Introduction
The highly specialized pancreatic β-cells are
located in the pancreas in structures called islets of Langerhans
(1). They play a unique and
important role in the physiology of organisms, as they synthesize
the hormone insulin (2). This
protein travels through the bloodstream to reach target peripheral
tissues to promote the uptake, utilization and storage of
nutrients, mainly glucose (3).
Therefore, β-cells are responsible for producing and secreting
insulin under tight regulation, which helps maintain circulating
glucose concentrations in the physiological range (1).
Insulin release is carefully regulated through a
feedback system that involves both insulin-sensitive tissues and
β-cells. The demand for insulin depends on physiological and
pathological conditions, such as aging, pregnancy and obesity.
Therefore, β-cells must adapt to these conditions by activating
different mechanisms, such as hyperplasia and hypertrophy, as well
as increasing insulin synthesis and secretion (2). In situations of metabolic stress, the
demand for insulin increases, and β-cells initially release more
insulin to respond to this demand. However, β-cell functions tend
to decline in response to this stress, resulting in decreased
glucose tolerance (3). Since
insulin plays a fundamental role in regulating glucose metabolism,
any alteration in its secretion, action, or both can generate a
series of metabolic imbalances characterized by chronic
hyperglycemia known as diabetes mellitus (DM) (2,3).
DM represents a serious public health problem
worldwide (4). In recent decades,
the health and economic burdens of DM have increased worldwide,
significantly impacting low- and middle-income countries (5). The 10th edition of the IDF Diabetes
Atlas reports a continued global increase in the prevalence of
diabetes; by 2021, 537 million adults (20–79 years) were living
with diabetes, and this number is projected to increase to 643
million by 2030 and 783 million by 2045 (6). It has also been reported that by
2021, diabetes was responsible for 6.7 million deaths and that
there were also 541 million adults with glucose intolerance, which
places them at high risk of suffering from diabetes (Fig. 1).
Diabetes can be divided into two main categories: i)
Type 1 diabetes (T1D) and ii) type 2 diabetes (T2D) (7). T1D involves an autoimmune process
that gradually and selectively destroys the β-cells of the
pancreas, leading to total insulin deficiency (7). While T2D encompasses ~90% of all DM
cases, with most presenting with a prediabetic state (7). Tissue sensitivity to the actions of
insulin is reduced, mainly in muscle, liver and adipose tissue
(8). This leads to failure in the
internalization of glucose in response to normal concentrations of
this hormone, which is known as insulin resistance (IR) (8). The clinical manifestations of the
disease occur when β-cells are unable to produce sufficient
compensatory insulin to counteract the resistance of the body to
insulin (9–12). Increased blood glucose can lead to
autoxidation or cross-linking with proteins present in the serum
(protein glycosylation) to give rise to a series of highly reactive
structures termed Amadori compounds (13). Prior to the clinical manifestations
of T2D, the β-cell mass decreases by up to 60% due to the increased
rate of apoptosis (11,12). In this sense, it should be noted
that apoptosis is a form of cell death that occurs in both
physiological and pathological situations in multicellular
organisms and constitutes a common mechanism of cell replacement,
tissue remodeling and renewal of damaged cells (14). Although it is a complex process, it
is characterized by cell shrinkage, chromatin condensation, DNA
fragmentation and the formation of apoptotic bodies (Fig. 2) (14). Apoptotic cells are rapidly
phagocytosed by neighboring cells or macrophages, thus preventing
an inflammatory reaction (14,15).
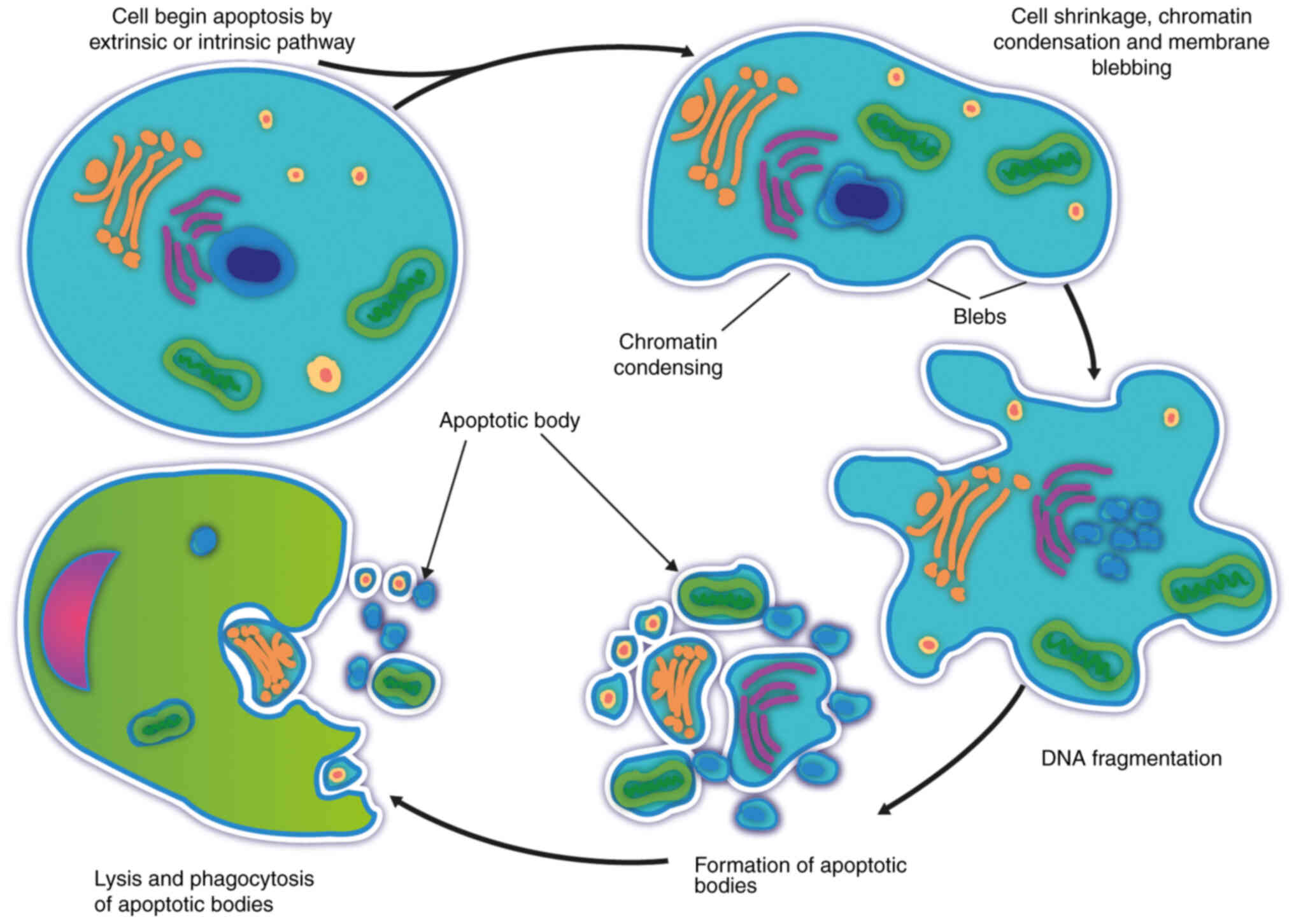 | Figure 2.Cell death by apoptosis. Apoptosis is
a physiological mechanism of programmed cell death that begins with
early chromatin condensation, leading to the formation of
perinuclear chromatin masses and decreased nuclear size, cytoplasm
compaction and cytoskeletal alterations, leading to cellular
shrinkage. In the same way, intranuclear DNA fragmentation occurs,
which leads to fragmentation of the cell, forming small vesicles
called apoptotic bodies still surrounded by a membrane, which
changes its composition, resulting in the translocation of
phosphatidylserine to its surface, which serves as a recognition
signal for macrophages; thus, the bodies are quickly
phagocytosed. |
Various potential mechanisms have been identified to
explain the increase in β-cell apoptosis during T2D, among which
chronic hyperglycemia is a key factor (12). The process of cell death by
apoptosis involves the participation of a series of regulatory
factors. One of the most crucial and widely studied factors in this
process is p53, which functions as the ‘guardian of the genome’
since it plays a fundamental role in the supervision of cellular
stress and in inducing apoptosis in tissues where stressors cause
severe and irreversible damage (16).
Therefore, the present review focuses on the role of
p53 in reducing pancreatic β-cells in the context of hyperglycemia.
It was previously shown that an increase in glucose promotes the
mobilization of p53 toward the mitochondria, increasing the
production of reactive oxygen species (ROS) by modifying the
mitochondrial membrane potential (17). Therefore, a series of experimental
evidence corresponding to the post-translational modifications of
p53 induced by hyperglycemia has been obtained (Table I). These effects are closely
related to the regulation of apoptosis in the pancreatic β-cell
line RINm5F by high glucose. In addition, the regulation that this
protein undergoes through its ubiquitination by murine protein
double minute 2 (Mdm2) will be analyzed (10,11,18).
 | Table I.Characteristics of the main articles
reviewed. |
Table I.
Characteristics of the main articles
reviewed.
| First author/s,
year | Methodology | Results | (Refs.) |
|---|
| Ortega-Camarillo
et al, | - Apoptosis index:
FC (PS exposure) | - High glucose
increased apoptosis | (17) |
| 2006 | and DNA
fragmentation | and translocation
of p53 protein |
|
|
| - p53 localization:
WB/IF | to the mitochondria
of RINm5F |
|
|
| - ROS production:
FC (DCDHF-DA) | cells |
|
|
| - ψm: JC-1 confocal
microscopy | - Alterations in ψm
and an increase |
|
|
| - NADPH-oxidase
inhibitors | in NADPH-oxidase
and mito- |
|
|
|
| chondrial ROS were
also observed. |
|
|
|
| - Increased glucose
stimulated p53 |
|
|
|
| mobilization to
mitochondria, ROS |
|
|
|
| production and
pancreatic β-cell death |
|
| Flores-López et
al, | - P-p53, p-p38MAPK:
WB mito- | - Localization of
p53 and its phospho- | (11) |
| 2013 |
chondria/cytosol | rylation in
mitochondria was repressed |
|
|
| - Apoptosis | in RINm5F cells
cultured at high |
|
|
| - Caspase-3;
Bcl-2/Bax ratio and | glucose and with
the p38 MAPK |
|
|
| cyt c: WB
mitochondria/cytosol | inhibitor. |
|
|
| - DNA
fragmentation | - Deletion of p38
MAPK also prevented |
|
|
| - P38 MAPK
inhibitor | mitochondrial
cytochrome c egress and |
|
|
|
| decreased apoptosis
of RINm5F cells |
|
| Flores-López et
al, | -
Phosphorylation | - The localization
and phosphorylation | (18) |
| 2012 | -
O-N-Acetylglucosaminylation and | of p53 at Ser 15
and Ser 392 in mito |
|
|
|
Poly-ADP-ribosylation of P53: IP; | chondria were
associated with an |
|
|
| WB | increase in ROS,
decrease in the Bcl-2/ |
|
|
|
| Bax ratio and
increase in apoptosis. |
|
|
|
| - High glucose
increased PARP concen- |
|
|
|
| tration and
Poly-ADP-ribosylation of |
|
|
|
| p53 in the nuclear
fraction. |
|
|
|
| -
N-acetylglucosaminylation of p53 |
|
|
|
| increases first in
the cytosol and at |
|
|
|
| 24 h in the
mitochondria, together |
|
|
|
| with p53
mobilization to this organelle. |
|
|
|
| - High glucose
induces apoptosis in |
|
|
|
| RINm5F cells in a
time-dependent |
|
|
|
| manner without any
changes of |
|
|
|
| mRNA p53
expression, although |
|
|
|
| alterations occur
in intracellular distri- |
|
|
|
| bution and
phosphorylation. |
|
| Barzalobre- | - p53-Mdm2 complex:
IP; WB; IF | - High glucose
reduced Mdm2 (mRNA | (10) |
| Gerónimo et
al, | - Ub-p53: WB | and protein) but
increased Mdm2 and |
|
| 2015 | - pAkt: WB | Akt
phosphorylation. |
|
|
|
| - It observed
p53-Mdm2 complex form- |
|
|
|
| ation, but p53
ubiquitination was |
|
|
|
| suppressed.
