Introduction
The Global Initiative for Chronic Obstructive Lung
Disease defines chronic obstructive pulmonary disease (COPD) as ‘a
common, preventable and treatable disease characterized by
persistent airflow limitation due to airway and/or alveolar
abnormalities, typically caused by significant exposure to toxic
particles or gases’ (1). The main
pathological features of COPD include chronic and abnormal
inflammatory responses mediated by the NF-κB signaling cascade in
the lungs (2) and
epithelial-mesenchymal transition (EMT) (3). As the third leading cause of death
worldwide (4), the global
mortality rate of COPD remains high, especially with the growing
global population and increasing life expectancy. Moreover, the
COVID-19 pandemic has notably impacted the quality of care and
experience for patients with COPD (5). Some studies have shown significantly
increased mortality in patients with COPD infected with COVID-19
(up to 32.2%) (5,6). Therefore, there is a need to explore
timely diagnostic and therapeutic approaches for COPD.
COPD is often considered a smoking-induced disease,
with smoking (including secondhand smoke) being the most notable
risk factor. A total of ~1.5 million of the 3 million global deaths
from COPD each year are attributed to smoking (7), and 8 million individuals die annually
from smoking-related diseases (8).
Other factors associated with COPD include air pollution (9), occupational exposure (10), environmental tobacco smoke
(11), infections and low
socioeconomic status (12).
Patients with advanced COPD often succumb to severe respiratory
distress and acute exacerbations. Current treatments, such as
bronchodilators and anti-inflammatory drugs, focus on symptom
relief and exacerbation management rather than targeting disease
progression and mortality, with the underlying mechanisms of
inflammation in COPD development and treatment remaining unclear
(13). Thus, the exploration into
COPD mechanisms and the development of novel therapies are crucial,
with an urgent need to develop treatments targeting immune
dysfunction and underlying inflammation to slow COPD
progression.
Ovarian tumor protease (OTU) domain-containing
protein 1 (OTUD1) is a member of the OTU subfamily of
deubiquitinating enzymes with an N-terminal disordered alanine,
proline, glycine-rich region, catalytic OTU domain and
ubiquitin-interacting motif. OTUD1 can cleave various ubiquitin
linkages and is involved in regulating multiple cellular functions
(14). Studies have shown that
OTUD1 inhibits the progression of non-small cell lung cancer by
mediating KLF4 stabilization, and that the deubiquitinase OTUD1,
upregulated by VE-822, inhibits the progression of lung
adenocarcinoma in vitro and in vivo by
deubiquitinating and stabilizing FHL1 (15,16),
suggesting new potential targets for treating non-small cell lung
cancer and lung adenocarcinoma. In recent years, increasing
evidence has demonstrated that OTUD1 inhibits the NF-κB pathway,
thereby negatively regulating inflammation and serving an important
role in inflammatory diseases (17,18).
Studies have shown that OTUD1 negatively regulates inflammatory
responses by inhibiting the activation of transforming growth
factor-β-activated kinase 1 (TAK1)-mediated MAPK and NF-κB
signaling pathways, providing protection against sepsis-induced
lung injury, which may be related to the ability of OTUD1 to
deubiquitinate TIPE2 (14,19). Furthermore, OTUD1 specifically
reverses K63-linked ubiquitination of RIPK1 and inhibits NEMO
recruitment, suppressing RIPK1-mediated NF-κB activation and
preventing intestinal inflammation (17). Studies have also shown that RIP2
mediates ischemic brain injury by promoting inflammatory responses,
whereas OTUD1 improves post-ischemic brain injury by specifically
cleaving K63 ubiquitination of RIP2 and inhibiting RIP2-induced
NF-κB activation (18,20).
In the present study, bioinformatics analysis
predicted the presence of N6-methyladenosine (m6A) methylation
sites in the 3′UTR of OTUD1. Related literature has indicated that
methyltransferase-like 3 (METTL3) can promote m6A methylation
(21) and COPD progression,
whereas METTL3 silencing can reduce OTUD1 mRNA m6A methylation,
thereby enhancing OTUD1 protein expression (22). YTH m6A RNA binding protein 2
(YTHDF2), a member of the YT521-B homology domain family 2, is an
m6A ‘reader’ that recognizes m6A modifications and triggers a
series of downstream biological responses. Increasing evidence has
indicated that YTHDF2 serves notable roles in various mechanisms
across both cancerous and non-cancerous conditions in patients
(23–25). Therefore, the present study aimed
to investigate the role of OTUD1 in COPD pathogenesis and its
regulation by METTL3-mediated m6A methylation. Specifically, the
study aimed to characterize OTUD1 expression patterns in COPD
progression using public datasets (GSE38974 and GSE69818) and
cigarette smoke extract (CSE)-exposed BEAS-2B cells; elucidate the
functional impact of OTUD1 on inflammation and pyroptosis through
overexpression and knockdown models, with a focus on NF-κB
signaling and inflammasome-related pathways; and delineate the
mechanism by which METTL3-driven m6A methylation, in collaboration
with the reader YTHDF2, suppresses OTUD1 expression under CSE
exposure. The findings aim to uncover novel therapeutic targets for
COPD by linking epigenetic regulation of OTUD1 to inflammatory
dysregulation in smoke-induced lung injury.
Materials and methods
Cell culture
The normal human lung epithelial cell line, BEAS-2B,
was obtained from The Cell Bank of Type Culture Collection of The
Chinese Academy of Sciences. BEAS-2B cells were cultured in a cell
incubator with 5% CO2 at 37°C using Dulbecco's Modified
Eagle's medium (DMEM; cat. no. SH30243.01; Hyclone; Cytiva)
supplemented with 10% fetal bovine serum (cat. no. 16000e044;
Gibco; Thermo Fisher Scientific, Inc.). Additionally, to avoid
bacterial contamination, 1% penicillin with streptomycin (Beijing
Solarbio Science & Technology Co., Ltd.) was added to the DMEM
and the medium was replaced every 2–3 days.
Cell transfection
Cells were transfected with plasmids using
Lipofectamine™ 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's instructions.
Briefly, cells were seeded at a density of 2×105
cells/well in 6-well plates 24 h prior to transfection to reach
70–80% confluence at the time of transfection. The concentrations
of small interfering (si)RNAs and the pcDNA3.1 plasmid (Invitrogen;
Thermo Fisher Scientific, Inc.) expressing OTUD1 (NM_001145373.3,
CDS: 384–1,892) used for transfection were 50 nM and 2 µg per well,
respectively. The empty pcDNA3.1 vector was used as a negative
control for OTUD1 overexpression (vector group). The siRNAs were
synthesized by Shanghai GenePharma Co., Ltd. Transfection was
performed at 37°C in a humidified atmosphere with 5%
CO2. Following transfection for 12 h, the cells were
incubated for 48 h before being subjected to subsequent
experiments. The sequences were as follows: OTUD1 siRNA (siOTUD1)-1
sense, 5′-AUCAUAGUGUCCGUUACUGAG-3′, antisense,
5′-CUCAGUAACGGACACUAUGAU-3′; siOTUD1-2 sense,
5′-UUUUAGACAAGGAUAUGGCCA-3′, antisense,
5′-UGGCCAUAUCCUUGUCUAAAA-3′; siOTUD1-3 sense,
5′-UUUGAAAGUGCUAUUACCCUG-3′, antisense,
5′-CAGGGUAAUAGCACUUUCAAA-3′; METTL3 siRNA (siMETTL3)-1 sense,
5′-UCUAACUCAGGAUCUGUAGCU-3′, antisense,
5′-AGCUACAGAUCCUGAGUUAGA-3′; siMETTL3-2 sense,
5′-UGUGUUUAUUGAUAAUUCGUC-3′, antisense 5′-GACGAAUUAUCAAUAAACACA-3′;
siYTHDF2-1 sense, 5′-UUAAUCCAUCCUUUUGAUGUA-3′, antisense,
5′-UACAUCAAAAGGAUGGAUUAA-3′; siYTHDF2-2 sense,
5′-ACUUUUGGAACAUUGCUUGCA-3′, antisense,
5′-UGCAAGCAAUGUUCCAAAAGU-3′; siRNA-negative control (siNC) sense,
5′-UUCUCAGAACGUGUCACAU-3′, antisense:
5′-AUGUGACACGUUCUGAGAA-3′.
Preparation of CSE and cell
treatment
The aim of the present study was to simulate the
microenvironment of cigarette smoke exposure in cell culture. The
CSE solution was prepared on the day of the experiment. First,
commercial cigarettes (each containing 2.5 mg nicotine and 12 mg
tar; purchased from Shanghai, China) were drawn into a flask
containing 10 ml DMEM at a rate of 1 cigarette every 3 min using a
vacuum pump. The pH of the CSE solution was adjusted to 7.4, and
the OD (A320-A540) was maintained between 0.9 and 1.2. The CSE
solution was then filtered through a 0.22-µm filter to remove
bacteria and other microorganisms. This solution was considered to
be 100% CSE and was further diluted with DMEM to a working
concentration of 10% for subsequent experiments, which were
performed within 1 h of preparation as needed. After transfection
with siRNA, cells were treated with the prepared CSE solution to
simulate the cell culture environment. The treatment was performed
at 37°C for 24 h. Pyrrolidine dithiocarbamate (PDTC; 10 µM; cat.
no. 52202ES50; Shanghai Yeasen Biotechnology Co., Ltd.), an NF-κB
inhibitor that inhibits IκB phosphorylation, blocks NF-κB
translocation into the nucleus and reduces the expression of
downstream cytokines (26), was
used to treat siOTUD1-transfected human BEAS-2B cells at 1 h
post-transfection for 24 h at 37°C.
