Introduction
Liver cancer is one of the most prevalent and
life-threatening cancer types worldwide. Due to its high incidence
and global mortality rates, as well as the limited therapeutic
options at advanced stages, liver cancer remains a major clinical
challenge (1–3).
Estrogen receptor (ER)-mediated signaling is known
to contribute to the pathogenesis of liver cancer (4,5).
ER-α36, an isoform of ER-α, has been reported to play a critical
role in the development of numerous tumors, including liver cancer
(6,7). ER-α36 is transcribed from a promoter
located in the first intron of the classic ER-α gene. Unlike
classical ER-α, ER-α36 is mainly localized on the plasma membrane,
where it mediates rapid estrogen signaling (8). ER-α36 expression in primary liver
cancer is upregulated compared with that in adjacent non-tumor
tissues, contrasting with the expression pattern of classical ER-α
(7,9). In addition, ER-α36-mediated rapid
estrogen signaling has been shown to promote the growth of
PLC/PRF/5 and HepG2 liver cancer cells, in culture and in
tumorspheres, through the epidermal growth factor
receptor/Src/extracellular signal-regulated kinase axis (10). Tamoxifen, an ER-α antagonist, has
not provided survival benefits in patients with breast cancer
(11), and ER-α36 has been
indicated to contribute to tamoxifen resistance in breast cancer
and glioblastoma cells (11,12).
These findings suggest that ER-α36, rather than classical ER-α, is
the key ER isoform involved in the development and progression of
liver cancer. However, the mechanism underlying the potential
contribution of ER-α36 to liver tumorigenesis remains unclear.
Autophagy is a catabolic pathway that maintains
cellular homeostasis and supports organelle renewal by removing
misfolded proteins, cytotoxic aggregates and damaged organelles
(13). During this process,
cytoplasmic components are sequestered into autophagosomes, which
subsequently fuse with lysosomes to form autolysosomes. Within
autolysosomes, cellular contents, including protein substrates,
receptors and other autophagosome-associated proteins, are degraded
by lysosomal acidic hydrolases (14). Thus, lysosomes function as the
terminal degradation component of the autophagic process. Studies
have shown that lysosomal dysfunction, including lysosomal membrane
permeabilization (LMP), loss of acidity, changes in lysosome
localization and defects in lysosome-associated signaling molecules
(15–18), can impair the degradative capacity
of the lysosomal-autophagy pathway. These dysfunctions have
profound implications for the development and progression of
numerous diseases, including cancer (19). Tumor cells rely heavily on
increased lysosomal function to meet their proliferation and
metabolism requirements, which renders them particularly sensitive
to lysosomal dysregulation (20,21).
For example, it has been found that the re-localization of
lysosomes to the tumor cell periphery facilitates the exocytosis of
cathepsins, neuraminidase-1 and heparinase, thereby promoting tumor
cell invasion, metastasis and angiogenesis (20,22).
In addition, lysosomal acidity has been shown to influence cell
growth through the maintenance of iron homeostasis (23). Thus, the lysosome is an important
regulatory hub for multiple pathways involved in cell proliferation
(21). However, the exact
mechanism by which lysosomal function contributes to the
proliferation of liver cancer cells remains unclear.
In the present study, the role of ER-α36 in
regulating the malignant proliferation of liver cancer cells was
investigated in vivo and in vitro. In addition, the
role of ER-α36 in autophagy and lysosomal distribution was
examined.
Materials and methods
Chemicals and antibodies
17β-estradiol (E2) was purchased from Sigma-Aldrich;
Merck KGaA. The ER-α36 antibody was kindly provided by Dr Zhaoyi
Wang (Shenogen Pharma Group). Antibodies against p62 (also known as
sequestosome 1; cat. no. 18420-1-AP), galectin-3 (Gal-3; cat. no.
60207-1-Ig), lysosome-associated membrane protein 1 (LAMP1; cat.
no. 21997-1-AP), AKT (cat. no. 10176-2-AP), phosphorylated-(p-)AKT
(cat. no. 66444-1-Ig) and β-actin (cat. no. 60008-1-IG) were
purchased from Proteintech Group, Inc. The microtubule-associated
protein 1 light chain 3b (LC3B) antibody (cat. no. A19665) was
purchased from ABclonal Biotech Co., Ltd. Antibodies for ubiquitin
(Ub; cat. no. AF0306) were purchased from Beyotime Biotech Inc. The
Ki67 antibody (cat. no. bs-2130R) was purchased from Biosynthesis
Biotechnology Co., Ltd. Chloroquine (CQ; cat. no. HY-17589A) and
the AKT inhibitor MK-2206 (cat. no. HY-108232) were obtained from
MedChemExpress.
Cell culture, stably transfected cell
line preparation and cell treatment
The HepG2 (cat. no. STCC10114) and Huh7 (cat. no.
STCC10102) human liver cancer cell lines, which endogenously
express ER-α36, were obtained from Shenogen Pharma Group (7). The authenticity of both cell lines
was confirmed by STR analysis. The cells were maintained in DMEM
(Thermo Fisher Scientific, Inc.) with 10% FBS; Zhejiang Tianhang
Biotechnology Co., Ltd.) at 37°C in a humidified incubator with 5%
CO2.
Stable ER-α36 knockdown cell lines and control cell
lines were established as previously described (10,24).
In brief, short hairpin RNA (shRNA) targeting human ER-α36 was
constructed using the pRNAT-U6.1/Neo vector (provided by Dr Zhaoyi
Wang) (25,26). The construct was validated by DNA
sequencing. The target sequence was as follows: shER-α36:
5′-GTTCAGTACCTATTGGCA-3′ (nucleotides 4,261-4,278; sequence ID:
BX640939.1). The empty vector and ER-α36 shRNA expression vector (2
µg/ml) were transfected into the liver cancer cells for 6 h at
37°C, using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) as the transfection reagent. The
transfected cells were selected with 600 µg/ml G418 (Beyotime
Institute of Biotechnology) for 3 weeks. After this, cells from ≥25
clones were pooled. The empty vector transfected cells were named
HepG2/Vector and Huh7/Vector, and those transfected with ER-α36
shRNA were named HepG2/Sh36 and Huh7/Sh36.
For treatment, the cells were maintained in phenol
red-free DMEM with 1% charcoal-stripped fetal calf serum (FCS;
Zhejiang Tianhang Biotechnology Co., Ltd.) for 48 h, and then
treated with MK-2206 (100 nM) for 6 h, followed by E2 (1 nM) for
either 30 min (detection of the phosphorylation levels of AKT) or
24 h (detection of LAMP1 expression) at 37°C in an atmosphere
containing 5% CO2 (10). An equal volume of alcohol was used
as the vehicle control. The cells were treated with CQ (20 µM) for
24 h at 37°C; the same volume of DMSO was used as the vehicle
control.
Western blot analysis
Cells and liver tissues were lysed with RIPA lysis
buffer (Beyotime Institute of Biotechnology). Protein
concentrations were measured using a BCA Protein Assay Kit. The
proteins (20 µg/lane) were separated by 10% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis, transferred to
polyvinylidene fluoride membranes. The membranes were blocked with
5% skim milk powder in Tris-buffered saline containing 0.1%
Tween-20 (TBST) for 1 h at room temperature. After blocking,
membranes were incubated overnight at 4°C with primary antibodies:
anti-ER-a36 (1:1,000), anti-LC3B (1:1,000), anti-p62 (1:1,000),
anti-Ub (1:1,000), anti-LAMP1 (1:1,000), anti-p-AKT (1:1,000),
anti-AKT (1:1,000) and anti-β-actin (1:2,000). After washing with
TBST, the membranes were incubated with HRP Conjugated AffiniPure
Goat Anti-rabbit/mouse IgG (H + L) secondary antibodies (cat. no.
BA1055/BA1051, 1:5,000, Wuhan Boster Biological Technology CO.,
LTD.) at room temperature for 1 h. Following incubation, membranes
were washed in TBST. The target proteins were detected using
enhanced chemiluminescence (ECL) detection reagent (BeyoECL Moon
kit; Beyotime Institute of Biotech Inc.). Signal intensities were
captured using ECL western-blotting analysis system (GE
Healthcare). And quantitative analysis was performed using ImageJ
version 1.53t software (National Institutes of Health).
