Introduction
Sepsis is a systemic inflammatory response secondary
to infection, and is the leading cause of mortality in intensive
care units, predominantly due to septic shock (1). Sepsis affects >3 million
individuals worldwide annually (2). Septic cardiomyopathy, characterized
by reduced left ventricular systolic and diastolic functions
(3,4), is a considerable and fatal
complication of sepsis, with mortality rates ranging between 40 and
50% (5,6). Despite extensive research identifying
apoptosis, oxidative stress, inflammation and calcium signaling as
key factors (5,7,8), the
precise pathophysiological mechanisms underlying septic
cardiomyopathy remain incompletely understood. This gap in
understanding hampers the development of targeted therapeutics for
this condition (9,10).
Mitochondria constitute the most voluminous
organelles within cardiomyocytes, representing ~35% of the total
cardiomyocyte volume (11).
Mitochondria are implicated in septic cardiomyopathy, serving as
the primary targets of cellular damage (12). The primary role of mitochondria
lies in ATP generation through oxidative phosphorylation (OXPHOS),
which is the central energy source for cardiomyocytes (13). Furthermore, the production of
reactive oxygen species (ROS) and the induction of apoptosis are
intrinsically linked to mitochondrial function (14,15).
Mitochondrial integrity and efficiency are maintained by a robust
quality control system encompassing biosynthesis, fission, fusion
and mitophagy (11). Dysfunctional
mitochondria exhibit a compromised aerobic oxidation capacity and a
reliance on glycolysis, culminating in an inadequate energy supply
and consequent cardiac dysfunction (16). Additionally, mitochondrial damage
is often associated with increased levels of ROS, and ROS signaling
is a key factor in the pathogenesis of septic cardiomyopathy
(5).
α-ketoglutarate (AKG), an intermediary in the
tricarboxylic acid (TCA) cycle, is instrumental in the adaptation
of cellular metabolism (17). AKG
is involved in numerous metabolic processes, including the
biosynthesis of amino acids, nucleotides, lipids and carnitine
(18). Beyond its role in energy
metabolism, AKG is involved in maintaining mitochondrial
homeostasis, exerting antioxidant and anti-inflammatory effects,
and promoting cellular proliferation (19,20).
Notably, AKG has been demonstrated to enhance energy
supplementation and mitigate oxidative stress during surgical
procedures, as indicated by the levels of oxidatively modified
proteins (21). In animal models
of diabetic cardiomyopathy and pressure overload cardiomyopathy,
AKG has been demonstrated to exert effective cardioprotective
effects by ameliorating cardiac remodeling and improving cardiac
function (22,23). However, to the best of our
knowledge, the specific impact of AKG in the context of septic
cardiomyopathy remains to be elucidated.
While the mechanisms underlying mitochondrial damage
have been investigated in various forms of cardiomyopathy, such as
dilated cardiomyopathy (24),
takotsubo cardiomyopathy (25) and
cardiomyopathy induced by antitumor drugs (26), their relevance in septic
cardiomyopathy remains largely unexplored. To address this,
cardiomyocytes and animal models of septic cardiomyopathy induced
by lipopolysaccharide (LPS), a constituent of Gram-negative
bacterial cell membranes (27),
were used in the present study. Echocardiography was employed to
assess cardiac function. For the examination of mitochondrial
ultrastructure, transmission electron microscopy was utilized. The
evaluation of mitochondrial function was carried out by means of
ATP production assays and Seahorse assays. Moreover, the levels of
reactive oxygen species were measured through staining with
dihydroethidium and the chloromethyl derivative CM-H2DCFDA. The
assessment of apoptosis was conducted using a TUNEL assay.
Additionally, western blotting was used to analyze the expression
of mitochondrial-associated proteins.
Materials and methods
Animals and treatment
Approval for the present study was obtained from the
Shaanxi Provincial People's Hospital Ethics Committee (Xi'an,
China) and the present study adhered to the National Institutes of
Health's Guide for the Care and Use of Laboratory Animals (28). In the present study, a total of 32
male C57BL/6 mice (age, 8 weeks; weight, 18–22 g), obtained from
GemPharmatech Co. Ltd., were acclimated for 1 week at a temperature
of 25°C, 55% humidity with a 12 h light/dark cycle and ad
libitum access to food and water.
The mice were randomly divided into four groups
(n=8/group). The two groups received a single intraperitoneal
injection of LPS (10 mg/kg; MilliporeSigma) dissolved in PBS, to
establish a model of septic cardiomyopathy (29). The remaining groups were
pre-treated with 2% AKG (MilliporeSigma) in the drinking water for
9 weeks prior to LPS administration (23). Animals were anesthetized by
isoflurane inhalation (4% induction and 2% maintenance), blood
samples were collected by cardiac puncture, and then cardiac tissue
was collected following cervical dislocation.
Echocardiography
Cardiac function was evaluated by transthoracic
echocardiography using a Vevo 2100 ultrasound system (VisualSonics,
Inc.) equipped with an MS550D transducer, as described previously
(25). For imaging, 2D images of
the left ventricle (LV) were captured at the papillary muscle
level. M-mode tracings, encompassing both the anterior and
posterior LV walls, were subsequently recorded. Key parameters,
including LV end-diastolic dimension (LVEDD), LV end-systolic
dimension (LVESD), ejection fraction and fractional shortening,
were measured in a blinded manner. These measurements were derived
from an average of five cardiac cycles to ensure accuracy and
reliability.
Western blotting
Western blotting was performed as described
previously (24). Following
protein transfer, polyvinylidene fluoride membranes were blocked
using 5% non-fat milk for 1 h at room temperature. This was
followed by an overnight incubation at 4°C with primary antibodies
against atrial natriuretic protein (ANP; cat. no. ab225844; 1:500;
Abcam), brain natriuretic peptide (BNP; cat. no. ab239510; 1:1,000;
Abcam), β-major histocompatibility complex (β-MHC; cat. no.
ab172967; 1:1,000; Abcam), BCL2 interacting protein 3 (Bnip3; cat.
no. ab10433; 1:1,000; Abcam), LC3 (cat. no. 4599S; 1:1,000; Cell
Signaling Technology, Inc.), Fis1 (cat. no. ab71498; 1:1,000;
Abcam), dynamin-related protein 1 (DRP1; cat. no. 8570S; 1:1,000;
Cell Signaling Technology, Inc.), NADH dehydrogenase (ubiquinone)
1α subcomplex subunit 12 (Ndufa12; cat. no. ab192617; 1:4,000;
Abcam), NADH:ubiquinone oxidoreductase subunit AB1 (Ndufab1; cat.
no. ab181021; 1:1,000; Abcam), mitochondrial NADH-ubiquinone
oxidoreductase chain 1 (MT-ND1; cat. no. ab181848; 1:5,000; Abcam),
succinate dehydrogenase (SDHA; cat. no. 11998; 1:4,000; Cell
Signaling Technology, Inc.), complex IV (COX IV; cat. no.
11242-1-AP; 1:3,000; Proteintech Group, Inc.), oxoglutarate
dehydrogenase (OGDH; cat. no. ab137773; 1:5,000; Abcam), pyruvate
dehydrogenase (PDH; cat. no. 3205; 1:3,000; Cell Signaling
Technology, Inc.), hypoxia inducible factor-1α (HIF-1α; cat. no.
36169; 1:1,000; Cell Signaling Technology, Inc.), NADPH oxidase 2
(NOX2; cat. no. bs-3889R; 1:1,000; BIOSS), NOX4 (cat. no. bs-1091R;
1:1,000; BIOSS), Bax (cat. no. 2772S; 1:3,000; Cell Signaling
Technology, Inc.) and Bcl-2 (cat. no. 3498S; 1:1,000; Cell
Signaling Technology, Inc.), GAPDH (cat. no. 10494-1-AP; 1:8,000;
Proteintech Group, Inc.), α-tubulin (cat. no. 11224-1-AP; 1:4,000;
Proteintech Group, Inc.) and HSP90 (cat. no. 11224-1-AP; 1:2,000;
Proteintech Group, Inc.). Following incubation with the primary
antibody, the membranes were treated with horseradish
peroxidase-conjugated secondary antibodies (anti-Mouse secondary
antibodies; cat. no. 62-6520; 1:10,000; Thermo Fisher Scientific,
Inc. and anti-Rabbit secondary antibodies; cat. no. VJ313046;
1:10,000; Thermo Fisher Scientific, Inc.) for 1 h at room
temperature. Signals were visualized using Clarity™
Western ECL Substrate (Bio-Rad Laboratories, Inc.). Densitometry
analysis was performed using ImageJ software (version 4.5.2;
National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was isolated from LV myocardial tissues
using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). From the extracted RNA, 1 µg was
reverse-transcribed into cDNA using a RT kit (cat. no. RR047A;
Takara Bio, Inc.) at 37°C for 15 min and 85°C for 5 sec. qPCR was
performed using TB Green™ Premix Ex Taq™ II
(Tli RNaseH Plus; cat. no. RR820A; Takara Bio, Inc.) on a Bio-Rad
CFX96 Real-time PCR Detection System (Bio-Rad Laboratories, Inc.).