Phosphorylation of p53 |
|
|
|
| Ser15 and ATM was
increased by high |
|
|
|
| glucose. Therefore,
the reduction of |
|
|
|
| pancreatic β-cells
mass is favored by |
|
|
|
| stabilization of
p53 due to reduced |
|
|
|
| expression of Mdm2
and low p53 |
|
|
|
|
ubiquitination. |
|
| Barzalobre- | - Animals drinking
water containing | - Carbohydrate in
drinking water | (9) |
| Geronimo et
al, | either 40% sucrose
or 40% fructose. | promotes apoptosis
and mobilization |
|
| 2023 | - Glucose tolerance
test was per- | of p53 from the
cytosol to the mito- |
|
|
| formed at week
15. | chondria of rat
pancreas β-cells before |
|
|
| - After 4 months
was measured: | blood glucose
rises. An increase in |
|
|
| - Apoptosis:
TUNEL. | p53, miR-34a and
Bax mRNA |
|
|
| - Bax and p53: WB,
IF and qPCR. | (P<0.001) was
detected in the sucrose |
|
|
| - Insulin,
triacylglycerol, serum | group. In addition
to hypertrigly- |
|
|
| glucose and fatty
acids in pan- | ceridemia,
hyperinsulinemia, glucose |
|
|
| creatic tissue were
measured | intolerance,
insulin resistance, accumu- |
|
|
|
| lation of visceral
fat and increased |
|
|
|
| pancreatic fatty
acids in the sucrose |
|
|
|
| group. Carbohydrate
consumption |
|
|
|
| increases p53 and
its mobilization into |
|
|
|
| β-cell mitochondria
and increases the |
|
|
|
| rate of apoptosis,
which occurs before |
|
|
|
| serum glucose
levels increase. |
|
For the present narrative literature review, PubMed
(https://pubmed.ncbi.nlm.nih.gov/) and
Web of Science (https://clarivate.com/products/scientific-and-academic-research/research-discovery-and-workflow-solutions/webofscience-platform/)
were searched using the keywords ‘apoptosis’, ‘pancreatic β-cell
apoptosis and hyperglycemia’, ‘p53 and pancreatic β-cell
apoptosis’, ‘p53 and hyperglycemia’, ‘posttranslational
modifications of p53’, and ‘p53 and ROS’. The most relevant
articles in English were selected and reviewed, and articles that
analyzed the role of p53 in the death of pancreatic β-cells due to
high glucose levels were identified. Also included is the
description of death by apoptosis, as well as other mechanisms
involved in the damage of β-cells due to high glucose levels.
Apoptotic pathways are the crossroads where
signals and regulatory mechanisms converge
Apoptosis constitutes an essential mechanism for the
selective elimination of cells and is integrally involved in
various biological events (19).
This process involves the activation of caspases, a family of
proteases with a cysteine residue in their active site that is key
in the transduction and execution of apoptotic signals in response
to various stimuli (14). They
exist as zymogens (proenzymes) in the cytoplasm, endoplasmic
reticulum, mitochondria and nuclear matrix of most cells (20). The apoptosis process can be
triggered by two fundamental pathways, the extrinsic pathway and
the intrinsic pathway, with both pathways including the activation
of caspases (14,20).
The extrinsic pathway of apoptosis is induced by
death receptors belonging to the superfamily of tumor necrosis
factor (TNF) receptors. Two of the most characterized receptors are
the Fas receptor, also termed CD95 or apoptosis-1 protein, and the
TNF receptor (TNFR). These receptors share a homologous sequence
called the death domain (DD), which can initiate a cascade of
events that lead to apoptosis upon receiving extracellular signals
(Fig. 3) (19). The ligands that bind to these
receptors belong to the TNF family and include the Fas ligand and
TNF. The activated Fas receptor recruits adapter molecules such as
the Fas-associated DD (FADD), which also contains a DD that binds
to the Fas receptor analog domain, and a death effector domain
(DED) that binds to a Fas receptor analog domain of procaspase-8
(14,19). This enzyme, also termed FADD-like
IL-1β-converting enzyme (FLICE), undergoes autocatalytic activation
by binding to FADD, becoming active caspase-8 (21). The complex formed by Fas, FADD and
FLICE is called the death-inducing signaling complex. Caspase-8
activates effector caspases 3, 6 and 7, which in turn release
various cellular proteins involved in proteolysis and DNA
degradation. In the case of TNF receptors (TNFR-I and TNFR-II),
they recruit the adapter molecule TRADD (TNFR-associated death
domain) (21). Thus, the complex
formed by these components activates both initiator caspases (8 and
10) and effector caspases (3, 6 and 7), similar to what is observed
with the Fas receptor (22,23).
 | Figure 3.Pathways of apoptosis. There are two
main signaling pathways by which a cell becomes apoptotic. The
extrinsic pathway begins by binding specific ligands to death
receptors on the cell surface, and this interaction leads to the
activation of caspases 8 and 10. The intrinsic pathway begins at
the mitochondrial level and leads to the formation of the
apoptosome, a protein complex that leads to the activation of
caspase-9. Caspases 8, 10 and 9 are known as regulatory caspases,
and when active, they begin an activation cascade of other
molecules, including caspases 3, 6 and 7, which are known as
effector caspases that initiate the proteolytic cascade that leads
to death by apoptosis. TNFR1, tumor necrosis factor receptor 1;
FADD, Fas-associated DD; TRADD, TNFR-associated death domain;
APAF-1, apoptotic protease activating factor 1; Cyt c, cytochrome
c; dATP, deoxyadenosine triphosphate; ROS, reactive oxygen species;
DED, death effector domain; PTP, permeability transition pore; Δψm,
transmembrane potential. |
The intrinsic pathway of apoptosis is triggered by
internal signals that focus on mitochondria (23). In the context of the intrinsic
pathway, there are contact zones between the mitochondrial
membranes, termed ‘dense zones’, whose protein components interact
to form the mitochondrial permeability transition pore (PTP), which
includes proteins of different cellular parts: The cytoplasm
(hexokinase), outer membrane [voltage-gated anion channel (VDAC)],
inner membrane [adenine nucleotide translocase (ANT)] and
mitochondrial matrix [cyclophilin D (CpD)] (24). Under physiological conditions, the
different components of the PTP are scattered (25). Thus, VDAC contributes to the
permeabilization of the outer membrane, ANT specifically controls
the passage of the different phosphorylated and unphosphorylated
forms of adenine nucleotides through the inner membrane, and CpD is
a peptidyl-propyl isomerase that is crucial for protein folding
(26). When an apoptotic stimulus
reaches the mitochondria, these protein components can assemble,
forming a pore with a radius of 1.0–1.3 nm, allowing the
nonselective passage of molecules <1.5 kDa (25). Its opening leads to the
permeabilization of mitochondrial membranes, which contributes to
mitochondrial dysregulation and the loss of transmembrane potential
(Δψm) due to an increase in matrix osmolarity (24). In addition, the entry of water
causes swelling of mitochondria and rupture of the outer membrane,
releasing molecules from the intermembrane space into the cytoplasm
(25). A total of 79 peptide
components that are released during the opening of the PTP have
been characterized (25,26). These components include molecules
with known pro-apoptotic activity, such as cytochrome c (cyt c),
Smac/DIABLO, apoptotic protease activating factor 1 (Apaf-1) and
apoptosis-inducing factor (AIF), and some members of the caspase
family, such as caspase-2 and caspase-9 (27,28).
Once released into the cytoplasm, these agents can activate
different apoptotic pathways. AIF triggers chromatin condensation
and DNA fragmentation independent of caspase activation (29). The pro-apoptotic proteins Bax and
Bak have been shown to accelerate the opening of VDAC channels and
allow cytochrome c egress, while the anti-apoptotic proteins Bcl-2
and Bcl-XL close these channels by directly binding to Bax and Bak
(30,31). Cytochrome c released into the
cytosol favors the activation of Apaf-1, which can oligomerize in
the presence of cytochrome c and deoxyadenosine triphosphate (dATP)
(23). The oligomers recruit
procaspase-9 using the recruitment domain of Apaf-1, thus forming
the apoptosome (23). Mature
caspase-9 is released from the complex and activates effector
caspases 3, 6 and 7, leading to the initiation of the proteolytic
cascade essential for apoptosis (15,27,32).
Glucolipotoxicity and p53 activation
Among the mechanisms involved in the induction of
damage to pancreatic β-cells, those related to the combination of
hyperglycemia and hyperlipidemia are the most studied. This is
justified by the exacerbated increase in the number of obese
people, including infants, worldwide. An increase in free fatty
acids (FFAs) in the circulation combined with IR and hyperglycemia,
which are frequently observed in individuals with obesity,
constitutes a state known as glucolipotoxicity and is involved in
the pathogenesis of T2D (33).
Excessive and chronic accumulation of fatty acids
inhibits insulin secretion in response to glucose and increases the
intracellular production of lipid intermediates, which are toxic to
β-cells and activate apoptosis (34). In these circumstances, the activity
of carnitine palmitoyl transferase 1 decreases due to the increase
in malonyl-CoA, resulting in the subsequent esterification of FFAs
and the generation of complex lipids such as sphingolipids and
ceramides, which accumulate in pancreatic β-cells and trigger cell
death (33).
An increase in FFAs promotes p53 activity. It has
been shown that in pancreatic β-cells (NIT-1 cells), palmitic acid
inhibits cell proliferation by increasing the expression of p53
(34) and activates apoptosis via
the p38 MAPK/p53 and NFκB pathway (35). The apoptosis of β-cells induced by
exposure to FFAs has also been related to the expression of miR-34
(36), which inhibits Akt and Mdm2
activation and p53 degradation (10,37).
Glucolipotoxicity also promotes the inflammatory response in
β-cells, which is manifested by the increased activity of
proinflammatory cytokines such as IFN-γ and TNF-α, which contribute
to the dysfunction and death of β-cells (38). Alterations in the expression of the
transcription factors Mafa and PDX-1 and of the insulin gene have
also been observed after exposure to FFAs and glucose (35,37).
Endoplasmic reticulum stress
Alterations in protein folding and the accumulation
of poorly folded proteins in the endoplasmic reticulum generate a
state known as endoplasmic reticulum stress. This response begins
with the activation of protein kinase RNA-like ER kinase (PERK),
activating transcription factor-6 and inositol-requiring enzyme-1,
whose activation decreases the translation of proteins and
increases the folding capacity of the endoplasmic reticulum and the
degradation of poorly folded proteins (38). If this process is not successful,
then the apoptosis process begins (38).
Excessive weight gain can lead to a state of insulin
resistance, during which insulin-dependent tissues are unable to
internalize glucose molecules (3).
