Introduction
Post-translational modifications (PTMs) refer to the
covalent alterations of proteins that occur during or after their
biosynthesis, typically catalyzed by enzymes (1). These modifications encompass the
addition of functional groups (such as phosphorylation, methylation
and acetylation), the conjugation of other proteins or peptides
(such as ubiquitination), chemical modifications of amino acid (AA)
residues (such as deamidation) and structural changes (such as
disulfide bridge formation) (2,3).
Ubiquitination, one of the most prevalent PTMs, involves a highly
conserved protein composed of 76 amino acids with a molecular
weight of approximately 8.5 kDa, which is ubiquitously present in
all eukaryotic cells (4,5). Ubiquitin is conjugated to its target
proteins through a complex enzymatic reaction cascade, consisting
of an activating enzyme (E1), a conjugating enzyme (E2) and a
specific ligase (E3), playing a pivotal role in the regulation of
eukaryotic cellular processes (6,7).
Ubiquitin fold modifier 1 (UFM1), a member of the
ubiquitin-like (UBL) protein family (8,9), is
composed of 85 amino acids and has a molecular weight of 9.1 kDa.
Despite limited sequence homology, UFM1 and ubiquitin exhibit
analogous tertiary structures and conceptually similar enzymatic
pathways, involving E1, E2 and E3 enzymes, which ultimately lead to
the covalent attachment of UFM1 to the lysine residues of
substrates through its C-terminal glycine. Akin to other UBLs, UFM1
is synthesized as a precursor protein that requires proteolytic
cleavage to generate its active mature form (10).
UFM1 possesses the ability to form polymeric chains
due to the presence of five lysine residues; however, these chains
predominantly connect through lysine 69 (10). In contrast to ubiquitination, which
involves numerous enzymes with overlapping functions, UFMylation is
characterized by a more restricted set of cellular mechanisms,
demonstrating high specificity. The maturation, activation,
attachment and removal of UFM1 are facilitated by only a few
ubiquitously expressed enzymes, specifically UBA5 (E1), UFC1 (E2),
UFL1 (E3) and UFSP1 and UFSP2 (10–14).
Several proteins associated with UFMylation serve
various scaffolding and targeting functions, including UFM1 binding
protein-1 (UFBP1), cyclin-dependent kinase 5 regulatory
subnit-associated protein 3 and retinal nerve fibre layer/ODR4.
These proteins ensure that enzymatic activities are accurately
directed and compartmentalized within relevant cellular
environments (15). Compared with
the more complex and redundant mechanisms governing ubiquitination,
this unique regulatory framework underscores the specialized role
of UFM1 in cellular processes (16).
UFMylation, a post-translational modification
identified a decade ago (17),
remains incompletely understood and its biological significance
warrants further investigation. Initial studies on UFM1 substrates
identified UFBP1 and ASC1 through in vitro assays (18,19);
however, the majority of UFM1-modified proteins remain
uncharacterized. With the advent of advanced screening technologies
and purification methods, new UFM1 substrates have been identified,
providing molecular-level insights into the structure and function
of UFMylation (9,20,21).
These discoveries have expanded its mechanistic roles in key
cellular processes, including the DNA damage response, endoplasmic
reticulum homeostasis, ribosomal quality control and hematopoietic
differentiation, establishing UFMylation as a crucial regulator of
cellular homeostasis (21,22). Its dysregulation has been linked to
the development of various human diseases, for example, severe
anemia (18), skeletal diseases
such as Sohat spondyloepiphyseal dysplasia (SEMD) (23) and neurological diseases such as
early-onset cerebellar atrophy and early-onset intractable epilepsy
(24,25). The present review refined the
current understanding of the enzymatic reactions underlying
UFMylation, elucidated its catalytic mechanisms and explored newly
emerging regulatory pathways that contribute to cellular
homeostasis. Furthermore, it comprehensively elucidated the
specific roles and novel molecular mechanisms of the UFM1
conjugation system in various cancers and antitumor immunity, for
instance, the loss of UFL1 in T cells inhibits the UFMylation of
programmed cell death protein 1 (PD-1), thereby promoting the
production of effector cytokines in CD8+ T cells and
enhancing their antitumor efficacy. These findings may provide new
strategies for identifying cancer diagnostic biomarkers and
clinical therapeutic targets in the future.
Roles of different proteases in
ubiquitin-like modifications
UFM1, like most UBL proteins, is expressed as a
precursor that must be proteolytically cleaved to generate the
active mature form (26). However,
its precursor features a unique C-terminal Ser84-Cys85 dipeptide
sequence, unlike the conserved C-terminal di-Gly (Gly-Gly) motif
found in most UBLs. The proteases UFSP1 and UFSP2, purified from
tissue extracts using His-GST-UFM1-Escontin as a substrate, have
been identified as the enzymes responsible for processing the UFM1
precursor and facilitating the removal of UFM1 modifications
(27).
UFSP1, an ~25 kDa family member protein, is present
in flies, mice and humans but absent in plants and nematodes. By
contrast, UFSP2, which is >40 kDa, is found in most
multicellular organisms, including Caenorhabditis elegans
and Arabidopsis thaliana (28). Despite sharing the same catalytic
mechanism, UFSP1 and UFSP2 exhibit significant structural
differences, particularly in the R-helix domain responsible for
recognizing and binding the UFM1 precursor. In UFSP1, the R loop
connecting β3 and β4, along with Trp98, is stabilized by
interactions with water molecules and residues connecting the α6
helices and β7 strands. Conversely, in UFSP2, the R loop connecting
β9, β10 and Trp342 shows no significant interactions apart from
hydrophobic interactions between Trp342 and Val395 (29).
Thus, a crucial function of UFSPs is to
proteolytically process the UFM1 precursor, producing mature UFM1.
The removal of the C-terminal serine and cysteine residues exposes
the C-terminal glycine of UFM1, enabling its maturation and
subsequent binding to substrates (30,31).
Upon maturation, UFM1 undergoes activation through a
trans-binding mechanism involving UBA5 dimerization. UBA5, the sole
E1 enzyme for UFMylation, is primarily localized in the cytoplasm.
Unbound UFM1-UBA5 dimers exhibit weak dimerization; however, UFM1
binding stabilizes the UBA5 dimer conformation and enhances its
affinity for ATP (32).
Specifically, two UFM1 molecules and two UBA5 molecules form an
interlocking structure, with each UFM1 molecule interacting with
one UFM1-interacting sequence (UIS) molecule at one end, while
Gly83 at the other end forms a high-energy thioester bond with
Cys250 of UBA5. ATP binding to the protomers in the UBA5 dimer
enables the charging of UFM1, forming an activated complex carrying
two UFM1 molecules (33). The
dimeric UBA5 is essential not only for UFM1 activation but also for
transferring UFM1 to UFC1. This highlights the critical
interdependence of the two UBA5 monomers for adenylation and UIS
domain structure (13,32).
Following activation, UFM1 is transferred from UBA5
to Cys116 of UFC1 through a trans-thiolation reaction, which
involves interactions between the UBA5-UFM1 dimer and UFC1
(34). Unlike UBA5, UFC1 is
primarily localized in the nucleus, with only partial cytoplasmic
localization. Intriguingly, while the charging process of UBA5
requires ATP, the transfer of UFM1 from UBA5 to UFC1 does not
(35). During this
trans-thiolation reaction, one thioester bond (UBA5-UFM1) is
cleaved and another (UFC1-UFM1) is formed, suggesting that energy
transfer can occur bidirectionally, with both directions being
energetically equivalent (36).
The reaction direction is determined by the concentrations of UBA5,
UFC1 and UFM1; overexpression of UBA5 can reverse the energy
transfer from activated UFM1 in UFC1 back to UBA5, indicating the
reversibility of E2 transfer. In the normative direction (E1-E2),
UFC1 binds to a tetrameric complex composed of two UFM1 molecules
and two UBA5 molecules, employing a similar trans-binding mechanism
that necessitates interaction with one UBA5 monomer and the receipt
of activated UFM1 from another (37).
The final step involves the transfer of UFM1 from
UFC1 to covalently link to the substrate's lysine residue, mediated
by the E3 enzyme UFL1 (38). UFL1
deficiency results in the loss of UFMylation and mice lacking UFL1
exhibit hematopoietic failure and embryonic lethality, suggesting
its role as the primary or sole E3 ligase. Although UFL1′s role in
UFMylation is well established, it does not directly mediate the
process. Instead, it forms a functional heterodimeric E3 ligase
complex with adapter proteins, such as UFBP1 and CDK5RAP3,
classifying it as a scaffold-type E3 ligase (16,39).
Scaffold-type E3 ligases recognize substrates and facilitate UFM1
transfer from UFC1 to the substrate. The E2 enzyme confers linkage
specificity at K69 during dual UFM1 formation in the absence of E3
ligase, indicating that this specificity is determined by the E2
enzyme. The E2-E3 complex can exert various effects on the
substrate, ranging from proteasome-dependent protein proteolysis to
the regulation of protein function, structure, assembly, or
localization (40,41). Following these processes, UFSP2 can
recycle the UFM1 linked to the substrate, allowing it to enter the
next UFMylation cycle (42)
(Fig. 1).
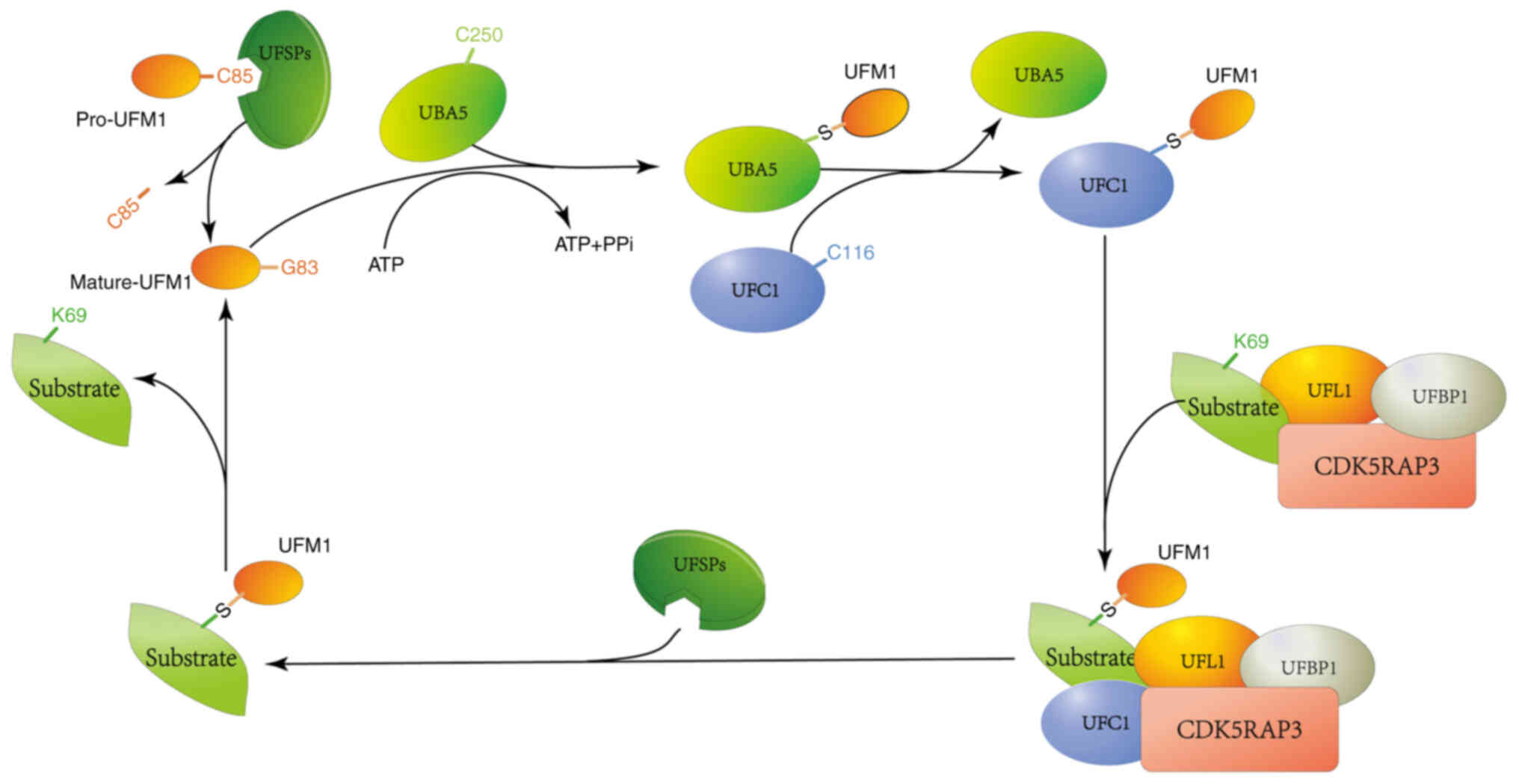 | Figure 1.The role of different proteases in
UFMylation. The UFM1 precursor undergoes proteolytic processing by
UFSP1, which cleaves the Ser84-Cys85 dipeptide to expose Gly83,
yielding mature UFM1. Subsequently, Gly83 of UFM1 forms a
high-energy thioester bond with Cys250 of UBA5, stabilizing the
dimeric conformation of UBA5 and enhancing its affinity for ATP.
