Introduction
Chicken ovalbumin upstream promoter-transcription
factor II [COUP-TFII; also known as nuclear receptor subfamily 2
group F member 2 (NR2F2)] is a member of the steroid/thyroid
nuclear receptor superfamily (1,2).
COUP-TFII is an orphan nuclear receptor that regulates the
transcription of numerous genes in multiple physiological and
pathological conditions (3), and
can adjust cellular processes, such as angiogenesis, metabolism and
differentiation, as a transcriptional regulator (3–5).
While it can act as either a transcriptional activator or
repressor, the latter appears to be more common (6).
Using conventional knockout and tissue-specific
conditional knockout mice, studies have revealed that COUP-TFII
serves an important role in heart development, angiogenesis and
vein identity determination (7–10).
Dysregulation of COUP-TFII has been implicated in the pathogenesis
of both cardiovascular diseases (CVDs) and colorectal cancer (CRC)
(3). In CVDs, COUP-TFII serves a
role in vascular remodeling and energy metabolism (3), while in CRC, it may function as
either a tumor suppressor or promoter, depending on the cellular
context (3,11). However, the understanding of the
detailed molecular mechanisms linking COUP-TFII to these diseases
remains incomplete.
CVDs and CRC are common causes of mortality and
morbidity worldwide (12). Growing
evidence suggests a shared pathophysiology between CVDs and CRC,
despite these being separate disease entities (13–15).
In addition, CVDs and CRC have overlapping modifiable risk factors,
such as diet, physical inactivity and chronic inflammation
(16). The development of
therapeutic modalities has improved the survival rates of patients
with CRC, thus increasing the numbers of CRC survivors who are at
higher risk for a first or subsequent CVD event; this is partly
attributable to the shared pathophysiology of the two diseases, and
the cardiotoxic effect of anticancer treatment (15). It is reasonable to hypothesize that
therapeutic strategies to improve modifiable CRC risk factors will
also have a beneficial effect on CVD prevention and vice versa.
The present review aims to summarize the structure
and regulatory mechanisms of COUP-TFII, to review its mechanistic
roles in CVDs (Table I) and CRC
(Table II) by focusing on
published papers identified using PubMed (https://pubmed.ncbi.nlm.nih.gov/) with the key words
‘COUP-TFII (NR2F2)’ and ‘CVDs’, ‘COUP-TFII (NR2F2)’ and ‘CRC’, or
‘CVDs’ and ‘CRC’, and to provide future research directions.
 | Table I.Roles and molecular mechanisms of
COUP-TFII in the vasculature and heart. |
Table I.
Roles and molecular mechanisms of
COUP-TFII in the vasculature and heart.
| First author/s,
year | Functions of
COUP-TFII | Molecular
mechanisms | Pathological
implications | (Refs.) |
|---|
| Pereira et
al, 1999; Wu et al, 2013; You et al, 2005; Chen
et al, 2012; Al Turki et al, 2014; Sissaoui et
al, 2020 | Vascular
development and patterning; expressed in venous ECs, atria,
coronary arteries, aorta and VSMCs; acts as a molecular switch
between venous and arterial differentiation | Binds to
cis-regulatory elements (e.g., HEY2 and FOXC1) to
repress arterial markers; recruits HDACs in venous ECs to actively
repress HEY2; interacts with ERG to promote
vein-specific gene expression; the −161K enhancer functions as a
bimodal switch based on COUP-TFII occupancy | Loss or forced
expression disrupts normal vascular identity, leading to aberrant
arterial marker expression in veins and vice versa | (7,8,10,42,44,45) |
| Cui et al,
2015 | Adult vascular
endothelium maintenance-maintains venous identity | Direct binding to
the BMP4 promoter to repress its expression; regulates
inflammatory (e.g., CXCL10/11 and CCL5) and
antithrombotic (for example, TFPI2) genes to prevent
EndMT | Dysregulation can
trigger EndMT, inflammation and altered antithrombotic
responses | (46) |
| Qin et al,
2010 | Angiogenesis -
promotes vessel stability and maturation | Directly
upregulates Ang-1 expression by binding with Sp1 to the Ang-1
promoter | Supports the
formation of stable blood vessels under physiological conditions;
disruption may impair vessel maturation | (20) |
| Dougherty et
al, 2023; Cao et al, 2019; Talati and Hemnes, 2015;
Poels et al, 2020; Rodríguez-García et al, 2017 | Pathological
angiogenesis and metabolic dysregulation; involvement in
atherosclerosis and PAH | Knockdown of
COUP-TFII activates AKT/STAT signaling and increases DKK1
expression; upregulates glycolytic genes (e.g., HK2, LDHA
and PFKFB3), enhancing oxidative stress and lactate
production | Elevated glycolysis
and lipogenesis destabilize atherosclerotic plaques and promote
endothelial proliferation, contributing to PAH pathogenesis;
overall, dysregulation leads to a pro-inflammatory,
hyperproliferative and migratory endothelial phenotype | (56–60) |
| Periera et
al, 1999; Lin et al, 2012; Al Turki et al, 2014;
Nakamura et al, 2011; Qiao et al, 2018; Upadia et
al, 2018; Cornea et al, 2008; Botto et al, 2001;
Perilhou et al, 2008; Vilhais-Neto et al, 2010;
Gruber and Epstein, 2004 | Heart
development-essential for proper cardiac morphogenesis;
conventional knockout leads to embryonic lethality (embryonic day
10) with poorly developed atria, cardinal veins and hemorrhagic
vessels; hypomorphic mutants display atrioventricular septal,
valvular defects and abnormal coronary morphogenesis | Regulates
transcriptional activation (missense variants affect activator
function while repressor function is preserved); interacts with
dosage-sensitive transcription factors (GATA4, TBX5 and NKX2-5);
environmentally responsive (modulated by high glucose and retinoic
acid via pathways such as the Foxo1 pathway) | Congenital heart
defects (atrial/ventricular septal defects, aortic stenosis,
coarctation of the aorta, DORV and VSD) | (7,9,44,61–68) |
| Wu et al,
2015; Kittleson et al, 2005; Hannenhalli et al,
2006 | Adult heart -
mitochondrial and metabolic regulation; maintains mitochondrial
function and metabolic homeostasis in cardiomyocytes | Overexpression
causes defects in the ETC; downregulates genes for mitochondrial
fusion and quality control (Mfn1, Mfn2, Opa1 and Pink1); suppresses
the PGC-1α-Sod2 pathway, leading to the accumulation of damaged
mitochondria; alters fatty acid and glucose metabolism | Mitochondrial
dysfunction and oxidative stress, leading to heart failure;
development of metabolic inflexibility in heart failure | (69–71) |
| Wu et al,
2015; Miao et al, 2022; Tan et al, 2020; Dhalla et
al, 2020; Dixon et al, 2012; Li et al, 2020 Ma
et al, 2020; Tang et al, 2021; Wang et al,
2022 | Elevated COUP-TFII
expression is observed in DIHF | Promotes
ferroptosis (an iron-dependent, non-apoptotic cell death marked by
lipid ROS accumulation) and mitochondrial dysfunction; knockdown
reduces mitochondrial damage and ferroptosis in palmitic
acid-treated neonatal rat cardiomyocytes; direct regulation of
mitochondrial metabolic regulator genes (for example, PPARα and
PGC-1α) | Exacerbation of
diabetic heart failure via increased oxidative stress,
mitochondrial damage and ferroptosis | (69,72–79) |
| Table II.Comparative summary of COUP-TFII
functions in CRC in different molecular contexts. |
Table II.
Comparative summary of COUP-TFII
functions in CRC in different molecular contexts.
| First author/s,
year |
Context/condition | Role of
COUP-TFII | Molecular
mechanisms/marker | (Refs.) |
|---|
| Shin et al,
2009; | High expression
levels in tissue | Tumor
suppressor | Associated with
improved | (80,81) |
| Yun et al,
2017 | microarray (early
studies) |
| survival, ↓ LN
metastasis |
|
| Yun and Park,
2020 | Overexpression in
SNU-C4 cells | Tumor
suppressor | ↑ p53, ↑ PTEN, ↓
Akt signaling | (82) |
| Yun et al,
2020 | Knockdown in HT-29
cells | Tumor
suppressor | ↑ Akt → ↓ GSK-3β →
↑ | (83) |
|
|
|
| β-catenin, ↑ MMP7,
↑ FOXC1 |
|
| Bau et al,
2014; | High expression
levels in EMT- | Tumor promoter | Directly activates
Snail1 | (84,86–88) |
| Cano et al,
2000; | prone CRC
cells |
| transcription → EMT
(by |
|
| Li et al,
2007; |
|
| downregulating
E-cadherin and |
|
| Kudo- Saito et
al, |
|
| enhancing
immunosuppression) |
|
| 2009 |
|
|
|
|
| Wang et al,
2017 | miR-21
overexpression | Tumor promoter | ↑ COUP-TFII →
inhibits | (90) |
|
|
|
| Smad7 → TGF-
β-induced EMT |
|
| Bao et al,
2019 | COUP-TFII
regulation of miR-34a | Tumor promoter | COUP-TFII → ↓
tumor- | (91) |
|
|
|
| suppressor miR-34a
→ ↑ cell |
|
|
|
|
| migration, ↓
chemosensitivity |
|
Literature search
A comprehensive literature search was conducted
using PubMed (https://pubmed.ncbi.nlm.nih.gov/) to identify relevant
studies on COUP-TFII in CVDs or in CRC. The search covered the
literature from January 1900 to January 2025. The inclusion
criteria were: i) Original research articles and reviews
investigating COUP-TFII expression and function in CVDs or CRC; ii)
studies using human tissue samples, cell lines or animal models;
iii) original articles and reviews under the key words ‘CVDs’ and
‘CRC’; and iv) articles published in English. The exclusion
criteria were: i) Studies lacking specific data on COUP-TFII; and
ii) non-English publications. All authors screened the articles,
and any discrepancies were resolved through discussion.
Structure and regulatory mechanisms of
COUP-TFII
Structural characteristics
As shown in Fig.
1A, COUP-TFII is composed of six regions containing three main
domains: An N-terminal domain containing a transcriptional
activation function (AF) motif [1–78 amino acids (aa)], a
DNA-binding domain (DBD; 79–151 aa) and a C-terminal ligand binding
domain (LBD; 177–411 aa), separated by a hinge region (D) (2,3,17).
 | Figure 1.Structure and transcriptional
regulatory mechanism of COUP-TFII. (A) Schematic structure of the
human COUP-TFII protein. The numbers represent the positions of
amino acids. (B) Transcriptional regulatory mechanism of COUP-TFII.
COUP-TFII binds to the 5′-AGGTCA-3′ motif or palindromic sequences
with various spacings (DR site), either directly (homodimer) or
indirectly, through heterodimer formation with other proteins (such
as RXR) to regulate downstream target gene expression. COUP-TFII
can also bind to Sp1 sites via interaction with Sp1 to
cooperatively activate gene expression. COUP-TFII, chicken
ovalbumin upstream promoter-transcription factor II; RXR, retinoid
X receptor; Sp1, specificity protein 1; TSS1, transcription start
site 1; DBD, DNA-binding domain; LBD, ligand-binding domain; AF,
activation function; DR, direct repeat. |
The N-terminal A/B region containing AF1 motif is
required for the recruitment of coactivators or corepressors. DBD,
located in the C region, contains two zinc-finger motifs that
enable COUP-TFII to bind to specific DNA sequences referred to as
direct repeat (DR) motifs composed of the AGGTCA sequence. These
sequences are commonly found in the regulatory regions of target
genes involved in developmental and metabolic pathways. To regulate
a variety of target genes, the DBD of COUP-TFII recognizes and
binds to DR elements and palindromic sequences with variable
spacing (DR-1, DR-2 and DR-4) within the promoter regions of target
genes (2,3,17).
The LBD, located in the E region, facilitates
interactions with coactivators such as steroid receptor
coactivator-1 (SRC-1) and peroxisome proliferator activated
receptor γ coactivator-1α (PGC-1α), and corepressors such as
nuclear receptor corepressor (NCoR) and silencing mediator for
retinoid or thyroid hormone receptor (SMRT) (2,3,17).