ELISA
Secreted IL-1β and IL-18 levels in the supernatant
were determined by ELISA. The cultured cells were centrifuged at
1,000 × g for 20 min at 4°C, and after removing the cell pellets,
the secreted IL-1β and IL-18 levels in the supernatant were
quantified using human IL-1β and IL-18 ELISA kits (cat. nos. EK101B
and EK118; Hangzhou Lianke Biology Technology Co., Ltd.), according
to the manufacturer's instructions. The concentration of IL-1β and
IL-18 was detected at 450 nm using a microplate reader (Bio-Rad
Laboratories, Inc.).
Pyroptosis assay
To detect cell pyroptosis, cultured cells
(~1×106 cells) were resuspended in PBS. Subsequently,
anti-active caspase-1 (1:1,000; cat. no. MA5-32137; Invitrogen;
Thermo Fisher Scientific, Inc.) was added and incubated for 1 h at
25°C. The cells were then washed with PBS three times to remove
non-combined caspase-1, followed by incubation with 3 µM propidium
iodide solution (cat. no. P3566; Invitrogen; Thermo Fisher
Scientific, Inc.) at room temperature for 15 min in the dark. Cell
pyroptosis was assessed by flow cytometry (NovoCyte D3000; Agilent
Technologies, Inc.) and analyzed using FlowJo 10.8.1 software (BD
Biosciences), and the rate of cell pyroptosis was calculated.
Reporter gene assay
The OTUD1 3′UTR sequence was cloned into the pGL3
vector (Promega Corporation). BEAS-2B cells were co-transfected
with siNC, siMETTL3-1 or siMETTL3-2 and the pGL3-OTUD1 3′UTR
luciferase reporter plasmid, using Lipofectamine 3000 as the
transfection reagent. The cells were cultured in 5% CO2
at 37°C for 24 h. Subsequently, the cells were lysed with lysis
buffer (cat. no. 89901; Invitrogen; Thermo Fisher Scientific,
Inc.), and the luciferase activity was measured by adding the
Renilla-Firefly Luciferase Dual Assay Kit (MedChemExpress)
luciferase substrate to the cell lysates. The luminescence was
detected using a luminometer, and the luciferase activity was
normalized to Renilla luciferase activity to assess the
effects of METTL3 knockdown on OTUD1 3′UTR-mediated reporter
expression.
RNA immunoprecipitation (RIP) and
methylated RNA immunoprecipitation (meRIP) assay
The RIP assay was performed using the Magna
MeRIP™ m6A Kit (MilliporeSigma). To isolate RNA-protein
complexes, cells were cultured until they reached 70–80%
confluence. RNA-protein complexes were crosslinked by adding 1%
formaldehyde for 10–15 min at room temperature, and then quenched
with 125 mM glycine for 5–10 min at 4°C and rinsed with cold PBS.
The cells (1×107) were lysed with a lysis buffer
containing protease, RNase and phosphatase inhibitors, and the
cells were homogenized, as required. Then, the RNA-protein
complexes were conjugated with antibodies (5 µg): Anti-YTHDF2 (cat.
no. ab220163; Abcam) or anti-IgG (cat. no. ab172730; Abcam) at 4°C
for 1 h. After which, the agarose beads and 50 µl protein A/G were
added and incubated at 4°C for 1 h. The beads were subsequently
rinsed with lysis buffer to remove unbound material, and RNA was
extracted using TRIzol® (cat. no. 15596026CN;
Invitrogen; Thermo Fisher Scientific, Inc.) and treated with DNase
to remove DNA contamination. Finally, RNA was analyzed by reverse
transcription-quantitative PCR (RT-qPCR).
For meRIP, the aforementioned RNA-protein complexes
were conjugated with 5 µg anti-m6A antibody (cat. no. ab208577;
Abcam) at 4°C for 1 h. The other processes were the same as those
performed for the RIP experiment.
Actinomycin D treatment
A stock solution of actinomycin D (1 mg/ml in DMSO;
MedChemExpress) was prepared. The stock solution was diluted to the
desired working concentration (5 µg/ml) in culture medium. For
treatment, the diluted actinomycin D solution was added to the
culture medium at the desired concentration. It was ensured that
the final concentration of DMSO did not exceed 0.1%. Cells were
treated with actinomycin D at 37°C for 4 h. The treatment was
applied after transfection to ensure that any potential effects of
actinomycin D on transcription could be accurately assessed in the
context of the transfected cells.
RT-qPCR
To quantify mRNA expression levels, RT-qPCR was
performed. Briefly, total RNA was extracted using TRIzol reagent
and quantified using a spectrophotometer. RNA was
reverse-transcribed using the PrimeScript™ RT reagent
kit (Takara Bio, Inc.) according to the manufacturer's protocol.
The RT reaction was carried out using 1 µg RNA in a total volume of
20 µl, with the following reaction conditions: 37°C for 15 min,
followed by 85°C for 5 sec to deactivate the reverse transcriptase
enzyme. qPCR was carried out using SYBR Green qPCR Master Mix
(Thermo Fisher Scientific, Inc.) on a C1000 thermal cycler (Bio-Rad
Laboratories, Inc.) with the following cycling conditions: Initial
denaturation at 95°C for 5 min; 40 cycles at 95°C for 30 sec,
annealing at 60°C for 30 sec and extension at 72°C for 30 sec; and
a final extension step at 72°C for 5 min to ensure complete
amplification. The primers used in the present study were
self-designed based on the target gene sequences retrieved from
GenBank (https://www.ncbi.nlm.nih.gov/genbank/). Primer design
was carried out using Primer3 (version 0.4.0) (https://bioinfo.ut.ee/primer3-0.4.0/) to
ensure appropriate melting temperatures and specificity. Primer
sequences were synthesized by Sangon Biotech Co., Ltd. The Cq value
was defined as the number of cycles needed for the fluorescent
signal in the reaction tube to reach the set threshold. The
relative mRNA levels normalized to GAPDH were calculated using the
2−ΔΔCq method (27).
The primer sequences for RT-qPCR were designed as follows: GAPDH
forward (F), 5′-CACCATCTTCCAGGAGCGAG-3′, reverse (R),
5′-TGATGACCCTTTTGGCTCCC-3′; OTUD1 F, 5′-TTTGGCTCAGTTGGCTCAGT-3′, R,
5′-CGCGTTTCCTTTGCACTTGA-3′; OTUD1 3′UTR F,
5′-CUUACACCCUGGGAAUAAUUG-3′, R, 5′-GATTAAGGCATTACACCTAC-3′; METTL3
F, 5′-GTGATCGTAGCTGAGGTTCGT-3′, R 5′-GGGTTGCACATTGTGTGGTC-3′;
YTHDF2, F, 5′-CAGGCAAGGCCCAATAATGC-3′, R,
5′-AAGTAGGGCATGGCTGTGTC-3′.
Western blotting
The total protein was extracted from BEAS-2B cells
using RIPA buffer (Beijing Solarbio Science & Technology Co.,
Ltd.), and the BCA protein assay kit (cat. no. A55864; Thermo
Fisher Scientific, Inc.) was used to determine the protein levels.
Subsequently, the samples were boiled at 95°C for 10 min and
proteins (20–40 µg/lane) were separated by SDS-PAGE on 10% gels at
a constant voltage (80 V) until the dye front reached the bottom of
the gel. The separated proteins were then transferred to a 0.2-µm
PVDF membrane. For transfer, a constant current of 400 mA was
applied for 1 h at 4°C to ensure efficient transfer of proteins.
The membranes were then blocked with 5% non-fat milk in TBST (20 mM
Tris-HCl, 150 mM NaCl, 0.1% Tween-20, pH 7.4) for 1 h at room
temperature to prevent non-specific binding of antibodies, followed
by incubation with the following primary antibodies: NF-κB p65
(1:1,000; cat. no. ab32536; Abcam), NF-κB phosphorylated (p-)p65
(1:1,000; cat. no. ab76302; Abcam), active caspase-1 p20 (1:1,000;
cat. no. abs154965; Absin Bioscience, Inc.), gasdermin D N-terminal
domain (GSDMD-N; 1:1,000; cat. no. ab215203; Abcam), NLR family
pyrin domain containing 3 (NLRP3; 1:1,000; cat. no. ab283819;
Abcam), METTL3 (1:1,000; cat. no. ab195352; Abcam), YTHDF2
(1:1,000; cat. no. ab220163; Abcam), OTUD1 (1:1,000; cat. no.
29921-1-AP; Proteintech Group, Inc.), and GAPDH (1:5,000; cat. no.
60004-1-Ig; Proteintech Group, Inc.) at 4°C overnight with gentle
agitation. The membrane was washed three times with TBST and then
incubated with horseradish peroxidase-conjugated secondary
antibodies (1:1,000; cat. nos. A0208 and A0216; Beyotime Institute
of Biotechnology) diluted in TBST containing 2% BSA (cat. no.
9048-46-8; Absin Bioscience, Inc.) for 1 h at 25°C. Finally, the
expression levels of proteins were measured using a
chemiluminescent imaging system (Tanon 5200; Tanon Science and
Technology Co., Ltd.).
Methylation site prediction
The present study employed a bioinformatics pipeline
to predict methylation sites in the OTUD1 gene. The full-length
mRNA sequence of Homo sapiens OTUD1 (accession no.