Colony formation assay
After seeding (2×103 cells/well), cells
were treated with MK-2206 (100 nM) for 6 h alone or in combination
with E2 (1 nM) for 24 h and cultured for 2 weeks at 37°C. The cell
colonies were washed with PBS and fixed with 4% paraformaldehyde
for 15 min at 37°C. After washing with PBS, cells were stained with
1% crystal violet for 30 min at 37°C. Colonies containing >50
cells were counted. Colonies count was performed using ImageJ v
2.14.0 (FIJI) software (National Institutes of Health).
Immunofluorescence (IF) staining
After seeding (5×103 cells/well), the
cells were fixed in 4% paraformaldehyde for 15 min at room
temperature. The tumor tissue was fixed in 4% paraformaldehyde for
24 h at room temperature, dehydrated in graded ethanol, embedded in
paraffin, and sectioned into 4 µm slices. After paraffin was
removed with xylene at room temperature, the slides were gradually
hydrated using ethanol. The slides were incubated in 3% hydrogen
peroxide/methanol buffer to quench endogenous peroxidase activity.
Antigen retrieval was performed by immersing the slides in
ethylenediamine tetra acetic acid buffer (pH 8.0) and boiling for 5
min. Then, the samples were blocked with 5% BSA (BioFroxx) for 1 h
at room temperature, incubated with primary antibodies: anti-LC3B
(1:500), anti-p62 (1:500), anti-LAMP1 (1:500) and anti-Gal-3
(1:500) overnight at 4°C, followed by incubation with secondary
antibodies conjugated to fluorescein: ABflo®
488-conjugated Goat anti-mouse/rabbit IgG (H+L) (cat. no.
AS076/AS053, 1:200, ABclonal Biotech Co.) or ABflo®
594-conjugated Goat anti-mouse/rabbit IgG (H+L) (cat. no.
AS054/AS039, 1:200, ABclonal Biotech Co.) for 1 h at room
temperature. The nuclei were stained with Hoechst 33342 for 10 min
at room temperature. Images were captured using a Leica TCS SP8
confocal microscope (Leica Microsystems. Inc.), and image analysis
was performed using ImageJ v 2.14.0 (FIJI) software (National
Institutes of Health).
pmCherry-enhanced green fluorescence
protein (EGFP)-LC3 puncta assay
After seeding (1×104 cells/well), cells
were infected with the pmCherry-EGFP-LC3b adenovirus (MOI: 20;
Beyotime Biotech Inc.) for 12 h at 37 °C. Then, the infection
medium was replaced with fresh complete medium and cells were
cultured for 24 h at 37 °C. The nuclei were stained with Hoechst
33342 for 10 min at room temperature. Images were captured using a
Leica TCS SP8 confocal microscope (Leica Microsystems. Inc.), and
image analysis was performed using ImageJ v 2.14.0 (FIJI) software
(National Institutes of Health).
Lyso-Tracker Red staining
After seeding (5×103 cells/well), cells
were incubated in Lyso-Tracker Red (50 nM, Beyotime Institute of
Biotech Inc.) for 15 min at 37 °C. The nuclei were stained with
Hoechst 33342 for 10 min at room temperature, and images were
captured using an Olympus BX53 fluorescence microscope (Olympus
Co., Ltd).
Animal experiments
All experimental procedures were performed in
compliance with the National Institutes of Health guidelines for
the Care and Use of Laboratory Animals and approved by the Ethics
Committee of Jianghan University (Wuhan, China; approval no.
JHDXLL2024-086). A total of 10 male BALB/c-nu mice (5 weeks old,
16–18 g) were purchased from Sipeifu (Beijing) Animal Technology
Co., Ltd., and housed in specific pathogen-free conditions at a
temperature of 24±2°C and relative humidity of 60%. The mice were
exposed to a 12-h light/dark cycle, and were given free access to
mouse feed sterilized and water sterilized by autoclaving. The
HepG2/Sh36 and HepG2/Vector cells (5×106 cells/mouse)
were injected into the livers of BALB/c-nu mice (n=4/group) to
establish 28-day orthotopic liver xenograft tumor models. The mice
were monitored daily for food and water intake, weight, body
posture, behavior, distress and response to external stimuli.
Several humane endpoints were established, including a >20% loss
of body weight, severe dehydration, refusal of food, severe pain or
distress, or a moribund state. However, no mice were humanely
sacrificed or found dead prior to the designated end of the study.
After 28 days, the mice were anesthetized with an intraperitoneal
injection of pentobarbital sodium at a dose of 50 mg/kg body weight
and sacrificed by cervical dislocation. Death was confirmed when no
breathing or heartbeat was detected for >5 min. The livers were
then harvested from the mice for further investigation.
H&E Staining
The tumor tissue was fixed in 4% paraformaldehyde
for 24 h at room temperature, dehydrated in graded ethanol,
embedded in paraffin, and sectioned into 4 µm slices. After
paraffin was removed with xylene at room temperature, the slides
were gradually hydrated using ethanol. The slides were dyed with
hematoxylin for 30 sec at room temperature and eosin for 1 min at
room temperature. Images were captured using a light
microscope.
Immunohistochemistry (IHC) assay
The tumor tissue was fixed in 4% paraformaldehyde
for 24 h at room temperature, dehydrated in graded ethanol,
embedded in paraffin, and sectioned into 4 µm slices. After
paraffin was removed with xylene at room temperature, the slides
were gradually hydrated using ethanol. The slides were incubated in
3% hydrogen peroxide/methanol buffer to quench endogenous
peroxidase activity. Antigen retrieval was performed by immersing
the slides in ethylenediamine tetra acetic acid buffer (pH 8.0) and
boiling for 5 min. The slides were blocked with 5% BSA (BioFroxx)
for 1 h at room temperature, incubated with primary antibodies:
anti-LC3B (1:200), anti-p62 (1:200), anti-LAMP1 (1:200) and
anti-Ki67 (1:200) overnight at 4°C, followed by incubation with
secondary antibodies (goat anti-rabbit/mouse HRP-labeled polymer;
cat. no. PV-9000, Beijing Zhong Shan-Golden Bridge Biological
Technology CO., Ltd.) for 1 h at 37°C. The slides were stained with
3,3′-Diaminobenzidine tetrahydrochloride (Beijing Zhong Shan
-Golden Bridge Biological Technology CO., Ltd.) and counterstained
with hematoxylin for 30 s at room temperature to visualize the cell
nuclei. Then, the slides were photographed by a light microscope.
Quantification of the positive staining was performed by measuring
the integral optical density using ImageJ v 2.14.0 (FIJI) software
(National Institutes of Health), with normalization to the stained
area.
Statistical analysis
Data are presented as the mean ± standard error of
mean of at least three independent experiments. The data were
analyzed using GraphPad Prism 8.0 software (Dotmatics) with one-way
analysis of variance (ANOVA) followed by Bonferroni's post hoc
tests. P<0.05 was considered to indicate a statistically
significant difference.