The amplification protocol involved an initial denaturation at 95°C
for 30 sec, followed by 40 cycles of denaturation at 95°C for 5 sec
and annealing/extension at 60°C for 30 sec. Gene expression levels
were quantified relative to the housekeeping gene GAPDH for
normalization using the 2−ΔΔCq method (30). The sequences of the primers used
for qPCR were: ANP forward, 5′-AAGAACCTGCTAGACCACCTGGAG-3′ and
reverse, 5′-TGCTTCCTCAGTCTGCTCACTCAG-3′; BNP forward,
5′-GGAAGTCCTAGCCAGTCTCCAGAG-3′ and reverse,
5′-GCCTTGGTCCTTCAAGAGCTGTC-3′; β-MHC forward,
5′-CAGAACACCAGCCTCATCAACCAG-3′ and reverse,
5′-TTCTCCTCTGCGTTCCTACACTCC-3′; and GAPDH forward,
5′-AGGTCGGTGTGAACGGATTTG-3′ and reverse,
5′-TGTAGACCATGTAGTTGAGGTCA-3′.
Histological analysis
TUNEL staining of the LV tissue sections was
performed according to the manufacturer's protocol. Briefly, heart
tissues were fixed in 4% paraformaldehyde at room temperature
overnight. Subsequently, tissues were embedded in paraffin and cut
into slices with a thickness of 4 µm. After dewaxing and hydration,
slices were stained with TUNEL reagent (cat. no. C1088; Beyotime
Institute of Biotechnology) at room temperature for 1 h. Then,
antifade mounting medium with DAPI (cat. no. P0131; Beyotime
Institute of Biotechnology) solution was incubated at room
temperature for 10 min for nuclear counterstaining. TUNEL-stained
sections were imaged using an Olympus DP-72 fluorescence microscope
(Olympus Corporation), focusing on the identification of apoptotic
cells within the myocardium. Images were analyzed using ImageJ. The
number of cardiomyocyte nuclei exhibiting green fluorescence
(indicating apoptosis) was counted. To ensure objectivity, all
analyses were carried out under double-blinded conditions. For each
histological section, five random visual fields were selected for
measurement. The average value from these fields was then used for
statistical analysis.
Transmission electron microscopy (TEM)
analysis
TEM was carried out as described previously
(24). Fresh apical myocardial
tissue from the LV was first fixed using 2.5% glutaraldehyde at 4°C
for 2 h. After fixation, a graded series of ethanol/acetone
solutions was employed for dehydration, with the final solution
being absolute acetone. The dehydrated samples were then
infiltrated with Epon 812 resin. Ultrathin sections of the embedded
tissues were prepared using an ultramicrotome (LKB-V/NOVA; Leica
Microsystems GmbH) and stained with acidified uranyl acetate for
enhanced contrast. Observation of the prepared samples was carried
out using a Hitachi Model H-7650 TEM (Hitachi, Ltd.). ImageJ was
used for quantitative analysis of the TEM images. Specifically, the
count of mitochondria was determined from five images at a
magnification of ×10,000 per LV sample. Additionally, the
proportion of mitochondria exhibiting structural impairments, such
as incomplete outer membranes or dissolved cristae, was assessed
from another set of five images at a magnification of ×30,000 per
LV sample. The count of aberrant mitochondria is presented as a
percentage of the total mitochondrial count.
Determination of ROS levels
ROS levels in myocardial tissues were quantified
using two distinct methods as described previously (24). Initially, O2− content
was assessed using 5-µm frozen myocardial sections. These sections
were incubated with dihydroethidium (DHE; 5 µM) for 1 h at 37°C.
Fluorescence images were captured at a magnification of ×400 (five
fields per heart) using an Olympus DP-72 fluorescence microscope
(Olympus Corporation), with excitation and emission wavelengths set
at 488 and 610 nm, respectively. In the second method,
cardiomyocytes were isolated via enzyme digestion using the
Langendorff perfusion system. Briefly, the aorta was retrogradely
perfused with collagenase II (cat. no. LS004177; Worthington
Biochemical Corporation) at room temperature for 15 min to digest
the heart. Subsequently, the heart was minced into small pieces and
centrifuged at 1,400 × g for 3 min at room temperature. Finally,
the isolated cardiomyocytes were transferred onto the cell culture
dish. The isolated cells were then incubated with chloromethyl
derivative CM-H2DCFDA (DCF; 5 µM) for 30 min at 37°C.
Fluorescence imaging was performed using a Leica TCS SP8 STED 3X
confocal microscope (Leica Microsystems GmbH), with a ×40 1.3 NA
oil immersion objective lens, with excitation at 488 nm and
emission at 525 nm, using standardized scanning parameters. The
intensity of DCF fluorescence was quantified using ImageJ, with an
average of 80 cells analyzed per heart.
H&E staining
Briefly, the heart was fixed in 4% paraformaldehyde
at room temperature overnight. Subsequently, it was embedded in
paraffin and sliced into 4 µm slices. After deparaffinization and
hydration, heart sections were stained with hematoxylin for 5 min
and eosin for 1 min at room temperature using the H&E Staining
kit (cat. no. 0105; Beyotime Institute of Biotechnology). The
fluorescence images were captured using an Olympus DP-72
fluorescence microscope (Olympus Corporation) from 3–5 random
fields.
ATP, lactate and malondialdehyde (MDA)
assay
Fresh LV tissue and plasma was harvested to
determine the content of ATP, lactate and MDA using commercial
kits. ATP kits (cat. no. A095-2-1; Nanjing Jiancheng Bioengineering
Institute), lactate kits (cat. no. E-BC-Ko44-M; Wuhan Elabscience
Biotechnology Co., Ltd.) and MDA kits (cat. no. R21869; Shanghai
Yuanye Biotechnology Co., Ltd.) were carried out according to the
manufacturer's protocol as described previously (25).
Measurement of circulating AKG
content
Plasma levels of AKG were quantified using a
commercial kit (cat. no. G0861W; Grace Biotech Co., Ltd.) according
to the manufacturer's protocol.
Isolation and culture of neonatal rat
ventricular myocytes (NRVMs)
A total of 24 Neonatal Sprague-Dawley rats (age, 1–2
days; weight, 6–7 g) were purchased from the Laboratory Animal
Center of Xi'an Jiaotong University (Xi'an, China), and humanely
euthanized via cervical dislocation. Subsequently, their hearts
were excised and subjected to enzymatic digestion using 1 ml 0.2%
collagenase II for 5–6 min at 37°C for six cycles. The digested
tissue was centrifuged at 2,200 × g for 5 min at room temperature
and the resultant cardiomyocytes were resuspended in F12 medium
(Thermo Fisher Scientific, Inc.) supplemented with 15% FBS (Thermo
Fisher Scientific, Inc.). This suspension was incubated for 60 min
at 37°C to facilitate differential adhesion, allowing for the
separation of cardiomyocytes from non-myocyte cells. To further
purify the culture, the cardiomyocytes were then treated with
5-bromo-2-deoxyuridine, an agent used to inhibit the proliferation
of non-cardiomyocyte cells (31).
After a 48-h incubation period at 37°C, the NRVMs given the
following treatments (Control, 2 mM AKG, 0.5 µg/ml LPS or a
combination of 2 mM AKG and 0.5 µg/ml LPS) were used for TUNEL
staining, western blotting and Seahorse assays. The control group
was treated with F12 medium containing 10% FBS. These treatments
were administered in F12 medium containing 10% FBS for a duration
of 24 h at 37°C as previously described (7,23).
Seahorse assay
The respiratory capacity of mitochondria in NRVMs
was evaluated using the Agilent Seahorse XF24 Extracellular Flux
Analyzer (Agilent Technologies, Inc.). NRVMs were initially seeded
onto Seahorse XF24 cell culture microplates and subjected to the
aforementioned treatments. The Seahorse XF sensor cartridge was
prepared by hydrating its probe plate with calibration solution in
a CO2-free incubator maintained at 37°C overnight.