NM_001145373.3, including 5′-UTR, CDS and 3′-UTR regions) was
retrieved from the NCBI database (https://www.ncbi.nlm.nih.gov/nuccore/NM_001145373.3).
CpG island and methylation region analyses were performed using the
MethPrimer platform (https://www.urogene.org/methprimer/), which integrates
sequence features (CpG dinucleotide density and GC content;
thresholds: Length >200 bp, GC content >50%,
observed/expected CpG ratio >0.6) with a machine learning model
(random forest algorithm) to identify potential methylation sites,
supplemented by cross-species conservation annotations. The ‘full
prediction’ mode was selected for whole-sequence scanning, with RNA
secondary structure analysis disabled, and a confidence threshold
>0.7. Bioinformatics analysis using the SRAMP prediction server
predicted the presence of an m6A methylation modification site in
the OTUD1 3′UTR (http://www.cuilab.cn/m6asiteapp/old), and the
predicted sites were validated against conserved regions in the
MethDB (http://www.methdb.net/) and MethBank
(https://ngdc.cncb.ac.cn/methbank/)
databases, and CpG island-gene domain correlations were visualized
via the UCSC Genome Browser (https://genome.ucsc.edu/).
Statistical analysis
The relevant data from two datasets in the Gene
Expression Omnibus database [GSE38974 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE38974)
and GSE69818 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE69818)]
were integrated for subsequent analyses of differences in gene
expression. Each experiment was performed in triplicate and data
are presented as the mean ± standard deviation of three independent
biological replicates. Statistical analyses were conducted using
GraphPad Prism 7.0 software (Dotmatics). To compare differences in
mean values among multiple groups, one-way analysis of variance
followed by Tukey's post hoc test was applied. For comparisons
between two groups, an unpaired two-tailed Student's t-test was
used. P<0.05 was considered to indicate a statistically
significant difference.
Results
Cigarette smoke stimulation inhibits
OTUD1 expression
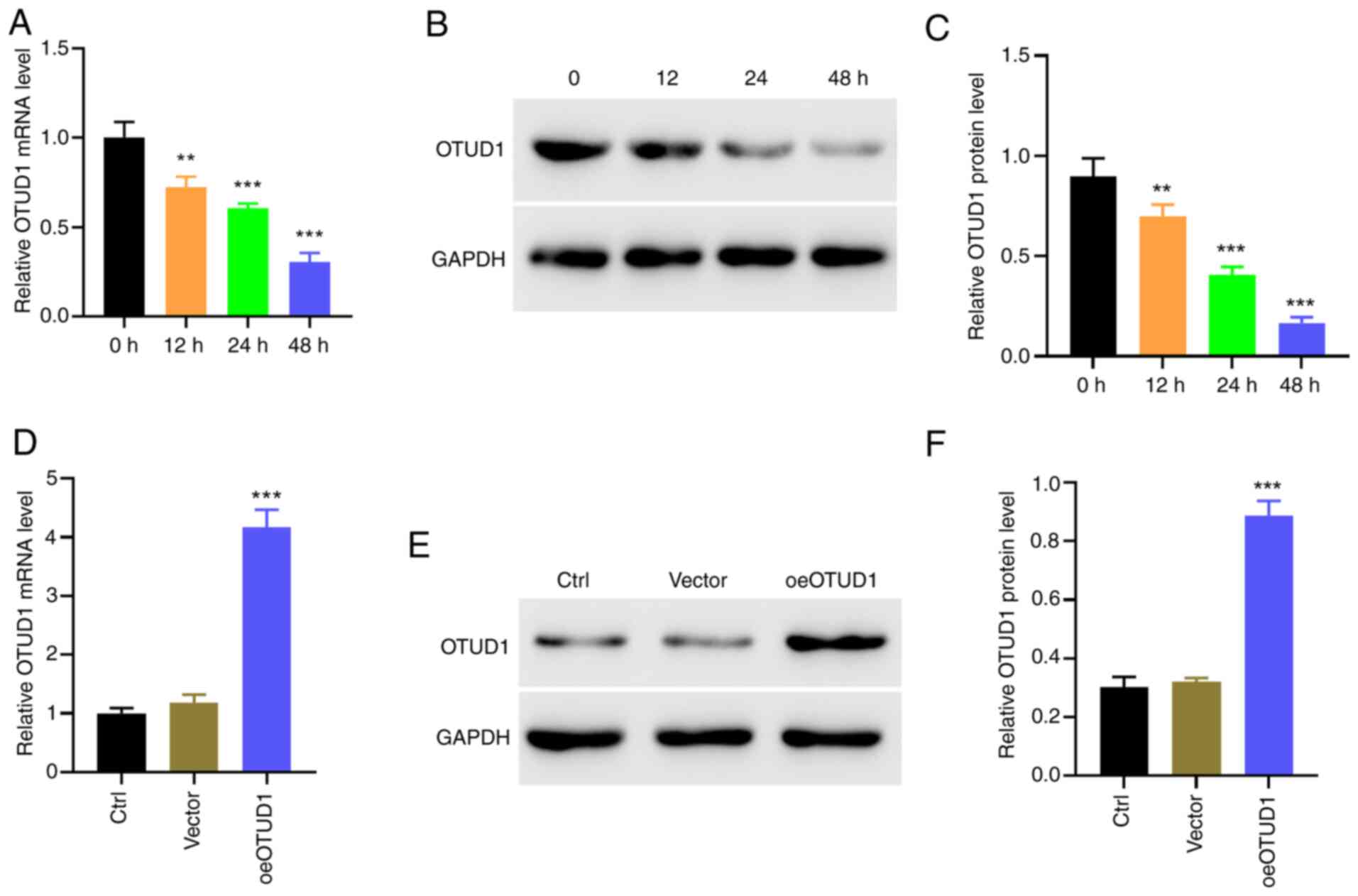
Analysis of COPD datasets GSE38974 (28) and GSE69818 (29) revealed that the expression levels
of the OTUD1 gene were lower in advanced COPD compared with those
in early COPD (Fig. S1).
Subsequently, OTUD1 expression was detected in BEAS-2B cells
treated with 10% CSE for 0, 12, 24 and 48 h using western blotting
and RT-qPCR. RT-qPCR and western blotting results indicated that
10% CSE stimulation inhibited the expression levels of OTUD1
(Fig. 1A-C), demonstrating that
cigarette smoke stimulation can decrease OTUD1 gene expression in
human BEAS-2B cells.
OTUD1 reduces cellular damage caused
by cigarette smoke stimulation by inhibiting the inflammatory
response
To further verify that OTUD1 has a protective effect
on cells stimulated with 10% CSE, an OTUD1 overexpression (oeOTUD1)
plasmid was constructed and transfected into human BEAS-2B cells,
after which, RT-qPCR and western blotting were used to verify the
transfection efficiency of oeOTUD1 (Fig. 1D-F). Subsequently, human BEAS-2B
cells were treated with 10% CSE, and pyroptosis was detected by
flow cytometry (Fig. 2A and B).
The results showed that pyroptosis was significantly increased in
the 10% CSE-treated group compared with that in the vector group,
whereas pyroptosis was reduced in the 10% CSE-treated oeOTUD1 group
compared with that in the group treated with 10% CSE alone. To
investigate whether the protective effect of OTUD1 on human BEAS-2B
cells was related to inflammation, the levels of pro-inflammatory
factors, NF-κB pathway proteins (p65 and p-p65) and proteins
related to inflammasomes (NLRP3, p20 and GSDMD-N) were further
detected. Common pro-inflammatory cytokines IL-1β and IL-18 were
selected, and the levels of IL-1β and IL-18 in cell supernatants
were detected by ELISA. The results showed that 10% CSE stimulation
of human BEAS-2B cells resulted in a significant increase in IL-1β
and IL-18 compared with that in normal human BEAS-2B cells;
however, overexpression of OTUD1 could partially inhibit the
elevation of IL-1β and IL-18 induced by cigarette smoke (Fig. 2C and D). To some extent, these
findings verified that OTUD1 could serve a protective role in cells
by reducing inflammation under cigarette smoke stimulation
conditions. To further verify this hypothesis, NF-κB p65/NF-κB
p-p65, NLRP3, active caspase-1 p20 and GSDMD-N were detected by
western blotting. The results showed that the expression levels of
NF-κB p65/NF-κB p-p65, NLRP3, active caspase-1,p20 and GSDMD-N were
significantly increased in the cell group treated with 10% CSE,
whereas they were reduced in the oeOTUD1 group (Fig. 2E-I). These findings further
validated that OTUD1 expression may serve a protective role in
cells by inhibiting inflammation.
