
Biophys. 695:1086112020. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Wang Q, Yang X, Song Y, Sun X, Li W, Zhang
L, Hu X, Wang H, Zhao N, Zhuang R, et al: Astragaloside
IV-targeting miRNA-1 attenuates lipopolysaccharide-induced cardiac
dysfunction in rats through inhibition of apoptosis and autophagy.
Life Sci. 275:1194142021. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Wu B, Song H, Fan M, You F, Zhang L, Luo
J, Li J, Wang L, Li C and Yuan M: Luteolin attenuates
sepsis-induced myocardial injury by enhancing autophagy in mice.
Int J Mol Med. 45:1477–1487. 2020.PubMed/NCBI
|
|
173
|
Han W, Wang H, Su L, Long Y, Cui N and Liu
D: Inhibition of the mTOR pathway exerts cardioprotective effects
partly through autophagy in CLP rats. Mediators Inflamm.
2018:47982092018. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Sang Z, Zhang P, Wei Y and Dong S:
miR-214-3p attenuates sepsis-induced myocardial dysfunction in mice
by inhibiting autophagy through PTEN/AKT/mTOR pathway. Biomed Res
Int. 2020:14090382020. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Hsieh CH, Pai PY, Hsueh HW, Yuan SS and
Hsieh YC: Complete induction of autophagy is essential for
cardioprotection in sepsis. Ann Surg. 253:1190–1200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Yu T, Liu D, Gao M, Yang P, Zhang M, Song
F, Zhang X and Liu Y: Dexmedetomidine prevents septic myocardial
dysfunction in rats via activation of α7nAChR and PI3K/Akt-mediated
autophagy. Biomed Pharmacother. 120:1092312019. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Zhang E, Zhao X, Zhang L, Li N, Yan J, Tu
K, Yan R, Hu J, Zhang M, Sun D and Hou L: Minocycline promotes
cardiomyocyte mitochondrial autophagy and cardiomyocyte autophagy
to prevent sepsis-induced cardiac dysfunction by Akt/mTOR
signaling. Apoptosis. 24:369–381. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Yuan X, Chen G, Guo D, Xu L and Gu Y:
Polydatin alleviates septic myocardial injury by promoting
SIRT6-mediated autophagy. Inflammation. 43:785–795. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Gao J, Chen X, Shan C, Wang Y, Li P and
Shao K: Autophagy in cardiovascular diseases: Role of noncoding
RNAs. Mol Ther Nucleic Acids. 23:101–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Leng Y, Zhang Y, Li X, Wang Z, Zhuang Q
and Lu Y: Receptor interacting protein kinases 1/3: The potential
therapeutic target for cardiovascular inflammatory diseases. Front
Pharmacol. 12:7623342021. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Hsieh YC, Athar M and Chaudry IH: When
apoptosis meets autophagy: Deciding cell fate after trauma and
sepsis. Trends Mol Med. 15:129–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Nishida K, Yamaguchi O and Otsu K:
Crosstalk between autophagy and apoptosis in heart disease. Circ
Res. 103:343–351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Galluzzi L, Bravo-San Pedro JM, Vitale I,
Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D,
Annicchiarico-Petruzzelli M, et al: Essential versus accessory
aspects of cell death: Recommendations of the NCCD 2015. Cell Death
Differ. 22:58–73. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Speir M and Lawlor KE: RIP-roaring
inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation
and autoinflammatory disease. Semin Cell Dev Biol. 109:114–124.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Kunchithapautham K and Rohrer B: Apoptosis
and autophagy in photoreceptors exposed to oxidative stress.
Autophagy. 3:433–441. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Nagata S, Hanayama R and Kawane K:
Autoimmunity and the clearance of dead cells. Cell. 140:619–630.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Humphries F, Yang S, Wang B and Moynagh
PN: RIP kinases: Key decision makers in cell death and innate
immunity. Cell Death Differ. 22:225–236. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Feoktistova M, Makarov R, Yazdi AS and
Panayotova-Dimitrova D: RIPK1 and TRADD regulate TNF-induced
signaling and ripoptosome formation. Int J Mol Sci. 22:124592021.
