Introduction
Pancreatic cancer is known for its dire survival
outcomes, with only about 12% of patients surviving beyond five
years (1). This disease is
expected to become the second leading cause of cancer-related
deaths in the United States by 2040 (2). The late stage at diagnosis and high
rate of recurrence after surgery significantly hinder long-term
survival rates for those affected (3). Surgical resection is possible for
merely 20% of these patients, as half of all cases are diagnosed
after the cancer has metastasized to distant organs (4,5).
Presently, the cornerstone of treatment for pancreatic cancer is
combination therapy. This approach integrates traditional
modalities like surgery, chemotherapy, and radiotherapy with more
novel strategies such as immunotherapy and targeted therapy
(6). Given this context, the
development of novel anticancer agents with varied mechanisms of
action is crucial to enhance the efficacy of combination therapy in
treating pancreatic cancer.
Rupatadine, primarily known as a selective
antagonist for the histamine H1 receptor and platelet-activating
factor (PAF) for treating allergic rhinitis, has garnered attention
for its potential anticancer effects (7–9). Its
anticancer properties first stem from its ability to induce
lysosomal membrane permeabilization, releasing lysosomal hydrolases
into the cytoplasm, which damages mitochondria and initiates
apoptosis. Further research has demonstrated Rupatadine's
capability to disrupt the TGFβ1 signaling pathway, a critical
promoter of cancer cell invasion, angiogenesis, and immune evasion
within the tumor microenvironment, thus playing a significant role
in hindering tumor progression and metastasis. Studies have shown
that Rupatadine's influence on the TGF-β signaling pathway may also
involve modulation of associated pathways such as PAF, NF-κB, and
p65, thereby broadening its therapeutic potential (10–13).
Despite extensive research exploring Rupatadine as a
potential anticancer agent across various cancer types (14–17),
its specific impact on pancreatic cancer has not been thoroughly
examined. This study is designed to rigorously assess Rupatadine's
anticancer efficacy through comprehensive in vitro and in
vivo experiments utilizing pancreatic cancer models. The
principal aim of this study is to elucidate the molecular and
cellular mechanisms by which Rupatadine influences pancreatic tumor
cells. Furthermore, this research seeks to enhance our
understanding of Rupatadine's pharmacological attributes and lay
the groundwork for its potential clinical applications in
oncology.
Materials and methods
Cell culture
The AsPC-1 (passage 11) and MIA PaCa-2 (passage 13)
pancreatic cancer cell lines were obtained from the Korea Cell Line
Bank (KCLB). KCLB routinely authenticates cell lines via short
tandem repeat (STR) profiling to confirm their identity and screen
for contamination. All cell lines were cultured under standard
conditions and used at the specified passage numbers to ensure
experimental consistency. Cultivation of AsPC-1 cells was conducted
using Roswell Park Memorial Institute (RPMI) medium (Hyclone).
Concurrently, MIA PaCa-2 cells were cultured in Dulbecco's modified
Eagle's medium (DMEM)/High (Hyclone). Both media were enriched with
a supplement comprising 10% fetal bovine serum (FBS; Hyclone), and
1% Penicillin-Streptomycin (Gibco BRL). The cell lines were
incubated at a temperature of 37°C in a humidified environment,
with a 5% CO2 atmosphere to ensure optimal growth
conditions.
Cell viability assay
The assessment of cell viability for both AsPC-1 and
MIA PaCa-2 cell lines was performed utilizing the Ez-cytox Cell
Viability Assay Kit (Itsbio) according to the manufacturer's
instructions. All experiments were performed in triplicate (n=3) to
ensure reproducibility and statistical significance.
Western blot analysis
Lysis of AsPC-1and MIA PaCa-2 cells, as well as
mouse tissue samples, was carried out using the EzRIPA Lysis kit
(ATTO Corporation). Protein concentration was determined using the
Bradford assay reagent (Bio-Rad). For protein visualization,
Western blot analysis was performed using primary antibodies at a
1:1,000 dilution, obtained from Cell Signaling Technology. This was
followed by incubation with horseradish peroxidase (HRP)-conjugated
secondary antibodies at a 1:2,000 dilution, sourced from Vector
Laboratories. Detection of specific immune complexes was achieved
using the Western Blotting Plus Chemiluminescence Reagent
(Millipore). The primary antibodies used targeted hPARP (Cell
Signaling Technology, #9542), MCL-1 (Cell Signaling Technology,
#5453), cleaved-caspase-3 (Cell Signaling Technology, #9664),
TGF-βR1 (Abcam, ab31013), p-SMAD2/3 (Cell Signaling Technology,
#8828s), SMAD2/3 (Cell Signaling Technology, #5678s), human
E-cadherin (Abcam, ab76055), Vimentin (Abcam, ab92547), Snail,
(Cell Signaling Technology, #3879), PUMA (Cell Signaling
Technology, #98672), and β-actin (Sigma-Aldrich, A5541). All
experiments were performed in triplicate (n=3) to ensure
reproducibility and statistical significance.