Although COUP-TFII lacks a known endogenous ligand,
the LBD modulates transcription through allosteric interactions
with coactivators or corepressors. Various classes of molecules can
interact with the LBD of COUP-TFII, including endogenous lipids and
metabolites (such as retinoic acid derivatives, fatty acids and
phospholipids), synthetic small molecules, steroid hormones,
peptides and coregulatory proteins (such as NCoR, SMRT and SRC-1).
The AF2 motif, located within the LBD, is pivotal for the
recruitment of coactivators or corepressors and modulating
transcriptional activity. The F region refers to the C-terminal
region of the COUP-TFII protein (2,3,17).
COUP-TFII is a ligand-regulated nuclear receptor.
Kruse et al (18)
demonstrated that COUP-TFII adopts an auto-repressed conformation,
as revealed by crystallographic analysis, due to the interaction
between the co-factor binding sites and the AF2 motif, preventing
co-factor recruitment in the absence of ligands. Ligands, such as
retinoic acids, can activate COUP-TFII by releasing it from its
auto-repressed conformation (18).
The findings of this integrative approach can enhance the
translational value of the structural analyses of COUP-TFII in the
absence or presence of ligands in drug development. However, to the
best of our knowledge, the physiological relevance of ligand
regulation remains unclear, with further studies being required to
identify endogenous ligands and validate tissue-specific
effects.
COUP-TFII can function as either a negative or
positive transcriptional regulator via direct binding to DNA
response elements (DR sites) or interaction with other
transcription factors [such as retinoid × receptor and specificity
protein 1 (Sp1)], respectively (3,17).
COUP-TFII can inhibit the transcription of target genes via the
recruitment of corepressors through the formation of homodimers or
heterodimers (Fig. 1B) (2,3).
Alternatively, it can also bind to Sp1 sites to activate the
transcription of target genes [such as neuropilin 2 and
angiopoietin-1 (Ang-1); Fig. 1B]
(19,20). The experimental evidence for this
mechanism was mainly obtained in in vitro studies and tumor
models (19,20). Qin et al (20) used a conditional COUP-TFII
knockout strategy in xenograft models to demonstrate that COUP-TFII
positively regulated tumor angiogenesis by directly binding to Sp1
binding sites in the promoter regions of angiogenesis-related
genes. These findings were supported by robust promoter-reporter
assays, chromatin immunoprecipitation (ChIP) and loss-of-function
experiments, providing convincing mechanistic evidence. Therefore,
in cancer biology contexts, the experimental quality of these
studies (19,20) is high. However, beyond oncogenic
models, their physiological relevance has yet to be validated.
Further studies using cardiovascular- or angiogenesis-specific
models are necessary to confirm the functional relevance of
COUP-TFII-Sp1 interactions in non-cancer conditions.
Regulatory mechanisms of COUP-TFII
expression and activity
The regulatory mechanisms of COUP-TFII expression
and activity are not completely understood, because its specific
ligand has not yet been fully characterized. The expression and
activity of COUP-TFII are tightly regulated by various mechanisms,
such as transcriptional and epigenetic regulation, and by
interactions with signaling pathways.
Transcriptional regulation of COUP-TFII
expression
Upstream regulators of COUP-TFII
expression
COUP-TFII expression has been reported to be
modulated by sonic hedgehog (Shh) during developmental processes,
because an Shh-responsive element was identified in the COUP-TFII
promoter (21). This finding was
based on in vitro promoter assays, and although revealing a
potential regulatory element, in vivo results were lacking.
Thus, the physiological significance of this regulation has yet to
be further validated. COUP-TFII expression has also been reported
to be regulated by retinoic acid (22,23),
estradiol (24) or MAPK pathways
(25). Retinoic acid and its
derivatives regulate COUP-TFII expression via nuclear
receptor-mediated pathways. This regulation is important to
maintain tissue homeostasis and differentiation (22,23).
However, since the study by Soosaar et al (23) was solely based on in vitro
experiments using a combination of cell culture and biochemical
assays, their findings may not be generalized. In addition,
retinoic acid has been reported to suppress COUP-TFII expression in
some contexts (such as the rat glioblastoma C3 and human
glioblastoma U373 cell lines), suggesting that the cellular context
and specificity of receptor isoforms influence the outcome of
retinoic acid signaling (23).
COUP-TFII expression is also regulated by other factors, including
Oct4 (26), microRNA
(miRNA/miR)-302 (26) and Ets-1
(27). COUP-TFII expression has
been revealed to be repressed by Oct4, which binds to the COUP-TFII
promoter, and by miR-302, which interacts with its 3′-untranslated
region (3′-UTR) (26). Oct4, as a
key pluripotency factor, interacts with COUP-TFII to regulate its
transcriptional activity, particularly in stem cell maintenance and
differentiation (26). The
findings of Rosa and Brivanlou (26) were based on overexpression and
knockdown experiments without addressing potential off-target
effects; however, in stem cell models, the results were
comprehensive. Furthermore, the suppressive effect of Oct4 on
COUP-TFII may not be conserved in non-pluripotent cell types,
suggesting that this regulation may be limited by tissue
specificity (26). Petit et
al (27) revealed that Ets-1,
together with SRC-1, could activate COUP-TFII expression in
luciferase reporter assays. This study is insightful for providing
explainable potential mechanisms. However, its conclusions are
derived from in vitro assays, and direct binding of Ets-1 to
the endogenous promoter in physiological conditions has yet to be
fully demonstrated. COUP-TFII expression is repressed by
hypoxia-inducible factor (HIF)-1α in hypoxic endometrial stromal
cells (28) and by the
artery-specific Δ-like canonical Notch ligand 4 -
Notch-hairy/enhancer of split-related with YRPW motif protein 2
(Hey2) signaling axis in endothelial progenitor cells under hypoxic
conditions (29). Both studies
(28,29) emphasized the importance of hypoxic
signaling; however, since the results are confined to specific cell
types, they do not address whether similar regulation occurs in
other cell lineages.
miRNAs are known to be important
post-transcriptional regulators in cancer (30). In particular, Yun and Park
(11) reviewed regulation of
COUP-TFII expression by miRNAs. For example, miRNA-27b
downregulates COUP-TFII expression through interaction with its
3′-UTR, leading to the inhibition of proliferation and invasion of
gastric cancer cells in vitro, and metastasis of gastric
cancer cells to the liver in vivo (31). A study by Feng et al
(31) demonstrated both in
vitro and in vivo effects, which reinforces its
conclusions. However, the mechanistic association between COUP-TFII
suppression and phenotypic outcomes was not directly validated by
rescue experiments, leaving room for alternative explanations.
Additionally, miR-302, miR-302a, miR-382, miR-101, miR-27a and
miR-194 have been reported to downregulate COUP-TFII expression,
and to have diverse functions in different cell types, such as
embryonic stem cells, mesenchymal C3H10T1/2 cells, colorectal
cancer cells and prostate cancer cells (26,32–37).
These studies used luciferase reporter assays, reverse
transcription-quantitative PCR and western blot analyses to
demonstrate miRNA-mediated repression of COUP-TFII. However, the
studies have several limitations. For example, Rosa and Brivanlou
(26) based their findings on
embryonic stem cells, limiting their applicability to cancer. For
miR-382 and miR-101 (34–36), COUP-TFII downregulation was
observed, but indirect effects could not be excluded. In the case
of miR-27a (36), opposing roles
in different tissues suggest context dependence. The study by Jeong
et al (37) demonstrated
that miR-194 functions as a key regulator of COUP-TFII in
mesenchymal stem cells, directing their fate toward differentiation
into osteoblasts and adipocytes.
Overall, although evidence supports the miRNA-based
regulation of COUP-TFII, context specificity, functional redundancy
and cell type-dependent expression complicate the identification of
consistent regulatory mechanisms.
Epigenetic regulation of COUP-TFII
expression
Methylation of DNA, usually at CpG island sites in
promoters of genes, has been reported to inhibit transcription by
interfering with the binding of transcription factors (38,39).
Using the CpGPLOT program from the European Bioinformatics
Institute website (http://www.ebi.ac.uk/emboss/cpgplot), Al-Rayyan et
al (40) revealed that the CpG
island is located in the promoter and within exon 1 of COUP-TFII,
and demonstrated that transcription of COUP-TFII might be regulated
by DNA methylation. The authors also revealed that methylation of
the COUP-TFII promoter led to its reduced expression, which
contributed to resistance to antiestrogen treatment in
endocrine-resistant breast cancer cells (40). This study provides important
evidence associating epigenetic repression of COUP-TFII with
therapeutic resistance, using bioinformatics prediction (CpGPLOT),
bisulfite sequencing and expression analysis. However, the findings
were based on a specific endocrine-resistant breast cancer cell
line (MCF-7-derived), which limits the generalizability.
Furthermore, while COUP-TFII repression was associated with
resistance, functional rescue assays were not performed, and thus,
the causal relationship is undetermined.
A total of four isoforms (Iso1, Iso2, Iso3 and Iso4)
are encoded by the NR2F2 gene. NR2F2-Iso1 is a full-length
protein composed of an N-terminal AF1 motif, a DBD, a hinge region
and an LBD containing a ligand-dependent AF2 motif. NR2F2-Iso2,
NR2F2-Iso3 and NR2F2-Iso4 are different from NR2F2-Iso1 in their
N-terminal sequences and do not have a DBD (41). Davalos et al (41) demonstrated that when neural crest
cells (NCCs) differentiated into melanocytes, NR2F2-Iso2 expression
was silenced by DNA methylation, whereas during the progression of
metastatic melanoma, this process was reversed. Thus, the study
suggested that DNA methylation and demethylation serve important
roles in the progression of metastatic melanoma, and their
regulation of NR2F2 activity through NR2F2-Iso2 contributes to the
acquisition of NCC-like and epithelial-to-mesenchymal transition
(EMT)-like features in transformed melanocytes (41).
Davalos et al (41) combined genome-wide methylation
profiling with functional assays to reveal that epigenetic
reactivation of NR2F2-Iso2 promoted melanoma progression. While the
study provided supportive evidence, it depended on melanoma cell
lines and xenografts, which may not fully reflect the heterogeneity
of human tumors. Additionally, since the association between
NR2F2-Iso2 and EMT-like features was based on correlative data and
functional assays, the direct molecular targets remain unknown.
The role of NR2F2 isoforms appears to be
context-dependent. In contrast to their oncogenic role in melanoma,
some truncated isoforms lacking the DBD exhibit growth-suppressive
effects in breast and lung cancer. These discrepancies suggest that
isoform function is influenced by cell type, co-regulators and
signaling context. Therefore, the pro-metastatic role of NR2F2-Iso2
should not be generalized across all types of cancer. Further
research should be performed to define isoform-specific mechanisms
and interactions in diverse tumor types.
Interactions with signaling pathways
MAPK pathway
Activation of the MAPK pathway has been reported to
increase COUP-TFII expression in certain cellular contexts. For
example, in breast cancer cell lines with elevated MAPK activity,
COUP-TFII levels are increased (25), suggesting that this pathway can
upregulate COUP-TFII as part of a broader cellular response to
growth signals. However, the precise relationship between MAPK
signaling and COUP-TFII expression is complex, and appears to vary
depending on the specific cell type and environmental context. This
study examined the association between MAPK signaling and COUP-TFII
upregulation in breast cancer models using pharmacological
inhibitors, suggesting an association between pathway activation
and COUP-TFII expression (25).
However, it lacks evidence regarding direct transcriptional
regulation by MAPK effectors, such as ERK, ETS like-1 protein or
c-fos. The study also used a limited number of cell lines, and
failed to investigate potential pathway interactions, such as
crosstalk with PI3K/AKT or hormonal signaling.
Notch signaling
Notch signaling serves an antagonistic role in
regulating COUP-TFII expression (10,29).
In endothelial cells (ECs), activation of Notch signaling leads to
a reduction in COUP-TFII levels, which in turn promotes arterial
differentiation (10,42). Conversely, COUP-TFII can inhibit
Notch signaling to maintain venous identity, thereby influencing
cell fate decisions during vascular development (10,42).
This reciprocal regulation emphasizes the role of COUP-TFII in
establishing and preserving vascular heterogeneity. Taken together,
these findings are supported by studies (10,42)
using genetic mouse models, lineage tracing and molecular assays
that demonstrate a causal association between Notch activation and
COUP-TFII downregulation in ECs during vascular development.
