Introduction
Stroke: From epidemiology to clinical
challenges
Stroke, also known as cerebrovascular accident, is
an acute neurological disorder caused by the disruption of cerebral
blood circulation and is one of the leading causes of mortality and
disability worldwide (1). The 2021
global burden of disease (GBD) analysis revealed that stroke ranked
first in age-standardized disability-adjusted life years,
significantly impacting global health across 19 of the 21 GBD
regions (2). The incidence and
mortality rates of stroke exhibit marked geographical variation
globally (3). In developed
countries, improvements in lifestyle and the optimization of
emergency care and secondary prevention measures have led to a
steady decline in stroke-related mortality rates (4). However, in low- and middle-income
countries, particularly in parts of Asia and Africa, stroke
incidence and mortality remain high due to limited early diagnostic
capabilities and inadequate medical resources, resulting in
significant disability rates (5).
Data from 2019 indicate that stroke mortality rates in Asian
countries such as China and India continue to rise, closely
associated with risk factors such as hypertension, diabetes,
smoking and the rapid pace of urbanization (6).
Stroke is primarily classified into two types:
Ischemic and hemorrhagic. Ischemic stroke results from cerebral
arterial embolism or stenosis, leading to interrupted blood supply
to the brain, accounting for the majority of cases (7). Hemorrhagic stroke, on the other hand,
is caused by the rupture of cerebral blood vessels, often
associated with hypertension and vascular abnormalities (8). This acute neurological disorder can
cause severe impairments in the acute phase, such as hemiplegia,
speech disorders and dysphagia. Over the long term, it can lead to
sequelae including motor, cognitive and other functional deficits,
markedly reducing the quality of life of patients while imposing
substantial burdens on families and society (9).
In China, stroke is one of the leading causes of
disease-associated mortality. Data from the Chinese Center for
Disease Control in 2020 indicated that there were ~3.4 million new
cases annually, with related deaths reaching ~2.3 million. The
direct and indirect economic costs of new cases amount to tens of
billions of yuan (10). In
conclusion, stroke poses a severe threat to global health and
places immense pressure on social and economic systems. With the
aging population and the prevalence of unhealthy lifestyles,
strengthening stroke prevention, early screening and innovative
treatment approaches have become urgent public health priorities
(11).
Key role of mitochondrial function in
the stroke pathophysiology
Mitochondria are central to cellular energy
metabolism and are crucial for the survival and functional
maintenance of neurons. In the pathological process of stroke,
mitochondria are responsible not only for energy generation but
also for regulating cellular calcium homeostasis, redox balance and
cell death signaling (12).
However, stroke-induced local ischemia, hypoxia and reperfusion
injury markedly disrupt mitochondrial structure and function,
triggering a series of pathological responses (13).
In the early stages of ischemic stroke, the
interruption of blood flow leads to severe hypoxia in brain tissue,
inhibiting mitochondrial oxidative phosphorylation and causing a
substantial reduction in energy production (14). The lack of energy impairs the
ability of neurons to maintain basic physiological functions,
leading to cellular dysfunction and death (15). Simultaneously, under hypoxic
conditions, mitochondria excessively produce reactive oxygen
species (ROS), which not only damage cellular membranes, proteins
and DNA but also further damage mitochondrial structure and
function, creating a cycle that accelerates the pathological
process (16).
During the reperfusion phase, although the
restoration of blood flow provides oxygen and nutrients to brain
tissue, the rapid influx of oxygen generates excessive ROS, causing
reperfusion injury (17). At this
stage, excessive calcium ions (Ca2+) rapidly enter the
cells and accumulate within the mitochondria, causing mitochondrial
Ca2+ overload. This impairs energy metabolism, activates
calcium signaling and triggers apoptosis and necrosis (18). Furthermore, the interaction between
oxidative stress and Ca2+ overload creates a cycle,
further exacerbating neuronal damage (19). In conclusion, mitochondrial
dysfunction is a driver of the pathological onset and progression
of stroke. It exacerbates neuronal damage through disruptions in
energy metabolism, oxidative stress, Ca2+ overload and
inflammation signaling networks (20). A deeper understanding of the
multiple roles of mitochondria in the pathological process of
stroke will help uncover disease mechanisms and provide important
directions for the development of innovative therapeutic
strategies.
Mitochondrial calcium dysregulation in
stroke pathology
Ca2+ are vital intracellular signaling
molecules involved in regulating physiological processes such as
muscle contraction, neurotransmission and cell proliferation
(21). In the pathological process
of stroke, disruption of intracellular calcium homeostasis is a
critical factor leading to neuronal injury. Mitochondria play a
central role in maintaining calcium balance by regulating calcium
influx and efflux. However, during stroke, particularly in the
ischemia/reperfusion phase, mitochondrial Ca2+ overload
often occurs, resulting in dysfunction and triggering apoptosis or
necrosis (22).
Mitochondrial Ca2+ transport is mediated
by the mitochondrial calcium uniporter (MCU) complex for calcium
influx and the sodium-calcium exchanger (NCLX) for calcium efflux.
Dysregulation of these calcium channels is closely associated with
stroke pathology (23), but their
molecular mechanisms and therapeutic potential remain to be
elucidated. The present study focuses on the molecular mechanisms
of mitochondrial Ca2+ transport, with an emphasis on the
regulatory properties of the MCU complex and NCLX and their roles
in neuronal injury. By elucidating these mechanisms, this research
aims to provide new insights into stroke pathology and offer a
theoretical foundation for developing innovative therapeutic
strategies targeting mitochondrial Ca2+ regulation.
Molecular mechanisms of mitochondrial
calcium transport
Mitochondria are not only the primary energy
producers in cells but also play a critical role in regulating
intracellular calcium homeostasis. The influx and efflux of
Ca2+ are essential mechanisms for maintaining
mitochondrial function and cellular physiological activities. As a
key signaling molecule, Ca2+ is indispensable in various
physiological processes, including signal transduction, energy
metabolism, redox balance and cell survival.
Molecular mechanisms of mitochondrial
calcium influx Mitochondrial calcium influx and its function
Mitochondrial Ca2+ influx is primarily
mediated by the MCU complex, which serves as the core channel for
mitochondrial Ca2+ transport (24). The influx of Ca2+ is
driven by the Ca2+ concentration gradient between the
cytoplasm and mitochondrial matrix, as well as the mitochondrial
membrane potential (25). Under
normal physiological conditions, the MCU complex maintains the
dynamic balance of mitochondrial calcium homeostasis. However,
under pathological conditions, excessive Ca2+ influx can
lead to mitochondrial Ca2+ overload, disrupting the
mitochondrial membrane potential, activating excessive ROS
production and ultimately inducing apoptosis or necrosis (26).