Immunocytochemistry analysis
AsPC-1and MIA PaCa-2 were seeded into 8-well plate
(SPL Life Science) at the concentration of 8,000 cells/well. After
cultured overnight, the cells were fixed with 4% paraformaldehyde
at room temperature for 10 min. Then the cells were blocked with
incubation buffer (Normal Hores serum in PBS) for 1 h at room
temperature. Following that, the cells were also stained with above
antibodies and incubated at 4°C protected from light. After 1 h
incubation, the cells were washed twice and stained with 100 ng/ml
DAPI for 10 min at room temperature. The immunocytochemistry
staining utilized antibodies against E-cadherin (Santa Cruze
Biotechnology, sc-8426), Vimentin (Santa Cruze Biotechnology,
sc-373717), Snail (Genetex, GTX125918) and GAPDH (Santa Cruze
Biotechnology, sc-365062). The final observation of the samples was
conducted using a fluorescence imaging system (EVOS U5000;
Invitrogen; Thermo Fisher Scientific, Inc.). All experiments were
performed in triplicate (n=3) to ensure reproducibility and
statistical significance.
Immunohistochemical analysis
For the immunohistochemical analysis, tissue
sections that were formalin-fixed and paraffin-embedded underwent
deparaffinization, followed by a rehydration process using a series
of ethanol solutions. Epitope retrieval was then carried out in
accordance with standard methodologies. The immunohistochemical
staining utilized antibodies against BIM (Cell Signaling
Technology, #2933s), Mcl-1 (Santa Cruze Biotechnology, sc-377487),
E-cadherin (Santa Cruze Biotechnology, sc-8426), and Snail
(Genetex, GTX125918). Post-staining, the samples were analyzed for
antibody expression using a laser-scanning microscope (Eclipse
TE300; Nikon).
TUNEL assay
The detection of apoptosis in pancreatic cancer
cells was carried out using TUNEL analysis, employing the In Situ
Apoptosis Detection Kit (Takara Bio Inc.), with the protocol
adhering to the manufacturer's instructions. In summary, the
procedure involved incubating sample slides with 50 µl of TUNEL
reaction mixture and TdT labeling reaction mix for a duration of 1
h at 37°C, ensuring the environment was dark. Following the
incubation, the slides were washed three times using
phosphate-buffered saline (PBS). The final observation of the
samples was conducted using a fluorescence imaging system (EVOS
U5000; Invitrogen; Thermo Fisher Scientific, Inc.).
Wound healing assay
The AsPC-1 and MIA PaCa-2 cell lines were cultured
in 6-well plates until they reached full confluence. Subsequently,
the growth medium was substituted with serum-free media, and the
cells underwent a 72-h incubation period. Post-incubation, the cell
monolayers were mechanically disrupted to create a wound, followed
by the application of a test agent (rupatadine). An exact region of
the wound was imaged using phase-contrast microscopy prior to the
administration of these agents. This same region was imaged again
after a 72-h period. The area of the wound was quantitatively
assessed using image analysis techniques, both initially and after
the 72-h interval. The rate of wound closure was calculated based
on the change in wound area over time, using the formula: [(initial
wound area-final wound area)/initial wound area] ×100, to express
this change as a percentage. All experiments were performed in
triplicate (n=3) to ensure reproducibility and statistical
significance.
Animals study design
Our animal study design involved five-week-old male
BALB/c nude mice (Orient Bio). Our animal experiments were approved
by the Institutional Animal Care and Use Committee (IACUC) of the
Catholic University of Korea (approval No. CUMC-2022-0014-01) and
were conducted in accordance with institutional and national
guidelines for the care and use of laboratory animals. For the
xenograft pancreatic cancer model, 1×106 AsPC-1 cells
were subcutaneously injected into the mice. In the evaluation of
rupatadine's in vivo efficacy, mice were grouped randomly,
with each group containing five mice. After two weeks
post-implantation of the tumor model, the mice were treated with
rupatadine (3 mg/kg) administered intraperitoneally, three times a
week for three weeks. Twenty-one days after initial treatment, the
animals were euthanized, and the tumors were surgically removed for
subsequent analysis. Animals were euthanized using compressed
carbon dioxide (CO2) gas at a displacement rate of
30–70% of the chamber volume per minute for 2–3 min. Loss of
consciousness was confirmed by observing lack of response and faded
eye color, and death was verified by cessation of respiration,
cardiac arrest, and pupil dilation. Throughout the experiment,
animal health was monitored daily, and humane endpoints-such as
weight loss >20%, severe lethargy, or ulceration-were predefined
and strictly observed.