However, the majority of the studies (10,42)
focused on embryonic or early postnatal stages, leaving its role in
adult or pathological conditions unclear. It is also uncertain
whether this regulatory axis applies to other vascular beds, such
as lymphatic or tumor endothelium. While mutual antagonism between
COUP-TFII and Notch in development processes has been established,
its dynamics in disease contexts require further investigation
across diverse tissues.
Wnt/β-catenin pathway
COUP-TFII expression has been reported to be
upregulated by the Wnt/β-catenin pathway (43). ChIP assays have demonstrated that
β-catenin/T-cell factor 7-like 2 or transcription factor 7-like 2
(TCF7L2) complexes bind directly to the COUP-TFII promoter, leading
to increased transcription. This interaction is notable in the
context of cell differentiation. For example, activation of Wnt
signaling, leading to the upregulation of COUP-TFII via
β-catenin/TCF7L2 complexes, results in the suppression of adipocyte
differentiation by inhibiting the expression of the adipogenic
transcription factor peroxisome proliferator-activated receptor
(PPAR)γ (43). Okamura et
al (43) demonstrated that
β-catenin/TCF7L2 directly bound to the COUP-TFII promoter in ChIP
and reporter assays in preadipocyte models, and confirmed
Wnt-mediated regulation through gain- and loss-of-function
approaches. However, the focus of the study on adipocytes limited
the generalizability to other cell types. It also remains undefined
whether upregulation of COUP-TFII is a primary Wnt response, or a
downstream effect. Broader in vivo and multi-lineage studies
are needed to fully define the mechanisms by which Wnt signaling
modulates COUP-TFII expression.
COUP-TFII in CVDs
Roles and molecular mechanisms of
COUP-TFII in the vasculature: From developmental patterning to
pathological angiogenesis
COUP-TFII is predominantly expressed in venous ECs,
the atria, coronary arteries, the aorta (44) and vascular smooth muscle (8). COUP-TFII is important for proper
vascular development (7). An in
vivo study revealed that genetic ablation of COUP-TFII induces
the ectopic expression of arterial markers in venous ECs, whereas
forced expression of COUP-TFII in arterial cells suppresses
arterial gene expression, underscoring its role as a molecular
switch in vascular differentiation (10). Mechanistically, COUP-TFII binds to
cis-regulatory elements at key loci, such as HEY2 and
FOXC1 which are required for arterial differentiation. By
binding to their regulatory regions, COUP-TFII inhibits the
activation of the arterial differentiation program (42). A study by Sissaoui et al
(45) demonstrated that in venous
ECs, COUP-TFII recruited histone deacetylases (HDACs) to actively
repress the expression of HEY2, a key mediator of arterial
identity. Furthermore, COUP-TFII interacted with ETS-related gene
(ERG) to induce the expression of several vein-specific
genes, such as family with sequence similarity 174 member B, a
disintergrin and metalloprotease with thrombospondin motifs 18 and
LIM homeobox 6 (45).
The working model proposed by Sissaoui et al
(45) suggests that a regulatory
element (the −161K enhancer) acts as a bimodal switch depending on
the cellular context: In venous ECs, COUP-TFII occupies this
enhancer to block ERG-mediated activation of HEY2, whereas
in arterial ECs lacking COUP-TFII, ERG binds to the −161K enhancer
and promotes arterial gene expression. In the study by Sissaoui
et al (45),
CRISPR-CRISPR-associated protein 9 enhancer deletion,
ChIP-sequencing and reporter assays were effectively used to define
the COUP-TFII-HDAC-ERG axis and demonstrate the role of COUP-TFII
as a repressor of arterial identity in venous ECs. However, the
focus on embryonic tissues and human umbilical vein ECs limits the
relevance to adult vasculature and pathological angiogenesis. In
pathological settings such as tumors, COUP-TFII expression can
persist in vessels with arterial features, suggesting that its
regulatory role may be more flexible than previously suggested
(20). Broader, context-specific
studies are needed to clarify its function in vascular
remodeling.
Cui et al (46) revealed the role and molecular
mechanism of COUP-TFII in adult vascular endothelium, demonstrating
that knockdown of COUP-TFII in adult vascular ECs upregulated the
expression levels of inflammatory genes [such as C-X-C motif
chemokine ligand (CXCL)10, CXCL11 and C-C motif chemokine ligand
(CCL)5], while downregulating the expression levels of
antithrombotic genes [such as tissue factor pathway inhibitor 2
(TFPI2)]. This dysregulation could trigger
endothelial-to-mesenchymal transition (EndMT). Furthermore, the
expression levels of arterial markers (such as ephrin-B2 and hes
family bHLH transcription factor 1/4), expression of genes involved
in angiogenesis [such as endothelial PAS domain protein 1/HIF-2α,
ephrin-A1, tyrosine kinase with immunoglobulin like and EGF like
domains-2 (Tie-2) and ephrin-A4] and the osteogenic potential
leading to calcium deposition of adult ECs were suppressed by
COUP-TFII overexpression. Cui et al (46) demonstrated that the actions of
COUP-TFII in adult ECs may be mediated by its negative regulation
of bone morphogenic protein 4 (BMP4) through direct binding to the
BMP4 promoter. These results suggest that COUP-TFII serves a key
role in maintaining the venous identity in adult ECs, as well as
during development (46). This
study focused on adult ECs, a less explored context for COUP-TFII.
Cui et al (46) used in
vitro knockdown/overexpression and ChIP assays to reveal that
COUP-TFII suppressed pro-inflammatory, angiogenic and osteogenic
gene programs, reinforcing their conclusions. However, reliance on
cultured ECs limits in vivo relevance, while other pathways
regulating BMP4 were not fully excluded. Their findings suggest
anti-inflammatory and anti-osteogenic roles of COUP-TFII in adult
ECs, which are inconsistent with reports of its pro-angiogenic and
immunosuppressive effects in tumor models (17,20).
These discrepancies underscore context-dependent functions of
COUP-TFII and emphasize the need for further investigations in
normal vs. diseased adult vasculature.
Angiogenesis, the formation of new blood vessels,
relies on coordinated interactions among shear stress, vascular
growth factors such as VEGF and Ang-1 (a ligand for Tie-2),
intracellular signaling pathways (such as Notch) and intercellular
contacts (47–54).
Knockdown experiments in mouse aortic vascular
smooth muscle cells and C3H10T1/2 fibroblasts have revealed that
COUP-TFII positively regulates Ang-1 expression levels (20). This regulation occurs through the
direct binding of COUP-TFII, in cooperation with Sp1, to the Ang-1
promoter. The upregulation of Ang-1 by COUP-TFII suggests a
pro-angiogenic role, promoting vessel stability and maturation
(20). However, this study
(20) was limited by its
dependence on in vitro fibroblast and smooth muscle cell
models, which may not reflect the in vivo angiogenic
environment. Although ChIP assays verified COUP-TFII binding to the
Ang-1 promoter, its functional relevance in ECs in vivo
remains unvalidated. Furthermore, the role of COUP-TFII in
angiogenesis appears to be context-dependent, while it can promote
Ang-1 expression, another study has revealed that it may suppress
VEGF receptor expression and inhibit sprouting, particularly in
tumors (55). These conflicting
findings suggest that COUP-TFII may balance vascular stabilization
and angiogenic modulation in a tissue- and context-specific
manner.
Dysregulation of COUP-TFII can lead to pathological
angiogenesis, contributing to various vascular diseases, such as
atherosclerosis and pulmonary arterial hypertension (PAH) (56). For instance, in multiple types of
human ECs (e.g., lung microvascular, coronary artery and pulmonary
artery ECs), knockdown of COUP-TFII activates the AKT and STAT
signaling pathways. This activation is accompanied by an increase
in Dickkopf-related protein 1 expression, resulting in a
pro-inflammatory, proliferative, hypermigratory and
apoptosis-resistant phenotype. These changes are further
characterized by the induction of EndMT, increased oxidative
stress, enhanced lactate production and the upregulation of
glycolytic genes, such as hexokinase 2, lactate dehydrogenase A and
6-phosphofructo-2-kinase/fructose-2,6-bisphophatase 3 (PFKFB3)
(56). The strengths of this study
include the use of multiple types of ECs, and the integration of
molecular, metabolic and phenotypic analyses (56). However, key limitations include the
reliance on in vitro knockdown models, which may not fully
reflect the complexity of the in vivo vascular
environment.
Suppression of COUP-TFII has been associated with
the induction of glycolytic and lipogenic pathways (57,58).
Elevated PFKFB3 levels, in particular, destabilize atherosclerotic
plaques and contribute to the progression of atherosclerosis
(59). Similarly, enhanced
endothelial glycolysis and proliferation, driven by COUP-TFII
suppression, may underlie the pathogenesis of PAH (57,60).
These studies are notable for their mechanistic data associating
metabolic shifts with vascular dysfunction and their use of
relevant EC models (57,60). However, limitations include a lack
of in vivo causal data, and uncertainty regarding whether
the metabolic changes are a primary driver, or a secondary effect,
of the diseases. Furthermore, other studies suggest that COUP-TFII
loss may induce protective metabolic adaptations in other contexts,
indicating potential tissue- or disease-specific variability that
warrants further investigation (3,5).
Role of COUP-TFII in the heart
Knockout mouse models and human genetic analyses
have been used to elucidate the role of COUP-TFII in the heart
during development (7,9,44,61–63).
Conventional COUP-TFII knockout mice die at embryonic day 10,
exhibiting poorly developed atria and cardinal veins with enlarged
hemorrhagic vessels, while hypomorphic mutants are born with
atrioventricular septal and valvular defects, as well as abnormal
coronary morphogenesis (7,9). EC-specific COUP-TFII knockout mice
exhibit embryonic lethality due to vascular defects, including
hypoplastic endocardial cushions, hemorrhage, and thin, dilated
vessels (9). This study provided
in vivo evidence for the key role of COUP-TFII in vascular
development, using a genetically controlled, cell type-specific
knockout model (9). However,
limitations include the early lethality of the models, which
restricts analysis to embryonic stages, and the lack of mechanistic
investigation of downstream pathways. To the best of our knowledge,
while no direct conflicting results have been reported, some
studies have suggested that the function of COUP-TFII may differ in
postnatal or under pathological conditions (17,20,55),
indicating potential stage-specific roles that require further
research.
Human genetic studies have revealed that COUP-TFII
missense variants or 15q terminal deletions encompassing COUP-TFII
are associated with atrial or ventricular septal defects, either in
isolation or in combination with other congenital heart defects,
such as aortic stenosis and coarctation of the aorta (44,61–63).
In vitro experimental data indicate that all six COUP-TFII
missense variants (Gln75dup, Asp170Val, Asn205Ile, Glu251Asp,
Ser341Tyr and Ala412Ser), identified in patients with
atrioventricular septal defects, have a measurable impact on the
transcriptional activator function of COUP-TFII in at least one of
two assays: Nerve growth factor-induced protein A or apolipoprotein
B (APOB) promoter-driven luciferase assays in HEK293 cells. By
contrast, the repressor function of COUP-TFII appears to remain
intact, as revealed by APOB promoter-driven luciferase assays in
HEPG2 cells. The promoter-specific effects of these individual
mutations likely reflect the complexity of the protein-protein
interactions involving COUP-TFII, which vary, depending on the
tissue, developmental stage and genomic context (44). The strengths of this study include
the integration of genetic and functional data; however,
limitations derive from the use of non-cardiac,
overexpression-based assays that may not accurately reflect in
vivo cardiac transcriptional networks (44). In addition, the mutation-specific
and promoter-dependent effects underscore the complexity of
COUP-TFII regulation, while for the majority of variants, no in
vivo confirmation of pathogenicity has been provided.