The UBA5-UFM1 dimer then interacts with the UFC1, facilitating the
transfer of UFM1 from UBA5 to UFC1. This reaction involves the
cleavage of one thioester bond (Gly83-Cys250) and the formation of
another (Gly83-Cys116). Finally, UFM1 is covalently conjugated to a
lysine residue (K69) on its substrate through an E3 ligase complex,
comprising the ligase UFL1 and its adaptor protein. The cycle is
completed when UFSP2 cleaves UFM1 from the substrate, allowing it
to re-enter the UFMylation pathway. UFM1, ubiquitin fold modifier
1; UFSP1, UFM1 specific peptidase 1; UBA5, ubiquitin-activating
enzyme 5; UFC1, ubiquitin fold modifier 1; UFSP2, UFM1 specific
peptidase 2. |
The functions of UFMylation in cells
The UFMylation pathway has been demonstrated to
regulate a wide range of cellular activities. These include DNA
damage repair, endoplasmic reticulum stress response, ribosome
modification, hematopoiesis, immune regulation (20) and neurodevelopment (43–47).
UFMylation and the maintenance of genomic
integrity in the cell nucleus
The genome of the cell is continually exposed to
attacks from both exogenous and endogenous DNA-damaging factors,
such as radiation, carcinogens and reactive free radicals. To
maintain genomic stability, cells have evolved a complex DNA damage
response (DDR) system. This system is responsible for sensing DNA
damage, halting the cell cycle and initiating repair processes.
Failure to detect or repair DNA damage can lead to genomic
instability, a hallmark of tumorigenesis (30).
Double-strand breaks (DSBs) represent the most toxic
form of DNA damage and their repair is primarily initiated by ATM
kinase (48). The activation of
ATM is mediated through the meiotic recombination 11 homolog
(MRE11)-radiation sensitive 50-Nijmegen breakage syndrome 1 (NBS1)
(MRN) complex, which phosphorylates Ser1981 and acetylates Lys3106.
Once activated, the ATM kinase rapidly phosphorylates local
chromatin, providing a scaffold for the assembly of higher-order
complexes that facilitate DNA repair. Subsequent studies have
revealed that the activation of ATM also involves the UFMylation
pathway. Following DNA damage, MRE11 undergoes UFMylation at
Lys282, a modification that is crucial for the assembly of the MRN
complex. This UFMylation event is essential for optimal ATM
activation, homologous recombination-mediated repair and the
maintenance of genomic integrity (49).
DNA damage induces the UFMylation of MRE11, which
facilitates the recruitment of the MRN complex to the damage site.
This process helps relieve the self-inhibition of ATM kinase at
DSBs, thereby promoting DSB repair and enhancing chromosomal
stability (45,46). In addition to MRE11, the UFMylation
of histone H4 has also been implicated in ATM activation. The MRN
complex recruits UFL1 to DNA DSBs, where UFL1 catalyzes the release
of histone H4 at Lys31, enhancing the recruitment of the SUV39H1
complex to DSBs (50). SUV39H1
induces H3K9me3 modification at DSBs, which can spread over
thousands of bases, forming a temporary repressive heterochromatic
domain (39). Subsequently, Tip60
is recruited to bind H3K9me3 and this interaction enhances its
acetyltransferase activity. This leads to the acetylation of Tip60
and subsequent activation of ATM at DSBs (51,52).
Related studies have identified the
nuclear-localized UFMylated protein P53, with endogenous UFMylation
being detectable in both human cancer cells and primary mouse
embryonic fibroblasts. In vitro UFMylation assays have
demonstrated that the UFMylation components UBA5, UFC1, UFL1, UFM1
and DDRGK1 (also known as UFM1-binding protein 1, UFBP1) are all
necessary for the modification of P53 (53). The absence of the amino (N)
terminal region of P53 prevents its binding to UFL1, indicating
that this region is crucial for UFL1 interaction. Notably, this
region can also bind to mouse double minute 2 homologue (MDM2), the
primary E3 ligase responsible for P53 degradation. Consequently,
UFL1 competes with MDM2 for binding to P53, thus contributing to
its stability (54,55). Notably, UFMylated P53 can be
detected in cells lacking UBA5 overexpression, provided that UFC1,
UFL1 and DDRGK1 are overexpressed. Conversely, UFMylated P53 levels
decrease in cells where UFC1, UFL1 and DDRGK1 have been knocked
out, while knocking out UBA5 has no significant effect. This
suggests that while UBA5 is essential for P53 UFMylation, it is not
a limiting factor in the cellular context (47).
Furthermore, UFMylation has been reported to play an
unanticipated role in maintaining telomere length.
UFMylation-modified MRE11 promotes the recruitment of PP1-α to the
MRN complex and facilitates the dephosphorylation of NBS1. This
process aids in releasing the MRN complex from telomeres and allows
Apollo to bind to TRF2. In the absence of UFMylation-modified
MRE11, NBS1 remains phosphorylated, leading to reduced MRN
recruitment at the telomeres (56,57).
The absence of MRN at the telomeres favors the formation of the
TRF2/SNM1 complex, ultimately resulting in the shortening of the
telomeric leading strand length (44).
As a co-activator of nuclear receptors, the
UFMylation modification of ASC1 is a pivotal step in the
transactivation of estrogen receptor α (ERα) by 17β-estradiol (E2).
In the absence of E2, the UFM1-specific protease UFSP2 binds to
ASC1, maintaining it in a non-UFMylated state (43,58).
However, in the presence of E2, ERα binds to ASC1, displacing UFSP2
and leading to ASC1 activation. The subsequent poly-UFMylation of
ASC1 enhances the recruitment of P300, SRC1 and ASC1 to the
promoters of ERα target genes. Overexpression of ASC1 or knockdown
of UFSP2 promotes ERα-mediated tumor formation in vivo,
which can be attenuated by treatment with the anti-breast cancer
drug tamoxifen (18,43). These findings underscore the
critical role of UFMylation in regulating genomic stability and its
potential implications in breast cancer progression.
UFMylation and endoplasmic reticulum
homeostasis
To maintain endoplasmic reticulum (ER) homeostasis,
cells have evolved a sophisticated protein quality control system
comprising various pathways. These pathways include
ribosome-associated quality control (RQC), endoplasmic
reticulum-associated degradation (ERAD), the unfolded protein
response (UPR) and ER-phagy (59–61).
In eukaryotic cells, a primary cause of ribosome
stalling due to erroneous translation is naturally occurring faulty
mRNA. These aberrant translation products can interfere with
functional proteins and are detrimental to the cell. The RQC
pathway effectively eliminates these stalled products in the
cytoplasm when ribosomes become inoperative (62,63).
This pathway employs a series of coordinated factors that sense
translation stalling, rescue stalled ribosomes and degrade abnormal
nascent polypeptides. Specifically, ribosome stalling during
protein translocation induces the attachment of the ubiquitin-like
modifier UFM1 to two conserved lysine residues near the COOH
terminus of the ER 60S ribosomal subunit RPL26 (uL24) (64,65).
Notably, RPL26-UFMylation facilitates the degradation of stalled
nascent chains. However, unlike the ERAD or previously established
RQC mechanisms utilizing proteasomal degradation, ribosomal
UFMylation directs translocation-stalled ER proteins for lysosomal
degradation. In summary, UFMylation plays a critical role in
regulating protein homeostasis within quality control pathways
(66).
ERAD, a protein degradation system located in the
ER, removes unfolded or misfolded proteins for proteasomal
degradation (67). Proteins that
fail to achieve their native conformation are targeted for
degradation through the ERAD pathway via a series of closely
coupled steps: substrate recognition, retrotranslocation to the
cytosol and ubiquitination for proteasomal degradation. HRD1
(HMG-CoA reductase degradation protein, also known as SYVN1) serves
as one of the key ubiquitin ligases for ERAD. HRD1 has been shown
to target various proteins, including the tumor suppressor tumor
protein p53, programmed death-ligand 1 (PD-L1), peroxisome
proliferator-activated receptor 1β, inositol-requiring enzyme 1
alpha (IRE1α), cyclic amp-responsive element-binding, lipoprotein
lipase, proopiomelanocortin, sigma non-opioid intracellular
receptor 1, ATP citrate lyase, nuclear factor erythroid 2-related
factor 2 and the rate-limiting acyltransferases
glycerol-3-phosphate acyltransferase, monoacylglycerol and
diacylglycerol O-acyltransferase 2 The regulation of these proteins
by HRD1 affects cellular physiological and pathological processes
through the degradation of misfolded proteins (68).
Under normal conditions, cells regulate the ERAD
clearance of misfolded ER-resident proteins through the UFMylation
modification of HRD1. However, during ER stress, the accumulation
of misfolded proteins within the ER triggers stress signals that
cause the dissociation of the DDRGK1-UFL1 complex from HRD1. This
dissociation diminishes HRD1 UFMylation, thereby inhibiting its
function and initiating the UPR to mitigate ER stress. In this
model, the removal of UFL1, HRD1, or the expression of HRD1-K610R
in HRD1 knockout cells facilitates the activation of the UPR,
particularly the IRE1α-XBP1 signaling pathway, which fine-tunes the
folding capacity of the ER (68).
Although ER-phagy is a form of selective autophagy,
it involves additional selectivity in the degradation of specific
ER proteins and subdomains (60).
Misfolded proteins in the ER are typically targeted for degradation
via the ERAD pathway; however, some misfolded proteins cannot enter
this pathway due to their inability to bind to the ERAD machinery
or their tightly packed structure that prevents unfolding. These
proteins ultimately accumulate in the ER and are delivered as cargo
to isolation vesicles known as autophagosomes, which subsequently
fuse with lysosomes in a process termed ER-phagy (69,70).
In summary, ER subdomains containing these abnormal proteins
exhibit affinity for ATG8 family proteins or FIP200, ensuring
selectivity for the autophagic degradation of the ER. These
proteins are expressed in specific tissues and localized to
different ER subdomains, such as sheet-like ER, tubular ER and
three-way junctions, facilitating the formation of autophagosomes
and undergoing ER-phagy (71–73).
In macro-ER-phagy, isolation membranes or
phagophores form along the ER marked for degradation (74). The localization of the E3 ligase
complex at the ER is crucial for the autophagic degradation of the
ER, with UFL1 and UFBP1 playing significant roles. The UFM1
substrate modification of NADH-cytochrome B5 reductase 3 (CYB5R3)
is dependent on the E3 ligase complex UFL1 and UFBP1, which are
located on the ER membrane (75,76).
The UFM1 binding site, Lys214, is situated at the interface between
the NADH and FAD domains of CYB5R3, resulting in the disruption of
the FAD domain and rendering CYB5R3 inactive. UFMylated CYB5R3 is
recognized by UFBP1, which is essential for further UFMylation of
CYB5R3 and enhances the E3 ligase activity of the UFL1-UFBP1
complex towards CYB5R3. Ultimately, the interaction of the UFL1
complex with UFMylated CYB5R3 and UFBP1 leads to the autophagic
degradation of ER subdomains (9,64,66,77).
In plasma cells, the IRE1α/XBP1 axis within the UPR
pathway upregulates Ufbp1 expression, which is critical for the
expansion of the ER network (75,78).
Simultaneously, UFBP1 plays a pivotal role in suppressing the PERK
pathway. This suppressive relationship is demonstrated by the
observation that PERK deletion restores the defects in B-cell to
plasma cell development caused by UFBP1 deficiency. Moreover, the
expression of UFBP1 and other molecules involved in the UFMylation
pathway in immature B cells is independent of IRE1α/XBP1 (75,79).
Thus, UFBP1 markedly regulates different branches of the UPR
pathway to promote plasma cell development and function.
Specifically, the IRE1α/XBP1 axis upregulates UFBP1 and genes in
the UFMylation pathway in plasma cells, while UFBP1 deficiency
impairs ER expansion and hinders immunoglobulin production
(53,80). Structural and functional analyses
reveal that Lys267 in UFBP1 is a critical lysine residue. Although
not essential for plasma cell development, it is vital for
immunoglobulin production and stimulating ER expansion in
IRE1α-deficient plasma cells. In summary, UFBP1 exerts differential
impacts on the development and function of plasma cells by
regulating distinct steps in the UPR pathway (79,81).
Relevant studies have combined genetic disruption of
UFMylation and de-UFMylation with affinity capture liquid
chromatography-tandem mass spectrometry (LC-MS/MS) techniques,
applying three consecutive filtration steps: i) Low-confidence hits
(<3 unique peptides in UFSP2 knockout), ii) proteins whose
abundance did not increase after UFSP2 knockout and iii) proteins
whose abundance increased following UBA5 knockout, to identify the
conserved ribosomal protein RPL26. Subsequently, genetic
manipulation using small interfering siRNA depletion or
CRISPR-Cas9-mediated cell line models was employed to investigate
the UFMylation process of RPL26 (65,77).
Although the absence of UFL1 or its interaction with UFBP1
eliminates RPL26 UFMylation, the UFMylation of RPL26 at two
distinct lysine residues (K132 and K134) is markedly increased in
UFSP2-/- cell lines (77,82). This modification is continuous;
overexpression of RPL26 K134R in UFSP2-/- cells abolishes both
monomeric and dimeric RPL26, while monomeric RPL26 is still present
in cells overexpressing RPL26 K132R. In conditions where UFMylation
is not dominant, such as in UFSP haploinsufficient cell lines
(UFSP1-/+/UFSP2-/-), only K134 modifications are observed,
indicating that the modification of RPL26 is highly specific.
Moreover, replacing endogenous RPL26 with a mutant allele lacking
both residues (RPL26K132R/K134R) abolishes the modification of
RPL26 (63,64,83).
This further indicates that the ribosomal RPL26 undergoes a dynamic
cycle of UFMylation and de-UFMylation catalyzed by enzymes attached
to the cytoplasmic surface of the ER, providing a functional link
between the UFMylation process and ER protein homeostasis (64,66).