Subsequently, during the assay, specific mitochondrial inhibitors
were sequentially added to the wells: Oligomycin A (1.5 µM; cat.
no. 103672-100; Agilent) to inhibit ATP synthase, trifluoromethoxy
carbonylcyanide phenylhydrazone (1 µM; cat. no. 103672-100;
Agilent) to uncouple the proton gradient and antimycin A (0.5 µM;
cat. no. 103672-100; Agilent) to inhibit the mitochondrial
respiratory chain. The oxygen consumption rate, a key indicator of
mitochondrial respiration, was measured following these additions,
according to the manufacturer's protocol.
Statistical analysis
Data are presented as the mean ± standard error of
the mean (n=6-8 repeats/group). To determine the normality and
homogeneity of variance of the data, Shapiro-Wilk's and Levene's
tests were respectively employed. For comparisons among multiple
groups, one-way ANOVA was carried out, followed by Tukey's post hoc
test. All statistical analyses were carried out using GraphPad
Prism (version 9.0; Dotmatics). P<0.05 was considered to
indicate a statistically significant difference.
Results
AKG supplementation improves LV
remodeling and cardiac dysfunction in LPS-induced septic
cardiomyopathy
In the present study, the cardiotoxic effects of LPS
in wild-type mice were investigated, focusing on LPS-induced
cardiac dysfunction and LV remodeling. Echocardiographic
assessments were carried out following exposure to vehicle control,
AKG, LPS or a combination of AKG and LPS. Notably, the LV ejection
fraction (LVEF) and LV fractional shortening (LVFS) in the LPS
group were reduced by 33 and 32%, respectively, compared with those
in the control group. Concurrently, there was a 40 and 60% increase
in LVEDD and LVESD (Fig. 1A and
B). These findings indicated cardiac dysfunction characteristic
of septic cardiomyopathy induced by LPS.
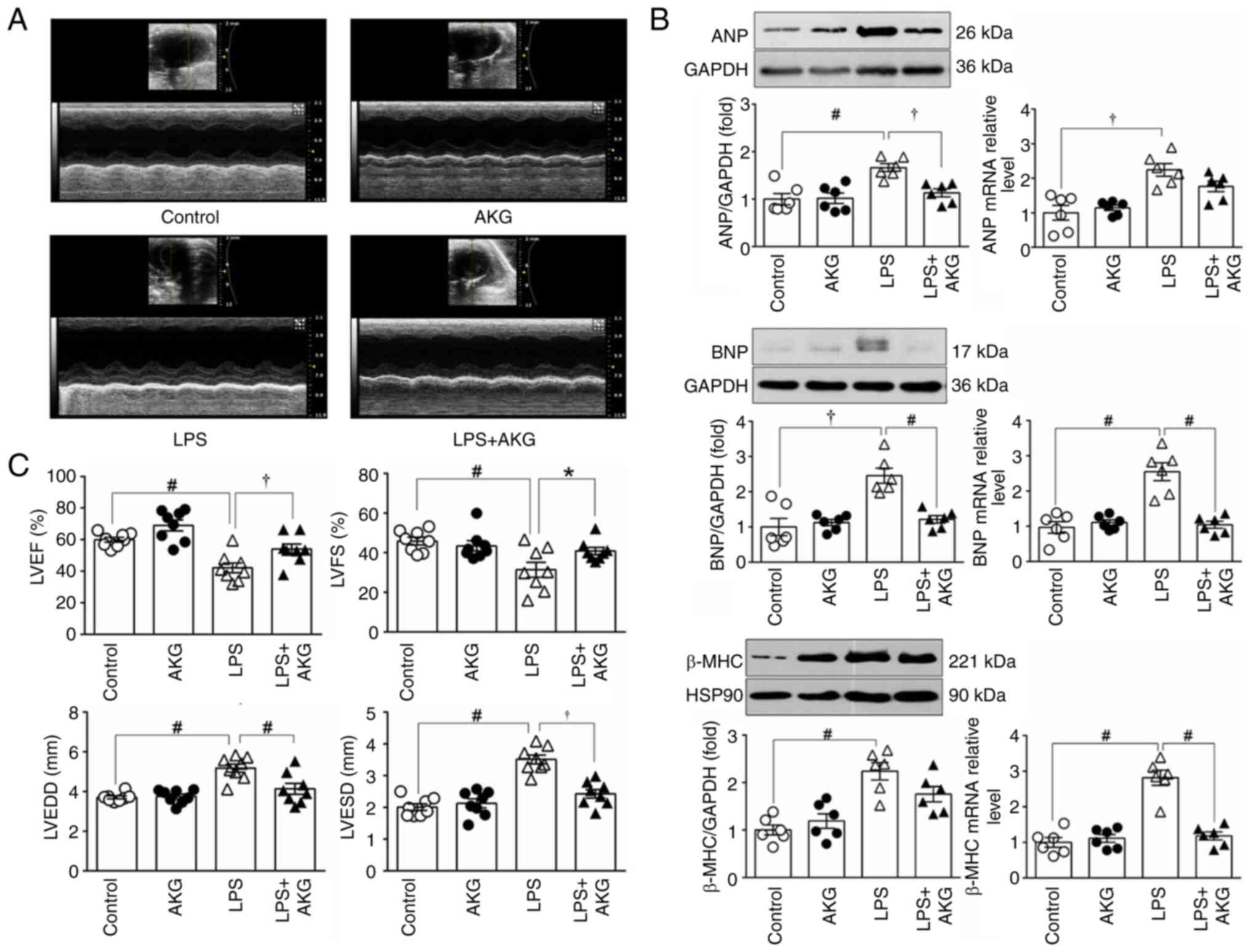 | Figure 1.AKG improves cardiac dysfunction in
an LPS-induced mouse model of septic cardiomyopathy. (A)
Representative 2D echocardiographic images of the LV short-axis
from mice treated with control, AKG, LPS and AKG + LPS. (C) LVEF,
LVFS, LVEDD and LVESD based on the LV ultrasound images
(n=8/group). (B) Protein and mRNA expression levels of ANP, BNP and
β-MHC in LV myocardium (n=6/group), GAPDH was used as the loading
control for ANP and BNP, while HSP90 was used as the loading
control for β-MHC. Data are presented as the mean ± SEM.
*P<0.05, †P<0.01, #P<0.001. AKG,
α-ketoglutarate; LPS, lipopolysaccharide; LV, left ventricle; LVEF,
LV ejection fraction; LVFS, LV fractional shortening; LVEDD, LV
end-diastolic dimension; LVESD, LV end-systolic dimension; ANP,
atrial natriuretic peptide; BNP, brain natriuretic peptide; β-MHC,
β-major histocompatibility complex; HSP90, heat shock protein
90. |
The cardioprotective effect of AKG supplementation
was evident. AKG treatment led to a 22 and 21% improvement in LVEF
and LVFS, along with a 20 and 32% reduction in LVEDD and LVESD,
respectively, compared with the LPS group (Fig. 1A and B). There were no significant
differences in the cardiac function indices between the AKG group
and the control group. This suggested that AKG supplementation did
not affect cardiac function when administered alone (Fig. 1B).
Cardiomyocyte remodeling was evaluated by assessing
the protein and transcriptional levels of ANP, BNP and β-MHC in the
LV myocardium. LPS-treated mice exhibited a notable increase in the
mRNA and protein expression levels of these markers, indicative of
cardiomyocyte remodeling. However, AKG supplementation may
partially mitigate the LPS-induced remodeling (Fig. 1C).
As shown in Fig.
S1, the circulating AKG concentration was decreased in the LPS
group compared with that in the control group, but the difference
was not significant. Although the concentration of circulating AKG
increased following AKG supplementation in the LPS group, the
differences were not statistically significant.
AKG exposure reverses mitochondrial
morphological damage and facilitates mitophagy and fission in
LPS-treated mice
Given that mitochondria constitute ~35% of the
volume of cardiomyocytes and serve a key role in cellular function
(11), a detailed analysis of
mitochondrial ultrastructure was performed in heart tissues from
mice in the different groups using TEM. LPS treatment induced
notable mitochondrial morphological damage, evidenced by increased
proportions of mitochondria with incomplete outer membranes or
dissolved cristae, compared with those in hearts from the control
group (Fig. 2A and B).
Additionally, quantitative TEM analysis revealed a reduction in the
mitochondrial number. This was further corroborated by assessing
the protein levels of Ndufa12 and Ndufab1, which serve as
mitochondrial markers (Fig.
2A-C).
