Introduction
Acute myocardial infarction (AMI) is the most severe
manifestation of coronary artery disease; it results from the acute
interruption of myocardial blood flow and leads to ischemic
necrosis of the heart muscle. It has a marked impact on global
health, affecting >7 million individuals annually. The
prevalence of AMI in China is increasing, and the prevalence of AMI
in men is higher than that in women. It has been predicted that the
number of patients with AMI in China will be >23 million by
2030, and data indicate that AMI is increasingly affecting younger
individuals (1,2). Cells do not instantly die after
myocardial ischemia, and prompt and efficient myocardial
reperfusion can mitigate AMI damage and limit infarct size.
However, ischemia-reperfusion (I/R), a process where blood supply
returns to tissue after a period of ischemia, can cause myocardial
I/R (MI/R), subsequently leading to I/R injury (IRI) and
cardiomyocyte death. IRI proceeds via several harmful mechanisms,
including inflammation, oxidative stress, excessive intracellular
calcium, and the rapid restoration of physiological pH (3). The abrupt reperfusion of an ischemic
heart can lead to ventricular arrhythmias, myocardial stunning,
microvascular obstruction and fatal M/IR injury. Despite the
possibility of additional myocardial injury, reperfusion remains
the most important and successful treatment for AMI (4). Thus, the prevention and alleviation
of IRI remains a critical issue in clinical practice.
Reactive oxygen species (ROS) are the primary type
of free radicals that contribute to M/IR injury. They do this by
inducing the opening of mitochondrial permeability transition
pores, serving as chemoattractants for neutrophils, and causing the
sarcoplasmic reticulum to become dysfunctional (5). In addition, ROS facilitate
intracellular calcium overload, damage the myocardial cell membrane
through lipid peroxidation, induce enzyme denaturation, cause
direct oxidative damage to DNA, and ultimately lead to the
apoptosis of cardiomyocytes. Therefore, managing the
microenvironment of the damaged myocardium by clearing away excess
ROS is crucial for the effective treatment of IRI (6). In the myocardium affected by IRI,
recruited and activated macrophages serve as reservoirs that
generate and release large amounts of ROS into the
microenvironment, directly exerting cytotoxic effects on
cardiomyocytes (7).
Bromodomain-containing protein 4 (BRD4), a member of
the bromodomain and extra-terminal (BET) protein family, functions
as a sensor for ROS. It is crucial for controlling the cell cycle,
inflammatory reactions and gene transcription, and is associated
with a number of age-related conditions, including tumors,
diabetes, pulmonary fibrosis, cardiovascular disorders and ischemic
brain damage (8,9). Src homology region 2-containing
protein tyrosine phosphatase 2 (SHP2) is a non-receptor protein
tyrosine phosphatase, extensively distributed in the cytoplasm of
mammals. It is a member of the protein tyrosine phosphatase class,
encoded by the ptpn11 gene, and contains two Src homology domains.
SHP2 plays a crucial role in regulating the oxidative stress
response by modulating various signaling pathways that influence
cellular reactions to oxidative stress. During MI/R, the regulatory
effect of SHP2 controls the extent of cardiomyocyte apoptosis and
injury, as it contributes to regulation of the inflammatory
response triggered by MI/R. SHP2 has been shown to modulate the
expression and release of inflammatory factors through pathways
such as the JAK/STAT pathway, thereby regulating the severity of
the inflammatory response (10–13).
Additionally, SHP2 has been demonstrated to inhibit the activation
of MAPK, which regulates the dynamic chromatin targeting of BRD4
(14). Therefore, it is speculated
that SHP2 may affect MI/R injury through the BRD4 signaling
pathway. Since SHP2 is known to play multiple regulatory roles in
MI/R injury, and to affect the extent of myocardial damage by
modulating key processes such as oxidative stress, cellular
apoptosis and inflammation, the aim of the present study was to
further explore the specific mechanisms of SHP2 and its potential
in the treatment of MI/R.
Materials and methods
Bioinformatics analysis
GEOquery (version 2.64.2; Bioconductor), limma
(version 3.52.2; Bioconductor), ggplot2 (version 3.3.6; http://ggplot2.tidyverse.org/) and ComplexHeatmap
(version 2.13.1; http://jokergoo.github.io/ComplexHeatmap/reference/ComplexHeatmap.html)
packages were used together with R software (version 4.2.1; R Core
Team). The GEOquery software was used to obtain the GSE108940
dataset from the Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) database. The
impute software (impute 1.0; Bioconductor) was used to impute
missing values from the data. The normalizeBetweenArrays function
of the limma package was then used to normalize the data, and
probes that corresponded to multiple RNAs were eliminated. Only the
probe with the greatest signal value was retained for probes that
corresponded to the same RNA. The ggplot2 software was used to
generate box plots. The data was subjected to principal component
analysis (PCA), and the ggplot2 tool was used to visualize the
findings. In addition, the ggplot2 tool was used to visualize the
findings of differential analysis, with a threshold set at
|LogFC|>1 and adjusted P-value (P.adj)<0.05. Row grouping
based on Euclidean distance and row normalization were carried out,
and the ComplexHeatmap package was used to visualize the
results.
ClusterProfiler (version 4.4.4), GOplot (version
1.0.2), ggplot2 (version 3.3.6) and org.Hs.eg.db (ID conversion
software) packages were used together with R software (version
4.2.1; R Foundation) to further analyze the GSE108940 dataset,
using mouse (Mus musculus) as the species for the analysis.
The ID conversion software was used to transform the RNA list to
Entrez IDs. The clusterProfiler package was used to perform an
enrichment analysis, and the GOplot package was used to compute the
z-score values for each enrichment item using the supplied
numerical data. When selecting gene clusters and pathways with
biological significance among the differentially expressed genes, a
threshold of P.adj<0.05 was applied. The identified
differentially expressed genes were subjected to Gene Ontology (GO)
(http://geneontology.org/) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) (http://geneontology.org/) pathway enrichment analyses
via the ClusterProfiler package. The ggplot2 software was used to
visualize the findings of the enrichment analysis.
The following packages were also used with R
software (version 4.2.1) to perform correlation analyses: car
(version 3.1-0), stats (version 4.2.1) and ggplot2 (version 3.3.6).
Considering the type and distribution of the data, Spearman's rank
correlation coefficient was used to perform the analysis. Scatter
plots for spleen tyrosine kinase (SYK), BRD4, NADPH oxidase 4
(NOX4), TANK binding kinase 1 (TBK1), interferon regulatory factor
3 (IRF3) and NLR family pyrin domain containing 3 (NLRP3) gene
co-expression were generated using the ggplot2 package.
Establishment of animal models
Myeloid-specific SHP2 knockout (KO) mice were
selected from the offspring of Lyz2-Cre+/− mice and
SHP2Flox/+C57BL/6 mice using the Cre-LoxP system. The
mice were purchased from Henan Skbes Biotechnology Co., Ltd. The
6-week-old male myeloid-specific SHP2 KO mice and wild-type (WT)
mice (18±1 g) were kept at 22±2°C with free access to food and
water and a 12-h light/dark cycle and 50% relative humidity. An
MI/R model was established in the myeloid-specific SHP2 KO mice and
the WT mice. To establish the model, the mice were anesthetized
using isoflurane gas (3% for initiation and 1.5% for maintenance).
The surgical instruments were sterilized in an autoclave and
aseptic technique was followed. A longitudinal incision was made in
the left anterior chest skin, and blunt dissection of the
subcutaneous tissue was performed. The third and fourth intercostal
spaces were identified, and the thoracic cavity was opened. Sterile
cotton wool was placed in the thoracic cavity to cover the lung
tissue to prevent damage to the lung tissue and potential fatality.
The third rib was carefully cut, and the thoracic cavity was gently
opened to expose the heart. The pericardium covering the heart
surface was carefully removed using forceps to expose the heart and
the left anterior descending branch. The heart was gently
compressed, and an 8-0 sterile suture was inserted ~1 mm away from
the bifurcation point of the left anterior descending branch,
running parallel to it. The suture was placed on the surface of the
heart with a polytetrafluoroethylene tube tied around it to secure
the suture in place. The heart was quickly placed back into the
chest, and the mouse was observed to ensure that an appropriate
increase (20–50% increase in base frequency) in respiratory rate
occurred. In addition, changes in heart rate and the color of the
distal ventricular myocardium were monitored to confirm successful
ischemia. Following 30 min of ischemia, the ligature was released
by untying the suture and removing the polytetrafluoroethylene tube
to allow blood flow to be restored to the ischemic area. The
restoration of the pale myocardial tissue to a reddish color
indicated successful reperfusion and model establishment. Four
groups of mice were established, namely the sham-WT,
sham-SHP2MAC-KO [macrophage-knockout (MAC-KO)], model-WT
and model-SHP2MAC-KO groups (each n=6 mice). The mice in
the sham groups underwent thoracotomy under anesthesia without any
further treatment. The experiment was approved by the Animal Ethics
Committee of Hebei North College (Zhangjiakou, China; approval no.