In eukaryotes, secretory pathway proteins are
primarily synthesized by ribosomes bound to the ER and inserted
into the ER lumen or integrated into membranes via the Sec61
translocon (63,84,85).
This process is highly sensitive to disturbances such as
translation stalling or defects in protein modifications, folding
and assembly, all of which can lead to defective polypeptides that
clog the translocon. The clogging of translocons triggers ribosome
filtration and activates transport-associated quality control
(TAQC) to degrade the clogged substrates (86,87).
Wang et al (88) identified
a membrane protein named SAYSD1 through whole-genome CRISPR-Cas9
screening, which aids in the TAQC process by binding to SEC61.
SAYSD1 also directly recognizes ribosomes and UFM1, associating
with stalled nascent chains to ensure their transport to lysosomes
for degradation via the TRAPP complex. Similar to UFM1 deficiency,
depletion of SAYSD1 leads to the accumulation of proteins stalled
in the ER, inducing ER stress. Notably, disrupting UFM1- and
SAYSD1-dependent TAQC in Drosophila results in the
accumulation of collagen that is stalled during translocation,
leading to collagen deposition defects, abnormal basement membrane
integrity and reduced stress tolerance (89). Thus, SAYSD1 functions as a sensor
for UFM1, coordinating the ribosomal UFMylation of blocked
translocon sites to maintain ER homeostasis during animal
development (88) (Fig. 2).
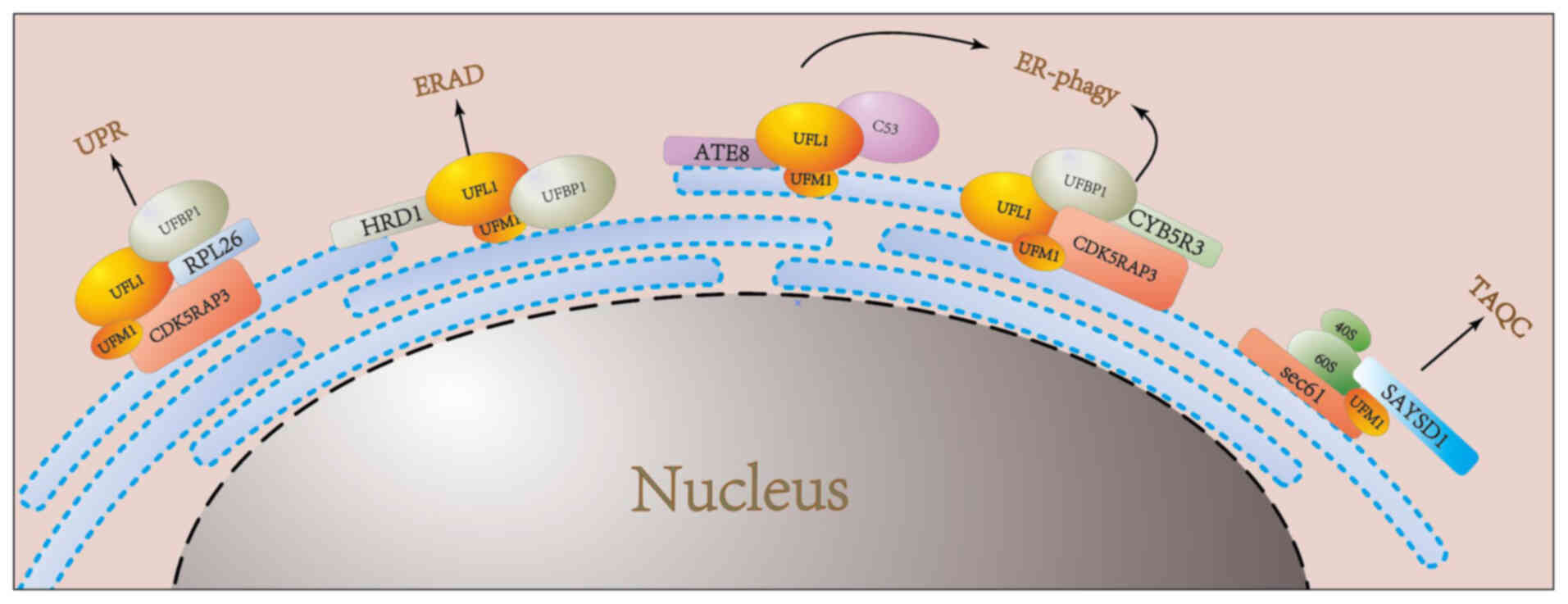 | Figure 2.UFMylation regulates the process of
the endoplasmic reticulum. In the UPR pathway, ribosomal protein
RPL26 undergoes a dynamic cycle of UFMylation and de-UFMylation,
catalyzed by Ufbp1 and UFL1 at the ER membrane. This regulatory
cycle is essential for maintaining ER homeostasis. Under
physiological conditions, cells modulate the ERAD of misfolded
ER-resident proteins through the UFMylation of HRD1, a process
mediated by the DDRGK1-UFL1 complex. During ER-phagy, the
UFL1-UFBP1 complex facilitates the UFMylation of CYB5R3, promoting
the autophagic degradation of specific ER subdomains. Additionally,
these ER subdomains exhibit affinity for ATG8 family proteins or
FIP200, ensuring the selective degradation of ER components. In the
TAQC pathway, SAYSD1 acts as a UFM1 sensor, directly recognizing
ribosomes and facilitating their transport to lysosomes for
degradation via the TRAPP complex. ER, endoplasmic reticulum; UPR,
unfolded protein response; RPL26, ribosomal protein L26; UFL1, UFM1
specific ligase 1; ERAD, ER–associated degradation; HRD1, HMG-CoA
reductase degradation 1; DDRGK1, DDRGK domain containing 1; UFBP1,
Ufm1 binding protein 1; CYB5R3, cytochrome B5 reductase 3; ATG8,
autophagy-related protein 8; FIP200, focal adhesion kinase family
interacting protein of 200kD; SAYSD1, SAYSVFN motif domain
containing 1; TRAPP, trafficking protein particle complex. |
UFMylation and hematopoiesis
Hematopoiesis, a complex process regulated by
specific transcription factors and cytokines, supports the
self-renewal, differentiation and survival of hematopoietic
progenitor cells at various stages of maturation (90,91).
The UFMylation system plays a pivotal role in modulating the
expression levels and activation of transcription factors that
facilitate erythropoiesis. It is crucial to identify UFM1 targets
for understanding how UFM1 protein modification governs erythroid
development, as defects in this process can lead to pathological
conditions, such as leukemia and myelodysplastic syndromes.
Germline deletions of UBA5, UFL1, or UFBP1 in mice
result in embryonic lethality and disrupted hematopoietic
development (92,93). UBA5 and other genes involved in
UFMylation are expressed in both primitive and definitive
erythrocytes, with the highest levels of UBA5 found in primitive
erythroid lineages. Mice deficient in UBA5 exhibit severe anemia
due to defects in megakaryocyte and erythrocyte differentiation,
ultimately resulting in death in utero. This deficiency
impairs the differentiation of common bone marrow progenitors into
megakaryocytes and erythroid progenitors. Notably, transgenic
expression of UBA5 in the erythroid lineage can rescue embryos from
anemia, extending their survival (92).
UFL1 is essential for embryonic development,
hematopoietic stem cell (HSC) survival and erythroid
differentiation. Knockout of UFL1 leads to reduced HSC survival
rates and embryonic death due to severe anemia (94). UFL1 is considered to function as an
E3 ligase for UFM1, promoting the UFMylation of DDRGK1 and ASC1.
Notably, both UBA5 and UFL1 knockout mice display extensive
phenotypic similarities, including defects in embryonic
erythropoiesis and increased mortality (93). Specifically, UBA5 deficiency
disrupts the development of megakaryocyte-erythroid progenitors
(MEP) in the fetal liver without affecting granulocyte-monocyte
progenitors (GMP). Conversely, conditional deletion of RCAD in
adult mice causes defects in MEP progenitors but does not affect
GMP. Additionally, a significant number of multinucleated erythroid
cells are observed in the embryos of both UBA5 and RCAD-deficient
mice, suggesting that UBA5 and RCAD may function in similar
cellular processes or signaling pathways during erythroid
development (90).
UFBP1 is also critical for embryonic development and
hematopoiesis; its deficiency results in defects in erythroid
development and embryonic death in mice. In adult mice, UFBP1
deficiency leads to impaired hematopoiesis, resulting in
pancytopenia and increased mortality (18). At the cellular level, the absence
of UFBP1 heightens endoplasmic reticulum stress and activates the
UPR, causing cell death in hematopoietic progenitor/stem cells
(19). Furthermore, UFBP1
deficiency suppresses the expression of erythroid transcription
factors GATA-1 and KLF1, inhibiting the transition from
colony-forming units-erythroid to proerythroblasts. Similar
phenotypic defects are observed in UFBP1-deficient mice when
compared with UFL1 and UBA5-deficient mice, indicating that UFBP1
serves as a crucial downstream effector in the regulation of
hematopoietic development mediated by the UFM1 system through
UFMylation. These findings underscore the significant role of
UFMylation in erythroid development and differentiation (18) (Table
I).
 | Table I.Phenotypic comparison of Uba5, Ufl1
and Ufbp1 knockout mice. |
Table I.
Phenotypic comparison of Uba5, Ufl1
and Ufbp1 knockout mice.
| First author,
year | Gene | KO phenotype | CKO phenotype | Biochemical
function | (Refs.) |
|---|
| Tatsumi et
al, 2011 | UBA5 | Embryo death and
impaired hematopoietic development. Fewer erythroid progenitor
cells. |
| UFM1 E1enzyme | (92) |
| Zhang et al,
2015 | UFL1 | Embryo death and
impaired hematopoietic development. Fewer erythroid progenitor
cells. | Animal death around
3 weeks after initial injection of tamoxifen. Pancytopenia and
severe anemia. Reduced number of erythroid progenitor cells in bone
marrow. Increased percentage and number of GMPs in bone marrow.
Impaired development from CFU-E to proerythroblast. Loss of HSC
function. | UFM1 E3 ligase | (93) |
| Cai et al,
2015 | UFBP1 | Embryo death and
impaired hematopoietic development. Impaired development of
hematopoietic progenitor cells. | Animal death around
3 weeks after initial injection of tamoxifen. Pancytopenia and
severe anemia. Reduced number of erythroid progenitor cells in bone
marrow. Increased percentage and number of GMPs in bone marrow.
Impaired development from CFU-E to proerythroblast. Loss of HSC
function. | UFM1 specific
protease | (18) |
UFMylation and skeletal development
Clinical studies have identified diseases
genetically linked to components of the UFM1 pathway, emphasizing
their crucial roles in development and tissue homeostasis. The
majority of mutations in UFM1 pathway components are associated
with a skeletal pathology known as SEMD (23,95).
A clinical case study of affected Italian families reported a
missense mutation, specifically UFSP2 D418A, in the UFSP2 gene. The
patients exhibited systemic skeletal dysplasia with infantile
onset, delayed skeletal development, demineralization, atrophy,
restricted mobility and joint destruction (95). Subsequently, heterozygous variants
of the UFSP2 gene, including Y290H, D426A and H428R, were
identified through exome sequencing, with the SEMD phenotype
observed in individuals harboring mutations in the catalytic
histidine (H420R) and the tyrosine residue (Y290H) that form the
oxocation pore (96). The Y282H
mutation resulted in a markedly milder phenotype. This finding
supports a new perspective that varying clinical outcomes may be
associated with the loss of catalytic UFSP2 activity linked to
different mutants, which has been confirmed in vitro
(24).
Furthermore, loss-of-function mutations in DDRGK1
(UFBP1, C20orf116) have also been demonstrated to cause SEMD.
DDRGK1-/- mice exhibit delayed mesenchymal condensation in limb
buds and early embryonic lethality. To further investigate this,
conditional knockout mice were generated by crossing Prx1-Cre
transgenic mice with DDRGKfl/fl mice to delete DDRGK1 expression in
limb mesenchymal cells. The mutant mice displayed progressively
severe shortening of the limbs, joint abnormalities, disorganized
growth plate tissue, reduced proliferation zones and enlarged
hypertrophic zones. These data highlight the importance of DDRGK1
in the development of growth plates (97). By contrast, conditional knockout of
DDRGK1 using osteoblast-specific osteocalcin-Cre and leptin
receptor-Cre lines did not result in bone phenotypes, indicating
that the effect on limb development is cartilage-specific. These
findings suggest that DDRGK1 is necessary postnatally for the
maintenance of normal growth plate morphology (98). Collectively, these discoveries
underscore the physiological role of DDRGK1 in the development and
maintenance of growth plate cartilage. Moreover, these genetic
mouse models recapitulate the clinical phenotypes of short stature
and joint abnormalities observed in patients with Sohat-type SEMD
(98).
UFMylation and neurodevelopment
UFMylation plays a crucial role in neurodevelopment.
Abnormalities in the UFMylation system can manifest as severe
infantile-onset encephalopathy, with or without seizures, severe
congenital neuropathies, or early-onset cerebellar atrophy
accompanied by ataxia (17,99).
