Introduction
Osteogenesis imperfecta (OI), also known as brittle
bone disorder, is a connective tissue disorder caused by an
abnormal structure and insufficient quantity of type I collagen or
incorrect modification and folding after translation (1,2). OI
affects ~1 in 15,000-20,000 individuals worldwide (3). The main clinical features of OI
include skeletal fragility, mild injury, multiple nontraumatic
fractures, skeletal deformities, blue/grey sclera, dentin
hypoplasia, progressive hearing loss in adults and short stature
(4). OI is characterized by strong
genetic heterogeneity and a wide range of clinical phenotypic
variations (3). Based on the
different clinical manifestations, genetic bases and genetic
patterns, OI is currently classified into ≥23 subtypes, of which
the OI–I-IV subtypes account for >90% of cases and are directly
caused by pathogenic variants in the COL1A1 and
COL1A2 genes, with COL1A1 variants being the most
common (5,6).
The COL1A1 gene is located at 17q21.33 and
has a total length of 17,530 bp, containing 51 exons and 50 introns
(7). The human COL1A1 gene
has been identified to produce a transcript with an mRNA length of
4,395 bp (NM:000088), encoding the type I collagen α1 strand [pro α
(I); NP_000079.2]. Thousands of pathogenic variants in
COL1A1, including missense, nonsense, insertion, deletion,
duplication and splice-site variants, have been reported. The
frequency of splice-site variants is relatively high, accounting
for ~15% of all recorded variants (5).
The present study used whole-exome sequencing (WES)
technology to identify a heterozygous variant, c.3531+1G>T, at
the splicing site in intron 47 of COL1A1 in a family with
OI. Essawi et al (8) first
reported this variant in the Palestinian population. However, to
the best of our knowledge, no functional studies have been
conducted on this variant to date. By extracting RNA from
lymphocyte strains and performing reverse transcription (RT)-PCR,
as well as conducting minigene experiments, the present study aimed
to investigate whether this variant affects splicing. To the best
of our knowledge, this is the first study on the effect of the
c.3531+1G>T variant in the COL1A1 gene.
Materials and methods
Compliance with ethical standards
The present study fully complied with the tenets of
The Declaration of Helsinki and was approved by the Ethics Board of
the Women and Children's Hospital, Xiamen University, China
(approval no. KY-2022-090-K02; Xiamen, China). Written informed
consent to participate in the present study was provided by the
participants, legal guardians or next of kin (when the participants
were minors <18 years old or elderly participants).
Subjects and clinical description
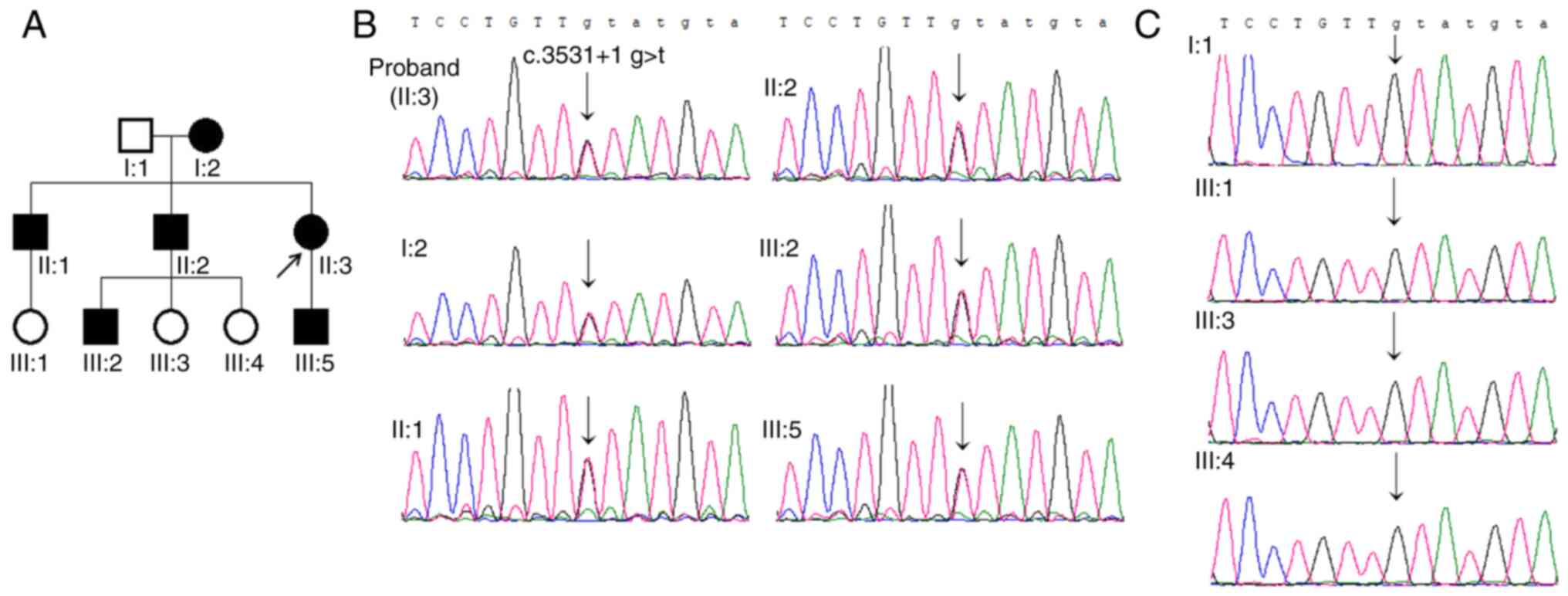
In the family investigated, six affected individuals
had a history of recurrent fractures without any other
complications and four individuals were unaffected, as shown in the
family pedigree (Fig. 1A). The
proband (II:3) was born with a light blue sclera and fell at the
age of 12 years, resulting in a fracture at the lower end of the
right elbow bone. The proband had no other fractures since then.
The proband was 34 years old and 156 cm tall. One healthy,
age-matched female from a non-related family participated in the
current study.
WES
A QIAamp Blood Mini Kit (Qiagen GmbH) was used to
extract DNA from the peripheral venous blood of the proband.
Subsequently, the genomic DNA concentration was determined on Qubit
3.0 (cat. no. Q33216; Thermo Fisher Scientific, Inc.) using the
Qubit dsDNA BR Assay Kit 100 assays (cat. no. Q32850; Thermo Fisher
Scientific, Inc.). The concentration of genomic DNA was 131 ng/µl.
