Introduction
Bone is a dynamic organ constantly undergoing
remodelling, which consists of two coordinated processes: Bone
resorption by osteoclasts and bone formation by osteoblasts. The
coupling imbalance between bone remodelling, indicated by low
bone-forming osteoblast activity and high bone-resorbing osteoclast
activity, is the basic mechanism of osteoporosis. Osteoporosis is a
bone disorder in which bones become weak, brittle and fragile,
leading to increased susceptibility to fracture (1). A direct association of autoimmune
diseases [such as rheumatoid arthritis and systemic lupus
erythematosus (SLE)] and inflammatory diseases (such as systemic
sclerosis and sarcoidosis) with impaired bone health has been
reported, which can be explained by inflammation, immune
dysregulation and the use of medications (2–4).
Chronic inflammatory responses promote osteoclastic activity and
accelerate bone breakdown, resulting in bone loss (5). T helper 17 (Th17) cells that produce
IL-17 are overactive in various autoimmune and inflammatory
disorders, which induces the receptor activator of nuclear factor
κ-Β ligand (RANKL) and inhibits osteoprotegerin (OPG) production by
osteoblasts (6). The long-term use
of glucocorticoids and methotrexate adversely affect bone
metabolism and increase the risk of fractures (7,8).
Chloroquine (CQ) and hydroxychloroquine (HCQ) are
medications used to prevent and treat malaria caused by
Plasmodium parasites. CQ and HCQ interfere with haemoglobin
proteolysis in the parasite causing the accumulation of toxic
byproducts to kill the parasite (9). Apart from their anti-plasmodial
property, CQ and HCQ have diverse pharmacological effects on other
medical conditions, including autoimmune diseases (10), cancer (11), viral infections (12), metabolic syndrome (9) and skeletal disorders (13,14),
because of their anti-inflammatory and immunomodulatory properties.
Chronic therapy with CQ or HCQ is necessary to manage autoimmune
diseases (10). Therefore,
investigating the effects and underlying molecular mechanism of CQ
or HCQ on the skeleton could yield valuable information to prevent
osteoporosis with minimal side effects.
The present review aims to summarize the role of CQ
and HCQ in protecting the bones from becoming porous. It also
highlights the underlying molecular mechanisms that govern the
action of CQ and HCQ. The present review may aid researchers and
interprofessional healthcare teams in recognizing the
skeletal-protecting potential of CQ and HCQ, and their mechanisms
of action.
Evidence acquisition
The research question set for the present review was
as follows: ‘What are the effects of CQ and HCQ on bone health and
their underlying mechanism of actions?’. To identify relevant
studies to be included in the present review, a comprehensive
literature search was caried out using two electronic databases
[PubMed (https://pubmed.ncbi.nlm.nih.gov/) and Web of Science
(https://www.webofknowledge.com/)] in
June 2024 using the search string ‘(chloroquine OR
hydroxychloroquine) AND (bone OR osteoporosis OR fracture OR
osteoblast OR osteoclast OR osteocyte)’. All available primary
studies examining the effects of CQ or HCQ on bone health from the
inception of the databases were considered. Articles without
relevant results or primary data (including reviews, letters to the
editor, perspectives, books and book chapters) were not considered.
Conference abstracts and proceedings were excluded because of the
preliminary nature of the data and the potential of data
duplication from full-text articles. Articles not written in
English were also excluded.
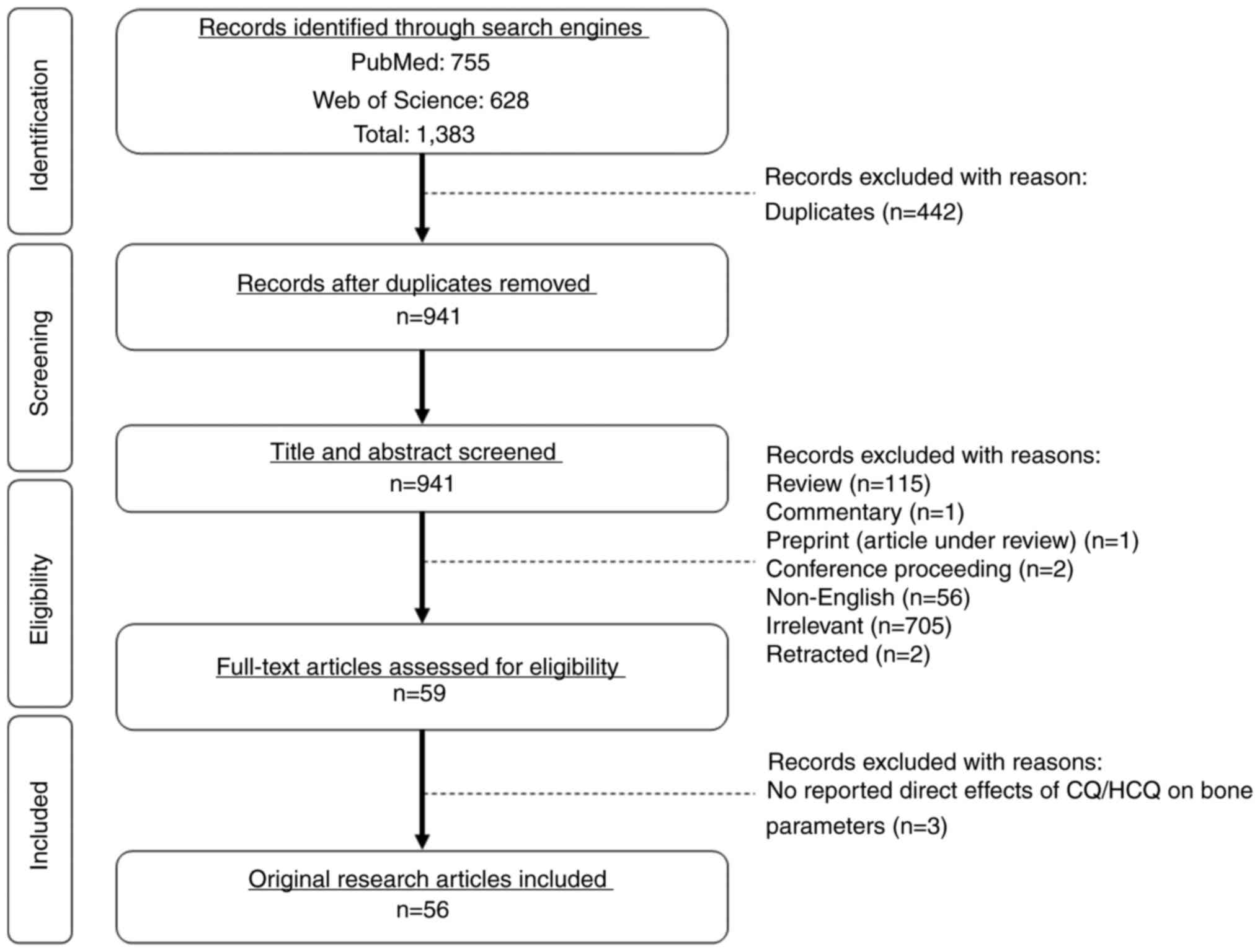
The literature search using the two electronic
databases identified 1,383 records (PubMed, 755; Web of Science,
628). Duplicates (n=442) were removed. Preliminary screening of the
title and abstract identified reviews (n=115), commentary (n=1),
preprint (articles under review; n=1), conference proceedings
(n=2), non-English articles (n=56), irrelevant articles (n=705) and
retracted articles (n=2) to be excluded. Full-text articles were
screened for eligibility based on the inclusion and exclusion
criteria. All original research articles reporting bone-related
changes and understanding of the mechanisms resulting from the
monotherapy of CQ and HCQ or combined therapy with other drugs were
included. Three articles were excluded after screening of full-text
articles because they did not report the direct effects of CQ and
HCQ on bone parameters as study outcomes. A total of 56 articles
adhering to the inclusion criteria were included in the present
review (Fig. 1).
Effects of CQ and HCQ on bone health in
humans
Accumulating evidence has revealed heterogenous
outcomes for the effects of CQ and HCQ on bone health, whereby
positive, negative and negligible effects have been observed
(Table I). The literature search
using the search string identified an early small-scale study
investigating the effects of CQ on serum bone markers in patients
with rheumatoid arthritis and seronegative spondyloarthropathies.
The level of serum osteocalcin (OCN) was increased, while the level
of alkaline phosphatase (ALP) remained unchanged after CQ therapy
at a dose of 250 mg/day (15).
Previous studies involving humans have shifted to elucidate the
effects of HCQ treatment on bone health. In a retrospective
nationwide cohort study involving patients with rheumatoid
arthritis with and without new-onset cardiovascular disease
(n=2,534), who were aged ≥40 years, the adjusted hazard ratio (HR,
0.12; 95% CI, 0.03–0.47) of developing osteoporotic vertebral
fracture was lower in patients receiving HCQ in a Taiwanese cohort
(13). In Japan, a cross-sectional
study was conducted on outpatients with SLE (n=246) to examine the
association of HCQ use with T-score and vertebral fractures. HCQ
use was associated with high lumbar (coefficient, 0.44; 95% CI,
0.077–0.81) and femoral (coefficient, 0.33; 95% CI, 0.020–0.63)
T-scores. In addition, HCQ use was associated with a lower
incidence of vertebral fractures (odds ratio, 0.098; 95% CI,
0.013–0.73) (14).
 | Table I.Studies investigating the effects of
CQ on bone health in humans. |
Table I.
Studies investigating the effects of
CQ on bone health in humans.
| First author/s,
year | Study
population | Treatment | Findings | (Refs.) |
|---|
| Ekenstam et
al, 1986 | Patients with
rheumatoid arthritis (n=36; aged 20–69 years) and seronegative
spondyloarthropathies (n=23; aged 26–67 years) | CQ (250
mg/day) | Serum OCN was
markedly increased after 3 and 6 months, but ALP was unchanged
after 3 and 9 months. | (15) |
| Hong et al,
2019 | Patients with
rheumatoid arthritis and new-onset CVD or without CVD (n=2,534;
aged ≥40 years) | HCQ (dose
unspecified) | Lower risk of
osteoporotic vertebral fracture in patients with rheumatoid
arthritis who were receiving HCQ (HR, 0.12; 95% CI,
0.03–0.47). | (13) |
| Nakajima et
al, 2023 | Patients with SLE
(n=246; median age, 47 years) | HCQ (dose
unspecified) | The use of HCQ was
associated with high lumbar (coefficient, 0.44; 95% CI, 0.077–0.81)
and femoral (coefficient, 0.33; 95% CI, 0.020–0.63) T-scores. The
use of HCQ was associated with a lower incidence of vertebral
fractures (OR, 0.098; 95% CI, 0.013–0.73). | (14) |
| Both et al,
2018 | Patients with
rheumatoid arthritis (n=63; aged ≥18 years) | HCQ (dose
unspecified) | Serum β-CTx was
lowered after 6 months. | (16) |
| Yu et al,
2021 | Patients with
rheumatoid arthritis (n=76; aged 59.05±10.95 years) | Iguratimod (25 mg;
twice a day) + methotrexate (10 mg; once per week) + HCQ (200 mg;
twice a day) for 48 weeks | BMD at L1-L4, left
femoral neck and left total hip were increased. Rheumatoid factor,
CRP, ESR and anti-CCP were reduced. | (17) |
| Park et al,
2024 | Patients with
juvenile-onset SLE (n=29; aged 11.5–14.8 years) | Cumulative HCQ
(4.64 or 10.1 g/kg) | Patients with a
higher cumulative HCQ dose exhibited low BMD. A higher cumulative
HCQ dose was associated with a low lumbar spine BMD Z-score. | (18) |
| Heidari et
al, 2012 | Patients with
rheumatoid arthritis (n=19; aged 54.5±7.7 years) | Low-dose
methotrexate (≥15 mg/week) alone or a combination of HCQ and/or
sulfasalazine and prednisolone (5 mg) | BMD of the femoral
neck and lumbar spine decreased. | (19) |
| Carbone et
al, 2020 | Women with
rheumatoid arthritis (n=363; aged 50–79 years) | HCQ (dose
unspecified) | No significant
association between HCQ use and fracture incidence (HR, 1.0; 95%
CI, 0.7–1.5). No significant association between combination
therapy of HCQ and methotrexate and fracture incidence (HR, 0.9;
95% CI, 0.5–1.6). | (20) |
Both et al (16) recruited patients with rheumatoid
arthritis (n=63) aged ≥18 years, who were receiving HCQ as
treatment. The levels of a serum bone resorption marker [β
C-terminal telopeptide (β-CTx)] and an inflammatory marker (CRP) at
baseline and after 6 months were measured. Blood samples were
collected at random moments throughout the day without fasting. HCQ
treatment for 6 months decreased the levels of β-CTx after adjusted
for the decrease in serum CRP, indicating that HCQ had a direct
effect on bone resorption, and the decrease in β-CTx was
independent of the inflammatory response reduction (16). Yu et al (17) studied the effects of combined
therapy of disease-modifying antirheumatic drugs (iguratimod,
methotrexate and HCQ) on bone mineral density (BMD) in patients
with rheumatoid arthritis (n=76; age, 59.05±10.95 years). BMD at
the lumbar spine (L1-L4), left femoral neck and left total hip
increased after 48 weeks of treatment. The combined therapy also
reduced rheumatoid factor, CRP, the erythrocyte sedimentation rate
and anti-cyclic citrullinated peptide (17).
However, two human studies reported negative effects
of HCQ on bone health. A recent study by Park et al
(18) found that patients with
juvenile-onset SLE and low BMD had a higher cumulative HCQ dose
(10.10 g/kg), whereas those with non-low BMD had a lower cumulative
HCQ dose (4.64 g/kg). In addition, the cumulative HCQ dose was
negatively associated with the lumbar spine BMD Z-score in patients
with juvenile-onset SLE (18).