COUP-TFII may also function as an environmentally
responsive factor by mediating the effects of known non-genetic
congenital heart disease risk factors, such as high glucose
(64) and retinoic acid levels
(65). Insulin and glucose have
been revealed to suppress COUP-TFII expression via the Foxo1
pathway in hepatocytes and pancreatic cells (66). Furthermore, COUP-TFII serves an
important role in retinoic acid signaling during development
(67). However, to the best of our
knowledge, direct evidence for its role in the developing heart is
scarce. Thus, the translational relevance of these findings remains
uncertain. Further studies are required to determine how glucose
and retinoic acid levels may alter COUP-TFII expression in the
developing heart. Additionally, COUP-TFII has been reported to
interact with other dosage-sensitive transcription factors that are
key for heart formation, including GATA binding protein 4 (GATA4),
T-box transcription factor 5 and NK2 homeobox 5 (68). Specifically, a novel heterozygous
mutation (p.G83X) in the COUP-TFII gene was identified in a family
affected by congenital double outlet right ventricle and
ventricular septal defect. Functional investigations demonstrated
that the COUP-TFII G83X-mutant protein exhibited no transcriptional
activity. Furthermore, the mutation abrogated the synergistic
transcriptional activation of COUP-TFII with GATA4 (62). These functional assays provide
mechanistic insights, but are limited by the use of overexpression
systems in non-cardiac cells. No in vivo validation or
comprehensive mutational analysis was carried out, and the
interaction between COUP-TFII and environmental signals in human
cardiogenesis has yet to be fully elucidated.
The role and molecular mechanisms of COUP-TFII in
the adult heart have been investigated using COUP-TFII
overexpression mouse models (69),
and two independent cohorts of patients with non-ischemic dilated
cardiomyopathy (DCM) (70) and
idiopathic DCM (dataset no. GSE406) (71). Using COUP-TFII-overexpressing mice
and rescue experiments through the removal of COUP-TFII in
calcineurin-transgenic (CnTg/Cre/F+) mice, this study
has demonstrated that COUP-TFII overexpression induced defects in
the electron transport chain (ETC), downregulation of genes
involved in mitochondrial fusion and quality control [such as
mitofusin (Mfn)1 and Mfn2, optic atrophy 1 (Opa1), and PTEN-induced
kinase 1 (Pink1)], accumulation of damaged mitochondria, and
reduced mitochondrial reactive oxygen species (ROS) scavenging
capacity, leading to oxidative stress and heart failure.
Mitochondrial dysfunction induced by COUP-TFII overexpression may
be mediated through the suppression of the PGC-1α-superoxide
dismutase 2 (Sod2) pathway (69).
Thus, alterations in the expression of key genes involved in both
fatty acid and glucose metabolism by COUP-TFII may contribute to
the development of metabolic inflexibility in heart failure
(69). These findings are
supported by gain- and loss-of-function models and transcriptomic
data from two independent DCM cohorts (70,71).
However, the limitations of these studies include the use of
non-physiological overexpression levels, which may not accurately
reflect COUP-TFII dysregulation in human disease, and a lack of
longitudinal data to determine causality in patients (70,71).
In addition, other COUP-TFII targets relevant to calcium handling,
fibrosis or apoptosis were not explored and potential
context-dependent roles of COUP-TFII in different tissues remain
unresolved.
Increased COUP-TFII expression has been observed in
diabetes-induced heart failure (DIHF) (72). Although metabolic disturbances,
oxidative stress-induced cardiomyocyte death, inflammation and
mitochondrial dysfunction are known to contribute to cardiac
remodeling in DIHF, the pathophysiology and underlying mechanisms
of DIHF remain unclear (73,74).
A previous study demonstrated that the overexpression of COUP-TFII
exacerbated DIHF by promoting ferroptosis and mitochondrial
dysfunction, as evidenced by reduced mitochondrial damage and
ferroptosis after COUP-TFII knockdown in palmitic acid (PA)-treated
neonatal rat cardiomyocytes (NRCMs) (72). Ferroptosis, an iron-dependent,
non-apoptotic form of cell death, is characterized by the
accumulation of lipid ROS and mitochondrial overload (75). Although ferroptosis contributes to
the development of CVDs and its suppression may alleviate diabetic
myocardial ischemia-reperfusion injury (76–78)
and cardiomyopathy (79), the role
of mitochondria in ferroptosis remains controversial. Further
studies are needed to validate the association between PA-induced
ferroptosis and mitochondrial dysfunction in NRCMs. Taken together,
the aforementioned studies suggest that COUP-TFII directly
regulates mitochondrial metabolic regulator genes, including PPARα
and PGC-1α (69), and its
upregulation is associated with heart failure, possibly as a result
of mitochondrial dysfunction (69,72).
While the study effectively associates COUP-TFII with ferroptosis
through lipid ROS and mitochondrial stress, its reliance on
neonatal rat models limits its translational relevance (69). Additionally, the mechanistic role
of mitochondria in ferroptosis is still open to debate (75–79),
and the causal relationship between COUP-TFII upregulation and DIHF
in human hearts is still unconfirmed. Further in vivo
validation and exploration of the interaction of COUP-TFII with key
mitochondrial regulators such as PPARα and PGC-1α are needed.
COUP-TFII in CRC
Tumor-suppressive role of COUP-TFII in
CRC
Several studies have demonstrated that COUP-TFII
acts as a tumor suppressor in CRC (80–83).
A retrospective study using tissue microarray assays have indicated
that high COUP-TFII expression is associated with improved survival
rates and reduced lymph node metastasis in patients with CRC;
however, the initial study was limited by small sample sizes and
short follow-up periods (80).
These findings have subsequently been confirmed in larger cohorts
with extended follow-up durations (81), supporting the hypothesis that
COUP-TFII exerts tumor-suppressive effects in CRC.
Functional studies in human CRC cells have provided
mechanistic insights. In SNU-C4 cells, COUP-TFII overexpression
suppressed proliferation and invasion by upregulating the tumor
suppressors p53 and PTEN, and by inhibiting Akt activity,
respectively (82,83). Conversely, COUP-TFII knockdown in
HT-29 CRC cells enhanced cell proliferation and invasive
capabilities through the activation of the Akt pathway, leading to
glycogen synthase kinase-3 β inactivation, β-catenin stabilization,
and the upregulation of MMP7 and FOXC1 (83); however, the use of only two CRC
cell lines (SNU-C4 and HT-29) limits the generalizability. These
results underscore the function of COUP-TFII as a negative
regulator of tumor aggressiveness in CRC. However, additional in
vivo validation studies are necessary to confirm these findings
and fully elucidate the mechanism by which COUP-TFII modulates CRC
progression.
Tumor-promoting role of COUP-TFII in
CRC
By contrast, a growing body of evidence indicates a
tumor-promoting role of COUP-TFII under certain conditions. For
example, patients with CRC with high COUP-TFII expression have an
increased risk of metastasis and reduced overall survival, compared
with patients with CRC with low COUP-TFII expression (84,85).
Mechanistically, COUP-TFII directly binds to the promoter region of
the Snail1 gene, a master regulator of EMT, leading to the
suppression of E-cadherin and increased immunosuppressive signaling
(84,86–88).
Additionally, miR-21, an oncomiR highly expressed in various types
of cancer (89), increases
COUP-TFII expression, which in turn promotes TGF-β-induced EMT by
inhibiting Smad7 (90). COUP-TFII
can also regulate the expression of miR-34a (91), a well-known tumor suppressor miRNA
that inhibits tumor cell migration and promotes chemosensitivity
(92–95). Notably, COUP-TFII-regulated miR-21
inhibits PTEN and elevates the levels of prostaglandin
E2, a pro-inflammatory and pro-tumorigenic mediator, by
suppressing 15-hydroxyprostaglandin dehydrogenase, further
contributing to tumor progression and inflammation (3,90).
Although these findings are supported by cell and animal models,
limitations remain, including reliance on overexpression systems,
and unclear relevance across different types of cancer. A
conflicting report of the anti-inflammatory roles of COUP-TFII in
other tissues suggests context-dependent effects (96), emphasizing the need for further
validation in human tumors and diverse inflammatory settings.
Dual functions of COUP-TFII as either a
tumor suppressor or a tumor promoter
COUP-TFII exhibits context-dependent dual roles in
CRC, functioning either as a tumor suppressor or as a tumor
promoter, depending on the molecular and cellular environment
(3,11). To illustrate these dichotomous
roles, Table II summarizes the
molecular and phenotypic outcomes associated with COUP-TFII
expression in CRC.
The findings presented in Table II highlight the dualistic and
context-dependent role of COUP-TFII in CRC, where its functional
role is modulated by dynamic interactions with transcriptional
regulators, non-coding RNAs and oncogenic signaling pathways.
As aforementioned, discrepancies in reported
findings are possibly due to differences in study conditions, such
as experimental models, patient cohorts and methodological
approaches. Additionally, the majority of the aforementioned
studies relied on in vitro and animal models, which may not
fully recapitulate the pathophysiology of human CRC. Another
limitation is the variability in COUP-TFII detection methods, such
as differences in antibody specificity and RNA expression analysis
techniques, which could contribute to inconsistent results. Future
studies with standardized methodologies, large patient cohorts and
human experimental data are needed to clarify the role of COUP-TFII
in CRC.
Interrelation between CVDs and CRC
CVDs and CRC are among the leading causes of
mortality worldwide (12). CRC is
the third most common malignancy worldwide, accounting for 11% of
all cancer diagnoses (97).
Although CVDs and CRC are distinct disease entities, emerging
evidence suggests that they may share common features (98,99).
Using an ischemic cardiomyopathy C57BL/6J-ApcMin/J
(APCmin) mouse model, researchers identified a potential
causal association between heart failure and CRC by observing the
association of presence of heart failure with enhanced tumor
formation, possibly via cardiac excreted factors, such as SerpinA3,
fibronectin and paraoxonase 1 (100). This study provided valuable
insights into a potential causal association between heart failure
and CRC, whereas its interpretation is still limited due to the use
of a familial CRC model that does not fully recapitulate sporadic
CRC, and the lack of mechanistic validation using targeted
interventions, such as knockdown or neutralization of candidate
cardiac excreted factors. In a clinical study, patients aged >65
years with stage I–III CRC were revealed to have a higher risk of
developing new-onset CVDs compared to a matched cohort of Medicare
patients without cancer (13),
suggesting the possibility of shared vulnerability or cancer
treatment-related cardiovascular complications. However, the
observational nature of the study lacks causal inference, while the
residual confounding effect of common risk factors (such as age,
diabetes and smoking) cannot be excluded. Two meta-analyses have
demonstrated that use of angiotensin converting enzyme
inhibitors/angiotensin receptor blockers and renin-angiotensin
system (RAS) inhibitors was associated with a decreased risk of
CRC, respectively (101,102). These findings indicate that
hypertension might be a causal factor for CRC. Although these
findings are promising, the analyses are limited by heterogeneity
in study design, population characteristics and potential
indication bias, since patients receiving RAS inhibitors may be
healthier or more closely monitored than non-users. Furthermore,
emerging evidence indicates that chronic inflammation and oxidative
stress, the underlying pathophysiological mechanisms for both
diseases, may contribute to the association between CVDs and CRC
(16,103,104). Additional mechanisms that have
been suggested include the cardiotoxicity of certain anticancer
agents, shared molecular pathways between the two diseases and
innate immunity (103,105–107). However, while these mechanisms
are supported by both experimental and clinical evidence (16,103–107), the evidence remains largely
correlative and prospective studies with mechanistic endpoints are
needed to clarify their roles and causality.
COUP-TFII as a common molecular
regulator of CVDs and CRC
Despite the controversial roles of COUP-TFII in
CRC, growing evidence (3,5,20,46,69,82,89,90)
has associated chronic inflammation, oxidative stress, angiogenesis
and metabolic dysregulation with both CVDs and CRC, with COUP-TFII
serving as a common molecular regulator. Fig. 2 illustrates the molecular
mechanisms by which COUP-TFII functions as a common regulator in
both CVDs and CRC.
