Introduction
Of the global population ~7% is estimated to reside
at altitudes >1,500 meters, whereas ~140 million individuals
inhabit regions >2,500 meters above sea level (1) and >40 million tourists visit
high-altitude destinations annually (2). The most notable environmental
characteristic of plateaus is the presence of low pressure and low
oxygen; this hypobaric hypoxia prompts a cascade of physiological
responses. For individuals who ascend rapidly to high altitudes,
alveolar tissue hypoxia ensues upon reaching the plateau. This
hypoxia results in the physiological phenomenon of hypoxic
pulmonary vasoconstriction (HPV), which facilitates the
redistribution of pulmonary blood flow to optimize
ventilation/perfusion matching; however, when hypoxia persists
without resolution, severe HPV induces acute pulmonary hypertension
(PH) and may even precipitate high-altitude pulmonary edema. This
phenomenon has been observed in individuals who are not
acclimatized to high-altitude environments, and can manifest at any
point between 1 and 5 days after ascending to an altitude of 2,500
meters above sea level (3).
However, the current pharmacological and non-pharmacological
preventive measures lack sufficient specificity and targeting
(4). Therefore, it is imperative
to identify novel strategies to enhance the prevention and
management of high altitude-induced lung injury.
Mitophagy, defined as the selective autophagic
degradation of damaged mitochondria, represents a crucial process
for maintaining mitochondrial quality control. Notably, there is
growing evidence to suggest that the process of mitophagy is
implicated in the pathogenesis of numerous lung diseases (5). As evidenced by previous studies,
autophagy in pulmonary artery smooth muscle cells has been
demonstrated to facilitate pulmonary vascular remodeling (6–8).
However, the mechanism of mitophagy in HPV remains unclear. Prior
research has demonstrated that diminished iron availability
enhances HPV (9). While reduced
serum iron is attributed to elevated erythropoiesis, it has been
hypothesized that pulmonary edema and PH at high altitude may be
associated with low serum iron (10,11).
Notably, crosstalk exists between iron homeostatic imbalance and
mitophagy, and iron chelators have been demonstrated to act as
mitophagy inducers (12,13). Therefore, it could be noteworthy to
determine whether reduced iron bioavailability under acute hypoxia
promotes pulmonary edema and PH by altering mitophagy.
Hypoxia-inducible factor 1α (HIF1α) is a principal
transcription factor that mediates the acute hypoxic response and
is expressed in nearly all cells of diverse animal organs (14). It has been demonstrated that
HIF1α-mediated mitophagy serves a role in the development of
ischemic diseases affecting the heart and kidney (15,16).
Nevertheless, it remains unclear as to whether a reduction in iron
bioavailability under acute hypoxia leads to an increase in HIF1α
expression, thereby triggering mitophagy, and contributing to the
development of pulmonary edema and acute PH. The current study
aimed to explore the role and mechanism of iron in acute
hypoxia-induced lung injury.
Materials and methods
Animal experiments
The experimental protocols and animal care
procedures employed in the present study were approved by the
Medical Ethics Committee of Qinghai Provincial People's Hospital
(approval no. 2023-95; Xinning, China). Male Sprague-Dawley rats
(weight, 200–250 g; age, 7–8 weeks) were procured from Jiangsu
Huachuang Xinnuo Pharmaceutical Science and Technology Co.,
Ltd.
The rat model of hypoxia was created by exposing the
animals to a hypobaric hypoxia chamber (cat. no. DYC-300; Guizhou
Fenglei Oxygen Chamber Co., Ltd.). Previous studies have shown that
acute mountain sickness usually occurs within a few hours to 24 h
upon entering high-altitude environments, with symptoms peaking
within 24–72 h and then gradually subsiding (17–20).
Thus, the present study centered on the first 3 days of acute
hypoxia. Meanwhile, for an improved comparison of the physiological
changes under acute hypoxia, it additionally included the
observation results of hypoxic treatments at 7 days and 4 weeks.
The initial animal models were exposed to hypoxia or normoxia for
0, 1, 2, 3 and 7 days and for 4 weeks (n=60; 5/group; each group
comprised different animals). The iron intervention animal models
were exposed to hypoxia for 3 days (n=40; 10/group). The air
pressure and oxygen content corresponded to an altitude of 6,000
meters above sea level. The pressure was 52.9 kPa, the oxygen
concentration was 10% (hypoxia), the temperature was 22.0°C, the
relative humidity was 50% and there was a 12-h light/dark cycle
(21–23).
The pharmacological intervention rat model was
generated as follows: Male Sprague-Dawley rats were administered
deferoxamine (DFO; 200 mg/kg; cat. no. HY-B0988; MedChemExpress) in
the hypoxia + DFO (HD) group or iron sucrose (20 mg/kg; cat. no.
HY-B2068; MedChemExpress) in the hypoxia + iron sucrose (HI) group
via daily intraperitoneal injection (24). The rats were then placed in a
hypoxic (10%) environment for 3 days, as previously described
(19–21). The control groups received an
equivalent volume of solvent via intraperitoneal injection under
either hypoxic (10% O2; HC group) or normoxic (21%
O2; NC group) conditions.
Invasive hemodynamic monitoring
The rats (n=3/group) were anesthetized with
isoflurane gas (induction, 2%; maintenance, 1.5–2.0%; inhalation)
and the mean pulmonary artery pressure (mPAP) was determined using
a right heart catheter. The catheter was positioned in the right
external jugular vein and advanced into the pulmonary artery via
the right ventricle. The position of the insertion was determined
by analyzing the waveforms produced by a biosignal acquisition
system (MP150; Biopac Systems, Inc.). After right cardiac
catheterization, the rats were sacrificed via cervical dislocation
under isoflurane anesthesia, and their death was confirmed by
observing the arrest of cardiac and respiratory functions and the
disappearance of the pain response. The lung tissue was then
excised and its wet weight was precisely measured. Subsequently,
the excised lung tissue underwent a 72-h drying process at 65°C;
after the completion of this period, the tissue was re-weighed to
obtain the dry weight. The lung wet/dry weight ratio was calculated
to reflect the water content of the lung tissue.
Detection of hematological parameters
and serum iron
Blood samples (1 ml) (n=3/group in the hypoxia study
at different time points; n=5/group in the pharmacological
intervention rat model) were collected from rats via cardiac
puncture under 1.5% isoflurane inhalation anesthesia, followed by
euthanasia through cervical dislocation while maintaining
anesthesia. Aliquots were transferred to EDTA-coated anticoagulant
tubes for complete blood count analysis following gentle inversion
(5–10 cycles), or were transferred to clot activator tubes for
centrifugation (4°C, 1,006 × g, 10 min) to obtain serum. Complete
blood count was performed using a YAN-305A hematology analyzer
(Shanghai Yuyan Scientific Instrument Co., Ltd.) with
species-specific calibration. Serum iron levels were quantified
using a commercial kit (cat. no. A039-1-1; Nanjing Jiancheng
Bioengineering Institute) according to the manufacturer's
guidelines. Raw data were processed for statistical analysis.
Transmission electron microscopy
Following anesthesia and euthanasia of the rats, the
heart and lungs were simultaneously removed and placed in 1X PBS.
The main pulmonary artery and its branches were meticulously
isolated from the right ventricle. Subsequently, the artery was
stabilized with 3% glutaraldehyde for 2 h at 4°C, followed by
post-fixation with 1% osmium tetroxide for 1 h at 4°C. A graded
series of acetone treatments was employed for dehydration, followed
by infiltration with Epon 812 and embedding. The semithin sections
(50 nm) were subjected to methylene blue staining at room
temperature for 2 min, whereas the ultrathin slides were
meticulously sliced using a single diamond knife and were
dual-stained with uranyl acetate at room temperature for 30 min and
lead citrate at room temperature for 15 min. Subsequently, the
slides were subjected to observation under a JEM-1400-FLASH
transmission electron microscope (JEOL Ltd.).
Morphological analysis of the lung
vessels
The lung tissue and main pulmonary artery from 3
rats/group were fixed in 4% paraformaldehyde at room temperature
for 12 h, dehydrated in a graded series of ethanol, cleared with
xylene (2×10 min) and embedded in paraffin via triple wax immersion
(56°C, 1 h/cycle). Sections (5 µm) were then stained with
hematoxylin at room temperature for 5 min and eosin at room
temperature for 3 min following standard hematoxylin and eosin
(H&E) staining protocols. An image was acquired using a BA400
digital light microscope with a digital interface (Motic
Incorporation, Ltd.). The rat lung tissue was evaluated for
evidence of lung injury in accordance with the established criteria
for pathological assessment of lung injury, as outlined by the
American Thoracic Society (25).