Heidari et al (19)
revealed that the BMD of the femoral neck and lumbar spine in
patients with rheumatoid arthritis (n=19; age, 54.5±7.7 years)
decreased after treatment with low-dose methotrexate (≤15 mg/week)
alone or in combination with HCQ or sulfasalazine and prednisolone
(5 mg) (19).
One study by Carbone et al (20) revealed the negligible effects of
HCQ on bone health. A total of 363 women with rheumatoid arthritis
aged 50–79 years who were treated with HCQ alone or combination
therapy of HCQ and methotrexate were enrolled. Fracture incidence
following HCQ use was estimated over a median follow-up length of
6.46 years. No significant association was identified between HCQ
use and fracture incidence (HR, 1.0; 95% CI, 0.7–1.5). A similar
relationship was noted between HCQ and methotrexate use and
fracture incidence (HR, 0.9; 95% CI, 0.5–1.6) (20).
Collectively, the findings from the majority of the
aforementioned studies indicated the potential protective effects
of CQ and HCQ in improving bone health (13–17).
One study reported that HCQ alone had an inhibitory effect on bone
resorption, which was not influenced by the reduction of
inflammation (16). On the
contrary, another study indicated that inflammatory indicators
decreased in response to combination drug therapy consisting of
iguratimod, methotrexate and HCQ (17). Therefore, the suppression of the
inflammatory reaction observed by Yu et al (17) may be at least partially attributed
to iguratimod and/or methotrexate. Several methodological
limitations (such as single-arm studies, small-scale samples, short
study duration and lack of standardisation) may lead to fallacious
conclusions, as well as less accurate and reliable results. The
dose of HCQ that was administered was also not mentioned in some of
the aforementioned studies (13,14,16,20).
Two studies reported negative outcomes (18,19),
which may be attributed to the small sample size and lack of
control group.
In vivo studies: Effects of CQ and
HCQ on bone health in animals
The effects of CQ and HCQ on bone health have been
examined using healthy animals and animal models, including
osteoporotic, knockdown, arthritic, fractured and osteotomy models
(Table II). An early study
examined the quantitative distribution of CQ radioactivity in the
bone tissues of animals. Adult and 8-day-old Sprague-Dawley rats,
as well as 1-day-old leghorn chickens received intraperitoneal
(i.p.) injections of [3-14C]CQ. The radioactivity of CQ
recovered in the bone was 11.6%, indicating the high affinity of CQ
for bone (21).
| Table II.Effects of CQ or HCQ on bone health
in animals. |
Table II.
Effects of CQ or HCQ on bone health
in animals.
| First author/s,
year | Animal
model(s) | Treatment (dose,
route of administration and duration) | Findings | (Refs.) |
|---|
| Fischer and Fitch,
1975 | Adult and 8-day-old
male and female Sprague-Dawley rats 1-day-old leghorn chickens |
[3-14C]CQ (96.4 µg; single i.p.
injection, 30 min) | Affinity of CQ and
its metabolites for bone ↑ | (21) |
| Aoki et al,
2020 |
Dexamethasone-induced mice | CQ (50 mg/kg/day;
i.p.; 7 days) | TRAP activity ↓,
ALP ↔ | (23) |
| Lin et al,
2016 |
Dexamethasone-induced mice Ovariectomised
mice | CQ (2 mg/kg; every
2 days; i.p.; 8 weeks) | BV/TV ↑, Tb.N ↑,
Oc.N ↓, Ob.N ↔, Ob.S ↔ BV/TV ↑, Tb.N ↑, Oc.N ↓, Ob.N ↔, Ob.S ↔ | (24) |
| Xiong et al,
2023 | Ovariectomised
rats | CQ (10 mg/kg; i.p.;
twice a week; 8 weeks) CQ (10 mg/kg; i.p.; twice a week; 8 weeks) +
quercetin (50 mg/kg/day; oral; 8 weeks) | P1NP ↔, CTX ↓,
Beclin 1 ↓, LC3II/I ratio ↓, p62/SQSTM1 ↑, Oc.N ↓ P1NP ↓, CTX ↓,
Beclin 1 ↓, LC3II/I ratio ↓, p62/SQSTM1 ↑, Oc.N ↓ | (26) |
| Xiu et al,
2014 | Ovariectomised
mice | CQ (50 mg/kg/day;
i.p.; 28 days) | Oc.N ↓, BV/TV
↑ | (27) |
| Liang et al,
2019 | Ovariectomised
mice | CQ (50 mg/kg/day;
i.p.; 1 month) CQ (50 mg/kg/day; i.p.; 1 month) + icariin (50
mg/kg/day; i.p.; 1 month) | BV/TV ↓, Tb.N ↓,
Tb.Th ↓, SMI ↔, Tb.Pf ↑, Oc.N ↑ BV/TV ↔, Tb.N ↔, Tb.Th ↔, SMI ↔,
Tb.Pf ↔, Oc.N ↔ | (28) |
| Ke et al,
2020 | Ovariectomised
rats | CQ (12.5 mg/kg/day;
i.p.; 60 days) CQ (12.5 mg/kg/day; i.p.; 60 days) + curcumin (110
mg/kg/day; oral; 60 days) | BV/TV ↔, Tb.N ↔,
Tb.Sp ↔, Tb.Ar ↔, TRAP ↔, ALP ↔ BV/TV ↑, Tb.N ↑, Tb.Sp ↓, Tb.Ar ↑,
TRAP ↓, ALP ↑ | (29) |
| Al-Bari et
al, 2012 | RANKL-induced
mice | CQ (50–200
mg/kg/day; 3 days) | BV/TV ↑, Tb.Th ↑,
Tb.N ↑, Tb.Sp ↓ | (30) |
| Mahmoud et
al, 2021 | D-galactose-induced
osteoporotic rats | CQ (10 mg/kg/day;
oral; 4 weeks) | BMD ↑, serum
calcium ↓, serum phosphate ↑, ALP ↓, OCN ↓, OPG ↑, RANKL ↓, CTSK ↓,
TRAP ↓, ERK ↓, percentage of bone loss ↓ | (33) |
| Alam et al,
2021 | Female ADO2
mice | CQ (1–10 mg/kg/day;
oral; 6 months) | BMC ↔, BMD ↔, BV/TV
↔, Tb.Th ↔, Tb.N ↔, Tb.Sp ↔, CTX ↔, TRAP ↑, P1NP ↔ | (34) |
| Jiang et al,
2023 |
Foxo1-overexpressing mice | CQ (2 mg/kg; every
2 days; i.p.) | BV/TV ↓, Tb.N ↓,
Tb.Sp ↑, Runx-2 ↓, OSX ↓, ALP ↓ | (36) |
| Li et al,
2023 | Arthritic mice | HCQ (80 mg/kg/day;
oral; 15 days) | Oc.N ↓, Oc.S ↓,
CTX-1 ↓ | (37) |
| Topak et al,
2023 | Rats subjected to
osteotomy at the right femur | HCQ sulphate
(0.45–3 mg; oral; 5 days) | Radiological score
↓, ALP ↓, callus/diaphysis ratio ↓, callus development ↓, BMD ↓,
CAT ↑, SOD ↑, GPx ↑, MDA ↑ | (38) |
| Önaloğlu et
al, 2024 | Rats subjected to
open diaphyseal femur fractures | HCQ sulphate (160
mg/kg/day; oral; 2 or 4 weeks) | MDA ↑, ALP ↓, OCN
↔, OPN ↔, CTSK ↑, TRAP ↑, histological healing scores ↔,
radiological scores ↔, total callus diameter to femoral bone
diameter ↓ | (39) |
| Tekçe et al,
2024 | Rats subjected to a
closed fracture at the right femur | HCQ (3–10 mg/kg;
two times per day; 15 days) | Histological
scoring ↔ | (40) |
Dexamethasone is a glucocorticoid medication used to
relieve inflammation (including swelling, heat, redness and pain)
that causes secondary osteoporosis (22). The continuous administration of
dexamethasone (2 mg/kg) for 35 days resulted in lower ALP activity
and higher tartrate-resistant acid phosphatase (TRAP) activity in
mice. The i.p. injection of CQ (50 mg/kg) for 7 days (between days
28 and 35) caused a reduction in TRAP activity without altering ALP
activity (23). TRAP is a key
enzyme produced by osteoclasts to break down bone matrix proteins
during bone resorption. Thus, lower TRAP activity suggests reduced
osteoclast activity and bone resorption. In another
dexamethasone-induced osteoporotic mouse model, i.p. injection of
dexamethasone (1 mg/kg) for 8 weeks reduced the bone volume/tissue
volume (BV/TV), trabecular number (Tb.N), osteoblast number and
osteoblast surface, and increased the osteoclast number (Oc.N). The
injection of CQ (2 mg/kg; i.p.) every 2 days mitigated
dexamethasone-challenged bone loss (increased BV/TV and Tb.N) by
reducing Oc.N without affecting the parameters linked to
osteoblasts and bone formation (24).
Bilateral ovariectomy removes the ovaries and
eliminates the secretion of oestrogen in animals, representing a
preclinical model for postmenopausal osteoporosis (25). The administration of CQ (10 mg/kg;
i.p.) twice a week for 8 weeks reduced cross-linked C-telopeptide
of type 1 collagen (CTX; a bone resorption marker), but did not
change the level of propeptide of type I procollagen (P1NP; a bone
formation marker) in ovariectomised rats (26). Lin et al (24) investigated the effects of CQ (2
mg/kg; i.p.) administered every 2 days for 8 weeks on bone
parameters using ovariectomised mice. Treatment with CQ resulted in
a reduction in Oc.N, but no change was observed in osteoblast
counts in ovariectomy-induced bone loss (24). In another study, CQ prevented
ovariectomy-induced bone loss as indicated by increased BV/TV and
reduced Oc.N in C57BL/6 mice (27). However, a study by Liang et
al (28) demonstrated
different findings, where i.p. injection of CQ (50 mg/kg/day) for 1
month reduced BV/TV, Tb.N and trabecular thickness, and increased
the trabecular pattern factor but caused no change in the structure
model index of ovariectomised mice (28). Another animal study reported that
the administration of CQ at 12.5 mg/kg for 60 days was insufficient
to induce any changes in bone markers (ALP and TRAP) and bone
microstructure [BV/TV, Tb.N, trabecular separation (Tb.Sp) and
trabecular bone area] in ovariectomised rats (29).
Using the ovariectomised animal model, several
studies have demonstrated that the presence of CQ might enhance or
weaken the bone-protecting effects of other compounds. For example,
CQ acted synergistically with curcumin (a compound isolated from
turmeric root) in ameliorating bone loss by increasing ALP
expression and decreasing TRAP expression, resulting in good bone
microarchitecture in ovariectomised rats (29). On the contrary, the combination of
CQ with quercetin (a polyphenol flavonoid found in several fruits,
vegetables, leaves, seeds and grain) (26) and icariin (a natural flavonoid
glycoside extracted from Herba epimedii with a
bone-protecting property) (28)
weakened their actions on attenuating bone loss in ovariectomised
mice. The co-administration of CQ with quercetin (50 mg/kg; oral
administration) reduced P1NP in ovariectomised rats compared with
those treated with CQ only (26).
When CQ was co-administered with icariin, CQ diminished the
icariin-induced increase of bone mass in ovariectomised mice
(28).
RANKL is a cytokine essential for osteoclast
differentiation (30). A study
looked at 8-week-old mice that were injected (i.p.) with RANKL (1
mg/kg) at 24 h intervals for 3 days to stimulate osteoclasts for
bone resorption. The injection of CQ (50–200 mg/kg) was given 1 h
before every RANKL injection. All bone microstructural parameters,
including BV/TV, Tb.Th, Tb.N and Tb.Sp, were measured after the
last injection of RANKL. The findings revealed marked increases in
BV/TV and Tb.N but a reduction in Tb.Sp in RANKL-induced bone loss
in mice (30).
D-galactose is a monosaccharide sugar that forms
lactose when combined with glucose. It causes pathological changes
that resemble natural ageing, including reduced bone mass (31). The prolonged administration of
D-galactose increases advanced glycation end products, receptors
for advanced glycation end products and nicotinamide adenine
dinucleotide phosphate oxidase, leading to the excessive
accumulation of reactive oxygen species (ROS) and mitochondrial
damage (32). In
D-galactose-induced osteoporotic rats, treatment with 10 mg/kg CQ
for 4 consecutive weeks increased tibial BMD, femoral BMD, serum
phosphate levels and OPG, but decreased serum calcium, ALP, OCN,
RANKL, cathepsin K (CTSK), TRAP and the percentage of bone loss
(33).
In a study using female mice with autosomal dominant
osteopetrosis type II mutation (ADO2) that developed osteopetrosis,
the administration of CQ (1–10 mg/kg/day) via drinking water for 6
months increased osteoclast activity, as evidenced by increased
TRAP expression. However, no change was observed in the improvement
of the osteoporotic bone phenotype of ADO2 mice (34). Foxo1 is a transcription factor
abundantly found in bone, indicating its role as a mediator for
skeletal development. Foxo1 is highly expressed in bone tissues,
and its silencing impairs skeleton formation (35,36).
The effects of CQ (2 mg/kg given every 2 days) via injection (i.p.)
on bone have been investigated in Foxo1-overexpressing mice.
Foxo1-overexpressing mice treated with CQ exhibited lower BV/TV and
Tb.N but higher Tb.Sp in the femur as compared to the wild-type
controls. The presence of CQ also downregulated the expression
levels of several osteogenic markers, including Runt-related
transcription factor 2 (Runx-2), osterix and ALP. The findings of
the study reiterated that CQ reversed the favourable osteogenic
effects of Foxo1 overexpression (36).