Results
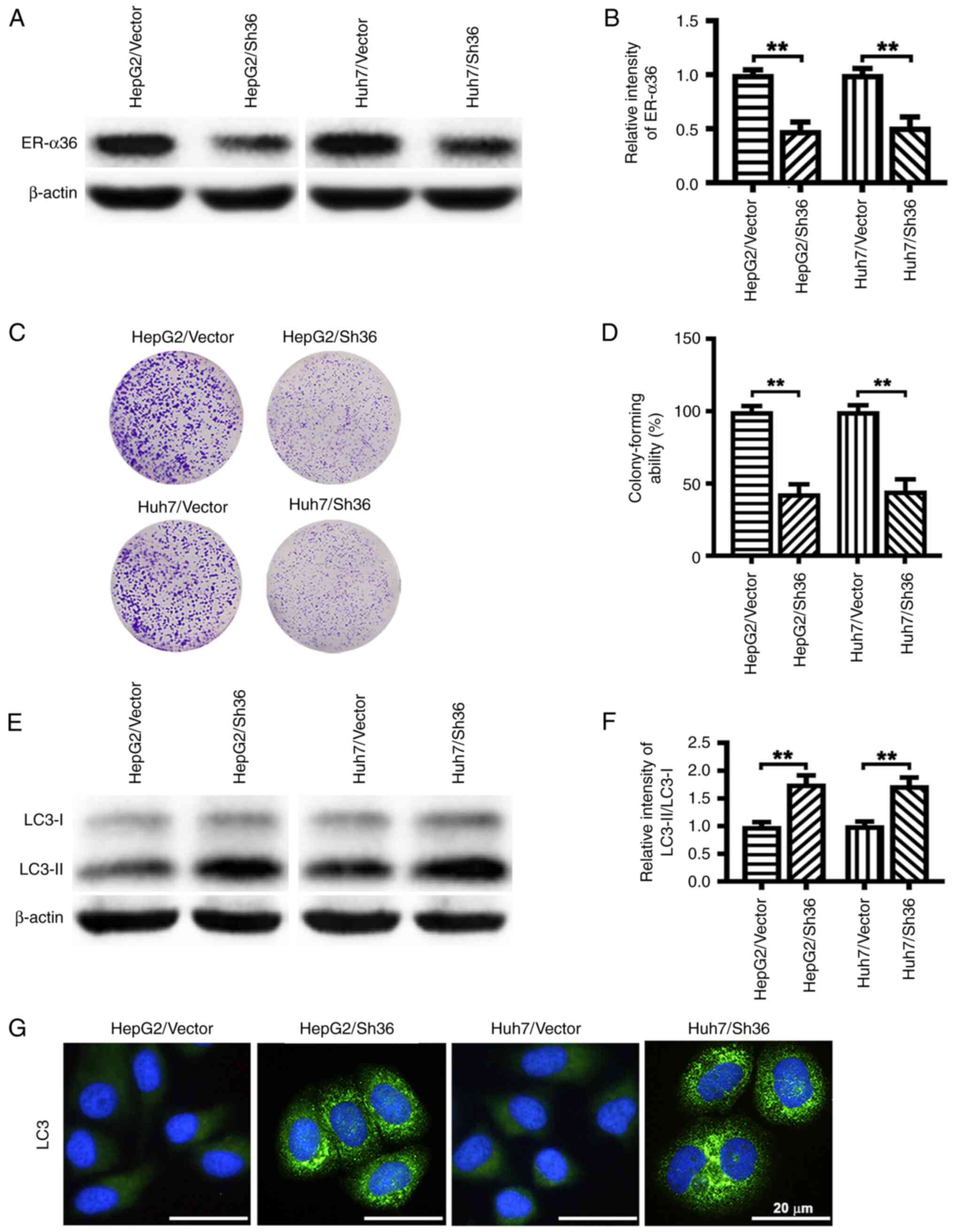
ER-α36 knockdown attenuates the
proliferation of liver cancer cells and promotes the accumulation
of autophagosomes
To test the role of ER-α36 in the proliferation and
autophagy of liver cancer cells, HepG2/Sh36 and Huh7/Sh36 cells
were prepared by transfection with an ER-α36 specific shRNA
expression vector, and HepG2/Vector and Huh7/Vector control cells
were prepared by transfection with an empty expression vector. The
successful knockdown of ER-α36 expression was verified by western
blotting (Fig. 1A and B). The
results of the colony formation assay revealed that ER-α36
knockdown attenuated the colony-forming ability of the liver cancer
cells (Fig. 1C and D). The
knockdown of ER-α36 expression also increased the LC3-II/LC3-I
ratio as revealed by western blot analysis (Fig. 1E and F), and increased the number
of LC3-II puncta as shown by IF staining (Fig. 1G). These findings suggest that
ER-α36 knockdown promoted autophagosome formation in the liver
cancer cells.
ER-α36 knockdown blocks autophagic
flux and autophagic degradation
The effect of ER-α36 knockdown on autophagic flux
was assessed. Western blotting revealed that the expression of p62,
a marker of autophagic flux, was increased in the HepG2/Sh36 and
Huh7/Sh36 cells compared with that in the respective empty
vector-transfected cells, suggesting that ER-α36 downregulation
impaired autophagic flux (Fig. 2A and
B). The transfected liver cancer cells were co-transfected with
pmCherry-EGFP-LC3b to monitor autophagy. In this system,
autophagosomes appear as yellow puncta, due to a combination of GFP
and mCherry fluorescence, whereas autolysosomes, formed by
autophagosome-lysosomal fusion, are identified by mCherry red
fluorescence only. This occurs because when autophagosomes fuse
with lysosomes, GFP is quenched by the acidic environment, whereas
mCherry remains fluorescent. The liver cancer cells with ER-α36
knockdown exhibited significantly increased numbers of yellow
puncta, indicative of autophagosomes, with occasional red dots
representing autolysosomes, suggesting that ER-α36 knockdown
blocked autophagic flux, thereby resulting in the accumulation of
autophagosomes (Fig. 2C and D).
Additionally, when the autophagosome-lysosome fusion inhibitor CQ
was used to treat the cells, western blotting revealed that ER-α36
knockdown increased the LC3-II/LC3-I ratio in the absence of CQ,
but caused no further increase in the presence of CQ. This suggests
that ER-α36 knockdown increased the LC3-II/LC3-I ratio due to
impaired autophagosome degradation (Fig. 2E and F). Furthermore, ER-α36
downregulation led to the accumulation of ubiquitinated proteins,
suggesting that substrate degradation was inhibited (Fig. 2G and H). Taken together, these
findings indicate that ER-α36 knockdown impaired autophagic flux by
inhibiting the degradation of autophagosomes.
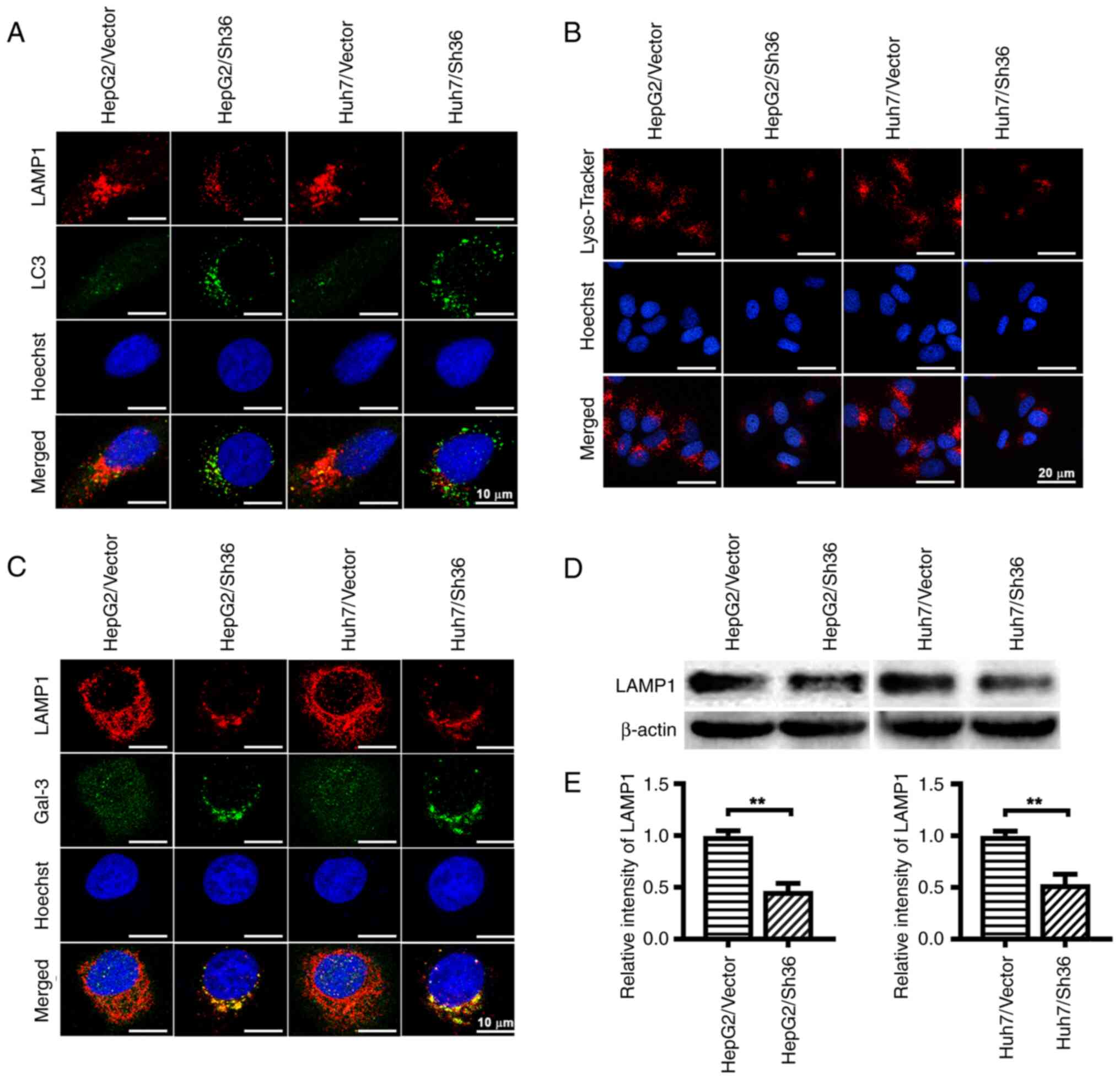
ER-α36 knockdown induces lysosomal
damage and LMP
To determine whether the autophagic degradation
defect induced by ER-α36 knockdown was due to impaired
autophagosome-lysosome fusion or defective lysosomal degradation,
IF staining of LC3 (green) and LAMP1 (red) was performed to assess
their co-localization (yellow) in liver cancer cells with different
levels of ER-α36 expression. Fewer red LAMP1 puncta and yellow
co-localization signals were observed in the HepG2/Sh36 and
Huh7/Sh36 cells compared with those in the corresponding vector
control cells (Fig. 3A),
suggesting that ER-α36 knockdown blocked autophagosome-lysosome
fusion, thereby contributing to the impairment of autophagic flux
in liver cancer cells.