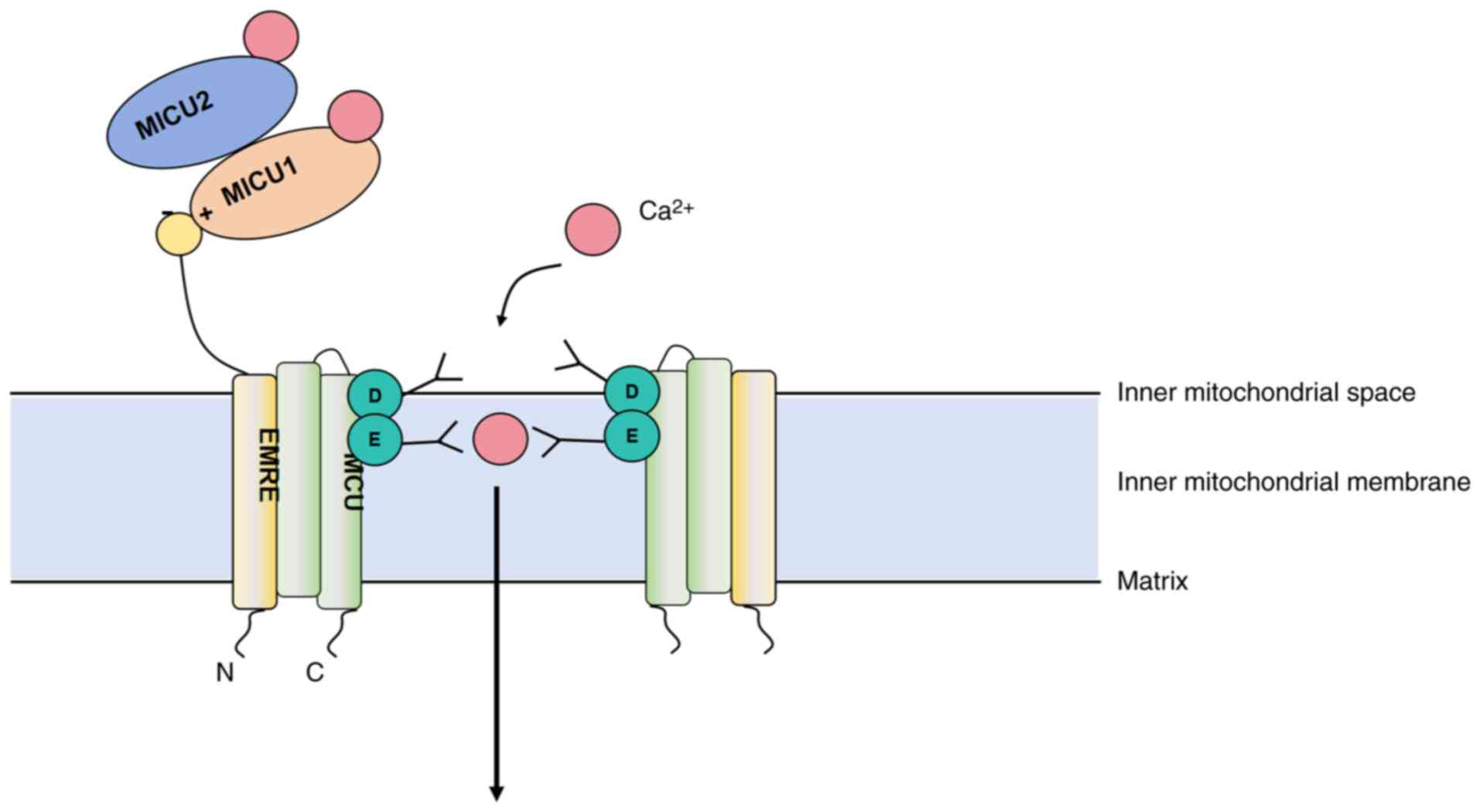
Composition and regulation of the MCU
complex
The MCU complex is located in the inner
mitochondrial membrane and is responsible for the influx of
Ca2+. It consists of the core protein MCU and auxiliary
proteins such as mitochondrial calcium uptake (MICU)1, MICU2,
essential MCU regulator (EMRE), and mitochondrial calcium uniporter
regulator 1 (MCUR1) (27), which
work together to regulate the amount and rate of Ca2+
influx, allowing Ca2+ to enter the mitochondrial matrix
and participate in the regulation of key cellular functions,
including energy metabolism, signal transduction and stress
responses (28). MCU is a
transmembrane protein with its N-terminus facing the mitochondrial
matrix and its C-terminus facing the cytoplasm. The transmembrane
region forms the Ca2+ channel, and its opening and
closing are regulated by factors such as mitochondrial membrane
potential, intracellular Ca2+ levels and redox status
(29). In addition to serving as a
Ca2+ channel, MCU also plays a role in regulating
mitochondrial energy metabolism. Its activation promotes ATP
synthesis, enhances the tricarboxylic acid cycle and oxidative
phosphorylation. Moreover, by facilitating the influx of
Ca2+, MCU activates enzymes involved in fatty acid
oxidation and glucose metabolism, thus supporting cellular energy
production (29). The structure of
the MCU complex is presented in Fig.
1.
The activity of the MCU complex is finely regulated
by MICU1, MICU2, EMRE and MCUR1. MICU1 and MICU2 are located on the
cytoplasmic side of the MCU and sense the Ca2+
concentration in the mitochondrial matrix, regulating the opening
of the MCU channel to control Ca2+ influx. When the
intracellular Ca2+ concentration is low, they bind to
the MCU complex and inhibit the opening of the channel; when the
Ca2+ concentration rises, they dissociate or undergo
structural changes, promoting the opening of the channel (30). EMRE enhances the Ca2+
transport activity of the MCU channel, and its absence leads to a
decrease in MCU complex function (31). MCUR1 interacts with the MCU complex
to maintain its stability and function; the absence of MCUR1 causes
abnormal Ca2+ accumulation within the mitochondria,
affecting cellular energy metabolism (27). The coordinated action of these
regulatory factors ensures that the MCU complex efficiently and
accurately regulates mitochondrial Ca2+ concentrations
under various physiological and pathological conditions, preventing
excessive accumulation that could lead to mitochondrial damage and
cell death.
In summary, mitochondrial Ca2+ influx is
primarily mediated by the finely regulated MCU complex, which plays
a central role in maintaining cellular calcium homeostasis and
energy metabolism (26). Under
ischemic conditions, dysregulation of MCU-mediated Ca2+
uptake contributes to mitochondrial calcium overload, oxidative
stress and neuronal injury, highlighting its significance in the
pathophysiology of stroke (32).
Future research should focus on elucidating the fine regulatory
mechanisms of MCU complex components, particularly their
spatiotemporal interactions, and developing precise therapeutic
strategies, such as selective MCU inhibitors, to mitigate
Ca2+ overload while preserving physiological
Ca2+ signaling.