In these circumstances, the demand for insulin increases, and
β-cells must synthesize and secrete more insulin. This demand also
falls on the endoplasmic reticulum, which must process more insulin
molecules (8). Exposure to
saturated FFAs such as palmitate affects the response capacity of
the endoplasmic reticulum and increases the levels of poorly folded
proteins, resulting in stress in the endoplasmic reticulum
(38). If the demand for
continuous insulin and ER stress are not resolved, then apoptosis
is activated due to the PERK-dependent increase in the
transcription factor CHOP, increased expression of the
pro-apoptotic proteins Puma and DP5, and inhibition of the
anti-apoptotic protein MCL-1 (39). One study has reported that
endoplasmic reticulum stress markers are also increased in patients
with T2D (38). The participation
of p53 in stress-induced apoptosis of the endoplasmic reticulum has
been related to the regulation of the expression of pro-apoptotic
and anti-apoptotic proteins.
Implication of cytokines and inflammation in
pancreatic β-cell dysfunction
Obesity constitutes a chronic low-intensity
inflammatory state that activates the innate immune system, which
in turn can alter glucose tolerance, contribute to the development
of insulin resistance, and progress to diabetes and cardiovascular
disease (40,41). An increase in the plasma
concentration of C-reactive protein in the acute phase predicts the
presence of coronary heart disease, metabolic syndrome and T2D
(40).
In addition, Hotamisligil (41) was one of the first researchers to
establish that adipocytes express TNF-α and that this expression is
related to obesity and resistance to systemic insulin. Other
proinflammatory molecules, such as resistin, IL-6, serum amyloid
A3, acid glycoprotein A1 and monocyte chemotactic protein-1, are
also expressed in adipose tissue and potentially induce themselves
in response to obesity and diabetes. These proinflammatory
molecules contribute to alterations in pancreatic β-cells (42). Diabetes is characterized by the
infiltration of inflammatory cells into the pancreas, which favors
the progressive destruction of β-cells (7). T cells and infiltrated macrophages
produce and secrete inflammatory cytokines such as IL-1β, IFN-γ and
TNF-α, which contribute to the loss of β-cells due to apoptosis
activation (43,44). This process highlights the
participation of the NF-κB nuclear factor, which regulates the
expression of genes involved in the response to stress, growth,
survival and cellular apoptosis (45). Previous studies have shown that IκB
transgenic mice-/- (NF-κB inhibitor protein), which are specific to
β-cells, do not develop streptozotocin-induced diabetes (45).
In adipose tissue, in addition to storing energy,
hormones such as adiponectin, leptin, resistin, and visfatin and
other mediators such as interleukins that regulate lipid and
glucose metabolism are produced, which also affect the function of
β-cells (46). Therefore, there is
a close association between the levels of adipokines secreted by
adipose tissue and the proper functioning of pancreatic β-cells.
The inhibitory effects of leptin on insulin synthesis, either
directly in the pancreas or indirectly mediated by the autonomic
nervous system, have been previously reported (47). Adiponectin is an anti-inflammatory
and anti-apoptotic cytokine. A decrease in adiponectin plasma
levels in obese individuals has been associated with insulin
resistance, metabolic syndrome and T2D (47). Adiponectin exerts its effects
through its interaction with its receptors (adipoR1 and adipoR2).
In addition to having anti-inflammatory properties, it contributes
to the activation of AMP-activated protein kinase in the muscle and
liver and increases the use of glucose and the oxidation of fatty
acids (40,47).
Apoptosis and oxidative stress
Increased apoptosis in β-cells plays a precise role
in the initiation and progression of T2D (48). Among the possible mechanisms that
explain the increase in apoptosis in these cells, the production of
ROS of mitochondrial origin stands out (49). In T2D, IR is primarily compensated
for by enhanced insulin secretion by β-cells. However, over time,
this mechanism can fail and lead to a loss of metabolic regulation,
resulting in chronic exposure of β-cells to elevated levels of
glucose (33). Notably, the
increase in intracellular levels of Ca2+ necessary for
the exocytosis of insulin also contributes to the formation of ROS.
Overproduction of free radicals decreases the activity of the
glycolytic enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH),
resulting in increased concentrations of glycolytic metabolites,
such as glyceraldehyde-3-phosphate or fructose-6-phosphate
(35–38,48,49).
Finally, GAPDH inhibition increases the levels of metabolites of
the glycolytic pathway. Consequently, the substrate for the polyol
pathway increases, and the enzyme aldose reductase consumes
available NADPH and decreases the antioxidant mechanisms of β-cells
(50). Furthermore, pancreatic
β-cells have a low capacity to respond to oxidative stress as the
expression of antioxidant enzymes is also low (51). In addition, the reaction of ROS
with biomolecules causes an autocatalytic chain reaction with
subsequent cellular damage and activates mechanisms that emphasize
the death of β-cells and decreased cell mass (48).
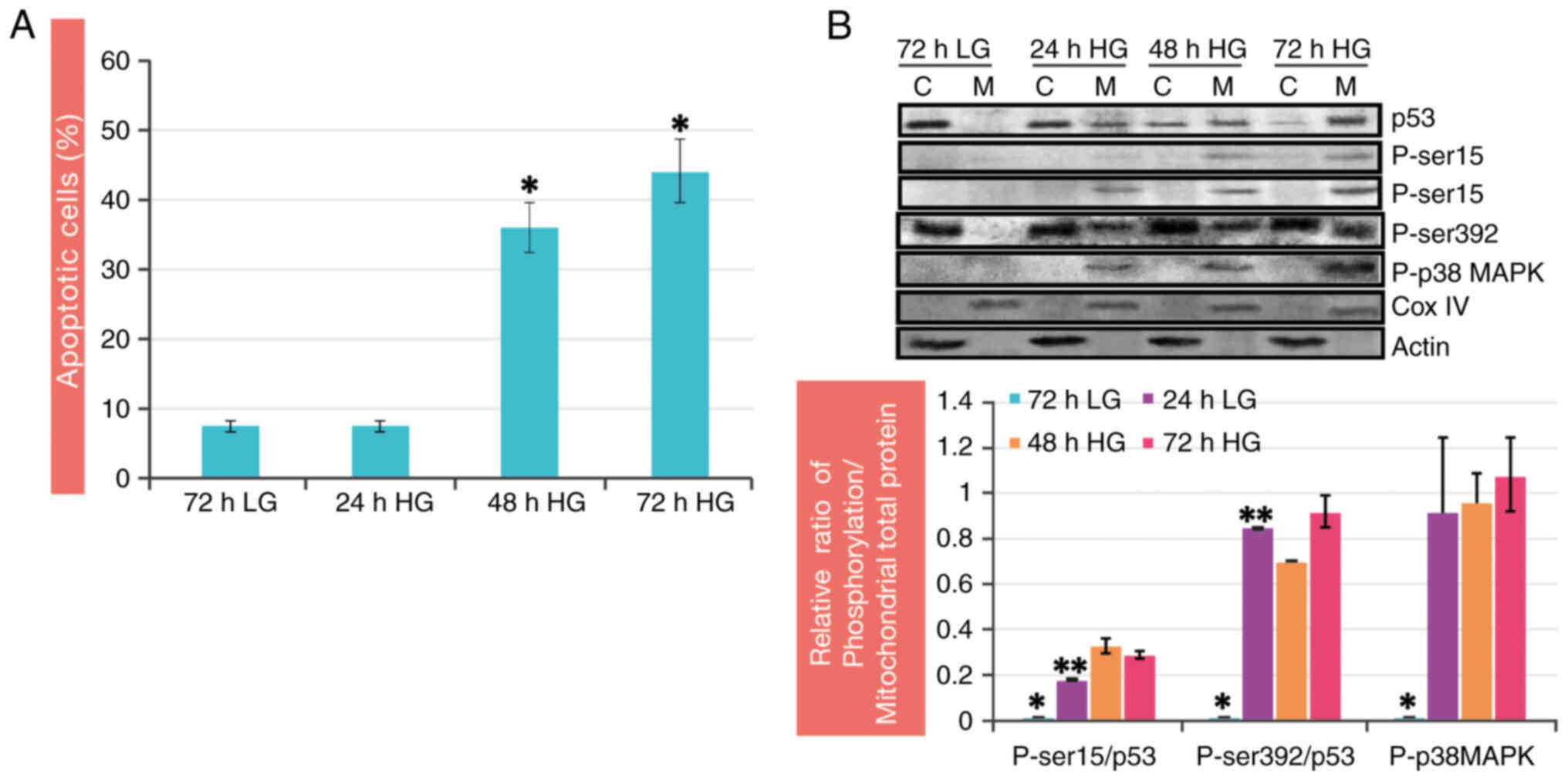
It was previously showed that by culturing RINm5F
pancreatic β-cells in the presence of a high concentration of
glucose (30 mM), the percentage of viable cells significantly
decreased after 48 h compared with that of the control (low
glucose, LG). These findings were consistent with the substantial
increase in the percentage of apoptotic cells (44%) at 72 h
compared with that in the control group (Fig. 4A) (11). Oxidative stress has been identified
as another critical mediator in the cell death process with the
ability to trigger or modulate apoptosis, and it has been found
that the pro-apoptotic role of ROS is manifested through
alterations in mitochondrial function, causing the release of
cytochrome c (32,52).
ROS and oxidative stress in pancreatic β-cells
exposed to high concentrations of glucose are characterized by an
alteration in the potential of the mitochondrial membrane and a
modification of its permeability (17). This alteration allows the release
of pro-apoptotic proteins such as cytochrome c into the cytosol
after 24 h of culture with high glucose. In addition, the
activation of caspase-3 is promoted (11,17).
The balance between pro- (Bax) and anti-apoptotic (Bcl-2) proteins,
among others, plays an important role in the permeability of
mitochondrial membranes and the release of other pro-apoptotic
proteins (53,54). In this sense, it was shown that the
Bcl-2/Bax ratio in the mitochondria decreases significantly after
24 h of culture with high glucose, which indicates the cytotoxicity
induced by the high concentration of glucose in β-cells (11,17).
These results are in accordance with previous reports that
hyperglycemia induces apoptosis in β-cells, causing a decrease in
Bcl-2 expression and an increase in Bax expression, aspects
associated with cytochrome c release and caspase-3 activation
(18,55). High glucose decreased ERK 1/2
phosphorylation. This observation is related to an increase in
apoptosis and a decrease in the rate of cell proliferation
(18). ERK 1/2 phosphorylation has
been shown to modulate genes related to proliferation,
differentiation, migration and death depending on the duration,
magnitude and cellular location (56). In β-cells, activation of ERK 1/2
favors the synthesis and secretion of insulin in response to
glucose and inhibits apoptosis (57); therefore, its inactivation impacts
cellular functions and the β-cell mass.
During the process of apoptosis, protein
translocation between the nucleus, cytoplasm and mitochondria
occurs, which promotes the regulation of this process of cell death
(17). The p53 protein is part of
this group of proteins and is known to be a regulator of apoptosis
when the cell presents irreparable damage to its structure
(58,59).
Dual role of the p53 protein: The guardian
of the genome and mediator of apoptosis in pancreatic β-cells
The p53 protein is known as the ‘guardian of the
genome’, and its activation leads to a series of crucial biological
consequences, ranging from the regulation of the cell cycle and DNA
recombination to chromosome segregation, cell aging and the
induction of apoptosis (60,61).
In response to different extracellular and intracellular stimuli,
such as DNA damage, p53 is activated, triggering various biological
responses such as cell cycle arrest or cell death. After cell
damage, the levels of p53 in cells rapidly increase. This increase
is mainly attributed to post-translational modifications that alter
the half-life of the protein and to the increase in the translation
of its mRNA, which results in the regulation of its target genes
(60,61).