20230610112) and followed the 3R principles.
Echocardiographic assessment of mouse
hearts
Echocardiography was performed on all mice in each
group 4 weeks after model establishment. An ultrasound probe was
used at a frequency of 10 MHz, with adjustment of the scanning
speed and image depth to obtain clear images. The mice were
anesthetized by the inhalation of 3% isoflurane gas for induction
and 1.5% isoflurane to maintain anesthesia. When anesthetized, the
mice were secured on a temperature-controlled platform in a supine
position. The body was slightly tilted to the left. After ensuring
stable breathing and a regular heart rate, the ultrasound probe was
gently placed on the left side of the sternum, with a slight
adjustment in the direction of the probe to obtain a clear
long-axis view of the left ventricle. M-mode ultrasound was used to
visualize the motion of the left ventricle. The following data were
recorded: Left ventricular internal diameter at end-diastole
(LVIDd), LVID at end-systole (LVIDs), left ventricular anterior
wall thickness at end-diastole (LVAWd), LVAW at end-systole
(LVAWs), ejection fraction (EF) and fractional shortening (FS).
Euthanasia and slice production
Each mouse was placed into an individually
ventilated cage, which was then covered securely prior to
euthanasia. The cage was filled with CO2 at a
displacement rate of 30–70% of the volume/min. After confirming
that the mouse was motionless, not breathing and had dilated
pupils, the CO2 flow was discontinued and the mouse
observed for another 2 min to confirm death. The heart was
collected, rinsed free of blood with iced saline, and then
transferred to a sterile Eppendorf tube and fixed by immersion in
4% paraformaldehyde solution. The fixed tissue was trimmed and then
dehydrated using 80, 90, 95 and 100% alcohol concentration
gradients. The dehydrated tissues were rendered transparent by
treatment with 100% alcohol for 30 min, benzyl alcohol for 10 min,
xylene I for 10 min and xylene II for 10 min, and then soaked in
paraffin wax for ≥1 h at 60°C. The paraffin-embedded tissues were
sectioned to a thickness of 4–5 µm using a microtome. Sections were
transferred to distilled water at 40°C for spreading, and then
mounted onto carrier slices using a cotton swab. After labeling and
numbering, the sections were dried in a thermostat.
Masson's staining
Tissues were fixed in 4% paraformaldehyde at 4°C for
24 h, routinely dehydrated and embedded. Sections (thickness, 4 µm)
were dewaxed and rehydrated. The sections were incubated in the
staining solution overnight at room temperature, followed by
rinsing with running water for 10 min. Nuclei were stained using
Regaud's hematoxylin staining solution for 10 min, and after
staining, the nuclei were rinsed briefly (10–15 sec) with distilled
water. Differentiation was performed using acidic ethanol followed
by rinsing with distilled water to stop differentiation. Next, the
nuclei were stained with Lichun red compound acidic compound red
solution for 10 min, then washed with 2% glacial acetic acid
aqueous solution for a few moments, treated with phosphomolybdic
acid solution for ~5 min and then washed with 0.2% glacial acetic
acid aqueous solution for 1 min. Staining was then performed with
aniline blue for 2 min, before washing with 0.2% glacial acetic
acid aqueous solution for 1 min. Afterwards, dehydration was
performed first with 95% ethanol and then with anhydrous ethanol,
before clearing with xylene and mounting with neutral gum. Results
were observed using a light microscope. The fibrosis area was then
calculated using imageJ (National Institutes of Health).
Cell culture and grouping
Myeloid-specific SHP2 KO mice and WT mice were
euthanized, and their bone marrow-derived macrophages (BMMs) were
collected. In brief, the femurs and tibias were isolated, cleaned
of adherent muscle tissue and washed with 9% physiological saline.
The bone ends were cut off to expose the bone marrow cavity, which
was washed with physiological saline. The bone marrow flush was
filtered through a 70-µm cell filter, and the resulting filtrate
was centrifuged at 300 × g for 5 min at room temperature. After
discarding the supernatant, the cell pellet was treated with red
blood cell lysis buffer (ACK Lysing Buffer; Thermo Fisher
Scientific). Following lysis, the suspension was centrifuged at 200
× g for 5 min at room temperature, and the supernatant was
discarded.
The lysed cell pellet was resuspended in complete
culture medium containing (cat. no. 11875093; Thermo Fisher
Scientific, Inc.) macrophage colony-stimulating factor (M-CSF)
(cat. no. P6015; Beyotime Institute of Biotechnology) and seeded
into a 12-well cell culture plate. The medium was supplemented with
10% fetal bovine serum and 1% penicillin-streptomycin solution. The
cells were cultured in the M-CSF-containing complete culture
medium, with a change of medium every 2 days. After 6 days, the
BMMs had differentiated into mature macrophages and were used for
subsequent experiments.
BMMs in the logarithmic growth phase were
transfected with BRD4 overexpression vector (BRD4-mimic) or a BRD4
short hairpin RNA (shRNA)-carrying vector using
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. The sequence of the
BRD4-shRNA was 5′-AACAATTTGTAAACATAGT-3′ BRD4-shRNA, and
HBLV-U6-Scramble-ZsGreen-Puro (Hanbio Biotechnology Co.) was used
as the vector backbone. BMMs from the myeloid-specific SHP2 KO
group were cultured in a low-sugar, low-serum (3% FBS) medium (cat.
no. 11054020; Thermo Fisher Scientific, Inc.) with 1% O2
to model the I/R environment. The following BMM groups were
established: WT (transfected with empty vector), KO, model-WT,
model-KO, model-WT + BRD4-shRNA, model-KO + BRD4-shRNA, model-WT +
BRD4-mimic and model-KO + BRD4-mimic.
The HL-1 mouse cardiomyocyte cell line was purchased
from Wuhan Procell Life Technology Co., Ltd. These cells were
cultivated in DMEM (cat. no. 11320033; Thermo Fisher Scientific,
Inc.) supplemented with 1% penicillin-streptomycin solution (cat.
no. 15140122; Thermo Fisher Scientific, Inc.) and 10% fetal bovine
serum (cat. no. A5670701; Thermo Fisher Scientific, Inc.). A
co-culture system comprising HL-1 cells and BMMs was established.
The co-culture was performed using a Transwell system to physically
separate the HL-1 cells from the BMMs and to exchange signaling
molecules through the co-culture substrate for indirect cell-cell
interactions. The ratio of HL-1 cells to BMMs was 1:1, and 50,000
cells were inoculated per well. The co-culture was performed at
37°C, and subsequent experiments were performed after 48 h of
co-culture.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from tissues and cells using
TRIzol® reagent (cat. no. 15596026CN; Thermo Fisher
Scientific, Inc.). RNA purity and concentration were measured, and
cDNA was synthesized using a reverse transcription kit
(PrimeScript™ RT reagent Kit; Takara Bio). Genomic DNA
removal reaction system: gDNA Eraser, 1 µl; 5X gDNA Eraser Buffer,
2 µl; RNA, 1 µg; RNAse FreeH2O, replenish to 10. A
Pipette gun was used to gently blow the reaction system to mix it
well and then incubation was performed at 42°C for 2 min to remove
genomic DNA. The reverse transcription reaction system was
configured, with genomic DNA removal on ice. Reverse transcription
reaction system product: 10 µl; PrimeScript RT Enzyme MIX I, 1 µl;
5X gDNA Eraser Buffer II, 4 µl; RT Peimer Mix, 1 µl; RNAse
FreeH2O, 4 µl. Reverse transcription was performed at
37°C for 15 min and 85°C for 5 sec.
The qPCR amplification system comprised SYBR FAST
qPCR Master Mix (cat.no. D7260; Beyotime Institute of
Biotechnology) (10 µl), forward primer (10 µmol/l, 0.5 µl), reverse
primer (10 µmol/l, 0.5 µl), cDNA template (1 µl) and
ddH2O (8 µl), in a total volume of 20 µl. The reaction
conditions were 95°C for 3 min for initial denaturation, followed
by 40 cycles of 95°C for 5 sec, 56°C for 10 sec and 72°C for 25
sec, and a melting curve analysis from 65 to 95°C. Relative
expression levels were calculated using the 2−ΔΔCt
method with GAPDH as the reference gene (15).
Primer sequences for the target genes (5′-3′) were
as follows: SHP2 forward, CTTGGTGAGTGAGTAGAT and reverse,
CTTACGGCAGAACATTAG; BRD4 forward, GTGGGAGGAAAGAAACAGGGACA and
reverse, AGGAGGAGGATTCGGCTGAGG; GAPDH forward, TCCCTCAAGATTGTCAGCAA
and reverse, AGATCCACAACGGATACATT.