Regarding the establishment and monitoring of tumor
formation before initiating Rupatadine treatment, tumors were
allowed to reach a predetermined size, specifically a volume of 100
mm3, confirmed through visual inspection before the
commencement of drug administration. The endpoint for the study was
matched based on the time-point rather than the stage, ensuring
that all groups were assessed over the same duration post-treatment
initiation.
Rupatadine pharmacokinetics in mouse
plasma and tissues
Mice received an intravenous dose of 10 mg/kg
rupatadine. Plasma samples were collected at intervals from 5 to
360 min and processed by mixing with four volumes of acetonitrile,
followed by centrifugation. Tissue samples at 5 and 60 min were
prepared similarly. Rupatadine concentrations were measured using
high-performance liquid chromatography (HPLC) with a Waters Arc
system, a C18 column, and a mobile phase of methanol and 0.3 M
sodium acetate (pH 4.4) at a 20:80 ratio, with a flow rate of 1
ml/min. The retention time for rupatadine was 5 min.
Statistical analysis
Statistical analysis was performed using SPSS 11.0
(SPSS Inc.), with data presented as mean ± standard deviation (SD).
Normality was assessed using the Shapiro-Wilk test. The
Kruskal-Wallis test with Dunn's post hoc test was used for
multiple-group comparisons, while the Mann-Whitney U test and
Student's t-test were applied for nonparametric and normally
distributed two-group comparisons, respectively. Statistical
significance was set at P<0.05.
Results
In vitro anticancer effects of
rupatadine
Rupatadine is a chemical compound characterized by a
cationic amphiphilic structure, consisting of a hydrophobic ring
system and a side chain with a cationic amine group (Fig. 1A). To further evaluate the
dose-dependent effects of rupatadine, dose-response curves were
generated for AsPC-1 and MIA-PaCa-2 pancreatic cancer cells, as
well as human pancreatic stellate cells (HPSC) (Fig. 1B). The IC50 values for AsPC-1 cells
were 23.5 µM (24 h) and 24.9 µM (48 h), while for MIA-PaCa-2 cells,
the IC50 values were 30.9 µM (24 h) and 35.1 µM (48 h). In the
MIA-PACA cell line, rupatadine displayed lower cell viability
compared to treatments with irinotecan (3.125–200 ng/ml),
5-fluorouracil (1–100 µM), and paclitaxel (1–10 nM) at both 24 and
48 h. This suggests that rupatadine may possess superior anticancer
effects relative to these well-established chemotherapeutic agents
(Fig. S1). In contrast,
rupatadine exhibited significantly lower cytotoxicity in human
pancreatic stellate cells (HPSC), with IC50 values of 66.8 µM at 24
h and 132.0 µM at 48 h, indicating a selectivity for pancreatic
cancer cells over normal pancreatic cells. Data are presented as
mean ± SD from three independent experiments. This data suggests
that rupatadine preferentially inhibits pancreatic cancer cells
while having a reduced effect on normal pancreatic cells.