Since lysosomal dysfunction can lead to autophagic
flux blockage and the accumulation of lysosomal substrates
(27), lysosomal integrity was
examined. A significant reduction in the Lyso-Tracker-staining of
lysosomes was observed in the HepG2/Sh36 and Huh7/Sh36 cells
compared with those in the respective vector control cells
(Fig. 3B), suggesting that ER-α36
downregulation compromised the acidic environment within lysosomes.
An increase in the pH of the lysosomal lumen is a known indicator
of LMP (28). As shown in Fig. 3C, IF staining revealed prominent
Gal-3 puncta in the HepG2/Sh36 and Huh7/Sh36 cells, whereas they
were only faintly visible in the vector control cells. In addition,
western blotting revealed that LAMP1 expression in the HepG2/Sh36
and Huh7/Sh36 cells was decreased compared with that in the vector
control cells (Fig. 3D and E).
These data indicate that ER-α36 knockdown disrupted lysosomal
integrity.
ER-α36 knockdown suppresses the
malignant proliferation of liver cancer cells and induces LMP
To investigate the function of ER-α36 in the
malignant proliferation of liver cancer cells in vivo, nude
mice were intrahepatically injected with HepG2/Vector cells or
HepG2/Sh36 cells to establish an orthotopic liver xenograft tumor
model for 28 days. While tumors were formed in the livers of all
mice, the tumor volumes formed by the HepG2/Sh36 cells were
significantly smaller than those formed by the HepG2/Vector cells.
The liver weight, body weight and ratio of liver weight to body
weight exhibited no significant differences between the two groups
(Fig. 4A and B). Furthermore, a
reduced number of pathological karyomitoses and Ki67-positive cells
were detected in the tumors formed from the HepG2/Sh36 cells by
H&E and IHC, suggesting that ER-α36 is involved in the
malignant proliferation of HepG2 cells (Fig. 4C and D). Western blot analysis and
IHC also revealed an increased LC3-II/I ratio and p62 expression
level, and decreased LAMP1 expression in the tumor tissues formed
by the HepG2/Sh36 cells compared with those in the tumors formed by
the HepG2/Vector cells (Fig. 4D, F and
G). In addition, IF staining demonstrated an increase in the
number of Gal-3 puncta in the tumors formed by the HepG2/Sh36 cells
compared with those formed by the HepG2/Vector cells (Fig. 4E). These results indicate that
ER-α36 knockdown suppressed the tumor proliferation of HepG2 cells
and induced LMP in vivo.
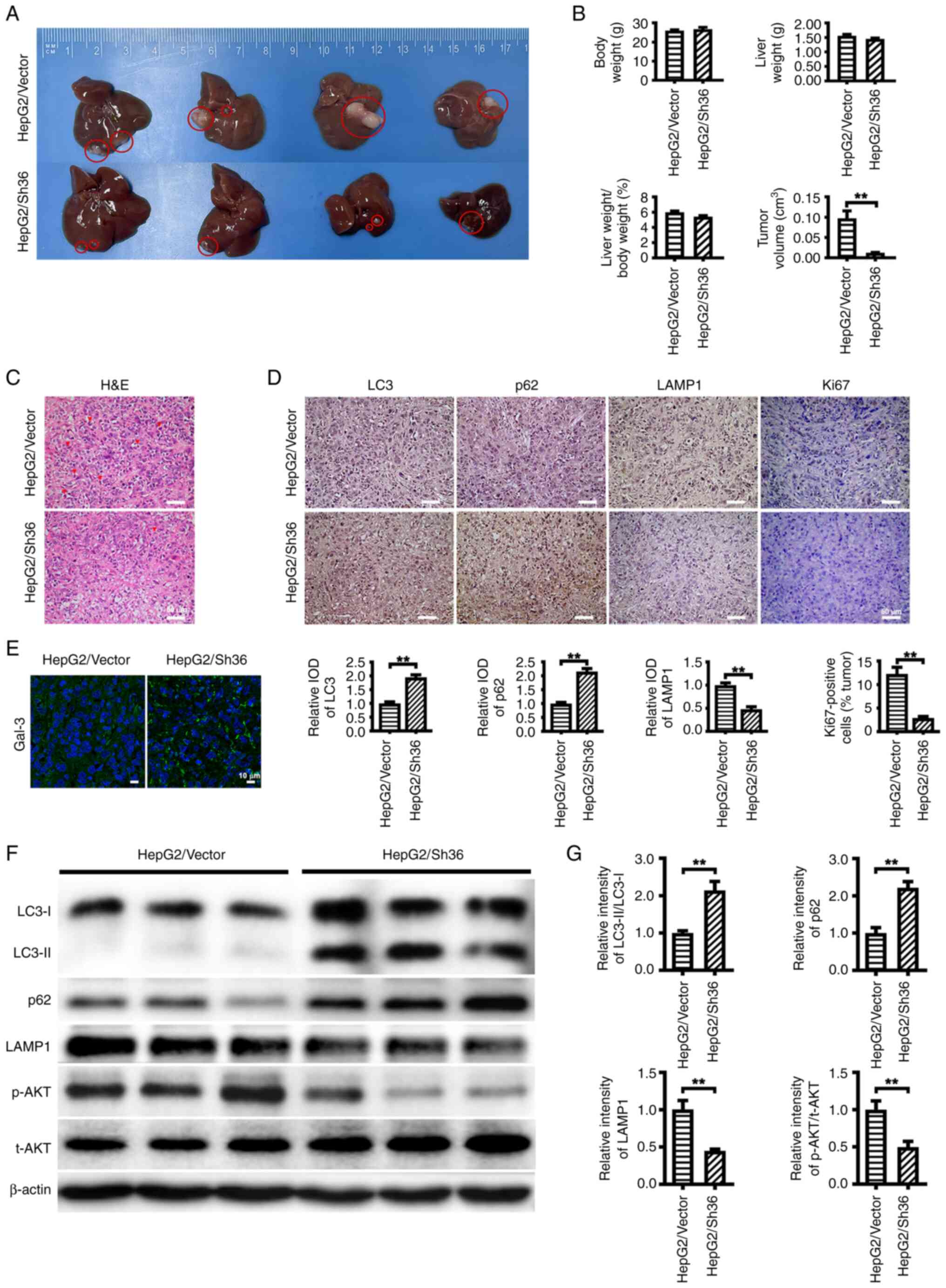 | Figure 4.ER-α36 knockdown suppresses the
malignant proliferation of liver cancer cells and induces lysosomal
membrane permeabilization. (A) Representative images of liver
tumors from nude mice intrahepatically injected with HepG2 cells
expressing different ER-α36 levels, harvested at 28 days
post-injection. (B) Quantitative analysis of liver tumor volume,
liver weight, body weight and the ratio of liver weight to body
weight. (C) Representative H&E staining images showing
pathological karyomitosis changes. Scale bar, 50 µm. (D)
Immunohistochemical staining of LC3, p62, LAMP1 and Ki67 with
corresponding IOD values. Scale bar, 50 µm. (E) Immunofluorescence
staining of Gal-3 in the tumors formed from the transfected HepG2
cells. Scale bar, 10 µm. (F) Western blotting results showing the
levels of LC3-II and LC3-II, p62, LAMP1, p-AKT and t-AKT in the
tumors, and (G) quantitative analyses of the LC3-II/LC3-I and