The p53 gene on chromosome 17 (17p13.1) is
composed of 11 exons, of which exon 1 contains a noncoding
sequence. In exon 2, there are two putative transcription start
sites, and exon 11 contains a stop codon and a large noncoding
sequence. The p53 protein has 393 amino acids divided into
functional domains (Fig. 5)
(60). The transactivation domain
(TAD) is located at its N-terminal end and is essential for
interactions with coactivators and transcriptional corepressors
(60,61). TADs are composed of two homologous
subdomains, TAD1 (residues 1–40) and TAD2 (residues 41–61), which
share conserved Φ-XX-Φ-Φ sequence motifs (where Φ represents
hydrophobic amino acids and X represents any amino acid), which are
common to numerous transcriptional regulatory proteins (62). The TAD is a proline-rich region
(PRD; residues 63–97) followed by a highly conserved DNA-binding
domain (DBD; residues 102–292), characterized by specific binding
to DNA sequences (63). Following
the DBD, a binding region harboring an integrated nuclear
localization signal (NLS; residues 301–323) is presented, followed
by the tetramerization domain (TET; residues 323–356), which
includes a nuclear export signal. Finally, there is a mostly
disordered C-terminal regulatory domain (residues 363–393)
(60). This last domain is
characterized by its basic nature; it contains two additional NLSs,
and it is a zone of important protein-protein interactions that
regulate the activity of p53, which binds to DNA in tetrameric form
(60,61). The formation of this quaternary
structure depends on the correct activation of p53 and
post-translational modifications of the TET (63). The C-terminal region is essential
for the formation of tetramers (64).
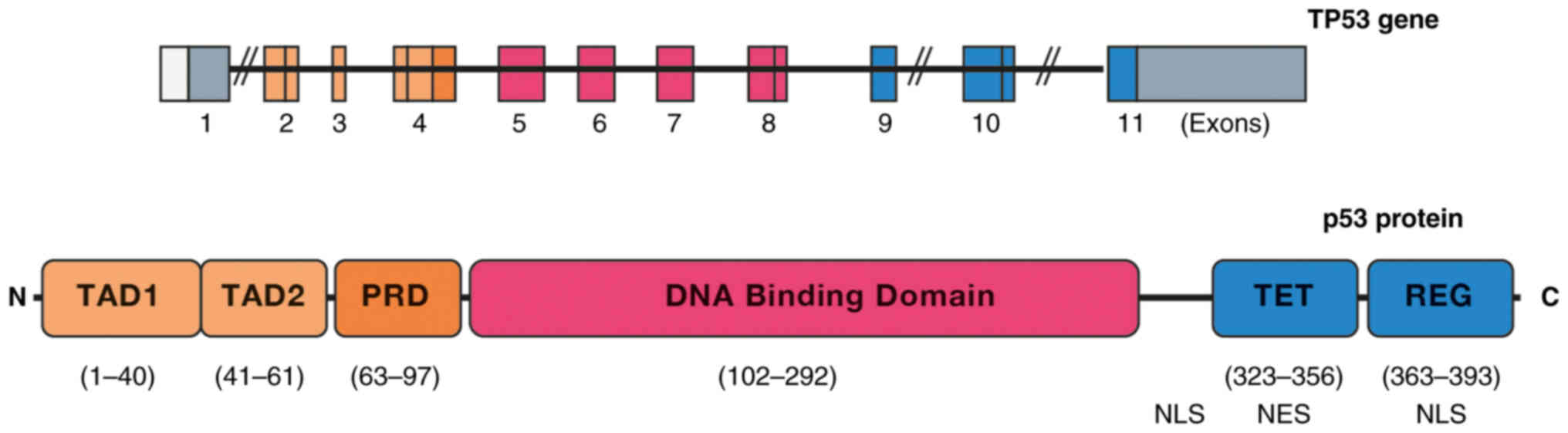 | Figure 5.Structures of the TP53 gene and p53
protein. A schematic representation of the 11 exons of the TP53
gene is shown, of which one is not encoded. TP53 encodes a protein
of 393 amino acids that has five domains: TAD, PRD, DNA-binding
domain, TET and REG. TAD, transactivation domain; PRD, proline-rich
region; TET, tetramerization domain; REG, regulatory domain; NLS,
nuclear localization signal; NES, leucine-rich nuclear export
signal. |
One of the essential functions of p53 is to monitor
cell stress and prevent the proliferation of damaged cells by
inducing apoptosis (58). As a
nuclear transcription factor, p53 can activate the transcription of
pro-apoptotic genes such as bax and puma (65,66).
Moreover, p53 can repress the expression of anti-apoptotic genes
such as bcl-2 and bcl-XL (30). In
addition, p53 also regulates the expression of death receptors. An
example is the death receptor Fas, which mediates apoptosis through
its interaction with the FasL ligand and, in turn, is under the
regulation of p53 (60).
Another prominent function of p53 is its role in
regulating apoptosis through transcription-independent mechanisms
(67,68). In response to multiple apoptotic
stimuli, a fraction of p53 translocates into mitochondria, where it
establishes physical interactions capable of inhibiting the
anti-apoptotic proteins Bcl-2 and Bcl-XL (69). Likewise, it contributes to the
activation of the pro-apoptotic Bax and Bak proteins that induce
permeabilization of the outer mitochondrial membrane by acting on
VDAC, allowing the release of apoptotic activators into the
cytoplasm (27,70). During the characterization of p53,
the possibility of its involvement in the regulation of cell
organelle functions was raised, which has been supported by the
observation of the mobilization of p53 toward the outer
mitochondrial membrane during apoptosis in response to stress from
γ radiation (62,71,72).
Exposure to high concentrations of glucose has been
linked to the apoptotic death of β-cells, a phenomenon that has
been associated with the translocation of the p53 protein into
mitochondria (17). The excessive
consumption of carbohydrates induces metabolic alterations,
negatively impacting the cellular functions of β-cells and
triggering their death (9). The
role of p53 in the death of pancreatic β-cells has recently been
analyzed using a model in which Sprague Dawley rats were subjected
to carbohydrate supplementation. For four months, the animals
received drinking water containing either 40% sucrose or 40%
fructose. Carbohydrate ingestion induced both apoptosis and the
mobilization of p53 from the cytosol to the mitochondria of rat
pancreatic β-cells, even before blood glucose levels rose (9). In addition, the ability of p53
knockout mice to regenerate the β-cell population and rescue the
diabetic phenotype has been demonstrated (73,74).
This finding emphasizes the critical importance of this protein in
the progression of diabetes. However, there is at least one report
that contradicts the participation of p53 in β-cell death (75).
The p53 protein is a transcription factor that is
responsible for monitoring DNA damage (60). Depending on the severity of the
damage, p53 induces apoptosis or arrests the cell cycle until DNA
repair mechanisms are activated. Activation of p53 occurs in
response to various types of stress, leading to its stabilization
and accumulation (63). p53 is
also known to be involved in the induction of apoptosis by acting
directly on mitochondria, where it forms complexes with the Bcl-2
and Bcl-XL proteins through its DBD and precedes the release of
cytochrome c and the activation of caspase-3 (76).
The apoptosis of β-cells exposed to high glucose
concentrations has been demonstrated to occur due to the exit of
cyt c into the cytosol and a decrease in the Bcl-2/Bax ratio in
mitochondria (11,17). These events are related to the
mobilization of p53 to mitochondria, increased ROS and decreased
mitochondrial membrane potential (11). Thus, it was concluded that high
glucose conditions regulate the intracellular distribution of p53
and favor its mitochondrial localization (11,17).
In addition, it promotes post-translational modifications of p53
that prevent its degradation and increase its biological activity
(9–11,17,18).
Post-translational modifications can
regulate p53 in β-cells under hyperglycemic conditions
Under physiological conditions, the p53 protein
remains in a latent state, reaching expression levels in some cell
types that are undetectable by immunohistochemical or Western blot
techniques (60). This protein,
with a half-life close to 15 min in some cell types, is found in an
inactive conformation that is diffusely located in the cell,
frequently being cytoplasmic (77). Although the p53 protein is found at
low concentrations in non-stressed cells, it is rapidly stabilized
and activated in response to various stimuli (59). These stimuli, which are associated
with cellular stress, such as direct damage to DNA by UV or
ionizing radiation, redox changes, hypoxia or thermal shock,
increase the levels of the p53 protein in the cell (78). This increase results from both
greater protein stability and biochemical activation through
post-translational modifications.
The functions of p53 are regulated by at least 10
different types of post-translational modifications, the most
frequent being phosphorylation, ubiquitination,
poly-ADP-ribosylation, acetylation, sumoylation, methylation,
glycosylation and O-GlcNAcylation at serine (Ser)149 of p53
(79–82). Various enzymes catalyze these
modifications and contribute to p53 activation through a wide
variety of mechanisms (83,84).
A number of these modifications are related to an increase in their
ability to arrest the cell cycle or induce apoptosis (58,59).
The permanence and functions of p53 in β-cells under hyperglycemic
conditions are controlled by different post-translational
modifications, including phosphorylation, O-GlcNAcylation and
poly-ADP-ribosylation, depending on the intracellular environment
(17,18).
p53 protein can be regulated by phosphorylation in
response to hyperglycemia: Implications for cell stability and
functions. It has been argued that the levels of the p53 protein
are intrinsically linked to the balance between its synthesis and
degradation, and it is evident that any effect on these events will
have different consequences in the cell (59,60).
A previous study has confirmed that high glucose concentrations do
not modify p53 transcription (10).
By contrast, it has been suggested that its
mobilization and increase in mitochondria result from protein
stabilization, which depends on post-translational modifications
such as phosphorylation (11). The
p53 protein contains multiple phosphorylation-prone sites on
(Ser)/threonine (Thr) residues distributed throughout the protein,
with enrichment at the N-terminus (57). Most of these sites can become
phosphorylated under cellular stress conditions, thus regulating
the functions of p53 (60).
Phosphorylation of p53 at the N-terminus, which occurs in the
cytosol, has been related to an increase in the transcription of
the pro-apoptotic protein Bax (85–87).
On the other hand, phosphorylation at the C-terminal end of Ser 392
(or Ser 389 in mice) is related to the formation of p53 tetramers,
DNA binding and the induction of apoptosis, in addition to
contributing to the suppression of tumors (88).
It has been previously reported that p53 is
phosphorylated at Ser15 and Ser392 in the mitochondria of
pancreatic β-cells cultured under high-glucose conditions (11). Phosphorylation at these residues
likely contributes to protein stability and is an essential
requirement for the interaction of p53 with pro- and/or
anti-apoptotic proteins (11). In
studies in which RBL-2H3 cells (mast cells) were treated with
eugenol (anti-allergenic), it was reported that p53 phosphorylation
at the Ser15 residue in mitochondria facilitates the interaction of
p53 with Bcl-2 and Bcl-xL, which in turn induces changes in the Δψm
and the release of cytochrome c (88). It is relevant to mention that the
observation of p53 phosphorylation at residue Ser 392 in
mitochondria in response to hyperglycemia seems to be the only one
thus far. However, there are at least two studies linking the
phosphorylation of this serine by p38 MAPK kinase with Bax
activation and apoptosis in myocytes cultured with high glucose
(89–91). Notably, p38 MAPK plays an important
role in regulating insulin secretion (92). Since p53 has multiple
phosphorylation sites, multiple kinases, including those activated
by oxidative stress, such as p38 MAPK, regulate this process
(93,94). Under high glucose conditions, the
subcellular redistribution of p38 MAPK (from the cytosol to
mitochondria) is stimulated, but its activation is only observed in
the mitochondrial fraction (Fig.