As the disease progresses, non-specific findings such as mild
myelination delay, hyperintense white matter signals, thinned
thalami and corpus callosum, cerebellar hypoplasia, or generalized
brain atrophy can be observed (24). Individuals with the most severe
forms of the disease may die shortly after birth or in infancy,
while those with milder symptoms can survive >20 years. Whole
exome sequencing analysis has identified rare autosomal recessive
variants in UBA5 in five children from four unrelated families, all
of whom exhibited similar severe intellectual disabilities,
microcephaly, motor disorders, or early-onset refractory epilepsy
(25,100). To date, 24 variants in UBA5 have
been linked to neurological disorders. Of these variants, ~67% are
located in the adenylylation domain of the UBA5 protein, 19% in the
UFC1-binding domain and C-terminus and 14% in the N-terminus
(101). Most missense mutations
lead to mild impairment of UBA5 function; however, two homozygous
inherited N-terminal variants have resulted in severe disease,
leading to the mortality of affected individuals within 16 days and
16 weeks, respectively (102,103). In zebrafish, UBA5 gene knockout
at early stages exhibits peripheral nerve and cerebellar axonal
damage, with mitochondrial damage identified in peripheral and
central nervous systems as well as in skeletal muscles (104). In Caenorhabditis elegans,
knockout of UBA5 and its human ortholog in the UFM1 cascade alters
cholinergic neurotransmission without affecting glutamatergic
transmission (105).
NCAM, a molecule that plays a role in neuronal
development and synaptic plasticity in the adult brain, has been
shown to interact with the UFMylation system. NCAM140, a subtype of
NCAM, interacts with UFC1 through its cytoplasmic domain and
co-localizes with UFC1 at the surface of B35 neuroblastoma cells
(101). Overexpression of UFM1
also leads to increased endocytosis of NCAM140. Whole exome
sequencing analysis revealed rare autosomal recessive variants in
UBA5 in five children from four unrelated families, presenting
similar severe intellectual disabilities, microcephaly, motor
disorders, or early-onset refractory epilepsy (106). Biochemical and cellular studies
of UBA5 mutant proteins in fibroblasts from affected individuals
indicate that pathogenic mutations impair UFMylation, resulting in
abnormal endoplasmic reticulum structure. In C. elegans,
knockout of UBA5 and its human ortholog in the UFM1 cascade affects
cholinergic neurotransmission without affecting glutamatergic
neurotransmission (107).
Furthermore, silencing of UBA5 in zebrafish induced abnormal
movements while reducing locomotor ability, suggesting a link
between UFMylation and seizures (105,108). These clinical, biochemical and
experimental findings demonstrate that UBA5 mutations can lead to
early-onset encephalopathy due to abnormal protein UFMylation
(105). Collectively, these
studies highlight the significant role of UFMylation in
neurodevelopment (Table II).
| Table II.Summary of verified UFMylation
substrates and enzymes. |
Table II.
Summary of verified UFMylation
substrates and enzymes.
| First author,
year | UFMylation
substrate | Functional
sites | Function | (Refs.) |
|---|
| Bakkenist and
Kastan, 2003 | MRE11 | K282 | Promotes MRN
complex formation and recruitment, maintains telomere length. | (49) |
| Qin et al,
2020 | Histone 4 | K31 | Amplifies ATM
activation and maintains genomic. integrity | (50) |
| Liu et al,
2020 | P53 | K351, K357, K370,
K373 | Maintains P53
stability and suppresses tumor growth. | (47) |
| Wang et al,
2020 | ASC1 | K324, K325, K334,
K367 | Promotes ERα
transactivation and breast cancer progression. | (165) |
| Luo et al,
2023 | HRD1 | K610 | Regulating the
degradation of endoplasmic reticulum related proteins and clearing
misfolded ER resident proteins. | (68) |
| Ishimura et
al, 2022 | CYB5R3 | K214 | Promotes the
formation of autophagosomes and initiate endoplasmic reticulum
phagocytosis. | (76) |
| Zhu et al,
2019 | UFBP1 | K267 | Maintains ER
homeostasis, promotes self-renewal and differentiation of
hematopoietic progenitor cells. | (75) |
| Liu et al,
2020 | RPL26 | K132, K134 | Maintains ER
homeostasis. | (47) |
| Wang et al,
2023 | SAYSD1 |
| Maintains ER
homeostasis. | (88) |
| Tatsumi et
al, 2011 | UBA5 |
| Promotes
self-renewal and differentiation of hematopoietic progenitor cells,
promotes neuronal development and synaptic plasticity in the
brain. | (92) |
| Zhang et al,
2015 | UFL1 |
| Promotes
self-renewal and differentiation of hematopoietic progenitor
cells. | (93) |
| Di Rocco et
al, 2018 | UFSP2 | Y290H, D426A,
H428R | Maintains normal
bone shape. | (95) |
UFMylation and immune regulation
The UFM1 conjugation family has emerged as a
promising therapeutic target for modulating immune responses,
either by enhancing antiviral defense or mitigating excessive
inflammation in autoimmune diseases. Studies have revealed that
upon RNA virus infection (109,110), UFL1 is recruited to the
membrane-associated scaffold protein 14-3-3ε, where it undergoes
UFMylation. This process leads to the activation of retinoic
acid-inducible gene I, which in turn triggers mitochondrial
antiviral signaling protein (MAVS)-mediated signaling cascades.
These cascades ultimately culminate in the induction of type I and
III interferons (IFNs) (110). In
response to herpes simplex virus type 1 infection, UFL1 expression
is rapidly downregulated at both the mRNA and protein levels in
peritoneal macrophages. The conditional deletion of UFL1 in
macrophages results in increased viral loads in the serum and
peripheral immune cells. This increase is accompanied by a marked
reduction in proinflammatory cytokines, including interleukin-6
(IL-6), tumor necrosis factor-α (TNF-α) and IFN-β. During
Epstein-Barr virus (EBV) infection, the viral-encoded G
protein-coupled receptor BILF1 hijacks UFL1 to mediate MAVS
UFMylation. This process promotes MAVS mitochondrial
mislocalization and subsequent lysosomal degradation, effectively
suppressing EBV-induced activation of the NLRP3 inflammasome and
thereby dampening host antiviral immune responses (109). These findings demonstrate that
UFMylation plays a crucial role in the body's immune
regulation.
The role of UFMylation in common
malignancies
Numerous studies have reported the amplification,
deletion, or mutation of genes encoding UFMylation factors (UBA5,
UFC1, UFL1, UFSP2 and UFM1) in malignant tumors across various
tissues. These findings indicate that UFMylation may either promote
or inhibit tumorigenesis, depending on the cellular environment.
The loss or mutation of components within the UFMylation pathway is
associated with a range of diseases. Deepening our understanding of
UFMylation's role in cancer may pave the way for the development of
novel therapeutic strategies (43,53,103,107,111).
Breast cancer
Breast cancer ranks among the top three most common
cancers worldwide (112,113). A significant body of literature
indicates that estrogen receptor-positive (ER+) tumors constitute a
large portion of cases. PTMs of ERα are critical in regulating its
expression, subcellular localization and hormonal response
sensitivity (114,115). UFMylation plays a crucial role in
positively regulating ERα stability and transactivation, which is
key in breast cancer development (58,116). By inhibiting ubiquitination, ERα
stability is markedly enhanced, whereas silencing UBA5 decreases
ERα stability. Lys171 and Lys180 of ERα have been identified as
primary UFM1 receptor sites and substituting these lysine residues
with arginine (2KR mutation) markedly reduces ERα stability
(116). Furthermore, the 2KR
mutation abolishes ERα's 17β-estradiol-induced transcriptional
activity and the expression of downstream targets such as PS2,
cyclin D1 and C-MYC, highlighting the essential role of ERα's
transcriptional activation function. The 2KR mutation also prevents
MCF7 cells from forming anchorage-independent colonies (48,116,117). Notably, UFM1 and its associated
mechanisms (UBA5, UFC1, UFL1 and UFBP1) are markedly upregulated in
ERα-positive breast cancer cell lines and tissues. In summary,
these findings suggest that ERα enhances breast cancer development
by stabilizing and promoting its transactivation function through
UFMylation (116,118).
Metformin can induce ferroptosis in breast cancer
cell lines, thereby inhibiting tumor growth independently of
conventional AMP-activated protein kinase (AMPK) signaling
(119). Mechanistically,
metformin increases intracellular Fe2+ and lipid ROS
levels. Specifically, metformin suppresses the UFMylation process
of SLC7A11, a key regulator of iron metabolism, leading to
decreased protein stability of SLC7A11 and consequently inhibiting
cancer proliferation (120).
Recently, two additional UFMylation substrates,
PLAC8 and PD-L1, have been identified as playing significant roles
in the pathogenesis of breast cancer (121–123). Mao et al (121) reported that PLAC8 is generally
expressed at high levels in triple-negative breast cancer (TNBC).
This is because it is modified by UFM1 UFMylation at Lys103, which
maintains its protein stability. The stable PLAC8 interacts with
glycosylated PD-L1, thereby upregulating PD-L1 levels, promoting
cancer cell proliferation and inhibiting immune responses (123,124). Additionally, PD-L1 can be
modified by various deubiquitinating and ubiquitinating proteins,
such as USP22, CSN5 and ARIH1. Treatment of MDA-MB-231 and HCC-1937
cells with MG-132 and chloroquine demonstrated that MG-132
inhibited proteasome activity, while chloroquine inhibited
lysosomal activity. Notably, chloroquine rescued the reduction of
PD-L1 protein levels induced by PLAC8 knockout. Collectively, these
data suggest that PLAC8 may also stabilize PD-L1 protein by
regulating its ubiquitination (121).
Liver cancer
Hepatocellular carcinoma (HCC) is a leading cause of
cancer-related deaths in a number of regions of the world. Risk
factors for HCC include chronic hepatitis, alcohol addiction,
metabolic liver diseases and exposure to dietary toxins such as
aflatoxins and aristolochic acids. In most cases, acute hepatitis,
chronic hepatitis and cirrhosis caused by chronic hepatitis B or C
virus infections are significant contributors to the development of
hepatocellular carcinoma (125).
Reports indicate that the UFMylation system is associated with the
occurrence and progression of HCC. In the livers of mice re-fed
with dihydro-2,4,6-trimethy l-3,5-pyridinedicarboxylate (DDC) and
in humans with alcoholic hepatitis (AH) and non-alcoholic
steatohepatitis (NASH) characterized by the presence of
Mallory-Denk bodies (MDB), the UFMylation pathway is downregulated,
including protein quality control (126,127). Notably, feeding the methyl donor
betaine alongside DDC markedly prevents the increase in UFMylation
expression in DDC-pretreated mice (128,129). Betaine notably inhibited the
transcriptional silencing of UFM1, UBA5 and UFSP1 associated with
MDB formation and prevented the increase in FAT10 and LMP7
expression induced by DDC re-feeding. A similar downregulation of
UFMylation was observed in multiple biopsies from patients with AH
and NASH. Compared with normal subjects, patients with AH and NASH
exhibited markedly elevated levels of DNA methylation in the
promoter CpG regions of UFM1, UFC1 and UFSP1 (130–132). The mRNA levels of DNA
methyltransferase 1 (DNMT1) and DNA methyltransferase 3β (DNMT3B)
were markedly upregulated in patients with AH and NASH, indicating
that the maintenance of UFMylation methylation may be co-mediated
by DNMT1 and DNMT3B (133,134).
It has been reported that an HCC suppressor known as
B3GALT5-AS1 negatively regulates the proliferation, invasion and
metastasis of HCC cells by modulating miR-934 and UFM1. This
suppression of HCC cell proliferation, invasion and metastasis was
observed when pGL3-UFM1-WT and pGL3-UFM1-MUT plasmids were
co-transfected with miR-NC or miR-934 into HCC cell and compared
with the co-transfection group with pGL3-UFM1-WT and NC (135), the co-transfection of the
pGL3-UFM1-WT plasmid with miR-934 markedly reduced the luciferase
activity of the reporter plasmid, thereby indirectly elucidating
the interaction between UFM1 and miR-934 and confirming that UFM1
is a regulatory target of miR-934. Subsequently, the expression of
UFM1 was measured in three groups (NC, miR-934 inhibitor and
si-UFM1) after transfection, revealing that the UFM1 mRNA
expression levels in the miR-934 inhibitor and si-UFM1 groups were
markedly lower than those in the miR-934 inhibitor-only group.
Importantly, it was determined that si-UFM1 could reverse the
decreased cell proliferation and migration abilities induced by the
miR-934 inhibitor (136).
Additionally, UFL1 has been identified as a tumor
suppressor in HCC, playing a critical role in the pathogenesis of
HCC by preventing cell invasion, inhibiting NF-κB signaling and
increasing the stability of LZAP protein. However, the exact role
of CDK5RAP3 in HCC remains controversial (39,137). One study reports that CDK5RAP3
may act as an oncogene, promoting the migratory and invasive
characteristics of the SMMC-7721 and HEPG2 cell lines (138). By contrast, another group of
studies indicated that CDK5RAP3 functions as a tumor suppressor in
HEPG2 cells. The precise role, function and mechanism of CDK5RAP3
in HCC warrant further investigation (16,76).
Lung cancer
Lung cancer is one of the deadliest cancers and is
becoming increasingly prevalent worldwide (139). Notably, the mortality rate of
lung cancer exceeds that of other tumors. Lung cancer encompasses
various subtypes, including lung adenocarcinoma (LUAD), lung
squamous cell carcinoma (LUSC), large cell lung carcinoma and small
cell lung carcinoma (140,141).
Recently, UBA5 has been identified as playing a significant role in
the growth of LUAD cells, resistance to cisplatin and promoting
immune evasion while also participating in the macrophage M2
polarization process, thereby altering the tumor microenvironment
and facilitating tumor progression (142). In LUAD, both UBA5 mRNA and
protein levels are highly expressed. The pharmacological inhibition
of UBA5 using DKM 2–93 effectively inhibited LUAD growth,
indicating that the suppression of UBA5 hinders LUAD cell
proliferation (143).