Molecular mechanisms of mitochondrial calcium
efflux Function and regulation of NCLX. NCLX is the primary
channel for mitochondrial Ca2+ efflux. It facilitates
the extrusion of Ca2+ from the mitochondrial matrix by
exchanging it with cytosolic Na+, relying on the
concentration gradients of sodium and calcium (33). This mechanism is crucial for
maintaining cellular Ca2+ homeostasis and plays a vital
role in regulating mitochondrial membrane potential, cellular
stress responses and energy metabolism (34). The activity of NCLX is regulated by
the sodium gradient and sodium pump function. Under pathological
conditions such as ischemia/reperfusion injury and
neurodegenerative diseases, NCLX prevents calcium overload, thereby
protecting mitochondrial and cellular function (34).
Other potential calcium efflux
pathways
While NCLX is the primary efflux pathway, other
mechanisms may contribute to mitochondrial Ca2+ efflux
under specific conditions: i) Tricarboxylic acid transporter (TST)
and Na+/H+ exchanger (NHE): TST primarily
mediates metabolite translocation but may facilitate
Ca2+ efflux during changes in calcium signaling.
Similarly, NHE can regulate mitochondrial Ca2+ efflux
through exchange mechanisms under pathological conditions (35). ii) Pannexin 1 channels: Pannexin 1,
a membrane channel protein, may regulate Ca2+ movement
during cellular stress or injury. It is hypothesized to play a role
in mitochondrial Ca2+ efflux, particularly under
conditions such as inflammation or hypoxia (36). iii) Calcium-activated potassium
channels (KCa channels): KCa channels are activated by elevated
Ca2+ levels and may indirectly modulate Ca2+
efflux, especially during ionic imbalances or stress conditions
(36). Fig. 2 illustrates the pathways of
Ca2+ efflux.
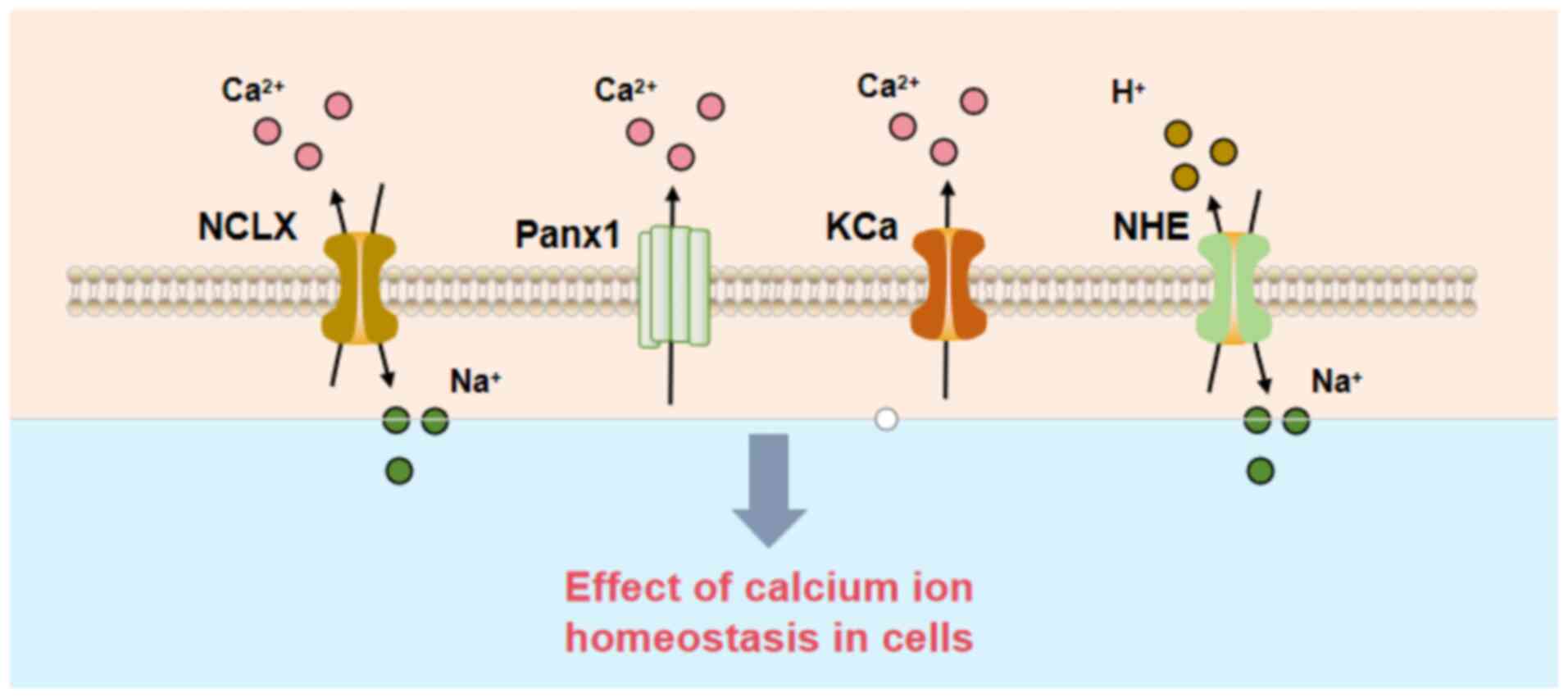 | Figure 2.Calcium efflux pathways.
Mitochondrial calcium efflux mechanisms, including the NCLX, Panx1,
KCa, and NHE, which cooperatively regulate intracellular calcium
homeostasis. NCLX, sodium-calcium exchanger; Panx1, pannexin 1;
KCa, calcium-activated potassium channels; NHE, sodium ion/hydrogen
ion exchanger; Ca2+, calcium ion; Na+, sodium
ion; H+, hydrogen ion. |
Mitochondrial calcium transport in
ischemia/reperfusion injury
Ischemia/reperfusion injury is a core pathological
process in stroke, characterized by the interplay of energy
metabolism dysfunction, oxidative stress and calcium homeostasis
imbalance (37,38). During the ischemic phase, blood
flow interruption leads to neuronal energy depletion and
Ca2+ signaling disruption. In the reperfusion phase,
although blood flow restoration provides oxygen and nutrients, it
is also accompanied by a massive influx of Ca2+ into the
cells, and Ca2+ overload further exacerbates
mitochondrial dysfunction (39).
The pathological mechanisms of mitochondrial Ca2+
transport in stroke are shown in Fig.
3.
 | Figure 3.Pathological mechanisms of
mitochondrial calcium transport in stroke. Schematic overview of
stroke-induced calcium overload leading to mitochondrial
dysfunction. Excessive Ca2+ influx via the MCU and
impaired efflux disrupt mitochondrial membrane potential, generate
ROS, trigger mPTP opening, and release apoptotic factors such as
AIF and Cyt c, resulting in neuronal death. MCU, mitochondrial
calcium uniporter; NCLX, sodium-calcium exchanger; ROS, reactive
oxygen species; AIF, apoptosis-inducing factor; Cyto C, cytochrome
c; mPTP, mitochondrial permeability transition pore. |
Mitochondrial calcium homeostasis imbalance
during ischemia. During ischemia, blood flow interruption leads
to severe hypoxia in brain tissue, inhibiting mitochondrial
oxidative phosphorylation and resulting in energy depletion that
weakens membrane ion pumps (such as Na+/K+
ATPase) (32). Elevated
extracellular Ca2+ concentrations drive a large influx
of Ca2+ into cells. The accumulation of Ca2+
further activates calcium-dependent enzymes (such as calpains and
phospholipase A2), which exacerbates membrane structural damage and
energy metabolism dysfunction, ultimately impairing mitochondrial
function (40).