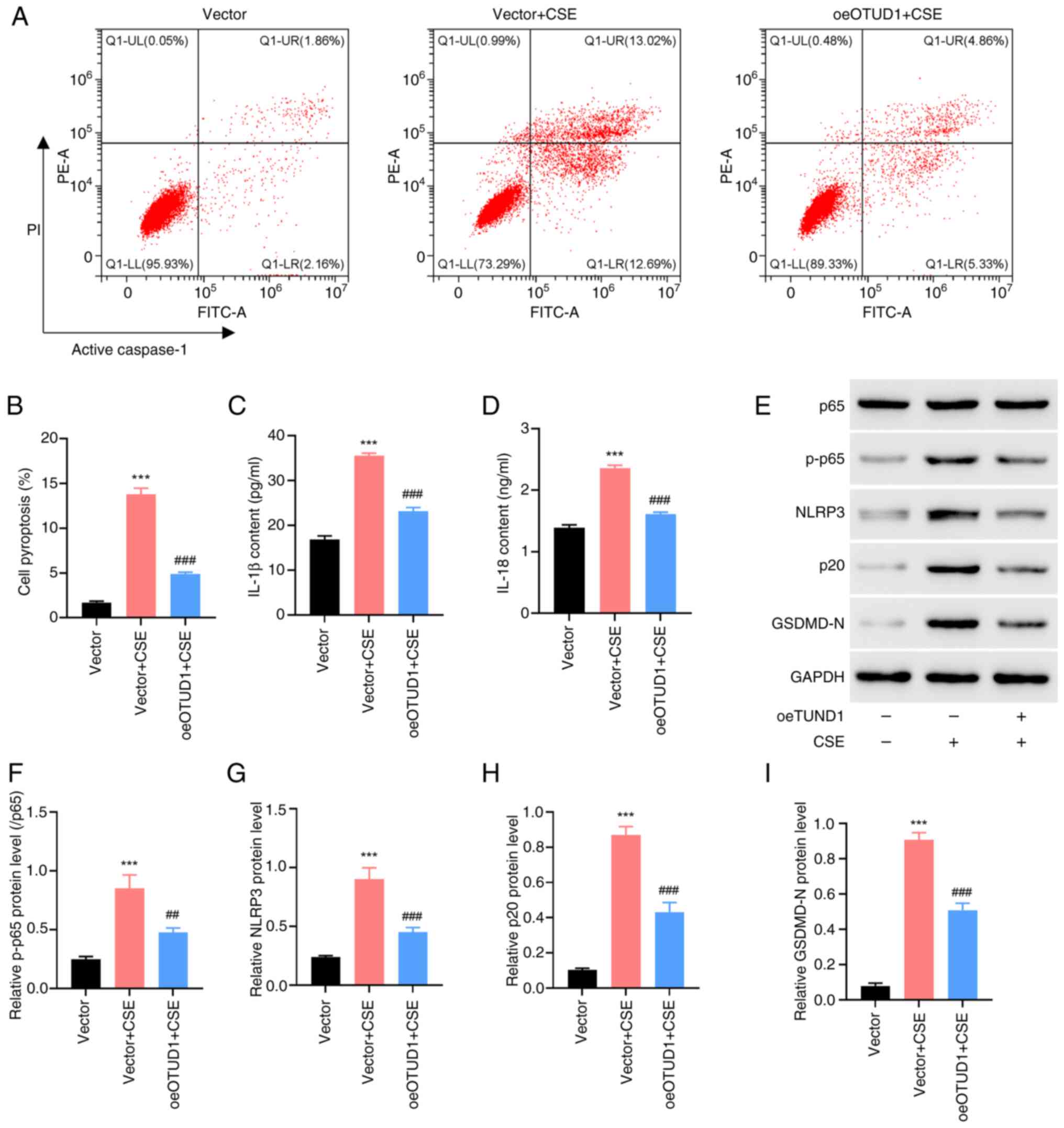 | Figure 2.oeOTUD1 ameliorates 10% CSE-induced
BEAS-2B cell pyroptosis. BEAS-2B cells were transfected with
oeOTUD1 vector and treated with 10% CSE for 24 h. (A) Flow
cytometry was performed to detect cell pyroptosis. (B) Quantitative
analysis of cell pyroptosis. ELISA was performed to detect changes
in (C) IL-1β and (D) IL-18 levels in the cell supernatants. (E)
Western blot analysis was performed to detect the protein
expression levels of (F) NF-κB p65 and NF-κB p-p65, (G) NLRP3, (H)
active caspase-1 p20 and (I) GSDMD-N. ***P<0.001 vs. Vector;
##P<0.01, ###P<0.001 vs. Vector + CSE.
Vector, empty pcDNA3.1 vector; CSE, cigarette smoke extract;
GSDMD-N, gasdermin D N-terminal domain; NLRP3, NLR family pyrin
domain containing 3; oe, overexpression; OTUD1, ovarian tumor
protease domain-containing protein 1; p-, phosphorylated. |
Reverse validation of the protective
effect of OTUD1 on cells through inhibition of the inflammatory
response in BEAS-2B cells
In the present study, reverse validation was
performed, whereby siOTUD1-transfected human BEAS-2B cells were
constructed, and the transfection efficiency of siOTUD1 was
verified by RT-qPCR and western blotting (Fig. 3A-C). The cells were divided into
the following three siRNA groups: i) siOTUD1-1; ii) siOTUD1-2; and
iii) siOTUD1-3. And the siOTUD1-1was used in subsequent
experiments, as it induced lower OTUD1 protein levels. The results
of the pyroptosis assay suggested that pyroptosis was significantly
increased after knockdown of OTUD1, whereas the addition of the
NF-κB inhibitor PDTC resulted in a decrease in pyroptosis compared
with that in the siOTUD1 group (Fig.
3D and E). Subsequently, ELISA was performed to detect the
levels of IL-1β and IL-18 in cell supernatants; the results were
consistent with the pyroptosis assay results, after silencing the
expression of OTUD1, the levels of the pro-inflammatory cytokines
IL-1β and IL-18 were significantly increased, whereas this increase
was inhibited by the addition of the NF-κB inhibitor PDTC (Fig. 3F and G). Furthermore, the
expression levels of NF-κB p65/NF-κB p-p65, NLRP3, active caspase-1
p20 and GSDMD-N were examined by western blotting, and the results
also showed that the expression of these proteins were markedly
increased after silencing the expression of OTUD1, whereas the
NF-κB inhibitor PDTC could attenuate the elevated inflammatory
response induced by OTUD1 knockdown (Fig. 3H-L). These findings indicated that
OTUD1 could serve a protective role in patients with COPD and that
this protective effect may be achieved through its
anti-inflammatory effects.
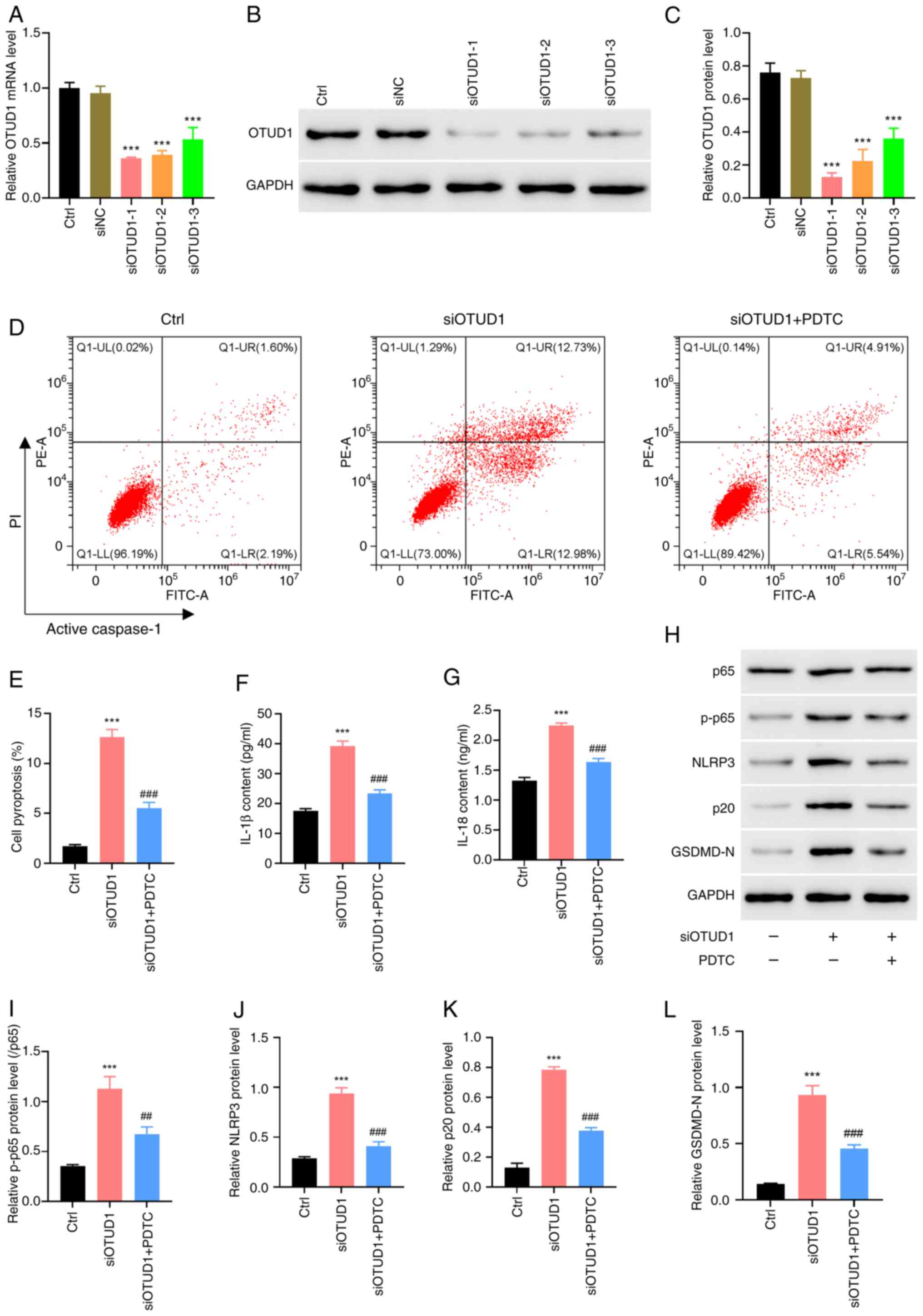 | Figure 3.siOTUD1 induces BEAS-2B cell
pyroptosis, which is inhibited by treatment with the NF-κB
inhibitor PDTC. A total of 24 h after transfection of BEAS-2B cells
with siOTUD1, changes in cytosolic mRNA and protein expression
levels of OTUD1 were detected using (A) reverse
transcription-quantitative PCR and (B) western blotting. (C)
Semi-quantitative analysis of OTUD1 protein expression. siOTUD1-1
was used for subsequent experiments. BEAS-2B cells transfected with
siOTUD1 were simultaneously co-treated with PDTC (100 µM) for 24 h.