Additionally, UBA5 positively correlates with
macrophage M2 polarization in LUAD. The knockdown of UBA5 directly
suppressed the in vitro polarization of macrophages to M2,
reduced the in vivo infiltration of macrophage M2 and
decreased lactate production in LUAD. These results suggest that
UBA5 regulates macrophage M2 polarization through lactate
secretion, thereby altering the immune microenvironment and
facilitating the escape of LUAD cells from immune surveillance
(144).
Despite its tumor-suppressive role in
hepatocellular carcinoma, UFL1 may act as an oncogene in lung
adenocarcinoma. UFL1 is upregulated in early lung adenocarcinoma
tissues and its overexpression promotes the proliferation of H1299
lung adenocarcinoma cells. UFL1 can bind to the regulatory domain
of P120-catenin, inhibiting the ubiquitin-mediated proteasomal
degradation of P120-catenin. Subsequently, P120-catenin promotes
the proliferation of lung adenocarcinoma through its interaction
with NLBP (145).
Colorectal cancer
In relevant studies on colorectal cancer, Zhou
et al (146) observed that
the knockdown of UFSP2 markedly promoted the growth rate of
colorectal cancer cells HT29 and HCT116 and their
anchorage-independent growth. Notably, the knockdown of UFSP2
expression markedly enhanced the growth of xenograft tumors derived
from UFSP2-depleted HT29 cells. These results demonstrate that
UFSP2 is a potential tumor suppressor in colorectal cancer.
Gastric cancer
Despite a decline in the incidence and mortality
rates of primary cancers in recent decades, gastric cancer remains
one of the three most common cancers worldwide. Most gastric cancer
patients exhibit nonspecific early symptoms and are often diagnosed
at advanced stages, which severely affects their prognosis.
Therefore, identifying new biomarkers is essential for early
diagnosing and treating gastric cancer (147,148). DDRGK1 is a target protein for
UFMylation and a key component of the UFMylation modification
system, playing a crucial role in cancer development (149). Shiwaku et al (150) found that the amino acid sequence
of CDK5RAP3 contains a ubiquitin-protein ligase binding domain. Wu
et al (151) revealed that
CDK5RAP3 can interact with DDRGK1 and UFL1 (RCAD), regulating the
stability of CDK5RAP2 and DDRGK1. Notably, patients with low
expressions of CDK5RAP3 and DDRGK1 had the worst prognoses, while
those with high expressions of these proteins exhibited the best
prognoses, with other patients falling in between. The predictive
accuracy of the combined expression of CDK5RAP3 and DDRGK1 was
higher than using either CDK5RAP3 or DDRGK1 alone and their
combined expression demonstrated superior predictive capability for
overall survival in cancer patients (152). Xi et al (153) discovered that DDRGK1 interacts
with IkBa and regulates its stability, thereby modulating the
transcriptional activity of NF-κB and the expression of its target
genes. Conversely, Wu et al found that the downregulation of
CDK5RAP3 increased cellular invasiveness and enhanced the
transcriptional activity of NF-κB (151). CDK5RAP3 binds to RelA to inhibit
its phosphorylation, increasing the association of HDAC with RERA
and thereby suppressing NF-κB transcriptional activity.
In summary, CDK5RAP3 and DDRGK1 interact and their
roles in the NF-κB pathway are similar. Additionally, Lin et
al (154) found that UFM1 was
downregulated in gastric cancer tissues. Patients with low UFM1
expression levels had poor prognoses, while UFM1 exhibits
inhibitory effects on oncogenicity, invasion and migration of
gastric cancer cells. Mechanistically, UFM1 suppresses gastric
cancer cells' epithelial-mesenchymal transition (EMT) by negatively
regulating the PI3K/AKT signaling pathway and increasing the
ubiquitination of PDK1, thereby exerting its tumor-suppressive
function (155).
Renal cell carcinoma
In renal cell carcinoma (RCC), the expressions of
UFL1 and UFBP1 are also downregulated and positively correlate with
P53 levels, a protein closely associated with various cancers. It
has been reported that P53 interacts with UFL1 and UfBP1 and is
modified by UFM1. UFMylation mediated by UFL1 and UFBP1 stabilizes
P53 by antagonizing MDM2-mediated ubiquitination and proteasomal
degradation, inhibiting cellular growth and tumor formation in
vivo (155).
In addition to analyzing RCC tissue microarrays
from 40 paired patient samples, studies using mouse xenograft
models indicate that UFL1 and UfBP1 can function as tumor
suppressors by regulating P53 stability. These results suggest that
UFMylation is a key post-translational modification for maintaining
P53 stability and tumor-suppressive function, implicating
UFMylation as a promising therapeutic target in cancer (47) (Table
III).
| Table III.Function of UFMylation in various
types of cancer. |
Table III.
Function of UFMylation in various
types of cancer.
| First author,
year | Cancer type | Gene | Function | (Refs.) |
|---|
| Yoo et al,
2022 | Breast cancer | ERa | Promotes breast
cancer development. | (116) |
| Yang et al,
2021 | Breast cancer | SLC7A11 | UFMylation
stabilizes SALC7A11 and metformin reduces the protein stability of
SLC7A11 by reducing UFM1. | (120) |
| Mao et al,
2022 | Breast cancer | PLAC8 | UFMylation of PLAC8
may influence tumor progression and immune response in triple
negative breast cancer cells by reducing PD-L1 ubiquitination. | (121) |
| Lim et al,
2016 | Breast cancer | PD-L1 | UFMylation of PD-L1
destabilizes PD-L1 by acting synergistically to promote its
ubiquitination. | (124) |
| Chen et al,
2021 | Hepatocellular
carcinoma | UFM1 | B3GALT5-AS1
regulates miR-934 and UFM1 to achieve negative regulation of HCC
cell proliferation, invasion, and metastasis. | (136) |
| Liu et al,
2014 | Hepatocellular
carcinoma | UFSP1 | Alcoholic hepatitis
and non-alcoholic steatohepatitis transcriptional down regulation
of FATylation and UFMylation. | (133) |
| Yang et al,
2019 | Hepatocellular
carcinoma | UFL1 | UFL1 act as
gatekeepers to prevent liver fibrosis and subsequent
steatohepatitis and Hepatocellular carcinoma development by
inhibiting the mTOR pathway. | (16) |
| Zhou et al,
2021 | Colon cancer | UfSP2 | UFSP2 is a
potential tumor suppressor in colon cancer. | (146) |
| Lin et al,
2018 | Gastric cancer | CDK5RAP3 and
DDRGK1 | CDK5RAP3
interacting with DDRGK1 suppresses the development of gastric
cancer by inhibiting the phosphorylation of AKT/GSK-3β and
negatively regulating Wnt/β-catenin signaling. | (152) |
| Lin et al,
2019 | Gastric cancer | UFM1 | UFM1 has inhibitory
effects on carcinogenicity, invasion, and migration of gastric
cancer cells. | (154) |
| Liu et al,
2020 | Renal cancer | P53 | UFMylation
stabilizes p53 by inhibiting its ubiquitination, which suppresses
cell growth and tumor formation. | (47) |
UFMylation in immunotherapy
Immune cells are central to antiviral defense and
antitumor immunity, yet the intricate tumor microenvironment
enables malignant cells to evade immune-mediated elimination
through diverse mechanisms (156). Among these, the upregulation of
inhibitory immune checkpoint receptors, such as PD-1, serves as a
key strategy to suppress T cell activation and cytotoxic function.
However, the limited clinical efficacy of anti-PD-1/PD-L1
immunotherapy underscores the urgent need for more effective
therapeutic strategies (21).
Recent findings highlight PD-L1 UFMylation as a crucial regulator
of PD-1/PD-L1 axis homeostasis in both human and murine tumor
cells, with its dysregulation compromising immune evasion (157). Loss of UFL1 in T cells abrogates
PD-1 UFMylation, thereby enhancing antitumor immunity.
Specifically, UFL1 deletion in T cells diminishes PD-1 UFMylation,
facilitates CD8+T cell-mediated tumor rejection and
promotes K48-linked ubiquitination and subsequent proteasomal
degradation of PD-1. In vitro and in vivo analyses
both demonstrate that UFL1 deficiency markedly reduces PD-1 protein
abundance while augmenting the production of effector cytokines,
including IFNγ, TNF and granzyme B, in CD8+T cells.
Furthermore, AMPK phosphorylates UFL1 at T536,
disrupting its interaction with PD-1 and attenuating PD-1
UFMylation (158). Notably,
conditional T cell-specific knockout (cKO) of UFL1 enhances
antitumor immunity. In murine models of Lewis lung carcinoma and
MC38 colorectal cancer, UFL1 cKO markedly improves responsiveness
to anti-CTLA-4 immunotherapy. However, UFL1 depletion in T cells
diminishes the antitumor efficacy of AMPK activators, suggesting
that the AMPK-UFL1 axis plays a pivotal role in regulating T
cell-mediated antitumor immunity (159).
Other tumors
Beyond the aforementioned tumor types, the UFM1
conjugation system has also been implicated in other human
malignancies. Sarcomas, a heterogeneous group of
mesenchymal-derived malignant neoplasms of connective tissue, are
broadly classified into soft tissue sarcomas and primary bone
sarcomas. These categories encompass a wide array of subtypes,
contributing to the remarkable diversity of this tumor group
(160). Among them, osteosarcoma
is the most prevalent primary bone malignancy in children,
adolescents and young adults (161). Notably, studies have reported
that UFBP1 suppresses the proliferation, migration and invasion of
human osteosarcoma cells (15,16,162). Mechanistically, as a component of
the E3 ligase complex in the UFM1 conjugation system, UFBP1
directly interacts with IκBα, regulating its stability and
attenuating NF-κB transcriptional activity (16). Notably, UFBP1 primarily binds to
the N-terminal domain of IκBα (1–106 aa), which harbors critical
phosphorylation sites (Ser32 and Ser36) and ubiquitination sites
(Lys21 and Lys22). By regulating the phosphorylation and stability
of IκBα, UFBP1 modulates NF-κB transcriptional activity and the
expression of its downstream target genes. Furthermore, UFBP1
depletion via siRNA markedly alters the expression of NF-κB target
genes, including cytokines, chemokines, adhesion molecules and
receptors, as well as enzymes involved in proliferation,
differentiation, apoptosis and stress responses. In summary, UFBP1
may promote the UFMylation-mediated degradation of IκBα, thereby
downregulating the expression of NF-κB pathway target genes and
suppressing osteosarcoma cell proliferation, migration and invasion
(153). In addition, RPL10
UFMylation has been identified in pancreatic cancer tissues and
cell lines (163), with this
modification being catalyzed by UFL1 at specific sites and reversed
by UFSP2-mediated de-UFMylation (164). Reduced UFMylation of RPL10
inhibits pancreatic cancer cell proliferation and stemness.
Moreover, transcription factor KLF4 positively regulates the
relationship between RPL10 inactivation and cellular stemness. Loss
of RPL10 is closely associated with tumorigenesis, primarily by
enhancing the expression of stemness-associated surface markers and
upregulating KLF4. Notably, mutations at key UFMylation sites in
RPL10 markedly impair pancreatic cancer cell proliferation and
stemness, further highlighting the functional relevance of this
post-translational modification in tumor development (163).
Conclusion
Protein PTM is a standard physiological process in
cells. Ubiquitination is a special modification of protein
post-translational modification, which plays a vital role in cell
life activities. UFM1 is a class ubiquitin modifier that attaches
to lysine residues on substrates after translation by a dedicated
enzyme system conserved in most eukaryotes. Despite structural
similarities between UFM1 and ubiquitin, the UFMylation machinery
employs unique mechanisms to ensure fidelity. Although the
physiological triggers and consequences of UFMylation are not fully
understood, its biological importance is reflected in frequent
mutations in the UFMylation pathway in human pathophysiology,
including musculoskeletal and neurodevelopmental disorders. Some of
these diseases can be explained by the increased endoplasmic
reticulum (ER) stress and disruption of translational homeostasis
observed upon loss of UFMylation. The role of UFM1 in these
processes may stem from its function in the ER, where ribosomes are
UFM1-glycosylated due to translational arrest. In addition,
UFMylation has been implicated in other cellular processes,
including DNA damage response and telomere maintenance.
To date, certain studies have shown that UFMylation
modification is also closely related to the development of tumors
(43,146,164,165). UFMylation modification seems
related to the stable expression of specific tumor suppressor
genes. For example, in clear cell renal cell carcinoma, UFMylation
of p53 promotes its stability, inhibiting tumor cell
proliferation.
UFMylation also plays a pivotal role in antitumor
immunity. Depletion of UFL1 in CD8+ T cells suppresses
PD-L1 expression, leading to the upregulation of downstream
effector cytokines such as IFN-γ and enhancing T-cell cytotoxicity.
As a potential therapeutic target, inhibition of UFMylation has
shown promise in cancer treatment. Notably, metformin disrupts the
UFMylation of ferroptosis-related proteases, destabilizing SLC7A11
and promoting ferroptosis, thereby suppressing tumor growth through
a mechanism independent of conventional AMPK signaling. However,
does UFMylation have a stabilizing effect on other tumor suppressor
proteins? At the same time, does UFMylation also participate in the
degradation of tumor-related genes and inhibit the occurrence and
development of tumors? In addition, is UFMylation also associated
with activation/inhibition of tumor-related signaling pathways?
None of this has been reported in much research. The revelation of
these mechanisms may expand the field of UFMylation and human
diseases.