Several studies have demonstrated the effects of HCQ
on bones. In a mouse model of arthritis, HCQ was administered by
oral gavage at a dose of 80 mg/kg daily for 15 days. Oc.N,
osteoclast surface (Oc.S) and CTX in the arthritic mice treated
with HCQ were decreased compared with those in mice treated with
normal saline (37). However, HCQ
has been reported to induce oxidative stress, thereby impairing
fracture healing (38). A study by
Topak et al (38) utilised
male Wistar albino rats with osteotomy at the mid-distal region of
the right femur, which were administered HCQ sulphate orally at
various doses (0.45–3 mg) for 5 days, and revealed that this
resulted in a decrease in the radiological score, ALP,
callus/diaphysis ratio, callus development and BMD.
Mechanistically, the circulating antioxidant enzyme activities
[catalase (CAT), superoxide dismutase (SOD) and glutathione
peroxidase (GPx)] were increased, which could be a response to
neutralise the overwhelming oxidative stress (as lipid peroxidation
was elevated) in rats treated with HCQ sulphate (38). In male rats subjected to open
diaphyseal femur fracture, the oral administration of HCQ sulphate
(160 mg/kg) increased malondialdehyde levels. In HCQ-treated
animals, no improvement was observed in the histological healing
scores, whereby the cartilage callus tissue disappeared and the
area of immature bone tissue was increased. The total callus
diameter at the femur was lower in the HCQ group compared with the
control group. Levels of ALP were decreased, whereas OCN and
osteopontin (OPN) levels were unchanged. Meanwhile, CTSK and TRAP
were increased in the HCQ group relative to the control group
(39). Another recent study
utilised male rats with a closed fracture created at the proximal
region of the femur. Treatment with HCQ (3–10 mg/kg) two times per
day (once in the morning and once in the evening) for 15 days did
not improve the histologic scoring compared with the negative
control (40).
In conclusion, bone is an important storage organ
for CQ. The aforementioned studies revealed that different doses,
treatment frequency and duration affect the skeletal actions of CQ.
Long-term daily administration adversely affects bone health. CQ
has more prominent effects on osteoclastic activity and bone
resorption compared with osteoblastic activity and bone formation.
In addition, CQ may serve as a modulator to reduce bone resorption
in osteoporotic and arthritic conditions whilst decreasing
osteoblastic activity and increasing osteoclastic activity during
osteopetrosis. Although CQ may appear to be a potential strategy in
the treatment of osteoporosis and osteopetrosis, CQ showed no
beneficial effect in inducing fracture healing.
In vitro studies: Effects of CQ and
HCQ on bone cells
Osteoblasts
In vitro studies reporting the effects of CQ
and HCQ on bone-related parameters using osteoblasts are summarised
in Table III. Incubation of CQ
at a concentration of 5 µM for 48 h did not exhibit a cytotoxic
effect on murine pre-osteoblast MC3T3-E1 cells (41). The presence of CQ (5–50 µM) has
been demonstrated to inhibit osteogenic differentiation (confirmed
by reduced ALP activity, and reduced OCN and Runx-2 expression) in
MC3T3-E1 cells, human dental pulp mesenchymal stem cells and bone
marrow mesenchymal stem cells (BMSCs) (28,42–45).
CQ (5–10 µM) suppressed mineralisation in BMSCs isolated from
normal mice (28,45). Similarly, BMSCs extracted from
CQ-injected mice exhibited less mineralised nodules and osteogenic
differentiation ability, as indicated by reduced expression of bone
morphogenetic protein-2 and Runx-2 (28). However, CQ (50 µM) augmented the
formation of mineralised nodules at the late stage of bone
formation (43).
| Table III.Studies investigating the effects of
CQ and HCQ in osteoblasts. |
Table III.
Studies investigating the effects of
CQ and HCQ in osteoblasts.
| First author/s,
year | Type of cells | Treatment | Findings | (Refs.) |
|---|
| Xu et al,
2021 | MC3T3-E1 cells | CQ (5 µM) | Cell viability ↔,
LC3II/I ratio ↔, apoptosis ↔, caspase-3 ↔, PARP ↔, ROS ↔, Nox4
↔ | (41) |
| Zhang et al,
2017 | MC3T3-E1 cells | CQ (10 µM) | ALP ↓,
mineralisation ↓, LC3II ↓, apoptosis ↑ | (45) |
| Zhang et al,
2023 | MC3T3-E1 cells | CQ (10 µM) | LC3II ↑, apoptosis
↑ | (76) |
| Li et al,
2024 | MC3T3-E1 cells | CQ (20 µM) | ALP ↓, LC3II/I
ratio ↑, p62/SQSTM1 ↑, apoptosis ↑, Bax ↑, Bcl-2 ↓, caspase-3 ↑,
p53 ↑, T-AOC ↓, SOD ↓, NO ↑ | (42) |
| Qiu et al,
2023 | MC3T3-E1 cells | CQ (50 µM) | ALP ↓,
mineralisation ↑, autophagosome ↓, autolysosome ↑, p62/SQSTM1 ↑,
Lamp1 ↑, Bcl-2 ↔ | (43) |
| Pantovic et
al, 2013 | Human dental pulp
mesenchymal stem cells | CQ (20 µM) | ALP ↓, OCN ↓,
Runx-2 ↓ | (44) |
| Liang et al,
2019 | BMSCs isolated from
BALB/c mice | CQ (5 µM) | ALP ↓,
mineralisation ↓ | (28) |
|
|
| CQ (5 µM) + icariin
(10 µM) | ALP ↓,
mineralisation ↓ |
|
|
| BMSCs isolated
from | N/A | Mineralisation ↓,
Runx-2 ↓, BMP-2 ↓, Beclin 1 ↔, LC3II/I ratio ↓, p62/SQSTM1 ↑ |
|
|
| BALB/c mice treated
with CQ |
|
|
|
|
| BMSCs isolated
from | N/A | Mineralisation ↓,
Runx-2 ↓, BMP-2 ↓, Beclin 1 ↓, LC3II/I ratio ↓, p62/SQSTM1 ↑ |
|
|
| BALB/c mice treated
with CQ and icariin |
|
|
|
| Zhang et al,
2020 | MC3T3-E1 cells | CQ (50 mM) +
lactoferrin (100 µg/ml) | P1NP ↓, autophagy
↓, p-p38 MAPK ↓, Nbr1 ↑ | (46) |
| Yi et al,
2020 | Cranial
osteoblasts | CQ (5 µM) +
triiodothyronine (100 nM) | Cell viability ↓,
ALP activity ↓, PCNA ↓, COL1 ↓, OCN ↓ | (47) |
| Zhao et al,
2021 | BMP-9-induced
BMSCs | CQ (10 µM) | ALP ↓,
mineralisation ↓ | (48) |
| Ni et al,
2020 | TNF-α-induced
MC3T3-E1 cells | CQ (10 µM) | Cell viability ↓,
LC3II/I ratio ↓, caspase-3 ↑ | (50) |
| Ni et al,
2020 | TNF-α-induced
MC3T3-E1 cells | CQ (10 µM) | LC3II ↑, p62/SQSTM1
↑, cleaved PARP ↑ | (77) |
| Liu et al,
2016 | Cadmium-induced rat
cranial osteoblasts | CQ (5 µM) | Cell viability ↓,
LC3II ↓, apoptosis ↑ | (51) |
| Both et al,
2018 | Human mesenchymal
stromal cells | HCQ (5 µg/ml) | ALP activity ↓,
alizarin red and von Kossa staining ↓, cell-surface attachment ↓,
cytoskeletal malformations (actin) ↔, apoptosis ↔ | (52) |
| Xu et al,
2020 | Co-culture of
MC3T3-E1 cells and BMMs | HCQ (dose
unspecified) + trehalose (50 mM) | Runx-2 ↑, OPN ↑,
ATF4 ↑, Beclin 1 ↓, LC3II/I ↑ | (49) |
| Yang et al,
2023 | BMSCs isolated from
C57BL/6 mice | HCQ (dose
unspecified) | Autolysosome ↓,
disruption of mitochondrial membrane potential ↑ | (79) |
| Liu et al,
2021 | BMSCs isolated from
BALB/c mice | HCQ (10 µM) | LC3II ↑, p62/SQSTM1
↑, cell senescence ↑ | (78) |
The presence of CQ weakened the efficiency of
icariin, lactoferrin (a glycoprotein derived from milk),
triiodothyronine (a thyroid hormone) and bone morphogenetic
protein-9 (BMP9) in attenuating bone loss. BMSCs treated with CQ
and icariin exhibited reduced ALP expression and mineralised nodule
formation compared with those treated with icariin alone. Likewise,
BMSCs isolated from mice co-treated with CQ and icariin exhibited
reduced osteogenic differentiation and mineralisation compared with
BMSCs isolated from mice administered icariin alone (28). In another study, lactoferrin was
found to increase the expression levels of P1NP (a bone formation
marker) in MC3T3-E1 cells, which was reduced following combined
therapy with CQ and lactoferrin (46). Yi et al (47) revealed that CQ reversed
triiodothyronine-promoted cell viability and ALP activity, as well
as proliferating cell nuclear antigen, type I collagen and OCN
expression in cranial osteoblasts. CQ also effectively inhibited
ALP activity and matrix mineralisation promoted by BMP9 in BMSCs
(48). Nonetheless, HCQ further
enhanced osteogenesis in trehalose-treated co-culture of MC3T3-E1
cells and bone marrow monocytes/macrophages (BMMs) as the
expression levels of Runx-2, activating transcription factor 4 and
OPN were increased (49).
TNF-α is an important inflammatory cytokine that
inhibits bone formation, enhances bone resorption and attenuates
osteoblast survival (50). Cadmium
is a toxic heavy metal known to be an environmental pollutant that
can induce apoptosis in osteoblasts (51). Incubation with CQ reduced the
viability of MC3T3-E1 cells induced by TNF-α (50) and rat cranial osteoblasts induced
by cadmium (51). The effects of
HCQ on osteoblasts have been evaluated in a study by Both et
al (52). HCQ at a
concentration of 5 µg/ml inhibited the differentiation of human
mesenchymal stromal cells into osteoblasts at day 7 and inhibited
osteoblast mineralisation at day 18. Human mesenchymal stromal
cells exhibited less cell surface attachment, but no cytoskeletal
malformations were observed after HCQ treatment (52).
Therefore, CQ alone did not exert cytotoxic effects
on osteoblasts. However, in vitro studies consistently
demonstrated that CQ and HCQ inhibited osteogenic differentiation
and mineralisation, with the exception that a higher dose of CQ (50
µM) increased mineralisation. The presence of CQ and HCQ might
strengthen or weaken the bone sparing effects of other compounds
(28,46–49).
Osteoclasts
Murine monocyte/macrophage cells (RAW264.7), BMMs or
osteoclast precursors can differentiate into osteoclasts under the
influence of macrophage colony-stimulating factor (M-CSF), RANKL or
inflammatory cytokines (30).
In vitro studies have consistently shown the inhibitory
effects of CQ on osteoclast differentiation and function (Table IV). Ke et al (29) reported that CQ (2 µM) treatment
caused no reduction in Oc.N, TRAP, MMP-9 and CTSK in BMMs incubated
with M-CSF and RANKL (29).
Al-Bari et al (30)
reported that CQ (1–10 µM) treatment inhibited the formation of
TRAP-positive multinucleated osteoclasts and resorption pits in a
dose-dependent manner using BMMs stimulated by M-CSF and RANKL. The
expression of nuclear factor of activated T-cells cytoplasmic 1
(NFATc1), the master transcription factor of osteoclast
differentiation, was also inhibited by CQ treatment (30). Two studies have shown that CQ (10
µM) reduced Oc.N and the resorption pit area in BMMs induced by
M-CSF and lipopolysaccharides (LPS) or RANKL (27,53).
Furthermore, the expression levels of MMP-9, TRAP and CTSK were
decreased in the presence of CQ (53). When using RAW264.7 cells stimulated
by RANKL, CQ (10 µM) treatment attenuated the formation of
osteoclasts, actin ring and resorption pits. In addition, CQ
treatment inhibited the RANKL-induced upregulation of osteoclast
differentiation factors, including MMP-9, TRAP, CTSK and dendritic
cell-specific transmembrane protein (54,55).
Likewise, CQ arrested M-CSF- and RANKL-induced osteoclast formation
from the precursor cells (56).
| Table IV.Effects of CQ or HCQ on
osteoclasts. |
Table IV.