The scoring indexes were based on the following criteria:
Neutrophil infiltration of lung tissue, hyaline membrane formation,
fibrin deposition in alveoli and thickening of alveolar septa. Each
index was scored on a 5-point scale, with the severity of injury
corresponding to the following categories: No injury, 0 points;
mild injury, 1 point; moderate injury, 2 points; severe injury, 3
points; and very severe injury, 4 points (22,23).
A total of five randomly selected fields of H&E-stained lung
tissue from each animal were scored and two experts scored the
sections in a double-blind manner.
Immunohistochemical staining
The protocols for fixation, embedding and sectioning
of the main pulmonary artery and alveolar tissue were the same as
those performed for H&E staining. Sections were deparaffinized
in xylene (2×10 min), rehydrated through a graded ethanol series (5
min/step), and subjected to microwave-mediated antigen retrieval in
citrate buffer (pH 6.0) at sub-boiling temperature for 10 min.
After cooling, endogenous peroxidase was blocked with 3%
H2O2 for 10 min at room temperature, followed
by blocking with 5% BSA (cat. no. SW3015; Beijing Solarbio Science
& Technology Co., Ltd.) for 30 min at room temperature. Primary
anti-HIF1α antibody (1:100; cat. no. ab179483; Abcam) was applied
overnight at 4°C, followed by an incubation with a HRP-conjugated
goat anti-rabbit secondary antibody (1:100; cat. no. GB23303; Wuhan
Servicebio Technology Co., Ltd.) for 1 h at room temperature;
Subsequently, DAB was employed to develop the color (cat. no.
GB1212; Wuhan Servicebio Technology Co., Ltd.), hematoxylin was
used to counterstain the nuclei at room temperature for 10 min and
neutral balsam was used to seal the mount. Observations were
conducted using a BA400 digital light microscope (Motic
Incorporation, Ltd.).
Cell culture
Human-derived primary pulmonary artery endothelial
cells (cat. no. 337714) were obtained from BeNa Culture Collection;
Beijing Beina Chunglian Institute of Biotechnology. The Research
Ethics Committee of Qinghai Provincial People's Hospital confirmed
that commercially procured human primary cells fall outside the
scope of required ethics approval. At the time of purchase, the
cells were in the first passage. The cells were cultured in
endothelial cell medium (cat. no. ECM 1001; ScienCell Research
Laboratories, Inc.) containing 1% vascular endothelial growth
factor at 37°C with 5% CO2. The cells were passaged, and
those between passages 3 and 5 were used in the present study. The
cells in the hypoxia group were cultured at 37°C under hypoxia (1%
O2) for 2 h.
Cellular pharmacological
interventions
Human-derived pulmonary artery endothelial cells
were treated with DFO (100 µmol/l; cat. no. HY-B0988;
MedChemExpress) in the HD group or with iron sucrose (50 µmol/l;
cat. no. HY-B2068; MedChemExpress) in the HI group as previously
described (26). The control
groups received an equivalent volume of solvent under either
hypoxic (1% O2, HC group) or normoxic (21%
O2, NC group) conditions.
Immunofluorescence assay
For immunofluorescence experiments conducted in
vivo (n=3/group), lung vascular tissue samples underwent
fixation, dehydration, embedding and sectioning following the
aforementioned standard H&E protocols. Subsequently, the
sections were dewaxed, rehydrated and subjected to
microwave-assisted antigen retrieval in citrate buffer (pH 6.0), as
per the immunohistochemistry methods. After blocking with
immunostaining buffer (cat. no. P0102; Beyotime Institute of
Biotechnology) for 1 h at room temperature, the following primary
antibodies were used to incubate the sections overnight at 4°C:
PTEN-induced putative kinase 1 (PINK1 1:100; cat. no. GB114934-100;
Wuhan Servicebio Technology Co., Ltd.), smooth muscle actin (SMA;
1:100; cat. no. A2547; MilliporeSigma) and HIF1α (1:100; cat. no.
ab179483; Abcam). Samples were washed three times with 1X PBS (10
min each), followed by incubation with a fluorescent secondary
antibody [Goat Anti-Rabbit Alexa Fluor® 488 for HIF1α
and Caspase 3 (cat. no. ab150077), Alexa Fluor 647 for PINK1 (cat.
no. ab150079) or Goat Anti-Mouse Alexa Fluor 647 for SMA (cat. no.
ab150115); 1:1,000; Abcam] for 1 h at room temperature. Nuclei were
counterstained with DAPI, and images were acquired using a confocal
laser microscope (FV1000; Olympus Corporation).
Pulmonary arterial endothelial cells were routinely
cultured to an appropriate density and were treated with hypoxia
and DFO or iron sucrose for 2 h. Cells were fixed with 4%
paraformaldehyde for 15 min at room temperature, permeabilized with
0.1% Triton X-100 for 10 min at room temperature and blocked with
immunostaining buffer (cat. no. P0102; Beyotime Institute of
Biotechnology) for 1 h at room temperature. Subsequently, the
sections were incubated with the primary antibodies against Caspase
3 (1:100; cat. no. 19677-1-AP; Proteintech Group, Inc.) and HIF1α
(1:100; cat. no. ab179483; Abcam) overnight at 4°C. The secondary
antibody incubation, nuclear counterstaining with DAPI and confocal
imaging (FV1000; Olympus Corporation) were performed according to
the aforementioned protocols.
To assess mitochondrial membrane potential (MMP) and
apoptosis of pulmonary arterial endothelial cells, mitochondrial
superoxide and Annexin V-FITC fluorescence assays were conducted in
accordance with the instructions provided in the MitoSOX and
Annexin V-FITC kits (cat. nos. S0061M and C1071M; Beyotime
Institute of Biotechnology). The images were examined using a
fluorescence microscope (THUNDER Imager; Leica Microsystems, Inc.)
and the immunofluorescence intensity of protein signals was
subsequently semi-quantified using ImageJ software (version 1.54f;
National Institutes of Health).
Western blot analysis
Proteins were extracted from lung tissues and
pulmonary arterial endothelial cells using RIPA lysis buffer (cat.
no. P0013B; Beyotime Institute of Biotechnology) and were
quantified using a BCA kit (Thermo Fisher Scientific, Inc.). A
20-µg protein sample from each group was then subjected to SDS-PAGE
on a 10% gel, followed by transfer to a polyvinylidene fluoride
membrane (PVDF; 0.22 µm; Merck KGaA). Following the transfer of the
proteins, 5% skimmed milk (BD Biosciences) was used to block the
PVDF membranes for 1 h at room temperature. The membranes were then
incubated overnight at 4°C with the following primary antibodies:
LC3 (1:1,000; cat. no. 14600-1-AP) and β-actin (1:5,000; cat. no.
66009-1-Ig) (both from Proteintech Group, Inc.). Subsequently, the
membranes were incubated with HRP-conjugated secondary antibodies
(1:5,000; cat. nos. E-AB-1003 and E-AB-1001; Wuhan Elabscience
Biotechnology Co., Ltd.) for 1 h at room temperature and
chemiluminescence signals were detected using an ECL kit (Abbkine
Scientific Co., Ltd.). Finally, ImageJ software (version 1.54f) was
employed to semi-quantify the intensity of the bands (27–29).
Cell proliferation assay
The Cell Counting Kit 8 (CCK8) assay (cat. no.
E-CK-A362; Wuhan Elabscience Biotechnology Co., Ltd.) was used to
investigate cell proliferation in accordance with the
manufacturer's instructions. The absorbance at 450 nm was measured
using a microplate reader (Biotek; Agilent Technologies, Inc.). In
addition, cell proliferation was detected using the Edu Cell
Proliferation Kit (cat. no. C0071L; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. Images
were captured using a fluorescence microscope (ECHO/RVL-100;
Discover Echo, Inc.) and ImageJ software (version 1.54f) was
employed for data analysis.
Statistical analysis
All data were processed using the statistical
software package SPSS 28.0 (IBM Corp.) and the graphical software
package GraphPad 10.0 (Dotmatics). Data with a normal distribution
are presented as the mean ± standard deviation. Statistical
comparisons of parameters (including lung wet/dry weight ratio,
serum iron and erythroid parameters) across different durations of
hypoxic treatment were performed using two-way ANOVA, followed by
Tukey's honestly significant difference post hoc test for multiple
comparisons. One-way ANOVA followed by the Tukey's post hoc test
was performed for multiple group comparisons when the data met the
assumptions of normality and homogeneity of variance. Non-normally
distributed ordinal data (lung injury scores) are presented as the
median (IQR), and were analyzed using Kruskal-Wallis test with
Dunn's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Hypoxic lung injury is most severe on
day 3 of acute hypoxia in rats
The lung dry/wet weight ratio was significantly
increased on days 2 and 3 of hypoxia compared with day 0 (Fig. 1C), indicating the presence of
pulmonary edema. Lung morphology results indicated that there were
no abnormal changes in lung tissue morphology during days 1–7 under
normoxia. However, under hypoxia, erythrocyte leakage from alveolar
spaces accompanied by inflammatory cell infiltration was observed.