In addition, 1% agarose gel electrophoresis was used to detect
genomic DNA integrity. Subsequently, the genomic DNA was randomly
fragmented using an ultrasonicator (cat. no. LE220; Covaris, Ltd.)
for 130 sec (2×65 sec) at a default temperature, duty cycle of 30%,
450 peak incident power (W) and 200 cycles per burst in a 96-well
microTUBE Plate. The temperature of the external cooler was below
10°C and 200–300 bp fragments were selected. Using the SureSelectXT
Reagent Kit (cat. no. G9611B; Agilent Technologies, Inc.), Agilent
V6 probe (cat. no. 5190-8863; Agilent Technologies, Inc.) and
Agencourt® AMPure® XP Beads (cat. no. A63881;
Beckman Coulter, Inc.), according to the manufacturers' protocols,
the target gene exon library was prepared. Detected using Qubit 3.0
and the ssDNA Assay Kit (cat. no. 12645ES76; Shanghai Yeasen
Biotechnology Co., Ltd.), the final library loading concentration
was 13.77 ng/µl. Finally, paired-end 150-bp sequencing was
performed on the MGISEQ-200RS platform (MGI Tech Co., Ltd.) using
the MGISEQ-200RS High Throughput (Rapid) Sequencing Kit (cat. no.
1000012555; MGITech Co., Ltd.). The quality control indices for
sequencing data were as follows: Average sequencing depth of the
target area of ≥150X; and proportion of sites with an average depth
of >20X in the target area >95%.
The sequenced fragments were compared with the UCSC
hg19 human reference genome using Burrows-Wheeler Aligner
[BWA-0.7.17 (r1188)] software (https://bio-bwa.sourceforge.net/) to remove
duplicates. The Genome Analysis Toolkit software (https://software.broadinstitute.org/gatk/) was used
for base quality correction and single nucleotide variant,
insertion/deletion and genotype detection. ExomeDepth (1.1.15)
(9) was used to detect copy number
variants at the exon level.
Detected variants were filtered according to the
following criteria: i) Frequency <1% according to the dbSNP
(https://www.ncbi.nlm.nih.gov/snp/),
1000 Genomes Project (Phase3, http://www.internationalgenome.org/), ESP6500 (V2;
http://genome.sph.umich.edu/wiki/NHLBI_Exome_Sequencing_Project),
and gnomAD databases (r2.1.1; http://gnomad-sg.org/); ii) amino acid alterations and
canonical splice site alterations; iii) homozygous, heterozygous or
compound heterozygous variants; iv) amino acid conservation across
species and pathogenicity prediction using in silico tools
[VarSome (https://varsome.com/), CADD (https://cadd.gs.washington.edu/snv),
MutationTaster (https://www.mutationtaster.org/), PolyPhen (http://genetics.bwh.harvard.edu/pph2/)
and SIFT (http://provean.jcvi.org/seq_submit.php)]; v)
genotype-phenotype analysis according to Exomiser (https://exomiser.readthedocs.io/) and Phenolyzer
(https://phenolyzer.wglab.org/); vi)
sequence variants interpreted following the guidelines of the
American College of Medical Genetics and Genomics (ACMG) and the
Association for Molecular Pathology (AMP) variant-interpretation
guidelines and the work groups from ClinGen (10–12).
In addition, records were checked for in the ClinVar database
(https://www.ncbi.nlm.nih.gov/clinvar/) and the
pathogenicity of splicing variants was predicted using VarCards2
(http://www.genemed.tech//varcards2/)
(13).
Sanger sequencing
A QIAamp Blood Mini Kit (Qiagen GmbH) was used to
extract DNA from the peripheral venous blood of all the family
members. Primers designed using Oligo 6 (Molecular Biology
Insights, Inc.) were used to amplify fragments of the candidate
variant site of COL1A1 for the proband and all the family
members, and an ABI 3130×l gene analyzer (Applied Biosystems;
Thermo Fisher Scientific, Inc.) was used for forward and reverse
sequencing.
Sequence analysis of COL1A1 cDNA
The present study established lymphocyte strains of
the peripheral blood of the proband, the father (I:1) and the
normal age-matched control according to the method introduced by
Neitzel (14). Total RNA was
extracted from the lymphocyte strains using TRIzol®
(Invitrogen; Thermo Fisher Scientific, Inc.) following the
manufacturer's recommended protocol. Subsequently, RNA was reverse
transcribed to cDNA using the RT kit (Invitrogen; Thermo Fisher
Scientific, Inc.), according to the manufacturer's protocol. The
COL1A1 cDNA was then amplified using two pairs of primers.
The forward primer in the first pair of primers was located in exon
45, whereas the reverse primer was located in exon 48 (1F:
5′-TAAAGGGTCACCGTGGCTTCT-3′, 1R: 5′-CAGGCTCTTGAGGGTGGTGT-3′). The
forward primer in the second pair of primers was located in exon
46, whereas the reverse primer was located in exon 49 (2F:
5′-CAAGGTCCCTCTGGAGCCTC-3′, 2R: 5′-GGTTGGGGTCAATCCAGTACTCT-3′). The
amplification was carried out using the TaKaRa LA Taq®
with GC Buffer (cat. no. RR02AG; Takara Biotechnology Co., Ltd.).
The reaction conditions were as follows: Pre-denaturation at 94°C
for 1 min; 35 cycles of denaturation at 94°C for 30 sec, annealing
at 63°C for 30 sec and extension at 72°C for 45 sec; and a final
extension at 72°C for 10 min and storage at 4°C. The PCR products
were separated by polyacrylamide gel electrophoresis on 6% gels and
were visualized with silver nitrate, and were then were separated
using 2% agarose gels. The gels were then cut, purified and Sanger
sequenced.
Minigene splicing assay
To verify the alternative splicing events, a
minigene splicing assay was performed. The 293T cell line was
selected to eliminate endogenous interference due to low expression
of type I collagen. 293T cells (cat. no. CL-0005; Procell Life
Science & Technology Co., Ltd.) were cultured at 37°C with 5%
CO2 in DMEM (cat. no. PM150210; Procell Life Science
& Technology Co., Ltd.) supplemented with 10% fetal bovine
serum (cat. no. 10091148; Gibco; Thermo Fisher Scientific, Inc.),
100 µg/ml streptomycin and 100 U/ml penicillin (cat. no. SV30010;
HyClone; Cytiva).