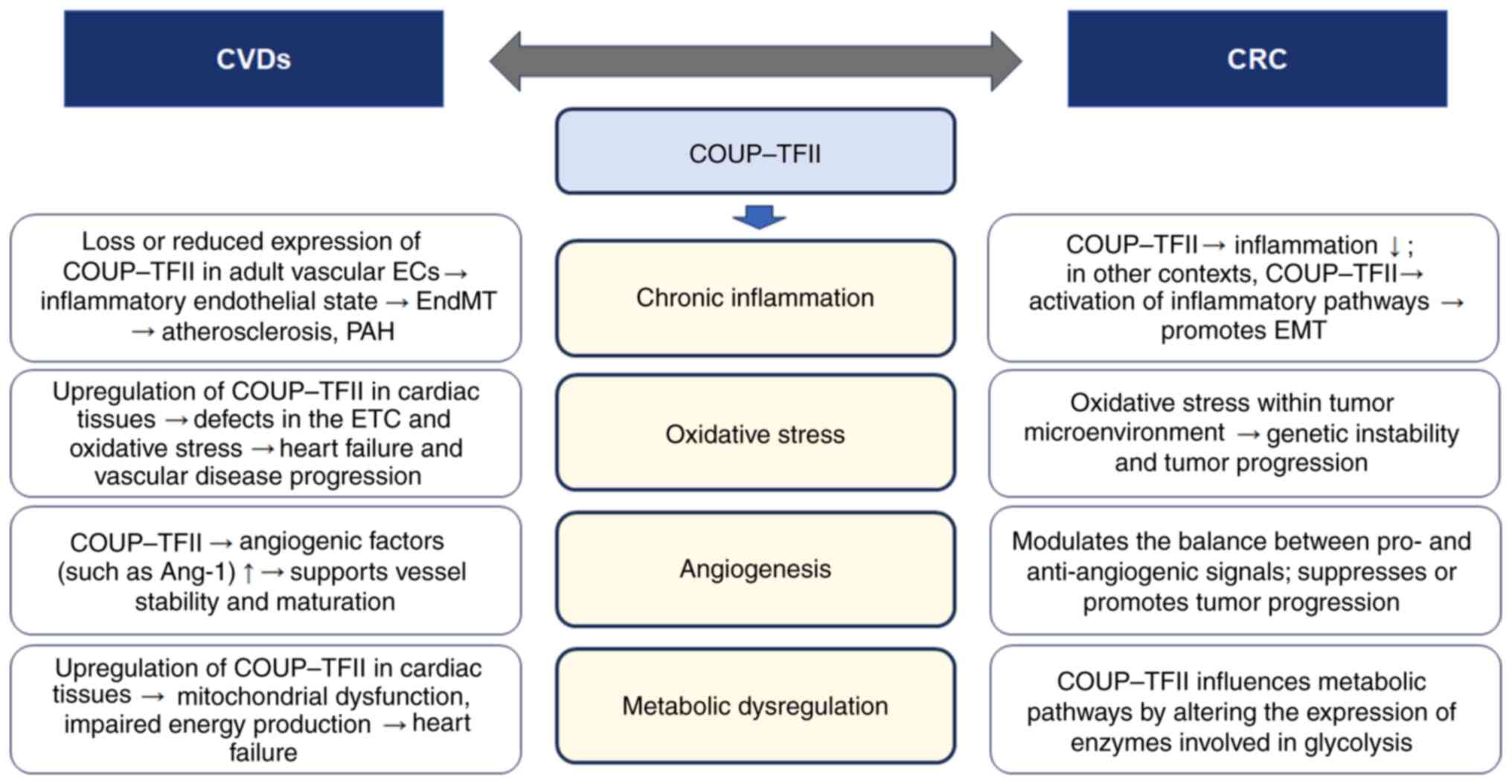 | Figure 2.Schematic illustration of the
molecular mechanisms by which COUP-TFII functions as a common
regulator in both CVDs and CRC. Although CVDs and CRC are distinct
disease entities, they share common pathophysiological features. In
particular, chronic inflammation, oxidative stress, angiogenesis
and metabolic dysregulation by COUP-TFII may serve roles in the
pathogenesis of both conditions. COUP-TFII, chicken ovalbumin
upstream promoter-transcription factor II; EC, endothelial cell;
EndMT, endothelial-to-mesenchymal transition; ETC, electron
transport chain; Ang-1, angiopoietin-1; EMT,
epithelial-to-mesenchymal transition; CVD, cardiovascular disease;
CRC, colorectal cancer; PAH, pulmonary arterial hypertension. |
Based on published reports (46,108) and the aforementioned
descriptions, chronic inflammation is involved in both CVDs and CRC
(Fig. 2). As previously mentioned
(46), COUP-TFII is key to
maintain endothelial homeostasis. In adult vascular ECs, it
represses the expression of pro-inflammatory cytokines (such as
CXCL10, CXCL11 and CCL5), while supporting the expression of
anti-thrombotic genes, such as TFPI2. Loss or reduced expression of
COUP-TFII can lead to an inflammatory endothelial state, triggering
EndMT, which contributes to atherosclerosis and other vascular
pathologies (46). In the tumor
microenvironment of CRC, chronic inflammation is a known driver of
cancer progression (108).
COUP-TFII appears to serve a dual role, depending on the cellular
context. In some CRC cells, overexpression of COUP-TFII promotes
tumor-suppressive signaling by upregulating p53 and PTEN while
inhibiting Akt (82), which can
help reduce inflammation. However, in other contexts, increased
COUP-TFII expression, especially when upregulated by miRNAs such as
miR-21, can promote EMT through activation of inflammatory
pathways, thereby enhancing tumor invasiveness (3,89,90).
Oxidative stress serves an important role in both
CVDs and CRC (Fig. 2). In the
heart and vasculature, COUP-TFII is involved in the regulation of
mitochondrial function. Upregulation in cardiac tissues has been
associated with defects in the ETC, and a reduction in the
expression of mitochondrial quality control genes (such as Mfn1,
Mfn2, Opa1 and Pink1). This leads to the accumulation of damaged
mitochondria and impaired ROS scavenging, culminating in elevated
oxidative stress, a key factor in heart failure and vascular
disease progression (69). While
the study by Wu et al (69)
is pivotal in terms of mechanistic exploration using transgenic
mouse models of DCM, some limitations should be addressed. First,
the cardiac-specific effects of COUP-TFII may not be directly
generalizable to other tissues, including the colon. Second,
although the mitochondrial dysfunction and oxidative stress were
associated with COUP-TFII levels, direct causality and
dose-response relationships were not fully established,
particularly with respect to downstream disease phenotypes. In CRC,
oxidative stress within the tumor microenvironment can promote
genetic instability and tumor progression (109). However, lack of direct evidence
associating COUP-TFII with ROS regulation in CRC leaves the
proposed relationship speculative. To the best of our knowledge,
the majority of the existing data, including the aforementioned
study by Yang et al (109), focus on general mechanisms of ROS
in cancer, and do not specifically implicate COUP-TFII. Therefore,
while it is plausible that regulation of metabolic and
mitochondrial pathways by COUP-TFII could indirectly influence ROS
levels in CRC, this hypothesis remains unvalidated in CRC-specific
in vivo models. Further research using CRC-specific
conditional knockout or overexpression models is essential to
clarify whether COUP-TFII modulates oxidative stress in the colon
epithelium or tumor microenvironment and whether this mechanism
contributes causally to colorectal tumorigenesis.
COUP-TFII regulates angiogenesis, affecting the
progression of both CVDs and CRC (Fig.
2). COUP-TFII positively regulates angiogenic factors such as
Ang-1 by binding to its promoter (often in cooperation with Sp1),
which supports vessel stability and maturation (46). This function is key for normal
vascular repair and to prevent pathological calcification (46). In CRC, COUP-TFII modulates the
balance between pro- and anti-angiogenic signals. By influencing
the expression of factors that control angiogenesis, COUP-TFII can
impact tumor vascularization (17,20).
Depending on the cellular context, this modulation may either
suppress or promote tumor progression.
COUP-TFII also regulates energy metabolism,
influencing both cardiac function and cancer cell proliferation
(Fig. 2). COUP-TFII regulates
various genes such as GLUTt4, PPARα, fatty acid binding
protein 3 and carnitine palmitoyltransferase 1 involved in both
fatty acid and glucose metabolism (3,5).
Upregulation of COUP-TFII in cardiac tissues has been associated
with the downregulation of key metabolic regulators, such as PGC-1α
and Sod2, leading to mitochondrial dysfunction, impaired energy
production and ultimately heart failure (69). In CRC, COUP-TFII can influence
metabolic pathways through its effects on the Akt signaling cascade
and by altering the expression of enzymes involved in glycolysis
(5). This metabolic dysregulation
provides the energetic and biosynthetic requirements for rapid
tumor cell proliferation, thereby contributing to cancer
progression.
Therapeutic implications of COUP-TFII in
CVDs
Vascular interventions using grafts are often
required to treat severe CVDs (110). However, traditional grafts face
challenges, such as susceptibility to infection, thrombogenicity
and poor long-term patency rates (111–113). Recent advances in vascular
biology underscore the key role of COUP-TFII in regulating
endothelial identity and angiogenesis, protecting against
atherosclerosis, and mitigating vascular calcification (114). Xing et al (114) demonstrated that COUP-TFII
overexpression following the localized delivery of COUP-TFII pDNA
nanocarriers (COUP-TFII@HPEI) in a rat abdominal artery replacement
model regulated stem/progenitor cell differentiation towards
endothelialization, and inhibited the calcification of
decellularized allografts. This presents an effective strategy to
enhance the applicability of decellularized allografts for vascular
interventions (114). However,
the limitations of this study include the use of a short-term
preclinical model, and the lack of long-term patency and immune
response data. Furthermore, the effect of COUP-TFII overexpression
on systemic metabolism and off-target tissues was not assessed. Wu
et al (69) revealed that
reducing the COUP-TFII expression in calcineurin
(CnTg/Cre/F+) transgenic mouse models that develop DCM
resulted in an increase in overall survival. Thus, inhibiting
COUP-TFII to restore the metabolic balance in the heart exhibits
potential as a therapeutic approach for heart failure, highlighting
the potential of COUP-TFII in CVD treatments. Nevertheless, there
are also limitations to this study, including its reliance on a
genetically engineered model with a strong calcineurin-dependent
phenotype, which may not generalize to other forms of heart
failure, and the use of global or tissue-specific knockouts without
temporal control of gene expression. Overall, these findings
emphasize the context-dependent effects of COUP-TFII: While its
overexpression may support vascular regeneration and graft
compatibility by enhancing endothelial reprogramming, it may
conversely exacerbate cardiac dysfunction when dysregulated in the
myocardium. This dichotomy emphasizes the importance of
spatiotemporal control in potential COUP-TFII-targeted therapies.
Additional studies are warranted to resolve these paradoxes,
validate the efficacy and safety in large-animal models or human
tissues, and establish optimal delivery strategies that minimize
off-target effects, while leveraging the therapeutic potential of
the gene of interest.
Therapeutic implications of COUP-TFII in
CRC
The role of COUP-TFII in CRC remains controversial.
Therefore, targeting COUP-TFII in CRC requires further exploration.
In tumor-promoting contexts of COUP-TFII, small molecules or
RNA-based therapies targeting COUP-TFII are anticipated to show
promise, according to previous preclinical models such as prostate
cancer xenograft models and patient-derived xenograft mice
(115). Qin et al
(20) revealed that the inhibition
of COUP-TFII via conditional ablation in a mouse xenograft model
resulted in impaired neoangiogenesis and suppressed tumor growth.
Although this study provided a mechanistic explanation for the
angiogenic role of COUP-TFII, its dependence on immune-deficient
mouse models may limit translational relevance, particularly in the
context of tumor-immune interactions (20). Additionally, Wang et al
(116) revealed that COUP-TFII
knockdown attenuated tumorigenesis and tumor progression in an
immunocompetent mouse glioblastoma model, indicating that COUP-TFII
may have a broader oncogenic function in various types of cancer.
However, the results are still based on a tumor type-specific model
and lack CRC-specific validation. Wang et al (115) identified a small molecular
inhibitor of COUP-TFII using a luminescence-based cell-based
high-throughput screening assay, and also revealed that this
inhibitor reduced prostate cancer cell proliferation, colony
formation, cell invasion and angiogenesis in xenograft mouse models
and patient-derived xenograft models. Although promising, the
selectivity, bioavailability and toxicity profiles of the compound
were not fully addressed, and its effects in CRC models remain
unknown. These findings support the rationale for developing
COUP-TFII-targeted agents, particularly in tumors with COUP-TFII
upregulation. Similarly, targeting COUP-TFII to inhibit
angiogenesis and metabolic reprogramming may suppress tumor
progression. To the best of our knowledge, no clinical trials
specifically targeting COUP-TFII have been carried out; however,
related nuclear receptor-targeting compounds, such as retinoic acid
receptor agonists (117) and HDAC
inhibitors (40), are being
investigated in cancer therapy and may provide insights into
potential COUP-TFII-targeted approaches.