Western blot analysis
BMMs and HL-1 cells from each group were collected,
and were lysed in pre-chilled RIPA lysis solution (cat. no. P0013B;
Betoyime Institute of Biotechnology) (89% RIPA + 10% 10X cocktail +
1% PMSF). After centrifuging the lysates for 20 min at 4°C and 300
× g, the supernatants were collected. A BCA protein assay kit was
used to measure the amount of protein in the supernatants. The
proteins (20 µg/lane) were separated using SDS-PAGE (SHP2, SYK,
STING, NOX4, IL-1β, GSDMD and GAPDH were used with 10% gels. BRD4
and NLRP3 used 8% gels.) and transferred to PVDF membranes. After
blocking for 90 min at room temperature using 5% skimmed milk, the
membranes were incubated at 4°C overnight with primary antibodies
targeting SHP2 (cat. no. ab300579; 1:1,000; Abcam), BRD4 (cat. no.
ab128874; 1:1,000; Abcam), SYK (cat. no. ab40781; 1:1,000; Abcam),
stimulator of interferon genes (STING) (cat. no. ab288157; 1:1,000;
Abcam), NOX4 (cat. no. AF1498; 1:1,000; Beyotime Institute of
Biotechnology), NLRP3 (cat. no. ab263899; 1:1,000; Abcam), IL-1β
(cat. no. ab234437; 1:1,000; Abcam), gasdermin D (GSDMD) (cat. no.
ab209845; 1:1,000; Abcam) and GAPDH (cat. no. ab181602; 1:1,000;
Abcam). After incubation with the primary antibody, the PVDF
membrane was removed from the antibody incubation cassette,
immersed in TBST and agitated on a shaker for 10 min. This step was
repeated three times. Antibodies were diluted using 5% skimmed milk
in TBST, secondary antibodies [Goat anti-rabbit IgG H&L (HRP);
cat. no. ab205718; 1:2,000; Abcam] were added and the membranes
were incubated for 1 h at room temperature. Finally, an ECL
detection kit (cat. no. P0018S; Beyotime Institute of
Biotechnology) was used to visualize the protein bands and an
imaging system was used to capture images of the bands. Greyscale
analysis was performed using GAPDH as a loading control. ImageJ was
used to analyze the bands (version 1.8.0; National Institutes of
Health).
Immunofluorescence staining
BMMs were washed three times with PBS containing 3%
BSA (cat. no. 9998; Cell Signaling Technology) and then fixed using
4% paraformaldehyde in PBS. The sample was fixed at 4°C for 48 h.
The cells were permeabilized by incubation at room temperature for
10 min in PBS containing 0.5% Triton X-100. Following
permeabilization, the cells were rinsed 3–6 times, each for 5 min,
with PBS containing 3% BSA. Next, the cells were incubated
overnight at 4°C with primary antibodies targeting SHP2 (cat. no.
ab131541; 1:100; Abcam) and BRD4 (cat. no. 63759; 1:100; Cell
Signaling Technology). After three rounds of washing with PBS
containing 0.3% BSA, the cells were incubated with secondary
antibodies [Donkey Anti-Rabbit IgG H&L (Alexa Fluor®
647); cat. no. ab150075; 1:200; and Goat anti-mouse IgG H&L
(Alexa Fluor® 488); cat. no. ab150113; 1:200; both
Abcam] for 1 h at room temperature. After three additional washes
with PBS containing 0.3% BSA, the cells were counterstained with
DAPI for nuclear visualization. After 15 min of incubation at room
temperature, 1X PBS was used for washing twice for 5 min each.
Finally, the cells were mounted with 50% glycerol (2 µl) and images
captured using a fluorescent microscope. Fluorescence intensity was
measured using Image J.
Flow cytometry
HL-1 cells from the co-culture experiment were
seeded at a density of 2×106 cells/well in a 6-well
plate. After allowing the cells to adhere, they were trypsinized;
then the cell suspension and supernatant were collected, mixed and
subjected to centrifugation. The sample was centrifuged at 400 × g
for 5 min at 4°C. Following removal of the supernatant, the cells
were resuspended in FITC and PI staining solution from an apoptosis
detection kit (cat. no. C1062S; Beyotime Institute of
Biotechnology) in accordance with the manufacturer's instructions.
After culture for 20 min in the dark at room temperature, cell
apoptosis was analyzed using flow cytometry (BD FACSCalibur; BD
Accuri C6; FlowJo, V10.10; BD Biosciences).
Statistical analysis
The statistical program Prism 9.0 (GraphPad;
Dotmatics) was used to analyze the data. Data are presented as the
mean ± standard deviation. For comparisons between two groups,
t-tests were used, and for comparisons among multiple groups,
two-way analysis of variance followed by Tukey's post hoc test was
used. P<0.05 was considered to indicate a statistically
significant difference.
Results
Bioinformatics analysis
The 12 samples from the GSE108940 dataset comprised
a sham group and an I/R group, each containing 6 samples derived
from the analysis of mouse heart tissue. Normalized signal
intensity (Fig. 1A) demonstrates
that the data was appropriately normalized. Marked differences
between the two groups were shown by PCA (Fig. 1B). A volcano plot revealed that
there were 173 significantly upregulated genes and 157
significantly downregulated genes in the I/R group. Expression of
SHP2 was downregulated, and expression of BRD4 and SYK was
upregulated in I/R (Fig. 1C). The
expression patterns of the 330 differentially expressed genes were
visualized using a heatmap (Fig.
1D). The input gene list was converted to Entrez IDs, and the
differentially expressed genes were subjected to GO and KEGG
analysis. The analysis yielded a bar plot (Fig. 1E), bubble plot (Fig. 1F), chord plot (Fig. 1G) and circle plot (Fig. 1H) for visualizing the biological
process (BP), cellular component (CC), molecular function (MF) and
KEGG enriched pathways. The analysis revealed the enrichment of
pathways including ‘TNF signaling pathway’, ‘rheumatoid arthritis’
and ‘IL-17 signaling pathway’. There was a significant positive
correlation between BRD4 and SYK (Fig.
1I; P<0.001), which may suggest a role for BRD4 in
regulating immune response and cell proliferation. Although the
correlation between BRD4 and NOX4 (Fig. 1J) was not significant (P=0.342),
this result may still reflect the potential function of BRD4 in
oxidative stress response. Further analysis showed that there was
also a significant positive correlation (P<0.001) between TBK1
and IRF3 (Fig. 1K), suggesting a
possible synergistic role of these two molecules in the antiviral
immune response. In addition, the positive correlation between IRF3
and NLRP3 (Fig. 1L; P<0.001)
further supported the key role of IRF3 in the regulation of
inflammatory response and cell death.