Wild-type hCOL1A1-Exon47-Exon48+c.3531+1G
(hCOL1A1-W) and mutant-type hCOL1A1-Exon47-Exon48+ c.3531+1T
(hCOL1A1-M) plasmids were constructed using the
pSPL3b[minigene]-Puro-SV40 vector by Suzhou Haixing Biosciences
Co., Ltd. Variants in minigene artefacts were confirmed using
Sanger sequencing. Minigene plasmids were transfected using an
electroporator (Neon™ Transfection System; cat. no. MPK5000; Thermo
Fisher Scientific, Inc.), Neon Transfection System Pipette (cat.
no. MPP100; Thermo Fisher Scientific, Inc.) and Neon Transfection
System 100 µl Kit (cat. no. MPK10096; Thermo Fisher Scientific,
Inc.) with 2×106 cells/100 µl. The electrical conversion
parameters were as follows: Pulse temperature, ~23°C; Pulse
voltage, 1,100 v; pulse duration, 20 msec; pulse number or pulse
repetition frequency, 2. The EGFP plasmid (control plasmid) was
electro-converted using the same parameters to determine the
electrical conversion efficiency. After electroporation, the cells
were harvested after immediately culturing at 37°C with 5%
CO2 in pre-warmed DMEM supplemented with 10% fetal
bovine serum for 48 h. Cells were processed for total RNA isolation
using TRIzol and treated with RQ1 DNase (cat. no. M6101; Promega
Corporation) to remove DNA according to the manufacturer's
protocols. cDNA was synthesized using a RT kit (cat. no. R323-01;
Vazyme Biotech Co., Ltd.), according to the manufacturer's
protocol. Subsequently, PCR was performed to verify alternative
events. The primers used for amplifying fragments containing this
site were as follows: pSPL3b-SD6: 5′-TCTGAGTCACCTGGACAACC-3′;
pSPL3b-SA2: 5′-ATCTCAGTGGTATTTGTGAGC-3′. The amplification was
carried out using the 2X Taq Master Mix (Dye Plus) (cat. no. P112;
Vazyme Biotech Co., Ltd.). The reaction conditions were as follows:
Pre-denaturation at 95°C for 5 min; 35 cycles of denaturation at
95°C for 30 sec, annealing at 58°C for 30 sec, and extension at
72°C for 45 sec; and a final extension at 72°C for 10 min and
storage at 4°C. The target product was 652 bp in length.
Subsequently, the product was separated by electrophoresis on a
1.5% agarose gel, followed by ethidium bromide staining, cutting,
purification. Finally, according to the manufacturer's protocols of
the Ultra-Universal TOPO cloning kit (cat. no. C603; Vazyme Biotech
Co., Ltd.), TA cloning was performed and the purified fragment
underwent Sanger sequencing.
Results
Identification of candidate variants
via WES and Sanger sequencing
High-quality sequencing data were obtained via WES.
For the proband, 15 Gb data were generated with a 99.78% coverage
of the target region and an average sequencing depth of 144 reads.
After filtering and analysis, a heterozygous splice site variant
(c.3531+1G>T) in the COL1A1 gene was found in the
proband.
Sanger sequencing confirmed the presence of
heterozygous variants in the proband. In addition, all of the other
five affected individuals in the family (Fig. 1B) and none of the four unaffected
individuals carried the variant (Fig.
1C).
There were no records of this variant in the dbSNP,
1000 Genomes Project, ESP6500 and ExAC databases. ClinVar includes
this variant and annotates it as a pathogenic variant. The dbscSNV
ADA SCORE was 1 and dbscSNV RF SCORE was 0.906 from VarCards2.
However, to the best of our knowledge, no functional studies have
been conducted on this variant to date. Therefore further analyses
were conducted to assess its role in OI.
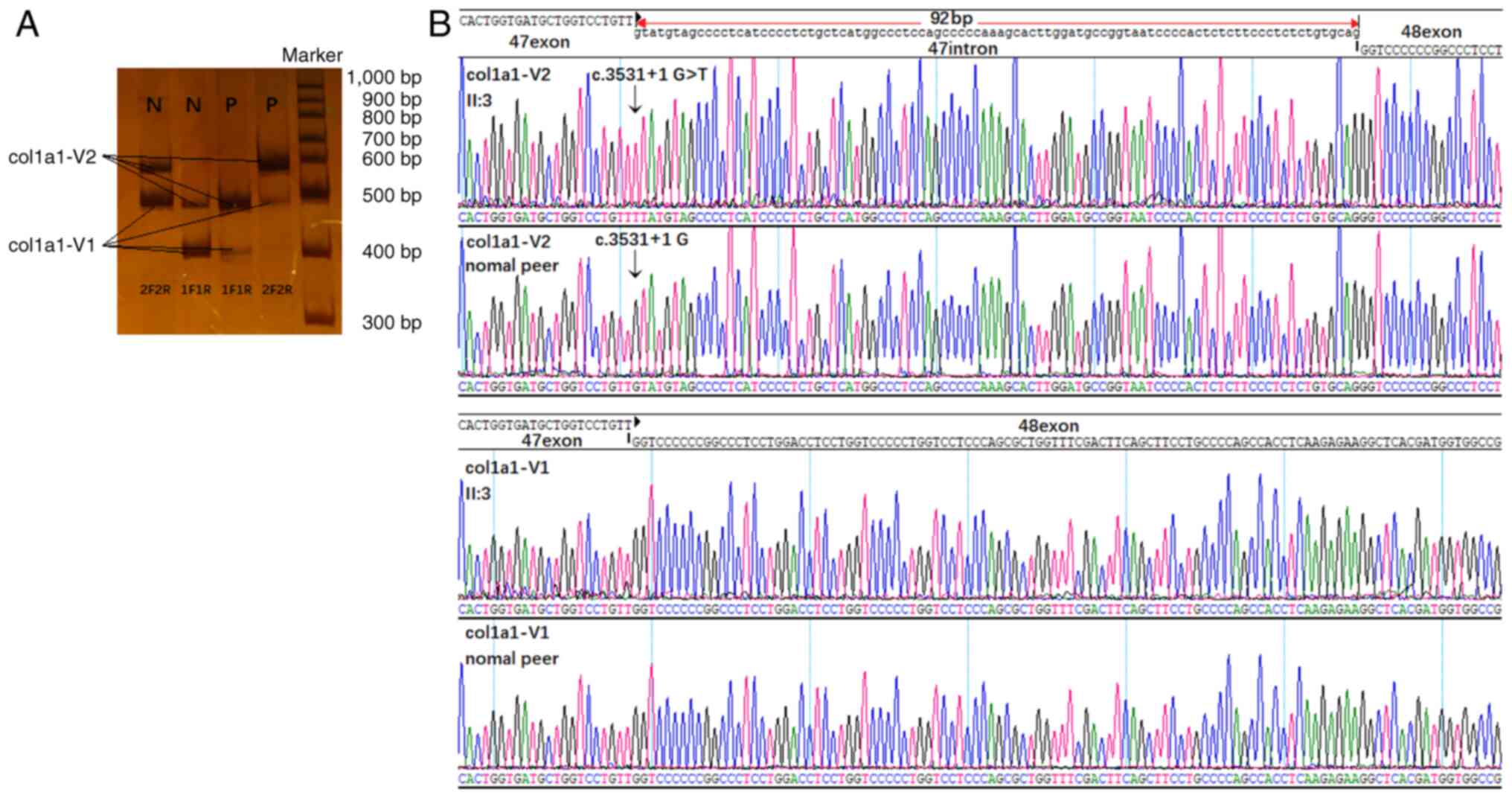
Sequence analysis of COL1A1 cDNA
COL1A1 cDNA samples of the lymphocyte strains
from both the proband and normal age-matched control were compared.