On the other hand, there is evidence that COUP-TFII
expression is associated with certain positive prognostic
indicators in CRC, suggesting its potential role as a tumor
suppressor (80,81). Therefore, extensive prospective
studies are necessary to reconcile these conflicting observations.
Kruse et al (18) reported
the potential of retinoic acids as COUP-TFII promoters using
multiple cell line experiments; however, the required concentration
was greater than the physiological levels of retinoic acids,
raising concerns regarding clinical feasibility. In
tumor-suppressive contexts, developing novel drugs that can enhance
COUP-TFII expression may provide novel therapeutic avenues for CRC
treatment.
In terms of synergy with existing treatments (for
example, doxorubicin or 5-fluorouracil), COUP-TFII modulation has
the potential to enhance the efficacy of standard chemotherapy and
targeted therapies (118). For
example, several studies have suggested that COUP-TFII modulates
Wnt/β-catenin and TGF-β signaling pathways, including EMT, both of
which are responsible for resistance to conventional CRC treatments
(84,89,118). In particular, the reduction of
COUP-TFII after doxorubicin treatment may contribute to EMT-induced
doxorubicin resistance in CRC cells (118), supporting the possibility that
COUP-TFII upregulation can lead to chemosensitivity to doxorubicin
in CRC. Taken together, combining COUP-TFII modulators with
conventional Wnt pathway inhibitors or immune checkpoint inhibitors
may improve treatment responses in resistant tumors by hindering
resistance mechanisms. Furthermore, given the role of COUP-TFII in
angiogenesis and metastasis, its inhibition could complement
anti-angiogenic therapies, such as VEGF inhibitors, by preventing
tumor vascularization and dissemination (17,20).
However, challenges remain in translating these findings into
clinical applications.
Future research directions
There are several challenges in research that
should be overcome in developing COUP-TFII-related therapies for
CVDs and CRC. One major concern is the lack of well-characterized
small-molecule inhibitors or modulators that can specifically
target COUP-TFII without affecting other nuclear receptors.
Additionally, the dual role of COUP-TFII as both an oncogene and
tumor suppressor in different contexts complicates therapeutic
strategies for CRC, underscoring the need to investigate potential
biomarkers to identify patients who would benefit from
COUP-TFII-targeted interventions. Furthermore, the development of
selective COUP-TFII modulators, optimization of drug delivery
systems and comprehensive preclinical validation are necessary
steps before clinical trials can be pursued.
Therefore, future research should focus on
elucidating the precise regulatory mechanisms of COUP-TFII,
identifying specific patient subgroups that would benefit from
COUP-TFII-targeted therapies, carrying out human clinical trials
based on previous preclinical experimental data, exploring
combination strategies to enhance treatment efficacy while
minimizing toxicity, and developing personalized treatments based
on these findings. Structural studies on the LBD of COUP-TFII may
help identify novel therapeutic ligands or modulators.
Investigating the interactions of COUP-TFII with key signaling
molecules, such as VEGF and PGC-1α, could further help identify
potential therapeutic targets. Additionally, understanding the
interaction between COUP-TFII-related CVDs and CRC will be key to
develop integrated therapeutic approaches. For example,
longitudinal studies examining the bidirectional risk between CVDs
and CRC could provide valuable insights into their shared
pathophysiological mechanisms. Biomarker-based approaches to
monitor COUP-TFII activity could also guide tailored treatment
strategies for patients with coexisting CVDs and CRC.
Conclusion
COUP-TFII is a versatile transcription factor with
key roles in both CVDs and CRC. Its structural features and
regulatory mechanisms enable it to affect diverse cellular
processes, from angiogenesis to metabolism. Future studies should
aim to uncover the tissue-specific roles of COUP-TFII in various
pathological contexts, develop selective modulators of COUP-TFII
for clinical applications, and explore the interplay between
COUP-TFII-mediated pathways in CVDs and CRC. Comprehensive
understanding of the dual roles of COUP-TFII in CRC and its
interrelation with CVDs, will provide opportunities for innovative
therapeutic strategies that target this nuclear receptor.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Research
Foundation of Korea (NRF) funded by the Korean Government (grant
no. 2022R1A2C1009739) and the Basic Science Research Program
through the NRF funded by the Ministry of Education (grant no.
2022R1I1A1A01072409).
Availability of data and materials
Not applicable.
Authors' contributions
THP and JIP were responsible for conception,
organization, searching the literature, writing of the first draft
and reviewing the manuscript. SHH, JGC, SJP and JYH searched the
literature and reviewed the manuscript. SHH and JIP designed the
figures. Data authentication is not applicable. All authors have
read and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AF
|
activation function
|
|
Ang-1
|
angiopoietin-1
|
|
APOB
|
apolipoprotein B
|
|
BMP4
|
bone morphogenic protein 4
|
|
CCL
|
C-C motif chemokine ligand
|
|
ChIP
|
chromatin immunoprecipitation
|
|
COUP-TFII
|
chicken ovalbumin upstream
promoter-transcription factor II
|
|
CVD
|
cardiovascular disease
|
|
CRC
|
colorectal cancer
|
|
CXCL
|
C-X-C motif chemokine ligand
|
|
DBD
|
DNA-binding domain
|
|
DCM
|
dilated cardiomyopathy
|
|
DIHF
|
diabetes-induced heart failure
|
|
DR
|
direct repeat
|
|
EC
|
endothelial cell
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
|
EndMT
|
endothelial-to-mesenchymal
transition
|
|
ERG
|
ETS-related gene
|
|
ETC
|
electron transport chain
|
|
HDAC
|
histone deacetylase
|
|
HIF
|
hypoxia-inducible factor
|
|
Mfn
|
mitofusin
|
|
miR
|
microRNA
|
|
NRCM
|
neonatal rat cardiomyocyte
|
|
Opa1
|
optic atrophy 1
|
|
PA
|
palmitic acid
|
|
PAH
|
pulmonary arterial hypertension
|
|
PFKFB3
|
6-phosphofructo-2-kinase/fructose-2,6-bisphophatase 3
|
|
PGC-1α
|
peroxisome proliferator activated
receptor γ coactivator-1α
|
|
Pink1
|
PTEN-induced kinase 1
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
RAS
|
renin-angiotensin system
|
|
ROS
|
reactive oxygen species
|
|
Shh
|
sonic hedgehog
|
|
SMRT
|
silencing mediator for retinoid or
thyroid hormone receptor
|
|
Sod2
|
superoxide dismutase 2
|
|
Sp1
|
specificity protein 1
|
|
SRC-1
|
steroid receptor coactivator-1
|
|
TCF7L2
|
T-cell factor 7-like 2 or
transcription factor 7-like 2
|
|
TFPI2
|
tissue factor pathway inhibitor 2
|
|
Tie-2
|
tyrosine kinase with immunoglobulin
like and EGF like domains-2
|
|
UTR
|
untranslated region
|
|
aa
|
amino acids
|
References
|
1
|
Wang LH, Tsai SY, Cook RG, Beattie WG,
Tsai MJ and O'Malley BW: COUP transcription factor is a member of
the steroid receptor superfamily. Nature. 340:163–166. 1989.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Tsai SY and Tsai MJ: Chick ovalbumin
upstream promoter-transcription factors (COUP-TFs): Coming of age.
Endocr Rev. 18:229–240. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Polvani S, Pepe S, Milani S and Galli A:
COUP-TFII in health and disease. Cells. 9:1012019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Pereira FA, Tsai MJ and Tsai SY: COUP-TF
orphan nuclear receptors in development and differentiation. Cell
Mol Life Sci. 57:1388–1398. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ashraf UM, Sanchez ER and Kumarasamy S:
COUP-TFII revisited: Its role in metabolic gene regulation.
Steroids. 141:63–69. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pereira FA, Qiu Y, Tsai MJ and Tsai SY:
Chicken ovalbumin upstream promoter transcription factor (COUP-TF):
Expression during mouse embryogenesis. J Steroid Biochem Mol Biol.
53:503–508. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Pereira FA, Qiu Y, Zhou G, Tsai MJ and
Tsai SY: The orphan nuclear receptor COUP-TFII is required for
angiogenesis and heart development. Genes Dev. 13:1037–1049. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wu SP, Cheng CM, Lanz RB, Wang T, Respress
JL, Ather S, Chen W, Tsai SJ, Wehrens XH, Tsai MJ and Tsai SY:
Atrial identity is determined by a COUP-TFII regulatory network.
Dev Cell. 25:417–426. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lin FJ, You LR, Yu CT, Hsu WH, Tsai MJ and
Tsai SY: Endocardial cushion morphogenesis and coronary vessel
development require chicken ovalbumin upstream
promoter-transcription factor II. Arterioscler Thromb Vasc Biol.
32:e135–e146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
You LR, Lin FJ, Lee CT, DeMayo FJ, Tsai MJ
and Tsai SY: Suppression of Notch signaling by the COUP-TFII
transcription factor regulates vein identity. Nature. 435:98–104.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yun SH and Park JI: Recent progress on the
role and molecular mechanism of chicken ovalbumin upstream
promoter-transcription factor II in cancer. J Int Med Res.
48:30000605209192362020. View Article : Google Scholar
|
|
12
|
GBD 2016 Causes of Death Collaborators, .
Global, regional, and national age-sex specific mortality for 264
causes of death, 1980–2016: A systematic analysis for the Global
Burden of Disease Study 2016. Lancet. 390:1151–1210. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kenzik KM, Balentine C, Richman J, Kilgore
M, Bhatia S and Williams GR: New-onset cardiovascular morbidity in
older adults with Stage I to III colorectal cancer. J Clin Oncol.
36:609–616. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Keramida K, Charalampopoulos G,
Filippiadis D, Tsougos E and Farmakis D: Cardiovascular
complications of metastatic colorectal cancer treatment. J
Gastrointest Oncol. 10:797–806. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brown JC, Caan BJ, Prado CM, Weltzien E,
Xiao J, Cespedes Feliciano EM, Kroenke CH and Meyerhardt JA: Body
composition and cardiovascular events in patients with colorectal
cancer: A population-based retrospective cohort study. JAMA Oncol.
5:967–972. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Whelton SP, Berning P, Blumenthal RS,
Marshall CH, Martin SS, Mortensen MB, Blaha MJ and Dzaye O:
Multidisciplinary prevention and management strategies for
colorectal cancer and cardiovascular disease. Eur J Intern Med.
87:3–12. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qin J, Tsai SY and Tsai MJ: The critical
roles of COUP-TFII in tumor progression and metastasis. Cell
Biosci. 4:582014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kruse SW, Suino-Powell K, Zhou XE,
Kretschman JE, Reynolds R, Vonrhein C, Xu Y, Wang L, Tsai SY, Tsai
MJ and Xu HE: Identification of COUP-TFII orphan nuclear receptor
as a retinoic acid-activated receptor. PLoS Biol. 6:e2272008.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lin FJ, Chen X, Qin J, Hong YK, Tsai MJ
and Tsai SY: Direct transcriptional regulation of neuropilin-2 by
COUP-TFII modulates multiple steps in murine lymphatic vessel
development. J Clin Invest. 120:1694–1707. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qin J, Chen X, Xie X, Tsai MJ and Tsai SY:
COUP-TFII regulates tumor growth and metastasis by modulating tumor
angiogenesis. Proc Natl Acad Sci USA. 107:3687–3692. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Krishnan V, Elberg G, Tsai MJ and Tsai SY:
Identification of a novel sonic hedgehog response element in the
chicken ovalbumin upstream promoter-transcription factor II
promoter. Mol Endocrinol. 11:1458–1466. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Qiu Y, Krishnan V, Pereira FA, Tsai SY and
Tsai MJ: Chicken ovalbumin upstream promoter-transcription factors
and their regulation. J Steroid Biochem Mol Biol. 56:81–85. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Soosaar A, Neuman K, Nornes HO and Neuman
T: Cell type specific regulation of COUP-TFII promoter activity.