Effects of CQ or HCQ on
osteoclasts.
| First author/s,
year | Types of cells | Treatment | Findings | (Refs.) |
|---|
| Al-Bari et
al, 2012 | BMMs stimulated by
M-CSF and RANKL | CQ (1–10 µM) | TRAP-positive cells
↓, resorption pit area ↓, NFATc1 ↓, TUNEL-positive cells ↑ (at high
dose) | (30) |
| Hu et al,
2022 | BMMs stimulated by
M-CSF and LPS | CQ (10 µM) | Oc.N ↓, MMP-9 ↓,
TRAP ↓, CTSK ↓, LC3II/I ratio ↑, p62/SQSTM1 ↑, TRAF3 ↑ | (53) |
| Xiu et al,
2014 | BMMs stimulated by
M-CSF and RANKL | CQ (10 µM) | Oc.N ↓, resorption
pit area ↓, TRAF3 ↑ | (27) |
| Hu et al,
2016 | BMMs stimulated by
M-CSF and RANKL | CQ (10 µM) | LC3II/I ratio ↑,
mTOR ↑, p-mTOR ↑ | (80) |
| Sun et al,
2021 | RAW264.7 cells
stimulated by RANKL | CQ (10 µM) | TRAP-positive cells
↓, resorption pit ↓, MMP-9 ↓, TRAP ↓, CTSK ↓, LC3II/I ratio ↑ | (54) |
| Wu et al,
2020 | RAW264.7 cells
stimulated by RANKL | CQ (10 µM) | TRAP-positive cells
↓, actin ring formation ↓, NFATc1 ↔, c-Fos ↔, DC-STAMP ↓ | (55) |
| Ito et al,
2005 | RAW264.7 cells
stimulated by RANKL | CQ (150 µM) | c-Fos ↔ | (68) |
| Yao et al,
2017 | Osteoclast
precursors stimulated by M-CSF and RANKL | CQ (dose
unspecified) | Oc.N ↓, TRAF3
↑ | (56) |
| Okusha et
al, 2020 | Murine monocytic
(RAW-D) cells stimulated by RANKL | CQ (10 µM) | c-fms ↑, RANK
↑ | (69) |
| Tran et al,
2020 | Murine monocytic
(RAW-D) cells stimulated by RANKL | CQ (1–30 µM) | c-fms ↑, RANK
↑ | (70) |
| Ke et al,
2020 | BMMs stimulated by
M-CSF and RANKL | CQ (2 µM) CQ (2 µM)
+ curcumin (5–15 µM) | Cell proliferation
↔, Oc.N ↔, TRAP ↔, MMP-9 ↔, CTSK ↔ Cell proliferation ↓, Oc.N ↓,
TRAP ↓, MMP-9 ↓, CTSK ↓ | (29) |
| Zhao et al,
2020 | BMMs stimulated by
M-CSF and RANKL | CQ (1 µmol/l) | TRAP-positive cells
↓, lacunar resorption area ↓, CTSK ↔, TRAP ↓, LC3II ↑, p62/SQSTM1
↔, autophagosome ↑ | (58) |
|
|
| CQ (1 µmol/l) + OPG
(40 ng/ml) | TRAP-positive cells
↔, lacunar resorption area ↑, CTSK ↑, TRAP ↑, LC3II ↑, p62/SQSTM1
↑, autophagosome ↑ |
|
| Tong et al,
2019 | BMMs stimulated by
M-CSF and RANKL | CQ (10 µmol/l) | TRAP-positive cells
↓, lacunar resorption area ↓, autophagosome ↑, Atg5 ↔, Atg7 ↔,
Atg12 ↔, Atg13 ↔ | (57) |
|
|
| CQ (10 µmol/l) +
OPG (40 ng/ml) | TRAP-positive cells
↓, lacunar resorption area ↓, autophagosome ↓, Atg12 ↓, Beclin 1 ↑,
p62/SQSTM1 ↑, LC3II ↓, p-mTOR ↓, Raptor ↓, GβL ↑, p-AMPK ↑, TSC2 ↓,
Rheb ↓, p-p70S6K ↓ |
|
| Aoki et al,
2020 |
Dexamethasone-induced BMMs stimulated by
M-CSF and RANKL | CQ (5 µM) | TRAP activity
↓ | (23) |
| Lin et al,
2016 |
Dexamethasone-induced BMMs stimulated by
M-CSF and RANKL | CQ (10 µM) | TRAP-positive cells
↓, bone resorption area ↓ | (24) |
| Ke et al,
2018 | IL-17A-induced
RAW264.7 cells stimulated by M-CSF and RANKL | CQ (30 µM) | Oc.N ↓ | (60) |
| Rieman et
al, 2001 | Human osteoclasts
isolated from fresh osteoclastoma tissue | CQ (25 µmol/l) | Lysosomal pH ↑,
proteolytic processing of CTSK ↓ | (61) |
| Su et al,
2018 | Titanium wear
particle-induced KG-1a cells | CQ (100 µM) | RANKL ↓ | (62) |
| Voronov et
al, 2013 | BMMs isolated from
mice carrying the R740S mutation stimulated by M-CSF and RANKL | CQ (10 µM) | TRAP-positive cells
↓, RCAN1 ↑ | (63) |
| Alam et al,
2021 | BMMs isolated from
ADO2+/+ mice stimulated by M-CSF and RANKL | CQ (30 nM) | Osteoclast
formation ↔, osteoclast survival ↓, CTX ↓, resorption pit area
↔ | (34) |
|
| BMMs isolated from
ADO2+/− mice stimulated by M-CSF and RANKL | CQ (30 nM) | Osteoclast
formation ↓, osteoclast survival ↔, CTX ↑, resorption pit area
↑ |
|
|
| BMMs isolated from
ADO2+/+ mice stimulated by M-CSF and RANKL | DCQ (100 nM) | Osteoclast
formation ↓, osteoclast survival ↓, CTX ↑, resorption pit area
↑ |
|
|
| BMMs isolated from
ADO2+/− mice stimulated by M-CSF and RANKL | DCQ (100 nM) | Osteoclast
formation ↓, osteoclast survival ↔, CTX ↑, resorption pit area
↑ |
|
| Both et al,
2018 | PBMC-sorted
monocytes | HCQ (5 µg/ml) | Multinuclear
osteoclasts ↓, surface resorption ↓, CTSK ↓, TM7SF4 ↑, lysosomal
membrane permeabilization ↑, DAPI staining ↔, Bax/Bcl-2 ↔, caspase
3 ↔ | (16) |
| Huang et al,
2023 | PBMCs isolated from
patients with rheumatoid arthritis stimulated by M-CSF and
RANKL | HCQ (dose
unspecified) | Cell viability ↓,
CTR ↓, TRAP ↓, CTSK ↓, MMP-9 ↓, apoptosis ↑, CCL20 ↓, CXCL8 ↓,
HIF-1α ↔, IL-1 ↓, IL-6 ↓, IL-17 ↓, TNF-α ↓, mTOR ↔, LC3I/II ratio
↔, Atg5 ↔, Beclin 1 ↔ | (64) |
| Xu et al,
2020 | Co-culture of
MC3T3-E1 cells and BMMs | HCQ (dose
unspecified) + trehalose (50 mM) | Oc.N ↓, OPG ↑,
OPG/RANKL ↑ | (49) |
| Lee et al,
2004 | Co-culture of
fibroblast-like synoviocytes and PBMCs isolated from healthy
volunteers | HCQ (1–20 µM) | Cell proliferation
↔, Oc.N ↔ | (65) |
Similar to the findings observed in animal studies,
CQ might enhance or weaken the inhibition of osteoclastogenesis
when given as combined therapy with other agents in vitro.
The combination of CQ and curcumin conferred synergistic effects in
reducing osteoclast differentiation in BMM stimulated by M-CSF and
RANKL (29). BMMs stimulated by
M-CSF and RANKL were treated with either 1 or 10 µmol/l CQ, and
reduced osteoclast formation, lacunar resorption area and TRAP
expression levels were observed (57,58).
The co-treatment with a lower dose of CQ (1 µmol/l) and OPG (40
ng/ml) reduced the ability of OPG to reduce the lacunar resorption
area, CTSK and TRAP expression levels (58). However, co-treatment with a higher
dose of CQ (10 µmol/l) and OPG (40 ng/ml) improved the ability of
OPG to inhibit osteoclastogenesis, as indicated by the further
reduction of TRAP-positive cells and lacunar resorption area
(57).
Dexamethasone increases osteoclast formation by
stimulating the proliferation and differentiation of osteoclast
precursors into mature osteoclasts (23,24).
The TRAP activity, osteoclast formation and bone resorption area
were decreased in dexamethasone-induced osteoclasts after
incubation with CQ (5–10 µM) (23,24).
IL-17A is a pro-inflammatory cytokine produced by Th17 cells, which
induces osteoclastogenesis and bone resorption (59). The increased mature osteoclast
formation of RAW264.7 cells induced by IL-17A was suppressed by 30
µM CQ (60). Osteoclastoma, also
known as a giant cell tumour of the bone, is a rare and benign
(non-cancerous) tumour developed predominantly at the ends of the
femur or the upper end of the tibia leading to local bone
destruction (61). The presence of
CQ (25 µmol/l) increased lysosomal pH and prevented the proteolytic
processing of CTSK in human osteoclasts extracted from fresh
osteoclastoma tissue (61). In
addition, the shedding of titanium particles from wear implants
into hard or soft tissues may stimulate the inflammatory response
and osteoclast differentiation (62). In macrophage (KG-1a) cells, the
upregulation of RANKL protein induced by a pure titanium particle
was inhibited by the addition of 100 µM CQ (62).
Another study utilised two different cell culture
models to resemble osteoporotic conditions. Heterozygous mice with
a point mutation (R740S) in the a3 subunit of V-ATPase (+/R740S)
displayed mild osteopetrosis and impaired bone resorption. The
TRAP-positive cells were reduced in BMMs isolated from mice
carrying the R740S mutation stimulated by M-CSF and RANKL and
exposed to CQ (10 µM). The mechanism underlying the
anti-osteoclastogenic action of CQ was modulated by the increased
level of the regulator of calcineurin, followed by NFATc1
inhibition (63). Alam et
al (34) isolated BMMs from
the femurs and tibias of ADO2+/+ and ADO2+/−
mice, displaying normal and impaired osteoclastic bone resorption,
respectively. Exposure to CQ (30 nM) caused reductions in CTX
expression levels and osteoclast survival in normal mice, as well
as a reduction in osteoclast formation and an increase in CTX and
resorption pit formation in ADO2+/− mouse-derived cells.
Treatment with desethylchloroquine (DCQ; a CQ metabolite) at 100 nM
resulted in decreased osteoclast formation and survival but
increased CTX expression levels and resorption pit formation in
BMMs extracted from normal mice. Similar concentrations of DCQ
decreased osteoclast formation, and increased CTX expression levels
and resorption pit formation, without affecting osteoclast survival
in ADO2+/− mouse-derived cells (34). The findings of these studies
indicate that osteoclast activity was enhanced by CQ and DCQ under
osteoporotic conditions but with fewer numbers of osteoclasts. Such
observations might be due to CQ and DCQ reducing the less efficient
osteoclast, leaving a subpopulation of highly active
osteoclasts.
Four studies have examined the effects of HCQ on
osteoclast formation and activity. Using human peripheral blood
mononuclear cell (PBMC)-sorted monocytes to differentiate into
osteoclasts, the presence of HCQ (5 µg/ml) was revealed to inhibit
the formation of multinuclear osteoclasts, surface resorption and
expression of CTSK while increase transmembrane 7 superfamily
member 4 (TM7SF4, an osteoclast fusion marker) expression (16). In another study using PBMCs from
patients with rheumatoid arthritis, HCQ reduced cell viability and
osteoclastogenic gene expression (calcitonin receptor; TRAP; CTSK
and MMP-9) when culturing in a medium containing M-CSF and RANKL
(64). Xu et al (49) further demonstrated that HCQ reduced
osteoclast formation in a co-culture system containing MC3T3-E1
cells and BMMs (49). However, one
study reported contradictory outcomes, whereby HCQ (1–20 µM) did
not inhibit cell proliferation and osteoclast formation in the
co-culture of fibroblast-like synoviocytes and PBMCs in the
presence of M-CSF (65).
The effects of CQ and HCQ on osteoclasts have been
established. CQ and HCQ exhibit positive effects on bone health by
modulating osteoclast function and activity. They inhibit
osteoclast proliferation and differentiation in an osteoclastogenic
environment with or without an inducer but promote
osteoclastogenesis during impaired bone resorption.
Osteocytes
To the best of our knowledge, only one study has
evaluated the effects of CQ on osteocytes (Table V). Pre-incubation with 50 µM CQ did
not exert a cytotoxic effect on osteocytic MLO-Y4 cells; however,
CQ exacerbated hydrogen peroxide-induced cell death (66).
| Table V.Effects of CQ in osteocytes. |
Table V.
Effects of CQ in osteocytes.
| First author/s,
year | Types of cells | Treatment | Findings | (Refs.) |
|---|
| Kar et al,
2019 | MLO-Y4 cells
H2O2-induced MLO-Y4 cells | CQ (50 µM) | No cytotoxicity
Cell death ↑ | (66) |
Mechanism of action of CQ and HCQ
M-CSF and receptor activator of
nuclear factor κ-Β (RANK)/RANKL/OPG pathways
The M-CSF and RANK/RANKL/OPG pathways are the most
implicated mechanisms for monocyte/macrophage-to-osteoclast
differentiation regulated by osteoblasts. M-CSF is a cytokine
constitutively secreted by osteoblasts, which causes haematopoietic
stem cells to differentiate into osteoclastic lineage in the
presence of RANKL. RANK is a homotrimeric transmembrane receptor
expressed on osteoclast precursors and mature osteoclasts. RANKL,
produced by osteoblasts, binds to RANK as its receptor, eventually
promoting osteoclast proliferation and maturation. Upon the
interaction between RANKL and RANK, tumour necrosis factor
receptor-associated factor 6 is recruited, stimulating a series of
downstream targets, including NF-κB, NFATc1, MAPK and PI3K, which
are responsible for osteoclast formation, activation and survival
(67). The decoy receptor for
RANKL, OPG, is also secreted by osteoblasts to prevent RANK/RANKL
ligation and its subsequent reactions (67).
In D-galactose-induced bone loss, oral treatment
with CQ (10 mg/kg) daily for 4 weeks replenished serum OPG levels
and blunted the elevation of RANKL. Mechanistically, the elevated
ERK expression in osteoporotic rats was mitigated by CQ
administration (33). In RAW264.7
cells stimulated by RANKL, incubation with CQ (10–150 µM) did not
cause any change in the expression of Fos proto-oncogene (c-Fos)
(55,68). Lysosomes contribute to the
proteolytic degradation of M-CSF receptor and RANK proteins,
whereas the blocking of lysosomes by CQ enhances their expression
in osteoclasts (69,70). In combination with other agent, HCQ
co-treatment with trehalose further increased the expression levels
of OPG and the OPG/RANKL ratio compared to trehalose alone in a
co-culture of MC3T3-E1 cells and BMMs (49) (Fig.