Furthermore, under the context of calcium
homeostasis disruption, mitochondrial membrane potential decreases,
and Ca2+ transport mechanisms become dysregulated,
leading to excessive calcium influx and mitochondrial
Ca2+ overload, further impairing mitochondrial function
(41).
Fluctuations in calcium signaling
during reperfusion
Although reperfusion restores blood flow and
provides oxygen and glucose, the rapid reintroduction of oxygen
triggers a burst of ROS, exacerbating oxidative damage to the
mitochondrial membrane and transport proteins. At the same time,
the fluctuations in calcium signaling further disrupt membrane
permeability, causing a rapid imbalance of Ca2+ between
the mitochondrial matrix and the cytoplasm (42).
Notably, a feedback loop exists between ROS
accumulation and Ca2+ overload: ROS oxidize the MCU
complex and NCLX, impairing Ca2+ transport regulation
and further exacerbating Ca2+ accumulation, while
Ca2+ overload enhances ROS production, continuously
amplifying mitochondrial damage and increasing the risk of neuronal
death (43).
Cellular apoptosis triggered by
abnormal calcium transport
Ca2+ overload activates apoptosis and
necrosis through multiple signaling pathways. For example,
mitochondrial Ca2+ overload induces the opening of the
mitochondrial permeability transition pore, leading to the release
of apoptotic factors such as cytochrome c (Cyt c) and
apoptosis-inducing factor (AIF). Cyt c activates the intracellular
apoptotic signaling pathway and promotes apoptosis through the
caspase cascade reaction (32,44),
while AIF triggers DNA fragmentation and nuclear membrane collapse,
resulting in irreversible damage (45).
Therapeutic strategies targeting
mitochondrial calcium transport
Current therapeutic strategies primarily focus on
regulating mitochondrial Ca2+ influx via the MCU
complex, enhancing Ca2+ efflux through the NCLX channel
and employing multi-target combination therapies to comprehensively
restore mitochondrial calcium homeostasis. These approaches
demonstrate significant therapeutic potential and will be discussed
in detail below.
Intervention strategies targeting the MCU
complex
Research progress on MCU
inhibitors
The MCU complex is the primary Ca2+
influx channel on the mitochondrial inner membrane. However,
excessive activation of the MCU complex can lead to mitochondrial
Ca2+ overload, triggering oxidative stress, apoptosis
and tissue damage (44).
Therefore, MCU inhibitors effectively prevent mitochondrial
dysfunction and cell death caused by Ca2+ overload by
limiting mitochondrial Ca2+ influx.
Ru360, a classic MCU inhibitor, markedly reduces
oxidative stress and apoptotic responses during
ischemia/reperfusion injury, thereby improving neurological
recovery. However, its clinical application is limited by poor
water solubility and insufficient specificity (46). By contrast, DS16570511 demonstrates
improved selectivity and pharmacokinetic properties in animal
models. It effectively inhibits mitochondrial Ca2+
accumulation, reduces oxidative stress levels in brain tissue and
enhances neuronal survival. These findings lay a foundation for
further optimization of MCU inhibitors (47).
Potential benefits of moderate MCU
activation in stroke recovery
Although MCU inhibition is the primary focus of
current research, the potential benefits of moderate MCU activation
during the recovery phase of stroke also warrant attention. During
the subacute and recovery stages of stroke, a moderate increase in
mitochondrial calcium may play a positive role by promoting
neuronal metabolic recovery and signal transmission. A study has
shown that moderate MCU activation enhances mitochondrial
responsiveness to calcium signaling, stimulates ATP production and
improves energy metabolism in damaged neurons, thereby facilitating
functional recovery (48).
However, the precise molecular mechanisms of MCU activation, its
optimal activation range, and its safety and efficacy in clinical
applications require further investigation.
Therapeutic strategies to regulate
mitochondrial calcium efflux
Development and application of NCLX
regulators
The development of NCLX activators aims to promote
mitochondrial Ca2+ efflux, thereby preventing
Ca2+ overload, mitigating damage to mitochondrial
membrane potential and energy metabolism and enhancing neuronal
survival (49). A recent study
demonstrated the critical role of NCLX in regulating glycolysis in
astrocytes (50). In adult mice
downregulation or loss of NCLX function reduces lactate output,
thereby impairing neuronal function and synaptic plasticity,
ultimately leading to deficits in learning and memory. These
findings suggest that NCLX may serve as a potential therapeutic
target for Alzheimer's disease and other cognitive
impairment-related disorders (51,52).
However, although NCLX-targeting therapeutic
strategies have shown potential in basic research, their
translation from the laboratory to clinical application still faces
numerous challenges (53). Future
research should prioritize the development of novel, highly
specific NCLX-targeted strategies to overcome the limitations of
existing compounds. Additionally, efforts must focus on elucidating
the mechanisms of action and targeting specificity while rigorously
evaluating their safety and efficacy in clinical settings,
facilitating their translation from laboratory research to
therapeutic application.
Target screening for protecting
mitochondrial calcium efflux pathways
In addition to directly activating NCLX, protecting
the function of proteins related to mitochondrial Ca2+
efflux is also an important research direction. For example,
enhancing the expression of calcium-binding proteins or modulating
their function can indirectly promote the efficiency of calcium
efflux (54). Furthermore, a
structural-functional study of NCLX have shown that some small
molecules can bind to key sites on NCLX to prevent functional
impairment caused by oxidative stress or pathological environments
(55). A deeper understanding of
the regulatory mechanisms of the NCLX pathway could help to
identify more potential therapeutic targets.
Multi-target combination therapy for
comprehensive regulation of mitochondrial calcium homeostasis
Antioxidant combination therapy targeting calcium
homeostasis
During stroke, oxidative stress is closely related
to calcium homeostasis imbalance. Antioxidants combined with drugs
that regulate mitochondrial Ca2+ transport may have a
synergistic effect. For instance, antioxidants such as vitamin C,
resveratrol and N-acetylcysteine can reduce ROS production, thereby
mitigating oxidative stress-induced damage to MCU and NCLX
(56). When these antioxidants are
used in combination with MCU inhibitors or NCLX activators, they
can provide dual protection by reducing calcium overload and
lowering oxidative stress, which effectively alleviates
stroke-induced damage (57).
Gene therapy and
mitochondrial-targeted drug delivery systems
Gene therapy holds potential for the treatment of
mitochondrial calcium homeostasis. By using gene editing
technologies, such as CRISPR-Cas9 (58), to regulate the expression of key
proteins such as MCU and NCLX, mitochondrial calcium dynamics can
be precisely controlled. Moreover, mitochondrial-targeted drug
delivery systems (such as the MITO-Porter nano-delivery system)
developed in previous years can deliver drugs specifically to
mitochondria, increasing drug concentrations at the target site and
enhancing therapeutic effects (59). The combination of targeted delivery
technology and gene therapy is expected to enable precise
regulation of mitochondrial Ca2+ homeostasis.