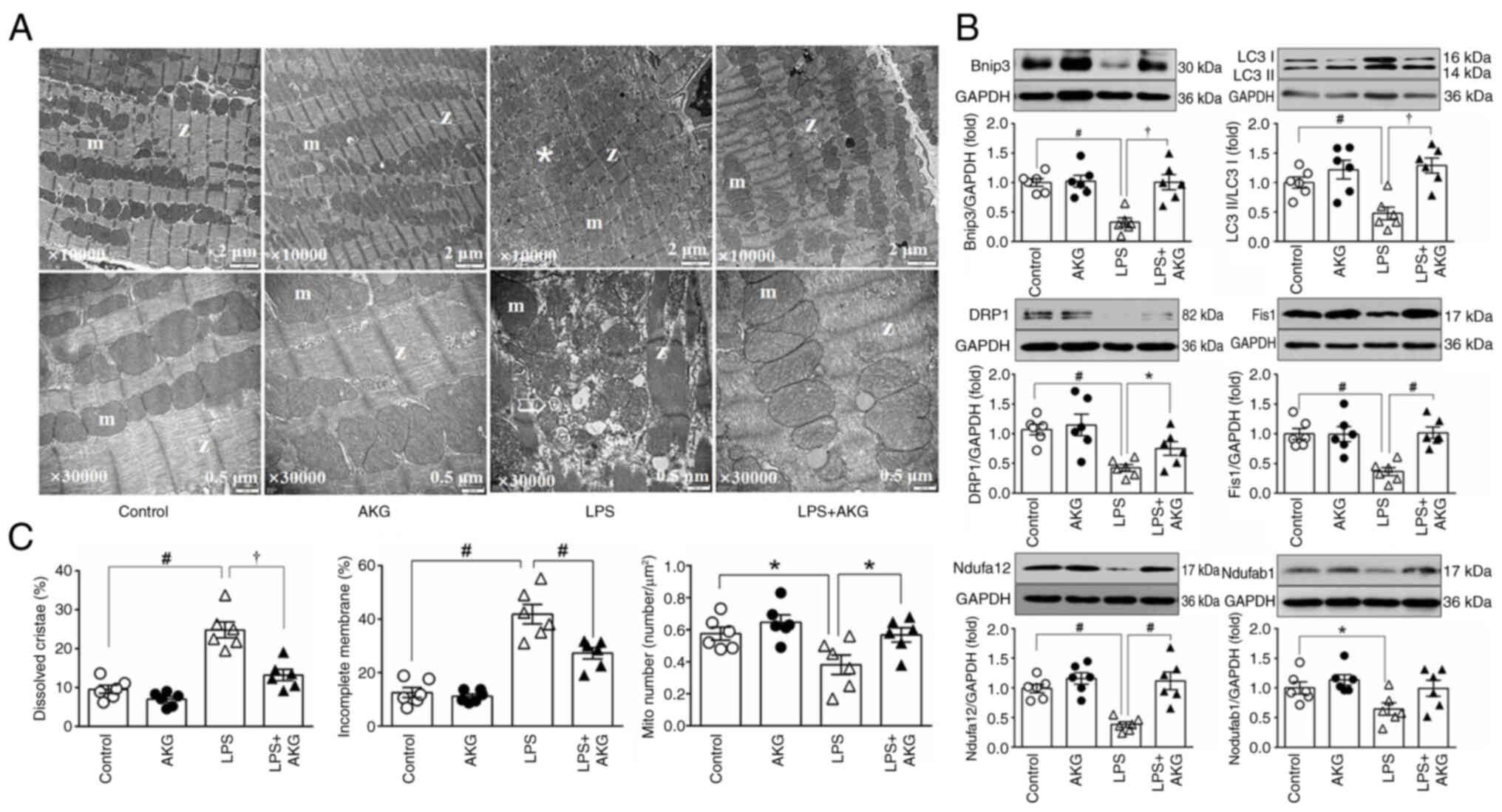 | Figure 2.AKG alleviates myocardial
mitochondrial morphological damage induced by LPS. (A) Electron
microscopy images of left ventricular myocardium. Magnification:
Top, ×10,000; bottom, ×30,000. *Mitochondria with dissolved
cristae; the arrow indicates mitochondria with incomplete outer
membranes (n=6/group). (B) Representative immunoblotting images and
semi-quantitative analysis of protein expression of markers of
mitochondrial mitophagy (Bnip3 and LC3), fission (DRP1 and Fis1)
and mitochondrial content (Ndufab1 and Ndufa12; n=6/group). (C)
Quantitative analysis of mitochondria. The percentage of
mitochondria with an incomplete outer membrane and dissolved
cristae, and the number of mitochondria per unit area (n=6/group).
Data are presented as the mean ± SEM. *P<0.05,
†P<0.01, #P<0.001. AKG,
α-ketoglutarate; m, mitochondria; Z, Z line; Mito number, the
number of mitochondria per unit area; Bnip3, BCL2 interacting
protein 3; DRP1, dynamin-related protein 1; Nduf, NADH:ubiquinone
oxidoreductase subunits; LPS, lipopolysaccharide. |
Mitochondrial quality control, encompassing
processes such as biosynthesis, fusion, fission and mitophagy, is
essential for maintaining mitochondrial integrity (11). In hearts from the group subjected
to LPS, there was a marked decrease in the expression levels of
proteins indicative of mitochondrial fission (DRP1 and Fis1) and
mitophagy (LC3 II/I and Bnip3) compared with the control group
(Fig. 2C). These findings
suggested impaired mitochondrial quality control mechanisms
following LPS exposure.
By contrast, AKG treatment mitigated these adverse
effects. It restored mitochondrial morphology by reducing the
fraction of mitochondria with structural impairments, and increased
the overall mitochondrial count, as evidenced by the increased
expression levels of mitochondrial content markers (Ndufa12;
Fig. 2A-C). Furthermore, AKG
positively influenced mitochondrial quality control, demonstrated
by the upregulated expression levels of proteins associated with
mitochondrial fission and mitophagy (Fig. 2C). This suggested that AKG
supplementation counteracted the mitochondrial dysfunction observed
in LPS-induced cardiomyopathy.
AKG restores myocardial mitochondrial
energy metabolism in septic mice
Generation of ATP through OXPHOS is the primary role
of mitochondria and this was further investigated in the context of
LPS-induced mitochondrial damage (13). Specifically, the changes in
mitochondrial energy metabolism in mouse hearts treated with LPS
were assessed.
Initially, immunoblotting was used to analyze heart
samples for the presence of specific marker proteins. These
included proteins associated with mitochondrial respiratory
electron transport chain complexes I, II and IV (MT-ND1, SDHA and
COX IV), and key enzymes involved in acetyl-CoA production and the
TCA cycle (PDH and OGDH). A notable decrease in the expression
levels of these proteins was observed, indicating impaired
mitochondrial aerobic oxidation. Conversely, the protein levels of
HIF-1α, indicative of anaerobic glycolysis, were revealed to be
elevated following LPS treatment (Fig.
3A and B).
 | Figure 3.AKG recovers myocardial mitochondrial
energy metabolism in an LPS-induced mouse model of septic
cardiomyopathy. Western blotting was carried out using protein
extracted from LV tissues, and protein expression was
semi-quantified and normalized to GAPDH (n=6/group). (A)
Representative blots of marker proteins for mitochondrial energy
metabolism: Mitochondrial complex I (MT-ND1), complex II (SDHA),
COX IV, tricarboxylic acid cycle (OGDH), a key enzyme for
acetyl-CoA synthesis (PDH) and anaerobic glycolysis (HIF-1α). (B)
Semi-quantitative analysis of mitochondrial energy metabolism
marker proteins. (C) ATP content in the LV tissues, and lactate
content in the plasma and LV tissues, as determined by an ELISA
(n=6/group). Data are presented as the mean ± SEM. *P<0.05,
†P<0.01, #P<0.001. LV, left ventricle;
MT-ND1, mitochondrial NADH-ubiquinone oxidoreductase chain 1;
HIF-1α, hypoxia inducible factor-1α; OGDH, ketoglutarate
dehydrogenase; PDH, pyruvate dehydrogenase; SDHA, succinate
dehydrogenase; AKG, α-ketoglutarate; LPS, lipopolysaccharide; COX
IV, complex IV; prot, protein. |
Furthermore, to directly assess mitochondrial
functionality, the levels of ATP and lactate were measured using an
ELISA. Analysis revealed a significant reduction in ATP levels in
the hearts of mice treated with LPS, accompanied by an increase in
lactate content in LV tissues and plasma (Fig. 3C). AKG supplementation led to a
partial increase in the abundance of proteins associated with
aerobic oxidation and ATP production, while concurrently decreasing
the levels of HIF-1α and lactate (Fig.