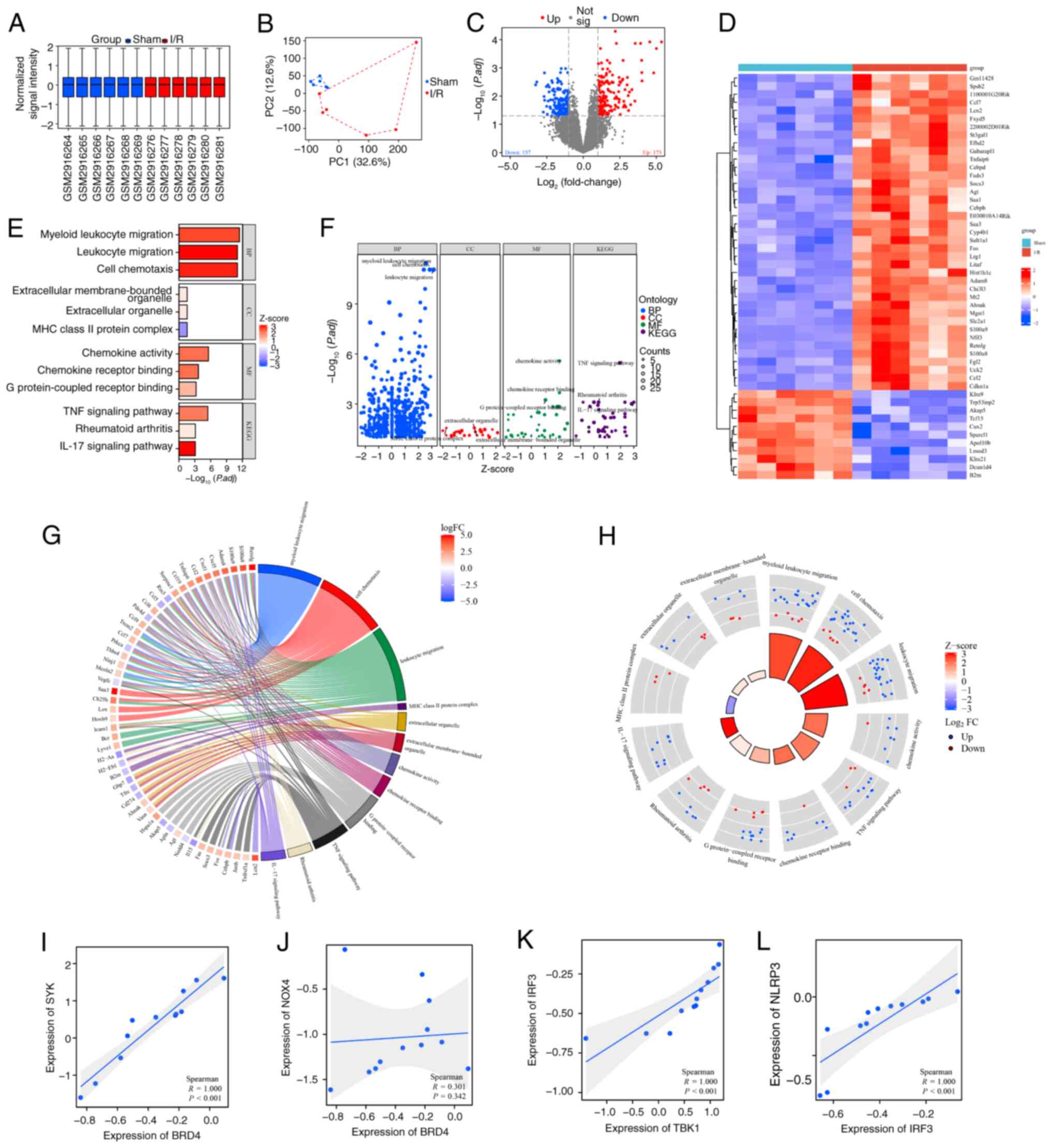 | Figure 1.Bioinformatics analysis of the murine
GSE108940 dataset. (A) Box plot showing the normalized signal
intensity of the samples. (B) Principal component analysis. (C)
Volcano plot showing the analysis of differentially expressed
genes. (D) Heatmap visualization of the differentially expressed
genes in which blue denotes upregulation and green denotes
downregulation. (E) Bar, (F) bubble, (G) chord and (H) circle plots
visualizing the results of Gene Ontology and KEGG enrichment
analysis. Scatter plots showing the correlation of (I) BRD4 and
SYK, (J) BRD4 and NOX4, (K) TBK1 and IRF3 and (L) IRF3 and NLRP3.
I/R, ischemia reperfusion; PC, principal component; P.adj, adjusted
P-value; KEGG, Kyoto Encyclopedia of Genes and Genomes; BP,
biological process; CC, cellular component; MF, molecular function;
BRD4, bromodomain-containing protein 4; SYK, spleen tyrosine
kinase; NOX4, NADPH oxidase 4; TBK1, TANK binding kinase 1; IRF3
interferon regulatory factor 3; NLRP3, NLR family pyrin domain
containing 3. |
Effects of myeloid-specific SHP2 KO on
cardiac function in M/IR injury in mice
The effect of myeloid-specific SHP2 gene KO on
cardiac function and structure during M/IR injury were
investigated. RT-qPCR analysis revealed that the relative mRNA
expression level of SHP2 in the sham-SHP2MAC-KO group
was significantly lower than that in the sham-WT group. In
addition, the relative mRNA expression level of SHP2 in the
model-SHP2MAC-KO group was significantly lower compared
with that in the model-WT group (Fig.
S1A). This demonstrates that SHP2 was successfully knocked out.
Subsequently, ultrasound results revealed that the sham-WT and
sham-SHP2MAC-KO groups did not vary significantly in
terms of LVIDd, LVIDs, LVAWd, LVAWs, EF or FS. The model-WT and
model-SHP2MAC-KO groups exhibited a substantial increase
in LVIDd and LVIDs values, and significant reductions in EF, FS,
LVAWd and LVAWs values compared with those of the sham-WT and
sham-SHP2MAC-KO groups, respectively. In addition, the
model-SHP2MAC-KO group had significantly higher LVIDd
and LVIDs values and significantly lower LVAWd, LVAWs, EF and FS
values compared with those in the model-WT group (Fig. 2). These findings suggest that the
cardiac dysfunction associated with myocardial ischemia-reperfusion
damage is exacerbated by myeloid-specific SHP2 deletion.
 | Figure 2.Exacerbation of cardiac dysfunction in
myeloid-specific SHP2 KO mice subjected to myocardial
ischemia-reperfusion. (A) Ultrasound cardiogram images of mice from
the sham-WT, sham-SHP2MAC-KO, model-WT, and
model-SHP2MAC-KO groups. (B) Statistical analysis of
various echocardiographic parameters in the four groups. Data are
expressed as the mean ± standard deviation. *P<0.05 and
**P<0.01. ns, not significant; SHP2, Src homology region
2-containing protein tyrosine phosphatase 2; WT, wild type; EF,
ejection fraction; FS, fractional shortening; LVIDd, left
ventricular internal diameter at end-diastole; LVIDs at
end-systole; LVAWd, left ventricular anterior wall thickness at
end-diastole; LVAWs, LVAW at end-systole; MAC-KO,
macrophage-knockout. |
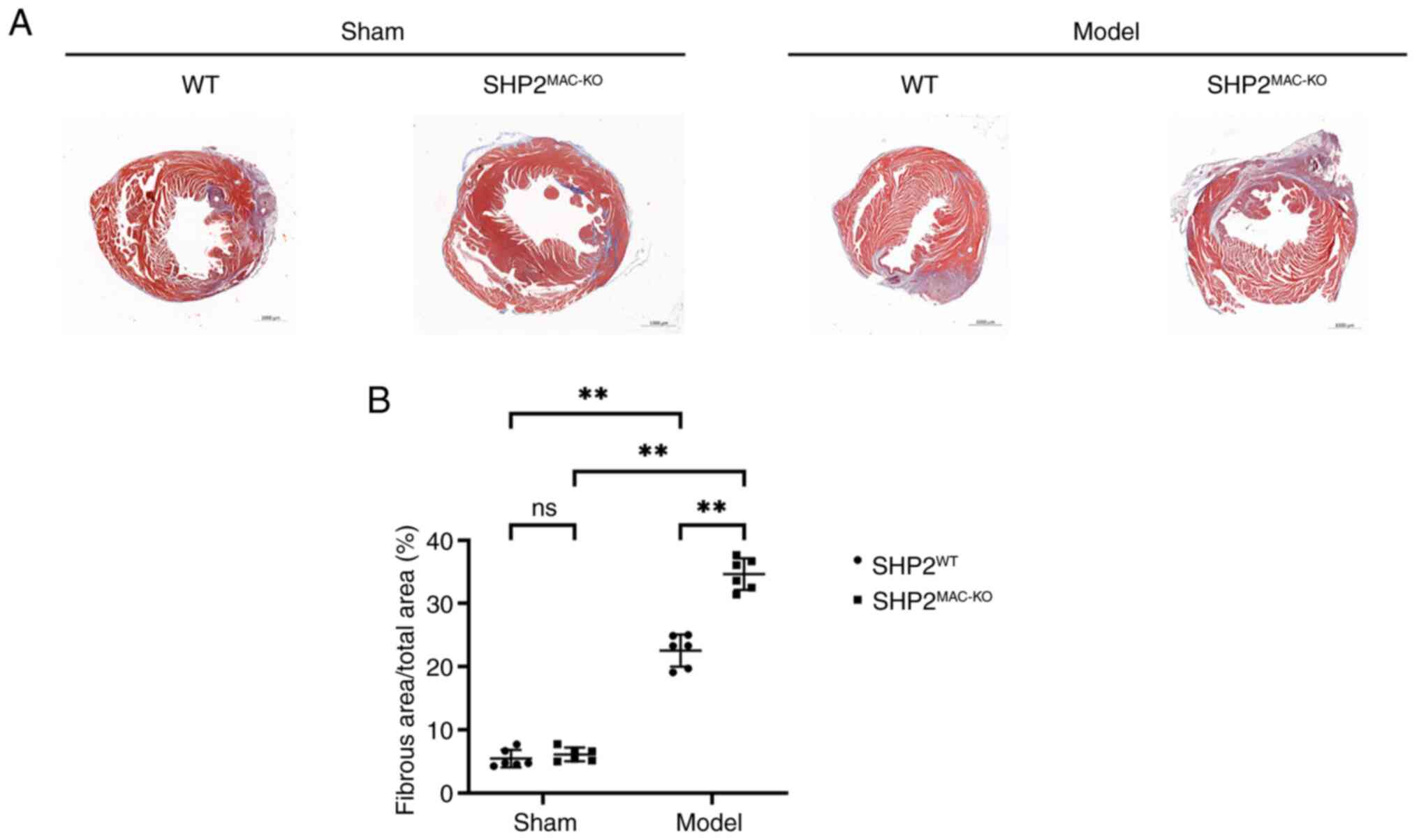
Effects of myeloid-specific SHP2 KO on
myocardial pathological injury in mice
Masson's staining results showed that the sham-WT
and sham-SHP2MAC-KO groups had normal cardiac tissue
structure, regular cardiomyocyte morphology and a minimal area of
myocardial infarction. The model-WT group exhibited nuclear
dissolution and fragmentation in some cardiomyocytes, in addition
to swollen cardiomyocytes, disorganized myocardial fibers with
intervening cavities, the acute infiltration of inflammatory cells
around necrotic cardiomyocytes, and an increased infarct area.