The target product (col1a1-V1) had lengths of 395 and 454 bp when
amplified using the first (1F1R) and second (2F2R) primer pairs,
respectively. Both the proband and the healthy individual had an
additional product (col1a1-V2) that was slightly longer than the
target product (~490 or 550 bp; Fig.
2A). In the healthy individual, the expression of col1a1-V1 was
high, whereas that of col1a1-V2 was low. By contrast, the proband
showed low expression of col1a1-V1 and high expression of
col1a1-V2. In-depth Sanger sequencing of these bands revealed that
col1a1-V1 did not contain intron 47 (Fig. 2B), while the slightly longer
col1a1-V2 retained it (Fig.
2B).
Minigene identification
The construction of a minigene plasmid is shown in
Fig. 3A. Minigene splicing assays
showed that plasmid hCOL1A1-W, underwent three types of splicing
(col1a1-V1, V2 and V3; Fig. 3B and
C). Col1a1-V1 was a result of normal splicing. Col1a1-V2
retained the entire intron 47, with the first base being G.
Col1a1-V3 removed the entire intron 47 and partially removed exon
48, leading to deletion of the sequence from nucleotide c.3532 to
c.3603 (c.3532_3603del). The agarose gel electrophoresis bands
corresponding to col1a1-V1 and V3 from hCOL1A1-W were very light
and cannot be observed in Fig. 3C.
The sequencing results have been detailed in the Supplementary
Materials (Fig. S1).
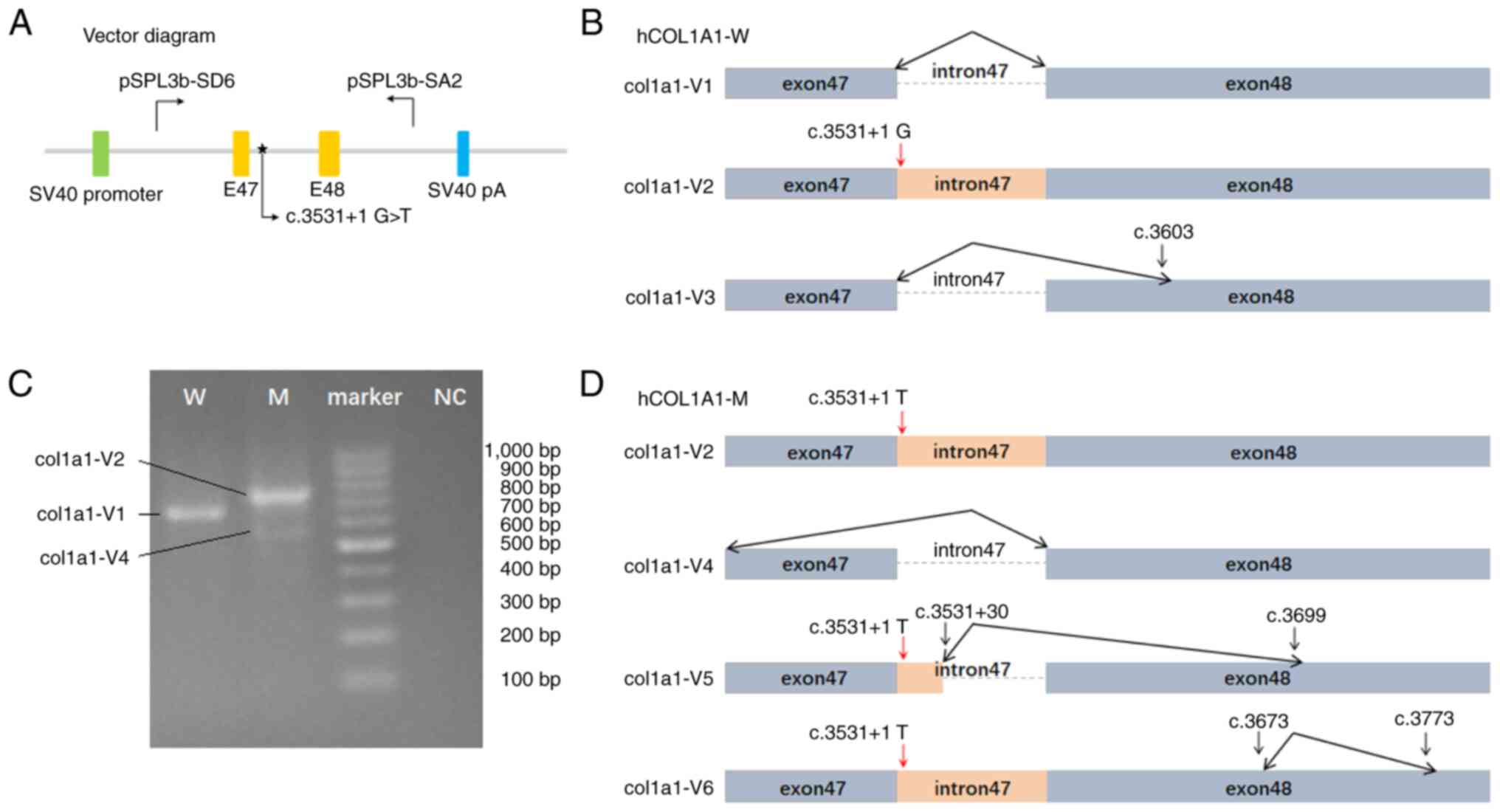 | Figure 3.(A) Vector diagram. Plasmids were
prepared according to the diagram. The W and M plasmids both
contained the human intron 47 sequence, but the W plasmid had a G,
while the M plasmid had a T at position c.3531+1. pSPL3b-SD6
referred to the forward primer used in PCR and pSPL3b-SA2 to the
reverse primer. (B) W exhibited three splicing patterns: Col1a1-V1,
col1a1-V2 and col1a1-V3. In col1a1-V1, the intron 47 was removed.