View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Ofengeim D and Yuan J: Regulation of rip1
kinase signalling at the crossroads of inflammation and cell death.
Nat Rev Mol Cell Biol. 14:727–736. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
191
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
192
|
Lu ZY, Cheng MH, Yu CY, Lin YS, Yeh TM,
Chen CL, Chen CC, Wan SW and Chang CP: Dengue nonstructural protein
1 maintains autophagy through retarding caspase-mediated cleavage
of beclin-1. Int J Mol Sci. 21:97022020. View Article : Google Scholar : PubMed/NCBI
|
|
193
|
Korsmeyer SJ, Wei MC, Saito M, Weiler S,
Oh KJ and Schlesinger PH: Pro-apoptotic cascade activates BID,
which oligomerizes BAK or BAX into pores that result in the release
of cytochrome c. Cell Death Differ. 7:1166–1173. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
194
|
Almeida RD, Manadas BJ, Carvalho AP and
Duarte CB: Intracellular signaling mechanisms in photodynamic
therapy. Biochim Biophys Acta. 1704:59–86. 2004.PubMed/NCBI
|
|
195
|
Fefelova N, Wongjaikam S, Siri-Angkul N,
Gwathmey J, Chattipakorn N, Chattipakorn S and Xie LH: Abstract
15737: Deficiency of mitochondrial calcium uniporter protects mouse
hearts from iron overload by attenuating ferroptosis. Circulation.
142 (Suppl 3):A157372020. View Article : Google Scholar
|
|
196
|
Yin Z, Ding G, Chen X, Qin X, Xu H, Zeng
B, Ren J, Zheng Q and Wang S: Beclin1 haploinsufficiency rescues
low ambient temperature-induced cardiac remodeling and contractile
dysfunction through inhibition of ferroptosis and mitochondrial
injury. Metabolism. 113:1543972020. View Article : Google Scholar : PubMed/NCBI
|
|
197
|
Kang R, Zhu S, Zeh HJ, Klionsky DJ and
Tang D: BECN1 is a new driver of ferroptosis. Autophagy.
14:2173–2175. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
198
|
Shao W, Yeretssian G, Doiron K, Hussain SN
and Saleh M: The caspase-1 digestome identifies the glycolysis
pathway as a target during infection and septic shock. J Biol Chem.
282:36321–36329. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
199
|
Rogers C, Fernandes-Alnemri T, Mayes L,
Alnemri D, Cingolani G and Alnemri ES: Cleavage of DFNA5 by
caspase-3 during apoptosis mediates progression to secondary
necrotic/pyroptotic cell death. Nat Commun. 8:141282017. View Article : Google Scholar : PubMed/NCBI
|
|
200
|
Mandal P, Feng Y, Lyons JD, Berger SB,
Otani S, Delaney A, Tharp GK, Maner-Smith K, Burd EM, Schaeffer M,
et al: Caspase-8 collaborates with caspase-11 to drive tissue
damage and execution of endotoxic shock. Immunity. 49:42–55.e6.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
201
|
Russo AJ and Rathinam VAK: Lipid
peroxidation adds fuel to pyr(optosis). Cell Host Microbe. 24:8–9.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
202
|
Bruni A, Bornstein S, Linkermann A and
Shapiro AMJ: Regulated cell death seen through the lens of islet
transplantation. Cell Transplant. 27:890–901. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
203
|
Zhou W, Chen C, Chen Z, Liu L, Jiang J, Wu
Z, Zhao M and Chen Y: NLRP3: A novel mediator in cardiovascular
disease. J Immunol Res. 2018:57021032018. View Article : Google Scholar : PubMed/NCBI
|