4B). The localization and phosphorylation of p53 coincide with
the phosphorylation of p38 MAPK in mitochondria during high glucose
treatment (11). These findings
were confirmed by colocalization studies of the phosphorylated
proteins by confocal microscopy, where a high colocalization
coefficient was observed for both p53 and p38 MAPK in mitochondria
(11). To verify the participation
of p38 MAPK in p53 phosphorylation, a specific inhibitor of p38
MAPK (SB203580) was added to cultured RINm5F cells exposed to high
glucose. The results demonstrated that the inhibitor prevented p53
phosphorylation at the Ser 392 and Ser 15 residues at all time
points tested. Similarly, a decrease in the release of cytochrome c
and the rate of apoptosis were observed (11). These data corroborate that p38 MAPK
activation under these conditions is responsible for p53
phosphorylation at the Ser 392 and Ser 15 residues in mitochondria.
Previous studies in other cell types have revealed that under
conditions of hyperglycemia, oxidative stress and UV radiation, p38
MAPK regulates apoptosis by phosphorylating p53 at Ser 392
(85,94,95)
and Ser 15 (96). In addition, p38
MAPK inhibition affects p53 mobilization to mitochondria (11). Phosphorylation likely has a
protective effect on p53, preventing its interaction and
recognition by Mdm2, which in turn preserves its stability and
degradation, allowing its mobilization to mitochondria. This would
trigger the activation of apoptosis, as well as its association
with anti-apoptotic proteins (10,11).
The interaction between p53 and Bcl-2 in the
mitochondrial fraction of cells treated with 30 mM glucose was
observed after 24 h. Inhibition of p53 phosphorylation at Ser392
and Ser15 by a p38 MAPK inhibitor affected the association of p53
with Bcl-2. These results indicate that this phosphorylation is
very important for promoting the association of p53 with Bcl-2 and
for p53 to remain in the mitochondria (11).
Furthermore, p53 phosphorylation was detected at
Ser15 in the nuclear fraction. Previous studies have confirmed that
this phosphorylation at Ser15 caused by UV radiation or hydrogen
peroxide is due to ataxia telangiectasia mutated kinase (ATM)
activation. It should be noted that phosphorylation of this residue
when the protein is in the cell nucleus helps to stabilize and
activate the transcriptional functions of p53, which are related to
the regulation of apoptosis (97).
The results of the present study showed an increase in ATM
phosphorylation in the cytosol and nucleus in response to high
glucose exposure, probably because of the oxidative stress that
prevails under these conditions (17,98).
In this context, ATM-deficient mice exhibit oxidative stress and
damage to DNA, proteins and lipids via ROS (98). In addition, the administration of
antioxidants such as N-acetyl cysteine has been shown to be
effective in reducing oxidative damage, reversing neurological
deficits and preventing premature aging in these animal models
(99). ATM plays a crucial role by
phosphorylating numerous substrates needed for DNA repair, cell
cycle regulation and apoptosis, including the p53 protein (97). In addition, p53 phosphorylation at
Ser15 in the nuclear fraction appears to be related to ATM
activation (98). Furthermore,
streptozotocin-induced p53 activation by ATM is a critical event in
DNA damage in β-cells in vitro (100). However, ATM activation is likely
also associated with the phosphorylation of other proteins and
indirectly regulates p53. ATM can phosphorylate p53 through Chk2
activation (101,102) or phosphorylate Mdm2 at Ser395,
thus preventing p53 ubiquitination and degradation; thus, ATM could
be an adjacent mechanism for p53 stabilization (84,101). Additionally, the relevance of ATM
activation in glucose metabolism has been noted, although the
precise underlying mechanisms have not yet been determined. ATM
inhibition affects the incorporation of glucose into muscle and
adipose tissue through the mobilization of GLUT4, which is why it
has been linked to the development of insulin resistance and T2D
(100).
O-GlcNAcylation. O-GlcNAcylation is a regulatory
mechanism of p53 stability and function in response to glucose. It
is a noncanonical glycosylation process involving the attachment of
single O-linked N-acetylglucosamine (O-GlcNAc) moieties to Ser and
Thr residues of cytoplasmic, nuclear and mitochondrial proteins
(102,103). O-GlcNAcylation is another
post-translational modification of p53 and depends on glucose
availability. In this mechanism, two enzymes participate: OGT
(O-linked N-acetylglucosamine transferase), which adds
N-acetylglucosamine residues to several proteins at the hydroxyl
group of Ser and Thr, and O-GlcNAcase (N-acetylglucosaminidase),
which eliminates the N-acetylglucosamine residues from the proteins
(104). Physiologically,
disruption of O-GlcNAc homeostasis has been implicated in the
pathogenesis of a plethora of human diseases, including cancer,
diabetes and neurodegeneration (105). O-GlcNAcylation has been linked to
T2D since the expression of OGT RNA in the pancreas and brain is
increased (2 Studies have reported that the protein level of p53 is
elevated in the liver of patients with T2DM, partly because hepatic
p53 is stabilized by O-GlcNAcylation and plays an essential role in
the physiological regulation of glucose homeostasis (79,80).
O-GlcNAcylation has been shown to stabilize p53 and prevent its
degradation (81).
In a high-glucose environment, p53 O-GlcNAcylation
is related to its stability and stops its degradation.
O-GlcNAcylation does not interfere with phosphorylation, but it
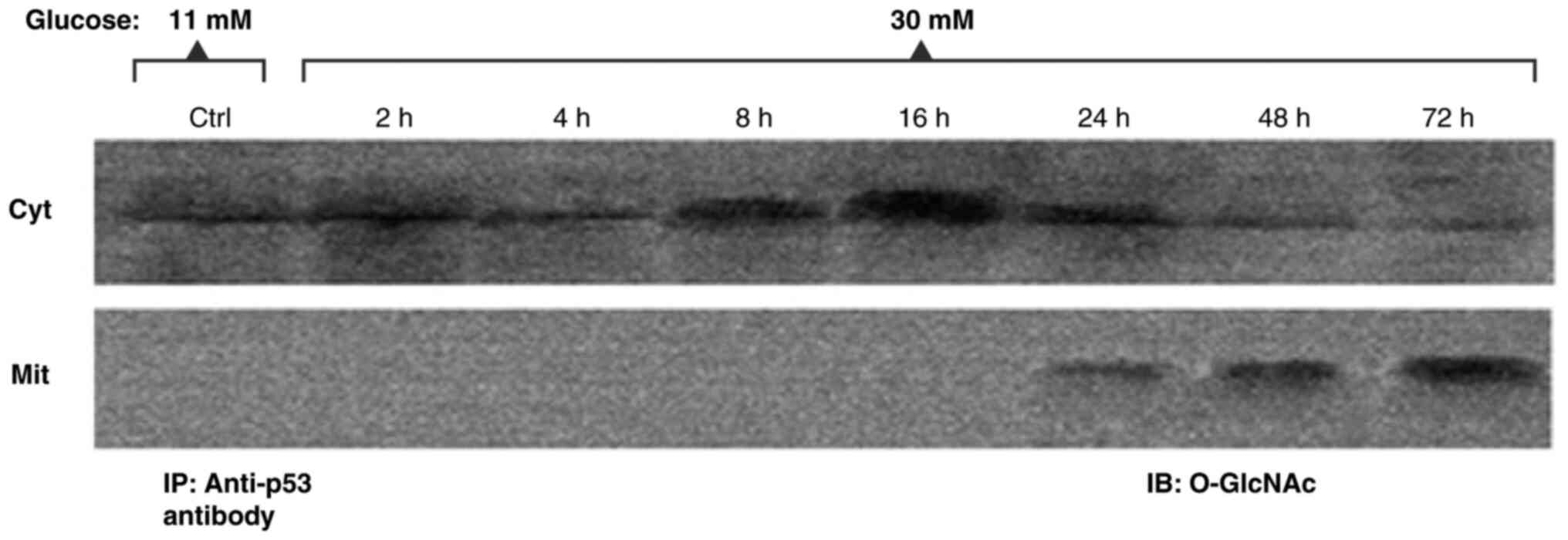
stimulates p53 apoptotic function (105–107). In the studies presented in the
present review, it was observed that O-GlcNAcylation precedes
apoptosis and increases as apoptosis signals appear in RINm5F cells
under hyperglycemic conditions (Fig.
6). In addition, the O-GlcNAcylation of p53 promotes its
mobilization to mitochondria, which can contribute to the release
of pro-apoptotic factors (18).
O-GlcNAcylation of p53 at Ser149 increases its stability upon
interference with Thr155 phosphorylation and weakens the
interaction of p53 with Mdm2, along with the ubiquitination and
thus proteolysis of p53, which translates into greater stability of
p53. Thus, it is hypothesized that O-GlcNAcylation stabilizes p53
and may create a signal for its mobilization to the
mitochondria.
Interaction between PARP and p53: Modulation of DNA
repair and apoptosis in response to cell damage and high glucose
conditions. When DNA damage occurs in cells, poly (ADP-ribose)
polymerase (PARP), a nuclear protein of 116 kDa with two zinc
finger motifs that bind to DNA and specifically detect nicks or
breaks, is activated (108,109). PARP catalyzes the transfer of
ADP-ribose units from NAD+, an essential cofactor in ATP
synthesis and redox potential balance, to glutamic acid and
aspartic acid residues of several nuclear proteins (108). This post-translational
modification is important for the activation of a series of
cellular processes, including DNA repair and replication, gene
transcription, the inflammatory response and cell death, and occurs
mainly in response to DNA damage generated by genotoxic agents, as
well as different stimuli to DNA damage, such as infection and
stress (109–111). In vivo immunoprecipitation
of PARP and p53 has shown that both proteins interact, and that
this PARP-p53 interaction may affect the function of the p53
protein, as it is ADP-ribosylated by PARP in response to DNA damage
or cellular aging (112).
The amino-terminal region of the PARP protein
contains a target sequence of caspases 3 and 7 that is responsible
for PARP proteolysis into two fragments of 89 and 24 kDa. This
proteolytic breakdown is currently considered a marker of
caspase-dependent apoptosis (112). Fragments generated by the action
of caspases 3 and 7 contribute to the inactivation of the intact
enzyme (113). PARP fragmentation
coincides with the induction of apoptosis in β-cells by
hyperglycemia (10). These results
agree with previous reports showing that hyperglycemia induces the
apoptosis of β-cells and pancreatic cell lines, such as RINm5F
(11,17). Short-term treatment of RINm5F cells
with high glucose (2–16 h) induces an increase in PARP protein
(18). Immunoprecipitation assays
revealed an association between PARP and p53, as well as
poly-ADP-ribosylation of p53 in the nuclear fraction, which
promotes the function of p53 in DNA repair mechanisms in the early
stages of cell damage (10,18).
Poly-ADP-ribosylation of p53 under high-glucose conditions also
likely contributes to its stability and mobilization in
mitochondria (18). After 24 h,
PARP degrades and becomes inactivated, which leads to a decrease in
poly-ADP-ribosylation of p53. PARP fragmentation and inactivation
coincided with increased Bax in mitochondria, cytochrome c release,
and increased apoptosis of RINm5F cells due to high glucose
conditions (18).
Dual regulation of p53: Interaction between
Mdm2 and ubiquitination activity in the context of hyperglycemia
and cellular stress
Cell survival largely depends on the p53
concentration. Mdm2 is one of the regulatory mechanisms of p53.
Mdm2 is an E3 ubiquitin ligase that binds to p53 and adds ubiquitin
residues to it for proteasomal degradation. However, the fate of
p53 depends on Mdm2 activation and the addition of ubiquitins,
after which mono- or polyubiquitylation is regulated by Mdm2
(114). As previously cited, the
formation of the p53-Mdm2 complex is determined by the
post-translational modifications of both proteins. The
ubiquitylation, SUMOylation and phosphorylation of Mdm2 disrupts
the formation of the p53-Mdm2 dimer and stabilizes p53.