FEBS Lett. 39:95–100. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Riggs KA, Wickramasinghe NS, Cochrum RK,
Watts MB and Klinge CM: Decreased chicken ovalbumin upstream
promoter transcription factor II expression in tamoxifen-resistant
breast cancer cells. Cancer Res. 66:10188–10198. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
More E, Fellner T, Doppelmayr H,
Hauser-Kronberger C, Dandachi N, Obrist P, Sandhofer F and
Paulweber B: Activation of the MAP kinase pathway induces chicken
ovalbumin upstream propter-transcription factor II (COUP-TFII)
expression in human breast cancer cell lines. J Endocrinol.
176:83–94. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rosa A and Brivanlou AH: A regulatory
circuitry comprised of miR-302 and the transcription factors OCT4
and NR2F2 regulates human embryonic stem cell differentiation. EMBO
J. 30:237–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Petit FG, Salas R, Tsai MJ and Tsai SY:
The regulation of COUP-TFII gene expression by Ets-1 is enhanced by
the steroid receptor co-activators. Mech Age Dev. 125:719–732.
2004. View Article : Google Scholar
|
|
28
|
Fu JL, Hsiao KY, Lee HC, Li WN, Chang N,
Wu MH and Tsai SJ: Suppression of COUP-TFII upregulates angiogenin
and promotes angiogenesis in endometriosis. Hum Reprod.
33:1517–1527. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Diez H, Fischer A, Winkler A, Hu CJ,
Hatzopoulos AK, Breier G and Gessler M: Hypoxia-mediated activation
of Dll4-Notch-Hey2 signaling in endothelial progenitor cells and
adoption of arterial cell fate. Exp Cell Res. 313:1–9. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ruan K, Fang X and Ouyang G: MicroRNAs:
Novel regulators in the hallmarks of human cancer. Cancer Lett.
285:116–126. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Feng Q, Wu X, Li F, Ning B, Lu X, Zhang Y,
Pan Y and Guan W: miR-27b inhibits gastric cancer metastasis by
targeting NR2F2. Protein Cell. 8:114–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu S, Wilson KD, Ghosh Z, Han L, Wang Y,
Lan F, Ransohoff KJ, Burridge P and Wu JC: MicroRNA-302 increases
reprogramming efficiency via repression of NR2F2. Stem Cells.
31:259–268. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kang IH, Jeong BC, Hur SW, Choi H, Choi
SH, Ryu JH, Hwang YC and Koh JT: MicroRNA-302a stimulates
osteoblastic differentiation by repressing COUP-TFII expression. J
Cell Physiol. 230:911–921. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhou B, Song J, Han T, Huang M, Jiang H,
Qiao H, Shi J and Wang Y: MiR-382 inhibits cell growth and invasion
by targeting NR2F2 in colorectal cancer. Mol Carcinog.
55:2260–2267. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang W, Liu J, Qiu J, Fu X, Tang Q, Yang
F, Zhao Z and Wang H: MicroRNA-382 inhibits prostate cancer cell
proliferation and metastasis through targeting COUP-TFII. Oncol
Rep. 36:3707–3715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin SC, Kao CY, Lee HJ, Creighton CJ,
Ittmann MM, Tsai SJ, Tsai SY and Tsai MJ: Dysregulation of
miRNAs-COUP-TFII -FOXM1-CENPF axis contributes to the metastasis of
prostate cancer. Nat Commun. 7:114182016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeong BC, Kang IH, Hwang YC, Kim SH and
Koh JT: MicroRNA-194 reciprocally stimulates osteogenesis and
inhibits adipogenesis via regulating COUP-TFII expression. Cell
Death Dis. 5:e15322014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kass SU, Pruss D and Wolffe AP: How does
DNA methylation repress transcription? Trends Genet. 13:444–449.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Momparler RL and Bovenzi V: DNA
methylation and cancer. J Cell Physiol. 183:145–154. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Al-Rayyan N, Litchfield LM, Ivanova MM,
Radde BN, Cheng A, Elbedewy A and Klinge CM: 5-Aza-2-deoxycytidine
and trichostatin A increase COUP-TFII expression in
antiestrogen-resistant breast cancer cell lines. Cancer Lett.
347:139–150. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Davalos V, Lovell CD, Von Itter R,
Dolgalev I, Agrawal P, Baptiste G, Kahler DJ, Sokolova E, Moran S,
Piqué L, et al: An epigenetic switch controls an alternative NR2F2
isoform that unleashes a metastatic program in melanoma. Nat
Commun. 14:18672023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen X, Qin J, Cheng CM, Tsai MJ and Tsai
SY: COUP-TFII is a major regulator of cell cycle and Notch
signaling pathways. Mol Endocrinol. 26:1268–1277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Okamura M, Kudo H, Wakabayyashi K, Tanaka
T, Nonaka A, Uchida A, Tsutsumi S, Sakakibara I, Naito M, Osborne
TF, et al: COUP-TFII acts downstream of Wnt/beta-catenin signal to
silence PPARgamma gene expression and repress adipogenesis. Proc
Natl Acad Sci USA. 106:5819–5824. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Al Turki S, Manickaraj AK, Mercer CL,
Gerety SS, Hitz MP, Lindsay S, D'Alessandro LC, Swaminathan GJ,
Bentham J, Arndt AK, et al: Rare variants in NR2F2 cause congenital
heart defects in humans. Am J Hum Genet. 94:574–585. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sissaoui S, Yu J, Yan A, Li R, Yukselen O,
Kucukural A, Zhu LJ and Lawson ND: Genomic characterization of
endothelial enhancers reveals a multifunctional role for NR2F2 in
regulation of arteriovenous gene expression. Cir Res. 126:875–888.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cui X, Lu YW, Lee V, Kim D, Dorsey T, Wang
Q, Lee Y, Vincent P, Schwarz J and Dai G: Venous endothelial marker
COUP-TFII regulates the distinct pathologic potentials of adult
arteries and veins. Sci Rep. 5:161932015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Folkman J: Angiogenesis in cancer,
vascular, rheumatoid and other disease. Nat Med. 1:27–31. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Adams RH and Alito K: Molecular regulation
of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol.
8:464–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Suri C, Jones PF, Patan S, Bartunkova S,
Maisonpierre PC, Davis S, Sat TN and Yancopoulos GD: Requisite role
for angiopoitin-1, a ligand for the TIE2 receptor, during embryonic
angiogenesis. Cell. 87:1171–1180. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jones N, Iljin K, Dumont DJ and Alitalo K:
The receptors: New modulators of angiogenic and lymphangiogenic
responses. Nat Rev Mol Cell Biol. 2:257–267. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ferrara N, Geber HP and LeCouter J: The
biology of VEGF and its receptors. Nat Med. 9:669–676. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Dickson MC, Martin JS, Cousins FM,
Kulkarni AB, Karlsson S and Akhurst RJ: Defective haematopoiesis
and vasculogenesis in transforming growth factor-beta 1 knock out
mice. Development. 121:1845–1854. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Larsson J, Goumans MJ, Sjőstrand LJ, van
Rooijen MA, Levéen P, Xu X, ten Dijke P, Mummery CL and Karlsson S:
Abnormal angiogenesis but intact hematopoietic potential in
TGF-beta type I receptor-deficient mice. EMBO J. 20:1663–1673.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hamada K, Sasaki T, Koni PA, Natsui M,
Kishimoto H, Sasaki J, Yajima N, Horie Y, Hasegawa G, Naito M, et
al: The PTEN/PI3K pathway governs normal vascular development and
tumor angiogenesis. Genes Dev. 19:2054–2065. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qin J, Chen X, Yu-Lee L, Tsai MJ and Tsai
SY: Nuclear receptor COUP-TFII controls pancreatic islet tumor
angiogenesis by regulating VEGF/VEGFR-2 signaling. Cancer Res.
70:8812–8821. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dougherty EJ, Chen LY, Awad KS, Ferreyra
GA, Demirkale CY, Keshavarz A, Gairhe S, Johnston KA, Hicks ME,
Sandler AB, et al: Inflammation and DKK1-induced AKT activation
contribute to endothelial dysfunction following NR2F2 loss. Am J
Physiol Lung Cell Mol Physiol. 324:L783–L798. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu
J, Wang J, Kovacs L, Ayon RJ, Liu Z, et al: PFKEB3-mediated
endothelial glycolysis promotes pulmonary hypertension. Proc Natl
Acad Sci USA. 116:13394–13403. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Talati M and Hemnes A: Fatty acid
metabolism in pulmonary arterial hypertension: Role in right
ventricular dysfunction and hypertrophy. Pulm Circ. 5:269–278.
2015. View
Article : Google Scholar : PubMed/NCBI
|
|
59
|
Poels K, Schnitzler JG, Waissi F, Levels
JHM, Stroes ESG, Daemen MJAP, Lutgens E, Pennekamp AM, De Kleijin
DPV, Seijkens TTP and Kroon J: Inhibition of PFKFB3 hampers the
progression of atherosclerosis and promotes plaque stability. Front
Cell Dev Biol. 8:5816412020. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rodríguez-García A, Samsó P, Fontova P,
Simon-Molas H, Manzano A, Castaño E, Rosa JL, Martinez-Outshoorn U,
Ventura F, Navarro-Sabaté À and Bartrons R: TGF-β1 targets Smad,
p38 MAPK, and PI3K/Akt signaling pathways to induce PFKFB3 gene
expression and glycolysis in glioblastoma cells. FEBS J.
284:3437–3454. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nakamura E, Makita Y, Okamoto T, Nagaya K,
Hayashi T, Sugimoto M, Manabe H, Taketazu G, Kajino H and Fujieda
K: 5.78 Mb terminal deletion of chromosome 15q in a girl,
evaluation of NR2F2 as candidate gene for congenital heart defects.
Eur J Med Genet. 54:354–356. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Qiao XH, Wang Q, Wang J, Liu XY, Xu YJ,
Huang RT, Xue S, Li YJ, Zhang M, Qu XK, et al: A novel NR2F2
loss-of-function mutation predisposes to congenital heart defect.
Eur J Med Genet. 61:197–203. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Upadia J, Gonzales PR and Robin NH: Novel
de novo pathogenic variant in the NR2F2 gene in a boy with
congenital heart defect and dysmorphic features. Am J Med Genet A.
176:1423–1426. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Cornea A, Gilboa SM, Besser LM, Botto LD,
Moore CA, Hobbs CA, Cleves MA, Riehle-Colarusso TJ, Waller DK and
Reece EA: Diabetes mellitus and birth defects. Am J Obstet Gynecol.
199:e1–e9. 2008.
|
|
65
|
Botto LD, Loffredo C, Scanlon KS, Ferencz
C, Khoury MJ, David Wilson P and Correa A: Vitamin A and cardiac
outflow tract defects. Epidemiology. 12:491–496. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Perilhou A, Tourrel-Cuzin C, Kharroubi I,
Henique C, Fauveau V, Kitamura T, Magnan C, Posric C, Prip-Buus C
and Vasseur-Cognet M: The transcription factor COUP-TFII is
negatively regulated by insulin and glucose via Foxo1- and
ChREBP-controlled pathways. Mol Cell Biol. 28:6568–6579. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Vilhais-Neto GC, Maruhashi M, Smith KT,
Vasseur-Cognet M, Peterson AS, Workman JI and Pourquié O: Rere
controls retinoic acid signaling and somite bilateral symmetry.
Nature. 463:953–957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Gruber PJ and Epstein JA: Developmental
gone awry. Congenital heart disease. Cir Res. 94:273–283. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wu SP, Kao CY, Wang L, Creighton CJ, Yang
J, Donti TR, Harmancey R, Vasquez HG, Graham BH, Bellen HJ, et al:
Increased COUP-TFII expression in adult hearts induces
mitochondrial dysfunction resulting in heart failure. Nat Commun.
6:82452015. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Kittleson MM, Minhas KM, Irizarry RA, Ye
SQ, Edness G, Breton E, Conte JV, Tomaselli G, Garcia JG and Hare
JM: Gene expression analysis of ischemic and nonischemic
cardiomyopathy: Shared and distinct genes in the development of
heart failure. Physiol Genomics. 21:299–307. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hannenhalli S, Putt ME, Gilmore JM, Wang
J, Parmacek MS, Epstein JA, Morrisey EE, Margulies KB and Cappola
TP: Transcriptional genomics associates FOX transcription factors
with human heart failure. Circulation. 114:1269–1276. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Miao W, Chen M, Chen M, Cui C, Zhu Y, Luo
X and Wu B: Nr2f2 overexpression aggravates ferroptosis and
mitochondrial dysfunction by regulating PGC-1α signaling in
diabetes-induced heart failure mice. Mediators Inflamm.