(D) Flow cytometry was performed to detect cell pyroptosis. (E)
Quantitative analysis of cell pyroptosis. ELISA was performed to
detect the changes in (F) IL-1β and (G) IL-18 levels in cell
supernatants. (H) Western blotting was performed to detect the
protein expression levels of (I) NF-κB p65 and NF-κB p-p65, (J)
NLRP3, (K) active caspase-1 p20 and (L) GSDMD-N. ***P<0.001 vs.
Ctrl; ##P<0.01, ###P<0.001 vs. siOTUD1.
GSDMD-N, gasdermin D N-terminal domain; NC, negative control;
NLRP3, NLR family pyrin domain containing 3; oe, overexpression;
OTUD1, ovarian tumor protease domain-containing protein 1; p-,
phosphorylated; si, small interfering. |
CSE stimulation promotes OTUD1
methylation by increasing METTL3 activity, which in turn inhibits
OTUD1 gene expression
Relevant studies have shown that METTL3 expression
is significantly elevated in patients with smoking-induced COPD and
COPD cell models, and that METTL3-mediated modification of m6A RNA
methylation regulates CSE-induced EMT by targeting OTUD1 mRNA,
ultimately serving a key regulatory role in the emergence of COPD
(30–32). Bioinformatics analysis predicted
the presence of an m6A methylation modification site in the OTUD1
3′UTR. meRIP-qPCR of OTUD1 methylation levels in human BEAS-2B
cells treated with 10% CSE demonstrated that the m6A methylation
level was significantly elevated in the 10% CSE-treated cells
compared with that in the control group (Fig. 4A), which indicated that 10% CSE
stimulation could inhibit OTUD1 expression by increasing m6A.
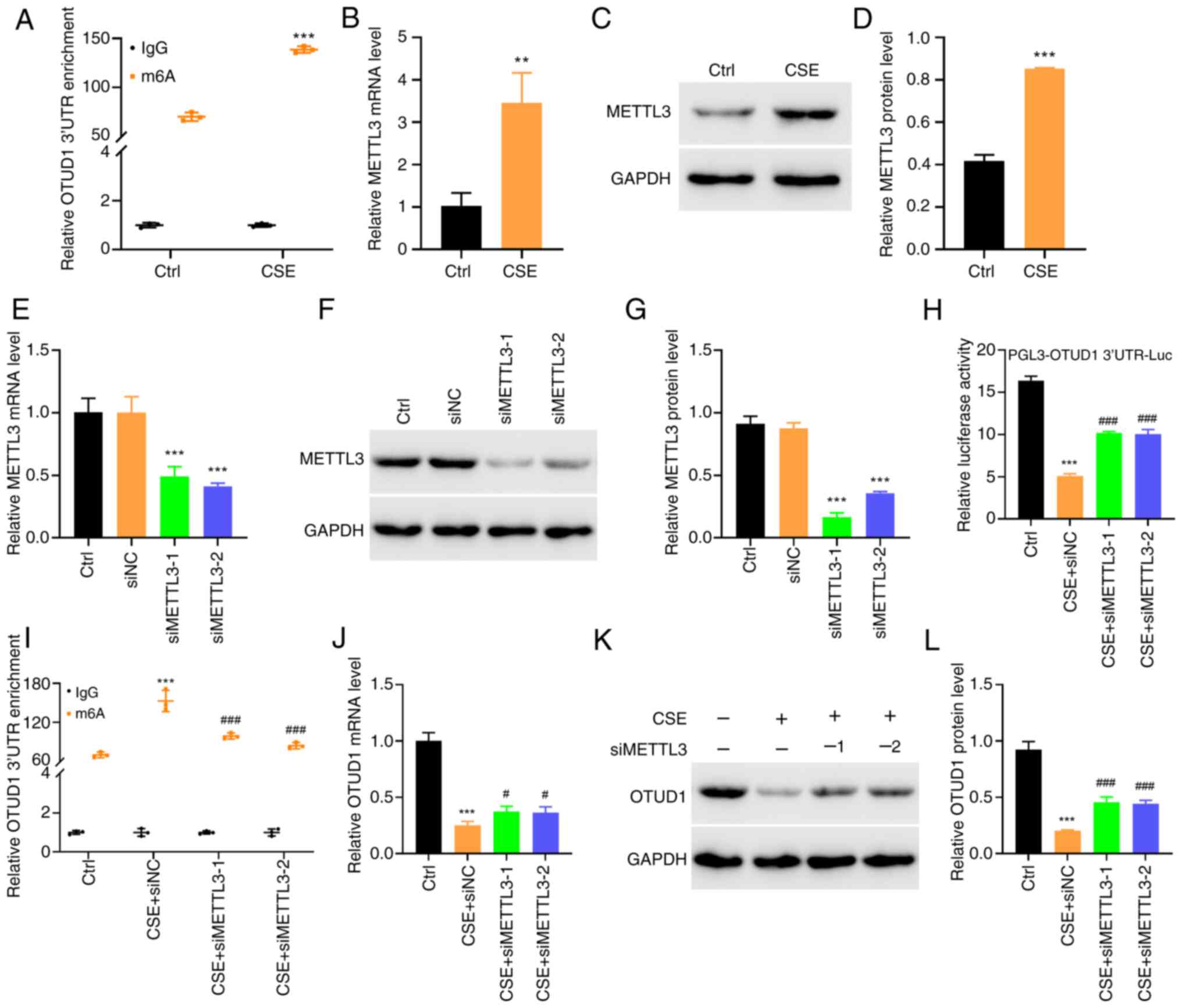 | Figure 4.CSE induces METTL3 expression,
promoting methylation of the OTUD1 3′UTR and inhibiting OTUD1
expression. (A) After treatment of BEAS-2B cells with 10% CSE for
24 h, the methylation level of the OTUD1 3′UTR was detected using
meRIP-qPCR. ***P<0.001 vs. Ctrl. (B) RT-qPCR and (C) western
blotting were used to detect changes in METTL3 mRNA and protein
expression in cells. (D) Semi-quantitative analysis of METTL3
protein expression. BEAS-2B cells were transfected with siMETTL3
for 24 h, and changes in METTL3 mRNA and protein expression were
detected by (E) RT-qPCR and (F) western blotting. (G)
Semi-quantitative analysis of METTL3 protein expression.
***P<0.001 vs. siNC. (H) BEAS-2B cells were transfected with the
pGL3-OTUD1 3′UTR luciferase reporter plasmid and siMETTL3, followed
by treatment with 10% CSE for 24 h, and OTUD1 transcriptional
activity was detected using a luciferase assay. (I) BEAS-2B cells
were transfected with siMETTL3 and treated with 10% CSE for 24 h,
and meRIP-qPCR was used to detect the methylation level of the
OTUD1 3′UTR. (J) RT-qPCR and (K) western blotting were used to
detect changes in OTUD1 mRNA and protein expression in cells. (L)
Semi-quantitative analysis of OTUD1 protein expression.
**P<0.01, ***P<0.001 vs. Ctrl; #P<0.05,
###P<0.001 vs. CSE + siNC. CSE, cigarette smoke
extract; m6A, N6-methyladenosine; meRIP-qPCR, methylated RNA
immunoprecipitation-quantitative PCR; METTL3,
methyltransferase-like 3; NC, negative control; OTUD1, ovarian
tumor protease domain-containing protein 1; RT-qPCR, reverse
transcription-quantitative PCR; si, small interfering. |
After human BEAS-2B cells were treated with 10% CSE,
METTL3 expression was detected by RT-qPCR and western blotting; the
results showed that the expression levels of METTL3 were elevated
in the 10% CSE group (Fig. 4B-D).
These findings indicated that cigarette smoke stimulation may
increase m6A methylation through increasing METTL3 activity levels,
which in turn inhibits the expression of OTUD1.
To provide further verification, 10% CSE-treated
human BEAS-2B cells transfected with siMETTL3 underwent meRIP-qPCR
to detect the m6A methylation level of OTUD1. RT-qPCR and western
blotting were used to detect the expression levels of OTUD1, and a
luciferase reporter gene was used to detect the 3′UTR activity of
OTUD1. The results showed that the methylation level of OTUD1 was
decreased, and the expression levels of OTUD1 and the 3′UTR
activity of OTUD1 were increased in cells transfected with siMETTL3
and stimulated with 10% CSE compared with those in the group
stimulated with 10% CSE alone (Fig.