This phenomenon was most pronounced on day 3 of acute hypoxia and
then gradually resolved, with a marked infiltration of inflammatory
cells reappearing after 4 weeks of hypoxia (Fig. 1A). Semi-quantitative analysis
further confirmed that hypoxia-induced lung tissue injury was most
pronounced on day 3 and peaked at 4 weeks compared with on day 0
under hypoxia (Fig. 1B).
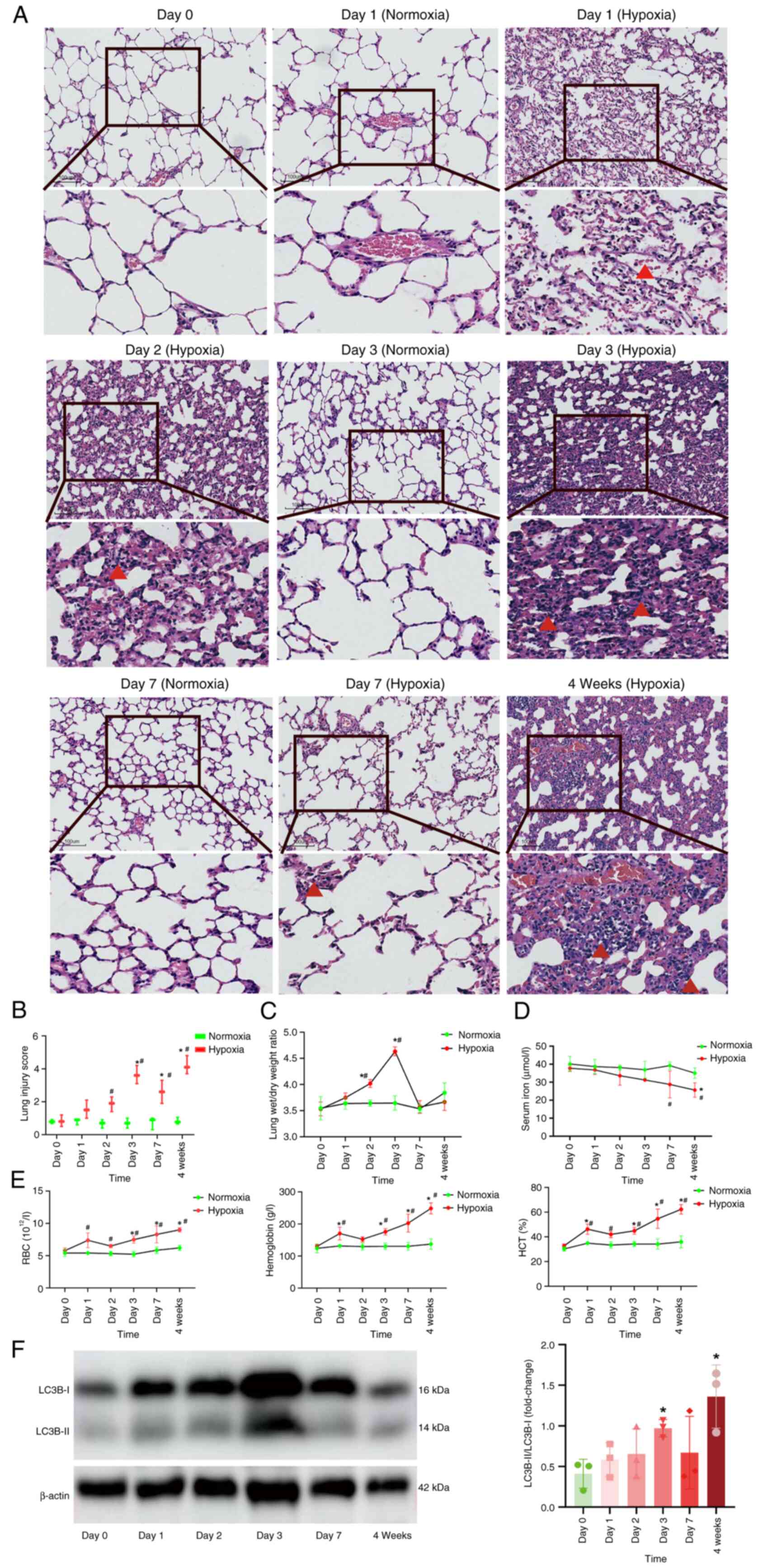 | Figure 1.Alterations in rats under hypoxia at
different time intervals. (A) Overview and magnified views of
hematoxylin and eosin staining micrographs of rat lung tissues
(magnification, ×100). Red triangles indicate erythrocytes in
alveolar spaces on day 1 under hypoxia and inflammatory cells on
days 2, 3 and 7 and week 4 under hypoxia. (B) Analysis of the
injury score of rat lung tissues, n=3. (C) Analysis of the wet/dry
weight ratio of rat lung tissues, n=3. (D) Serum iron concentration
in rats, n=3. (E) RBC, hemoglobin and HCT levels in rats, n=3. (F)
Representative western blotting images and analysis in rat lung
tissues, n=3. *P<0.05 vs. Day 0 group under hypoxia,
#P<0.05 vs. the normoxia group at the same time
point. RBC, red blood cell; HCT, hematocrit. |
Reduced serum iron concentration and
compensatory erythropoiesis occur in a rat model under acute
hypoxia
To elucidate the involvement of iron in acute
mountain sickness, serum iron and erythropoiesis levels were
quantified under normoxia/hypoxia at multiple timepoints. The
results demonstrated that, under normoxia, there were no
statistically significant differences in serum iron, red blood cell
(RBC), hemoglobin and hematocrit (HCT) levels among the groups. By
contrast, under hypoxia, as the duration of hypoxia increased,
serum iron concentration exhibited a gradually decreasing trend,
the serum iron concentration in the 4-week hypoxia group exhibited
statistically significant differences compared with that in the day
0 group. By contrast, RBC, HCT and hemoglobin levels were increased
with prolonged hypoxia exposure. RBC parameters in hypoxia groups
at day 3 and 7 and week 4 were significantly different compared
with those in the day 0 group under hypoxia, whereas hemoglobin and
HCT levels in hypoxia groups day 1, 3 and 7, and week 4 exhibited
significant changes relative to the day 0 control under hypoxia
(Fig. 1D and E). These findings
indicated that the oxygen content in the rats was diminished and
compensatory erythrocyte hyperplasia was exhibited. Given the
pivotal role of iron chelators in the initiation of mitophagy,
western blot analysis was performed to assess the expression levels
of the mitophagy protein LC3B. The results demonstrated that
LC3B-II/LC3B-I expression was significantly upregulated in response
to acute hypoxia, particularly on day 3 of hypoxia exposure
compared to day 0 under hypoxia, and peaked after 4 weeks of
hypoxic treatment (Fig. 1F).
Iron chelator treatment exacerbates
lung tissue injury and elevated mPAP under acute hypoxia, whereas
iron sucrose treatment attenuates lung tissue injury and elevated
mPAP induced by acute hypoxia
Given the reduced serum iron concentration observed
in rats during acute hypoxia, the present study aimed to intervene
in iron metabolism in a rat model using an iron chelator and iron
sucrose to elucidate the role of iron in acute high-altitude
sickness (Fig. 2A). The results of
right heart catheterization for mPAP measurements indicated that
the iron chelator DFO was associated with an elevation in mPAP
under acute hypoxia. By contrast, iron sucrose was associated with
a reduction in mPAP elevation under acute hypoxia (Fig. 2B and D). Morphological analysis
indicated the presence of alveolar tissue destruction and
inflammatory cell infiltration of the alveolar tissue under acute
hypoxia, which was accompanied by fracturing and disorganization of
elastic fiber septa in the main pulmonary artery (Fig. 2C). Quantitative data corroborated
the findings of H&E staining (Fig.
2E). Furthermore, the wet/dry weight ratio of the lungs was
increased under acute hypoxia (Fig.
2F). The administration of an iron chelator resulted in more
pronounced acute hypoxia-induced lung injury, whereas iron sucrose
demonstrated a protective effect against this injury. The serum
iron concentration analysis provided additional evidence supporting
the efficacy of the iron chelator and iron sucrose modeling
approaches (Fig. 2G).