Comparative analysis of mitochondrial
calcium-targeted strategies in stroke
Combination therapies offer a holistic approach but
require careful optimization to balance efficacy and safety.
Mechanistically, MCU inhibitors act rapidly to block
Ca2+ influx, making them vital during the ischemic phase
when Ca2+ surges are most damaging (46,47),
but their broad inhibition may impair beneficial Ca2+
signaling needed for recovery (48). Conversely, NCLX activators
fine-tune Ca2+ efflux, supporting mitochondrial
resilience and neuronal repair (49,50),
yet the scarcity of specific activators hinders their immediate
clinical use (53). Combination
therapies, integrating antioxidants or targeted delivery, provide a
multifaceted approach, addressing both Ca2+
dysregulation and oxidative stress (56,59),
but their complexity poses challenges in dosing and patient
compliance. Clinically, MCU inhibitors are prioritized in emergency
settings (46), NCLX activators
hold promise for rehabilitation (50) and combination therapies suit
complex cases with comorbidities such as cardiovascular disease
(60). Patient-specific factors,
such as stroke severity and concurrent conditions, further dictate
strategy selection, underscoring the need for personalized
diagnostics and scalable delivery systems to enhance translation
from bench to bedside (58,59).
In summary, MCU inhibitors excel in acute
neuroprotection, NCLX activators support long-term recovery and
combination therapies, including antioxidants and gene-based
approaches, holistically address Ca2+ and oxidative
stress, particularly in complex cases. Current research has
advanced MCU inhibitor specificity (e.g., DS16570511) and explored
novel delivery systems (e.g., MITO-Porter), but challenges persist,
including limited NCLX activator development, off-target effects
and clinical translation barriers due to stroke heterogeneity and
comorbidity interactions. Future efforts should prioritize specific
NCLX activators, optimize combination therapy regimens and develop
biomarkers for personalized treatment to overcome these hurdles and
improve stroke outcomes.
Diagnosis and dynamic monitoring of
mitochondrial calcium transport
Identifying specific biomarkers to monitor abnormal
mitochondrial Ca2+ transport during stroke has become a
hotspot in neuroscience research. Changes in mitochondrial
Ca2+ transport proteins, calcium signaling abnormalities
and dynamic monitoring technologies provide new directions for
early diagnosis and real-time monitoring (61).
Early diagnostic calcium signaling
biomarkers
Key proteins involved in mitochondrial
Ca2+ transport, including the MCU complex, NCLX and
other regulatory factors, play a crucial role in the pathogenesis
of stroke. Therefore, detecting their expression and activity
levels is an important approach to exploring stroke pathophysiology
and identifying diagnostic biomarkers (62). For instance, methods such as
Western blotting, immunohistochemical staining, fluorescence
staining and reverse transcription-quantitative PCR can be used to
assess changes in the expression of these proteins in stroke models
(63). Furthermore, flow cytometry
and electrophysiological techniques allow for real-time monitoring
of calcium signaling changes in cellular and animal models,
providing potential methods for the early detection of
mitochondrial Ca2+ transport abnormalities (64).
Development of dynamic monitoring
technologies for mitochondrial calcium transport
In previous years, real-time monitoring techniques
based on fluorescent probes and calcium imaging have made notable
progress in the biomedical field, becoming essential tools for
studying mitochondrial Ca2+ transport and
Ca2+ signaling changes (65). Compared with traditional methods,
these technologies offer unique advantages in accurately capturing
the temporal changes of mitochondrial Ca2+ transport.
For example, a study developed a copper nanocluster-based
fluorescent probe for real-time imaging and ratio detection of
calcium ions in neurons. This probe demonstrates a strong linear
response to Ca2+ concentrations in the range of 2–350
µM, enabling the rapid detection of dynamic calcium signal changes
in neurons (66). Additionally,
Thiabaud et al (67)
developed an innovative texaphyrin-based calcium sensor designed
for dynamic monitoring and detection of intracellular calcium
signals using multimodal imaging techniques. This technology shows
promising potential applications in neuroscience, cardiovascular
disease research and the exploration of calcium signaling-related
pathological mechanisms.
Future research directions and
challenges
Frontiers in mitochondrial calcium
transport mechanisms
Despite notable progress in understanding
mitochondrial Ca2+ transport mechanisms in recent years,
a number of unresolved questions remain (68). For example, the interaction network
of mitochondrial Ca2+ transport proteins and the
regulatory relationships between different Ca2+
transport channels within the cell are not fully understood,
especially regarding the regulation of the MCU complex. While
several key regulatory factors (such as MICU1/2 and EMRE) have been
identified, the precise mechanisms by which they fine-tune the
spatiotemporal patterns of mitochondrial Ca2+ influx
remain unclear (69). Furthermore,
how to balance Ca2+ influx and efflux under various
pathological conditions (such as ischemia/reperfusion injury) to
prevent mitochondrial calcium overload and cell death remains an
urgent issue. Future research will need to explore these regulatory
mechanisms in detail through high-throughput screening, proteomics
and single-molecule techniques.
Key challenges in translating from laboratory to
clinic. Currently, most research on mitochondrial
Ca2+ transport relies on animal models, particularly
mouse models. However, these models face challenges in clinical
translation. For instance, the changes in mitochondrial
Ca2+ transport and underlying pathological mechanisms in
animal models may differ from the manifestations of stroke in
humans, especially in studies of long-term sequelae and chronic
stroke (70). Future research
should focus on developing more refined animal models that better
simulate the complex pathological processes of human stroke, as
well as exploring effective ways to safely and efficiently
translate experimental findings into clinical applications.
Challenges of emerging technologies
and interdisciplinary integration
With advancements in technology, innovative tools
and methods have provided new perspectives for the study of
mitochondrial Ca2+ transport. Single-cell genomics
technology allows for in-depth investigation of the expression and
function of Ca2+ transport channels at the single-cell
level, aiding in the understanding of cellular heterogeneity and
individualized responses (63).
Super-resolution microscopy techniques, such as STED and SIM
microscopes, enable the observation of dynamic changes in
mitochondrial calcium at the subcellular level, revealing the
mechanisms of Ca2+ influx and efflux (71). Despite these advancements driving
our understanding of mitochondrial Ca2+ transport
mechanisms, challenges remain in overcoming issues related to the
integration of technologies, interdisciplinary collaboration and
extensive data analysis, all of which are crucial for effectively
supporting the development of novel therapeutic strategies.