3A-C). This suggested that AKG reversed the alterations in
mitochondrial energy metabolism induced by LPS treatment.
AKG reduces ROS content and inhibits
apoptosis in mice treated with LPS
Given the key role of mitochondria in the regulation
of ROS generation and apoptosis (15), the impact of AKG on these
parameters in LPS-induced cardiomyopathy was assessed.
To assess oxidative stress, DCF and DHE staining
were carried out to determine ROS levels in myocardial tissue
(Fig. 4A and B). Additionally, the
expression levels of key enzymes involved in ROS production, namely
NOX2 and NOX4, were examined (Fig.
4D). MDA, a biomarker of lipid peroxidation, was also measured
to provide further insights into oxidative stress (Fig. 4C). Compared with the control group,
a significant increase in ROS generation was observed in the
LPS-treated group. This elevation in ROS levels was mitigated in
the LPS + AKG group, suggesting that AKG reduced oxidative stress
in LPS-induced septic cardiomyopathy.
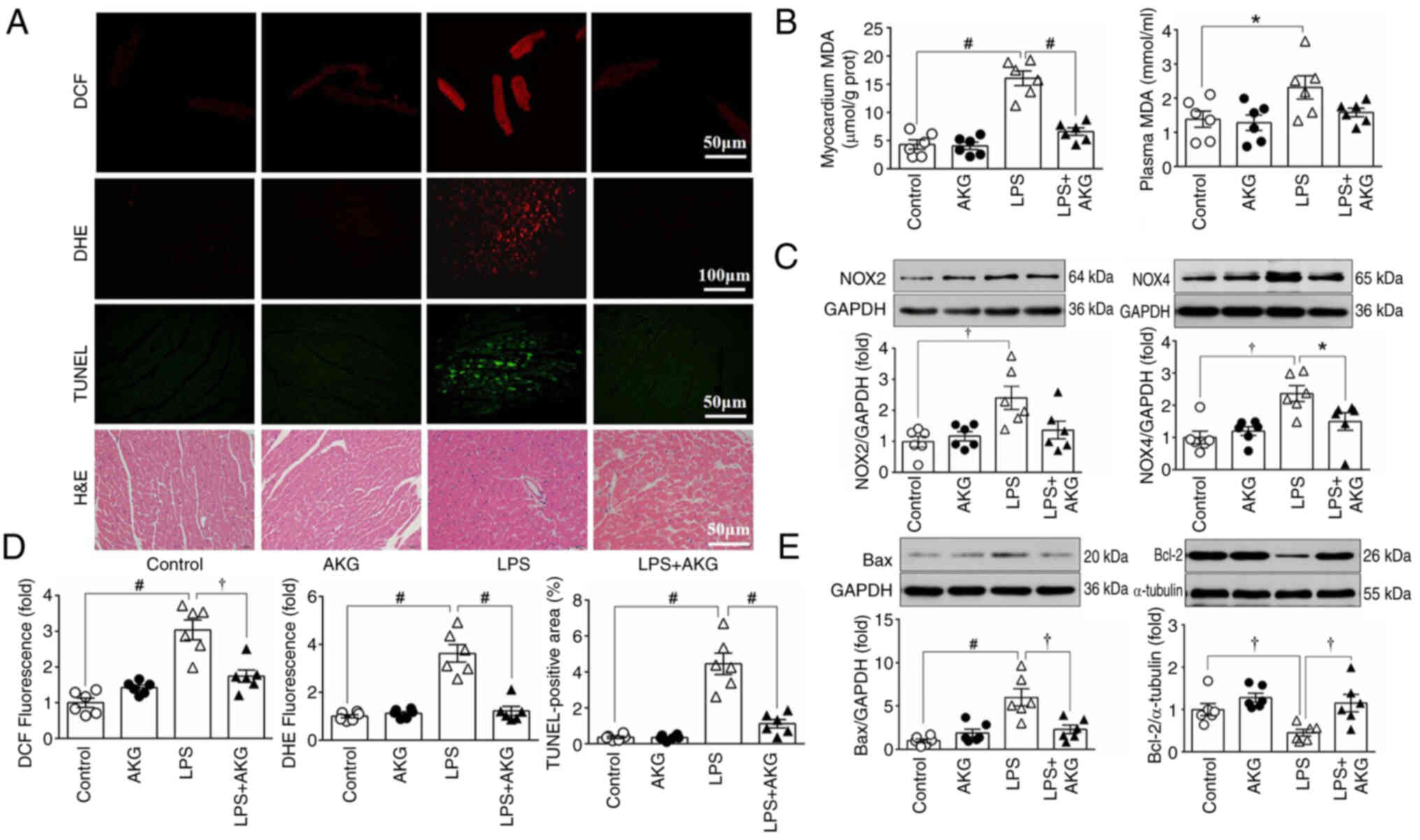 | Figure 4.AKG reduces myocardial ROS and
apoptosis in an LPS-induced mouse model of septic cardiomyopathy.
ROS levels were detected using various methods, including DCF
staining, DHE staining, MDA content determination using an ELISA,
and examination of NOX2/4 protein expression. Apoptosis was
assessed via TUNEL staining, and examination of the protein
expression levels of Bax and Bcl-2. (A) From top to bottom:
Representative images of DCF, DHE, TUNEL and H&E staining
(n=6/group). (B) Changes in myocardial and plasma levels of MDA
(n=6/group). (C) Western blot bands and analysis of marker proteins
for NOX2 and NOX4 (n=6/group). (D) Quantitative analysis of DCF,
DHE and TUNEL staining (n=6/group). (E) Representative blots and
analysis of marker proteins for Bax and Bcl-2 (n=6/group). Data are
presented as the mean ± SEM. *P<0.05, †P<0.01,
#P<0.001. AKG, α-ketoglutarate; LPS,
lipopolysaccharide; ROS, reactive oxygen species; DCF, chloromethyl
derivative CM-H2DCFDA; DHE, dihydroethidium; MDA,
malondialdehyde; NOX, NADPH oxidase; prot, protein. |
To evaluate apoptosis, TUNEL staining was performed
and the protein expression levels of Bax and Bcl-2 in myocardial
tissue were measured. Enhanced apoptosis was evident in the LPS
group compared with both the control and AKG groups (Fig. 4A, B and E). However, AKG
administration in the LPS-treated group led to a reduction in
apoptosis. These findings indicated that AKG supplementation
attenuated both oxidative stress and apoptosis induced by LPS in
the context of septic cardiomyopathy.
To examine the morphology of the myocardium, H&E
staining was performed. In the LPS group, the myocardium showed a
disorderly arrangement of myocytes, karyolysis and an increased
presence of inflammatory cells. However, AKG administration
improved the abnormal histological structure (Fig. 4A).
AKG reduces apoptosis, improves
mitochondrial energy metabolism and increases mitochondrial
turnover in vitro
The effects of AKG on NRVMs subjected to LPS
treatment were explored (Fig. 5).
The impact on apoptosis was initially assessed using TUNEL staining
and measurement of Bcl-2 protein levels. Compared with the control
group, a seven-fold increase in the optical density of
TUNEL-positive cells was observed in the LPS group, coupled with a
significant decrease in Bcl-2 protein levels, indicating increased
apoptosis due to LPS exposure. However, simultaneous administration
of AKG significantly mitigated LPS-induced apoptosis in NRVMs
(Fig. 5A, D and E).