Col1a1-V2 retained the entire intron 47. In col1a1-V3 the entire
intron 47 and part of exon 48 was deleted. The red arrow represents
the position of c.3531+1, where the base is a wild-type G. The
black arrow represents the position of the splicing. (C)
Electrophoresis results of the minigene splicing assays. The arrows
indicates the positions of the bands corresponding to col1a1-V1,
col1a1-V2 and col1a1-V4. The V2 and V3 bands from hCOL1A1-W cannot
be observed, and the V5 and V6 bands from hCOL1A1-M cannot be
observed. (D) M exhibited four splicing patterns, i.e. col1a1-V2,
col1a1-V4, col1a1-V5 and col1a1-V6. Col1a1-V2 retained the entire
intron 47. In col1a1-V4 the entire exon 47 and intron 47 was
deleted. Col1a1-V5 retained part of intron 47 and part of exon 48.
Col1a1-V6 retained the entire intron 47 and part of exon 48. The
red arrow represents the position of c.3531+1, where the base is a
mutant-type T. The black arrow represents the position of the
splicing. W, wild type plasmid hCOL1A1-W; M, mutation type plasmid
hCOL1A1-M; NC, blank control (water was used as a template). |
By contrast, plasmid hCOL1A1-M underwent four types
of splicing (col1a1-V2, V4, V5 and V6; Fig. 3C and D). Col1a1-V2, like wild-type
COL1A1, retained intron 47, with the first base being T. In
col1a1-V4, the entire exon 47 and intron 47 were removed. Col1a1-V5
partially retained intron 47 and exon 48, leading to the insertion
of the intron 47 sequence from positions c.3531+1 to c.3531+30
[r.3531_3532ins(u; 3531+2_3531+30)] and the deletion of the
sequence from nucleotides c.3532 to c.3699 (c.3532_3699del).
Col1a1-V6 retained the entire intron 47 and partly retained exon
48, leading to the deletion of the sequence from nucleotides c.3674
to c.3773 (c.3674_3773del). The agarose gel electrophoresis bands
corresponding to col1a1-V5 and V6 from hCOL1A1-M were very light
and cannot be observed in Fig. 3C.
These sequencing results have been detailed in Fig. S2.
Discussion
To date, 21 genes have been reported to cause OI
(Table I), most of which lead to
type I collagen synthesis disorders (15–19).
Type I collagen is the main structural protein of bones and
connective tissue, and is an important component that interacts
with cell surfaces and the extracellular matrix. The triple helix
structure of type I procollagen molecules consists of two α1 chains
and one α2 chain, encoded by the COL1A1 and COL1A2
genes, respectively (20).
Variants in these genes cause types I–IV OI. Extensive experimental
and clinical observational studies have shown an association
between clinical manifestations of OI and the sites of genetic
variants. Since assembly of the triple helix starts from the
C-terminus, variants in this region of the α1 and α2 chains can
lead to more unstable collagen structures and more severe
phenotypes (21,22). The type I phenotype of OI is
relatively mild and is mainly caused by nonsense, frameshift or
splice-site variants in COL1A1 and COL1A2, leading to
premature translation termination, halving type I collagen
synthesis (23). Types II–IV OI
constitute more severe phenotypes; these cases are mainly caused by
missense variants leading to amino acid substitutions, resulting in
structural changes in type I collagen (22–25).
Few pathogenic variants are caused by splicing variants,
insertions, deletions or duplications, all of which may result in
changes in the coding box sequence and carboxyl end peptide,
thereby altering the structure of type I collagen (22). The proband and their affected
family members in the present study were classified as having type
I OI due to their mild phenotype.
 | Table I.Genes associated with osteogenesis
imperfecta. |
Table I.
Genes associated with osteogenesis
imperfecta.
| Gene | Locus | Gene OMIM
number | Inheritance | OI type | Phenotype OMIM
number |
|---|
| COL1A1 | 17q21.33 | 120150 | AD | OI I | 166200 |
| COL1A1 | 17q21.33 | 120150 | AD | OI II | 166210 |
| COL1A1 | 17q21.33 | 120150 | AD | OI III | 259420 |
| COL1A1 | 17q21.33 | 120150 | AD | OI IV | 166220 |
| COL1A2 | 7q21.3 | 120160 | AD | OI II | 166210 |
| COL1A2 | 7q21.3 | 120160 | AD | OI III | 259420 |
| COL1A2 | 7q21.3 | 120160 | AD | OI IV | 166220 |
| IFITM5 | 11p15.5 | 614757 | AD | OI V | 610967 |
|
SERPINF1 | 17p13.3 | 172860 | AR | OI VI | 613982 |
| CRTAP | 3p22.3 | 605497 | AR | OI VII | 610682 |
| P3H1 | 1p34.2 | 610339 | AR | OI VIII | 610915 |
| PPIB | 15q22.31 | 123841 | AR | OI IX | 259440 |
|
SERPINH1 | 11q13.5 | 600943 | AR | OI X | 613848 |
| FKBP10 | 17q21.2 | 607063 | AR | OI XI | 610968 |
| SP7 | 12q13.13 | 606633 | AR | OI XII | 613849 |
| BMP1 | 8p21.3 | 112264 | AR | OI XIII | 614856 |
| TMEM38B | 9q31.2 | 611236 | AR | OI XIV | 615066 |
| WNT1 | 12q13.12 | 164820 | AR | OI XV | 615220 |
| CREB3L1 | 11p11.2 | 616215 | AR | OI XVI | 616229 |
| SPARC | 5q33.1 | 182120 | AR | OI XVII | 616507 |
| TENT5A | 6q14.1 | 611357 | AR | OI XVIII | 617952 |
| MBTPS2 | Xp22.12 | 300294 | XLR | OI XIX | 301014 |
| MESD | 15q25.1 | 607783 | AR | OI XX | 618644 |
| KDELR2 | 7p22.1 | 609024 | AR | OI XXI | 619131 |
| CCDC134 | 22q13.2 | 618788 | AR | OI XXII | 619795 |
| PHLDB1 | 11q23.3 | 612834 | AR | OI XXIII | 620639 |
Although the proband in the current study had only
one fracture at the age of 12 years old, with normal height, a pale
blue sclera, and no bone deformities or other complications, other
patients in the family had recurrent fractures (the number of
fractures was unknown). A patient with the same variant reported by
Willing et al (26) had
mild symptoms. However, Essawi et al (8) reported that a patient with OI caused
by this variant had already suffered 15 fractures before the age of
5 years, and the patient had a short stature and blue sclera.