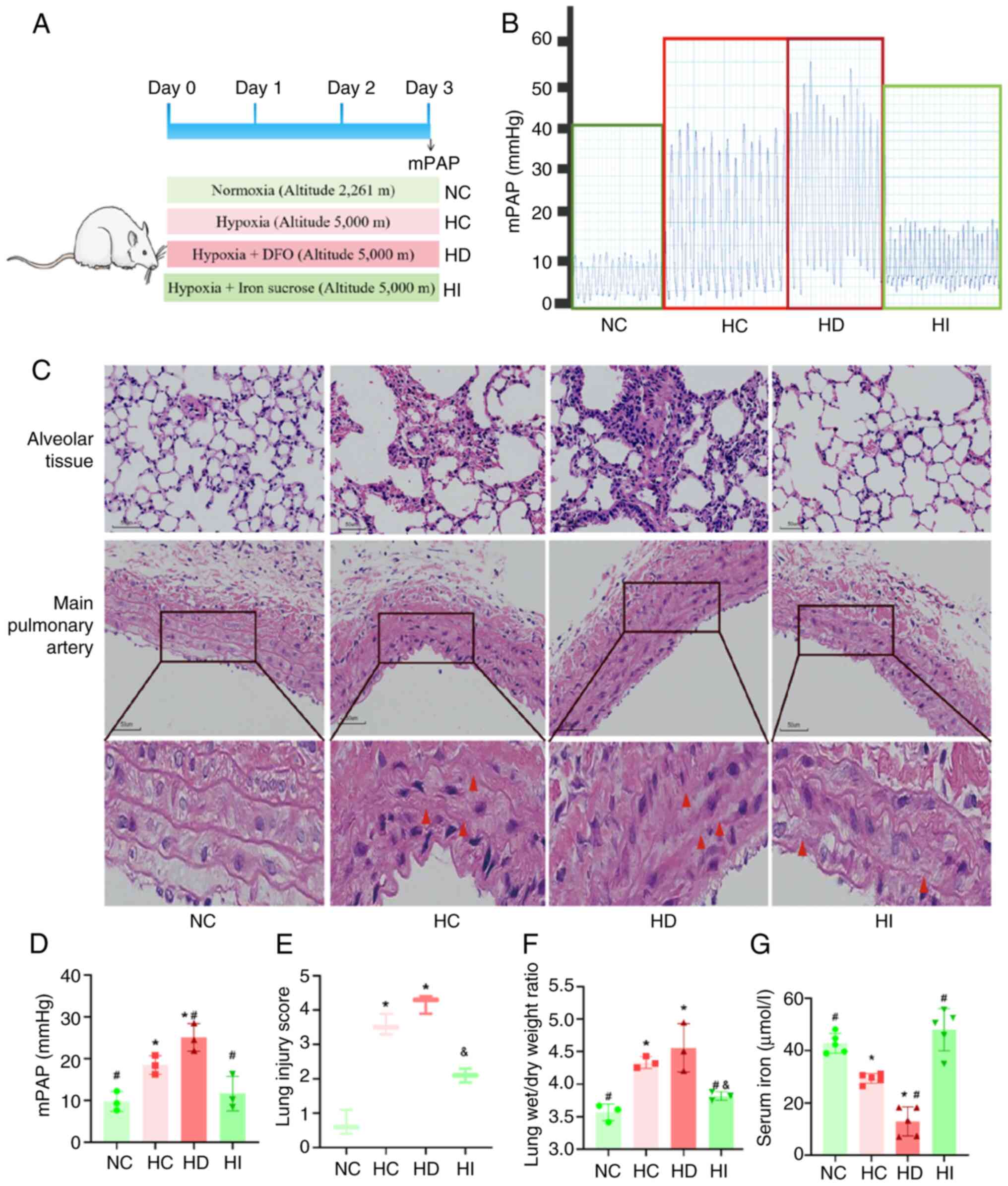 | Figure 2.Effects of iron on the pulmonary
vasculature and lung tissue under acute hypoxia. (A) Schematic
diagram of animal modeling. (B) Representative images of mPAP
waves. (C) Representative images of hematoxylin and eosin staining
of lung tissues and the main pulmonary artery in rats
(magnification, ×200). Red triangles indicate regions exhibiting
elastic fiber layer disruption and structural disorganization in
the pulmonary aorta. (D) Analysis of mPAP, n=3. (E) Analysis of the
injury score of lung tissues in rats, n=3. (F) Analysis of the
wet/dry weight ratio of lung tissues in rats, n=3. (G) Serum iron
concentration in rats, n=5. *P<0.05 vs. NC group;
#P<0.05 vs. HC group; &P<0.05 vs.
HD group. NC, normoxia control; HC, hypoxia control; HD, hypoxia +
deferoxamine; HI, hypoxia + iron sucrose; mPAP, mean pulmonary
artery pressure. |
Iron modulates the HIF1α activity of
the pulmonary vasculature and lung tissue under acute hypoxia
Given the pivotal role of iron as a cofactor for
HIF1α activation, the present study assessed HIF1α expression.
Immunohistochemistry and immunofluorescence staining of the main
pulmonary artery and alveolar tissues in rats demonstrated that
acute hypoxia resulted in an increase in HIF1α expression. In
addition, the iron chelator promoted an increase in HIF1α
expression, whereas iron sucrose inhibited the increase in HIF1α
expression under acute hypoxic conditions (Fig. 3A-E). Furthermore, alterations in
the expression of SMA in the pulmonary vasculature and lung tissue
in response to acute hypoxia were in accordance with the expression
of HIF1α (Fig. 3B-E).
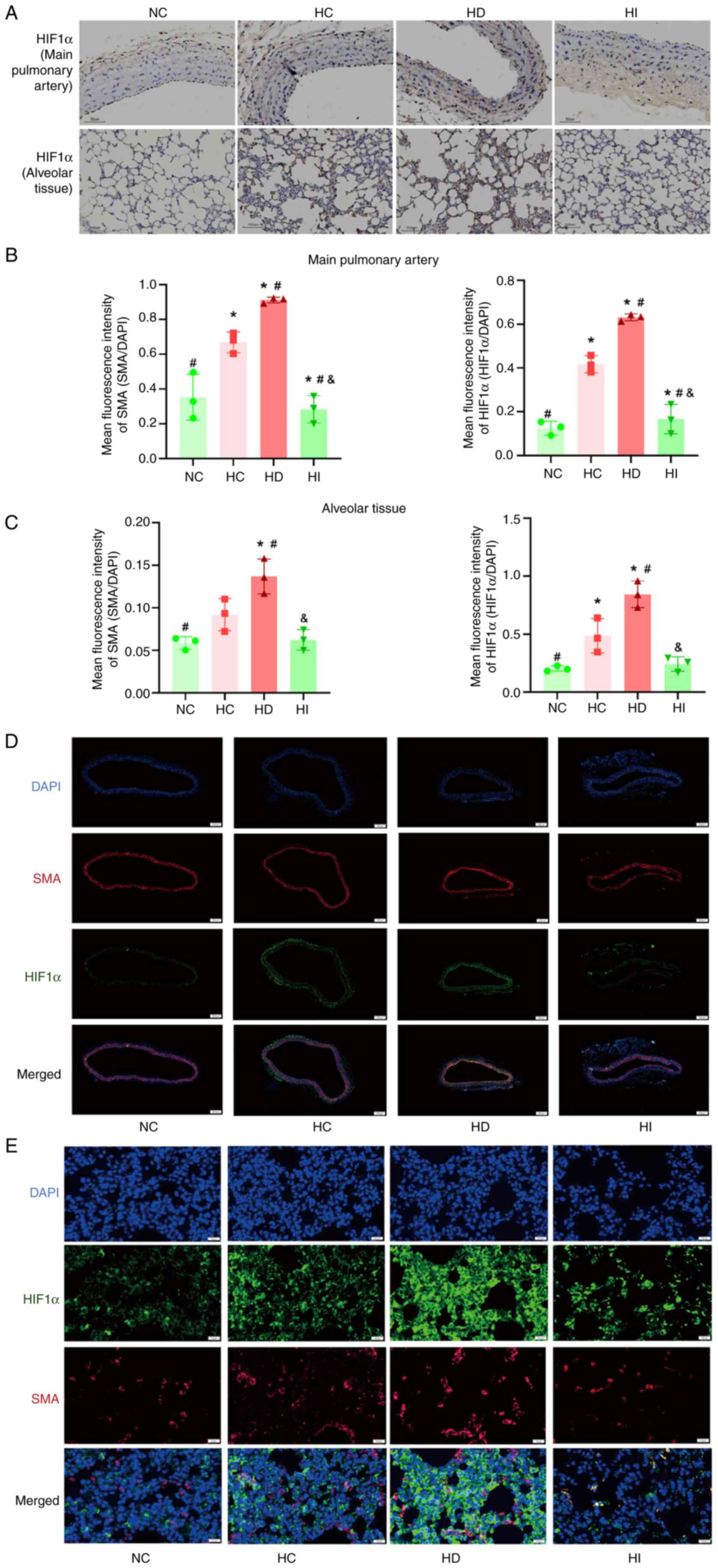 | Figure 3.Role of iron in the regulation of
HIF1α. (A) Representative HIF1α immunohistochemical staining of the
main pulmonary artery (magnification, ×200) and alveolar tissue
(magnification, ×100); brown regions indicate HIF1α-positive areas
detected by immunohistochemical staining. Analysis of
immunofluorescence staining of the (B) pulmonary vasculature and
(C) pulmonary tissue, n=3. Representative images of
immunofluorescence staining of the (D) pulmonary vasculature
(magnification, ×500) and (E) pulmonary tissue (magnification,
×400). *P<0.05 vs. NC group; #P<0.05 vs. HC group;
&P<0.05 vs. HD group. NC, normoxia control; HC,
hypoxia control; HD, hypoxia + deferoxamine; HI, hypoxia + iron
sucrose; HIF1α, hypoxia-inducible factor 1α; SMA, smooth muscle
actin. |
Iron modulates mitophagy in the
pulmonary vasculature under acute hypoxia
Given the pivotal role of iron chelators and hypoxia
in triggering mitophagy, the current study conducted a detailed
examination of this process. The results of transmission electron
microscopy of the main pulmonary artery demonstrated that acute
hypoxia resulted in mitochondrial swelling, accompanied by
increased mitophagy (Fig. 4A). The
results of immunofluorescence analysis of the pulmonary vasculature
demonstrated that acute hypoxia resulted in elevated expression of
PINK1, a pivotal protein for mitophagy (Fig. 4B and D); this was accompanied by an
increase in the expression of the mitophagy protein LC3B-II.