In summary, studies of the mechanisms and
biological functions of UFMylation-related signaling pathways will
reveal insights into basic cell biology and may provide new
therapeutic opportunities for human health. However, this study
also has certain limitations, e.g. numerous tumor-related tumor
suppressor proteins or oncoproteins may have UFMylation, which
seriously affects the occurrence and development of various major
human diseases; however, these proteins were not extensively
described in this study. Therefore, such correlation studies should
be further analyzed in the future.
Acknowledgements
Not applicable.
Funding
The present study was supported by program for the grants from
the Scientific Research project of Education Department of Yunnan
Province (grant no. 2023Y0787), Science and Technology Plan of
Yunnan Provincial Department of Science and Technology,
202401AY070001-283 and The Kunming Health Science and Technology
Personnel Training Project (grant no. 2022-SW-10).
Availability of data and materials
Not applicable.
Authors' contributions
YT and CY designed and revised the article. RQ, JZ,
YY, FM, XY and KZ wrote the first draft of this review. Data
authentication is not applicable. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Millar AH, Heazlewood JL, Giglione C,
Holdsworth MJ, Bachmair A and Schulze WX: The scope, functions, and
dynamics of posttranslational protein modifications. Annu Rev Plant
Biol. 70:119–151. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hsu YC, Hsieh YH, Liao CC, Chong LW, Lee
CY, Yu YL and Chou RH: Targeting post-translational modifications
of histones for cancer therapy. Cell Mol Biol (Noisy-le-grand).
30:69–84. 2015.
|
|
3
|
Hitosugi T and Chen J: Post-translational
modifications and the Warburg effect. Oncogene. 33:4279–4285. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Herhaus L and Dikic I: Expanding the
ubiquitin code through post-translational modification. EMBO Rep.
16:1071–1083. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hochstrasser M: Origin and function of
ubiquitin-like proteins. Nature. 458:422–429. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sahtoe DD and Sixma TK: Layers of DUB
regulation. Trends Biochem Sci. 40:456–467. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Karpiyevich M and Artavanis-Tsakonas K:
Ubiquitin-like modifiers: Emerging regulators of protozoan
parasites. Biomolecules. 10:14032020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Komatsu M, Chiba T, Tatsumi K, Iemura S,
Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T and Tanaka K: A
novel protein-conjugating system for Ufm1, a ubiquitin-fold
modifier. EMBO J. 23:1977–1986. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gerakis Y, Quintero M, Li H and Hetz C:
The UFMylation system in proteostasis and beyond. Trends Cell Biol.
29:974–986. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Banerjee S, Kumar M and Wiener R:
Decrypting UFMylation: How proteins are modified with UFM1.
Biomolecules. 10:14422020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Witting KF, van der Heden van Noort GJ,
Kofoed C, Ormeno CT, El Atmioui D, Mulder MPC and Ovaa H:
Generation of the UFM1 toolkit for profiling UFM1-specific
proteases and ligases. Angew Chem Int Ed Engl. 57:14164–14168.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lorenz S, Cantor AJ, Rape M and Kuriyan J:
Macromolecular juggling by ubiquitylation enzymes. BMC Biol.
11:652013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jing Y, Mao Z and Chen F: UFMylation
system: An emerging player in tumorigenesis. Cancers (Basel).
14:35012022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Clague MJ, Urbe S and Komander D: Breaking
the chains: Deubiquitylating enzyme specificity begets function.
Nat Rev Mol Cell Biol. 20:338–352. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lin M, Zheng X and Jin J: Nontraditional
translation is the key to UFMylation and beyond. J Biol Chem.
298:1024312022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang R, Wang H, Kang B, Chen B, Shi Y,
Yang S, Sun L, Liu Y, Xiao W, Zhang T, et al: CDK5RAP3, a UFL1
substrate adaptor, is crucial for liver development. Development.
146:dev1692352019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang S, Moy N and Yang R: The UFM1
conjugation system in mammalian development. Dev Dyn. 252:976–985.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cai Y, Pi W, Sivaprakasam S, Zhu X, Zhang
M, Chen J, Makala L, Lu C, Wu J, Teng Y, et al: UFBP1, a key
component of the ufm1 conjugation system, is essential for
ufmylation-mediated regulation of erythroid development. PLoS
Genet. 11:e10056432015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tandra V, Anderson T, Ayala JD, Weintraub
NL, Singh N, Li H and Li J: Ufmylation of UFBP1 is dispensable for
endoplasmic reticulum stress response, embryonic development, and
cardiac and intestinal homeostasis. Cells. 12:19232023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang X, Lv X, Ma J and Xu G: UFMylation:
An integral post-translational modification for the regulation of
proteostasis and cellular functions. Pharmacol Ther.
260:1086802024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ding LJ, Jiang X, Li T and Wang S: Role of
UFMylation in tumorigenesis and cancer immunotherapy. Front
Immunol. 15:14548232024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang X, Xu X and Wang Z: The
post-translational role of UFMylation in physiology and disease.
Cells. 12:25432023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang X, Zhou T, Wang X, Xia Y, Cao X,
Cheng X, Cao Y, Ma P, Ma H, Qin A and Zhao J: Loss of DDRGK1
impairs IRE1α UFMylation in spondyloepiphyseal dysplasia. Int J
Biol Sci. 19:4709–4725. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang G, Tang S, Wang H, Pan H, Zhang W,
Huang Y, Kong J and Wang Y, Gu J and Wang Y: UFSP2-related
spondyloepimetaphyseal dysplasia: A confirmatory report. Eur J Med
Genet. 63:1040212020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Colin E, Daniel J, Ziegler A, Wakim J,
Scrivo A, Haack TB, Khiati S, Denommé AS, Amati-Bonneau P, Charif
M, et al: Biallelic variants in UBA5 reveal that disruption of the
UFM1 cascade can result in early-onset encephalopathy. Am J Hum
Genet. 99:695–703. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sasakawa H, Sakata E, Yamaguchi Y, Komatsu
M, Tatsumi K, Kominami E, Tanaka K and Kato K: Solution structure
and dynamics of Ufm1, a ubiquitin-fold modifier 1. Biochem Biophys
Res Commun. 343:21–26. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Millrine D, Cummings T, Matthews SP, Peter
JJ, Magnussen HM, Lange SM, Macartney T, Lamoliatte F, Knebel A and
Kulathu Y: Human UFSP1 is an active protease that regulates UFM1
maturation and UFMylation. Cell Rep. 40:1111682022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kang SH, Kim GR, Seong M, Baek SH, Seol
JH, Bang OS, Ovaa H, Tatsumi K, Komatsu M, Tanaka K and Chung CH:
Two novel ubiquitin-fold modifier 1 (Ufm1)-specific proteases,
UfSP1 and UfSP2. J Biol Chem. 282:5256–5262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ha BH, Jeon YJ, Shin SC, Tatsumi K,
Komatsu M, Tanaka K, Watson CM, Wallis G, Chung CH and Kim EE:
Structure of ubiquitin-fold modifier 1-specific protease UfSP2. J
Biol Chem. 286:10248–10257. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Millrine D, Peter JJ and Kulathu Y: A
guide to UFMylation, an emerging posttranslational modification.
FEBS J. 290:5040–5056. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liang Q, Jin Y, Xu S, Zhou J, Mao J, Ma X,
Wang M and Cong YS: Human UFSP1 translated from an upstream
near-cognate initiation codon functions as an active UFM1-specific
protease. J Biol Chem. 298:1020162022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mashahreh B, Hassouna F, Soudah N,
Cohen-Kfir E, Strulovich R, Haitin Y and Wiener R: Trans-binding of
UFM1 to UBA5 stimulates UBA5 homodimerization and ATP binding.
FASEB J. 32:2794–2802. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Soudah N, Padala P, Hassouna F, Kumar M,
Mashahreh B, Lebedev AA, Isupov MN, Cohen-Kfir E and Wiener R: An
N-terminal extension to UBA5 adenylation domain boosts UFM1
activation: isoform-specific differences in ubiquitin-like protein
activation. J Mol Biol. 431:463–478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kumar M, Padala P, Fahoum J, Hassouna F,
Tsaban T, Zoltsman G, Banerjee S, Cohen-Kfir E, Dessau M,
Rosenzweig R, et al: Structural basis for UFM1 transfer from UBA5
to UFC1. Nat Commun. 12:57082021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tolmachova KA, Farnung J, Liang JR, Corn
JE and Bode JW: Facile preparation of UFMylation activity-based
probes by chemoselective installation of electrophiles at the
C-terminus of recombinant UFM1. ACS Cent Sci. 8:756–762. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kumari S, Banerjee S, Kumar M, Hayashi A,
Solaimuthu B, Cohen-Kfir E, Shaul YD, Rouvinski A and Wiener R:
Overexpression of UBA5 in cells mimics the phenotype of cells
lacking UBA5. Int J Mol Sci. 23:74452022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liess AKL, Kucerova A, Schweimer K, Yu L,
Roumeliotis TI, Diebold M, Dybkov O, Sotriffer C, Urlaub H,
Choudhary JS, et al: Autoinhibition mechanism of the
ubiquitin-conjugating enzyme UBE2S by autoubiquitination.
Structure. 27:1195–1210. e72019. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Tatsumi K, Sou YS, Tada N, Nakamura E,
Iemura S, Natsume T, Kang SH, Chung CH, Kasahara M, Kominami E, et
al: A novel type of E3 ligase for the Ufm1 conjugation system. J
Biol Chem. 285:5417–5427. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Peter JJ, Magnussen HM, DaRosa PA,
Millrine D, Matthews SP, Lamoliatte F, Sundaramoorthy R, Kopito RR
and Kulathu Y: A non-canonical scaffold-type E3 ligase complex
mediates protein UFMylation. EMBO J. 41:e1110152022. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Deshaies RJ and Joazeiro CA: RING domain
E3 ubiquitin ligases. Annu Rev Biochem. 78:399–434. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Deol KK, Lorenz S and Strieter ER:
Enzymatic logic of ubiquitin chain assembly. Front Physiol.
10:8352019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Jiang Q, Wang Y, Xiang M, Hua J, Zhou T,
Chen F, Lv X, Huang J and Cai Y: UFL1, a UFMylation E3 ligase,
plays a crucial role in multiple cellular stress responses. Front
Endocrinol (Lausanne). 14:11231242023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yoo HM, Kang SH, Kim JY, Lee JE, Seong MW,
Lee SW, Ka SH, Sou YS, Komatsu M, Tanaka K, et al: Modification of
ASC1 by UFM1 is crucial for ERα transactivation and breast cancer
development. Mol Cell. 56:261–274. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lee L, Oliva AB, Martinez-Balsalobre E,
Churikov D, Peter J, Rahmouni D, Audoly G, Azzoni V, Audebert S,
Camoin L, et al: UFMylation of MRE11 is essential for telomere
length maintenance and hematopoietic stem cell survival. Sci Adv.
7:eabc73712021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Qin B, Yu J, Nowsheen S, Wang M, Tu X, Liu
T, Li H, Wang L and Lou Z: UFL1 promotes histone H4 ufmylation and
ATM activation. Nat Commun. 10:12422019. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang Z, Gong Y, Peng B, Shi R, Fan D, Zhao
H, Zhu M, Zhang H, Lou Z, Zhou J, et al: MRE11 UFMylation promotes
ATM activation. Nucleic Acids Res. 47:4124–4135. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Liu J, Guan D, Dong M, Yang J, Wei H,
Liang Q, Song L, Xu L, Bai J, Liu C, et al: UFMylation maintains
tumour suppressor p53 stability by antagonizing its ubiquitination.
Nat Cell Biol. 22:1056–1063. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lee JH and Paull TT: Activation and
regulation of ATM kinase activity in response to DNA double-strand
breaks. Oncogene. 26:7741–7748. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bakkenist CJ and Kastan MB: DNA damage
activates ATM through intermolecular autophosphorylation and dimer
dissociation. Nature. 421:499–506. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Qin B, Yu J, Nowsheen S, Zhao F, Wang L
and Lou Z: STK38 promotes ATM activation by acting as a reader of
histone H4 ufmylation. Sci Adv. 6:eaax82142020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Fang Z and Pan Z: Essential role of
ubiquitin-fold modifier 1 conjugation in DNA damage response. DNA
Cell Biol. 38:1030–1039. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sun Y, Jiang X, Chen S, Fernandes N and
Price BD: A role for the Tip60 histone acetyltransferase in the
acetylation and activation of ATM. Proc Natl Acad Sci USA.
102:13182–13187. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liu J, Wang Y, Song L, Zeng L, Yi W, Liu
T, Chen H, Wang M, Ju Z and Cong YS: A critical role of DDRGK1 in
endoplasmic reticulum homoeostasis via regulation of IRE1α
stability. Nat Commun. 8:141862017. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Rusiecki R, Witkowski J and
Jaszczewska-Adamczak J: MDM2-p53 interaction inhibitors: The
current state-of-art and updated patent review (2010-Present).
Recent Pat Anticancer Drug Discov. 14:324–369. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wang S, Zhao Y, Aguilar A, Bernard D and
Yang CY: Targeting the MDM2-p53 protein-protein interaction for new
cancer therapy: Progress and challenges. Cold Spring Harb Perspect
Med. 7:a0262452017. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Adams KE, Medhurst AL, Dart DA and Lakin
ND: Recruitment of ATR to sites of ionising radiation-induced DNA
damage requires ATM and components of the MRN protein complex.