Therefore, the OI phenotype caused by this variant is variable,
ranging from mild to moderate.
Using WES and Sanger sequencing, the present study
reported a heterozygous splice variant, c.3531+1G>T, in
COL1A1 in patients from a family with OI. The results showed
that this variant resulted in a decrease in the normal transcript
(col1a1-V1) production and an increase in the abnormal transcript
(col1a1-V2) production. In col1a1-V2, intron 47 was transformed
into a coding sequence, resulting in a mutation of amino acid 1,178
from glycine to valine and the premature generation of a stop codon
at position 92 from this site, which might lead to protein
truncation (p.Gly1178Valfs*91). Multiple frameshift mutations have
been reported downstream of this site, which would lead to
truncated proteins and cause OI–I, such as c.3540del (p.Gly1181fs),
c.3540dup (p.Gly1181fs), c.3566del (p.Pro1189fs), c.3566dup
(p.Gly1190fs) and c.3567del (p.Gly1190fs) (26,27–32).
Variants causing haploinsufficiency of COL1A1 are considered
pathogenic (26,31,32).
The present study further investigated splicing patterns using the
minigene assay. The minigene experiment showed that hCOL1A1-M
underwent four types of splicing, while hCOL1A1-W only underwent
three types of splicing. Col1a1-V3, with a c.3532_3603del,
resulting in amino acid deletions from positions 1,178-1,201
(p.Gly1178_Leu1201del), would miss five repeats of GXY in pro α
(I). In col1a1-V4, the entire exon 47 and intron 47 were removed,
resulting in amino acid deletions from positions 1,141-1,177
(p.Arg1141_Val1177del), leading to 12 repeats of GXY being missed
in pro α (I). Col1a1-V5 retained part of intron 47 and part of exon
48, which resulted in an increase of 10 amino acids and a lack of
five repeats of GXY in pro α (I). Col1a1-V6 retained the entire
intron 47 and partially retained exon 48, resulting in the
premature generation of a stop codon at the 81st amino acid after
p.Val1177, forming a truncated protein. The reduction of GXY
repeats in Col1a1-V3, V4 and V5 could affect the formation of the
triple helix of collagen. These results indicated that the
c.3531+1G>T variant had an effect on intron 47 splicing. The
splice variant c.3531+1G>T contributed to the disappearance of
the classic donor splice sites of intron 47, which led to intron 47
retention or partial retention.
Several previously identified variants at this
splicing site, namely c.3531+1G>A, c.3531+1G>C,
c.3531+1G>T and c.3531+2T>C, have been reported to lead to OI
(6,8,26,31–34).
Of these, experimental studies have revealed that the
c.3531+1G>A variant leads to the skipping of exon 48 and reduced
mRNA levels (26). No such
experiments were conducted on the remaining variants. Previous
studies have revealed other COL1A1 variants that may cause
premature termination, leading to reduced mRNA stability in the
mutated alleles (26,31,35).
Ultimately, such a change would decrease type I collagen synthesis,
resulting in type I OI. Based on the ACMG and AMP guidelines for
interpreting sequence variants, this variant was classified as
pathogenic. Specific evidence for the pathogenicity of this variant
is as follows: i) PVS1: Null variant (canonical +/- 1 splice sites)
in a gene where the loss of function is a known mechanism of
disease; ii) PS2: In vivo and in vitro experiments in
the present study suggested that this variant may affect the normal
splicing of RNA; iii) PP1: It was consistent with co-segregation;
this variant was detected in all six affected individuals; iv) PP3:
Multiple lines of computational evidence support a deleterious
effect on the gene or gene product.
Notably, it was found that healthy individual also
expressed the col1a1-V2 transcript but at markedly lower level than
it in the proband. A minigene experiment also confirmed the
presence of col1a1-V2 as well as a transcript that produces a
truncated exon 48 from the wild-type gene. These results suggested
that the normal COL1A1 gene might generate alternative
transcripts. The minigene experiment revealed more transcripts than
the two identified in the cDNA from peripheral blood lymphocyte
strains. This difference might be due to the different sources of
cells or the markedly lower expression levels of other transcripts,
which prevented their detection through gel electrophoresis. The
difference in the expression level of transcripts between patients
and normal individuals indicates that variants at this splicing
site may lead to changes in selective splicing behavior and disrupt
the regulatory mechanism of selective splicing. The resulting
changes in expression level may, in turn, cause OI. However, the
alternative transcripts may indeed be insignificant in the process
of collagen synthesis and may never mature to exist in the
extracellular matrix. Studies have constructed plasmids and
transfected single cell lines for 24 or 48 h to extract RNA for
studying splice-site mutations (36–38).
This method has been validated to be helpful for the pathogenicity
assessment of splice-site mutations. Therefore, the present study
also harvested RNA 48 h after plasmid transfection into a single
cell line. However, due to the varying levels of gene expression in
different cell lines, more cell lines are needed to confirm this
finding. In addition, RNA extraction was performed only 48 h after
plasmid transfection into 293T cells, so the expression of
COL1A1 might not be comprehensive enough; the culture time
after transfection could be extended to 72 h. We aim to continue to
extract RNA from transfected cells 72 h later to observe the
expression of this gene. Due to the inability to obtain tissues
from the human body, we are currently unable to conduct western
blotting experiments to assess the stability of the protein. More
research is needed to detect changes in the expression of this
protein.
OI not only seriously affects the quality of life of
patients, but is also associated with an economic burden to
families. In addition, no effective cure for this disorder
currently exists. Prenatal diagnosis and in vitro
fertilization combined with pre-implantation genetic testing can
effectively prevent families with OI from having children with the
same variant again. In-depth research on the genetic pathogenesis
of OI will provide a theoretical basis for the prevention and
treatment of this disorder, which has important clinical value and
scientific significance.