p-AKT/t-AKT ratios, and p62 and LAMP1 expression levels. β-actin
was used as the internal loading control. Data are presented as the
mean ± SEM. **P<0.01. ER, estrogen receptor; H&E,
hematoxylin and eosin; LC3, microtubule-associated protein 1 light
chain 3; LAMP1, lysosome-associated membrane protein 1; Gal-3,
galectin-3; IOD, integrated optical density; p-, phosphorylated;
t-, total; Sh36, transfected with ER-α36 specific short hairpin RNA
expression vector; Vector, transfected with empty vector. |
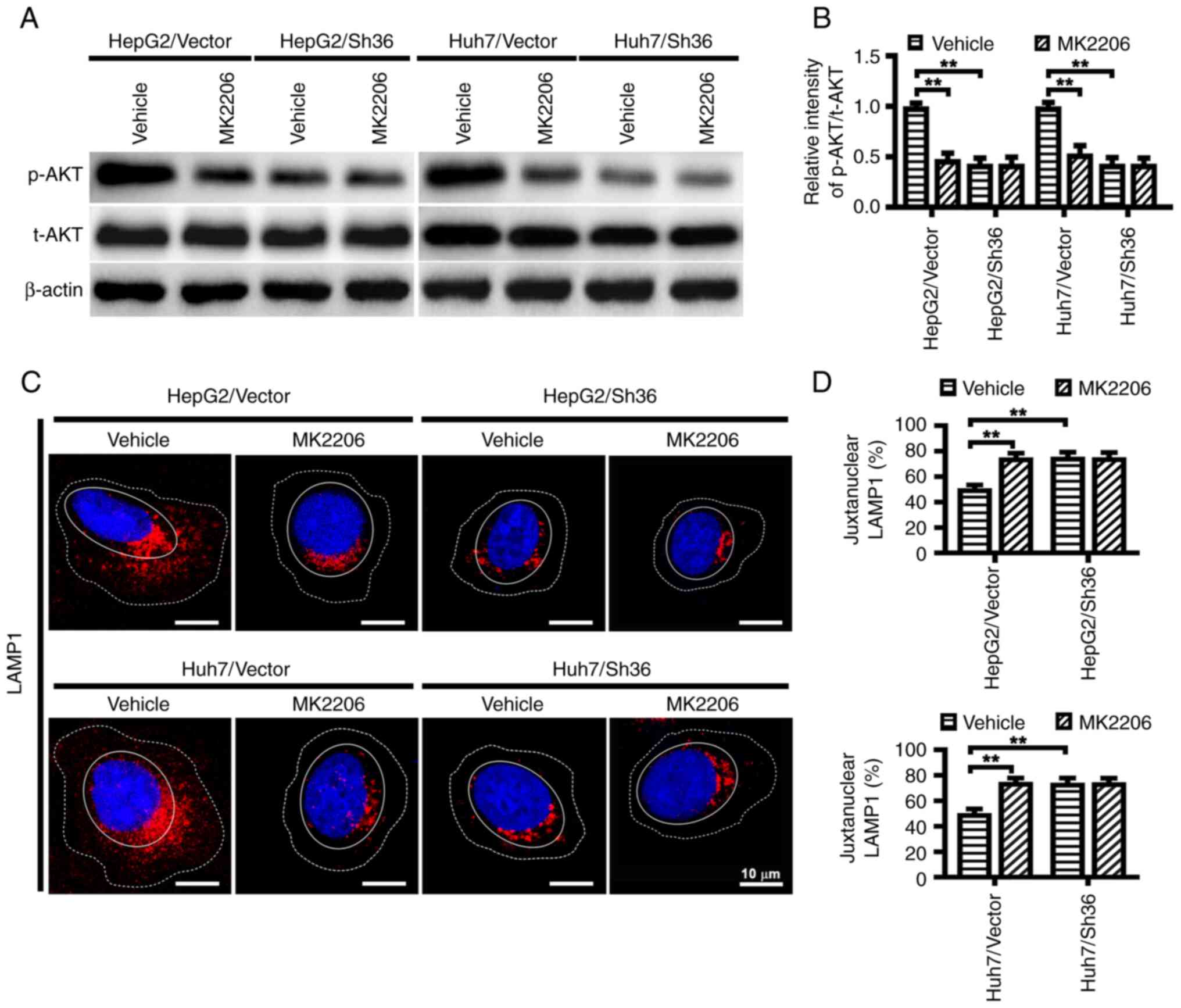
ER-α36 knockdown decreases AKT
phosphorylation and influences lysosomal localization in liver
cancer cells
AKT signaling, a typical event in ER-α36-mediated
rapid estrogenic signaling, has been reported to influence
lysosomal localization (10,29).
To determine whether ER-α36 mediated AKT activation is involved in
the regulation of lysosomal localization in liver cancer cells, the
phosphorylation levels of AKT in the HepG2 and Huh7 cells with and
without ER-α36 knockdown and in the tumors formed from them were
examined. Decreased AKT phosphorylation at Ser 473 was observed in
the liver cancer cells with ER-α36 knockdown and in the tumors
formed from them compared with that in the corresponding vector
controls (Figs. 4F and G, 5A and B). In addition, when compared with
the HepG2/Vector and Huh7/Vector cells, an accumulation of LAMP1
was observed at the juxtanuclear region in the HepG2/Sh36 and
Huh7/Sh36 cells (Fig. 5C and D),
consistent with earlier reports that the AKT inhibitor MK-2206
promotes the juxtanuclear clustering of lysosomes (29). However, in the present study,
MK-2206 did not further reduce AKT phosphorylation or influence
lysosome localization in HepG2/Sh36 and Huh7/Sih6 cells (Fig. 5C and D). These results indicate
that ER-α36 expression knockdown attenuated AKT phosphorylation,
which then led to the juxtanuclear clustering of lysosomes.
AKT is involved in the ER-α36
knockdown-induced changes in lysosomal localization and LMP in
liver cancer cells
To examine the effect of AKT on ER-α36 regulated LMP
and lysosomal localization, which influence the proliferation of
liver cancer cells, the transfected liver cancer cells were treated
with E2 and/or MK-2206. It was observed that treatment of the
HepG2/Vector and Huh7/Vector cells with E2 upregulated AKT
phosphorylation at Ser 473 and increased the colony-forming ability
of the cells, and these effects were abrogated by MK-2206 (Fig. 6A, B, E and F). This is consistent
with the previous finding that AKT signaling is involved in
ER-α36-mediated rapid estrogenic signaling and stimulation of cell
proliferation (10). The E2
treatment also upregulated LAMP1 expression (Fig. 6C, D and H), increased Lyso-Tracker
fluorescence intensity (Fig. 6G)
and re-localized lysosomes to the cell periphery in the
HepG2/Vector and Huh7/Vector cells (Fig. 6G), while MK-2206 abrogated all
these E2-induced effects. In addition, a diffuse distribution of
Gal-3 in the cytoplasm and nucleus was observed in E2-treated
HepG2/Vector and Huh7/Vector cells, while a punctate pattern was
observed in the HepG2/Vector and Huh7/Vector cells treated with the
combination of E2 and MK-2206 (Fig.
6H). By contrast, these phenomena were almost undetectable in
the HepG2/Sh36 and Huh7/Sh36 cells. These findings strongly suggest
that AKT is involved in the lysosomal localization and LMP changes
associated with ER-α36 knockdown.