Complex relationship between
mitochondrial Ca2+ transport and stroke comorbidities
(e.g., Alzheimer's and cardiovascular diseases)
Mitochondrial Ca2+ transport plays a
crucial role in the pathophysiology of stroke and is closely linked
to other neurodegenerative diseases, such as Alzheimer's disease
and cardiovascular diseases. Studies have shown that stroke
patients often experience comorbidities, including cognitive
impairment and cardiovascular diseases. These comorbidities may
interact through mitochondrial Ca2+ dysregulation,
thereby promoting neurodegenerative processes (72). For example, in Alzheimer's disease,
abnormal Ca2+ signaling is closely associated with
mitochondrial dysfunction, both contributing to neuronal damage
(73). Similarly, the increased
stroke risk in cardiovascular disease patients is closely linked to
the disruption of mitochondrial calcium homeostasis, with
Ca2+ overload exacerbating cardiovascular injury and
worsening the stroke process (60). Therefore, future research should
focus on exploring the role of mitochondrial Ca2+
transport in stroke and its comorbidities, particularly how
targeting Ca2+ transport could intervene in the onset
and progression of these pathological processes. Interdisciplinary
collaborative research will play an increasingly important role in
advancing this field.
Summary
Mitochondrial Ca2+ transport plays a
crucial role in the pathophysiology of stroke. During the
ischemia/reperfusion injury phase, disruptions in Ca2+
homeostasis lead to mitochondrial dysfunction, excessive ROS
production and the activation of cell death signals. Dysfunction of
the Ca2+ influx channel MCU complex and the efflux
channel NCLX has been shown to be one of the core drivers of stroke
pathogenesis. Targeting the regulation of these channels has become
a key direction for stroke therapy, including strategies such as
MCU inhibitors, NCLX activators and combined antioxidant
treatments. Furthermore, the dynamic monitoring of Ca2+
signaling provides new perspectives for early diagnosis and
precision treatment. Despite notable progress in basic research,
translating these findings into safe and effective clinical
treatments remains a challenge. Future research should continue to
focus on mitochondrial Ca2+ regulatory mechanisms,
explore innovative diagnostic tools and develop multidimensional
intervention strategies to improve stroke prognosis and enhance
patient quality of life.
Acknowledgements
Not applicable.
Funding
This research was funded by grants from the Basic Public Welfare
Research Program of Zhejiang Province (grant no. LGF20H270003), the
Scientific Research Project of Affiliated Hospital of Zhejiang
University of Traditional Chinese Medicine (grant no.
2023FSYYZQ15), the Jiaxing Key Laboratory of Integrated Traditional
Chinese and Western Medicine Rehabilitation Research on
Cerebrovascular Diseases [grant no. 2022(38)], the Jiaxing City
Science and Technology Bureau Project (grant no. 2024AD30075),
Zhejiang Provincial Science and Technology Plan Project of
Traditional Chinese Medicine (grant no. 2025ZL568) and the Special
Research Fund Project of Zhejiang Provincial Rehabilitation Medical
Association (grant no. ZKKY2023007).
Availability of data and materials
Not applicable.
Authors' contributions
YL and WJ performed the literature review and wrote
the manuscript. HZ, XD, KL and XY revised the manuscript. XW and HR
performed the literature review and contributed to the acquisition
and analysis of data. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Qin C, Yang S, Chu YH, Zhang H, Pang XW,
Chen L, Zhou LQ, Chen M, Tian DS and Wang W: Signaling pathways
involved in ischemic stroke: molecular mechanisms and therapeutic
interventions. Signal Transduct Target Ther. 7:2152022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
GBD 2021 Nervous System Disorders
Collaborators, . Global, regional, and national burden of disorders
affecting the nervous system, 1990–2021: A systematic analysis for
the global burden of disease study 2021. Lancet Neurol. 23:344–381.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Feigin VL, Krishnamurthi RV, Parmar P,
Norrving B, Mensah GA, Bennett DA, Barker-Collo S, Moran AE, Sacco
RL, Truelsen T, et al: Update on the global burden of ischemic and
hemorrhagic stroke in 1990–2013: The GBD 2013 study.
Neuroepidemiology. 45:161–176. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Prendes CF, Rantner B, Hamwi T, Stana J,
Feigin VL, Stavroulakis K and Tsilimparis N; GBD Collaborators
Study Group, : Burden of stroke in Europe: An analysis of the
global burden of disease study findings from 2010 to 2019. Stroke.
55:432–442. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li XY, Kong XM, Yang CH, Cheng ZF, Lv JJ,
Guo H and Liu XH: Global, regional, and national burden of ischemic
stroke, 1990–2021: An analysis of data from the global burden of
disease study 2021. EClinicalMedicine. 75:1027582024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mercy UC, Farhadi K, Ogunsola AS, Karaye
RM, Baguda US, Eniola OA, Yunusa I and Karaye IM: Revisiting recent
trends in stroke death rates, United States, 1999–2020. J Neurol
Sci. 451:1207242023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Famakin BM, Chimowitz MI, Lynn MJ, Stern
BJ and George MG; WASID Trial Investigators, : Causes and severity
of ischemic stroke in patients with symptomatic intracranial
arterial stenosis. Stroke. 40:1999–2003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boehme AK, Esenwa C and Elkind MS: Stroke
risk factors, genetics, and prevention. Circ Res. 120:472–495.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ananth CV, Brandt JS, Keyes KM, Graham HL,
Kostis JB and Kostis WJ: Epidemiology and trends in stroke
mortality in the USA, 1975–2019. Int J Epidemiol. 52:858–866. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Abissegue G, Yakubu SI, Ajay AS and
Niyi-Odumosu F: A systematic review of the epidemiology and the
public health implications of stroke in Sub-Saharan Africa. J
Stroke Cerebrovasc Dis. 33:1077332024. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao Y, Hua X, Ren X, Ouyang M, Chen C, Li
Y, Yin X, Song P, Chen X, Wu S, et al: Increasing burden of stroke
in China: A systematic review and meta-analysis of prevalence,
incidence, mortality, and case fatality. Int J Stroke. 18:259–267.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yang JL, Mukda S and Chen SD: Diverse
roles of mitochondria in ischemic stroke. Redox Biol. 16:263–275.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kaur MM and Sharma S: Mitochondrial repair
as potential pharmacological target in cerebral ischemia.
Mitochondrion. 63:23–31. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ham PB III and Raju R: Mitochondrial
function in hypoxic ischemic injury and influence of aging. Prog
Neurobiol. 157:92–116. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
An H, Zhou B and Ji X: Mitochondrial
quality control in acute ischemic stroke. J Cereb Blood Flow Metab.
41:3157–3170. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Andrabi SS, Parvez S and Tabassum H:
Ischemic stroke and mitochondria: Mechanisms and targets.
Protoplasma. 257:335–343. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Granger DN and Kvietys PR: Reperfusion
injury and reactive oxygen species: The evolution of a concept.
Redox Biol. 6:524–551. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Garbincius JF and Elrod JW: Mitochondrial
calcium exchange in physiology and disease. Physiol Rev.