Using WES technology, the present study identified a
heterozygous variant, c.3531+1G>T, at the splicing site of
intron 47 of the COL1A1 gene in a family with OI. The
results revealed that this variant could lead to changes in
splicing behavior and alter transcript expression levels. RT-PCR
experiments on lymphocyte strains from the proband and an
age-matched control showed that the c.3531+1G>T variant affected
the splicing of intron 47. Minigene splicing assays also indicated
that this variant site had an impact on intron 47 splicing. Thus,
this variant was considered pathogenic for the proband. To the best
of our knowledge, no functional study has yet been conducted
regarding this variant and this variation has not been reported in
the Chinese population. The present study is the first report of
c.3531+1G>T in COL1A1 associated with OI in a Chinese
family. It is also the first study regarding the effect of the
c.3531+1G>T variant on the COL1A1 gene. Moreover, the OI
phenotype caused by this variant is variable, ranging from mild
(only one fracture and normal height) to moderate (15 fractures and
short stature). These results may expand the spectrum of pathogenic
variants associated with OI.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82101955), the Fujian Provincial
Natural Science Foundation of China (grant no. 2023J05269), the
General Fund Project of Xiamen Natural Science Foundation (grant
no. 3502Z202373117), and the Fujian Provincial Health Technology
Project (grant no. 2020GGB064).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding authors. The sequencing data
generated in the present study may be found in the Genome Sequence
Archive (Genomics, Proteomics & Bioinformatics 2021) in
National Genomics Data Center (Nucleic Acids Res 2022), China
National Center for Bioinformation/Beijing Institute of Genomics,
Chinese Academy of Sciences under accession number GSA-Human:
HRA011257 or at the following URL: https://ngdc.cncb.ac.cn/gsa–human/browse/HRA011257.
Authors' contributions
YH, YZ, LZ and YS designed the study, performed the
laboratory experiments and bioinformatics analysis of WES. YZ, LZ,
YS, XY and JX collected data and performed Sanger sequencing
validation experiments. YH and YG confirm the authenticity of all
the raw data. YH wrote the manuscript. LM and YG analyzed and
interpreted the data, supervised the study and made revisions to
the manuscript. All authors have read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
The present study fully complied with the tenets of
The Declaration of Helsinki and was approved by the Ethics Board of
the Women and Children's Hospital and the Experimental Animal
Center of Xiamen University, China (approval no. KY-2022-090-K02).
Written informed consent to participate in this study was provided
by the participants, legal guardians or next of kin.
Patient consent for publication
The patients provide written informed consent,
agreeing to publish their clinical manifestations and data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Uttarilli A, Shah H, Bhavani GS, Upadhyai
P, Shukla A and Girisha KM: Phenotyping and genotyping of skeletal
dysplasias: Evolution of a center and a decade of experience in
India. Bone. 120:204–211. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nicol L, Morar P, Wang Y, Henriksen K, Sun
S, Karsdal M, Smith R, Nagamani SCS, Shapiro J, Lee B and Orwoll E:
Alterations in non-type I collagen biomarkers in osteogenesis
imperfecta. Bone. 120:70–74. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forlino A, Cabral WA, Barnes AM and Marini
JC: New perspectives on osteogenesis imperfecta. Nat Rev
Endocrinol. 7:540–557. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ablin DS: Osteogenesis imperfecta: A
review. Can Assoc Radiol J. 49:110–123. 1998.PubMed/NCBI
|
|
5
|
Marini JC, Forlino A, Bächinger HP, Bishop
NJ, Byers PH, Paepe A, Fassier F, Fratzl-Zelman N, Kozloff KM,
Krakow D, et al: Osteogenesis imperfecta. Nat Rev Dis Primers.
3:170522017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li L, Mao B, Li S, Xiao J, Wang H, Zhang
J, Ren X, Wang Y, Wu Y, Cao Y, et al: Genotypic and phenotypic
characterization of Chinese patients with osteogenesis imperfecta.
Hum Mutat. 40:588–600. 2019.PubMed/NCBI
|
|
7
|
Tromp G, Kuivaniemi H, Stacey A, Shikata
H, Baldwin CT, Jaenisch R and Prockop DJ: Structure of a
full-length cDNA clone for the prepro alpha 1(I) chain of human
type I procollagen. Biochem J. 253:919–922. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Essawi O, Symoens S, Fannana M, Darwish M,
Farraj M, Willaert A, Essawi T, Callewaert B, De Paepe A, Malfait F
and Coucke PJ: Genetic analysis of osteogenesis imperfecta in the
Palestinian population: Molecular screening of 49 affected
families. Mol Genet Genomic Med. 6:15–26. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Plagnol V, Curtis J, Epstein M, Mok KY,
Stebbings E, Grigoriadou S, Wood NW, Hambleton S, Burns SO,
Thrasher AJ, et al: A robust model for read count data in exome
sequencing experiments and implications for copy number variant
calling. Bioinformatics. 28:2747–2754. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
laboratory quality assurance committee. Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American college of medical genetics and
genomics and the association for molecular pathology. Genet Med.
17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Biesecker LG and Harrison SM: ClinGen
sequence variant interpretation working group. The ACMG/AMP
reputable source criteria for the interpretation of sequence
variants. Genet Med. 20:1687–1688. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abou Tayoun AN, Pesaran T, DiStefano MT,
Oza A, Rehm HL, Biesecker LG and Harrison SM: ClinGen sequence
variant interpretation working group (ClinGen SVI). Recommendations
for interpreting the loss of function PVS1 ACMG/AMP variant
criterion. Hum Mutat. 39:1517–1524. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang Z, Zhao G, Zhu Z, Wang Y, Xiang X,
Zhang S, Luo T, Zhou Q, Qiu J, Tang B, et al: VarCards2: An
integrated genetic and clinical database for ACMG-AMP
variant-interpretation guidelines in the human whole genome.