2).
 | Figure 2.Schematic of the effects of CQ and
HCQ on the M-CSF and RANK/RANKL/OPG pathways. The molecular changes
caused by CQ and HCQ are represented by black and grey arrows,
respectively. AP-1, activating protein-1; c-fms, macrophage
colony-stimulating factor receptor; c-Fos, Fos proto-oncogene; CQ,
chloroquine; DC-STAMP, dendritic cell-specific transmembrane
protein; HCQ, hydroxychloroquine; M-CSF, macrophage
colony-stimulating factor; NFATc1, nuclear factor of activated
T-cells cytoplasmic 1; OPG, osteoprotegerin; RANK, receptor
activator of nuclear factor κ-Β; RANKL, receptor activator of
nuclear factor κ-Β ligand; RCAN1, regulator of calcineurin; TRAF3,
tumour necrosis factor receptor-associated factor 3; TRAF6, tumour
necrosis factor receptor-associated factor 6; ↑,
increase/upregulate; ↓, decrease/downregulate; ↔, no change. |
Autophagy machinery
Autophagy, also known as auto-phagocytosis, is a
natural conserved process involved in the degradation and
correction of intracellular proteins for the eradication of damaged
or senescent macromolecules and organelles (71). Autophagy has two main regulators,
namely 5′AMP-activated protein kinase (AMPK) as an activator and
mTOR as an inhibitor blocking the auto-phagosomal membrane
formation (72).
Initiation/nucleation, elongation, maturation and degradation are
the four major stages in autophagy regulated by autophagy-related
proteins (Atg). Autophagy is initiated following an autophagic
stimulus such as oxidative stress, hypoxia and lack of nutrients.
The phagophore is formed by the combination of two multiprotein
complexes: The class III PI3K nucleation complex [comprising Beclin
1, vacuolar protein sorting (Vps)34, Vps15 and Atg14] and the
Unc-51-like kinases 1 (ULK1) initiation complex (comprising ULK1,
Atg13 and Atg101). The elongation step involves the elongation of
the phagophore enclosing cytoplasmic components that are
non-functional, forming an autophagosome. The elongation and
closure of autophagosomes are assisted by LC3 and Atg. LC3 exists
as LC3I under physiological conditions and is converted to LC3II
after conjugation with phospholipid phosphatidylethanolamine when
autophagy is activated, thereby gradually increasing the LC3II/I
ratio. Cytoplasmic components selected for degradation are tagged
by selective autophagy co-receptors, such as sequestosome-1
(p62/SQSTM1) and neighbour of Brca1 gene (Nbr1)]. These
co-receptors are recognised by LC3II, which directed them to the
interior of the autophagosome. The maturation step involves the
fusion of autophagosomes with lysosomes, creating an acidic
environment that facilitates the degradation of autophagosomal
contents. In the final step, autophagosome contents are degraded by
hydrolases from lysosomes, and the resulting molecules (lipids,
amino acids and nucleotides) are reused to recycle cell components
and produce energy (72,73).
Autophagy serves an important role in the
homeostasis of bone cells (including osteoblasts, osteoclasts and
osteocytes). Therefore, the dysfunction of autophagy is associated
with the progression of bone loss and subsequent osteoporosis
(74,75). The well-known autophagic inhibitor
CQ blocks the fusion of autophagosome with lysosome and disrupts
the acidic environment by inactivating acid hydrolases in
lysosomes, favouring reduced osteoblast activity (74). Pretreatment with CQ (5 µM) was
insufficient to cause any change in the LC3II/I ratio of MC3T3-E1
cells compared with control cells (41). Two studies have reported distinct
outcomes; in one study, CQ (10 µM) reduced LC3II expression levels,
while another study revealed increased LC3II expression in MC3T3-E1
cells, which may be due to different experimental conditions
(acidic environment and high glucose) (45,76).
Considering that only LC3II expression was assessed as an autophagy
marker, determining the effects of CQ on autophagy under these
conditions may be challenging. High concentrations of CQ (20–50 µM)
increased LC3II expression and the accumulation of p62/SQSTM1, but
decreased the formation of autophagosomes (42,43).
Larger autolysosomes were accumulated and the expression levels of
lysosome-associated membrane glycoprotein 1 (a lysosomal membrane
component) were increased in MC3T3-E1 cells treated with CQ,
indicating the impairment of auto-lysosomal degradation function
(43). BMSCs obtained from
CQ-injected mice exhibited unchanged Beclin 1 expression levels, a
lower LC3II/I ratio and increased p62/SQSTM1 (28). The in vitro findings are
consistent with those observed in an in vivo experimental
setting. An animal study by Xiong et al (26) demonstrated that the expression
levels of Beclin 1 and the LC3II/LC3I ratio were upregulated, and
the levels of p62/SQSTM1 remained unchanged, indicating the
activation of autophagy in ovariectomised rats compared with normal
controls. The administration of CQ (10 mg/kg; i.p.) inhibited
autophagy, thereby reducing Beclin 1 expression and the LC3II/I
ratio but increasing the accumulation of p62/SQSTM1 in
ovariectomised rats (26).
In the presence of other osteogenic agents, CQ
suppresses autophagic and osteoblastic activities, resulting in the
weakening of the bone-protecting effects of icariin (28), lactoferrin (46) and quercetin (26). BMSCs extracted from mice injected
with CQ and icariin exhibited decreased Beclin 1 expression and
LC3II/I ratios but increased p62/SQSTM1 expression (28). In a study by Zhang et al
(46), the presence of CQ reduced
lactoferrin-induced osteoblastic differentiation by suppressing
autophagic activity via increased expression of Nbr1 and reduced
activation of p38 MAPK (46). In
ovariectomised rats injected with CQ and fed quercetin, the
expression of Beclin 1 was further reduced compared with that in
the animals treated with quercetin only (26). In TNF-α-stimulated MC3T3-E1 cells,
the same researchers reported heterogenous outcomes on LC3II
accumulation (decreased or increased) after treatment with CQ (10
µM) in two different studies (50,77).
The discrepancy arises because of the different mechanisms of
action involved. Connective tissue growth factor enhanced autophagy
via protein kinase B and ERK activations; hence, LC3II did not
accumulate when CQ was added (50). On the contrary, the inhibition of
JAK2 suppressed autophagy, which was further inhibited upon CQ
treatment, leading to LC3II accumulation (77). p62/SQSTM1 was upregulated in
CQ-treated TNF-α-stimulated MC3T3-E1 cells, indicating the
suppression of autophagy (77). In
cadmium-stimulated MC3T3-E1 cells, CQ (5 µM) reduced the expression
levels of LC3II and decreased cell viability (51). Treatment with HCQ also caused
accumulation of LC3II and p62/SQSTM1, disrupted the mitochondrial
membrane potential and blocked the formation of autolysosomes in
BMSCs harvested from C57BL/6 mice, hindering the autophagic vacuole
degradation of compromised mitochondria, which led to cellular
senescence (78,79). HCQ treatment attenuated
trehalose-induced autophagy, as shown by reduced Beclin 1 and
increased LC3II in MC3T3-E1 cells (49). The aforementioned studies
reiterated that CQ and HCQ suppressed autophagy, thereby reducing
osteogenic differentiation. Co-treatment with CQ or HCQ further
suppressed the autophagic activity and weakened the effects of
other bone-protecting compounds. In addition, the inhibition of
autophagy by CQ exacerbated the inhibition of osteogenic
differentiation by an inflammatory mediator (TNF-α) and toxic heavy
metal (cadmium).
Using BMMs or RAW264.7 cells induced by M-CSF and/or
RANKL, the cell culture treated with CQ (1–10 µM) demonstrated
inhibition of autophagy as indicated by the increased LC3II/I
ratio, reduced transformation from autophagosomes to autolysosomes,
as well as unchanged p62/SQSTM1 accumulation and Atg protein
expression (54,57,58,80).
The reduction of RANKL-induced osteoclast formation and function
was mediated by the increase of tumour necrosis factor
receptor-associated factor 3 (TRAF3; an anti-osteoclastic factor)
(27,56) and mTOR protein levels (80). Furthermore, the LPS-inhibited TRAF3
protein level was recovered by autophagic inhibition by CQ, and a
higher LC3II/I ratio and p62/SQSTM1 expression were noted (53). BMMs co-treated with CQ (1 µM) and
OPG (40 ng/ml) inhibited autophagy as indicated by the high LC3II/I
ratio, p62/SQSTM1 expression and autophagosomes (58). The combination of a higher dose of
CQ (10 µM) with OPG further reduced osteoclast formation and
activity compared with CQ alone (57). However, this combination treatment
resulted in autophagy inhibition to a lower extent, evidenced by a
reduction in the number of autophagosomes, LC3II/I ratio and Atg12
levels, whereas the expression level of Beclin 1 and p62/SQSTM1 was
increased (57). In mTOR
signalling, the levels of phosphorylated mTOR and Raptor were
reduced, but the expression levels of G protein β-subunit-like
protein (a kinase domain of mTOR that stabilises the interaction
between Raptor and mTOR) were increased in M-CSF- and RANKL-induced
BMMs treated with CQ and OPG compared with those treated with CQ
only. In the AMPK pathway, phosphorylated AMPK was higher;
meanwhile, tuberous sclerosis complex 2, Ras homolog enriched in
the brain and phosphorylated p70 ribosomal protein S6 kinase were
reduced in the CQ and OPG treatment groups compared with the
CQ-only group (57). However,
Huang et al (64)
demonstrated that HCQ failed to exert any effect on autophagy in
PBMCs isolated from patients with rheumatoid arthritis stimulated
by M-CSF and RANKL (64).
Therefore, CQ inhibited the autophagolysosomal degradation of
TRAF3, thereby forming fewer osteoclasts in response to M-CSF,
RANKL and/or LPS. Furthermore, CQ increased the lysosomal pH via
the activation of mTOR (a negative regulator of autophagy), thereby
inhibiting osteoclastogenesis (57). Compared with CQ, the
pharmacological inhibition of autophagy by HCQ was not detected
(Fig. 3).
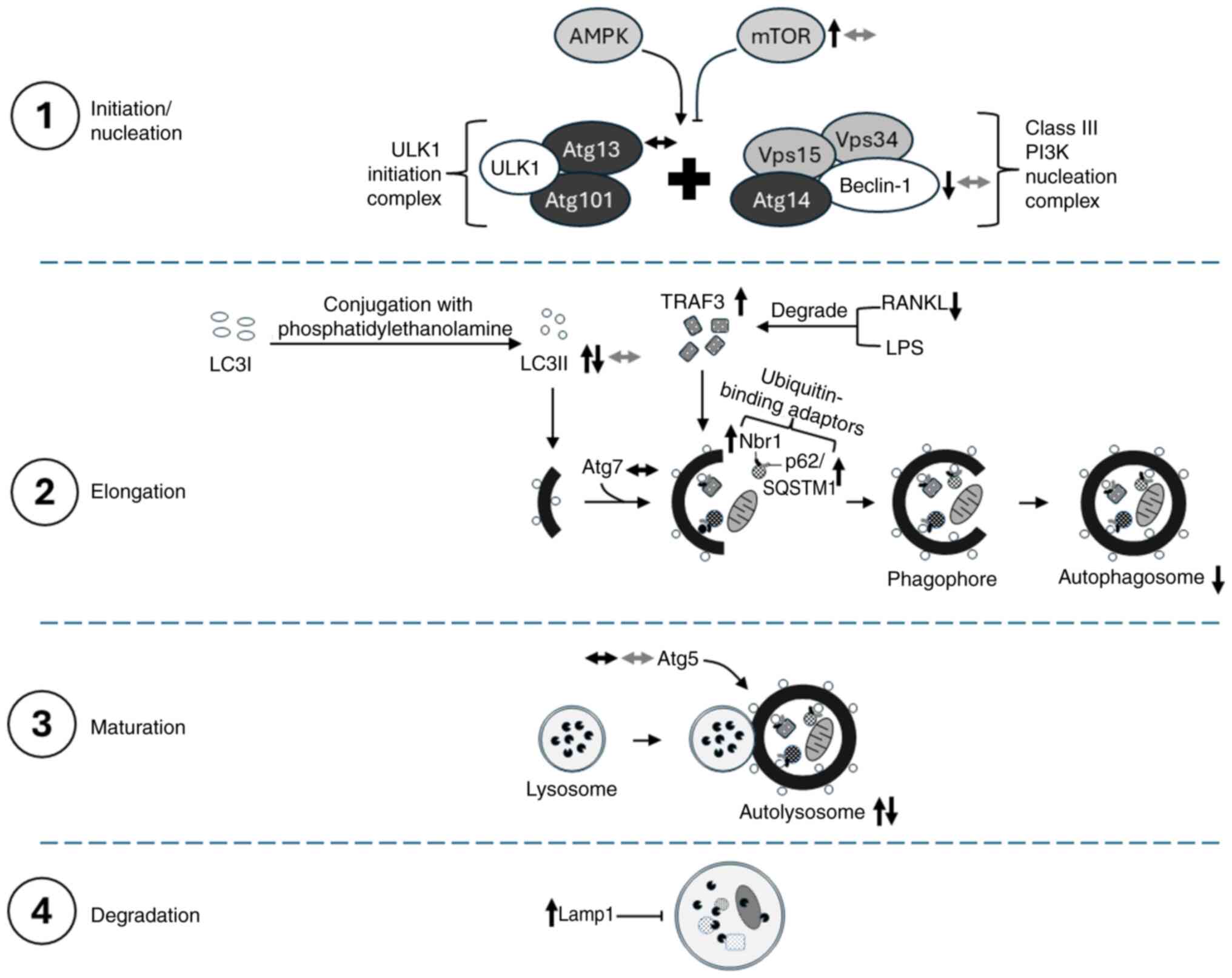 | Figure 3.Schematic of the effects of CQ and
HCQ on the regulation of autophagy machinery. The molecular changes
caused by CQ and HCQ are represented by black and grey arrows,
respectively. AMPK, 5′AMP-activated protein kinase; Atg,
autophagy-related proteins; CQ, chloroquine; HCQ,
hydroxychloroquine; lamp1, lysosome-associated membrane
glycoprotein 1; LPS, lipopolysaccharides; Nbr1, neighbour of Brca1
gene; p62/SQSTM1, sequestosome-1; RANKL, receptor activator of
nuclear factor κ-Β ligand; TRAF3, tumour necrosis factor
receptor-associated factor 3; ULK1, Unc-51-like kinases 1; Vps,
vacuolar protein sorting; ↑, increase/upregulate; ↓,
decrease/downregulate; ↔, no change. |
Apoptosis
Apoptosis is a self-destruction process of cells
triggered by cellular injury and oxidative stress, which is
regulated by two major mechanisms (81). In the mitochondria-dependent
(intrinsic) pathway, intracellular stimuli upregulate pro-apoptotic
(Bax) but downregulate anti-apoptotic (Bcl-2) molecules, leading to
the disruption of mitochondrial outer membrane permeabilization and
the release of cytochrome c. Subsequently, cytochrome
c interacts with apoptotic protease activating factor-1,
deoxyadenosine triphosphate and procaspase-9, forming an
apoptosome, which converts procaspase-9 to caspase-9. The
downstream effector caspases (caspase-3 and caspase-7) are
activated, acting as a molecular switch for apoptosis (81). In the death receptor-mediated
(extrinsic) pathway, Fas ligand and tumour necrosis factor-related
apoptosis-inducing ligand (TRAIL) are two death ligands recognised
by Fas and TRAIL receptors, respectively. Eventually, the
Fas-associated death domain and caspase-8 are recruited to form a
death-inducing signalling complex and activate downstream effector
caspase-3 to induce cell fragmentation and apoptosis (81).