Furthermore, the results indicated that DFO promoted hypoxia-driven
PINK1 and LC3B-II upregulation, whereas iron sucrose inhibited this
response (Fig. 4B-E).
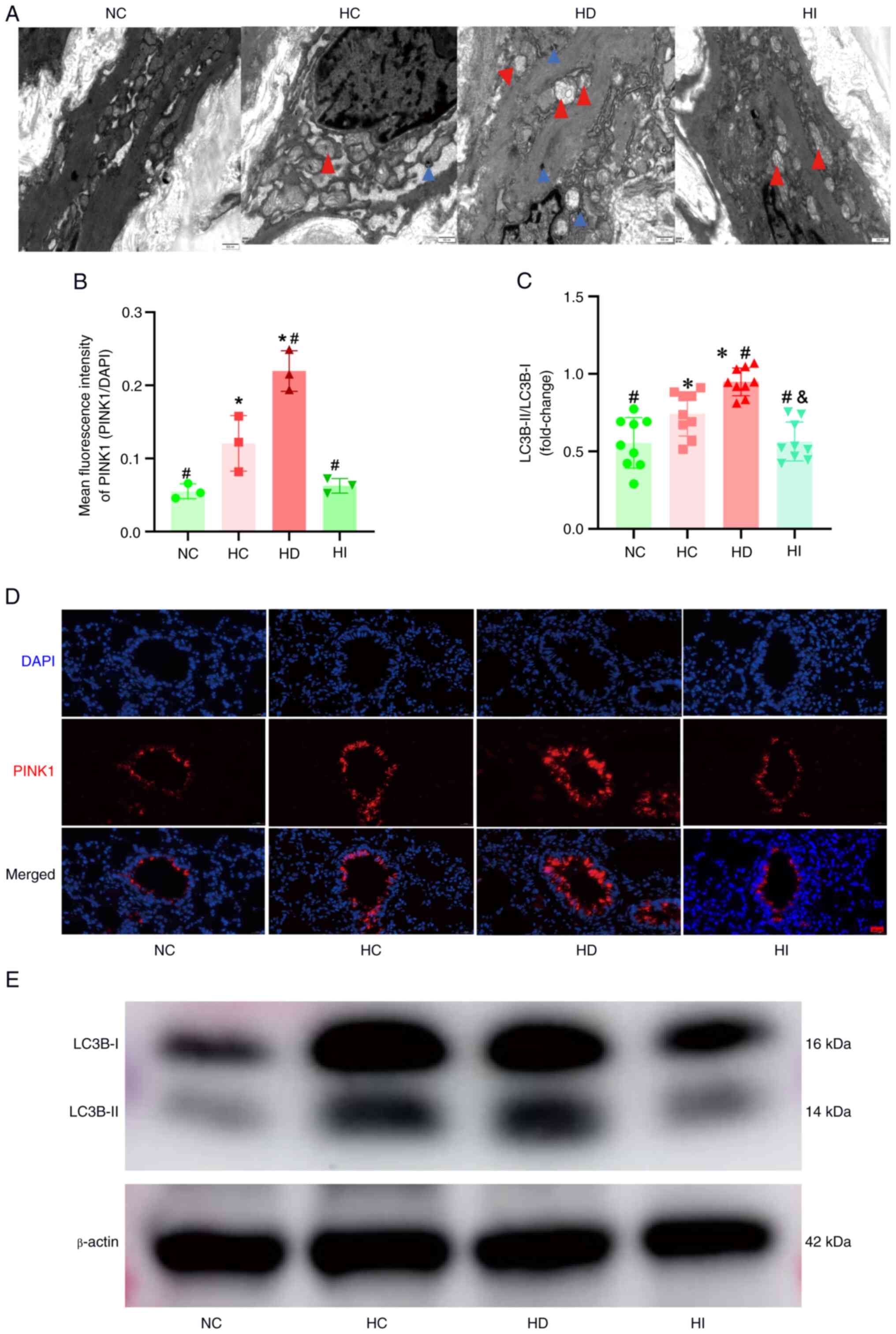 | Figure 4.Role of iron in mitophagy in the
pulmonary vasculature. (A) Representative images of transmission
electron microscopy of the main pulmonary artery (magnification,
×20,000). Red triangles represent swollen mitochondria, blue
triangles represent mitophagy. (B) Semi-quantitative analysis of
PINK1 immunofluorescence, n=3. (C) Semi-quantitative analysis of
LC3B-II band intensity, n=9. (D) Representative images of PINK1
immunofluorescence in the pulmonary vasculature (magnification,
×400). (E) Representative images of western blot analysis.
*P<0.05 vs. NC group; #P<0.05 vs. HC group;
&P<0.05 vs. HD group. PINK1, putative kinase
protein 1; NC, normoxia control; HC, hypoxia control; HD, hypoxia +
deferoxamine; HI, hypoxia combined + iron sucrose. |
Iron regulates the proliferation and
apoptosis of pulmonary artery endothelial cells under acute
hypoxia
To further validate the role of iron in acute HPV,
in vitro experiments were conducted to examine the effects
of iron on the proliferation of human pulmonary artery endothelial
cells. The Edu and CCK8 assay results demonstrated that the
proliferation of human pulmonary artery endothelial cells was
diminished under acute hypoxia (Fig.
5A-C). Furthermore, the administration of an iron chelator led
to a further reduction in endothelial cell proliferation under
hypoxic conditions, whereas the use of iron sucrose inhibited this
reduction (Fig. 5A-C).
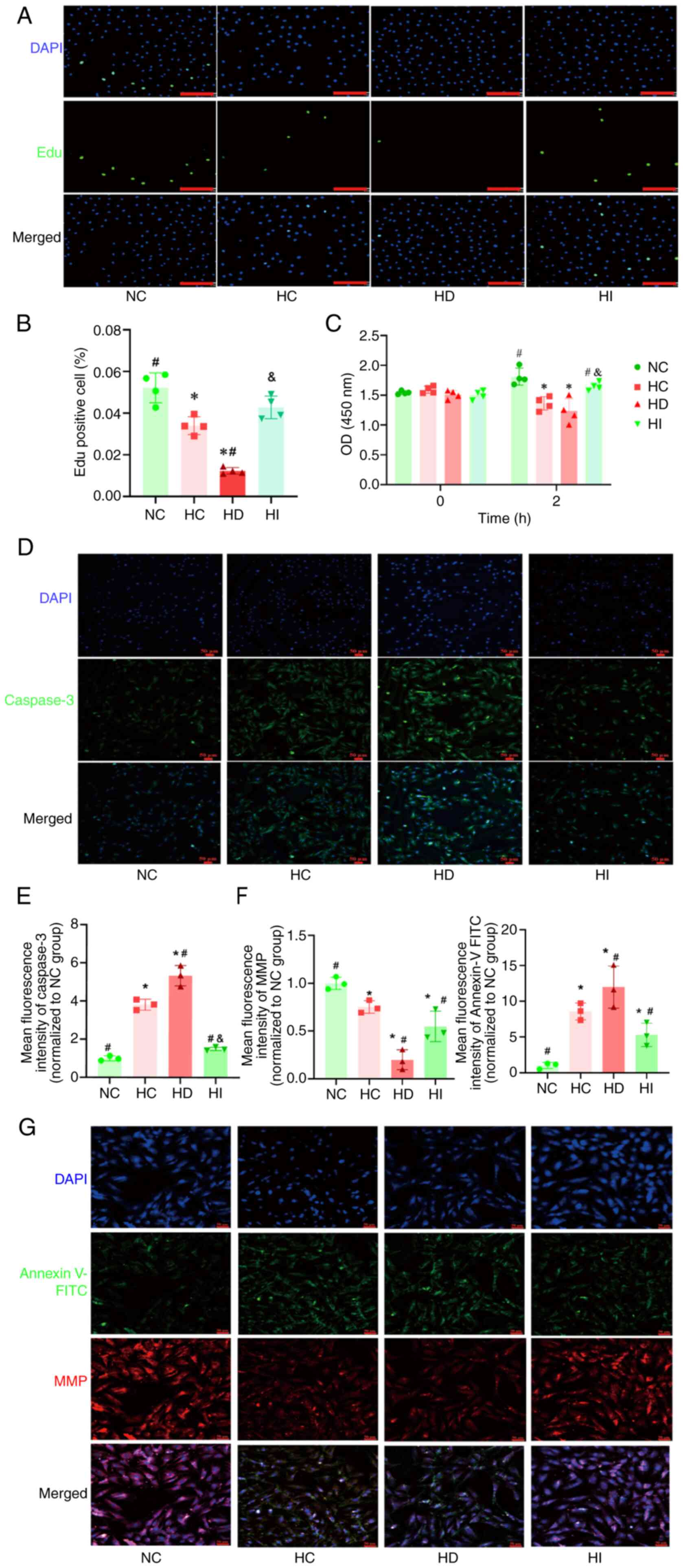 | Figure 5.Role of iron in the proliferation and
apoptosis of pulmonary artery endothelial cells under acute
hypoxia. (A) Representative images of Edu staining (scale bar, 170
µm). (B) Quantitative analysis of the Edu assay, n=4. (C)
Quantitative analysis of the Cell Counting Kit 8 assay, n=4. (D)
Representative images of Caspase 3 immunofluorescence staining
(magnification, ×200). (E) Semi-quantitative analysis of Caspase 3
immunofluorescence, n=3. (F) Semi-quantitative analysis of MMP and
Annexin V-FITC, n=3. (G) Representative images of MMP and Annexin
V-FITC (magnification, ×100). *P<0.05 vs. NC group,
#P<0.05 vs. HC group; &P<0.05 vs.