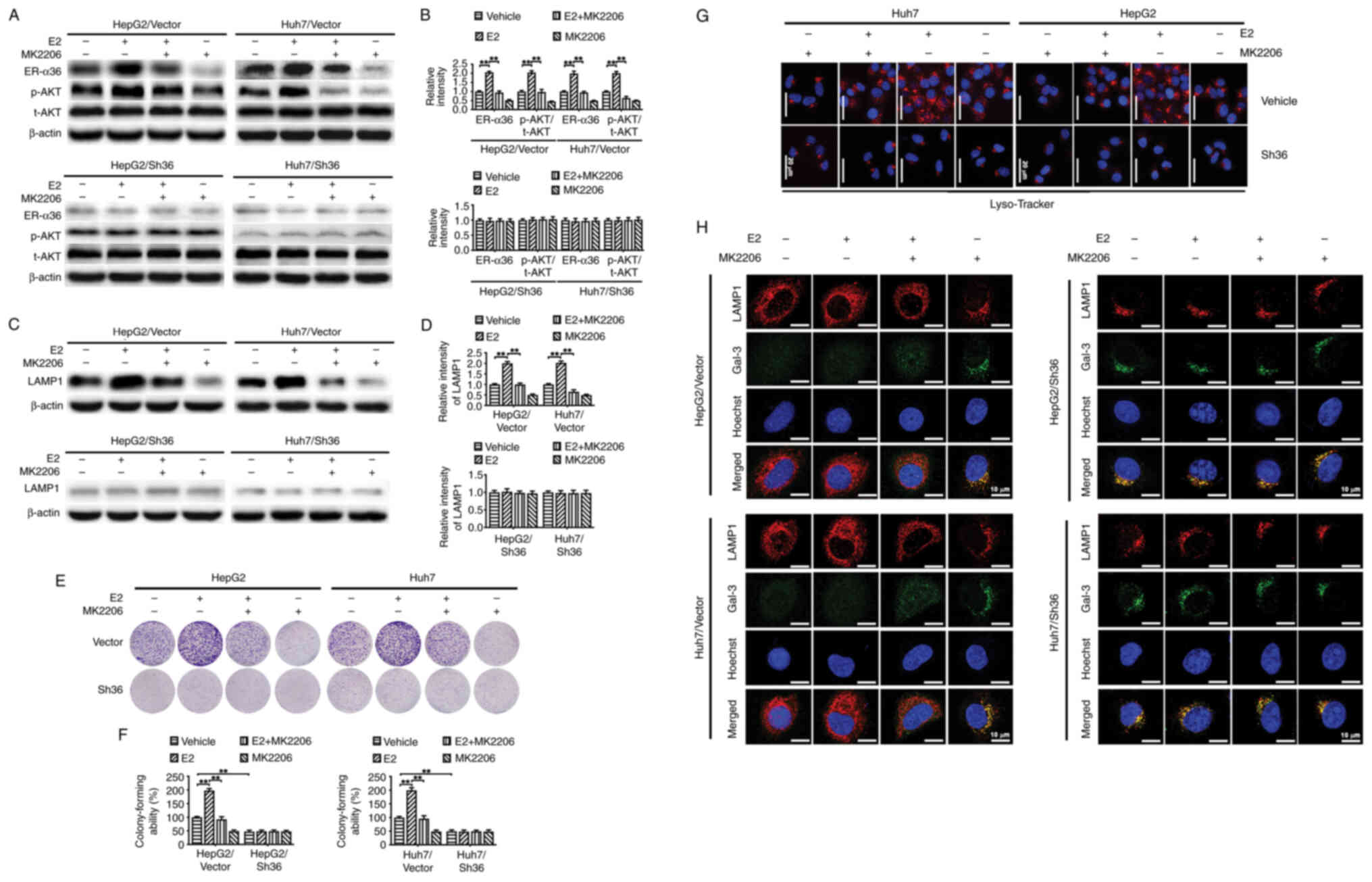 | Figure 6.AKT is involved in the lysosomal
localization and LMP induced by ER-α36 knockdown. Transfected liver
cancer cells with different levels of ER-α36 expression were
treated with MK-2206 (100 nM) for 6 h, followed by E2 (1 nM) for 30
min, and the same volume of alcohol was used as a vehicle control.
(A) Representative western blots of ER-α36, p-AKT and AKT, and (B)
quantitative analysis of ER-α36 expression and the p-AKT/t-AKT
ratio. In subsequent assays, the transfected liver cancer cells
were treated with MK-2206 (100 nM) for 6 h, followed by E2 (1 nM)
for 24 h. (C) Representative western blots of LAMP1 and (D)
quantitative analysis of LAMP1 expression. (E) Representative
images of colony formation by the liver cancer cells and (F)
quantitative analysis of colony forming ability. Data are presented
as the mean ± SEM. **P<0.01. (G) Fluorescence microscopy images
of the cells following staining with Lyso-Tracker Red. Scale bar,
20 µm. (H) Co-localization of Gal-3 (green) and LAMP1 (red) in the
cells as revealed by immunofluorescence staining. Scale bar, 10 µm.
ER, estrogen receptor; E2, 17β-estradiol; Gal-3, galectin-3; p-,
phosphorylated; t-, total; LAMP1, lysosome-associated membrane
protein 1; Sh36, transfected with ER-α36 specific short hairpin RNA
expression vector; Vector, transfected with empty vector. |
Discussion
The present study provides evidence that the
knockdown of ER-α36 impaired autophagic flux and inhibited the
malignant proliferation of HepG2 and Huh7 human liver cancer cells
in vivo and in vitro. ER-α36 knockdown also induced
LMP as well as lysosomal dysfunction, including pH elevation,
impaired protein degradation and the juxtanuclear clustering of
lysosomes. These findings suggest that the maintenance of normal
lysosomal function is one of the key roles of ER-α36 in liver
cancer cells.
Previous studies have suggested that ER-α36 is
involved in liver tumorigenesis (10), that ER-α36 mRNA levels gradually
increase from normal liver tissue to cirrhotic liver and liver
carcinoma tissues (7), and that
upregulated ER-α36 expression is associated with primary liver
cancer (9). In addition, the
knockdown of ER-α36 expression has been shown to attenuate the
metastasis of HepG2 and Huh7 cells by downregulating
epithelial-mesenchymal transition and the Src signaling pathway
(24). Furthermore, ER-α36 has
been demonstrated to be a prognostic factor in breast cancer
(30). However, the molecular
mechanism by which ER-α36 promotes cancer cell proliferation is
unclear.
Impaired autophagy has been linked to various
pathological conditions in humans, including liver dysfunction and
carcinogenesis (31). In
glioblastoma cells, ER-α36 has been reported to counteract
tamoxifen-mediated cell apoptosis by promoting autophagy via
inactivation of the AKT/mammalian target of rapamycin (mTOR)
signaling pathway (12).
Furthermore, ER-α36 promotes autophagy during the development of
acquired tamoxifen resistance (12,32),
and its expression level has been shown to correlate with that of
p62 in a three-dimensional culture model (12).
The present study also demonstrated that ER-α36
knockdown attenuated the malignant proliferation of liver cancer
cells and impaired autophagic flux. Specifically, ER-α36 knockdown
increased the LC3-II/LC3-I ratio and p62 expression levels in HepG2
and Huh7 cells, as well as in the tumor tissues formed by these
cells in mice, compared with those in the corresponding controls.
Furthermore, the use of a double-tagged LC3 construct
(pmCherry-EGFP-LC3b) revealed that autophagic flux in
ER-α36-knockdown cells was impaired compared with that in the
control cells. To further elucidate the dynamics of autophagy, CQ
was used as a late-stage autophagy inhibitor to determine whether
ER-α36 knockdown promotes or inhibits autophagy. CQ inhibits
autophagosome degradation; therefore, when CQ treatment is
administered, any observed changes in the LC3-II/LC3-I ratio
represent autophagosome synthesis, whereas in the absence of CQ,
they represent the combined effects of both autophagosome synthesis
and degradation (33–36). In the present study, ER-α36
knockdown increased the LC3-II/LC3-I ratio in the absence of CQ,
but not its presence. This suggests that ER-α36 knockdown induces
the aggregation of autophagosomes due to impaired autophagosome
degradation, indicating a blockade of autophagic flux.