The pro-apoptotic effects of CQ on osteoblasts were
observed at higher doses (10–20 µM) but not at a lower dose (5 µM)
(41,42,45,76).
The levels of Bax, caspase-3 and p53 were increased, whereas the
expression levels of Bcl-2 were reduced (42). Treatment with CQ at 50 µM did not
cause any change in Bcl-2 levels (43). In the presence of inflammatory
cytokines such as TNF-α, treatment with CQ elevated caspase-3 and
cleaved poly (adenosine diphosphate-ribose) polymerases, indicating
enhanced osteoblast apoptosis (50,77).
The cadmium-mediated apoptotic cell death in rat cranial
osteoblasts was also further potentiated by the autophagy inhibitor
CQ (51). On the contrary, HCQ (5
µg/ml) demonstrated no effect on apoptotic events in human
mesenchymal stromal cells (52).
Several studies have investigated the effects of CQ
and HCQ on osteoclast apoptosis. The survival of BMMs stimulated by
M-CSF and RANKL was not affected by a lower concentration of CQ,
but the number of terminal deoxynucleotidyl transferase dUTP
nick-end labelling-positive cells was increased at a higher
concentration of CQ (7.5–10 µM) (30). Two studies have reported the
differential effects of HCQ on apoptosis using PBMC-sorted
monocytes and PBMCs isolated from patients with rheumatoid
arthritis stimulated by M-CSF and RANKL, whereby the former
reported no apoptotic effects using 5 µg/ml HCQ (16) and the latter indicated an increase
in apoptosis with an unknown concentration of HCQ (64). The variation in the treatment dose
of HCQ may influence the outcomes observed in these studies.
Oxidative stress
Oxidative stress reflects an imbalance between the
production of ROS and reactive nitrogen species (RNS) with the
antioxidant defence capacity to detoxify them, inducing lipid
peroxidation, protein modifications and deoxyribonucleic acid
damage (82). Nicotinamide adenine
dinucleotide phosphate oxidase 4 (Nox4) is the main source of ROS
production, whereas RNS synthesis is initiated by the interaction
between nitric oxide (NO) and superoxide anions (82). SOD, GPx and CAT are antioxidant
enzymes that contribute to the first-line antioxidant defence in
the body (82). CQ inhibits
osteoblastic activity via the induction of oxidative stress.
Pretreatment with low-dose CQ (5 µM) had no effect on ROS formation
and Nox4 expression in MC3T3-E1 cells (41). A higher concentration of CQ (20 µM)
reduced the total antioxidant capacity and SOD levels but increased
NO levels, indicating the elevation of oxidative stress (42). To the best of our knowledge, to
date, the antioxidative effect of CQ or HCQ in osteoclasts is
limited.
Inflammation
Inflammation refers to the activation of the innate
and adaptive immune system that produces an array of inflammatory
cytokines, such as TNF-α, ILs, interferons and chemokines (64). These molecules perpetuate
inflammation to affect the differentiation and function of
osteoblasts and osteoclasts, leading to excessive bone degradation
and impaired osteoblast function (64). HCQ administration reduced the
generation of inflammatory cytokines in PBMCs isolated from
patients with rheumatoid arthritis stimulated by M-CSF and RANKL.
The levels of chemokine (C-C motif) ligand 20, CXC motif chemokine
ligand 8, IL-1, IL-6, IL-17 and TNF-α, were decreased, but the
expression levels of hypoxia-inducible factor 1α remained unchanged
(64). Nonetheless, the role of CQ
and HCQ in the inflammatory response in osteoblasts remains
unclear.
Perspectives
In vitro studies have revealed that CQ and
HCQ inhibit osteoblastic (28,42–48,52)
and osteoclastic (16,23,24,27,30,34,49,53–58,60–64)
activities simultaneously. The inhibition of osteoblast
differentiation and mineralisation by CQ and HCQ is mediated
through the suppression of autophagy, activation of oxidative
stress and osteoblast apoptosis. Autophagy dysfunction increases
ROS production and accelerates cell senescence, leading to
increased osteoblast apoptosis and decreased numbers of osteocytes
(83). In osteoclasts, CQ inhibits
osteoclastogenesis via the suppression of the RANK/RANKL/OPG system
(49,62,69,70),
autophagy (53,54,57,58,64,80)
and the inflammatory response (64). Although CQ treatment increases the
expression levels of RANK and c-fms (the receptors for RANKL and
M-CSF respectively), the OPG/RANKL ratio is increased (69,70),
and the expression levels of c-Fos remain unchanged (55,68),
thereby leading to the downstream inhibition of ERK and NFATc1
expression during osteoclastogenesis. Furthermore, in osteoclasts,
the induction of autophagy contributes to pro-osteoclastogenesis.
CQ inhibits autophagy by preventing TRAF3 degradation and limiting
RANKL-induced osteoclast formation (27,56).
CQ also increases the lysosomal pH, thereby causing the
accumulation of mTOR and phosphorylated-mTOR protein to
subsequently inhibit autophagy (57,61).
However, the inhibition of autophagy by HCQ has not been observed
in osteoclasts. In addition, CQ and HCQ enhance osteoclast
apoptosis in mature osteoclasts (64).
The inhibition of bone formation and bone
resorption by CQ and HCQ occur in parallel, but their net effect on
the skeletal system requires further investigation with a study
design focusing on the effects of CQ and HCQ on bone health. The
selection of the treatment dose, frequency and duration of CQ and
HCQ should be carefully considered. Based on the aforementioned
studies, the intervention dose of CQ was 2–200 mg/kg in mice and 10
mg/kg in rats (23,24,26–30,33,34,36).
Meanwhile, the effective HCQ dose in protecting the bone was 80
mg/kg (37). The calculated human
equivalent dose ranged between 9.73 and 973 mg for CQ and was ~389
mg for HCQ in an adult weighing 60 kg. In humans, 200–250 mg CQ or
HCQ was used, which was in line with the well-tolerated doses of CQ
and HCQ for rheumatic diseases (200–400 mg/day) and novel
coronavirus-19 (500 mg/day) (9).
In animals, the short-term continuous daily
administration of CQ (<28 days) was revealed to be protective to
bone (23,27), and the intermittent administration
of CQ was recommended to exert beneficial effects on bone for
long-term treatment (24,26). These synergistic effects may not be
observed in combination therapy with CQ and other osteoprotective
agents (28). Only a paucity of
animal studies has investigated the skeletal effects of HCQ
(37–40); thus, drawing a concrete conclusion
is challenging. The outcomes obtained from preclinical studies were
not reflected in the clinical setting. Firstly, the effectiveness
of CQ on bone has been widely established in vitro and in
vivo; however, the use of HCQ has been preferred in human
studies. HCQ is a metabolite derived from CQ by adding a hydroxyl
group, making it less toxic with fewer side effects and allowing it
to dissolve more easily in the body (84). Thus, HCQ is a safer medication for
patients (84).
Both CQ and HCQ have excellent oral absorption,
good bioavailability, high volume of distribution (extensive drug
sequestration by tissues), are hepatically metabolized, have a long
half-life and their metabolites are excreted through urine and
faeces (85). They can cross the
placenta but toxicity in the foetus has not been reported (85). Toxicity develops rapidly (1–3 h
after ingestion) in the case of overdose (85). The use of CQ and HCQ is associated
with several adverse events such as gastrointestinal discomfort,
allergic reactions, retinal damage, cardiomyopathy and skin
hyperpigmentation (86).
Although CQ and HCQ have protective effects on
inflammatory bone loss, the risk of using these agents should not
be overlooked. Dose optimisation and regular monitoring are the key
for the safe use of CQ and HCQ. Regular ophthalmologic monitoring
is important for long-term CQ and HCQ use to prevent retinal
toxicity, which can cause irreversible vision loss if not detected
early (87). The patient history
of ocular disease should be considered before prescribing these
medications (87). In addition,
regular cardiac monitoring (including electrocardiogram and
echocardiography) could be beneficial in detecting cardiac
complications early, ensuring the safe prolonged use of these drugs
(88). Dose optimisation for CQ
and HCQ focuses on balancing therapeutic benefits whilst minimising
toxicity. To further minimise risks, intermittent or pulsed dosing
strategies (given every other day) may be considered for long-term
use. Further research is necessary to confirm the efficacy of
intermittent or pulsed dosing in metabolic bone disorders.
Effective bone health monitoring (including
diagnostic assessments, regular follow-ups and lifestyle
evaluations) is key for the prevention, early detection and
management of bone disorders. Dual-energy X-ray absorptiometry is
the gold standard for measuring BMD. The assessment is recommended
every 1–2 years to monitor bone loss in women >65 years old, men
>70 years old and younger individuals with risk factors such as
oestrogen deficiency, smoking, low body mass, prior fractures or
chronic glucocorticoid use (89).
Micro-computed tomography, an advanced imaging technique that can
generate three-dimensional images, enables the high-resolution
evaluation of density, geometry and microarchitecture of
mineralised tissues (90). The
evaluation of bone turnover markers reflects the rate of bone
remodelling, which may provide additional information on bone
health and the effectiveness of osteoporosis treatments (91). Consistent follow-ups are essential
to ensure medication adherence and adequate calcium and vitamin D
intake, as well as to address lifestyle factors such as physical
activity, smoking and alcohol consumption (92). All these approaches are
non-invasive and suitable for human studies. Meanwhile, in
vivo samples enable the use of bone histomorphometry and a
universal testing machine to characterise bone cells, analyse bone
microstructure, assess the bone remodelling rate and evaluate bone
strength.
The currently available evidence has limitations
that need to be addressed. Several of the included studies used CQ
and HCQ as inhibitors for autophagy rather than directly
investigating the skeletal effects of CQ and HCQ (29,53,66,80).
In addition, in certain studies, a statistical test was not
performed to compare the CQ or HCQ group and a control group
because determining the effect of CQ or HCQ on bone was not the
primary objective of the study (28,49,68).
Clinical evidence supporting the use of CQ and HCQ for bone health
remains limited. The majority of the available data are derived
from observational studies in patients with autoimmune diseases
such as rheumatoid arthritis and SLE (13–20).
Notably, to the best of our knowledge, no large-scale randomised
clinical trials have specifically assessed the impact of CQ or HCQ
on osteoporosis, fracture prevention or long-term skeletal health.
The absence of targeted research makes it difficult to determine
their optimal dosing, duration and effectiveness in managing
metabolic bone disorders.
The future development trends in the use of CQ and
HCQ for bone health may focus on targeted delivery systems, the
development of safer analogues and the integration of precision
medicine approaches. Various nanotechnology platforms, including
liposomes, polymeric nanoparticles, dendrimers, polyelectrolyte
complexes, lipid-based nanoparticles and metallic nanoparticles,
have been explored to enhance the reformulation of CQ and HCQ
(93). These approaches aim to
improve target tissue exposure while increasing overall safety and
efficacy (93). In addition,
developing novel CQ and HCQ analogues through molecular
modifications can enhance their pharmacokinetic and pharmacodynamic
properties, minimise undesirable side effects and reduce costs
(94), while preserving their
bone-protective properties. Researchers are also encouraged to
explore precision medicine approaches by identifying genetic or
metabolic markers to monitor treatment efficacy and determine the
patients who would benefit from CQ- and HCQ-based therapies.
While CQ and HCQ show potential for modulating bone
metabolism, several gaps in knowledge remain. Further in
vitro studies are needed to elucidate dose-dependent effects on
osteoblasts, osteoclasts and osteocytes, particularly from low to
high concentrations of CQ and HCQ. Potential drug interactions and
synergistic effects of CQ and HCQ with osteoporosis medications
(such as bisphosphonates, denosumab, selective oestrogen receptor
modulators, vitamin D and calcium) and natural anti-osteoporotic
products should be investigated using cell culture models.
Long-term animal studies are necessary to evaluate the effects of
chronic CQ and HCQ use on bone microarchitecture, strength and
turnover markers. Additionally, the efficacy and safety of optimal
dosing regimens (continuous or intermittent administration) and
targeted delivery formulations should be thoroughly investigated in
animal models before clinical application. In clinical research,
large-scale trials with patient stratification should be conducted
to evaluate the efficacy and safety profile of CQ and HCQ, as well
as to identify genetic and metabolic biomarkers. These trials
should include a comparison between CQ and HCQ and standard
osteoporosis treatments such as bisphosphonates, denosumab or
teriparatide.
Conclusion
CQ and HCQ are potentially useful to strengthen the
bone. However, the interaction of CQ or HCQ with other compounds
may weaken their bone-conserving effects. Therefore, treatment
strategies need to be carefully selected for patients with multiple
comorbidities. CQ or HCQ are also contraindicated in patients with
hepatic and renal insufficiency (85,86).