4E-L). Therefore, by knocking down METTL3, it was further
verified that 10% CSE stimulation may promote OTUD1 methylation by
increasing METTL3 activity, which in turn suppresses OTUD1 gene
expression. Since transfection with siMETTL3-1 resulted in the
lowest METTL3 protein levels, siMETTL3-1 was used in the subsequent
experiments.
Inhibition of the m6A methylation
reader YTHDF2 mitigates the increased methylation of OTUD1 induced
by 10% CSE
To assess the transfection efficiency of siYTHDF2,
the expression levels of YTHDF2 were detected in human BEAS-2B
cells transfected with siYTHDF2 using RT-qPCR and western blotting
(Fig. 5A-C). siYTHDF2-1 was used
in the subsequent experiments as it induced lower YTHDF2 protein
levels than siYTHDF2-2. Actinomycin D, a transcription inhibitor,
forms stable complexes by intercalating into DNA, blocking
DNA-dependent RNA polymerase activity, and thereby inhibiting
transcription, making it a common tool for RNA stability assays
(16). BEAS-2B cells with YTHDF2
knockdown were treated with 10% CSE and 5 µg/ml actinomycin D for
0.5, 3 and 6 h, followed by RT-qPCR to assess OTUD1 transcription
levels. The results showed that OTUD1 expression was higher in the
group treated with 10% CSE combined with YTHDF2 knockdown compared
with that in the group treated with 10% CSE alone (Fig. 5D), thus indicating that YTHDF2
inhibition also suppressed m6A methylation, thereby partially
relieving the suppression of OTUD1 expression.
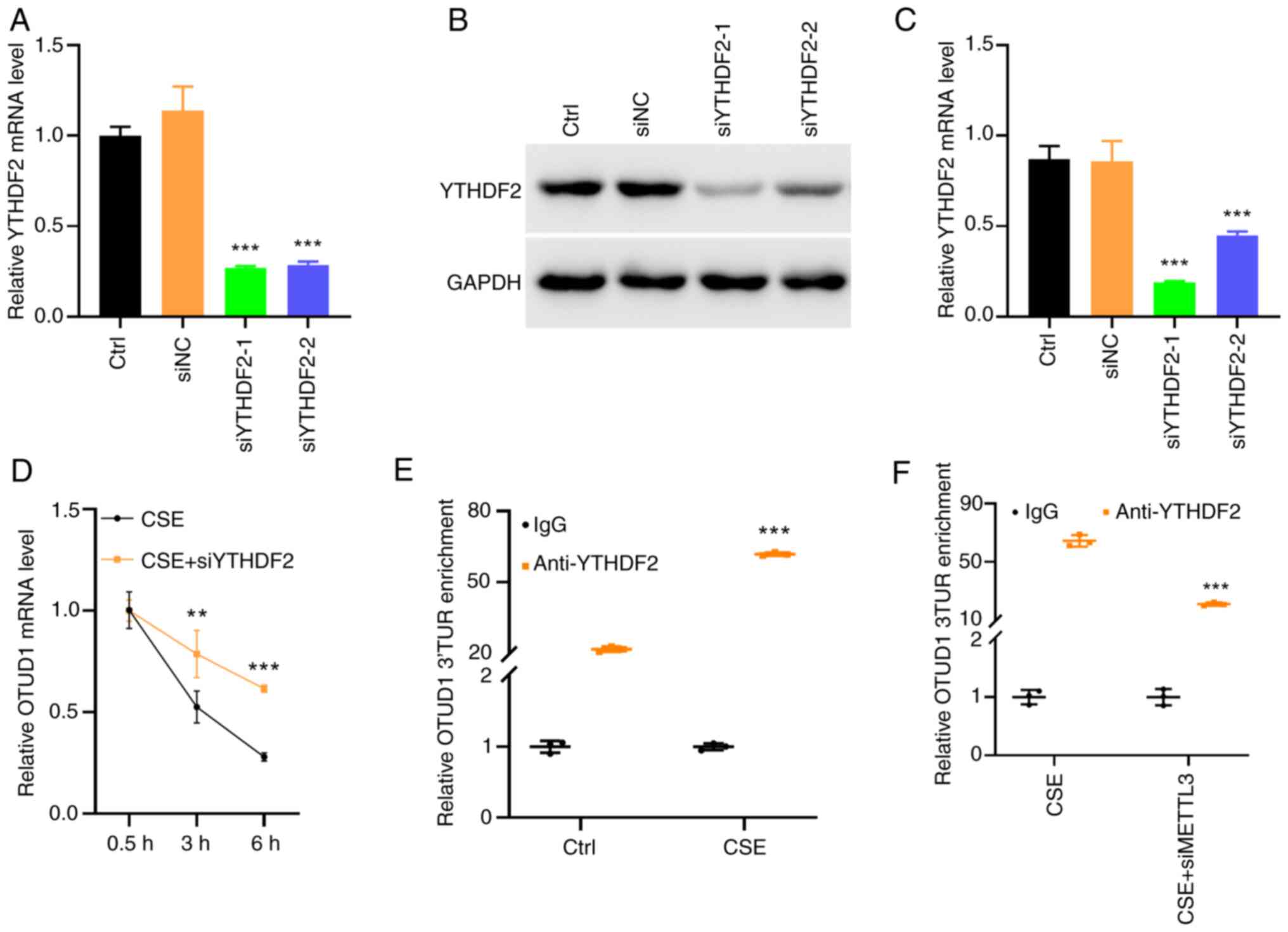 | Figure 5.YTHDF2 regulates OTUD1 transcription.
After transfecting BEAS-2B cells with siYTHDF2 for 24 h, changes in
YTHDF2 mRNA and protein expression levels were detected by (A)
RT-qPCR, (B) western blotting. (C) Smei-quantitative analysis of
YTHDF2 protein expression. ***P<0.001 vs. siNC. siYTHDF2-1 was
used for subsequent experiments. (D) After transfection of BEAS-2B
cells with siYTHDF2, cells were treated with 10% CSE and 5 µg/ml
actinomycin D for 0, 3 and 6 h, and collected for RT-qPCR to detect
changes in OTUD1 mRNA expression. **P<0.01, ***P<0.001 vs.
CSE. (E) BEAS-2B cells were treated with 10% CSE for 24 h, and the
RIP assay was used to detect the enrichment of YTHDF2 on the OTUD1
3′UTR. ***P<0.001 vs. Ctrl. (F) BEAS-2B cells were transfected
with siMETTL3 and treated with 10% CSE for 24 h, followed by RIP
assay to detect YTHDF2 enrichment on OTUD1 3′UTR. ***P<0.001 vs.
CSE. CSE, cigarette smoke extract; METTL3, methyltransferase-like
3; NC, negative control; OTUD1, ovarian tumor protease
domain-containing protein 1; RIP, RNA immunoprecipitation; RT-qPCR,
reverse transcription-quantitative PCR; si, small interfering;
YTHDF2, YTH m6A RNA binding protein 2. |
To validate this hypothesis, BEAS-2B cells were
treated with 10% CSE, and meRIP-qPCR was performed to detect the
binding of YTHDF2 to the 3′UTR of OTUD1. The results indicated that
cigarette smoke exposure increased the binding of YTHDF2 to the
OTUD1 3′UTR (Fig. 5E).
Furthermore, in BEAS-2B cells treated with 10% CSE alone or
combined with METTL3 knockdown, meRIP-qPCR was used to assess
YTHDF2 binding to the OTUD1 3′UTR. The results showed that METTL3
inhibition reduced the binding of YTHDF2 to the OTUD1 3′UTR
(Fig. 5F).
Discussion
Recent studies have shown that OTUD1 regulates the
NF-κB pathway primarily by inhibition, inducing protective effects
on sepsis-induced lung injury, inflammatory bowel disease and
inflammation-mediated ischemic brain injury (17,18).
Specifically, OTUD1 inhibits the activation of certain inflammatory
pathways, such as the NF-κB pathway, by removing ubiquitin from key
signaling proteins (33). This
deubiquitination activity prevents the activation of NF-κB, which
serves a central role in promoting inflammation. For example, in
the NF-κB pathway, OTUD1 serves a role in preventing the activation
of NF-κB by removing K63-linked ubiquitin chains from RIPK1. This
de-ubiquitination process reduces the ability of NEMO (a key
protein in the NF-κB signaling pathway) to bind to RIPK1, thus
suppressing the activation of NF-κB signaling. Essentially, OTUD1
acts as a negative regulator of this pathway (34). In addition, OTUD1 inhibits
TAK1-mediated MAPK and NF-κB pathways (35) to reduce inflammation in conditions
such as sepsis-induced lung injury and ischemic brain injury.
Furthermore, OTUD1 deubiquitinase regulates NF-κB- and
KEAP1-mediated inflammatory and oxidative stress responses
(34). Overall, OTUD1 acts as a
negative regulator of these pathways by inhibiting their
activation, which aids suppression of excessive inflammation. The
anti-inflammatory effects of OTUD1 may indicate a novel role in
COPD.