HD group. NC, normoxia control; HC, hypoxia control; HD, hypoxia +
deferoxamine; HI, hypoxia + iron sucrose; MMP, mitochondrial
membrane potential. |
The present study also employed immunofluorescence
to elucidate the effect of iron on the apoptosis of pulmonary
artery endothelial cells under acute hypoxia. The findings
demonstrated that acute hypoxia resulted in the upregulation of the
apoptotic protein Caspase 3 and Annexin V-FITC (Fig. 5D-G), accompanied by a reduction in
MMP (Fig. 5G). The administration
of an iron chelator led to a further exacerbation of these
hypoxia-induced abnormalities, whereas iron sucrose was observed to
exert a protective effect, ameliorating the aforementioned acute
hypoxia-induced abnormalities in pulmonary artery endothelial cells
(Fig. 5D-G).
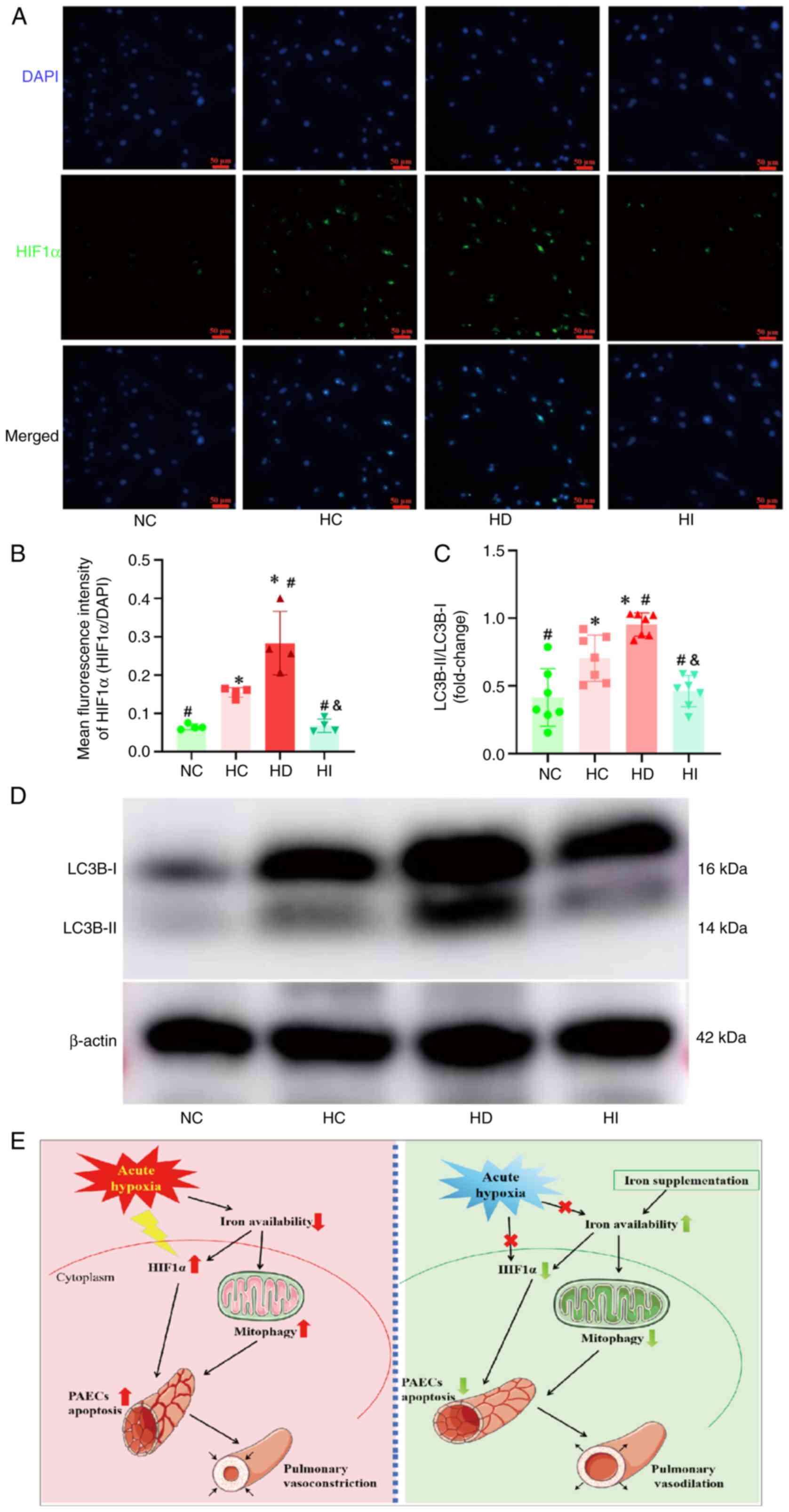
Iron modulates HIF1α expression and
mitophagy in pulmonary artery endothelial cells under acute
hypoxia
The expression of HIF1α was observed to be elevated
in pulmonary artery endothelial cells subjected to acute hypoxia
(Fig. 6A and B). The
administration of an iron chelator was revealed to facilitate HIF1α
expression, whereas the introduction of iron sucrose was
demonstrated to impede HIF1α expression in the context of acute
hypoxia. The results of western blot analysis of endothelial cells
demonstrated that acute hypoxia induced an increase in LC3B-II
expression, DFO promoted LC3B-II expression under acute hypoxia,
whereas iron sucrose suppressed its upregulation (Fig. 6C and D).
Discussion
A considerable number of individuals climb for
recreational, occupational and competitive sporting purposes. The
typical pulmonary manifestation of acute hypobaric hypoxia is HPV,
which, if left undiagnosed, can result in excessive HPV, leading to
PH and pulmonary edema. The onset of these disorders can occur at
any point between a few hours and 5 days after the individual has
ascended to a given altitude (4).
The present study employed a hypoxia administration protocol to
investigate the effects of prolonged hypoxia on pulmonary edema and
lung tissue injury in rats. The results demonstrated that the
degree of pulmonary edema and lung tissue injury was most
significant on day 3 of hypoxia. Additionally, it was shown that
serum iron exhibited a sustained decline, RBC levels demonstrated
an increase and the expression of mitophagy markers in lung tissues
was markedly elevated on day 3 of hypoxia. These findings indicated
that day 3 of plateau entry may be a period of high incidence of
acute altitude sickness. Furthermore, it may be observed that after
day 3, with the prolongation of the duration of hypoxia, the
performance of the acute plateau response improves; this may be
related to the physiological adaption of the body to hypoxia
(30).