Clinical trials are warranted to determine whether CQ and HCQ can
be safely integrated into the management of osteoporosis.
Acknowledgements
Not applicable.
Funding
This work was supported by a Fundamental Grant (grant no.
FF-2024-090) provided by the Faculty of Medicine, Universiti
Kebangsaan Malaysia and the Fundamental Research Grant Scheme
(FRGS) (grant no. FRGS/1/2023/SKK10/UKM/02/3) provided by the
Ministry of Higher Education, Malaysia.
Availability of data and materials
Not applicable.
Authors' contributions
SKW wrote the manuscript. Data authentication is
not applicable. The author read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The author declares that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
ADO2
|
autosomal dominant osteopetrosis type
II mutation
|
|
ALP
|
alkaline phosphatase
|
|
AMPK
|
5′AMP-activated protein kinase
|
|
Atg
|
autophagy-related protein
|
|
BMD
|
bone mineral density
|
|
BMM
|
bone marrow monocyte/macrophage
|
|
BMSC
|
bone marrow mesenchymal stem cell
|
|
BV/TV
|
bone volume/tissue volume
|
|
CAT
|
catalase, c-fms, macrophage
colony-stimulating factor receptor
|
|
c-Fos
|
Fos proto-oncogene
|
|
CQ
|
chloroquine
|
|
CTSK
|
cathepsin K
|
|
CTX
|
cross-linked C-telopeptide of type 1
collagen
|
|
DCQ
|
desethylchloroquine
|
|
GPx
|
glutathione peroxidase
|
|
HCQ
|
hydroxychloroquine
|
|
HR
|
hazard ratio
|
|
i.p.
|
intraperitoneal
|
|
LPS
|
lipopolysaccharide
|
|
M-CSF
|
macrophage colony-stimulating
factor
|
|
Nbr1
|
neighbour of Brca1 gene
|
|
NFATc1
|
nuclear factor of activated T-cells
cytoplasmic 1
|
|
NO
|
nitric oxide
|
|
Nox4
|
nicotinamide adenine dinucleotide
phosphate oxidase 4
|
|
OCN
|
osteocalcin
|
|
Oc.N
|
osteoclast number
|
|
OPG
|
osteoprotegerin
|
|
OPN
|
osteopontin
|
|
PBMC
|
peripheral blood mononuclear cell
|
|
P1NP
|
propeptide of type I procollagen
|
|
p62/SQSTM1
|
sequestosome-1
|
|
RANK
|
receptor activator of nuclear factor
κ-Β
|
|
RANKL
|
receptor activator of nuclear factor
κ-Β ligand
|
|
ROS
|
reactive oxygen species
|
|
Runx-2
|
Runt-related transcription factor
2
|
|
SLE
|
systemic lupus erythematosus
|
|
SOD
|
superoxide dismutase
|
|
Tb.N
|
trabecular number
|
|
Tb.Sp
|
trabecular separation
|
|
TRAF
|
tumour necrosis factor
receptor-associated factor
|
|
TRAP
|
tartrate-resistant acid
phosphatase
|
|
β-CTx
|
β C-terminal telopeptide
|
|
ULK1
|
Unc-51-like kinases 1
|
|
Vps
|
vacuolar protein sorting
|
|
Th17
|
T helper 17
|
|
RNS
|
reactive nitrogen species
|
References
|
1
|
Elahmer NR, Wong SK, Mohamed N, Alias E,
Chin KY and Muhammad N: Mechanistic insights and therapeutic
strategies in osteoporosis: A comprehensive review. Biomedicines.
12:16352024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wu S, Ye Z, Yan Y, Zhan X, Ren L, Zhou C,
Chen T, Yao Y, Zhu J, Wu S, et al: The causal relationship between
autoimmune diseases and osteoporosis: A study based on Mendelian
randomization. Front Endocrinol (Lausanne). 14:11962692023.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Avouac J, Koumakis E, Toth E, Meunier M,
Maury E, Kahan A, Cormier C and Allanore Y: Increased risk of
osteoporosis and fracture in women with systemic sclerosis: A
comparative study with rheumatoid arthritis. Arthritis Care Res
(Hoboken). 64:1871–1878. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Caffarelli C, Cameli P, Al Refaie A,
Giglio E, Manzana G, Mondillo C, Noacco Y, Olivieri C, Bargagli E
and Gonnelli S: Bone fragility and sarcoidosis: An underestimated
relationship. Front Med (Lausanne). 9:10260282022. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Amarasekara DS, Yu J and Rho J: Bone loss
triggered by the cytokine network in inflammatory autoimmune
diseases. J Immunol Res. 2015:8321272015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lin D, Li L, Sun Y, Wang W, Wang X, Ye Y,
Chen X and Xu Y: IL-17 regulates the expressions of RANKL and OPG
in human periodontal ligament cells via TRAF6/TBK1-JNK/NF-κB
pathways. Immunology. 144:472–485. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kao FC, Hsu YC, Tu YK, Chen TS, Wang HH
and Lin JC: Long-term use of immunosuppressive agents increased the
risk of fractures in patients with autoimmune diseases: An 18-year
population-based cohort study. Biomedicines. 11:27642023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Satoh K, Mark H, Zachrisson P, Rydevik B,
Byröd G, Kikuchi S, Konno S and Sekiguchi M: Effect of methotrexate
on fracture healing. Fukushima J Med Sci. 57:11–18. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wong SK: Repurposing new use for old drug
chloroquine against metabolic syndrome: A review on animal and
human evidence. Int J Med Sci. 18:2673–2688. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dima A, Jurcut C and Arnaud L:
Hydroxychloroquine in systemic and autoimmune diseases: Where are
we now? Joint Bone Spine. 88:1051432021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Verbaanderd C, Maes H, Schaaf MB, Sukhatme
VP, Pantziarka P, Sukhatme V, Agostinis P and Bouche G: Repurposing
drugs in oncology (ReDO)-chloroquine and hydroxychloroquine as
anti-cancer agents. Ecancermedicalscience. 11:7812017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hashem AM, Alghamdi BS, Algaissi AA,
Alshehri FS, Bukhari A, Alfaleh MA and Memish ZA: Therapeutic use
of chloroquine and hydroxychloroquine in COVID-19 and other viral
infections: A narrative review. Travel Med Infect Dis.
35:1017352020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hong WJ, Chen W, Yeo KJ, Huang PH, Chen DY
and Lan JL: Increased risk of osteoporotic vertebral fracture in
rheumatoid arthritis patients with new-onset cardiovascular
diseases: A retrospective nationwide cohort study in Taiwan.
Osteoporos Int. 30:1617–1625. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nakajima T, Doi H, Watanabe R, Murata K,
Takase Y, Inaba R, Itaya T, Iwasaki T, Shirakashi M, Tsuji H, et
al: Factors associated with osteoporosis and fractures in patients
with systemic lupus erythematosus: Kyoto Lupus Cohort. Mod
Rheumatol. 34:113–121. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ekenstam EA, Ljunghall S and Hällgren R:
Serum osteocalcin in rheumatoid arthritis and other inflammatory
arthritides: Relation between inflammatory activity and the effect
of glucocorticoids and remission inducing drugs. Ann Rheum Dis.
45:484–490. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Both T, Zillikens MC, Schreuders-Koedam M,
Vis M, Lam WK, Weel A, van Leeuwen J, van Hagen PM, van der Eerden
BCJ and van Daele PLA: Hydroxychloroquine affects bone resorption
both in vitro and in vivo. J Cell Physiol. 233:1424–1433. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yu JJ, Wu F, Ma RR, Wu TJ, Zhang Y and
Ying ZH: The effects of iguratimod combined with methotrexate and
hydroxychloroquine on bone mineral density in patients with
rheumatoid arthritis. Pharmazie. 76:507–510. 2021.PubMed/NCBI
|
|
18
|
Park SJ, Sim SY, Jeong DC, Suh BK and Ahn
MB: Factors affecting bone mineral density in children and
adolescents with systemic lupus erythematosus. Ann Pediatr
Endocrinol Metab. 29:191–200. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Heidari B, Monadi M and Ghazi Mirsaed MA:
Bone mineral density changes during treatment of rheumatoid
arthritis with disease-modifying-anti-rheumatic drugs. Caspian J
Intern Med. 3:354–357. 2012.PubMed/NCBI
|
|
20
|
Carbone L, Vasan S, Elam R, Gupta S,
Tolaymat O, Crandall C, Wactawski-Wende J and Johnson KC: The
association of methotrexate, sulfasalazine, and hydroxychloroquine
use with fracture in postmenopausal women with rheumatoid
arthritis: Findings from the women's health initiative. JBMR Plus.
4:e103932020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Fischer VW and Fitch CD: Affinity of
chloroquine for bone. J Pharm Pharmacol. 27:527–529. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chaichit S, Sato T, Yu H, Tanaka YK, Ogra
Y, Mizoguchi T and Itoh M: Evaluation of Dexamethasone-induced
osteoporosis in vivo using zebrafish scales. Pharmaceuticals
(Basel). 14:5362021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aoki S, Shimizu K and Ito K:
Autophagy-dependent mitochondrial function regulates osteoclast
differentiation and maturation. Biochem Biophys Res Commun.
527:874–880. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin NY, Chen CW, Kagwiria R, Liang R,
Beyer C, Distler A, Luther J, Engelke K, Schett G and Distler JH:
Inactivation of autophagy ameliorates glucocorticoid-induced and
ovariectomy-induced bone loss. Ann Rheum Dis. 75:1203–1210. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yousefzadeh N, Kashfi K, Jeddi S and
Ghasemi A: Ovariectomized rat model of osteoporosis: A practical
guide. EXCLI J. 19:89–107. 2020.PubMed/NCBI
|
|
26
|
Xiong Y, Huang CW, Shi C, Peng L, Cheng
YT, Hong W and Liao J: Quercetin suppresses ovariectomy-induced
osteoporosis in rat mandibles by regulating autophagy and the NLRP3
pathway. Exp Biol Med (Maywood). 248:2363–2380. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xiu Y, Xu H, Zhao C, Li J, Morita Y, Yao
Z, Xing L and Boyce BF: Chloroquine reduces osteoclastogenesis in
murine osteoporosis by preventing TRAF3 degradation. J Clin Invest.
124:297–310. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liang X, Hou Z, Xie Y, Yan F, Li S, Zhu X
and Cai L: Icariin promotes osteogenic differentiation of bone
marrow stromal cells and prevents bone loss in OVX mice via
activating autophagy. J Cell Biochem. 120:13121–13132. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ke D, Wang Y, Yu Y, Wang Y, Zheng W, Fu X,
Han J, Zhang G and Xu J: Curcumin-activated autophagy plays a
negative role in its anti-osteoclastogenic effect. Mol Cell
Endocrinol. 500:1106372020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Al-Bari MAA, Shinohara M, Nagai Y and
Takayanagi H: Inhibitory effect of chloroquine on bone resorption
reveals the key role of lysosomes in osteoclast differentiation and
function. Inflammation and Regeneration. 32:222–231. 2012.
View Article : Google Scholar
|
|
31
|
Wang S, Feng W, Liu J, Wang X, Zhong L, Lv
C, Feng M, An N and Mao Y: Alginate oligosaccharide alleviates
senile osteoporosis via the RANKL-RANK pathway in
D-galactose-induced C57BL/6J mice. Chem Biol Drug Des. 99:46–55.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Imerb N, Thonusin C, Pratchayasakul W,
Arunsak B, Nawara W, Ongnok B, Aeimlapa R, Charoenphandhu N,
Chattipakorn N and Chattipakorn SC: D-galactose-induced aging
aggravates obesity-induced bone dyshomeostasis. Sci Rep.
12:85802022. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mahmoud MAA, Saleh DO, Safar MM, Agha AM
and Khattab MM: Chloroquine ameliorates bone loss induced by
d-galactose in male rats via inhibition of ERK associated
osteoclastogenesis and antioxidant effect. Toxicol Rep. 8:366–375.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Alam I, Gerard-O'Riley RL, Acton D,
Hardman SL, Hong JM, Bruzzaniti A and Econs MJ: Chloroquine
increases osteoclast activity in vitro but does not improve the
osteopetrotic bone phenotype of ADO2 mice. Bone. 153:1161602021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Teixeira CC, Liu Y, Thant LM, Pang J,
Palmer G and Alikhani M: Foxo1, a novel regulator of osteoblast
differentiation and skeletogenesis. J Biol Chem. 285:31055–31065.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Jiang Y, Luo W, Zhou F, Gong P and Xiong
Y: The role of FOXO1-mediated autophagy in the regulation of bone
formation. Cell Cycle. 22:829–840. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li Q, Yue T, Du X, Tang Z, Cui J, Wang W,
Xia W, Ren B, Kan S, Li C, et al: HSC70 mediated autophagic
degradation of oxidized PRL2 is responsible for osteoclastogenesis
and inflammatory bone destruction. Cell Death Differ. 30:647–659.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Topak D, Gürbüz K, Doğar F, Bakır E,
Gürbüz P, Kılınç E, Bilal Ö and Eken A: Hydroxychloroquine induces
oxidative stress and impairs fracture healing in rats. Jt Dis Relat
Surg. 34:346–355. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Önaloğlu Y, Beytemür O, Saraç EY, Biçer O,
Güleryüz Y and Güleç MA: The effects of hydroxychloroquine-induced
oxidative stress on fracture healing in an experimental rat model.
Jt Dis Relat Surg. 35:146–155. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tekçe G, Arıcan M, Karaduman ZO, Turhan Y,
Sağlam S, Yücel MO, Coşkun SK, Tuncer C and Uludağ V: Radiologic
and histopathologic effects of favipiravir and hydroxychloroquine
on fracture healing in rats. Naunyn Schmiedebergs Arch Pharmacol.
397:7857–7864. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xu G, Li X, Zhu Z, Wang H and Bai X: Iron
overload induces apoptosis and cytoprotective autophagy regulated
by ROS generation in Mc3t3-E1 cells. Biol Trace Elem Res.