Phosphorylation of Mdm2 at Ser395 by ATM activation has been shown
to decrease the ability of Mdm2 to drive p53 degradation.
Furthermore, it was previously reported that Mdm2
phosphorylation by Akt is involved in the regulation of p53, as it
reduces transactivation and increases p53 ubiquitination (115,116). Akt is a critical regulator of
cell proliferation and survival (115). Phosphorylation of Mdm2 at Ser166
and Ser186 by Akt increases its E3 ligase activity, and can protect
cells against p53-induced apoptosis caused by hyperglycemia
(10). Notably, other kinases,
such as ERK 1/2, can phosphorylate Mdm2 at Ser166. However, with
increased glucose, ERK 1/2 decreases and apoptosis increases
(11).
Increased glucose decreases Mdm2 mRNA and protein
levels in the nucleus and cytosol, Mdm2 expression is regulated by
p53 (10). Previous studies have
shown that in cells cultured with high concentrations of glucose,
p53 is not found entirely in the nucleus but is mobilized to other
organelles, such as mitochondria (11,17).
Therefore, it cannot stimulate the expression of Mdm2. Furthermore,
DNA fragmentation due to hyperglycemia can also affect Mdm2 mRNA
expression (10).
The E3 ubiquitin ligase activity of Mdm2 is
situated within the RING-finger domain in the carboxyl-terminal
region, where the lysine acceptor is also found, whose main
function is to mark p53 for degradation (117). The central Mdm2 acidic domain
binds to the RING-finger domain and stimulates its catalytic
activation and ubiquitin release by the E2 enzyme (117). The interaction between the acidic
domain and the RING-finger domain is modulated by phosphorylated
ATM (10). In this model, an
increase in ATM phosphorylation due to hyperglycemia was also
observed, so it cannot be ruled out that stress and the
phosphorylation cascade induced by high glucose may phosphorylate
some residues present in or near the RING-finger domain and the
acidic core domain of Mdm2 and suppress p53 polyubiquitination and
degradation.
By contrast, ubiquitination of p53 depends on the
energy supply. Hyperglycemia increases ROS and mitochondrial
uncoupling, leading to decreased ATP levels. Therefore, if ATP
decreases, ubiquitins cannot condense the glycine residues of their
carboxyl-terminal region with the lysine residues of p53, and p53
degradation is inhibited (114,115).
Conclusions
The mechanisms involved in pancreatic β-cell loss
due to hyperglycemia are not yet fully known. Hyperglycemia is
characterized by exacerbated ROS production, which stimulates the
activation of phosphorylation cascades that may suppress Mdm2 E3
ubiquitin ligase activity and inhibit the p53-Mdm2 complex. Thus,
it prevents p53 degradation and promotes p53 recruitment to
mitochondria and β-cell death. Other changes initiated by
hyperglycemia, such as poly-(ADP-ribosylation) and O-GlcNAcylation,
also influence p53 stabilization and activation (Fig. 7). The studies described in the
present review aimed to elucidate the biochemical events related to
the loss of β-cells in patients with T2D. These findings offer a
reference point for the search for new therapeutic targets,
possibly for post-translational modifications of p53.
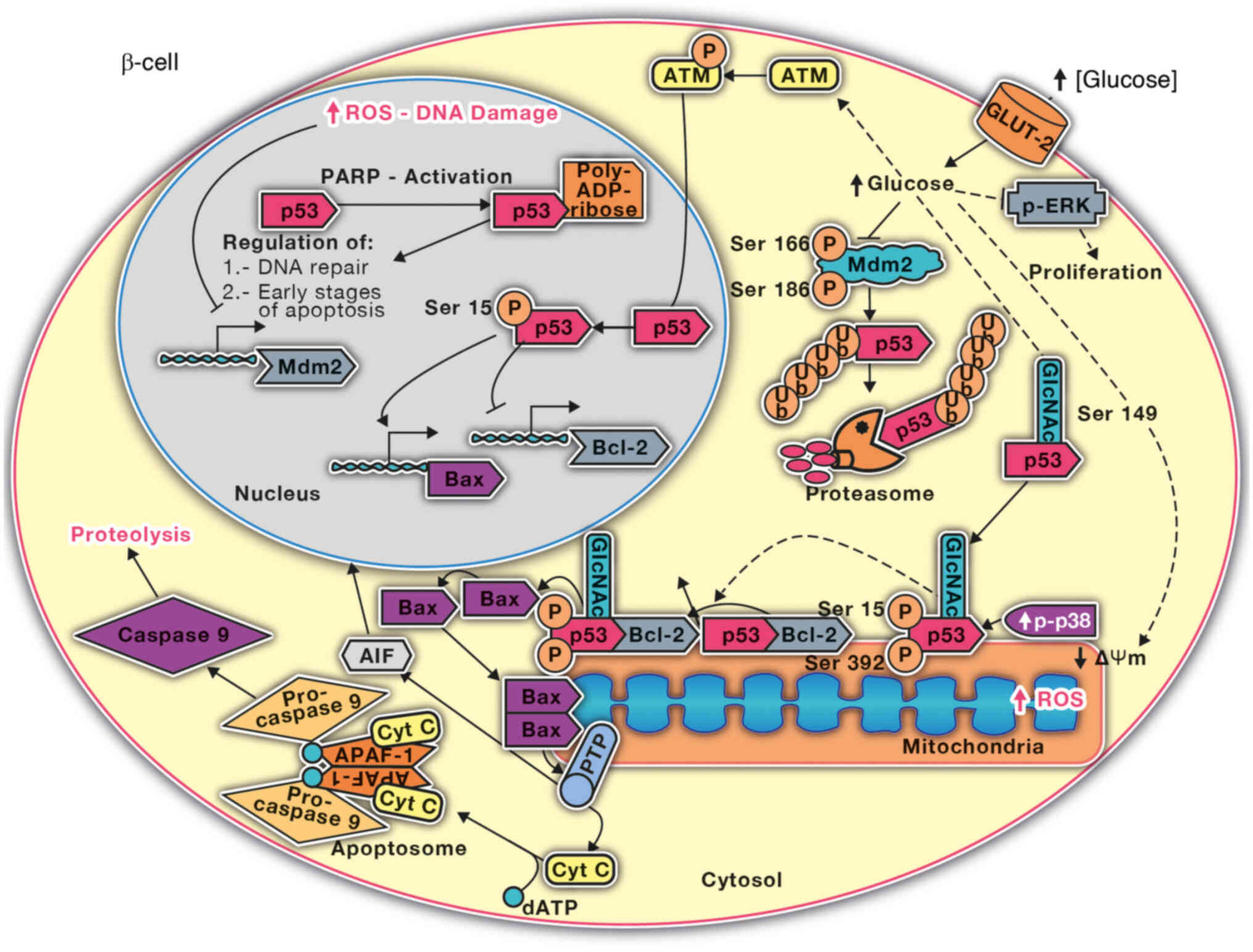 | Figure 7.Regulation of p53 due to high glucose
in pancreatic β-cells. Poly-ADP-ribosylation occurs in the early
stages of RINm5F cell culture with high glucose, which allows p53
to regulate DNA damage repair and the early stages of apoptosis.
p53 O-GlcNAcylation is increased in the cytosol and stabilizes p53,
enabling its migration to the mitochondria, where it is
phosphorylated via p38 MAPK. The phosphorylation of p53 likely
promotes its interaction with anti-apoptotic proteins (Bcl-2) and
the release of pro-apoptotic elements such as cytochrome c. These
events lead to the activation of caspases. High glucose conditions
also inhibit the phosphorylation and activation of Mdm2 and the
ubiquitination of p53, as well as its degradation by the
proteasome. ROS, reactive oxygen species; PARP, poly (ADP-ribose)
polymerase; Mdm2, murine protein double minute 2; ATM, ataxia
telangiectasia mutated kinase; p, phosphorylated; GlcNAc
N-Acetylglucosamine, dATP, deoxyadenosine triphosphate; Δψm,
transmembrane potential; cyt c, cytochrome c; Ub, ubiquitin; AIF,
apoptosis-inducing factor; APAF-1, apoptotic protease activating
factor 1; PTP, permeability transition pore. |
Acknowledgments
Not applicable.
Funding
This study was supported by the Fiscal Resources Program for
Research of the National Institute of Pediatrics (grant nos.
2022/067, 2019/072 and 2020/016), and Border Science 2023, National
Council of Humanities, Sciences and Technologies (grant nos.
CF-2023-I-811 and CF-2023-I-2755).
Availability of data and materials
Not applicable.
Authors' contributions
LAFL, AAR and COC conceptualized the study; LAFL,
COC, AAR, ACR, IDlMDlM and SEF performed the investigation; LAFL,
COC, IDlMDlM, SEF, IGT, RVR, AAR and GLV wrote, reviewed and edited
the manuscript; LAFL visualized the study; LAFL and COC performed
study supervision and analyzed the information. Data authentication
is not appliable. All the authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Marchetti P, Bugliani M, De Tata V,
Suleiman M and Marselli L: Pancreatic beta cell identity in humans
and the role of type 2 diabetes. Front Cell Dev Biol. 5:552017.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang P, Fiaschi-Taesch NM, Vasavada RC,
Scott DK, García-Ocaña A and Stewart AF: Diabetes mellitus-advances
and challenges in human β-cell proliferation. Nat Rev Endocrinol.
11:201–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abdul-Ghani MA, Tripathy D and DeFronzo
RA: Contributions of beta-cell dysfunction and insulin resistance
to the pathogenesis of impaired glucose tolerance and impaired
fasting glucose. Diabetes Care. 29:1130–1139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun H, Saeedi P, Karuranga S, Pinkepank M,
Ogurtsova K, Duncan BB, Stein C, Basit A, Chan JCN, Mbanya JC, et
al: IDF diabetes atlas: Global, regional and country-level diabetes
prevalence estimates for 2021 and projections for 2045. Diabetes
Res Clin Pract. 183:1091192022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X,
Song X, Ren Y and Shan PF: Global, regional, and national burden
and trend of diabetes in 195 countries and territories: An analysis
from 1990 to 2025. Sci Rep. 10:147902020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
International Diabetes Federation, .
Diabetes Atlas. (10th Edition). https://fmdiabetes.org/atlas-idf-10o-edicion-2021/September
13–2023
|
|
7
|
Harreiter J and Roden M: Diabetes
mellitus: Definition, classification, diagnosis, screening and
prevention (update 2023). Wien Klin Wochenschr. 135 (Suppl
1):S7–S17. 2023.(In German). View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rojas J, Bermudez V, Palmar J, Martínez
MS, Olivar LC, Nava M, Tomey D, Rojas M, Salazar J, Garicano C and
Velasco M: Pancreatic beta cell death: Novel potential mechanisms
in diabetes therapy. J Diabetes Res. 2018:96018012018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barzalobre-Geronimo R, Contreras-Ramos A,
Cervantes-Cruz AI, Cruz M, Suárez-Sánchez F, Goméz-Zamudio J,
Diaz-Rosas G, Ávalos-Rodríguez A, Díaz-Flores M and
Ortega-Camarillo C: Pancreatic β-cell apoptosis in normoglycemic
rats is due to mitochondrial translocation of p53-induced by the
consumption of sugar-sweetened beverages. Cell Biochem Biophys.