Oncogene. 25:3894–3904. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Vazifehmand R, Ali DS, Homaie FM,
Jalalvand FM, Othman Z, Deming C, Stanslas J and Sekawi Z: Effects
of HSV-G47Delta oncolytic virus on telomerase and telomere length
alterations in glioblastoma multiforme cancer stem cells under
hypoxia and normoxia conditions. Curr Cancer Drug Targets.
24:1262–1274. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ufmylation of ASC1 is essential for breast
cancer development. Cancer Discov. 4:OF102014. View Article : Google Scholar
|
|
59
|
Chen Q, Xiao Y, Chai P, Zheng P, Teng J
and Chen J: ATL3 is a tubular ER-Phagy receptor for
GABARAP-mediated selective autophagy. Curr Biol. 29:846–855.
e62019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Stephani M, Picchianti L, Gajic A,
Beveridge R, Skarwan E, de Medina Hernandez V, Mohseni A, Clavel M,
Zeng Y, Naumann C, et al: A cross-kingdom conserved ER-phagy
receptor maintains endoplasmic reticulum homeostasis during stress.
Elife. 9:e583962020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Simsek D, Tiu GC, Flynn RA, Byeon GW,
Leppek K, Xu AF, Chang HY and Barna M: The mammalian
Ribo-interactome reveals ribosome functional diversity and
heterogeneity. Cell. 169:1051–1065. e182017. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Komatsu M, Inada T and Noda NN: The UFM1
system: Working principles, cellular functions, and
pathophysiology. Mol Cell. 84:156–169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ishimura R, Ito S, Mao G, Komatsu-Hirota
S, Inada T, Noda NN and Komatsu M: Mechanistic insights into the
roles of the UFM1 E3 ligase complex in ufmylation and
ribosome-associated protein quality control. Sci Adv.
9:eadh36352023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Walczak CP, Leto DE, Zhang L, Riepe C,
Muller RY, DaRosa PA, Ingolia NT, Elias JE and Kopito RR: Ribosomal
protein RPL26 is the principal target of UFMylation. Proc Natl Acad
Sci USA. 116:1299–1308. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Scavone F, Gumbin SC, Da Rosa PA and
Kopito RR: RPL26/uL24 UFMylation is essential for
ribosome-associated quality control at the endoplasmic reticulum.
Proc Natl Acad Sci USA. 120:e22203401202023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Wang L, Xu Y, Rogers H, Saidi L, Noguchi
CT, Li H, Yewdell JW, Guydosh NR and Ye Y: UFMylation of RPL26
links translocation-associated quality control to endoplasmic
reticulum protein homeostasis. Cell Res. 30:5–20. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
French SW, Masouminia M, Samadzadeh S,
Tillman BC, Mendoza A and French BA: Role of protein quality
control failure in alcoholic hepatitis pathogenesis. Biomolecules.
7:112017. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Luo H, Jiao QB, Shen CB, Gong WY, Yuan JH,
Liu YY, Chen Z, Liu J, Xu XL, Cong YS and Zhang XW: UFMylation of
HRD1 regulates endoplasmic reticulum homeostasis. FASEB J.
37:e232212023. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wilkinson S: Emerging principles of
selective ER autophagy. J Mol Biol. 432:185–205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chino H and Mizushima N: ER-Phagy: Quality
control and turnover of endoplasmic reticulum. Trends Cell Biol.
30:384–398. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Picchianti L, de Medina Hernandez V, Zhan
N, Irwin NA, Groh R, Stephani M, Hornegger H, Beveridge R,
Sawa-Makarska J, Lendl T, et al: Shuffled ATG8 interacting motifs
form an ancestral bridge between UFMylation and autophagy. EMBO J.
42:e1120532023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Stephani M, Picchianti L and Dagdas Y: C53
is a cross-kingdom conserved reticulophagy receptor that bridges
the gap betweenselective autophagy and ribosome stalling at the
endoplasmic reticulum. Autophagy. 17:586–587. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Klebanovych A, Vinopal S, Draberova E,
Sladkova V, Sulimenko T, Sulimenko V, Vosecká V, Macůrek L, Legido
A and Dráber P: C53 Interacting with UFM1-protein ligase 1
regulates microtubule nucleation in response to ER stress. Cells.
11:5552022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Reggiori F and Molinari M: ER-phagy:
Mechanisms, regulation, and diseases connected to the lysosomal
clearance of the endoplasmic reticulum. Physiol Rev. 102:1393–1448.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhu H, Bhatt B, Sivaprakasam S, Cai Y, Liu
S, Kodeboyina SK, Patel N, Savage NM, Sharma A, Kaufman RJ, et al:
Ufbp1 promotes plasma cell development and ER expansion by
modulating distinct branches of UPR. Nat Commun. 10:10842019.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Ishimura R, El-Gowily AH, Noshiro D,
Komatsu-Hirota S, Ono Y, Shindo M, Hatta T, Abe M, Uemura T,
Lee-Okada HC, et al: The UFM1 system regulates ER-phagy through the
ufmylation of CYB5R3. Nat Commun. 13:78572022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Liang JR, Lingeman E, Luong T, Ahmed S,
Muhar M, Nguyen T, Olzmann JA and Corn JE: A genome-wide ER-phagy
screen highlights key roles of mitochondrial metabolism and
ER-resident UFMylation. Cell. 180:1160–1177. e202020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Raymundo DP, Doultsinos D, Guillory X,
Carlesso A, Eriksson LA and Chevet E: Pharmacological targeting of
IRE1 in cancer. Trends Cancer. 6:1018–1030. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Cai Y, Zhu G, Liu S, Pan Z, Quintero M,
Poole CJ, Lu C, Zhu H, Islam B, Riggelen JV, et al: Indispensable
role of the Ubiquitin-fold modifier 1-specific E3 ligase in
maintaining intestinal homeostasis and controlling gut
inflammation. Cell Discov. 5:72019. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Hwang J and Qi L: Quality control in the
endoplasmic reticulum: Crosstalk between ERAD and UPR pathways.
Trends Biochem Sci. 43:593–605. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lemaire K, Moura RF, Granvik M,
Igoillo-Esteve M, Hohmeier HE, Hendrickx N, Newgard CB, Waelkens E,
Cnop M and Schuit F: Ubiquitin fold modifier 1 (UFM1) and its
target UFBP1 protect pancreatic beta cells from ER stress-induced
apoptosis. PLoS One. 6:e185172011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
DaRosa PA, Penchev I, Gumbin SC, Scavone
F, Wachalska M, Paulo JA, Ordureau A, Peter JJ, Kulathu Y, Harper
JW, et al: UFM1 E3 ligase promotes recycling of 60S ribosomal
subunits from the ER. Nature. 627:445–452. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Makhlouf L, Peter JJ, Magnussen HM, Thakur
R, Millrine D, Minshull TC, Harrison G, Varghese J, Lamoliatte F,
Foglizzo M, et al: The UFM1 E3 ligase recognizes and releases 60S
ribosomes from ER translocons. Nature. 627:437–444. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
von der Malsburg K, Shao S and Hegde RS:
The ribosome quality control pathway can access nascent
polypeptides stalled at the Sec61 translocon. Mol Biol Cell.
26:2168–2180. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Brandman O and Hegde RS:
Ribosome-associated protein quality control. Nat Struct Mol Biol.
23:7–15. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Wan L, Juszkiewicz S, Blears D, Bajpe PK,
Han Z, Faull P, Mitter R, Stewart A, Snijders AP, Hegde RS and
Svejstrup JQ: Translation stress and collided ribosomes are
co-activators of cGAS. Mol Cell. 81:2808–2822. e102021. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Inada T: Quality controls induced by
aberrant translation. Nucleic Acids Res. 48:1084–1096. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Wang L, Xu Y, Yun S, Yuan Q,
Satpute-Krishnan P and Ye Y: SAYSD1 senses UFMylated ribosome to
safeguard co-translational protein translocation at the endoplasmic
reticulum. Cell Rep. 42:1120282023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Joazeiro CAP: Mechanisms and functions of
ribosome-associated protein quality control. Nat Rev Mol Cell Biol.
20:368–383. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Cai Y, Singh N and Li H: Essential role of
Ufm1 conjugation in the hematopoietic system. Exp Hematol.
44:442–446. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Hattangadi SM, Wong P, Zhang L, Flygare J
and Lodish HF: From stem cell to red cell: Regulation of
erythropoiesis at multiple levels by multiple proteins, RNAs, and
chromatin modifications. Blood. 118:6258–6268. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Tatsumi K, Yamamoto-Mukai H, Shimizu R,
Waguri S, Sou YS, Sakamoto A, Taya C, Shitara H, Hara T, Chung CH,
et al: The Ufm1-activating enzyme Uba5 is indispensable for
erythroid differentiation in mice. Nat Commun. 2:1812011.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang M, Zhu X, Zhang Y, Cai Y, Chen J,
Sivaprakasam S, Gurav A, Pi W, Makala L, Wu J, et al: RCAD/Ufl1, a
Ufm1 E3 ligase, is essential for hematopoietic stem cell function
and murine hematopoiesis. Cell Death Differ. 22:1922–1934. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang Y, Zhang M, Wu J, Lei G and Li H:
Transcriptional regulation of the Ufm1 conjugation system in
response to disturbance of the endoplasmic reticulum homeostasis
and inhibition of vesicle trafficking. PLoS One. 7:e485872012.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Di Rocco M, Rusmini M, Caroli F, Madeo A,
Bertamino M, Marre-Brunenghi G and Ceccherini I: Novel
spondyloepimetaphyseal dysplasia due to UFSP2 gene mutation. Clin
Genet. 93:671–674. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhang G, Tang S, Wang H, Pan H, Zhang W,
Huang Y, Kong J and Wang Y, Gu J and Wang Y: Corrigendum to
UFSP2-related spondyloepimetaphyseal dysplasia: A confirmatory
report. Eur J Med Genet. 63:1040212020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Franceschi R, Iascone M, Maitz S,
Marchetti D, Mariani M, Selicorni A, Soffiati M and Maines E: A
missense mutation in DDRGK1 gene associated to Shohat-type
spondyloepimetaphyseal dysplasia: Two case reports and a review of
literature. Am J Med Genet A. 188:2434–2437. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Weisz-Hubshman M, Egunsula AT, Dawson B,
Castellon A, Jiang MM, Chen-Evenson Y, Zhiyin Y, Lee B and Bae Y:
DDRGK1 is required for the proper development and maintenance of
the growth plate cartilage. Hum Mol Genet. 31:2820–2830. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ni M, Afroze B, Xing C, Pan C, Shao Y, Cai
L, Cantarel BL, Pei J, Grishin NV, Hewson S, et al: A pathogenic
UFSP2 variant in an autosomal recessive form of pediatric
neurodevelopmental anomalies and epilepsy. Genet Med. 23:900–908.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Muona M, Ishimura R, Laari A, Ichimura Y,
Linnankivi T, Keski-Filppula R, Herva R, Rantala H, Paetau A,
Pöyhönen M, et al: Biallelic variants in UBA5 link dysfunctional
ufm1 ubiquitin-like modifier pathway to severe infantile-onset
encephalopathy. Am J Hum Genet. 99:683–694. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhang J, Zhu H, Liu S, Quintero M, Zhu T,
Xu R, Cai Y, Han Y and Li H: Deficiency of murine UFM1-Specific E3
ligase causes microcephaly and inflammation. Mol Neurobiol.
59:6363–6372. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Arnadottir GA, Jensson BO, Marelsson SE,
Sulem G, Oddsson A, Kristjansson RP, Benonisdottir S, Gudjonsson
SA, Masson G, Thorisson GA, et al: Compound heterozygous mutations
in UBA5 causing early-onset epileptic encephalopathy in two
sisters. BMC Med Genet. 18:1032017. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nahorski MS, Maddirevula S, Ishimura R,
Alsahli S, Brady AF, Begemann A, Mizushima T, Guzmán-Vega FJ, Obata
M, Ichimura Y, et al: Biallelic UFM1 and UFC1 mutations expand the
essential role of ufmylation in brain development. Brain.
141:1934–1945. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Serrano RJ, Oorschot V, Palipana D,
Calcinotto V, Sonntag C, Ramm G and Bryson-Richardson RJ: Genetic
model of UBA5 deficiency highlights the involvement of both
peripheral and central nervous systems and identifies widespread
mitochondrial abnormalities. Brain Commun. 5:fcad3172023.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Cabrera-Serrano M, Coote DJ, Azmanov D,
Goullee H, Andersen E, McLean C, Davis M, Ishimura R, Stark Z,
Vallat JM, et al: A homozygous UBA5 pathogenic variant causes a
fatal congenital neuropathy. J Med Genet. 57:835–842. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Mignon-Ravix C, Milh M, Kaiser CS, Daniel
J, Riccardi F, Cacciagli P, Nagara M, Busa T, Liebau E and Villard
L: Abnormal function of the UBA5 protein in a case of early
developmental and epileptic encephalopathy with suppression-burst.
Hum Mutat. 39:934–938. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Duan R, Shi Y, Yu L, Zhang G, Li J, Lin Y,
Guo J, Wang J, Shen L, Jiang H, et al: UBA5 mutations cause a new
form of autosomal recessive cerebellar ataxia. PLoS One.
11:e01490392016. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Al-Saady ML, Kaiser CS, Wakasuqui F,
Korenke GC, Waisfisz Q, Polstra A, Pouwels PJW, Bugiani M, van der
Knaap MS, Lunsing RJ, et al: Homozygous UBA5 variant leads to
hypomyelination with thalamic involvement and axonal neuropathy.