102:893–992. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ludhiadch A, Sharma R, Muriki A and Munshi
A: Role of calcium homeostasis in ischemic stroke: A review. CNS
Neurol Disord Drug Targets. 21:52–61. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guicciardi ME, Trussoni CE, LaRusso NF and
Gores GJ: The spectrum of reactive cholangiocytes in primary
sclerosing cholangitis. Hepatology. 71:741–748. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patergnani S, Danese A, Bouhamida E,
Aguiari G and Giorgi C: Various aspects of calcium signaling in the
regulation of apoptosis, autophagy, cell proliferation, and cancer.
Int J Mol Sci. 21:83232020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fels JA and Manfredi G: Sex differences in
ischemia/reperfusion injury: The role of mitochondrial permeability
transition. Neurochem Res. 44:2336–2345. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Rahi V and Kaundal RK: Exploring the
intricacies of calcium dysregulation in ischemic stroke: Insights
into neuronal cell death and therapeutic strategies. Life Sci.
347:1226512024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fan M, Zhang J, Tsai CW, Orlando BJ,
Rodriguez M, Xu Y, Liao M, Tsai MF and Feng L: Structure and
mechanism of the mitochondrial Ca(2+) uniporter holocomplex.
Nature. 582:129–133. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chang X, Liu R, Li R, Peng Y, Zhu P and
Zhou H: Molecular mechanisms of mitochondrial quality control in
ischemic cardiomyopathy. Int J Biol Sci. 19:426–448. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Guo J, Wang Y, Shi C, Zhang D, Zhang Q,
Wang L and Gong Z: Mitochondrial calcium uniporter complex:
Unveiling the interplay between its regulators and calcium
homeostasis. Cell Signal. 121:1112842024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Patron M, Checchetto V, Raffaello A,
Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De
Stefani D and Rizzuto R: MICU1 and MICU2 finely tune the
mitochondrial Ca2+ uniporter by exerting opposite effects on MCU
activity. Mol Cell. 53:726–737. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ma J, Li J, Jin C, Yang J, Zheng C, Chen
K, Xie Y, Yang Y, Bo Z, Wang J, et al: Association of gut
microbiome and primary liver cancer: A two-sample Mendelian
randomization and case-control study. Liver Int. 43:221–233. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y,
Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, et al:
Architecture of the mitochondrial calcium uniporter. Nature.
533:269–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang C, Jacewicz A, Delgado BD, Baradaran
R and Long SB: Structures reveal gatekeeping of the mitochondrial
Ca(2+) uniporter by MICU1-MICU2. Elife. 9:e599912020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Delgado de la Herran H, Vecellio Reane D,
Cheng Y, Reane D, Cheng Y, Katona M, Hosp F, Greotti E,
Wettmarshausen J, Patron M, et al: Systematic mapping of
mitochondrial calcium uniporter channel (MCUC)-mediated calcium
signaling networks. EMBO J. 43:5288–5326. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Brookes PS, Yoon Y, Robotham JL, Anders MW
and Sheu SS: Calcium, ATP, and ROS: A mitochondrial love-hate
triangle. Am J Physiol Cell Physiol. 287:C817–833. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kostic M and Sekler I: Functional
properties and mode of regulation of the mitochondrial Na(+)/Ca(2+)
exchanger, NCLX. Semin Cell Dev Biol. 94:59–65. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Takeuchi A and Matsuoka S: Physiological
and pathophysiological roles of mitochondrial Na+-Ca2+ exchanger,
NCLX, in hearts. Biomolecules. 11:18762021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Alvear TF, Farias-Pasten A, Vergara SA,
Prieto-Villalobos J, Silva-Contreras A, Fuenzalida FA, Quintanilla
RA and Orellana JA: Hemichannels contribute to mitochondrial Ca(2+)
and morphology alterations evoked by ethanol in astrocytes. Front
Cell Dev Biol. 12:14343812024. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tano JY and Gollasch M: Calcium-activated
potassium channels in ischemia reperfusion: A brief update. Front
Physiol. 5:3812014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Dambrova M, Zuurbier CJ, Borutaite V,
Liepinsh E and Makrecka-Kuka M: Energy substrate metabolism and
mitochondrial oxidative stress in cardiac ischemia/reperfusion
injury. Free Radic Biol Med. 165:24–37. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han Y, Li X, Yang L, Zhang D, Li L, Dong
X, Li Y, Qun S and Li W: Ginsenoside Rg1 attenuates cerebral
ischemia-reperfusion injury due to inhibition of NOX2-mediated
calcium homeostasis dysregulation in mice. J Ginseng Res.
46:515–525. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bertero E, Popoiu TA and Maack C:
Mitochondrial calcium in cardiac ischemia/reperfusion injury and
cardioprotection. Basic Res Cardiol. 119:569–585. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Curcio M, Salazar IL, Mele M, Canzoniero
LMT and Duarte CB: Calpains and neuronal damage in the ischemic
brain: The swiss knife in synaptic injury. Prog Neurobiol.
143:1–35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Odagiri K, Katoh H, Kawashima H, Tanaka T,
Ohtani H, Saotome M, Urushida T, Satoh H and Hayashi H: Local
control of mitochondrial membrane potential, permeability
transition pore and reactive oxygen species by calcium and
calmodulin in rat ventricular myocytes. J Mol Cell Cardiol.
46:989–997. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu L, Tan JL, Chen ZY and Huang G:
Cardioprotection of post-ischemic moderate ROS against
ischemia/reperfusion via STAT3-induced the inhibition of MCU
opening. Basic Res Cardiol. 114:392019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Guan L, Che Z, Meng X, Yu Y, Li M, Yu Z,
Shi H, Yang D and Yu M: MCU Up-regulation contributes to myocardial
ischemia-reperfusion Injury through calpain/OPA-1-mediated
mitochondrial fusion/mitophagy Inhibition. J Cell Mol Med.
23:7830–7843. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiang C, Shen J, Wang C, Huang Y, Wang L,
Yang Y, Hu W, Li P and Wu H: Mechanism of aconitine mediated
neuronal apoptosis induced by mitochondrial calcium overload caused
by MCU. Toxicol Lett. 384:86–95. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Shintani-Ishida K, Inui M and Yoshida KI:
Ischemia-reperfusion induces myocardial infarction through
mitochondrial Ca2+ overload. J Mol Cell Cardiol. 53:233–239. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
de Jesús García-Rivas G,
Guerrero-Hernández A, Guerrero-Serna G, Rodríguez-Zavala JS and
Zazueta C: Inhibition of the mitochondrial calcium uniporter by the
oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from
irreversible injury in postischemic rat heart. FEBS J.
272:3477–3488. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kon N, Murakoshi M, Isobe A, Kagechika K,
Miyoshi N and Nagayama T: DS16570511 is a small-molecule inhibitor
of the mitochondrial calcium uniporter. Cell Death Discov.