Nucleic Acids Res. 52:D1478–D1489. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Neitzel H: A routine method for the
establishment of permanent growing lymphoblastoid cell lines. Hum
Genet. 73:320–326. 1986. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Dijk FS, Semler O, Etich J, Köhler A,
Jimenez-Estrada JA, Bravenboer N, Claeys L, Riesebos E, Gegic S,
Piersma SR, et al: Interaction between KDELR2 and HSP47 as a key
determinant in osteogenesis imperfecta caused by bi-allelic
variants in KDELR2. Am J Hum Genet. 107:989–999. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tuysuz B, Uludag Alkaya D, Geyik F,
Alaylıoğlu M, Kasap B, Kurugoğlu S, Akman YE, Vural M and Bilguvar
K: Biallelic frameshift variants in PHLDB1 cause mild-type
osteogenesis imperfecta with regressive spondylometaphyseal
changes. J Med Genet. 60:819–826. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Umair M, Hassan A, Jan A, Ahmad F, Imran
M, Samman MI, Basit S and Ahmad W: Homozygous sequence variants in
the FKBP10 gene underlie osteogenesis imperfecta in consanguineous
families. J Hum Genet. 61:207–213. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Umair M, Alhaddad B, Rafique A, Jan A,
Haack TB, Graf E, Ullah A, Ahmad F, Strom TM, Meitinger T and Ahmad
W: Exome sequencing reveals a novel homozygous splice site variant
in the WNT1 gene underlying osteogenesis imperfecta type 3. Pediatr
Res. 82:753–758. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hayat A, Hussain S, Bilal M, Kausar M,
Almuzzaini B, Abbas S, Tanveer A, Khan A, Siddiqi S, Foo JN, et al:
Biallelic variants in four genes underlying recessive osteogenesis
imperfecta. Eur J Med Genet. 63:1039542020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Rauch F and Glorieux FH: Osteogenesis
imperfecta. Lancet. 363:1377–1385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Malfait F, Symoens S, De Backer J,
Hermanns-Lê T, Sakalihasan N, Lapière CM, Coucke P and De Paepe A:
Three arginine to cysteine substitutions in the pro-alpha
(I)-collagen chain cause Ehlers-Danlos syndrome with a propensity
to arterial rupture in early adulthood. Hum Mutat. 28:387–395.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jovanovic M, Guterman-Ram G and Marini JC:
Osteogenesis imperfecta: Mechanisms and signaling pathways
connecting classical and rare OI types. Endocr Rev. 43:61–90. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marini JC, Forlino A, Cabral WA, Barnes
AM, San Antonio JD, Milgrom S, Hyland JC, Körkkö J, Prockop DJ, De
Paepe A, et al: Consortium for osteogenesis imperfecta mutations in
the helical domain of type I collagen: Regions rich in lethal
mutations align with collagen binding sites for integrins and
proteoglycans. Hum Mutat. 28:209–221. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wei S, Yao Y, Shu M, Gao L, Zhao J, Li T,
Wang Y and Xu C: Genotype-phenotype relationship and follow-up
analysis of a Chinese cohort with osteogenesis imperfecta. Endocr
Pract. 28:760–766. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Aliyeva L, Ongen YD, Eren E, Sarisozen MB,
Alemdar A, Temel SG and Sag SO: Genotype and phenotype correlation
of patients with osteogenesis imperfecta. J Mol Diagn. 26:754–769.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Willing MC, Deschenes SP, Scott DA, Byers
PH, Slayton RL, Pitts SH, Arikat H and Roberts EJ: Osteogenesis
imperfecta type I: Molecular heterogeneity for COL1A1 null alleles
of type I collagen. Am J Hum Genet. 55:638–647. 1994.PubMed/NCBI
|
|
27
|
Vandersteen AM, Lund AM, Ferguson DJ,
Sawle P, Pollitt RC, Holder SE, Wakeling E, Moat N and Pope FM:
Four patients with Sillence type I osteogenesis imperfecta and mild
bone fragility, complicated by left ventricular cardiac valvular
disease and cardiac tissue fragility caused by type I collagen
mutations. Am J Med Genet A. 164A:386–391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Malmgren B, Andersson K, Lindahl K,
Kindmark A, Grigelioniene G, Zachariadis V, Dahllöf G and Åström E:
Tooth agenesis in osteogenesis imperfecta related to mutations in
the collagen type I genes. Oral Dis. 23:42–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Van Dijk FS: Genetics of osteoporosis in
children. Endocr Dev. 28:196–209. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Swinnen FK, Coucke PJ, De Paepe AM,
Symoens S, Malfait F, Gentile FV, Sangiorgi L, D'Eufemia P, Celli
M, Garretsen TJ, et al: Osteogenesis imperfecta: The audiological
phenotype lacks correlation with the genotype. Orphanet J Rare Dis.
6:882011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Byers PH, Duvic M, Atkinson M, Robinow M,
Smith LT, Krane SM, Greally MT, Ludman M, Matalon R, Pauker S, et
al: Ehlers-Danlos syndrome type VIIA and VIIB result from
splice-junction mutations or genomic deletions that involve exon 6
in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet.
72:94–105. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Körkkö J, Ala-Kokko L, De Paepe A,
Nuytinck L, Earley J and Prockop DJ: Analysis of the COL1A1 and
COL1A2 genes by PCR amplification and scanning by
conformation-sensitive gel electrophoresis identifies only COL1A1
mutations in 15 patients with osteogenesis imperfecta type I:
Identification of common sequences of null-allele mutations. Am J
Hum Genet. 62:98–110. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Baralle D and Baralle M: Splicing in
action: Assessing disease causing sequence changes. J Med Genet.
42:737–748. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bardai G, Moffatt P, Glorieux FH and Rauch
F: DNA sequence analysis in 598 individuals with a clinical
diagnosis of osteogenesis imperfecta: Diagnostic yield and mutation
spectrum. Osteoporos Int. 27:3607–3613. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stover ML, Primorac D, Liu SC, McKinstry
MB and Rowe DW: Defective splicing of mRNA from one COL1A1 allele
of type I collagen in nondeforming (type I) osteogenesis
imperfecta. J Clin Invest. 92:1994–2002. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ito K, Patel PN, Gorham JM, McDonough B,
DePalma SR, Adler EE, Lam L, MacRae CA, Mohiuddin SM, Fatkin D, et
al: Identification of pathogenic gene mutations in LMNA and MYBPC3
that alter RNA splicing. Proc Natl Acad Sci USA. 114:7689–7694.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dong Z, Wang Y, Zhang J, Zhu F, Liu Z,
Kang Y, Lin M and Shi H: Analyzing the effects of BRCA1/2 variants
on mRNA splicing by minigene assay. J Hum Genet. 68:65–71. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang Y, Luo H, Pan L, Feng C, Guo Z, Zou
Y, Zeng B, Huang S, Yuan H, Wu P, et al: Reevaluating the
splice-altering variant in TECTA as a cause of nonsyndromic hearing
loss DFNA8/12 by functional analysis of RNA. Hum Mol Genet. Apr
27–2024.(Epub ahead of print). View Article : Google Scholar
|