Neuropediatrics. 52:489–494. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yiu SPT, Zerbe C, Vanderwall D, Huttlin
EL, Weekes MP and Gewurz BE: An Epstein-Barr virus protein
interaction map reveals NLRP3 inflammasome evasion via MAVS
UFMylation. Mol Cell. 83:2367–2386. e152023. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Snider DL, Park M, Murphy KA, Beachboard
DC and Horner SM: Signaling from the RNA sensor RIG-I is regulated
by ufmylation. Proc Natl Acad Sci USA. 119:e21195311192022.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Xie Z, Fang Z and Pan Z: Ufl1/RCAD, a Ufm1
E3 ligase, has an intricate connection with ER stress. Int J Biol
Macromol. 135:760–767. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
DeSantis CE, Ma J, Gaudet MM, Newman LA,
Miller KD, Sauer A, Jemal A and Siegel RL: Breast cancer
statistics, 2019. CA Cancer J Clin. 69:438–451. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2020. CA Cancer J Clin. 70:7–30. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Li Z, Wei H, Li S, Wu P and Mao X: The
role of progesterone receptors in breast cancer. Drug Des Devel
Ther. 16:305–314. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Jozwik KM and Carroll JS: Pioneer factors
in hormone-dependent cancers. Nat Rev Cancer. 12:381–385. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Yoo HM, Park JH, Kim JY and Chung CH:
Modification of ERα by UFM1 increases its stability and
transactivity for breast cancer development. Mol Cells. 45:425–434.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Yoo HM, Park JH, Jeon YJ and Chung CH:
Ubiquitin-fold modifier 1 acts as a positive regulator of breast
cancer. Front Endocrinol (Lausanne). 6:362015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Le Romancer M, Poulard C, Cohen P, Sentis
S, Renoir JM and Corbo L: Cracking the estrogen receptor's
posttranslational code in breast tumors. Endocr Rev. 32:597–622.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Schulten HJ: Pleiotropic effects of
metformin on cancer. Int J Mol Sci. 19:28502018. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang J, Zhou Y, Xie S, Wang J, Li Z, Chen
L, Mao M, Chen C, Huang A, Chen Y, et al: Metformin induces
ferroptosis by inhibiting UFMylation of SLC7A11 in breast cancer. J
Exp Clin Cancer Res. 40:2062021. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Mao M, Chen Y, Yang J, Cheng Y, Xu L, Ji
F, Zhou J, Zhang X, Li Z, Chen C, et al: Modification of PLAC8 by
UFM1 affects tumorous proliferation and immune response by
impacting PD-L1 levels in triple-negative breast cancer. J
Immunother Cancer. 10:e0056682022. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Feng X, Wei Z, Tao X, Du Y, Wu J, Yu Y, Yu
H and Zhao H: PLAC8 promotes the autophagic activity and improves
the growth priority of human trophoblast cells. FASEB J.
35:e213512021. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Hsu JM, Li CW, Lai YJ and Hung MC:
Posttranslational modifications of PD-L1 and their applications in
cancer therapy. Cancer Res. 78:6349–6353. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Lim SO, Li CW, Xia W, Cha JH, Chan LC, Wu
Y, Chang SS, Lin WC, Hsu JM, Hsu YH, et al: Deubiquitination and
stabilization of PD-L1 by CSN5. Cancer Cell. 30:925–939. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Kulsuptrakul J, Wang R, Meyers NL, Ott M
and Puschnik AS: A genome-wide CRISPR screen identifies UFMylation
and TRAMP-like complexes as host factors required for hepatitis A
virus infection. Cell Rep. 34:1088592021. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
French SW, Bardag-Gorce F, Li J, French BA
and Oliva J: Mallory-Denk body pathogenesis revisited. World J
Hepatol. 2:295–301. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Bardag-Gorce F, Oliva J, Villegas J,
Fraley S, Amidi F, Li J, Dedes J, French B and French SW:
Epigenetic mechanisms regulate Mallory Denk body formation in the
livers of drug-primed mice. Exp Mol Pathol. 84:113–121. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Li J, Li XM, Caudill M, Malysheva O,
Bardag-Gorce F, Oliva J, French BA, Gorce E, Morgan K, Kathirvel E,
et al: Betaine feeding prevents the blood alcohol cycle in rats fed
alcohol continuously for 1 month using the rat intragastric tube
feeding model. Exp Mol Pathol. 91:540–547. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Oliva J, Bardag-Gorce F, Li J, French BA,
Nguyen SK, Lu SC and French SW: Betaine prevents Mallory-Denk body
formation in drug-primed mice by epigenetic mechanisms. Exp Mol
Pathol. 86:77–86. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Liu H, Gong M, French BA, Li J, Tillman B
and French SW: Mallory-Denk body (MDB) formation modulates
Ufmylation expression epigenetically in alcoholic hepatitis (AH)
and non-alcoholic steatohepatitis (NASH). Exp Mol Pathol.
97:477–483. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Dong Y and Wang A: Aberrant DNA
methylation in hepatocellular carcinoma tumor suppression (Review).
Oncol Lett. 8:963–968. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Esteller M, Corn PG, Baylin SB and Herman
JG: A gene hypermethylation profile of human cancer. Cancer Res.
61:3225–3229. 2001.PubMed/NCBI
|
|
133
|
Liu H, Li J, Tillman B, French BA and
French SW: Ufmylation and FATylation pathways are downregulated in
human alcoholic and nonalcoholic steatohepatitis, and mice fed DDC,
where Mallory-Denk bodies (MDBs) form. Exp Mol Pathol. 97:81–88.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Li H, Rauch T, Chen ZX, Szabo PE, Riggs AD
and Pfeifer GP: The histone methyltransferase SETDB1 and the DNA
methyltransferase DNMT3A interact directly and localize to
promoters silenced in cancer cells. J Biol Chem. 281:19489–19500.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Fagerberg L, Hallstrom BM, Oksvold P,
Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S,
Danielsson A, Edlund K, et al: Analysis of the human
tissue-specific expression by genome-wide integration of
transcriptomics and antibody-based proteomics. Mol Cell Proteomics.
13:397–406. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Chen E, Zhou B, Bian S, Ni W and Chen Z:
The lncRNA B3GALT5-AS1 functions as an HCC suppressor by regulating
the miR-934/UFM1 axis. J Oncol. 2021:17764322021. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Yang S, Yang R, Wang H, Huang Y and Jia Y:
CDK5RAP3 deficiency restrains liver regeneration after partial
hepatectomy triggering endoplasmic reticulum stress. Am J Pathol.
190:2403–2416. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Sheng L, Li J, Rao S, Yang Z and Huang Y:
Cyclin-dependent kinase 5 regulatory subunit associated protein 3:
Potential functions and implications for development and disease.
Front Oncol. 11:7604292021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Bade BC and Cruz CS: Lung cancer 2020:
Epidemiology, etiology, and prevention. Clin Chest Med. 41:1–24.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Xia C, Dong X, Li H, Cao M, Sun D, He S,
Yang F, Yan X, Zhang S, Li N and Chen W: Cancer statistics in China
and United States, 2022: Profiles, trends, and determinants. Chin
Med J (Engl). 135:584–590. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Nabet BY, Hamidi H, Lee MC, Banchereau R,
Morris S, Adler L, Gayevskiy V, Elhossiny AM, Srivastava MK, Patil
NS, et al: Immune heterogeneity in small-cell lung cancer and
vulnerability to immune checkpoint blockade. Cancer Cell.
42:429–443. e42024. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Wang X, Wu Y, Gu J and Xu J:
Tumor-associated macrophages in lung carcinoma: From mechanism to
therapy. Pathol Res Pract. 229:1537472022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Hu J, Zhang L, Xia H, Yan Y, Zhu X, Sun F,
Sun L, Li S, Li D, Wang J, et al: Tumor microenvironment remodeling
after neoadjuvant immunotherapy in non-small cell lung cancer
revealed by single-cell RNA sequencing. Genome Med. 15:142023.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Xu D, Zhang D, Wei W and Zhang C: UBA5
inhibition restricts lung adenocarcinoma via blocking macrophage M2
polarization and cisplatin resistance. Exp Cell Res.
440:1141482024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Kim CH, Nam HS, Lee EH, Han SH, Cho HJ,
Chung HJ, Lee NS, Choi SJ, Kim H, Ryu JS, et al: Overexpression of
a novel regulator of p120 catenin, NLBP, promotes lung
adenocarcinoma proliferation. Cell Cycle. 12:2443–2453. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Zhou J, Ma X, Xu L, Liang Q, Mao J, Liu J,
Wang M, Yuan J and Cong YS: Genomic profiling of the UFMylation
family genes identifies UFSP2 as a potential tumour suppressor in
colon cancer. Clin Transl Med. 11:e6422021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Smyth EC, Nilsson M, Grabsch HI, van
Grieken NC and Lordick F: Gastric cancer. Lancet. 396:635–648.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Karimi P, Islami F, Anandasabapathy S,
Freedman ND and Kamangar F: Gastric cancer: Descriptive
epidemiology, risk factors, screening, and prevention. Cancer
Epidemiol Biomarkers Prev. 23:700–713. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Lin M, Lian NZ, Cao LL, Huang CM, Zheng
CH, Li P, Xie JW, Wang JB, Lu J, Chen QY, et al: Down-regulated
expression of CDK5RAP3 and UFM1 suggests a poor prognosis in
gastric cancer patients. Front Oncol. 12:9277512022. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Shiwaku H, Yoshimura N, Tamura T, Sone M,
Ogishima S, Watase K, Tagawa K and Okazawa H: Suppression of the
novel ER protein Maxer by mutant ataxin-1 in Bergman glia
contributes to non-cell-autonomous toxicity. EMBO J. 29:2446–2460.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Wu J, Lei G, Mei M, Tang Y and Li H: A
novel C53/LZAP-interacting protein regulates stability of C53/LZAP
and DDRGK domain-containing Protein 1 (DDRGK1) and modulates
NF-kappaB signaling. J Biol Chem. 285:15126–15136. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lin JX, Xie XS, Weng XF, Zheng CH, Xie JW,
Wang JB, Lu J, Chen QY, Cao LL, Lin M, et al: Low expression of
CDK5RAP3 and DDRGK1 indicates a poor prognosis in patients with
gastric cancer. World J Gastroenterol. 24:3898–3907. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Xi P, Ding D, Zhou J, Wang M and Cong YS:
DDRGK1 regulates NF-κB activity by modulating IκBα stability. PLoS
One. 8:e642312013. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Lin JX, Xie XS, Weng XF, Qiu SL, Yoon C,
Lian NZ, Xie JW, Wang JB, Lu J, Chen QY, et al: UFM1 suppresses
invasive activities of gastric cancer cells by attenuating the
expres7sion of PDK1 through PI3K/AKT signaling. J Exp Clin Cancer
Res. 38:4102019. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Zhou Y, Ye X, Zhang C, Wang J, Guan Z, Yan
J, Xu L, Wang K, Guan D, Liang Q, et al: Ufl1 deficiency causes
kidney atrophy associated with disruption of endoplasmic reticulum
homeostasis. J Genet Genomics. 48:403–410. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Momayyezi P, Bilev E, Ljunggren HG and
Hammer Q: Viral escape from NK-cell-mediated immunosurveillance: A
lesson for cancer immunotherapy? Eur J Immunol. 53:e23504652023.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Zhou J, Ma X, He X, Chen B, Yuan J, Jin Z,
Li L, Wang Z, Xiao Q, Cai Y, et al: Dysregulation of PD-L1 by
UFMylation imparts tumor immune evasion and identified as a
potential therapeutic target. Proc Natl Acad Sci USA.
120:e22157321202023. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Ma EH, Poffenberger MC, Wong AH and Jones
RG: The role of AMPK in T cell metabolism and function. Curr Opin
Immunol. 46:45–52. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
He C, Xing X, Chen HY, Gao M, Shi J, Xiang
B, Xiao X, Sun Y, Yu H, Xu G, et al: UFL1 ablation in T cells
suppresses PD-1 UFMylation to enhance anti-tumor immunity. Mol
Cell. 84:1120–1138. e82024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Zhu MMT, Shenasa E and Nielsen TO:
Sarcomas: Immune biomarker expression and checkpoint inhibitor
trials. Cancer Treat Rev. 91:1021152020. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Brown HK, Schiavone K, Gouin F, Heymann MF
and Heymann D: Biology of bone sarcomas and new therapeutic
developments. Calcif Tissue Int. 102:174–195. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Meng H, Ai H, Li D, Jiang X, Zhang H, Xu J
and Huang S: Bombyx mori UFBP1 regulates apoptosis and promotes
BmNPV proliferation by affecting the expression of ER chaperone
BmBIP. Int J Biol Macromol. 283:1376812024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Wang K, Chen S, Wu Y, Wang Y, Lu Y, Sun Y
and Chen Y: The ufmylation modification of ribosomal protein L10 in
the development of pancreatic adenocarcinoma. Cell Death Dis.
14:3502023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
MacLeod G, Bozek DA, Rajakulendran N,
Monteiro V, Ahmadi M, Steinhart Z, Kushida MM, Yu H, Coutinho FJ,
Cavalli FMG, et al: Genome-wide CRISPR-Cas9 screens expose genetic
vulnerabilities and mechanisms of temozolomide sensitivity in
glioblastoma stem cells. Cell Rep. 27:971–986.e9. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Wang S, Jia M, Su M, Hu X, Li J, Xu Y and
Qiu W: Ufmylation is activated in renal cancer and is not
associated with von hippel-lindau mutation. DNA Cell Biol.
39:654–660. 2020. View Article : Google Scholar : PubMed/NCBI
|