3:170452017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Wescott AP, Kao JPY, Lederer WJ and Boyman
L: Voltage-energized Calcium-sensitive ATP production by
mitochondria. Nat Metab. 1:975–984. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cohen HM, Salik O and Elrod JW: Signaling
pathways regulating mitochondrial calcium efflux-a commentary on
Rozenfeld et al: ‘Essential role of the mitochondrial
Na(+)/Ca(2+) exchanger NCLX in mediating PDE2-dependent neuronal
survival and learning’. Cell Calcium. 113:1027642023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Cabral-Costa JV, Vicente-Gutiérrez C,
Agulla J, Lapresa R, Elrod JW, Almeida Á, Bolaños JP and
Kowaltowski AJ: Mitochondrial sodium/calcium exchanger NCLX
regulates glycolysis in astrocytes, impacting on cognitive
performance. J Neurochem. 165:521–535. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Jadiya P, Cohen HM, Kolmetzky DW, Kadam
AA, Tomar D and Elrod JW: Neuronal loss of NCLX-dependent
mitochondrial calcium efflux mediates age-associated cognitive
decline. iScience. 26:1062962023. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Jadiya P, Kolmetzky DW, Tomar D, Di Meco
A, Lombardi AA, Lambert JP, Luongo TS, Ludtmann MH, Praticò D and
Elrod JW: Impaired mitochondrial calcium efflux contributes to
disease progression in models of Alzheimer's disease. Nat Commun.
10:38852019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Viejo L, Rubio-Alarcon M, Arribas RL,
Moreno-Castro M, Pérez-Marín R, Braun-Cornejo M, Estrada-Valencia M
and de Los Ríos C: Synthesis and biological assessment of
4,1-benzothiazepines with neuroprotective activity on the Ca(2+)
overload for the treatment of neurodegenerative diseases and
stroke. Molecules. 26:44372021. View Article : Google Scholar
|
|
54
|
Garbincius JF, Salik O, Cohen HM,
Choya-Foces C, Mangold AS, Makhoul AD, Schmidt AE, Khalil DY,
Doolittle JJ, Wilkinson AS, et al: TMEM65 regulates NCLX-dependent
mitochondrial calcium efflux. bioRxiv. Oct 9–2023.(Epub ahead of
print). doi: 10.1101/2023.10.06.561062.
|
|
55
|
Roy S, Dey K, Hershfinkel M, Ohana E and
Sekler I: Identification of residues that control Li+ versus Na+
dependent Ca2+ exchange at the transport site of the mitochondrial.
Biochim Biophys Acta Mol Cell Res. 1864.997–1008. 2017.PubMed/NCBI
|
|
56
|
Li Z, Bi R, Sun S, Chen S, Chen J, Hu B
and Jin H: The role of oxidative stress in acute ischemic
stroke-related thrombosis. Oxid Med Cell Longev. 2022:84188202022.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Choya-Foces C, Navarro E, Rios CL, López
MG, Egea J, Hernansanz-Agustín P and Martínez-Ruiz A: The
mitochondrial Na(+)/Ca(2+) exchanger NCLX is implied in the
activation of hypoxia-inducible factors. Redox Biol. 77:1033642024.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gupta D, Bhattacharjee O, Mandal D, Sen
MK, Dey D, Dasgupta A, Kazi TA, Gupta R, Sinharoy S, Acharya K, et
al: CRISPR-Cas9 system: A new-fangled dawn in gene editing. Life
Sci. 232:1166362019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yamada Y and Harashima H: MITO-porter for
mitochondrial delivery and mitochondrial functional analysis. Handb
Exp Pharmacol. 240:457–472. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Bick AG, Wakimoto H, Kamer KJ, Sancak Y,
Goldberger O, Axelsson A, DeLaughter DM, Gorham JM, Mootha VK,
Seidman JG and Seidman CE: Cardiovascular homeostasis dependence on
MICU2, a regulatory subunit of the mitochondrial calcium uniporter.
Proc Natl Acad Sci USA. 114:E9096–E9104. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sasaki H, Nakagawa I, Furuta T, Yokoyama
S, Morisaki Y, Saito Y and Nakase H: Mitochondrial calcium
uniporter (MCU) is Involved in an ischemic postconditioning effect
against ischemic reperfusion brain injury in mice. Cell Mol
Neurobiol. 44:322024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Verma M, Callio J, Otero PA, Sekler I,
Wills ZP and Chu CT: Mitochondrial calcium dysregulation
contributes to dendrite degeneration mediated by PD/LBD-associated
LRRK2 mutants. J Neurosci. 37:11151–11165. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Lin W, Wang Y, Chen Y, Wang Q, Gu Z and
Zhu Y: Role of calcium signaling pathway-related gene regulatory
networks in ischemic stroke based on multiple WGCNA and single-cell
analysis. Oxid Med Cell Longev. 2021:80604772021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Aliotta A, Bertaggia Calderara D and
Alberio L: Flow cytometric monitoring of dynamic cytosolic calcium,
sodium, and potassium fluxes following platelet activation.
Cytometry A. 97:933–944. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Greotti E and Pozzan T: Live mitochondrial
or cytosolic calcium imaging using genetically-encoded cameleon
indicator in mammalian cells. Bio Protoc. 10:e35042020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu Z, Jing X, Zhang S and Tian Y: A
copper nanocluster-based fluorescent probe for real-time imaging
and ratiometric biosensing of calcium ions in neurons. Anal Chem.
91:2488–2497. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Thiabaud GD, Schwalm M, Sen S, Barandov A,
Simon J, Harvey P, Spanoudaki V, Müller P, Sessler JL and Jasanoff
A: Texaphyrin-based calcium sensor for multimodal imaging. ACS
Sens. 8:3855–3861. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Vecellio Reane D, Serna JDC and Raffaello
A: Unravelling the complexity of the mitochondrial Ca(2+)
uniporter: Regulation, tissue specificity, and physiological
implications. Cell Calcium. 121:1029072024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Cui C, Yang J, Fu L, Wang M and Wang X:
Progress in understanding mitochondrial calcium uniporter
complex-mediated calcium signalling: A potential target for cancer
treatment. Br J Pharmacol. 176:1190–1205. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Woodruff TM, Thundyil J, Tang SC, Sobey
CG, Taylor SM and Arumugam TV: Pathophysiology, treatment, and
animal and cellular models of human ischemic stroke. Mol
Neurodegener. 6:112011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhang Y, Wang J, Xing S, Li L, Zhao S, Zhu
W, Liang K, Liu Y and Chen L: Mitochondria determine the sequential
propagation of the calcium macrodomains revealed by the
super-resolution calcium lantern imaging. Sci China Life Sci.
63:1543–1551. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Matuz-Mares D, González-Andrade M,
Araiza-Villanueva MG, Vilchis-Landeros MM and Vázquez-Meza H:
Mitochondrial calcium: Effects of its imbalance in disease.
Antioxidants (Basel). 11:8012022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Calvo-Rodriguez M and Bacskai BJ:
Mitochondria and calcium in Alzheimer's disease: From cell
signaling to neuronal cell death. Trends Neurosci. 44:136–151.
2021. View Article : Google Scholar : PubMed/NCBI
|