Compared with the model-WT group, the model-SHP2MAC-KO
group exhibited greater cardiomyocyte swelling and myocardial fiber
damage, an increased infiltration of acute inflammatory cells, and
larger infarct area (Fig. 3).
These results indicate that myeloid-specific SHP2 KO exacerbates
pathological damage in mouse M/IR injury.
Effects of SHP2 on the
BRD4/SYK/STING/NOX4/NLRP3 signaling pathway
Analysis of the BMMs by RT-qPCR showed that the mRNA
expression of SHP2 in the KO group was significantly lower than
that in the WT group, but no significant difference in the mRNA
expression of BRD4 between the KO and WT groups was detected. The
mRNA expression of SHP2 in the model-WT group was significantly
higher than that in the WT group, but the mRNA expression of SHP2
between the model-KO and KO groups was not significantly different.
The mRNA expression of BRD4 in the model-WT and model-KO groups was
significantly higher than that in the WT and KO groups,
respectively. In the Model-WT and Model-KO cells, transfection with
BRD4-shRNA did not induce any significant changes in the mRNA
expression of SHP2, while the relative mRNA expression of BRD4 was
significantly reduced in both types of cells following BRD4-shRNA
transfection, and the significant difference between the WT and KO
cells was eliminated. In the Model-WT and Model-KO cells,
transfection with BRD4-mimic did not cause any significant changes
in the mRNA expression of SHP2, but significantly increased the
mRNA expression of BRD4 in both cell types, resulting in no
significant difference between the WT and KO cells (Fig. S1B). Western blot examination of
the BMMs revealed that the expression of SHP2 in the KO group was
significantly lower than that of the WT group, but did not detect
any discernible changes in the expression levels of BRD4, SYK,
STING, NOX4 or NLRP3 between the KO and WT groups. The expression
of SHP2 was significantly higher in the model-WT group than in the
WT group, but did not differ significantly between the model-KO and
KO groups. The expression levels of BRD4, SYK, STING, NOX4 and
NLRP3 in the model-WT and model-KO groups were significantly higher
than those in the WT and KO groups, respectively. The expression
level of SHP2 in the model-WT and model-KO cells did not
significantly change following transfection with BRD4-shRNA,
whereas the expression levels of BRD4, SYK, STING, NOX4, and NLRP3
significantly decreased and the significant differences in
expression between the WT and KO cells were eliminated. Similarly,
the relative protein expression of SHP2 in the model-WT and
model-KO cells did not significantly change following transfection
with BRD4-mimic, but the expression levels of BRD4, SYK, STING,
NOX4 and NLRP3 significantly increased and the significant
differences between the WT and KO cells were eliminated.
Immunofluorescence staining of the BMMs revealed
that the fluorescence intensity of SHP2 in the KO group was
significantly lower than that of the WT group, while there was no
discernible difference in the fluorescence intensity of BRD4
between the KO and WT groups. The fluorescence intensity of SHP2 in
the model-WT group was significantly higher than that in the WT
group, but there was no discernible difference in the fluorescence
intensity of SHP2 between the model-KO and KO groups. The
fluorescence intensity of BRD4 in the model-WT and model-KO groups
was significantly higher than that in the WT and KO groups,
respectively (Fig. 4).
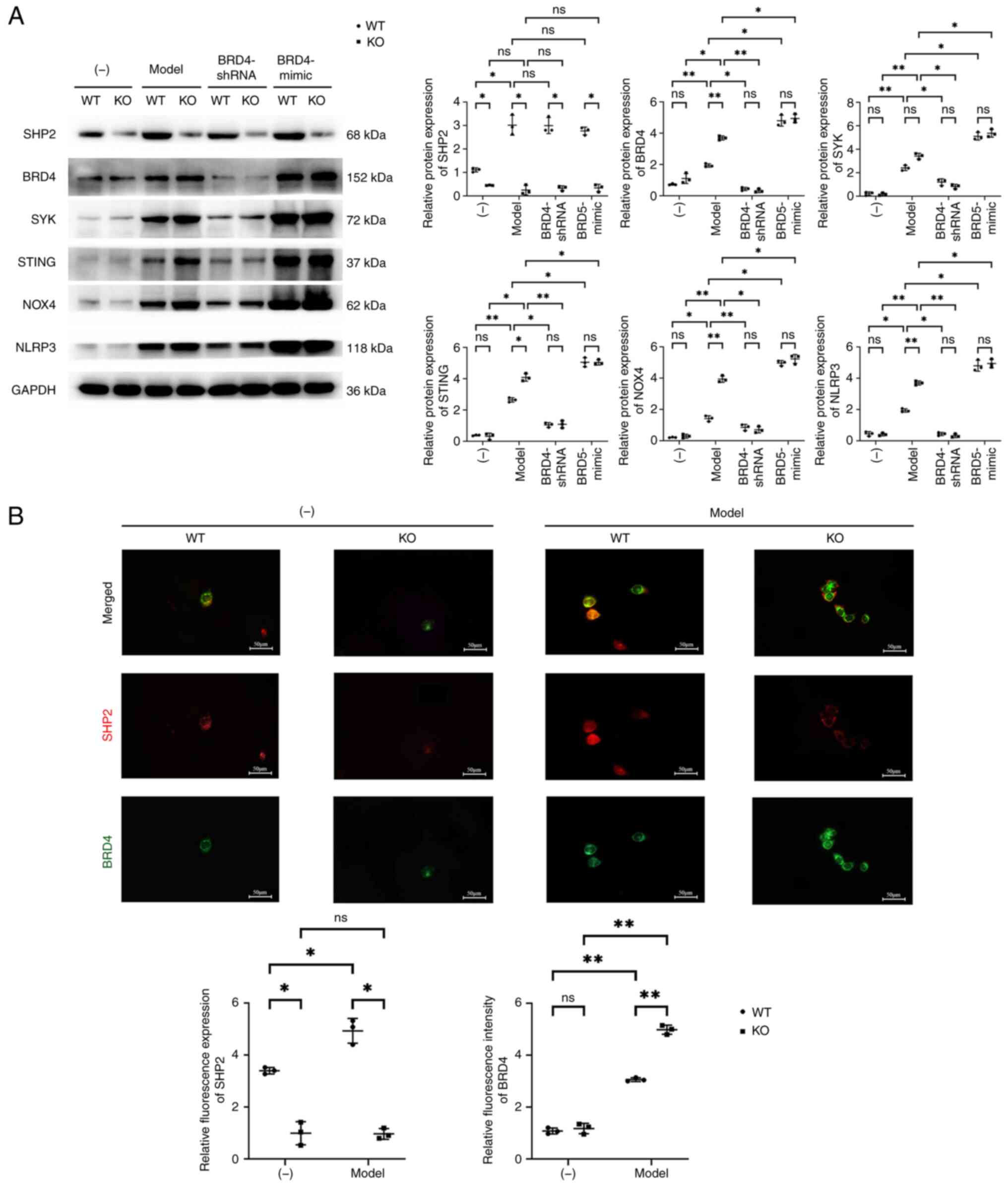 | Figure 4.Effects of SHP2 on
BRD4/SYK/STING/NOX4/NLRP3 signaling. (A) Representative western
blots and quantitative analysis of the relative protein expression