The colocalization of LC3 with LAMP1 in the HepG2
and Huh7 cells was observed to decrease following ER-α36 knockdown
in the present study, suggesting that ER-α36 downregulation
disrupts autophagosome-lysosome fusion. Intact lysosomes are highly
acidic, and this acidic environment is maintained by the integrity
of the lysosomal membrane (37).
Loss of lysosomal integrity results in elevated lysosomal pH, which
can impair autophagic degradation and block autophagic flux
(13,38–40).
In the present study, decreased LAMP1 expression was observed
following ER-α36 knockdown, suggesting a potential loss of
lysosomal membrane integrity that may affect lysosomal
function.
Gal-3 staining, a well-established indicator of
lysosomal injury, is commonly used to assess lysosomal integrity
(41). In the present study, the
accumulation of Gal-3-positive puncta was observed in the liver
cancer cells with ER-α36 knockdown in vivo and in
vitro, further supporting a role of ER-α36 in the regulation of
LMP. Together, these findings suggest that ER-α36 downregulation
impairs autophagic flux and inhibits liver cancer cell
proliferation.
The positioning of lysosomes within the cytoplasm
has been shown to influence the rate of autophagosome-lysosome
fusion, and thus regulate autophagic flux (42). For example, the knockout of
biogenesis of lysosome-related organelles complex-1, which
facilitates the kinesin-dependent movement of lysosomes toward the
cell periphery, results in the juxtanuclear clustering of lysosomes
and impairs both their encounter and fusion with autophagosomes
(43). The reduction in encounters
occurs due to the inability of lysosomes to move toward the
peripheral cytoplasm, where autophagosomes are predominantly formed
(44). Notably, the peripheral
localization of lysosomes has been associated with pathological
processes, including cancer cell growth, invasion and metastasis
(45), whereas the accumulation of
lysosomes at the juxtanuclear region is associated with reduced
proteinase secretion and a reduction in the invasiveness of tumor
cells (46). In the present study,
ER-α36 knockdown led to the juxtanuclear clustering of lysosomes,
which may impair the fusion of autophagosomes and lysosomes,
thereby inhibiting autophagic flux.
The lysosome functions as a central hub for
signaling networks (45). AKT
signaling has been shown to regulate lysosomal positioning
(29,47). Juxtanuclear clustering of lysosomes
has been shown to delay the activation of mTOR complex 1 (mTORC1),
mTORC2 and AKT following serum replenishment (48). By contrast, the peripheral
clustering of lysosomal mTORC1 brings it closer to activated AKT,
which predominantly localizes near to the plasma membrane during
the recovery phase following serum starvation (42). Notably, ER-α36-mediated rapid
estrogen signaling involves the AKT signaling pathway, which has
been implicated in liver tumorigenesis (10). In the present study, it was found
that ER-α36 downregulation induced a reduction in AKT
phosphorylation and the accumulation of LAMP1 in the juxtanuclear
region. In addition, the inhibition of AKT phosphorylation was
found to block ER-α36-mediated effects, including its promotion of
cell proliferation and induction of lysosomal dispersal to the cell
periphery. A previous study reported that decreased AKT expression
is associated with elevated cytosolic Ca2+ levels, which
induce LMP and lysosomal damage (49). In the present study, the role of
ER-α36-mediated rapid estrogen signaling in LMP was investigated,
and the results revealed that ER-α36 downregulation reduced AKT
phosphorylation and LMP in vivo and in vitro.
Treatment with E2 upregulated ER-α36 and LAMP1 expression,
activated AKT and increased the Lyso-Tracker staining of lysosomes
in empty vector-transfected HepG2 and Huh7 cells; all these effects
were attenuated by MK-2206. However, these phenomena were almost
undetectable in the HepG2 and Huh7 cells with ER-α36 knockdown,
suggesting AKT is involved in the lysosomal localization and LMP
changes associated with ER-α36 knockdown.
However, some important issues remain to be
explored. LMP often results in the translocation of lysosomal
cathepsins from the lysosomal lumen to the cytoplasm, a process
implicated in the regulation of various cell death pathways
(16). In the present study,
Lyso-Tracker Red staining and the assessment of lysosomal integrity
via LAMP1 and Gal-3 puncta assays provided evidence of LMP.
However, additional assays such as cathepsin release or dextran
leakage assays are necessary for further confirmation. Whether all
these mechanisms are involved in the ER-α36-induced proliferation
of liver cancer cells remains to be established.
In conclusion, the present study revealed that
ER-α36 knockdown induced LMP, disrupted lysosomal membrane
integrity, altered lysosomal localization and impeded autophagic
activity via the inactivation of AKT. These effects may all
contribute to the inhibition of liver cancer cell proliferation
in vivo and in vitro. Thus, lysosomal dysfunction may
be an underlying mechanism by which ER-α36 promotes liver cancer
development.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Natural Science
Foundation of China (grant no. 81872040) and the Research Fund of
Jianghan University (grant no. 2021jczx-002).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HuH, XW and ZWe were responsible for methodology,
investigation, managing, storing and maintaining data to ensure its
reliability and availability, analysis and writing the original
draft of the manuscript. AW, XF, HaH and ZWu were responsible for
the methodology, investigation and the acquisition of data. XS and
BS were responsible for analysis and interpretation of data,
supervision, reviewing and editing the manuscript. QC, XH and HZ
were responsible for the methodology, resources and the acquisition
of data. YL and ZF were responsible for conceptualization,
reviewing and editing the manuscript, supervision and funding
acquisition. ZF and HuH confirm the authenticity of all the raw
data. All authors have read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
Al experimental procedures were performed in
compliance with the National Institutes of Health guidelines for
the Care and Use of Laboratory Animals and were approved by the
Ethics Committee of Jianghan University (Wuhan, China; approval no.
JHDXLL2024-086).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
2
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49.
2024.PubMed/NCBI
|
|
3
|
Han B, Zheng R, Zeng H, Wang S, Sun K,
Chen R, Li L, Wei W and He J: Cancer incidence and mortality in
China, 2022. J Natl Cancer Cent. 4:47–53. 2024.PubMed/NCBI
|
|
4
|
Kasarinaite A, Sinton M, Saunders PTK and
Hay DC: The influence of sex hormones in liver function and
disease. Cells. 12:16042023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Iavarone M, Lampertico P, Seletti C,
Donato MF, Ronchi G, del Ninno E and Colombo M: The clinical and
pathogenetic significance of estrogen receptor-beta expression in
chronic liver diseases and liver carcinoma. Cancer. 98:529–534.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tu BB, Lin SL, Yan LY, Wang ZY, Sun QY and
Qiao J: ER-α36, a novel variant of estrogen receptor α, is involved
in EGFR-related carcinogenesis in endometrial cancer. Am J Obstet
Gynecol. 205:227.e221–226. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miceli V, Cocciadiferro L, Fregapane M,
Zarcone M, Montalto G, Polito LM, Agostara B, Granata OM and
Carruba G: Expression of wild-type and variant estrogen receptor
alpha in liver carcinogenesis and tumor progression. OMICS.
15:313–317. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Su X, Xu X, Li G, Lin B, Cao J and Teng L:
ER-α36: A novel biomarker and potential therapeutic target in
breast cancer. Onco Targets Ther. 7:1525–1533. 2014.PubMed/NCBI
|
|
9
|
Zhang J, Ren J, Wei J, Chong CC, Yang D,
He Y, Chen GG and Lai PB: Alternative splicing of estrogen receptor
alpha in hepatocellular carcinoma. BMC Cancer. 16:9262016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
You H, Meng K and Wang ZY: The ER-α36/EGFR
signaling loop promotes growth of hepatocellular carcinoma cells.
Steroids. 134:78–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li G, Zhang J, Jin K, He K, Zheng Y, Xu X,
Wang H, Wang H, Li Z, Yu X, et al: Estrogen receptor-α36 is
involved in development of acquired tamoxifen resistance via
regulating the growth status switch in breast cancer cells. Mol
Oncol. 7:611–624. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qu C, Ma J, Zhang Y, Han C, Huang L, Shen
L, Li H, Wang X, Liu J, Zou W, et al: Estrogen receptor variant
ER-α36 promotes tamoxifen agonist activity in glioblastoma cells.