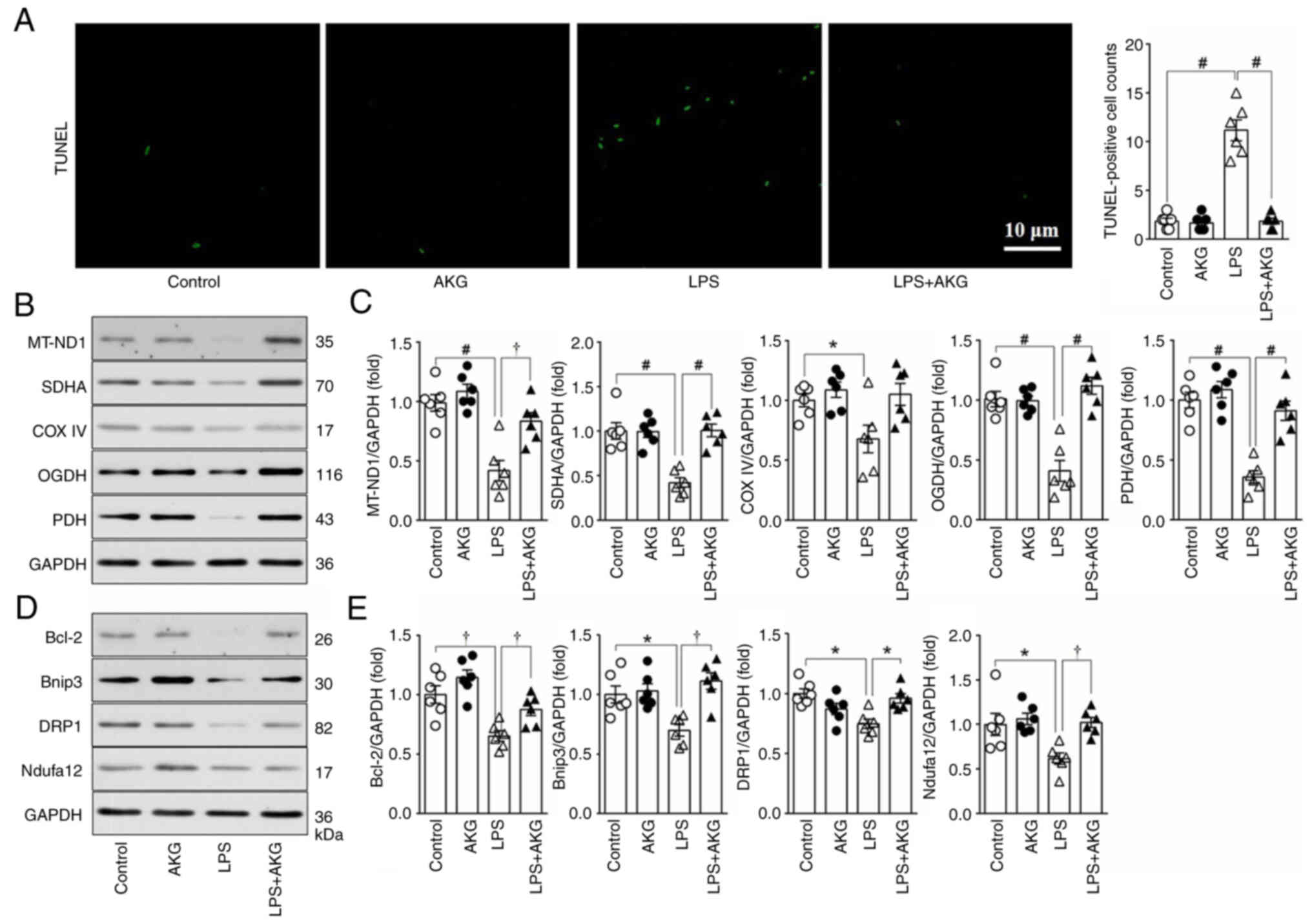 | Figure 5.Effect of AKG on apoptosis and
mitochondrial metabolism and dynamics in vitro. Neonatal rat
ventricular myocytes were treated with vehicle, AKG, LPS or AKG +
LPS. (A) Images and quantitative analysis of the TUNEL staining
(n=6/group). (B) Representative western blot images and (C)
semi-quantification of markers for mitochondrial metabolism
(MT-ND1, SDHA, COX IV, OGDH and PDH) (n=6/group). (D) Protein
expression levels of selected markers for apoptosis (Bcl-2),
mitochondrial quality control (Bnip3 and DRP1) and mitochondrial
number (Ndufa12). (E) Semi-quantitative analysis of protein
expression of Bcl-2, Bnip3, DRP1 and Ndufa12) (n=6/group). Data are
presented as the mean ± SEM. *P<0.05, †P<0.01,
#P<0.001. AKG, α-ketoglutarate; LPS,
lipopolysaccharide; MT-ND1, mitochondrial NADH-ubiquinone
oxidoreductase chain 1; SDHA, succinate dehydrogenase; COX IV,
complex IV; OGDH, ketoglutarate dehydrogenase; PDH, pyruvate
dehydrogenase; Bnip3, BCL2 interacting protein 3; DRP1,
dynamin-related protein 1; Nduf, NADH:ubiquinone oxidoreductase
subunits. |
Additionally, the expression levels of marker
proteins integral to mitochondrial aerobic oxidation, including
MT-ND1, SDHA, COX IV, OGDH and PDH, were explored. LPS treatment
resulted in a significant reduction in the expression levels of
these proteins. By contrast, AKG administration alleviated this
reduction, suggesting its efficacy in improving mitochondrial
aerobic oxidation when this is compromised by LPS (Fig. 5B and C).
Immunoblotting analysis revealed alterations in
mitochondrial turnover induced by LPS, evidenced by decreased
expression levels of proteins associated with mitophagy (Bnip3) and
fission (DRP1), along with a reduction in the mitochondrial content
marker Ndufa12. Notably, AKG supplementation significantly
counteracted these effects, suggesting its potential to restore
mitochondrial turnover disrupted by LPS treatment (Fig. 5D and E). These findings
collectively underscore the therapeutic potential of AKG in
mitigating mitochondrial dysfunction and apoptosis in LPS-induced
cardiomyopathy.
AKG improves mitochondrial respiration
damage induced by LPS in vitro
Whether AKG directly enhanced mitochondrial function
was subsequently assessed. A Seahorse XF mitochondrial stress test
analyzer was used to evaluate the mitochondrial respiratory
capacity in NRVMs (Fig. 6A).
 | Figure 6.Effect of AKG on mitochondrial
respiratory capacity in vitro. (A) OCR curve of the control,
AKG, LPS and AKG + LPS groups (n=5/group). (B) Non-mitochondrial
respiration, (C) basal respiration, (D) maximal respiration, (E)
ATP production, (F) proton leak and (G) spare respiration capacity
were calculated. Data are presented as the mean ± SEM. *P<0.05,
†P<0.01, #P<0.001. AKG,
α-ketoglutarate; LPS, lipopolysaccharide; OCR, oxygen consumption
rate; FCCP, trifluoromethoxy carbonylcyanide phenylhydrazone. |
Key parameters of mitochondrial respiratory
function, including basal and maximal respiratory capacity
(Fig. 6C and D), ATP production
(Fig. 6E) and spare respiratory
capacity (Fig. 6G), were assessed.
These parameters were found to be significantly compromised in
NRVMs treated with LPS, compared with those in the control group.
Notably, when AKG was administered in conjunction with LPS, an
improvement in mitochondrial respiration was observed.
This enhancement suggested that AKG supplementation
effectively counteracted the mitochondrial dysfunction induced by
LPS in NRVMs. In the present study, no significant differences in
non-mitochondrial respiration in NRVMs were observed.
Discussion
In the present study, focusing on the mechanism of
myocardial mitochondrial damage in septic cardiomyopathy, NRVMs and
animal models were used to investigate the effects of AKG
supplementation on LPS-induced myocardial injury. The results
revealed several novel insights: Firstly, LPS was revealed to
mediate cardiac remodeling and dysfunction; secondly, myocardial
mitochondrial damage in septic cardiomyopathy, as evidenced by
morphological abnormalities, impaired mitochondrial quality control
and reduced energy metabolism, was accompanied by increased
apoptosis and ROS production; and thirdly, the present study
demonstrated that AKG supplementation alleviated myocardial
mitochondrial damage and improved cardiac function in septic
cardiomyopathy. The present results highlight the therapeutic
potential of AKG in mitigating mitochondrial dysfunction and
associated cardiac impairments in this condition, providing a
possible direction for future research and treatment
approaches.
AKG, a key intermediate of the Krebs cycle, situated
between succinyl CoA and isocitrate, serves as a precursor for
glutamate and glutamine (32).
Notably, circulating AKG levels are elevated in patients with heart
failure, but are decreased in obese and diabetic individuals
(32,33). Previous animal research has
indicated a reduction in AKG levels in diabetic cardiomyopathy and
ischemic heart failure (34), and
supplementation with AKG ameliorated myocardial pathological
remodeling and enhanced cardiac function in these models (22,23).
Despite these findings, to the best of our knowledge, the specific
role of AKG in septic cardiomyopathy remains unexplored. The
present study addresses this gap by investigating the impact of AKG
in septic cardiomyopathy induced by LPS, shedding light on
potential therapeutic avenues in this context.
The present study demonstrated that AKG
supplementation mitigated cardiac dysfunction induced by LPS, as
evidenced by echocardiographic assessments. Furthermore, myocardial
remodeling was assessed based on the mRNA and protein expression
levels of ANP, BNP and β-MHC, which revealed that AKG effectively
reduced pathological myocardial remodeling. However, the precise
mechanism by which AKG improved cardiac function remains unclear.