Iron is an essential element for almost all living
organisms, as it is an important component of hemoglobin and
iron-sulfur proteins. Furthermore, it is essential for a number of
physiological processes, including immune surveillance, oxygen
transport and cell proliferation (31). Notably, long-term residents at high
altitudes respond to the diminished partial pressure of inhaled
oxygen by stimulating erythropoiesis. The increase in hemoglobin
levels necessitates a substantial intake of dietary iron
supplementation, which can result in an iron deficiency if not
replenished in a timely manner (32). The present study demonstrated that
rats subjected to acute hypoxia exhibited a reduction in serum iron
levels and an increase in hemoglobin concentration. In light of the
continuing debate surrounding the role of iron status in HPV
(9,10,33,34),
the current study revealed that low iron levels may intensify HPV,
whereas iron supplementation diminished it. This finding aligns
with the results of prior clinical investigations (9,34).
Nevertheless, the role of iron in high altitude-induced pulmonary
edema remains relatively understudied, with current research
largely focused on HPV. The present study demonstrated that iron
supplementation may be an effective intervention for acute hypoxic
pulmonary edema; however, the observed effect may be attributed to
its ability to improve acute HPV. Further in-depth studies are
required to elucidate the precise relationship between iron and
pulmonary edema.
HIF1α is a principal transcription factor that
mediates the acute hypoxic response; its expression is increased
following exposure to acute hypoxia (35). In addition, HIF1α serves a pivotal
role in the induction of mitophagy and may be linked to the
progression or inhibition of disease states (36). Furthermore, iron, which acts as a
crucial cofactor for proline hydroxylase, has been demonstrated to
regulate HIF1α expression under hypoxic conditions (37). Concurrently, iron chelators have
also been demonstrated to induce mitophagy (10,11).
It could be hypothesized that the combination of HIF1α and iron
chelators may intensify mitophagy during acute hypoxia. The
findings of the present study demonstrated that acute hypoxia
resulted in an elevation in HIF1α expression. Notably, an iron
chelator was observed to enhance HIF1α expression and mitophagy
under acute hypoxia. By contrast, iron sucrose was shown to
mitigate the aforementioned abnormalities. This provides a
comprehensive illustration of the relationship between iron, HIF1α
and mitophagy in acute high altitude-induced lung injury.
Beyond mitophagy, excessive pulmonary inflammation
and oxidative stress are established contributors to the
pathogenesis of acute lung injury (38). The present findings demonstrated
that acute hypoxia could induce inflammatory cell infiltration in
alveolar tissues, indicating an inflammatory response. This
response involves cytokine interactions, with HIF1α playing a
crucial regulatory role. Acute lung injury exhibits a
pro-inflammatory positive feedback loop, where inflammatory factors
upregulate HIF1α, which in turn amplifies inflammation and
exacerbates injury (39). Notably,
ferritin, an iron storage protein, serves as an inflammatory marker
(40,41). The reduction of iron in the
circulation during acute hypoxia may be due to the increased
inflammation in lung tissues and the elevation of ferritin levels;
future studies should elucidate the precise mechanisms underlying
iron-mediated inflammatory regulation in acute lung injury.
Additionally, a previous study has verified that the increase in
reactive oxygen species under hypoxia may contribute to the
progression of acute lung injury; this occurs through the
autoxidative disruption of cellular microstructures and the
structural or functional damage to pulmonary vascular endothelial
cells, epithelial cells and smooth muscle cells, resulting in
dysfunction of the alveolar-vascular endothelial barrier and
vasodilatory function (42). The
present study demonstrated that acute hypoxia can lead to an
imbalance between proliferation and apoptosis in pulmonary vascular
endothelial cells, thereby inducing acute lung injury. Notably,
excessive iron aggregation has been shown to promote oxidative
stress and exacerbate lung injury (43). Therefore, precisely controlling
circulating iron levels may help to mitigate lung injury.
Taken together, the present study identified the
most critical time point of acute lung injury during hypobaric
hypoxia. This finding offers a scientific foundation for the
clinical transportation of patients suffering from acute altitude
sickness. Simultaneously, the current study identified the changes
in iron levels under acute hypoxia and performed iron-intervention
experiments to determine the role and mechanism of serum iron
status in high altitude-induced hypoxic lung injury, thereby
providing a novel target for intervention in the treatment of acute
lung injury at high altitude (Fig.
6E). However, the present study has several limitations. First,
human trials were not conducted due to laboratory constraints and
ethical restrictions in human genetics. Future research will
include human trials, pending improved laboratory conditions and
ethics approval, to further elucidate the disease mechanism.
Second, the animal study design lacked an intervention treatment
group under normoxic conditions. While the hypoxia-based
intervention effectively demonstrated its effect on acute hypoxic
lung injury, this still represents a shortcoming of the study.
Finally, although the present study focused on mitophagy, the
pathogenesis of acute hypoxic lung injury is multifactorial,
involving mechanisms such as inflammation and oxidative stress.
Furthermore, the evaluation of lung injury encompasses diverse
indicators including pulmonary function tests. However, the present
study primarily concentrated on mitophagy-related mechanisms and
employed histopathological analysis along with wet/dry weight ratio
as principal evaluation metrics for lung injury, which constitutes
one of the limitations in this work. Future studies will employ
diverse methodologies to assess the modeling efficacy of lung
injury and explore these additional mechanistic pathways, aiming to
provide a more comprehensive understanding of high altitude-induced
acute hypoxic lung injury.
In conclusion, the present study demonstrated the
role of iron-mediated HIF1α activation and mitophagy in acute lung
injury. The onset of acute hypoxia was revealed to result in a
reduction in iron bioavailability, which subsequently triggered the
activation of HIF1α and facilitated mitophagy. This ultimately led
to the development of HPV and pulmonary edema. In the future, it
may be possible to increase iron bioavailability to prevent high
altitude-induced acute lung injury, and to inhibit HIF1α activity
and mitophagy in order to treat acute lung injury. Both of these
approaches provide new potential therapeutic targets for high
altitude-induced acute lung injury.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Key Laboratory of High
Altitude Medicine (Ministry of Education), Key Laboratory of
Application and Foundation for High Altitude Medicine Research in
Qinghai Province (Qinghai-Utah Joint Research Key Lab for High
Altitude Medicine), and Famous Doctor Studio of Hainan Tibetan
Autonomous Prefecture, Qinghai Province.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YG and RG conceived and designed the experiments.
YG, YH, HW and FZ performed experiments and analyzed data. YG, YH
and RG wrote and revised the manuscript. YG, FZ and RG confirm the
authenticity of all the raw data. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
The experimental protocols and animal care
procedures employed in the present study were approved by the
Medical Ethics Committee of Qinghai Provincial People's Hospital
(approval no. 2023-95). The Research Ethics Committee of Qinghai
Provincial People's Hospital confirmed that commercially procured
human primary cells fall outside the scope of required ethical
approval.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Penaloza D and Arias-Stella J: The heart
and pulmonary circulation at high altitudes: Healthy highlanders
and chronic mountain sickness. Circulation. 115:1132–1146. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mirrakhimov AE and Strohl KP:
High-altitude pulmonary hypertension: An update on disease
pathogenesis and management. Open Cardiovasc Med J. 10:19–27. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sydykov A, Mamazhakypov A, Maripov A,
Kosanovic D, Weissmann N, Ghofrani HA, Sarybaev AS and Schermuly
RT: Pulmonary hypertension in acute and chronic high altitude
maladaptation disorders. Int J Environ Res Public Health.
18:16922021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Luks AM, Swenson ER and Bärtsch P: Acute
high-altitude sickness. Eur Respir Rev. 26:1600962017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharma A, Ahmad S, Ahmad T, Ali S and Syed
MA: Mitochondrial dynamics and mitophagy in lung disorders. Life
Sci. 284:1198762021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu R, Xu C, Zhang W, Cao Y, Ye J, Li B,
Jia S, Weng L, Liu Y, Liu L and Zheng M: FUNDC1-mediated mitophagy
and HIF1α activation drives pulmonary hypertension during hypoxia.
Cell Death Dis. 13:6342022. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bao C, Liang S, Han Y, Yang Z, Liu S, Sun
Y, Zheng S, Li Y, Wang T, Gu Y, et al: The novel lysosomal
autophagy inhibitor (ROC-325) ameliorates experimental pulmonary
hypertension. Hypertension. 80:70–83. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Li Y, Chen Y, Yu X, Wang S, Sun
H, Zheng X, Zhang L, Wang Y and Zhu D: Circ-calm4 regulates
hypoxia-induced pulmonary artery smooth muscle autophagy by binding
Purb. J Mol Cell Cardiol. 176:41–54. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Smith TG, Talbot NP, Privat C, Rivera-Ch
M, Nickol AH, Ratcliffe PJ, Dorrington KL, León-Velarde F and
Robbins PA: Effects of iron supplementation and depletion on
hypoxic pulmonary hypertension: Two randomized controlled trials.