levels of SHP2, BRD4, SYK, STING, NOX4 and NLRP3 in murine BMMs,
including GAPDH as the loading control. (B) Immunofluorescence
staining images and quantitative analysis of the relative
fluorescence intensity of SHP2 and BRD4 in the BMMs. Data are
expressed as the mean ± standard deviation. *P<0.05 and
**P<0.01. ns, not significant; SHP2, Src homology region
2-containing protein tyrosine phosphatase 2; BRD4,
bromodomain-containing protein 4; SYK, spleen tyrosine kinase;
STING, stimulator of interferon genes; NOX4, NADPH oxidase 4;
NLRP3, NLR family pyrin domain containing 3; BMMs, bone
marrow-derived macrophages; WT, wild type; KO, knockout; shRNA,
short hairpin RNA; mimic, overexpression vector. |
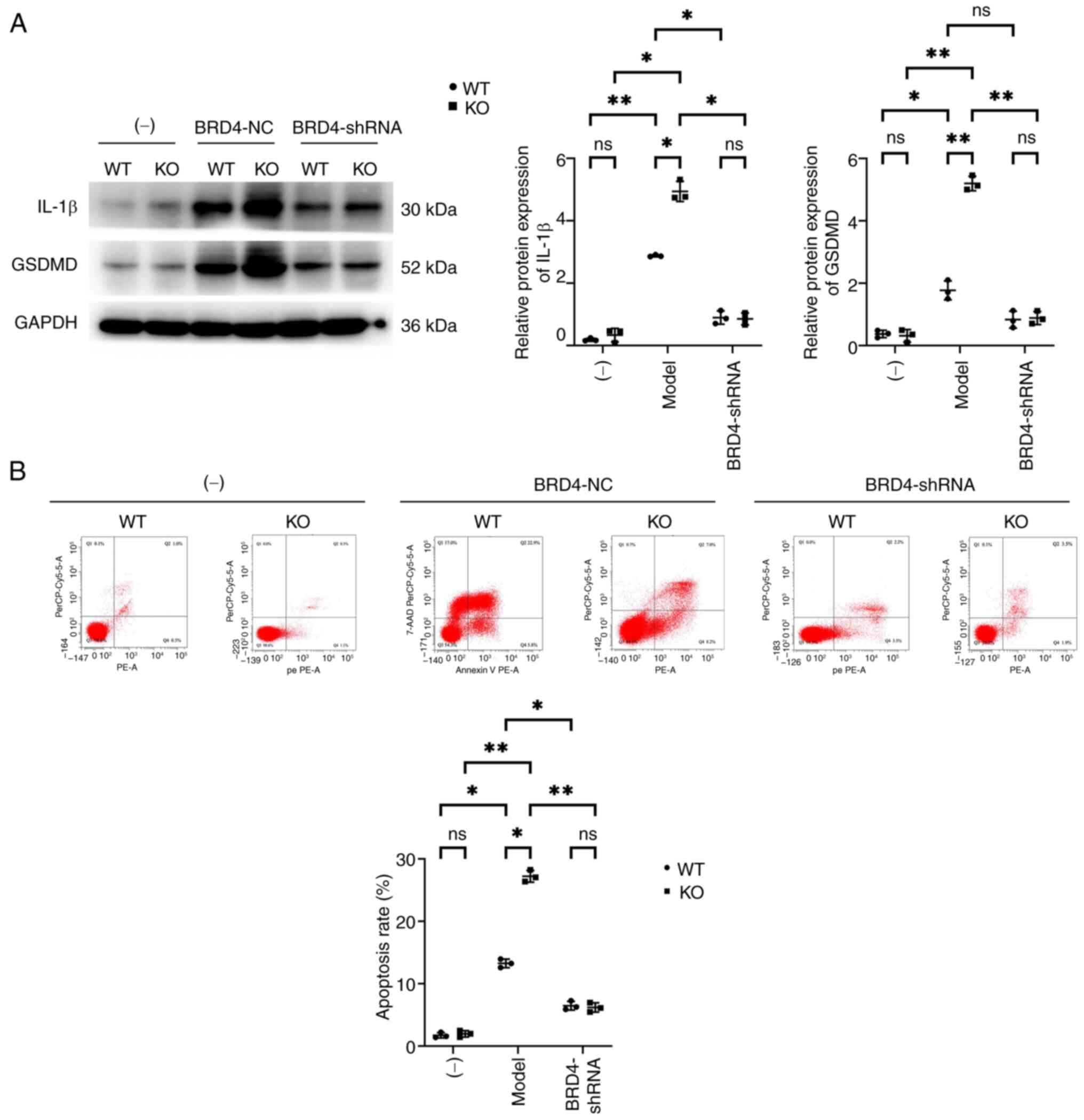
Effects of SHP2 on cardiomyocyte
apoptosis
Western blotting was used to detect the expression
of apoptotic proteins associated with BMMs. Western blotting
revealed that the expression levels of IL-1β and GSDMD in the KO
group were not significantly from those in the WT group. However,
the expression levels of GSDMD and IL-1β in the model-WT and
model-KO groups were significantly higher than those in the WT and
KO groups, respectively. In addition, the expression levels of
GSDMD and IL-1β in the model-KO group were elevated compared with
those in the model-WT group. The expression of IL-1β and GSDMD in
the model-WT and model-KO groups significantly decreased after
transfection with BRD4-shRNA, and the differences between the WT
and KO cells were abolished.
Flow cytometry revealed that the HL-1 cells from the
KO and WT groups had similarly low apoptotic rates. The apoptotic
rates of the cells in the model-WT and model-KO groups was
significantly increased compared with those in the WT and KO
groups, respectively. In addition, the apoptosis rate in the
model-KO group was significantly elevated compared with that in the
model-WT group. The apoptotic rate of cells in the model-WT and
model-KO groups significantly decreased after transfection with
BRD4-shRNA, and the difference between the groups was abolished
(Fig. 5). These results support
the hypothesis that myeloid SHP2 inhibits MI/R injury by regulating
BRD4/SYK/STING/NOX4/NLRP3 signaling (Fig. 6).
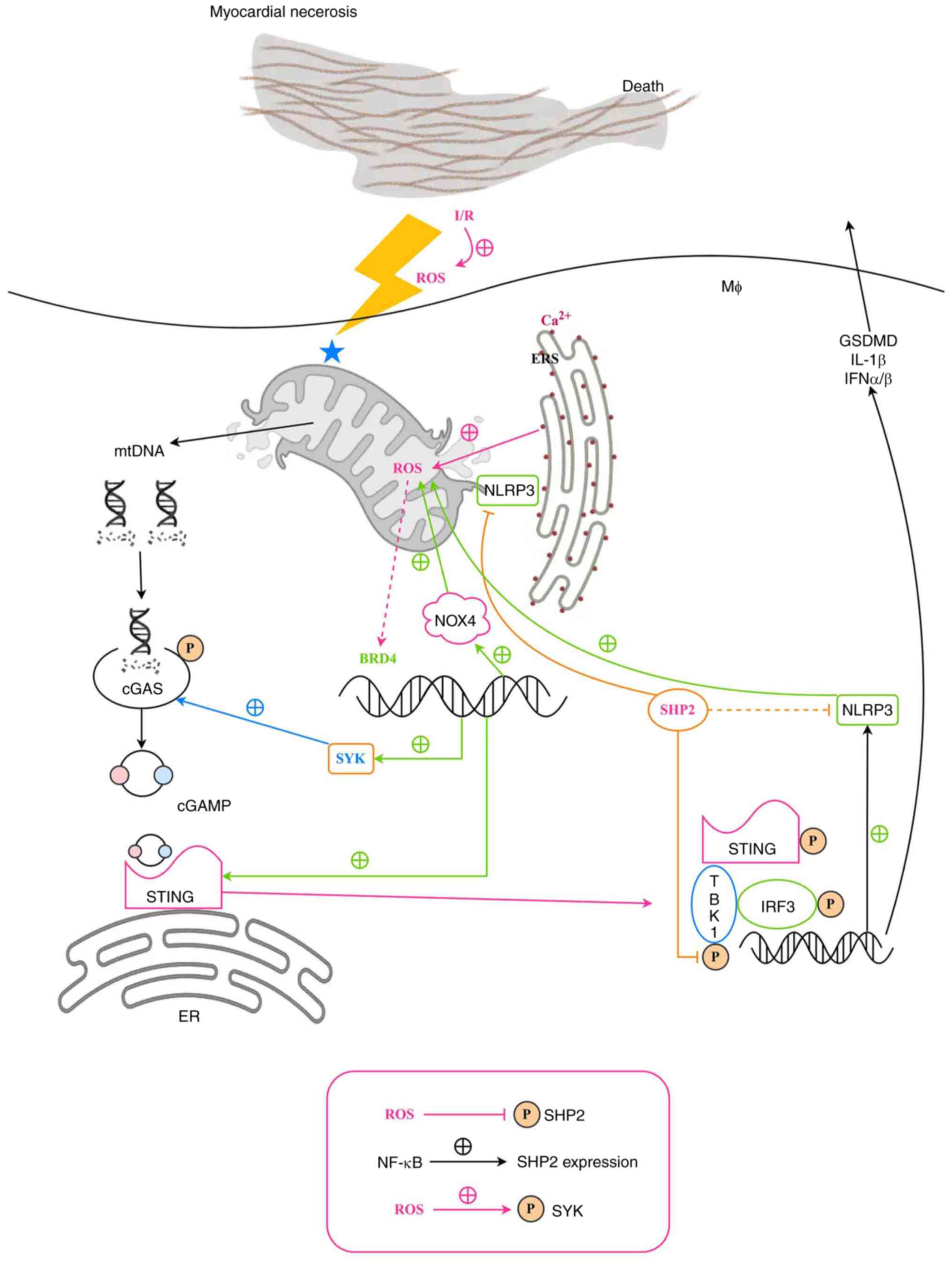 | Figure 6.Schematic illustration of the
proposed mechanism by which myeloid SHP2 regulates myocardial I/R
injury. BRD4, bromodomain-containing protein 4; cGAMP, cyclic
GMP-AMP; cGAS, cyclic GMP-AMP synthase; ER, endoplasmic reticulum;
ERS, ER stress; GSDMD, gasdermin D; IFN, interferon; I/R,
ischemia-reperfusion; IRF3, interferon regulatory factor 3; MØ,
macrophage; mtDNA, mitochondrial DNA; NLRP3, NLR family pyrin
domain containing 3; NOX4, NADPH oxidase 4; P, phosphorylation;
ROS, reactive oxygen species; SHP2, Src homology region
2-containing protein tyrosine phosphatase 2; STING, stimulator of
interferon genes; SYK, spleen tyrosine kinase; TBK1, TANK binding
kinase 1. |
Discussion
The most effective clinical intervention for
myocardial ischemia is prompt reperfusion, which partially restores
cardiac function. However, paradoxically, reperfusion can
exacerbate cell death and myocardial injury, a phenomenon known as
MI/R injury. This triggers excessive ROS production, increased
apoptosis and disrupted autophagy, which collectively intensify
cardiomyocyte damage and compound myocardial tissue injury. Such
effects diminish the therapeutic efficacy of reperfusion and may
cause irreversible myocardial damage and increased infarct size
(16–18). Addressing MI/R injury remains a key
challenge in clinical practice, underscoring the necessity of
developing effective strategies for its mitigation.