2022:83733892022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Tan Y, Zhang Z, Zheng C, Wintergerst KA,
Keller BB and Cai L: Mechanisms of diabetic cardiomyopathy and
potential therapeutic strategies: Preclinical and clinical
evidence. Nat Rev Cardiol. 17:585–607. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dhalla NS, Shah AK and Tappia PS: Role of
oxidative stress in metabolic and subcellular abnormalities in
diabetic cardiomyopathy. Int J Mol Sci. 21:24132020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferrptosis, an iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li W, Li W, Leng Y, Xiong Y and Xia Z:
Ferroptosis is involved in diabetes myocardial ischemia/reperfusion
injury through endoplasmic reticulum stress. DNA Cell Biol.
39:210–225. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Ma S, Sun L, Wu W, Wu J, Sun Z and Ren J:
USP22 protects against myocardial ischemia-reperfusion injury via
the SIRT-p53/SLC7A11-dependent inhibition of ferroptosis-induced
cardiomyocyte death. Front Physiol. 11:5513182020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Rad Biol Med. 162:339–352.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wang X, Chen X, Zhou W, Men H, Bao T, Sun
Y, Wang Q, Tan Y, Keller BB, Tong Q, et al: Ferroptosis is
essential for diabetic cardiomyopathy and is prevented by
sulforaphane via AMPK/NRF2 pathways. Acta Phama Sin B. 12:708–722.
2022. View Article : Google Scholar
|
|
80
|
Shin SW, Kwon HC, Rho MS, Choi HJ, Kwak JY
and Park JI: Clinical significance of chicken ovalbumin upstream
promoter-transcription factor II expression in human colorectal
cancer. Oncol Rep. 21:101–106. 2009.PubMed/NCBI
|
|
81
|
Yun SH, Park MG, Kim YM, Roh MS and Park
JI: Expression of chicken ovalbumin upstream promoter-transcription
factor II and liver X receptor as prognostic indicators for human
colorectal cancer. Oncol Lett. 14:4011–4020. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Yun SH and Park JI: COUP-TFII
overexpression inhibits cell proliferation and invasion via
increased expression of p53 and PTEN and decreased Akt
phosphorylation in human colorectal cancer SNU-C4 cells. Anticancer
Res. 40:767–777. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yun SH, Han SH and Park JI: COUP-TFII
knock-down promotes proliferation and invasion in colorectal cancer
cells via activation of Akt pathway and up-regulation of FOXC1.
Anticancer Res. 40:177–190. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bao Y, Gu D, Feng W, Sun X, Wang X, Zhang
X, Shi Q, Cui G, Yu H, Tang C and Deng A: COUP-TFII regulates
metastasis of colorectal adenocarcinoma cells by modulating Snail1.
Br J Cancer. 111:933–943. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Wang C, Zhou Y, Ruan R, Zheng M, Han W and
Liao L: High expression of COUP-TFII cooperated with negative Smad4
expression predicts poor prognosis in patients with colorectal
cancer. Int J Clin Exp Pathol. 8:7112–7121. 2015.PubMed/NCBI
|
|
86
|
Cano A, Pérez-Moreno MA, Rodrigo I, Blanco
MJ, del Barrio MG, Portillo F and Nieto MA: The transcription
factor snail controls epithelial-mesenchymal transitions by
repressing E-cadherin expression. Nat Cell Biol. 2:76–83. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li X, Deng W, Lobo-Ruppert SM and Ruppert
JM: Gli1 acts through Snail and E-cadherin to promote nuclear
signaling by beta-catenin. Oncogene. 26:4489–4498. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kudo-Saito C, Shirako H, Takeuchi T and
Kawakami Y: Cancer metastasis is accelerated through
immunosuppression during Snail-induced EMT of cancer cells. Cancer
Cell. 15:195–206. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Pfeffer SR, Yang CH and Pfeffer LM: The
role of miR-21 in cancer. Drug Dev Res. 76:270–277. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wang H, Nie L, Wu L, Liu Q and Guo X:
NR2F2 inhibits Smad7 expression and promotes TGF-β-dependent
epithelial-mesenchymal transition of CRC via transactivation of
miR-21. Biochem Biophys Res Commun. 485:181–188. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Bao Y, Lu Y, Feng W, Yu H, Guo H, Tao Y,
Shi Q, Chen W and Wang X: COUP-TFII promotes epithelial-mesenchymal
transition by inhibiting miR-34a expression in colorectal cancer.
Int J Oncol. 54:1337–1344. 2019.PubMed/NCBI
|
|
92
|
Zhang D, Qiu X, Li J, Zheng S, Li L and
Zhao H: TGF-β secreted by tumor-associated macrophages promotes
proliferation and invasion of colorectal cancer via miR-34a-VEGF
axis. Cell Cycle. 17:2766–2778. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Luo Y, Chen JJ, Lv Q, Qin J, Huang YZ, Yu
MH and Zhong M: Long non-coding RNA NEAT1 promotes colorectal
cancer progression by competitively binding miR-34a with SIRT1 and
enhancing the Wnt/β-catenin signaling pathway. Cancer Lett.
440–441. 11–22. 2019.
|
|
94
|
Li Y, Gong P, Hou JX, Huang W, Ma XP, Wang
YL, Li J, Cui XB and Li N: miR-34a regulates multidrug resistance
via positively modulating OAZ2 signaling in colon cancer cells. J
Immunol Res. 2018:74985142018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Wu QB, Sheng X, Zhang N, Yang MW and Wang
F: Role of microRNAs in the resistance of colorectal cancer to
chemoradiotherapy. Mol Clin Oncol. 8:523–527. 2018.PubMed/NCBI
|
|
96
|
Li X, Large MJ, Creighton CJ, Lanz RB,
Jeong JW, Young SL, Lessey BA, Palomino WA, Tsai SY and DeMayo FJ:
COUP-TFII regulates human endometrial stromal genes involved in
inflammation. Mol Endocrinol. 27:2041–2054. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Bray F, Ferlay J, Soerjomataram I, Siegel
RL, Torre LA and Jemal A: Global cancer statics 2018: GLOBOCAN
estimates of incidence and mortality worldwide for 36 cancers in
185 countries. CA Cancer J Clin. 68:394–424. 2018.PubMed/NCBI
|
|
98
|
Niederseer D, Stadlmayr A, Huber-Schőnauer
U, Plöderl M, Schmied C, Lederer D, Patsch W, Aigner E and Datz C:
Cardiovascular risk and known coronary artery disease are
associated with colorectal adenoma and advanced neoplasia. J Am
Coll Cardiol. 69:2348–2350. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Erichsen R, Sværke C, Sørensen HT, Sandler
RS and Baron JA: Risk of colorectal cancer in patients with acute
myocardial infarction and stroke: A nationwide cohort study. Cancer
Epidemiol Biomarkers Prev. 22:1994–1999. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Meijers WC, Maglione M, Bakker SJL,
Oberhuber R, Kieneker LM, de Jong S, Haubener BJ, Nagengast WB,
Lyon AR, van der Vegt B, et al: Heart failure stimulates tumor
growth by circulating factors. Circulation. 138:678–691. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Dai YN, Wang JH, Zhu JZ, Lin JQ, Yu CH and
Li YM: Angiotensin-converting enzyme inhibitors/angiotensin
receptor blockers therapy and colorectal cancer: A systematic
review and meta-analysis. Cancer Causes Control. 26:1245–1255.
2015.PubMed/NCBI
|
|
102
|
Chen X, Yi CH and Ya KG: Renin-angiotensin
system inhibitor use and colorectal cancer risk and mortality: A
dose-response meta analysis. J Renin Angiotensin Aldosterone Syst.
Jul 6–2020.(Epub ahead of print). View Article : Google Scholar
|
|
103
|
Koene RJ, Prizment AE, Blaes A and Konety
SH: Shared risk factors in cardiovascular disease and cancer.
Circulation. 133:1104–1114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang C, Cheng Y, Luo D, Wang J, Liu J,
Luo Y, Zhou W, Zhuo Z, Guo K, Zeng R, et al: Association between
cardiovascular risk factors and colorectal cancer: A systematic
review and meta-analysis of prospective cohort studies.
eClinicalMedicine. 34:1007942021. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Bertero E, Canepa M, Maack C and Ameri P:
Linking heart failure to cancer: Background evidence and research
perspectives. Circulation. 138:735–742. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Ridker PM, Everett BM, Thuren T, MacFadyen
JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Amker
SD, et al: Antiinflammatory therapy with canakinumab for
atherosclerotic disease. N Engl J Med. 377:1119–1131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Prizment AE, Folsom AR, Dreyfus J,
Anderson KE, Visvanathan K, Joshu CE, Platz EA and Pankow JS:
Plasma C-reactive protein, genetic risk score, and risk of common
cancers in the atherosclerosis risk in communities study. Cancer
Causes Control. 24:2077–2087. 2013.PubMed/NCBI
|
|
108
|
Qu D, Shen L, Liu S, Li H, Ma Y, Zhang R,
Wu K, Yao L, Li J and Zhang J: Chronic inflammation confers to the
metabolic reprogramming associated with tumorigenesis of colorectal
cancer. Cance Biol Ther. 18:237–244. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Yang L, Fang C, Zhang R and Zhou S:
Prognostic value of oxidative stress-related genes in colorectal
cancer and its correlation with tumor immunity. BMC Genomics.
25:82024. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kannan RY, Salacinski HJ, Butler PE,
Hamilton G and Seifalian AM: Current status of prosthetic bypass
grafts: A review. J Biomed Mater Res B Appl Biomater. 74:570–581.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Goncalves RC, Banfi A, Oliveira MB and
Mano JF: Strategies for re-vascularization and promotion of
angiogenesis in trauma and disease. Biomaterials. 269:1206282021.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kirkton RD, Santiago-Maysonet M, Lawson
JH, Tente WE, Dahl SLM, Niklason LE and Prichard HL: Bioengineered
human acellular vessels recellularize and evolve into living blood
vessels after human implantation. Sci Transl Med. 11:eaau69342019.
View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Lin RZ, Im GB, Luo AC, Zhu Y, Hong X,
Neumeyer J, Tang HW, Perrimon N and Melero-Martin JM: Mitochondrial
transfer mediates endothelial cell engraftment through mitophagy.
Nature. 629:660–668. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Xing M, Wang F, Chu R, Wang H, Sun Y, Qian
M, Jiang H, Midgley AC, Dai G and Zhao Q: Localized COUP-TFII pDNA
delivery modulates stem/progenitor cell differentiation to enhance
endothelialization and inhibit calcification of decellularized
allografts. Adv Sci (Weinh). 12:e24097442025. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang L, Cheng CM, Qin J, Xu M, Kao CY, Shi
J, You E, Gong W, Rosa LP, Chase P, et al: Small-molecule inhibitor
targeting orphan nuclear receptor COUP-TFII for prostate cancer
treatment. Sci Adv. 6:eaaz80312020. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Wang F, Zhang S, Sun F, Chen W, Liu C,
Dong H, Cui B, Li L, Sun C, Du W, et al: Anti-angiogenesis and
anti-immunosuppression gene therapy through targeting COUP-TFII in
an in situ glioblastoma mouse model. Cancer Gene Ther.
31:1135–1150. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Lin B, Chen GQ, Xiao D, Kollori SK, Cao X,
Su H and Zhang XK: Orphan receptor COUP-TF is required for
induction of retinoic acid receptor beta, growth inhibition, and
apoptosis by retinoic acid in cancer cells. Mol Cell Biol.
20:957–970. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Wang X, Jiang R, Cui E, Feng W, Guo H, Gu
D, Tang C, Xue T and Bao Y: COUP-TFII suppresses colorectal
carcinoma resistance to doxorubicin involving inhibition of
epithelial-mesenchymal transition (corrected). Am J Transl Res.
8:3921–3929. 2016.PubMed/NCBI
|