In the present study, the analysis of COPD datasets
(GSE38974 and GSE69818) revealed that OTUD1 expression was lower in
advanced COPD compared with that in the earlier stage. To assess
the role of OTUD1 in COPD progression, BEAS-2B cells were
stimulated with 10% CSE, and the results indicated that OTUD1
expression was decreased under CSE. To further confirm the role of
OTUD1, oeOTUD1 models were constructed in BEAS-2B cells. Under
similar CSE conditions, cells in the oeOTUD1 group exhibited
reduced pyroptosis, decreased levels of the pro-inflammatory
cytokines IL-1β and IL-18, and reduced expression levels of NF-κB
and inflammasome-related proteins compared with those in the
control group. These findings suggested that OTUD1 may reduce cell
damage and pyroptosis induced by 10% CSE through inhibiting
inflammation. To validate this further, siOTUD1 models were
constructed and cells were also treated with the NF-κB inhibitor
PDTC. The results showed that siOTUD1 aggravated cell pyroptosis,
increased the levels of the pro-inflammatory cytokines IL-1β and
IL-18, and enhanced the expression levels of NF-κB and
inflammasome-related proteins. However, the addition of PDTC, an
NF-κB inhibitor, reduced the levels of pyroptosis and inflammation.
These findings further confirm that OTUD1 may reduce cell damage
induced by CSE by inhibiting inflammatory responses.
METTL3 and m6A are frequently dysregulated in
various pathological processes, controlling the expression of
specific genes and regulating cellular functions (36–38).
Additionally, METTL3 and m6A are actively involved in the
pathogenesis of pulmonary diseases, including chronic conditions
such as pulmonary fibrosis, pulmonary arterial hypertension and
COPD, as well as acute diseases such as pneumonia, SARS-CoV-2
infection and sepsis-induced acute respiratory distress syndrome
(39,40). The RNA m6A modification mediated by
METTL3 represents a novel mechanism underlying the onset and
progression of these pulmonary diseases. Related research has
indicated that METTL3 expression is notably increased in patients
with smoking-induced COPD and COPD cell models (41,42).
Furthermore, METTL3 silencing has been reported to inhibit the EMT
process induced by CSE in human bronchial epithelial cells
(BEAS-2B) (22). Mechanistically,
METTL3 silencing reduced m6A methylation of OTUD1 mRNA in the
current study, thereby enhancing OTUD1 protein expression.
Bioinformatics analysis predicted the presence of m6A methylation
sites in the 3′UTR of OTUD1. To explore the association between
METTL3-mediated m6A methylation and OTUD1 expression, BEAS-2B cells
were treated with 10% CSE, and RT-qPCR and western blotting were
used to detect METTL3 expression. The results showed that METTL3
levels were increased following 10% CSE stimulation, leading to
elevated m6A methylation of OTUD1 and reduced OTUD1 expression.
Upon constructing siMETTL3 models, m6A methylation levels were
decreased whereas OTUD1 expression was increased, suggesting that
10% CSE stimulation promotes m6A methylation via increased METTL3
expression, thereby inhibiting OTUD1 expression. Subsequently,
interference models were constructed for the m6A methylation reader
YTHDF2. Following cigarette smoke exposure, siYTHDF2 knockdown
alleviated the decrease in OTUD1 expression caused by cigarette
smoke. These findings suggested that cigarette smoke exposure may
inhibit OTUD1 expression by promoting m6A methylation, a process
that requires the catalytic activity of METTL3 and the involvement
of the reader YTHDF2. Cigarette smoke also increased the binding of
YTHDF2 to the 3′UTR of OTUD1, which was partially suppressed by
METTL3 knockdown. In conclusion, cigarette smoke exposure could
increase the m6A methylation of OTUD1 involving METTL3 and YTHDF2,
thereby inhibiting OTUD1 expression.
While previous studies have suggested that OTUD1 has
tumor-suppressive properties, often linked to its ability to
increase apoptosis in cancer cells (43,44),
the findings of the present study indicated that OTUD1 may reduce
pyroptosis in the context of COPD. This apparent contradiction may
arise from the differing biological contexts in which OTUD1
functions. In cancer, the inhibition of NF-κB and other
inflammatory pathways by OTUD1 often results in increased apoptosis
of tumor cells, which is beneficial for limiting tumor progression
(45). However, in COPD, the role
of OTUD1 may shift towards protecting healthy lung cells from
excessive inflammatory responses, such as pyroptosis, which is
commonly induced by cigarette smoke exposure (46).
In the present study, oeOTUD1 reduced pyroptosis in
BEAS-2B cells treated with CSE, suggesting that OTUD1 serves a
protective role in the lungs under inflammatory stress conditions.
This protective effect is likely due to its ability to inhibit
excessive inflammation by suppressing NF-κB activation and
inflammasome-related proteins, such as NLRP3 and GSDMD-N. Through
this mechanism, OTUD1 may reduce cell damage and maintain cellular
integrity, which contrasts with its pro-apoptotic effect in the
context of cancer cells, where inflammation and immune evasion are
significant factors. Further investigation into the specific
mechanisms by which OTUD1 modulates inflammation and pyroptosis in
different disease contexts will help to clarify its dual role in
cellular responses.
In conclusion, in patients with COPD affected by
cigarette smoke exposure, OTUD1 expression may decrease as the
disease progresses. The OTUD1 gene reduces cell damage and
inflammation caused by cigarette smoke exposure by inhibiting
inflammatory responses. CSE (10%) was shown to inhibit OTUD1
expression by upregulating METTL3, which may enhance
METTL3-mediated methylation and promote YTHDF2 recognition of OTUD1
methylation. Although the present study revealed the potential role
of OTUD1 through database analysis and cellular experiments, these
findings have not yet been validated in patient samples. Future
research should use clinical data to analyze OTUD1 expression in
tissue samples obtained from patients with COPD and to assess its
association with disease severity, with the aim of enhancing the
clinical applicability of these findings.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the Natural Science
Foundation of China (grant no. 2023YFC3043507), Shanghai Pudong
Hospital and the Discipline Construction Promoting Project of
Shanghai Pudong Hospital (grant no. Zdzk2020-11).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JG, ZS, WT, ZW and YS designed the project. ZS, WT,
JX, WC and ZC performed experiments, data analysis and
interpretation. The manuscript was drafted by JG and YS. Study
supervision was carried out by ZW and YS. ZW and YS confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
COPD
|
chronic obstructive pulmonary
disease
|
|
OTUD1
|
ovarian tumor protease
domain-containing protein 1
|
|
CSE
|
cigarette smoke extract
|
|
m6A
|
N6-methyladenosine
|
|
METTL3
|
methyltransferase-like 3
|
|
YTHDF2
|
YTH m6A RNA binding protein 2
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
|
RIP
|
RNA immunoprecipitation
|
|
TAK1
|
transforming growth factor β-activated
kinase 1
|
|
EMT
|
epithelial-mesenchymal transition
|
|
GSDMD-N
|
gasdermin D N-terminal domain
|
|
NLRP3
|
NLR family pyrin domain containing
3
|
References
|
1
|
Agustí A, Celli BR, Criner GJ, Halpin D,
Anzueto A, Barnes P, Bourbeau J, Han MK, Martinez FJ, Montes de Oca
M, et al: Global initiative for chronic obstructive lung disease
2023 report: GOLD executive summary. Eur Respir J. 61:23002392023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Brandsma CA, Van den Berge M, Hackett TL,
Brusselle G and Timens W: Recent advances in chronic obstructive
pulmonary disease pathogenesis: From disease mechanisms to
precision medicine. J Pathol. 250:624–635. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Su X, Wu W, Zhu Z, Lin X and Zeng Y: The
effects of epithelial-mesenchymal transitions in COPD induced by
cigarette smoke: An update. Respir Res. 23:2252022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
GBD Chronic Respiratory Disease
Collaborators, . Prevalence and attributable health burden of
chronic respiratory diseases, 1990–2017: A systematic analysis for
the global burden of disease study 2017. Lancet Respir Med.
8:585–596. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Madawala S, Quach A, Lim JY, Varatharaj S,
Perera B, Osadnik C and Barton C: Healthcare experience of adults
with COPD during the COVID-19 pandemic: A rapid review of
international literature. BMJ Open Respir Res. 10:e0015142023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Rabbani G, Shariful Islam SM, Rahman MA,
Amin N, Marzan B, Robin RC and Alif SM: Pre-existing COPD is
associated with an increased risk of mortality and severity in
COVID-19: A rapid systematic review and meta-analysis. Expert Rev
Respir Med. 15:705–716. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
GBD 2017 Risk Factor Collaborators, .
Global, regional, and national comparative risk assessment of 84
behavioural, environmental and occupational, and metabolic risks or
clusters of risks for 195 countries and territories, 1990–2017: A
systematic analysis for the global burden of disease study 2017.
Lancet. 392:1923–1994. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
GBD 2019 Tobacco Collaborators, . Spatial,
temporal, and demographic patterns in prevalence of smoking tobacco
use and attributable disease burden in 204 countries and
territories, 1990–2019: A systematic analysis from the global
burden of disease study 2019. Lancet. 397:2337–2360. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Niu Y, Niu H, Meng X, Zhu Y, Ren X, He R,
Wu H, Yu T, Zhang Y, Kan H, et al: Associations between air
pollution and the onset of acute exacerbations of COPD: A
time-stratified case-crossover study in China. Chest. 166:998–1009.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pathak U, Gupta NC and Suri JC: Risk of
COPD due to indoor air pollution from biomass cooking fuel: A
systematic review and meta-analysis. Int J Environ Health Res.