Cancer Sci. 110:221–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Qi Z, Yang W, Xue B, Chen T, Lu X, Zhang
R, Li Z, Zhao X, Zhang Y, Han F, et al: ROS-mediated lysosomal
membrane permeabilization and autophagy inhibition regulate
bleomycin-induced cellular senescence. Autophagy. 20:2000–2016.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamamoto H, Zhang S and Mizushima N:
Autophagy genes in biology and disease. Nat Rev Genet. 24:382–400.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gros F and Muller S: The role of lysosomes
in metabolic and autoimmune diseases. Nat Rev Nephrol. 19:366–383.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xiang L, Lou J, Zhao J, Geng Y, Zhang J,
Wu Y, Zhao Y, Tao Z, Li Y, Qi J, et al: Underlying mechanism of
lysosomal membrane permeabilization in CNS injury: A literature
review. Mol Neurobiol. 62:626–642. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nie B, Liu X, Lei C, Liang X, Zhang D and
Zhang J: The role of lysosomes in airborne particulate
matter-induced pulmonary toxicity. Sci Total Environ.
919:1708932024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eriksson I and Öllinger K: Lysosomes in
cancer-at the crossroad of good and evil. Cells. 13:4592024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Li Z, Zhang Y, Lei J and Wu Y: Autophagy
in oral cancer: Promises and challenges (Review). Int J Mol Med.
54:1162024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ballabio A and Bonifacino JS: Lysosomes as
dynamic regulators of cell and organismal homeostasis. Nat Rev Mol
Cell Biol. 21:101–118. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cao M, Luo X, Wu K and He X: Targeting
lysosomes in human disease: From basic research to clinical
applications. Signal Transduct Target Ther. 6:3792021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Davidson SM and Vander Heiden MG: Critical
functions of the lysosome in cancer biology. Annu Rev Pharmacol
Toxicol. 57:481–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weber RA, Yen FS, Nicholson SPV, Alwaseem
H, Bayraktar EC, Alam M, Timson RC, La K, Abu-Remaileh M, Molina H
and Birsoy K: Maintaining iron homeostasis is the key role of
lysosomal acidity for cell proliferation. Mol Cell.
77:645–655.e647. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang R, Chen J, Yu H, Wei Z, Ma M, Ye X,
Wu W, Chen H and Fu Z: Downregulation of estrogen receptor-α36
expression attenuates metastasis of hepatocellular carcinoma cells.
Environ Toxicol. 37:1113–1123. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Deng H and Wang ZY: Estrogen
activation of the mitogen-activated protein kinase is mediated by
ER-α36 in ER-positive breast cancer cells. J Steroid Biochem Mol
Biol. 143:434–443. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang ZY and Yin L: Estrogen receptor
alpha-36 (ER-α36): A new player in human breast cancer. Mol Cell
Endocrinol. 418:193–206. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kendall RL and Holian A: The role of
lysosomal ion channels in lysosome dysfunction. Inhal Toxicol.
33:41–54. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chen Y, Zhu S, Liao T, Wang C, Han J, Yang
Z, Lu X, Hu Z, Hu J, Wang X, et al: The HN protein of Newcastle
disease virus induces cell apoptosis through the induction of
lysosomal membrane permeabilization. PLoS Pathog. 20:e10119812024.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu B, Liu DA, Guan L, Myint PK, Chin L,
Dang H, Xu Y, Ren J, Li T, Yu Z, et al: Stiff matrix induces
exosome secretion to promote tumour growth. Nat Cell Biol.
25:415–424. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li Q, Sun H, Zou J, Ge C, Yu K, Cao Y and
Hong Q: Increased expression of estrogen receptor α-36 by breast
cancer oncogene IKKε promotes growth of ER-negative breast cancer
cells. Cell Physiol Biochem. 31:833–841. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Pedro JM, Cadwell K, Cecconi F,
Choi AM, et al: Autophagy in major human diseases. EMBO J.
40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Koirala M and DiPaola M: Overcoming cancer
resistance: Strategies and modalities for effective treatment.
Biomedicines. 12:18012024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tanida I, Minematsu-Ikeguchi N, Ueno T and
Kominami E: Lysosomal turnover, but not a cellular level, of
endogenous LC3 is a marker for autophagy. Autophagy. 1:84–91. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ju JS, Varadhachary AS, Miller SE and
Weihl CC: Quantitation of ‘autophagic flux’ in mature skeletal
muscle. Autophagy. 6:929–935. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Rubinsztein DC, Cuervo AM, Ravikumar B,
Sarkar S, Korolchuk V, Kaushik S and Klionsky DJ: In search of an
‘autophagomometer’. Autophagy. 5:585–589. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ou M, Cho HY, Fu J, Thein TZ, Wang W,
Swenson SD, Minea RO, Stathopoulos A, Schönthal AH, Hofman FM, et
al: Inhibition of autophagy and induction of glioblastoma cell
death by NEO214, a perillyl alcohol-rolipram conjugate. Autophagy.
19:3169–3188. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nakamura S, Akayama S and Yoshimori T:
Autophagy-independent function of lipidated LC3 essential for TFEB
activation during the lysosomal damage responses. Autophagy.
17:581–583. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chan H, Li Q, Wang X, Liu WY, Hu W, Zeng
J, Xie C, Kwong TNY, Ho IHT, Liu X, et al: Vitamin D (3) and
carbamazepine protect against Clostridioides difficile infection in
mice by restoring macrophage lysosome acidification. Autophagy.
18:2050–2067. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gu H, Qiu H, Yang H, Deng Z, Zhang S, Du L
and He F: PRRSV utilizes MALT1-regulated autophagy flux to switch
virus spread and reserve. Autophagy. 20:2697–2718. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang J, Zeng W, Han Y, Lee WR, Liou J and
Jiang Y: Lysosomal LAMP proteins regulate lysosomal pH by direct
inhibition of the TMEM175 channel. Mol Cell. 83:2524–2539.e2527.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tanaka T, Warner BM, Michael DG, Nakamura
H, Odani T, Yin H, Atsumi T, Noguchi M and Chiorini JA: LAMP3
inhibits autophagy and contributes to cell death by lysosomal
membrane permeabilization. Autophagy. 18:1629–1647. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Korolchuk VI, Saiki S, Lichtenberg M,
Siddiqi FH, Roberts EA, Imarisio S, Jahreiss L, Sarkar S, Futter M,
Menzies FM, et al: Lysosomal positioning coordinates cellular
nutrient responses. Nat Cell Biol. 13:453–460. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jia R, Guardia CM, Pu J, Chen Y and
Bonifacino JS: BORC coordinates encounter and fusion of lysosomes
with autophagosomes. Autophagy. 13:1648–1663. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhao YG, Codogno P and Zhang H: Machinery,
regulation and pathophysiological implications of autophagosome
maturation. Nat Rev Mol Cell Biol. 22:733–750. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pu J, Schindler C, Jia R, Jarnik M,
Backlund P and Bonifacino JS: BORC, a multisubunit complex that
regulates lysosome positioning. Dev Cell. 33:176–188. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Steffan JJ, Dykes SS, Coleman DT, Adams
LK, Rogers D, Carroll JL, Williams BJ and Cardelli JA: Supporting a
role for the GTPase Rab7 in prostate cancer progression. PLoS One.
9:e878822014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Palma M, Riffo E, Farias A,
Coliboro-Dannich V, Espinoza-Francine L, Escalona E, Amigo R,
Gutiérrez JL, Pincheira R and Castro AF: NUAK1 coordinates growth
factor-dependent activation of mTORC2 and Akt signaling. Cell
Biosci. 13:2322023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Jia R and Bonifacino JS: Lysosome
positioning influences mTORC2 and AKT signaling. Mol Cell.
75:26–38. e232019. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Seo SU, Woo SM, Lee HS, Kim SH, Min KJ and
Kwon TK: mTORC1/2 inhibitor and curcumin induce apoptosis through
lysosomal membrane permeabilization-mediated autophagy. Oncogene.
37:5205–5220. 2018. View Article : Google Scholar : PubMed/NCBI
|