199:3781–3792. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li Y, Yu P, Gao Y, Ma Z, Wang H, Long Y,
Ma Z and Liu R: Effects of the combination of Epimedii Folium and
Ligustri Lucidi Fructus on apoptosis and autophagy in SOP rats and
osteoblasts via PI3K/AKT/mTOR pathway. Biomed Pharmacother.
173:1163462024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Qiu Y, Zhao Y, Long Z, Song A, Huang P,
Wang K, Xu L, Molloy DP and He G: Liquiritigenin promotes
osteogenic differentiation and prevents bone loss via inducing
auto-lysosomal degradation and inhibiting apoptosis. Genes Dis.
10:284–300. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Pantovic A, Krstic A, Janjetovic K, Kocic
J, Harhaji-Trajkovic L, Bugarski D and Trajkovic V: Coordinated
time-dependent modulation of AMPK/Akt/mTOR signaling and autophagy
controls osteogenic differentiation of human mesenchymal stem
cells. Bone. 52:524–531. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhang Z, Lai Q and Li Y, Xu C, Tang X, Ci
J, Sun S, Xu B and Li Y: Acidic pH environment induces autophagy in
osteoblasts. Sci Rep. 7:461612017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhang Y, Zhang ZN, Li N, Zhao LJ, Xue Y,
Wu HJ and Hou JM: Nbr1-regulated autophagy in Lactoferrin-induced
osteoblastic differentiation. Biosci Biotechnol Biochem.
84:1191–1200. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yi L, Zhong T, Huang Y and Huang S:
Triiodothyronine promotes the osteoblast formation by activating
autophagy. Biophys Chem. 267:1064832020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhao X, Huang B, Wang H, Ni N, He F, Liu
Q, Shi D, Chen C, Zhao P, Wang X, et al: A functional autophagy
pathway is essential for BMP9-induced osteogenic differentiation of
mesenchymal stem cells (MSCs). Am J Transl Res. 13:4233–4250.
2021.PubMed/NCBI
|
|
49
|
Xu X, Wang R, Wu R, Yan W, Shi T, Jiang Q
and Shi D: Trehalose reduces bone loss in experimental biliary
cirrhosis rats via ERK phosphorylation regulation by enhancing
autophagosome formation. FASEB J. 34:8402–8415. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ni Y, Zhang H and Li Z and Li Z:
Connective tissue growth factor (CCN2) inhibits TNF-α-induced
apoptosis by enhancing autophagy through the Akt and Erk pathways
in osteoblasts. Pharmazie. 75:213–217. 2020.PubMed/NCBI
|
|
51
|
Liu W, Dai N, Wang Y, Xu C, Zhao H, Xia P,
Gu J, Liu X, Bian J, Yuan Y, et al: Role of autophagy in
cadmium-induced apoptosis of primary rat osteoblasts. Sci Rep.
6:204042016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Both T, van de Peppel HJ, Zillikens MC,
Koedam M, van Leeuwen J, van Hagen PM, van Daele PLA and van der
Eerden BCJ: Hydroxychloroquine decreases human MSC-derived
osteoblast differentiation and mineralization in vitro. J
Cell Mol Med. 22:873–882. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Hu J, Zeng XY, Song CC and Zhang L:
Lipopolysaccharide promotes the osteoclastogenesis through the
autophagic degradation of TNF receptor-associated factor 3.
Scienceasia. 48:697–704. 2022. View Article : Google Scholar
|
|
54
|
Sun J, Chen W, Li S, Yang S, Zhang Y, Hu
X, Qiu H, Wu J, Xu S and Chu T: Nox4 Promotes RANKL-induced
autophagy and osteoclastogenesis via activating
ROS/PERK/eIF-2α/ATF4 pathway. Front Pharmacol. 12:7518452021.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wu CH, Ou CH, Yen IC and Lee SY:
4-Acetylantroquinonol B inhibits osteoclastogenesis by inhibiting
the autophagy pathway in a simulated microgravity model. Int J Mol
Sci. 21:69712020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yao Z, Lei W, Duan R, Li Y, Luo L and
Boyce BF: RANKL cytokine enhances TNF-induced osteoclastogenesis
independently of TNF receptor associated factor (TRAF) 6 by
degrading TRAF3 in osteoclast precursors. J Biol Chem.
292:10169–10179. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Tong X, Gu J, Song R, Wang D, Sun Z, Sui
C, Zhang C, Liu X, Bian J and Liu Z: Osteoprotegerin inhibit
osteoclast differentiation and bone resorption by enhancing
autophagy via AMPK/mTOR/p70S6K signaling pathway in vitro. J Cell
Biochem. 120:1630–1642. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhao H, Sun Z, Ma Y, Song R, Yuan Y, Bian
J, Gu J and Liu Z: Antiosteoclastic bone resorption activity of
osteoprotegerin via enhanced AKT/mTOR/ULK1-mediated autophagic
pathway. J Cell Physiol. 235:3002–3012. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Song L, Tan J, Wang Z, Ding P, Tang Q, Xia
M, Wei Y and Chen L: Interleukin-17A facilitates osteoclast
differentiation and bone resorption via activation of autophagy in
mouse bone marrow macrophages. Mol Med Rep. 19:4743–4752.
2019.PubMed/NCBI
|
|
60
|
Ke D, Fu X, Xue Y, Wu H, Zhang Y, Chen X
and Hou J: IL-17A regulates the autophagic activity of osteoclast
precursors through RANKL-JNK1 signaling during osteoclastogenesis
in vitro. Biochem Biophys Res Commun. 497:890–896. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rieman DJ, McClung HA, Dodds RA, Hwang SM,
Holmes MW, James IE, Drake FH and Gowen M: Biosynthesis and
processing of cathepsin K in cultured human osteoclasts. Bone.
28:282–289. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Su B, Li D, Xu J, Zhang Y, Cai Z, Kauther
MD and Ma R: Wear particles enhance autophagy through up-regulation
of CD147 to promote osteoclastogenesis. Iran J Basic Med Sci.
21:806–812. 2018.PubMed/NCBI
|
|
63
|
Voronov I, Ochotny N, Jaumouillé V, Owen
C, Manolson MF and Aubin JE: The R740S mutation in the V-ATPase a3
subunit increases lysosomal pH, impairs NFATc1 translocation, and
decreases in vitro osteoclastogenesis. J Bone Miner Res.
28:108–118. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Huang Y, Gao X, An Y, Zeng P, Chen C, Ma W
and Yao X: Inhibitory effect of jinwujiangu prescription on
peripheral blood osteoclasts in patients with rheumatoid arthritis
and the relevant molecular mechanism. Mediators Inflamm.
2023:48144122023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Lee CK, Lee EY, Chung SM, Mun SH, Yoo B
and Moon HB: Effects of disease-modifying antirheumatic drugs and
antiinflammatory cytokines on human osteoclastogenesis through
interaction with receptor activator of nuclear factor kappaB,
osteoprotegerin, and receptor activator of nuclear factor kappaB
ligand. Arthritis Rheum. 50:3831–3843. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kar R, Riquelme MA, Hua R and Jiang JX:
Glucocorticoid-induced autophagy protects osteocytes against
oxidative stress through activation of MAPK/ERK signaling. JBMR
Plus. 3:e100772019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wong SK, Chin KY and Ima-Nirwana S:
Berberine and musculoskeletal disorders: The therapeutic potential
and underlying molecular mechanisms. Phytomedicine. 73:1528922020.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Ito Y, Inoue D, Kido S and Matsumoto T:
c-Fos degradation by the ubiquitin-proteasome proteolytic pathway
in osteoclast progenitors. Bone. 37:842–849. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Okusha Y, Tran MT, Itagaki M, Sogawa C,
Eguchi T, Okui T, Kadowaki T, Sakai E, Tsukuba T and Okamoto K:
Rab11A functions as a negative regulator of osteoclastogenesis
through dictating lysosome-induced proteolysis of c-fms and RANK
surface receptors. Cells. 9:23842020. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tran MT, Okusha Y, Feng Y, Morimatsu M,
Wei P, Sogawa C, Eguchi T, Kadowaki T, Sakai E, Okamura H, et al:
The inhibitory role of Rab11b in osteoclastogenesis through
triggering lysosome-induced degradation of c-Fms and RANK surface
receptors. Int J Mol Sci. 21:93522020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Florencio-Silva R, Sasso GR, Simões MJ,
Simões RS, Baracat MC, Sasso-Cerri E and Cerri PS: Osteoporosis and
autophagy: What is the relationship? Rev Assoc Med Bras (1992).
63:173–179. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Hansen M, Rubinsztein DC and Walker DW:
Autophagy as a promoter of longevity: Insights from model
organisms. Nat Rev Mol Cell Biol. 19:579–593. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wong SK, Chin KY and Ima-Nirwana S: The
osteoprotective effects of kaempferol: The evidence from in vivo
and in vitro studies. Drug Des Devel Ther. 13:3497–3514. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Wang J, Zhang Y, Cao J, Wang Y, Anwar N,
Zhang Z, Zhang D, Ma Y, Xiao Y, Xiao L, et al: The role of
autophagy in bone metabolism and clinical significance. Autophagy.
19:2409–2427. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Yin X, Zhou C, Li J, Liu R, Shi B, Yuan Q
and Zou S: Autophagy in bone homeostasis and the onset of
osteoporosis. Bone Res. 7:282019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang P, Liao J, Wang X and Feng Z: High
glucose promotes apoptosis and autophagy of MC3T3-E1 osteoblasts.
Arch Med Sci. 19:138–150. 2023.PubMed/NCBI
|
|
77
|
Ni Y, Zhang H, Zhang J and Li Z and Li Z:
Inhibition of JAK2 by AG490 promotes TNF-α-induced apoptosis by
inhibiting autophagy in MC3T3-E1 cells. Pharmazie. 75:255–260.
2020.PubMed/NCBI
|
|
78
|
Liu F, Yuan Y, Bai L, Yuan L, Li L, Liu J,
Chen Y, Lu Y, Cheng J and Zhang J: LRRc17 controls BMSC senescence
via mitophagy and inhibits the therapeutic effect of BMSCs on
ovariectomy-induced bone loss. Redox Biol. 43:1019632021.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yang Q, Zou Y, Wei X, Ye P, Wu Y, Ai H,
Zhang Z, Tan J, Zhou J, Yang Y, et al: PTP1B knockdown alleviates
BMSCs senescence via activating AMPK-mediated mitophagy and
promotes osteogenesis in senile osteoporosis. Biochim Biophys Acta
Mol Basis Dis. 1869:1667952023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hu Y, Carraro-Lacroix LR, Wang A, Owen C,
Bajenova E, Corey PN, Brumell JH and Voronov I: Lysosomal pH plays
a key role in regulation of mTOR activity in osteoclasts. J Cell
Biochem. 117:413–425. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wong SK, Chin KY and Ima-Nirwana S:
Quercetin as an agent for protecting the bone: A review of the
current evidence. Int J Mol Sci. 21:64482020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wong SK, Mohamad NV, Ibrahim N, Chin KY,
Shuid AN and Ima-Nirwana S: The molecular mechanism of Vitamin E as
a bone-protecting agent: A review on current evidence. Int J Mol
Sci. 20:14532019. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Li X, Xu J, Dai B, Wang X, Guo Q and Qin
L: Targeting autophagy in osteoporosis: From pathophysiology to
potential therapy. Ageing Res Rev. 62:1010982020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Askarian F, Firoozi Z, Ebadollahi-Natanzi
A, Bahrami S and Rahimi HR: A review on the pharmacokinetic
properties and toxicity considerations for chloroquine and
hydroxychloroquine to potentially treat coronavirus patients.
Toxicol Res. 38:137–148. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Browning DJ: Pharmacology of chloroquine
and hydroxychloroquine. Hydroxychloroquine and Chloroquine
Retinopathy. Springer Nature. 35–63. 2014.
|
|
86
|
Stokkermans TJ, Falkowitz DM and Trichonas
G: Chloroquine and Hydroxychloroquine Toxicity. Treasure Island:
StatPearls Publishing; 2024
|
|
87
|
Ruamviboonsuk P, Lai TYY, Chang A, Lai CC,
Mieler WF and Lam DSC: Chloroquine and hydroxychloroquine retinal
toxicity consideration in the treatment of COVID-19. Asia Pac J
Ophthalmol (Phila). 9:85–87. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Cohen IV, Makunts T, Moumedjian T, Issa MA
and Abagyan R: Cardiac adverse events associated with chloroquine
and hydroxychloroquine exposure in 20 years of drug safety
surveillance reports. Sci Rep. 10:191992020. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
National Institute for Health and Care
Excellence, . Guidelines, in Osteoporosis: assessing the risk of
fragility fracture. https://www.nice.org.uk/guidance/cg146April
29–2024
|
|
90
|
Jehoon O, Kwon HJ, Cho TH, Woo SH, Rhee YH
and Yang HM: Micro-computed tomography with contrast enhancement:
An excellent technique for soft tissue examination in humans. PLoS
One. 16:e02542642021. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Greenblatt MB, Tsai JN and Wein MN: Bone
turnover markers in the diagnosis and monitoring of metabolic bone
disease. Clin Chem. 63:464–474. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
LeBoff MS, Greenspan SL, Insogna KL,
Lewiecki EM, Saag KG, Singer AJ and Siris ES: The Clinician's guide
to prevention and treatment of osteoporosis. Osteoporos Int.
33:2049–2102. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Stevens DM, Crist RM and Stern ST:
Nanomedicine reformulation of chloroquine and hydroxychloroquine.
Molecules. 26:1752020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Al-Bari MA: Chloroquine analogues in drug
discovery: New directions of uses, mechanisms of actions and toxic
manifestations from malaria to multifarious diseases. J Antimicrob
Chemother. 70:1608–1621. 2015. View Article : Google Scholar : PubMed/NCBI
|