Macrophages are the most abundant resident
leukocytes in the healthy heart. They are the primary inflammatory
cells that infiltrate infarcted myocardium and a key source of the
pro-inflammatory cytokine IL-1β (19). Previous studies have established an
association between SHP2 mutations and congenital cardiovascular
disorders (20). In the GSE108940
dataset, data for sham and I/R groups of mice were normalized and
significant differences between the two groups were revealed by
PCA. Further analysis revealed that there were 173 upregulated and
157 downregulated genes in the I/R group, and a heat map
illustrated the expression pattern of 330 differentially expressed
genes. Following the conversion of these genes to Entrez IDs, GO
and KEGG enrichment analysis was performed and the associated BP,
CC, MF and KEGG-enriched pathways were identified. This revealed
that these genes were enriched in various terms, including ‘TNF
signaling pathway’, ‘rheumatoid arthritis’ and ‘IL-17 signaling
pathway’. In addition, BRD4 expression exhibited a positive
correlation with SYK and NOX4 expression, although the latter
correlation was not statistically significant. Also, TBK1
expression positively correlated with IRF3, which in turn
positively correlated with the expression of NLRP3. In an in
vivo experiment, the present study found that myeloid-specific
SHP2 KO in a mouse model of MI/R suppressed EF, FS, LVAWd, and
LVAWs and increased LVIDd and LVIDs. In addition, the
myeloid-specific SHP2 KO was observed to increase cardiomyocyte
edema and fibrosis, promote the infiltration of acute inflammatory
cells, and increase the enlargement of the infarct area. These
findings indicate that myeloid-specific SHP2 KO exacerbates the
functional and pathological damage caused by MI/R.
BRD4, a member of the BET protein family, plays a
pivotal role in cellular senescence, apoptosis, inflammation and
gene transcription. Previous research has shown that the
downregulation of BRD4 attenuates ischemic brain injury by
inhibiting inflammation and apoptosis (21). Furthermore, BRD4 suppression delays
macrophage senescence via modulation of the NF-κB pathway, thereby
decelerating the progression of atherosclerosis. In MI/R injury,
the excessive production of ROS disrupts mitochondrial function and
damages mitochondrial DNA (mtDNA), aggravating myocardial injury.
ROS also activate BRD4, which upregulates SYK activity and, in
turn, increases cyclic GMP-AMP synthase (cGAS) expression,
underscoring the role of BRD4 in the regulation of the cGAS pathway
(22).
The cGAS/STING signaling pathway is as an important
component of the innate immune system. Initially discovered as a
cytoplasmic pattern recognition receptor for detecting pathogenic
DNA, cGAS was later found to bind to cytosolic free double-stranded
DNA, leading to the synthesis of the second messenger cyclic
GMP-AMP (cGAMP) from ATP and GTP. The cGAMP interacts with STING,
which exists in a homodimeric form on the endoplasmic reticulum,
promoting its translocation to perinuclear microsomes. This further
activates TBK1, leading to the phosphorylation of STING and
providing a binding site for IRF3. In the TBK1-STING-IRF3 complex,
TBK1 catalyzes the phosphorylation of IRF3, inducing the secretion
of NLRP3 and type I interferon (IFN) family member IFNβ (23,24).
SHP2 interacts with TBK1, suppressing its kinase activity via the
dephosphorylation of critical residues. In addition, type I
interferon synergizes with pro-inflammatory signals such as NF-κB,
which amplifies cytokine network responses and indirectly
stimulates IL-1β production (25).
NLRP3 is an intracellular pattern recognition
receptor that detects pathogenic molecules and endogenous damage
signals. Upon activation, NLRP3 forms the inflammasome complex by
combining with apoptosis-associated speck-like protein containing a
CARD and caspase-1. This complex often assembles at
mitochondrial-endoplasmic reticulum contact sites (MERCs), which
are microenvironments that facilitate signal transduction (26,27).
NLRP3 localizes to mitochondria via interaction with binding
partners such as thioredoxin-interacting protein in response to ROS
(28). Calcium ion release at
MERCs is pivotal in inflammasome activation, capturing calcium
signals to initiate downstream pathways. Signal 2 activators
recruit SHP2 to mitochondria via its RRWFH motif, with
translocation facilitated by Tom20/Tom40 and Tim23 complexes. SHP2
dephosphorylates ANT1 at Tyr191 within the mitochondrial matrix,
which regulates mitochondrial homeostasis, mitigating mtDNA leakage
and curbing ROS production, thereby dampening NLRP3 inflammasome
activation. Thus, SHP2 serves as a negative regulator of the NLRP3
inflammasome (29).
IL-1β is a central pro-inflammatory cytokine that
plays a pivotal role in infection and inflammation. Activation of
the NLRP3 inflammasome allows caspase-1 to cleave pro-IL-1β into
its mature, active form, which is secreted extracellularly to
mediate inflammation. The essential pyroptosis effector GSDMD is
cleaved by caspase-1 into two fragments: N-terminal fragment
GSDMD-N and C-terminal fragment GSDMD-C. The GSDMD-N fragment forms
membrane pores that lead to cellular content leakage and pyroptotic
cell death, a pro-inflammatory form of programmed cell death.
Pyroptosis is characterized by membrane rupture and the release of
inflammatory mediators through GSDMD-mediated pores (30,31).
The present study revealed that the myeloid-specific KO of SHP2
promoted the expression of BRD4, SYK, STING, NOX4 and NLRP3 in
BMMs, as well as IL-1β and GSDMD in HL-1 cells co-cultured with the
BMMs, and induced pyroptosis in HL-1 cells. Furthermore, the
transfection of BMMs with BRD4-shRNA and BRD4-mimic demonstrated
that SHP2 mediates the regulation of the BRD4 signaling pathway in
BMMs in response to MI/R injury.
In summary, the present study suggests that myeloid
SHP2 inhibits MI/R injury by regulating BRD4/SYK/STING/NOX4/NLRP3
signaling. This implies that molecules in this pathway may serve as
new therapeutic targets for the treatment of MI/R injury. In
addition, it explored the possibility that SHP2 in the myeloid
lineage may provide a comprehensive, multi-level protective
mechanism via the simultaneous regulation of multiple signaling
pathways, such as those involving BRD4, SYK, STING, NOX4 and NLRP3.
This concept of multi-pathway regulation greatly expands the
understanding of the pathophysiology of MI/R injury compared with
that of traditional single-pathway research, and suggests novel
targets for treatment. Furthermore, the concept of combining
systemic and local inflammation regulation offers a new perspective
for the treatment of IRI. However, numerous studies on SHP2 and the
aforementioned signaling pathways have been conducted in animal
models, which may limit the extrapolation of research results to
human physiological and pathological states. Notably, while mouse
models are widely used in the study of cardiovascular disease and
immune responses, their immune systems differ from those of humans.
Therefore, translating these findings into clinical treatments will
require further verification in human studies.