Our previous studies investigated mitochondrial damage mechanisms
in dilated cardiomyopathy (24),
takotsubo cardiomyopathy (25) and
antitumor drug-induced cardiomyopathy (26). Notably, mitochondrial abnormalities
have been recognized as a pivotal factor in the pathogenesis of
septic cardiomyopathy (7).
Building upon this understanding, the role of mitochondrial damage
mechanisms in septic cardiomyopathy and the therapeutic efficacy of
AKG were assessed.
In the present study, exploration of mitochondrial
morphology in septic cardiomyopathy using TEM revealed abnormal
mitochondrial ultrastructure in the myocardium from mice exposed to
LPS, notably characterized by an increased proportion of
mitochondria with incomplete outer membranes and dissolved cristae.
This structural damage facilitates the release of mitochondrial DNA
into the cytoplasm, exacerbating myocardial damage and systemic
inflammation (35). Additionally,
such compromised mitochondria may prompt the cytoplasmic release of
pro-apoptotic proteins such as cytochrome c, further
promoting apoptosis (36,37). Mitochondria are subject to rigorous
‘quality control’ processes, including biosynthesis, fission,
fusion and mitophagy, to maintain their quality and quantity
(38). In the present study, a
reduction in the expression levels of proteins associated with
mitochondrial fission and mitophagy was observed, which was
consistent with the decreased mitochondrial count detected by
electron microscopy. While moderate mitophagy and fission are key
for clearing damaged mitochondria and ensuring ATP production
(39), their insufficiency leads
to an accumulation of dysfunctional mitochondria. Of note, the
findings of the present study indicated that AKG supplementation
counteracted these detrimental changes. Both TEM and biochemical
assays suggested that AKG enhanced mitochondrial quality control,
facilitating the elimination of damaged and malfunctioning
mitochondria and thereby sustaining mitochondrial self-renewal and
function.
To investigate the impact of mitochondrial
morphological changes on the function of mitochondria, the present
study assessed mitochondrial function in septic cardiomyopathy.
Mitochondria primarily generate ATP through OXPHOS (13). LPS exposure reduced the levels of
marker proteins for mitochondrial electron transport chain
complexes (MT-ND1, SDHA and COX IV) and key enzymes of the
mitochondrial TCA cycle (OGDH) and acetyl CoA synthesis (PDH),
which are key for mitochondrial aerobic oxidation. Correspondingly,
there was a noticeable decrease in ATP levels in the myocardium
exposed to LPS, in agreement with other findings in septic
cardiomyopathy (8). Furthermore,
an increase in lactate levels in myocardium and plasma, along with
an increase in HIF-1α protein levels, indicated a shift towards
anaerobic glycolysis while inhibiting OXPHOS (40). Notably, AKG supplementation
effectively reversed these disturbances in mitochondrial energy
metabolism. In addition, mitochondrial function was indirectly
evaluated by measuring ROS levels and apoptosis in myocardial
tissues. Mitochondria are considerable producers of ROS, especially
during OXPHOS, with 11 potential ROS-generating sites, particularly
in complexes I and III (41). LPS
treatment led to an increase in myocardial ROS abundance and
enhanced apoptosis. By contrast, AKG supplementation significantly
mitigated these pathological changes, as evidenced by DCF, DHE and
TUNEL staining, along with the assessment of oxidative stress (MDA
and NOX2/4) and apoptosis-related proteins (Bax and Bcl-2). There
was no significant change in the expression of MDA in the plasma
after AKG administration, which might be influenced by the
metabolism of other organs. These results collectively suggested
that AKG effectively alleviated the dysfunction of myocardial
mitochondria in LPS-induced septic cardiomyopathy.
In an extension of the in vivo research,
in vitro experiments were performed to further elucidate the
effects of LPS and AKG supplementation on mitochondrial function in
NRVMs. In agreement with the results of the animal studies,
exposure of NRVMs to LPS resulted in increased apoptosis and a
reduction in the expression levels of proteins that are key for
mitochondrial aerobic oxidation and mitochondrial quality control.
Conversely, AKG supplementation effectively reversed these
detrimental effects. Analysis of the Seahorse metabolic flux
analyzer data demonstrated that AKG administration significantly
ameliorated LPS-induced disturbances in mitochondrial respiratory
capacity. This improvement was evidenced by the normalization of
key parameters such as basal respiration, maximal respiration and
ATP production, underscoring the potential of AKG in mitigating
mitochondrial dysfunction in septic cardiomyopathy.
A limitation of the present study is that it
primarily focused on the effects of short-term administration of
AKG, but the long-term effects and potential side effects of AKG
administration were not fully investigated. Another limitation is
that although the present study supported the protective effect of
AKG in septic cardiomyopathy induced by LPS, the precise molecular
mechanisms underlying these beneficial effects were not fully
elucidated. In addition, the present study investigated the effects
of AKG in male mice only; its effect in female mice was not
assessed.
In summary, the present study on LPS-induced septic
cardiomyopathy revealed that AKG supplementation enhanced cardiac
performance and rectified cardiac dysfunction. This effect was
achieved through restoration of mitochondrial ultrastructure,
augmentation of energy metabolism, and reduction of oxidative
stress and cellular apoptosis. The present study not only shed
light on the underlying mechanisms of myocardial mitochondrial
damage in septic cardiomyopathy but also suggested that AKG-based
therapeutic interventions may be a treatment option for this
condition. Thus, these findings offer insights and pave the way for
the development of novel AKG-based therapeutic strategies in the
management of septic cardiomyopathy.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by grants from the Key Research and
Development of Shaanxi Province (grant no. 2021ZDLSF02-03), the
National Natural Scientific Foundation of China (grant no.
82070858), the Youth Scientific Research and Innovation Team
Program of Shaanxi Province (grant no. 2022-SLRH-LJ-014), and the
Technology Talents Support Program of Shaanxi Provincial People's
Hospital (grant no. 2023JY-28).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
WW, BYX, JKW, GCG and ZWL conceived and designed the
experiments and wrote the manuscript. WW, BYX, QM, SS and BTL
performed the experiments and analyzed the data. ZWL, SS and JKW
made substantial contributions to manuscript revision and
supervision. WW, BYX and GCG confirm the authenticity of all the
raw data. All authors have read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Ethical
Committee of Shaanxi Provincial People's Hospital (approval no.
2021071; Xi'an, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AKG
|
α-ketoglutarate
|
|
Bnip3
|
BCL2 interacting protein 3
|
|
MT-ND1
|
mitochondrial NADH-ubiquinone
oxidoreductase chain 1
|
|
DRP1
|
dynamin-related protein 1
|
|
HIF-1α
|
hypoxia inducible factor-1α
|
|
OGDH
|
ketoglutarate dehydrogenase
|
|
PDH
|
pyruvate dehydrogenase
|
|
SDHA
|
succinate dehydrogenase
|
|
LPS
|
lipopolysaccharide
|
|
OXPHOS
|
oxidative phosphorylation
|
|
ROS
|
reactive oxygen species
|
|
LV
|
left ventricle
|
|
LVEDD
|
left ventricular end-diastolic
dimension
|
|
LVESD
|
left ventricular end-systolic
dimension
|
|
ANP
|
atrial natriuretic protein
|
|
BNP
|
brain natriuretic peptide
|
|
β-MHC
|
β-major histocompatibility complex
|
|
Ndufa12
|
NADH dehydrogenase (ubiquinone) 1α
subcomplex subunit 12
|
|
Ndufab1
|
NADH:ubiquinone oxidoreductase subunit
AB1
|
|
NOX2
|
NADPH oxidase 2
|
|
TEM
|
transmission electron microscopy
|
|
DHE
|
dihydroethidium
|
|
DCF
|
chloromethyl derivative CM-H2DCFDA
|
|
MDA
|
malondialdehyde
|
|
NRVM
|
neonatal rat ventricular myocyte
|
References
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fleischmann C, Scherag A, Adhikari NK,
Hartog CS, Tsaganos T, Schlattmann P, Angus DC and Reinhart K;
International Forum of Acute Care Trialists, : Assessment of global
incidence and mortality of hospital-treated sepsis. Current
estimates and limitations. Am J Respir Crit Care Med. 193:259–272.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Prescott HC and Angus DC: Enhancing
recovery from sepsis: A review. JAMA. 319:62–75. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sato R and Nasu M: A review of
sepsis-induced cardiomyopathy. J Intensive Care. 3:482015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hollenberg SM and Singer M:
Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol.