81:503–514. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Barzalobre-Gerónimo R, Flores-López LA,
Baiza-Gutman LA, Cruz M, García-Macedo R, Ávalos-Rodríguez A,
Contreras-Ramos A, Díaz-Flores M and Ortega-Camarillo C: Erratum
to: Hyperglycemia promotes p53-Mdm2 interaction but reduces p53
ubiquitination in RINm5F cells. Mol Cell Biochem. 406:3012015.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Flores-López LA, Díaz-Flores M,
García-Macedo R, Ávalos-Rodríguez A, Vergara-Onofre M, Cruz M,
Contreras-Ramos A, Konigsberg M and Ortega-Camarillo C: High
glucose induces mitochondrial p53 phosphorylation by p38 MAPK in
pancreatic RINm5F cells. Mol Biol Rep. 40:4947–4958. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Butler AE, Janson J, Bonner-Weir S, Ritzel
R, Rizza RA and Butler PC: Beta-cell deficit and increased
beta-cell apoptosis in humans with type 2 diabetes. Diabetes.
52:102–110. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
McFarland KF, Catalano EW, Day JF, Thorpe
SR and Baynes JW: Nonenzymatic glucosylation of serum proteins in
diabetes mellitus. Diabetes. 28:1011–1014. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomita T: Apoptosis in pancreatic β-islet
cells in type 2 diabetes. Bosn J Basic Med Sci. 16:162–179. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chandra J, Zhivotovsky B, Zaitsev S,
Juntti-Berggren L, Berggren PO and Orrenius S: Role of apoptosis in
pancreatic beta-cell death in diabetes. Diabetes. 50 (Suppl
1):S44–S47. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gottlieb TM and Oren M: p53 and apoptosis.
Semin Cancer Biol. 8:359–368. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ortega-Camarillo C, Guzmán-Grenfell AM,
García-Macedo R, Rosales-Torres AM, Avalos-Rodríguez A, Durán-Reyes
G, Medina-Navarro R, Cruz M, Díaz-Flores M and Kumate J:
Hyperglycemia induces apoptosis and p53 mobilization to
mitochondria in RINm5F cells. Mol Cell Biochem. 281:163–171. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Flores-López LA, Cruz-López M,
García-Macedo R, Gómez-Olivares JL, Díaz-Flores M,
Konigsberg-Fainstein M and Ortega-Camarillo C: Phosphorylation,
O-N-acetylglucosaminylation and poly-ADP-ribosylation of P53 in
RINm5F cells cultured in high glucose. Free Radic Biol Med. 53
(Suppl 2):S952012. View Article : Google Scholar
|
|
19
|
DeLong MJ: Apoptosis: A modulator of
cellular homeostasis and disease states. Ann N Y Acad Sci.
842:82–90. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nicholson DW and Thornberry NA: Caspases:
Killer proteases. Trends Biochem Sci. 22:299–306. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Peter ME and Krammer PH: Mechanisms of
CD95 (APO-1/Fas)-mediated apoptosis. Curr Opin Immunol. 10:545–551.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ichim G and Tait SWG: A fate worse than
death: Apoptosis as an oncogenic process. Nat Rev Cancer.
16:539–548. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Elmore S: Apoptosis: A review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Johnson N, Khan A, Virji S, Ward JM and
Crompton M: Import and processing of heart mitochondrial
cyclophilin D. Eur J Biochem. 263:353–359. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crompton M: Mitochondrial intermembrane
junctional complexes and their role in cell death. J Physiol.
529:11–21. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Andreeva L, Heads R and Green CJ:
Cyclophilins and their possible role in the stress response. Int J
Exp Pathol. 80:305–315. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Marchenko ND, Zaika A and Moll UM: Death
signal-induced localization of p53 protein to mitochondria. A
potential role in apoptotic signaling. J Biol Chem.
275:16202–16212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lemasters JJ, Qian T, He L, Kim JS, Elmore
SP, Cascio WE and Brenner DA: Role of mitochondrial inner membrane
permeabilization in necrotic cell death, apoptosis, and autophagy.
Antioxid Redox Signal. 4:769–781. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Susin SA, Lorenzo HK, Zamzami N, Marzo I,
Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler
M, et al: Molecular characterization of mitochondrial
apoptosis-inducing factor. Nature. 397:441–446. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh R, Letai A and Sarosiek K:
Regulation of apoptosis in health and disease: The balancing act of
BCL-2 family proteins. Nat Rev Mol Cell Biol. 20:175–193. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsujimoto Y and Shimizu S: VDAC regulation
by the Bcl-2 family of proteins. Cell Death Differ. 7:1174–1181.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Garrido C, Galluzzi L, Brunet M, Puig PE,
Didelot C and Kroemer G: Mechanisms of cytochrome c release from
mitochondria. Cell Death Differ. 13:1423–1433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Roche E: Diabetes tipo 2:
Gluco-lipo-toxicidad y disfunción de la célula β pancreática. Ars
Pharm. 44:313–332. 2003.
|
|
34
|
Chen SS, Jiang T, Wang Y, Gu LZ, Wu HW,
Tan L and Guo J: Activation of double-stranded RNA-dependent
protein kinase inhibits proliferation of pancreatic β-cells.
Biochem Biophys Res Commun. 443:814–820. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yuan H, Zhang X, Huang X, Lu Y, Tang W,
Man Y, Wang S, Xi J and Li J: NADPH oxidase 2-derived reactive
oxygen species mediate FFAs-induced dysfunction and apoptosis of
β-cells via JNK, p38 MAPK and p53 pathways. PLoS One. 5:e157262010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lu H, Hao L, Li S, Lin S, Lv L, Chen Y,
Cui H, Zi T, Chu X, Na L and Sun C: Elevated circulating stearic
acid leads to a major lipotoxic effect on mouse pancreatic beta
cells in hyperlipidaemia via a miR-34a-5p-mediated
PERK/p53-dependent pathway. Diabetologia. 59:1247–1257. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mayo LD and Donner DB: A
phosphatidylinositol 3-kinase/Akt pathway promotes translocation of
Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA.
98:11598–11603. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lytrivi M, Castell AL, Poitout V and Cnop
M: Recent insights into mechanisms of β-Cell Lipo- and
glucolipotoxicity in type 2 diabetes. J Mol Biol. 432:1514–1534.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cunha DA, Igoillo-Esteve M, Gurzov EN,
Germano CM, Naamane N, Marhfour I, Fukaya M, Vanderwinden JM,
Gysemans C, Mathieu C, et al: Death protein 5 and p53-upregulated
modulator of apoptosis mediate the endoplasmic reticulum
stress-mitochondrial dialog triggering lipotoxic rodent and human
β-cell apoptosis. Diabetes. 61:2763–2775. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saltevo J, Vanhala M, Kautiainen H,
Kumpusalo E and Laakso M: Association of C-reactive protein,
interleukin-1 receptor antagonist and adiponectin with the
metabolic syndrome. Mediators Inflamm. 2007:935732007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hotamisligil GS: Inflammation and
metabolic disorders. Nature. 444:860–867. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tilg H and Moschen AR: Inflammatory
mechanisms in the regulation of insulin resistance. Mol Med.
14:222–231. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhao YF and Chen C: Regulation of
pancreatic beta-cell function by adipocytes. Sheng Li Xue Bao.
59:247–252. 2007.PubMed/NCBI
|
|
44
|
Donath MY, Schumann DM, Faulenbach M,
Ellingsgaard H, Perren A and Ehses JA: Islet inflammation in type 2
diabetes: From metabolic stress to therapy. Diabetes Care. 31
(Suppl 2):S161–S164. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Eldor R, Yeffet A, Baum K, Doviner V, Amar
D, Ben-Neriah Y, Christofori G, Peled A, Carel JC, Boitard C, et
al: Conditional and specific NF-kappaB blockade protects pancreatic
beta cells from diabetogenic agents. Proc Natl Acad Sci USA.
103:5072–5077. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nolan CJ, Madiraju MSR,
Delghingaro-Augusto V, Peyot ML and Prentki M: Fatty acid signaling
in the beta-cell and insulin secretion. Diabetes. 55 (Suppl
2):S16–S23. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kadowaki T, Yamauchi T, Kubota N, Hara K,
Ueki K and Tobe K: Adiponectin and adiponectin receptors in insulin
resistance, diabetes, and the metabolic syndrome. J Clin Invest.
116:1784–1792. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bensellam M, Laybutt DR and Jonas JC: The
molecular mechanisms of pancreatic β-cell glucotoxicity: Recent
findings and future research directions. Mol Cell Endocrinol.
364:1–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Brownlee M: The pathobiology of diabetic
complications: A unifying mechanism. Diabetes. 54:1615–1625. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Du X, Matsumura T, Edelstein D, Rossetti
L, Zsengellér Z, Szabó C and Brownlee M: Inhibition of GAPDH
activity by poly(ADP-ribose) polymerase activates three major
pathways of hyperglycemic damage in endothelial cells. J Clin
Invest. 112:1049–1057. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Grankvist K, Marklund SL and Täljedal IB:
CuZn-superoxide dismutase, Mn-superoxide dismutase, catalase and
glutathione peroxidase in pancreatic islets and other tissues in
the mouse. Biochem J. 199:393–398. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gottlieb RA: Mitochondria: Execution
central. FEBS Lett. 482:6–12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Schellenberg B, Wang P, Keeble JA,
Rodriguez-Enriquez R, Walker S, Owens TW, Foster F, Tanianis-Hughes
J, Brennan K, Streuli CH and Gilmore AP: Bax exists in a dynamic
equilibrium between the cytosol and mitochondria to control
apoptotic priming. Mol Cell. 49:959–971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Adams JM and Cory S: The Bcl-2 protein
family: Arbiters of cell survival. Science. 281:1322–1326. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Huang Q, Bu S, Yu Y, Guo Z, Ghatnekar G,
Bu M, Yang L, Lu B, Feng Z, Liu S and Wang F: Diazoxide prevents
diabetes through inhibiting pancreatic beta-cells from apoptosis
via Bcl-2/Bax rate and p38-beta mitogen-activated protein kinase.
Endocrinology. 148:81–91. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Lawrence M, Shao C, Duan L, McGlynn K and
Cobb MH: The protein kinases ERK1/2 and their roles in pancreatic
beta cells. Acta Physiol (Oxf). 192:11–17. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ito K, Nakazato T, Yamato K, Miyakawa Y,
Yamada T, Hozumi N, Segawa K, Ikeda Y and Kizaki M: Induction of
apoptosis in leukemic cells by homovanillic acid derivative,
capsaicin, through oxidative stress: Implication of phosphorylation
of p53 at Ser-15 residue by reactive oxygen species. Cancer Res.
64:1071–1078. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Shen Y and White E: p53-dependent
apoptosis pathways. Adv Cancer Res. 82:55–84. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Green DR and Kroemer G: Cytoplasmic
functions of the tumor suppressor p53. Nature. 458:1127–1130. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Riley T, Sontag E, Chen P and Levine A:
Transcriptional control of human p53-regulated genes. Nat Rev Mol
Cell Biol. 9:402–412. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Jenkins LM, Durell SR, Mazur SJ and
Appella E: p53 N-terminal phosphorylation: A defining layer of
complex regulation. Carcinogenesis. 33:1441–1449. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
el-Deiry WS, Kern SE, Pietenpol JA,
Kinzler KW and Vogelstein B: Definition of a consensus binding site
for p53. Nat Genet. 1:45–49. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ahn J and Prives C: The C-terminus of p53:
The more you learn the less you know. Nat Struct Biol. 8:730–732.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: Release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Müller M, Wilder S, Bannasch D, Israeli D,
Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M
and Krammer PH: p53 activates the CD95 (APO-1/Fas) gene in response
to DNA damage by anticancer drugs. J Exp Med. 188:2033–2045. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Caelles C, Helmberg A and Karin M:
p53-dependent apoptosis in the absence of transcriptional
activation of p53-target genes. Nature. 370:220–223. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sansome C, Zaika A, Marchenko ND and Moll
UM: Hypoxia death stimulus induces translocation of p53 protein to
mitochondria. Detection by immunofluorescence on whole cells. FEBS
Lett. 488:110–115. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Schuler M, Bossy-Wetzel E, Goldstein JC,
Fitzgerald P and Green DR: p53 induces apoptosis by caspase
activation through mitochondrial cytochrome c release. J Biol Chem.