30:75–88. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xing Z, Yang T, Shi S, Meng X, Chai D, Liu
W, Tong Y, Wang Y, Ma Y, Pan M, et al: Combined effect of ozone and
household air pollution on COPD in people aged less than 50 years
old. Thorax. 79:35–42. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lamichhane DK, Leem JH and Kim HC:
Associations between ambient particulate matter and nitrogen
dioxide and chronic obstructive pulmonary diseases in adults and
effect modification by demographic and lifestyle factors. Int J
Environ Res Public Health. 15:3632018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shakeel I, Ashraf A, Afzal M, Sohal SS,
Islam A, Kazim SN and Hassan MI: The molecular blueprint for
chronic obstructive pulmonary disease (COPD): A new paradigm for
diagnosis and therapeutics. Oxid Med Cell Longev. 2023:22975592023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ming T, Liu H, Yuan M, Tian J, Fang Q, Liu
Y, Kong Q, Wang Q, Song X, Xia Z and Wu X: The deubiquitinase OTUD1
deubiquitinates TIPE2 and plays a protective role in sepsis-induced
lung injury by targeting TAK1-mediated MAPK and NF-κB signaling.
Biochem Pharmacol. 227:1164182024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ma X, Wang L, Shi G and Sun S: The
deubiquitinase OTUD1 inhibits non-small cell lung cancer
progression by deubiquitinating and stabilizing KLF4. Thorac
Cancer. 13:761–770. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang Q, Li J, Chen Z, Jiang K, Yang K,
Huang F, Huang A, Zhang X, Zhang J and Wang H: VE-822 upregulates
the deubiquitinase OTUD1 to stabilize FHL1 to inhibit the
progression of lung adenocarcinoma. Cell Oncol (Dordr).
46:1001–1014. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wu B, Qiang L, Zhang Y, Fu Y, Zhao M, Lei
Z, Lu Z, Wei YG, Dai H, Ge Y, et al: The deubiquitinase OTUD1
inhibits colonic inflammation by suppressing RIPK1-mediated NF-κB
signaling. Cell Mol Immunol. 19:276–289. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zheng S, Li Y, Song X, Wu M, Yu L, Huang
G, Liu T, Zhang L, Shang M, Zhu Q, et al: OTUD1 ameliorates
cerebral ischemic injury through inhibiting inflammation by
disrupting K63-linked deubiquitination of RIP2. J
Neuroinflammation. 20:2812023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang Y, Fei Y, Xu X, Yao J, Wang J, Liu C
and Ding H: Shikonin attenuates cerebral ischemia/reperfusion
injury via inhibiting NOD2/RIP2/NF-κB-mediated microglia
polarization and neuroinflammation. J Stroke Cerebrovasc Dis.
33:1076892024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhong R, Wen C, Qiu Y, Shen X, Sun Z, Peng
L, Liu T, Huang S and Peng X: Anti-inflammatory and
immunomodulatory effects of Glycyrrhiza uralensis fisch. On
ulcerative colitis in rats: Role of nucleotide-binding
oligomerization domain 2/receptor-interacting protein 2/nuclear
factor-kappa B signaling pathway. J Ethnopharmacol. 344:1194572025.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang C, Chen L, Peng D, Jiang A, He Y,
Zeng Y, Xie C, Zhou H, Luo X, Liu H, et al: METTL3 and
N6-methyladenosine promote homologous recombination-mediated repair
of DSBs by modulating DNA-RNA hybrid accumulation. Mol Cell.
79:425–442.e7. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Wang L, Yan F, Yang M, Gao H and
Zeng Y: Mettl3 mediated m6A methylation involved in
epithelial-mesenchymal transition by targeting SOCS3/STAT3/SNAI1 in
cigarette smoking-induced COPD. Int J Chron Obstruct Pulmon Dis.
18:1007–1017. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Song L, Liu H, Yang W, Yin H, Wang J, Guo
M and Yang Z: Biological functions of the m6A reader YTHDF2 and its
role in central nervous system disorders. Biochem Pharmacol.
230:1165762024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gao L, Lv G, Liu Z, Tian Y, Han F, Li L,
Wang G and Zhang Y: Alcohol-induced C/EBP β-driven VIRMA decreases
oxidative stress and promotes pancreatic ductal adenocarcinoma
growth and metastasis via the m6A/YTHDF2/SLC43A2 pathway. Oncogene.
44:1118–1132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shuai Y, Ma Z, Ju J, Li C, Bai X, Yue J,
Wang X, Yuan P and Qian H: The N6-methyladenosine writer METTL3
promotes breast cancer progression through YTHDF2-dependent
posttranscriptional silencing of GSDMD. Apoptosis. 30:226–238.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cau SBA, Guimaraes DA, Rizzi E, Ceron CS,
Souza LL, Tirapelli CR, Gerlach RF and Tanus-Santos JE: Pyrrolidine
dithiocarbamate down-regulates vascular matrix metalloproteinases
and ameliorates vascular dysfunction and remodelling in
renovascular hypertension. Br J Pharmacol. 164:372–381. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ezzie ME, Crawford M, Cho JH, Orellana R,
Zhang S, Gelinas R, Batte K, Yu L, Nuovo G, Galas D, et al: Gene
expression networks in COPD: microRNA and mRNA regulation. Thorax.
67:122–131. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Faner R, Cruz T, Casserras T,
López-Giraldo A, Noell G, Coca I, Tal-Singer R, Miller B,
Rodriguez-Roisin R, Spira A, et al: Network analysis of lung
transcriptomics reveals a distinct B-cell signature in emphysema.
Am J Respir Crit Care Med. 193:1242–1253. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xia H, Wu Y, Zhao J, Cheng C, Lin J, Yang
Y, Lu L, Xiang Q, Bian T and Liu Q: N6-Methyladenosine-modified
circSAV1 triggers ferroptosis in COPD through recruiting YTHDF1 to
facilitate the translation of IREB2. Cell Death Differ.
30:1293–1304. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang K, Sun X, Xu X, Lu J, Zhang B, Li Q,
Wang C, Ding S, Huang X, Liu X, et al: METTL3-mediated m6A
modification of OTUD1 aggravates press overload induced myocardial
hypertrophy by deubiquitinating PGAM5. Int J Biol Sci.
20:4908–4921. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jing H, Song J, Sun J, Su S, Hu J, Zhang
H, Bi Y and Wu B: METTL3 governs thymocyte development and thymic
involution by regulating ferroptosis. Nat Aging. 4:1813–1827. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen J, Wang T, Li X, Gao L, Wang K, Cheng
M, Zeng Z, Chen L, Shen Y and Wen F: DNA of neutrophil
extracellular traps promote NF-κB-dependent autoimmunity via
cGAS/TLR9 in chronic obstructive pulmonary disease. Signal
Transduct Target Ther. 9:1632024. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oikawa D, Gi M, Kosako H, Shimizu K,
Takahashi H, Shiota M, Hosomi S, Komakura K, Wanibuchi H, Tsuruta
D, et al: OTUD1 deubiquitinase regulates NF-κB- and KEAP1-mediated
inflammatory responses and reactive oxygen species-associated cell
death pathways. Cell Death Dis. 13:6942022. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Oikawa D, Shimizu K and Tokunaga F:
Pleiotropic roles of a KEAP1-associated deubiquitinase, OTUD1.
Antioxidants (Basel). 12:3502023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liu Y, Yang D, Liu T, Chen J, Yu J and Yi
P: N6-methyladenosine-mediated gene regulation and therapeutic
implications. Trends Mol Med. 29:454–467. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Su X, Lu R, Qu Y and Mu D: Diagnostic and
therapeutic potentials of methyltransferase-like 3 in liver
diseases. Biomed Pharmacother. 172:1161572024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Su X, Lu R, Qu Y and Mu D:
Methyltransferase-like 3 mediated RNA m6 A modifications
in the reproductive system: Potentials for diagnosis and therapy. J
Cell Mol Med. 28:e181282024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ledford H: How air pollution causes lung
cancer-without harming DNA. Nature. 616:419–420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Su X, Feng Y, Qu Y and Mu D: Association
between methyltransferase-like 3 and non-small cell lung cancer:
Pathogenesis, therapeutic resistance, and clinical applications.
Transl Lung Cancer Res. 13:1121–1136. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang Q, Shen J, Luo S, Yuan Z, Wei S, Li
Q, Yang Q, Luo Y and Zhuang L: METTL3-m6A methylation inhibits the
proliferation and viability of type II alveolar epithelial cells in
acute lung injury by enhancing the stability and translation
efficiency of Pten mRNA. Respir Res. 25:2762024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Ambrosino P, Vitacca M, Marcuccio G,
Spanevello A, Ambrosino N and Maniscalco M: A comparison of GOLD
and STAR severity stages in individuals with COPD undergoing
pulmonary rehabilitation. Chest. 167:387–401. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang Z, Fan Y, Xie F, Zhou H, Jin K, Shao
L, Shi W, Fang P, Yang B, van Dam H, et al: Breast cancer
metastasis suppressor OTUD1 deubiquitinates SMAD7. Nat Commun.
8:21162017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu H, Zhong L, Lu Y, Liu X, Wei J, Ding
Y, Huang H, Nie Q and Liao X: Deubiquitylase OTUD1 confers
Erlotinib sensitivity in non-small cell lung cancer through
inhibition of nuclear translocation of YAP1. Cell Death Discov.
8:4032022. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Zhou T, Wu Y, Qian D, Tang H, Liu X, Qiu
J, Wang D, Hong W, Meng X and Zheng Q: OTUD1 chemosensitizes
triple-negative breast cancer to doxorubicin by modulating P16
expression. Pathol Res Pract. 247:1545712023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xie W, Luo B and Zhang L: Effect of AMPK
on the apoptosis in NHBE cells of COPD induced by CSE. China J Mod
Med. 24:28–33. 2014.
|