18:424–434. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
van der Poll T, van de Veerdonk FL,
Scicluna BP and Netea MG: The immunopathology of sepsis and
potential therapeutic targets. Nat Rev Immunol. 17:407–420. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Haileselassie B, Mukherjee R, Joshi AU,
Napier BA, Massis LM, Ostberg NP, Queliconi BB, Monack D, Bernstein
D and Mochly-Rosen D: Drp1/Fis1 interaction mediates mitochondrial
dysfunction in septic cardiomyopathy. J Mol Cell Cardiol.
130:160–169. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhu XX, Wang X, Jiao SY, Liu Y, Shi L, Xu
Q, Wang JJ, Chen YE, Zhang Q, Song YT, et al: Cardiomyocyte
peroxisome proliferator-activated receptor α prevents septic
cardiomyopathy via improving mitochondrial function. Acta Pharmacol
Sin. 44:2184–2200. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kong W, Kang K, Gao Y, Liu H, Meng X, Yang
S, Yu K and Zhao M: Dexmedetomidine alleviates LPS-induced septic
cardiomyopathy via the cholinergic anti-inflammatory pathway in
mice. Am J Transl Res. 9:5040–5047. 2017.PubMed/NCBI
|
|
10
|
Brealey D, Brand M, Hargreaves I, Heales
S, Land J, Smolenski R, Davies NA, Cooper CE and Singer M:
Association between mitochondrial dysfunction and severity and
outcome of septic shock. Lancet. 360:219–223. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dorn GW II: Mitochondrial dynamics in
heart disease. Biochim Biophys Acta. 1833:233–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lelubre C and Vincent JL: Mechanisms and
treatment of organ failure in sepsis. Nat Rev Nephrol. 14:417–427.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dorn GW II, Vega RB and Kelly DP:
Mitochondrial biogenesis and dynamics in the developing and
diseased heart. Genes Dev. 29:1981–1991. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Martínez-Reyes I and Chandel NS:
Mitochondrial TCA cycle metabolites control physiology and disease.
Nat Commun. 11:1022020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mantzarlis K, Tsolaki V and Zakynthinos E:
Role of oxidative stress and mitochondrial dysfunction in sepsis
and potential therapies. Oxid Med Cell Longev. 2017:59852092017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carey BW, Finley LW, Cross JR, Allis CD
and Thompson CB: Intracellular α-ketoglutarate maintains the
pluripotency of embryonic stem cells. Nature. 518:413–416. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chang LC, Chiang SK, Chen SE and Hung MC:
Targeting 2-oxoglutarate dehydrogenase for cancer treatment. Am J
Cancer Res. 12:1436–1455. 2022.PubMed/NCBI
|
|
19
|
Asadi Shahmirzadi A, Edgar D, Liao CY, Hsu
YM, Lucanic M, Asadi Shahmirzadi A, Wiley CD, Gan G, Kim DE, Kasler
HG, et al: Alpha-ketoglutarate, an endogenous metabolite, extends
lifespan and compresses morbidity in aging mice. Cell Metab.
32:447–456.e6. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
TeSlaa T, Chaikovsky AC, Lipchina I,
Escobar SL, Hochedlinger K, Huang J, Graeber TG, Braas D and
Teitell MA: α-Ketoglutarate accelerates the initial differentiation
of primed human pluripotent stem cells. Cell Metab. 24:485–493.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Matzi V, Lindenmann J, Muench A,
Greilberger J, Juan H, Wintersteiger R, Maier A and Smolle-Juettner
FM: The impact of preoperative micronutrient supplementation in
lung surgery. A prospective randomized trial of oral
supplementation of combined alpha-ketoglutaric acid and
5-hydroxymethylfurfural. Eur J Cardiothorac Surg. 32:776–782. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dhat R, Mongad D, Raji S, Arkat S,
Mahapatra NR, Singhal N and Sitasawad SL: Epigenetic modifier
alpha-ketoglutarate modulates aberrant gene body methylation and
hydroxymethylation marks in diabetic heart. Epigenetics Chromatin.
16:122023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
An D, Zeng Q, Zhang P, Ma Z, Zhang H, Liu
Z, Li J, Ren H and Xu D: Alpha-ketoglutarate ameliorates pressure
overload-induced chronic cardiac dysfunction in mice. Redox Biol.
46:1020882021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wu W, Ziemann M, Huynh K, She G, Pang ZD,
Zhang Y, Duong T, Kiriazis H, Pu TT, Bai RY, et al: Activation of
Hippo signaling pathway mediates mitochondria dysfunction and
dilated cardiomyopathy in mice. Theranostics. 11:8993–9008. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wu W, Lu Q, Ma S, Du JC, Huynh K, Duong T,
Pang ZD, Donner D, Meikle PJ, Deng XL and Du XJ: Mitochondrial
damage in a takotsubo syndrome-like mouse model mediated by
activation of β-adrenoceptor-Hippo signaling pathway. Am J Physiol
Heart Circ Physiol. 324:H528–H541. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
She G, Du JC, Wu W, Pu TT, Zhang Y, Bai
RY, Zhang Y, Pang ZD, Wang HF, Ren YJ, et al: Hippo pathway
activation mediates chemotherapy-induced anti-cancer effect and
cardiomyopathy through causing mitochondrial damage and
dysfunction. Theranostics. 13:560–577. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abdulmahdi W, Patel D, Rabadi MM, Azar T,
Jules E, Lipphardt M, Hashemiyoon R and Ratliff BB: HMGB1 redox
during sepsis. Redox Biol. 13:600–607. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McGrath JC and Lilley E: Implementing
guidelines on reporting research using animals (ARRIVE etc.): New
requirements for publication in BJP. Br J Pharmacol. 172:3189–3193.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhao H, Zhang M, Zhou F, Cao W, Bi L, Xie
Y, Yang Q and Wang S: Cinnamaldehyde ameliorates LPS-induced
cardiac dysfunction via TLR4-NOX4 pathway: The regulation of
autophagy and ROS production. J Mol Cell Cardiol. 101:11–24. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han Y, Tian H and Gao X: NORAD regulates
proliferation and apoptosis in cardiomyocytes under high-glucose
treatment through miRNA-150-5p/ZEB1 axis. Eur Rev Med Pharmacol
Sci. 24:11259–11265. 2020.PubMed/NCBI
|
|
32
|
Chen PA, Xu ZH, Huang YL, Luo Y, Zhu DJ,
Wang P, Du ZY, Yang Y, Wu DH, Lai WY, et al: Increased serum
2-oxoglutarate associated with high myocardial energy expenditure
and poor prognosis in chronic heart failure patients. Biochim
Biophys Acta. 1842:2120–2125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Spallotta F, Cencioni C, Atlante S,
Garella D, Cocco M, Mori M, Mastrocola R, Kuenne C, Guenther S,
Nanni S, et al: Stable oxidative cytosine modifications accumulate
in cardiac mesenchymal cells from type2 diabetes patients: Rescue
by α-ketoglutarate and TET-TDG functional reactivation. Circ Res.
122:31–46. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lai L, Leone TC, Keller MP, Martin OJ,
Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, et
al: Energy metabolic reprogramming in the hypertrophied and early
stage failing heart: a multisystems approach. Circ Heart Fail.
7:1022–1031. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yan C, Duanmu X, Zeng L, Liu B and Song Z:
Mitochondrial DNA: Distribution, mutations, and elimination. Cells.
8:3792019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ghezzi D, Sevrioukova I, Invernizzi F,
Lamperti C, Mora M, D'Adamo P, Novara F, Zuffardi O, Uziel G and
Zeviani M: Severe X-linked mitochondrial encephalomyopathy
associated with a mutation in apoptosis-inducing factor. Am J Hum
Genet. 86:639–649. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tilokani L, Nagashima S, Paupe V and
Prudent J: Mitochondrial dynamics: Overview of molecular
mechanisms. Essays Biochem. 62:341–360. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chaanine AH, Joyce LD, Stulak JM, Maltais
S, Joyce DL, Dearani JA, Klaus K, Nair KS, Hajjar RJ and Redfield
MM: Mitochondrial morphology, dynamics, and function in human
pressure overload or ischemic heart disease with preserved or
reduced ejection fraction. Circ Heart Fail. 12:e0051312019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Shirihai OS, Song M and Dorn GW II: How
mitochondrial dynamism orchestrates mitophagy. Circ Res.
116:1835–1849. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Brand MD: Mitochondrial generation of
superoxide and hydrogen peroxide as the source of mitochondrial
redox signaling. Free Radic Biol Med. 100:14–31. 2016. View Article : Google Scholar : PubMed/NCBI
|