275:7337–7342. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Donahue RJ, Razmara M, Hoek JB and Knudsen
TB: Direct influence of the p53 tumor suppressor on mitochondrial
biogenesis and function. FASEB J. 15:635–644. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Mihara M, Erster S, Zaika A, Petrenko O,
Chittenden T, Pancoska P and Moll UM: p53 has a direct apoptogenic
role at the mitochondria. Mol Cell. 11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Wei H, Wang H, Wang G, Qu L, Jiang L, Dai
S, Chen X, Zhang Y, Chen Z, Li Y, et al: Structures of p53/BCL-2
complex suggest a mechanism for p53 to antagonize BCL-2 activity.
Nat Commun. 14:43002023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Hinault C, Kawamori D, Liew CW, Maier B,
Hu J, Keller SR, Mirmira RG, Scrable H and Kulkarni RN: Δ40 Isoform
of p53 controls β-cell proliferation and glucose homeostasis in
mice. Diabetes. 60:1210–1222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Kon N, Zhong J, Qiang L, Accili D and Gu
W: Inactivation of arf-bp1 induces p53 activation and diabetic
phenotypes in mice. J Biol Chem. 287:5102–5111. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Uhlemeyer C, Müller N, Rieck M, Kuboth J,
Schlegel C, Grieß K, Dorweiler TF, Heiduschka S, Eckel J, Roden M,
et al: Selective ablation of P53 in pancreatic beta cells fails to
ameliorate glucose metabolism in genetic, dietary and
pharmacological models of diabetes mellitus. Mol Metab.
67:1016502023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Douc-Rasy S and Bénard J: A new view on
p53 protein cytoplasmic sequestration. Bull Cancer. 90:380–382.
2003.(In French). PubMed/NCBI
|
|
77
|
Giaccia AJ and Kastan MB: The complexity
of p53 modulation: Emerging patterns from divergent signals. Genes
Dev. 12:2973–2983. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Harris SL and Levine AJ: The p53 pathway:
Positive and negative feedback loops. Oncogene. 24:2899–2908. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gonzalez-Rellan MJ, Fondevila MF,
Fernandez U, Rodríguez A, Varela-Rey M, Veyrat-Durebex C, Seoane S,
Bernardo G, Lopitz-Otsoa F, Fernández-Ramos D, et al:
O-GlcNAcylated p53 in the liver modulates hepatic glucose
production. Nat Commun. 12:50682021. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Labuschagne CF, Zani F and Vousden KH:
Control of metabolism by p53-cancer and beyond. Biochim Biophys
Acta Rev Cancer. 1870:32–42. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yang WH, Kim JE, Nam HW, Ju JW, Kim HS,
Kim YS and Cho JW: Modification of p53 with O-linked
N-acetylglucosamine regulates p53 activity and stability. Nat Cell
Biol. 8:1074–1083. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wen J and Wang D: Deciphering the PTM
codes of the tumor suppressor p53. J Mol Cell Biol. 13:774–785.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Liu Y, Tavana O and Gu W: p53
modifications: Exquisite decorations of the powerful guardian. J
Mol Cell Biol. 11:564–577. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lavin MF and Gueven N: The complexity of
p53 stabilization and activation. Cell Death Differ. 13:941–950.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Thompson T, Tovar C, Yang H, Carvajal D,
Vu BT, Xu Q, Wahl GM, Heimbrook DC and Vassilev LT: Phosphorylation
of p53 on key serines is dispensable for transcriptional activation
and apoptosis. J Biol Chem. 279:53015–53022. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Steegenga WT, van der Eb AJ and Jochemsen
AG: How phosphorylation regulates the activity of p53. J Mol Biol.
263:103–113. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Hoogervorst EM, Bruins W, Zwart E, van
Oostrom CTM, van den Aardweg GJ, Beems RB, van den Berg J, Jacks T,
van Steeg H and de Vries A: Lack of p53 Ser389 phosphorylation
predisposes mice to develop 2-acetylaminofluorene-induced bladder
tumors but not ionizing radiation-induced lymphomas. Cancer Res.
65:3610–3616. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Park BS, Song YS, Yee SB, Lee BG, Seo SY,
Park YC, Kim JM, Kim HM and Yoo YH: Phospho-ser 15-p53 translocates
into mitochondria and interacts with Bcl-2 and Bcl-xL in
eugenol-induced apoptosis. Apoptosis. 10:193–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Fiordaliso F, Leri A, Cesselli D, Limana
F, Safai B, Nadal-Ginard B, Anversa P and Kajstura J: Hyperglycemia
activates p53 and p53-regulated genes leading to myocyte cell
death. Diabetes. 50:2363–2375. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Malhotra A, Vashistha H, Yadav VS, Dube
MG, Kalra SP, Abdellatif M and Meggs LG: Inhibition of p66ShcA
redox activity in cardiac muscle cells attenuates
hyperglycemia-induced oxidative stress and apoptosis. Am J Physiol
Heart Circ Physiol. 296:H380–H388. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Sumara G, Formentini I, Collins S, Sumara
I, Windak R, Bodenmiller B, Ramracheya R, Caille D, Jiang H, Platt
KA, et al: Regulation of PKD by the MAPK p38delta in insulin
secretion and glucose homeostasis. Cell. 136:235–248. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Evans JL, Goldfine ID, Maddux BA and
Grodsky GM: Oxidative stress and stress-activated signaling
pathways: A unifying hypothesis of type 2 diabetes. Endocr Rev.
23:599–622. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen K, Albano A, Ho A and Keaney JF Jr:
Activation of p53 by oxidative stress involves platelet-derived
growth factor-beta receptor-mediated ataxia telangiectasia mutated
(ATM) kinase activation. J Biol Chem. 278:39527–39533. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Thornton TM and Rincon M: Non-classical
p38 map kinase functions: Cell cycle checkpoints and survival. Int
J Biol Sci. 5:44–51. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Nakagami H, Morishita R, Yamamoto K,
Yoshimura SI, Taniyama Y, Aoki M, Matsubara H, Kim S, Kaneda Y and
Ogihara T: Phosphorylation of p38 mitogen-activated protein kinase
downstream of bax-caspase-3 pathway leads to cell death induced by
high D-glucose in human endothelial cells. Diabetes. 50:1472–1481.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
She QB, Chen N and Dong Z: ERKs and p38
kinase phosphorylate p53 protein at serine 15 in response to UV
radiation. J Biol Chem. 275:20444–20449. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Fu X, Wan S, Lyu YL, Liu LF and Qi H:
Etoposide induces ATM-dependent mitochondrial biogenesis through
AMPK activation. PLoS One. 3:e20092008. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Barzilai A, Rotman G and Shiloh Y: ATM
deficiency and oxidative stress: A new dimension of defective
response to DNA damage. DNA Repair (Amst). 1:3–25. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Reliene R, Fischer E and Schiestl RH:
Effect of N-acetyl cysteine on oxidative DNA damage and the
frequency of DNA deletions in atm-deficient mice. Cancer Res.
64:5148–5153. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Uhlemeyer C, Müller N, Grieß K, Wessel C,
Schlegel C, Kuboth J and Belgardt BF: ATM and P53 differentially
regulate pancreatic beta cell survival in Ins1E cells. PLoS One.
15:e02376692020. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Borges HL, Linden R and Wang JYJ: DNA
damage-induced cell death: Lessons from the central nervous system.
Cell Res. 18:17–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Halaby MJ, Hibma JC, He J and Yang DQ: ATM
protein kinase mediates full activation of Akt and regulates
glucose transporter 4 translocation by insulin in muscle cells.
Cell Signal. 20:1555–1563. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Xu Y: Regulation of p53 responses by
post-translational modifications. Cell Death Differ. 10:400–403.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hart GW, Slawson C, Ramirez-Correa G and
Lagerlof O: Cross talk between O-GlcNAcylation and phosphorylation:
Roles in signaling, transcription, and chronic disease. Annu Rev
Biochem. 80:825–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jahangir Z, Ahmad W and Shabbiri K:
Alternate phosphorylation/O-GlcNAc modification on human insulin
IRSs: A road towards impaired insulin signaling in Alzheimer and
diabetes. Adv Bioinformatics. 2014:3247532014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Bond MR and Hanover JA: O-GlcNAc cycling:
A link between metabolism and chronic disease. Annu Rev Nutr.
33:205–229. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Akimoto Y, Hart GW, Wells L, Vosseller K,
Yamamoto K, Munetomo E, Ohara-Imaizumi M, Nishiwaki C, Nagamatsu S,
Hirano H and Kawakami H: Elevation of the post-translational
modification of proteins by O-linked N-acetylglucosamine leads to
deterioration of the glucose-stimulated insulin secretion in the
pancreas of diabetic Goto-Kakizaki rats. Glycobiology. 17:127–140.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Schraufstatter IU, Hyslop PA, Hinshaw DB,
Spragg RG, Sklar LA and Cochrane CG: Hydrogen peroxide-induced
injury of cells and its prevention by inhibitors of
poly(ADP-ribose) polymerase. Proc Natl Acad Sci USA. 83:4908–4912.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
D'Amours D, Desnoyers S, D'Silva I and
Poirier GG: Poly(ADP-ribosyl)ation reactions in the regulation of
nuclear functions. Biochem J. 342:249–268. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Oliver FJ, Menissier-de Murcia J and de
Murcia G: Poly(ADP-ribose) polymerase in the cellular response to
DNA damage, apoptosis, and disease. Am J Hum Genet. 64:1282–1288.
1999. View
Article : Google Scholar : PubMed/NCBI
|
|
111
|
Elkholi R and Chipuk JE: How do I kill
thee? Let me count the ways: p53 regulates PARP-1 dependent
necrosis. Bioessays. 36:46–51. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ba X and Garg NJ: Signaling mechanism of
poly(ADP-ribose) polymerase-1 (PARP-1) in inflammatory diseases. Am
J Pathol. 178:946–955. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chaitanya GV, Steven AJ and Babu PP:
PARP-1 cleavage fragments: Signatures of cell-death proteases in
neurodegeneration. Cell Commun Signal. 8:312010. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li M, Brooks CL, Wu-Baer F, Chen D, Baer R
and Gu W: Mono- versus polyubiquitination: Differential control of
p53 fate by Mdm2. Science. 302:1972–1975. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Feng J, Tamaskovic R, Yang Z, Brazil DP,
Merlo A, Hess D and Hemmings BA: Stabilization of Mdm2 via
decreased ubiquitination is mediated by protein kinase
B/Akt-dependent phosphorylation. J Biol Chem. 279:35510–35517.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ogawara Y, Kishishita S, Obata T, Isazawa
Y, Suzuki T, Tanaka K, Masuyama N and Gotoh Y: Akt enhances
Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem.
277:21843–21850. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Fang S, Jensen JP, Ludwig RL, Vousden KH
and Weissman AM: Mdm2 is a RING finger-dependent ubiquitin protein
ligase for itself and p53. J Biol Chem. 275:8945–8951. 2000.
View Article : Google Scholar : PubMed/NCBI
|