JAMA. 302:1444–1450. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Altamura S, Bärtsch P, Dehnert C,
Maggiorini M, Weiss G, Theurl I, Muckenthaler MU and Mairbäurl H:
Increased hepcidin levels in high-altitude pulmonary edema. J Appl
Physiol (1985). 118:292–298. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lan M, Wu S and Fernandes TM: Iron
deficiency and pulmonary arterial hypertension. Nutr Clin Pract.
37:1059–1073. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Schiavi A, Strappazzon F and Ventura N:
Mitophagy and iron: Two actors sharing the stage in age-associated
neuronal pathologies. Mech Ageing Dev. 188:1112522020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Brogyanyi T, Kejík Z, Veselá K, Dytrych P,
Hoskovec D, Masařik M, Babula P, Kaplánek R, Přibyl T, Zelenka J,
et al: Iron chelators as mitophagy agents: Potential and
limitations. Biomed Pharmacother. 179:1174072024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shimoda LA and Laurie SS: HIF and
pulmonary vascular responses to hypoxia. J Appl Physiol (1985).
116:867–874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Feng J, Zhan J and Ma S: LRG1 promotes
hypoxia-induced cardiomyocyte apoptosis and autophagy by regulating
hypoxia-inducible factor-1α. Bioengineered. 12:8897–8907. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao
WT, Ma HK, Jiang MD, Xu TT, Xu J, et al: HIF-1α-BNIP3-mediated
mitophagy in tubular cells protects against renal
ischemia/reperfusion injury. Redox Biol. 36:1016712020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Imray C, Wright A, Subudhi A and Roach R:
Acute mountain sickness: Pathophysiology, prevention, and
treatment. Prog Cardiovasc Dis. 52:467–484. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Beidleman BA, Fulco CS, Glickman EL,
Cymerman A, Kenefick RW, Cadarette BS, Andrew SP, Staab JE, Sils IV
and Muza SR: Acute mountain sickness is reduced following 2 days of
staging during subsequent ascent to 4300 m. High Alt Med Biol.
19:329–338. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang SL, Ibrahim NA, Jenarun G and Liew
HB: Incidence and determinants of acute mountain sickness in Mount
Kinabalu, Malaysia. High Alt Med Biol. 21:265–272. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hsu TY, Weng YM, Chiu YH, Li WC, Chen PY,
Wang SH, Huang KF, Kao WF, Chiu TF and Chen JC: Rate of ascent and
acute mountain sickness at high altitude. Clin J Sport Med.
25:95–104. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pu X, Lin X, Qi Y, Li Y, Li T, Liu Y and
Wei D: Effects of Fdft 1 gene silencing and VD3 intervention on
lung injury in hypoxia-stressed rats. Genes Genomics. 44:1201–1213.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zeng Y, Cao W, Huang Y, Zhang H, Li C, He
J, Liu Y, Gong H and Su Y: Huangqi Baihe Granules alleviate
hypobaric hypoxia-induced acute lung injury in rats by suppressing
oxidative stress and the TLR4/NF-κB/NLRP3 inflammatory pathway. J
Ethnopharmacol. 324:1177652024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dai C, Lin X, Qi Y, Wang Y, Lv Z, Zhao F,
Deng Z, Feng X, Zhang T and Pu X: Vitamin D3 improved
hypoxia-induced lung injury by inhibiting the complement and
coagulation cascade and autophagy pathway. BMC Pulm Med. 24:92024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dongiovanni P, Valenti L, Ludovica
Fracanzani A, Gatti S, Cairo G and Fargion S: Iron depletion by
deferoxamine up-regulates glucose uptake and insulin signaling in
hepatoma cells and in rat liver. Am J Pathol. 172:738–747. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Matute-Bello G, Downey G, Moore BB,
Groshong SD, Matthay MA, Slutsky AS and Kuebler WM; Acute Lung
Injury in Animals Study Group, : An official American Thoracic
Society workshop report: Features and measurements of experimental
acute lung injury in animals. Am J Respir Cell Mol Biol.
44:725–738. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang Y, Guo Y, Feng X, Yang P, Liu Y, Dai
X, Zhao F, Lei D, Li X, Liu Y and Li Y: Iron metabolism disorder
regulated by BMP signaling in hypoxic pulmonary hypertension.
Biochim Biophys Acta Mol Basis Dis. 1869:1665892023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhai K, Deng L, Wu Y, Li H, Zhou J, Shi Y,
Jia J, Wang W, Nian S, Jilany Khan G, et al: Extracellular
vesicle-derived miR-146a as a novel crosstalk mechanism for
high-fat induced atherosclerosis by targeting SMAD4. J Adv Res.
S2090-1232(24)00355-2. 2024.(Epub ahead of print). View Article : Google Scholar
|
|
28
|
Zhai K, Wang W, Zheng M, Khan GJ, Wang Q,
Chang J, Dong Z, Zhang X, Duan H, Gong Z and Cao H: Protective
effects of Isodon suzhouensis extract and glaucocalyxin A on
chronic obstructive pulmonary disease through SOCS3-JAKs/STATs
pathway. Food Front. 4:511–523. 2023. View Article : Google Scholar
|
|
29
|
Duan H, Wang W, Li S, Khan GJ, Ma Y, Liu
F, Zhai K, Hu H and Wei Z: The potential mechanism of Isodon
suzhouensis against COVID-19 via EGFR/TLR4 pathways. Food Sci Hum
Well. 13:3245–3255. 2024. View Article : Google Scholar
|
|
30
|
Naeije R: Physiological adaptation of the
cardiovascular system to high altitude. Prog Cardiovasc Dis.
52:456–466. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y, Lu Y and Jin L: Iron metabolism
and ferroptosis in physiological and pathological pregnancy. Int J
Mol Sci. 23:93952022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Muckenthaler MU, Mairbäurl H and Gassmann
M: Iron metabolism in high-altitude residents. J Appl Physiol
(1985). 129:920–925. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Patrician A, Dawkins T, Coombs GB, Stacey
B, Gasho C, Gibbons T, Howe CA, Tremblay JC, Stone R, Tymko K, et
al: Global research expedition on altitude-related chronic health
2018 iron infusion at high altitude reduces hypoxic pulmonary
vasoconstriction equally in both lowlanders and healthy andean
highlanders. Chest. 161:1022–1035. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Willie CK, Patrician A, Hoiland RL,
Williams AM, Gasho C, Subedi P, Anholm J, Drane A, Tymko MM,
Nowak-Flück D, et al: Influence of iron manipulation on hypoxic
pulmonary vasoconstriction and pulmonary reactivity during ascent
and acclimatization to 5050 m. J Physiol. 599:1685–1708. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Engebretsen BJ, Irwin D, Valdez ME,
O'Donovan MK, Tucker A and van Patot MT: Acute hypobaric hypoxia
(5486 m) induces greater pulmonary HIF-1 activation in hilltop
compared to madison rats. High Alt Med Biol. 8:312–321. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
He G, Nie JJ, Liu X, Ding Z, Luo P, Liu Y,
Zhang BW, Wang R, Liu X, Hai Y and Chen DF: Zinc oxide
nanoparticles inhibit osteosarcoma metastasis by downregulating
β-catenin via HIF-1α/BNIP3/LC3B-mediated mitophagy pathway. Bioact
Mater. 19:690–702. 2022.PubMed/NCBI
|
|
37
|
Chen Y, Li X, Wang Z, Yuan S, Shen X, Xie
X, Xing K and Zhu Q: Iron deficiency affects oxygen transport and
activates HIF1 signaling pathway to regulate phenotypic
transformation of VSMC in aortic dissection. Mol Med. 30:902024.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Y, Xiang D, Zhang H, Yao H and Wang Y:
Hypoxia-inducible factor-1: A potential target to treat acute lung
injury. Oxid Med Cell Longev. 2020:88714762020. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lin H and Jin F: Advancement of
pathological role of hypoxia-inducible factor 1 in acute lung
injury. Int J Respir. 39:1885–1889. 2019.
|
|
40
|
Mahroum N, Alghory A, Kiyak Z, Alwani A,
Seida R, Alrais M and Shoenfeld Y: Ferritin-from iron, through
inflammation and autoimmunity, to COVID-19. J Autoimmun.
126:1027782022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Kell DB and Pretorius E: Serum ferritin is
an important inflammatory disease marker, as it is mainly a leakage
product from damaged cells. Metallomics. 6:748–773. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu T and Zhao D: Research progress of
role of reactive oxygen species in acute lung injury acute
respiratory distress syndrome. Int J Respir. 39:1890–1894.
2019.
|
|
43
|
An HS, Yoo JW, Jeong JH, Heo M, Hwang SH,
Jang HM, Jeong EA, Lee J, Shin HJ, Kim KE, et al: Lipocalin-2
promotes acute lung inflammation and oxidative stress by enhancing
macrophage iron accumulation. Int J Biol Sci. 19:1163–1177. 2023.
View Article : Google Scholar : PubMed/NCBI
|