Supplementary Material
Supporting Data
Acknowledgements
The authors acknowledge that due to the complexity
of establishing the mouse MI/R model, Mr. Jianchun Fan, a student
of Hebei North College (Zhangjiakou, Hebei, China) performed the
surgery and provided guidance.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YL and ZJ were responsible for study concept and
design. HY, TW and TC contributed to manuscript writing and the
cell experiments. CG and FZ were responsible for the animal
experiments. YL and ZJ confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Ethics
Committee of Hebei North College (approval number, 20240610112).
The Medical Ethics Committee of the Third Hospital of Hebei Medical
University was aware of the collaboration with Hebei North College
and considered that since Hebei North College had already conducted
an ethical review, no additional ethical review was necessary.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X,
Huang Z, Lin M, Wu H and Xu D: Naringenin alleviates myocardial
ischemia/reperfusion injury by regulating the nuclear
factor-erythroid factor 2-related factor 2 (Nrf2)/System
xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis.
Bioengineered. 12:10924–10934. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tian H, Zhao X, Zhang Y and Xia Z:
Abnormalities of glucose and lipid metabolism in myocardial
ischemia-reperfusion injury. Biomed Pharmacother. 163:1148272023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang XJ, Liu X, Hu M, Zhao GJ, Sun D,
Cheng X, Xiang H, Huang YP, Tian RF, Shen LJ, et al:
Pharmacological inhibition of arachidonate 12-lipoxygenase
ameliorates myocardial ischemia-reperfusion injury in multiple
species. Cell Metab. 33:2059–2075.e10. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schanze N, Hamad MA, Nührenberg TG, Bode C
and Duerschmied D: Platelets in myocardial ischemia/reperfusion
injury. Hamostaseologie. 43:110–121. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bugger H and Pfeil K: Mitochondrial ROS in
myocardial ischemia reperfusion and remodeling. Biochim Biophys
Acta Mol Basis Dis. 1866:1657682020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shen S, He F, Cheng C, Xu B and Sheng J:
Uric acid aggravates myocardial ischemia-reperfusion injury via
ROS/NLRP3 pyroptosis pathway. Biomed Pharmacother. 133:1109902021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hao T, Qian M, Zhang Y, Liu Q, Midgley AC,
Liu Y, Che Y, Hou J and Zhao Q: An injectable dual-function
hydrogel protects against myocardial ischemia/reperfusion injury by
modulating ROS/NO disequilibrium. Adv Sci (Weinh). 9:e21054082022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu H, Wang L, Weng X, Chen H, Du Y, Diao
C, Chen Z and Liu X: Inhibition of Brd4 alleviates renal
ischemia/reperfusion injury-induced apoptosis and endoplasmic
reticulum stress by blocking FoxO4-mediated oxidative stress. Redox
Biol. 24:1011952019. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu W, Wu RD, Lv YG, Liu YM, Huang H and
Xu JQ: BRD4 blockage alleviates pathological cardiac hypertrophy
through the suppression of fibrosis and inflammation via reducing
ROS generation. Biomed Pharmacother. 121:1093682020. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu M, Fu D, Gao T, Jiang H, Yang P and Li
X: The low expression of miR-155 promotes the expression of SHP2 by
inhibiting the activation of the ERK1/2 pathway and improves cell
pyroptosis induced by I/R in mice. Aging (Albany NY). 16:4778–4788.
2024.PubMed/NCBI
|
|
11
|
Gao T, Liu M, Fu D, Xue Y, Liao J, Yang P
and Li X: Allicin treats myocardial infarction in I/R through the
promotion of the SHP2 axis to inhibit p-PERK-mediated oxidative
stress. Aging (Albany NY). 16:5207–5223. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sha M, Li H, Guo B and Geng X:
Myeloid-specific knockout of SHP2 regulates PI3K/PLCγ signaling
pathway to protect against early myocardial infarction injury.
Aging (Albany NY). 15:9877–9889. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lauriol J, Jaffré F and Kontaridis MI: The
role of the protein tyrosine phosphatase SHP2 in cardiac
development and disease. Semin Cell Dev Biol. 37:73–81. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Stratton MS, Bagchi RA, Felisbino MB,
Hirsch RA, Smith HE, Riching AS, Enyart BY, Koch KA, Cavasin MA,
Alexanian M, et al: Dynamic chromatin targeting of BRD4 stimulates
cardiac fibroblast activation. Circ Res. 125:662–677. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhao J, Zhang J, Liu Q, Wang Y, Jin Y,
Yang Y, Ni C and Zhang L: Hongjingtian injection protects against
myocardial ischemia reperfusion-induced apoptosis by blocking ROS
induced autophagic-flux. Biomed Pharmacother. 135:1112052021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li X and Jin Y: Inhibition of miR-182-5p
attenuates ROS and protects against myocardial ischemia-reperfusion
injury by targeting STK17A. Cell Cycle. 21:1639–1650. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu L, Zhang W, Huang C, Liang Q, Bao H,
Gong Z, Xu M, Wang Z, Wen M and Cheng X: FoxO4 promotes myocardial
ischemia-reperfusion injury: The role of oxidative stress-induced
apoptosis. Am J Transl Res. 10:2890–2900. 2018.PubMed/NCBI
|
|
19
|
Francisco J and Del Re DP: Inflammation in
myocardial ischemia/reperfusion injury: Underlying mechanisms and
therapeutic potential. Antioxidants (Basel). 12:19442023.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhou L, Yang S and Zou X: Farrerol
alleviates myocardial ischemia/reperfusion injury by targeting
macrophages and NLRP3. Front Pharmacol. 13:8792322022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu L, Yang C, Lavayen BP, Tishko RJ,
Larochelle J and Candelario-Jalil E: Targeted BRD4 protein
degradation by dBET1 ameliorates acute ischemic brain injury and
improves functional outcomes associated with reduced
neuroinflammation and oxidative stress and preservation of
blood-brain barrier integrity. J Neuroinflammation. 19:1682022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li X, Chen X, Zheng L, Chen M, Zhang Y,
Zhu R, Chen J, Gu J, Yin Q, Jiang H, et al: Non-canonical
STING-PERK pathway dependent epigenetic regulation of vascular
endothelial dysfunction via integrating IRF3 and NF-κB in
inflammatory response. Acta Pharm Sin B. 13:4765–4784. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qiao X, Zong Y, Liu Z, Wu Z, Li Y, Wang L
and Song L: The cGAS/STING-TBK1-IRF regulatory axis orchestrates a
primitive interferon-like antiviral mechanism in oyster. Front
Immunol. 12:6897832021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Oduro PK, Zheng X, Wei J, Yang Y, Wang Y,
Zhang H, Liu E, Gao X, Du M and Wang Q: The cGAS-STING signaling in
cardiovascular and metabolic diseases: Future novel target option
for pharmacotherapy. Acta Pharm Sin B. 12:50–75. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zheng Q, Hou J, Zhou Y, Yang Y, Xie B and
Cao X: Siglec1 suppresses antiviral innate immune response by
inducing TBK1 degradation via the ubiquitin ligase TRIM27. Cell
Res. 25:1121–1136. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tian L, Yu Q, Zhang L and Zhang J:
Accelerated fibrosis progression of diabetic nephropathy from high
uric acid's activation of the ROS/NLRP3/SHP2 pathway in renal
tubular epithelial cells under high glucose conditions. Altern Ther
Health Med. June 5–2024.(Epub ahead of print).
|
|
27
|
Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen
L, Shen Y, Wang X, Feng GS, Sun Y and Xu Q: Tyrosine phosphatase
SHP2 negatively regulates NLRP3 inflammasome activation via
ANT1-dependent mitochondrial homeostasis. Nat Commun. 8:21682017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin Q, Li S, Jiang N, Shao X, Zhang M, Jin
H, Zhang Z, Shen J, Zhou Y, Zhou W, et al: PINK1-parkin pathway of
mitophagy protects against contrast-induced acute kidney injury via
decreasing mitochondrial ROS and NLRP3 inflammasome activation.
Redox Boil. 26:1012542019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Qiu Z, He Y, Ming H, Lei S, Leng Y and Xia
ZY: Lipopolysaccharide (LPS) aggravates high glucose- and
hypoxia/reoxygenation-induced injury through activating
ROS-dependent NLRP3 inflammasome-mediated pyroptosis in H9C2
cardiomyocytes. J Diabetes Res. 2019:81518362019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li S, Sun Y, Song M, Song Y, Fang Y, Zhang
Q, Li X, Song N, Ding J, Lu M and Hu G:
NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in
astrocyte pathological injury in mouse model of depression. JCI
Insight. 6:e1468522021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Coll RC, Schroder K and Pelegrín P: NLRP3
and pyroptosis blockers for treating inflammatory diseases. Trends
Pharmacol Sci. 43:653–668. 2022. View Article : Google Scholar : PubMed/NCBI
|