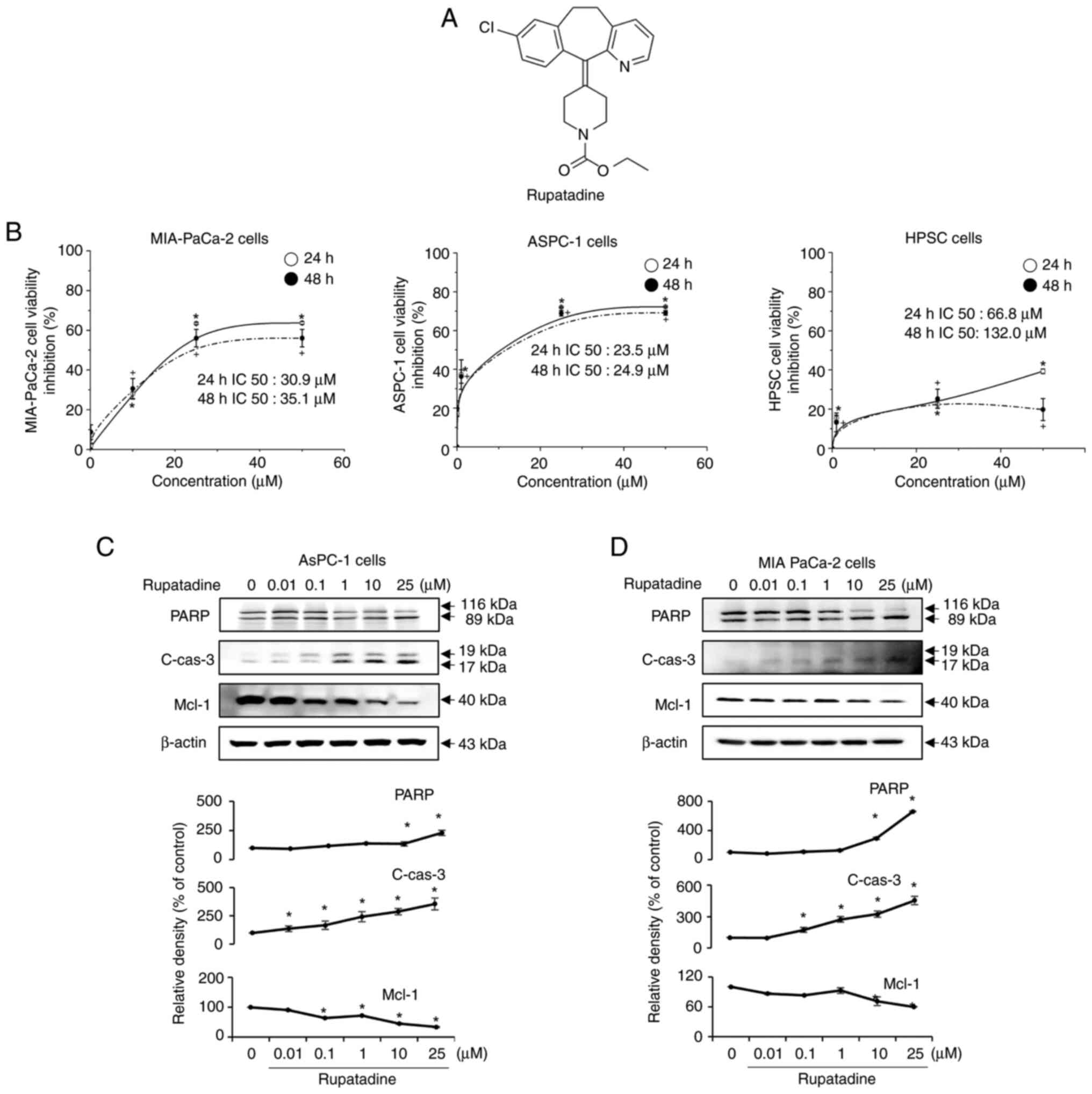 | Figure 1.In vitro anticancer effects of
rupatadine on pancreatic cancer cell lines. (A) Chemical structure
of rupatadine. As a cationic amphiphilic drug, the structure of
rupatadine, comprising a hydrophobic ring and a cationic amine
group, facilitates its penetration through the plasma membrane. (B)
Dose-response curves comparing the effects of rupatadine on AsPC-1
and MIA-PaCa-2 pancreatic cancer cells with those on HPSC (bottom).
For the cancer cell lines, a dose-dependent decrease in viability
is observed with rupatadine concentrations from 0 to 50 µM, with
IC50 values of 23.5 µM (24 h) and 24.9 µM (48 h) for
AsPC-1 cells (top, left) and 30.9 µM (24 h) and 35.1 µM (48 h) for
MIA-PaCa-2 cells (top, right). By contrast, HPSCs (bottom) exhibit
significantly higher IC50 values of 66.8 µM (24 h) and
132.0 µM (48 h), indicating lower sensitivity to rupatadine. (C)
Induction of apoptosis in AsPC-1 cells. Western blot analysis shows
a dose-dependent increase in the levels of pro-apoptotic markers
PARP and cleaved caspase-3 and a reduction in the anti-apoptotic
marker Mcl-1 in AsPC-1 pancreatic cancer cells following treatment
with rupatadine, ranging from 0 to 25 µM (P<0.05). (D) Induction
of apoptosis in MIA-PACA-2 cells. MIA-PACA-2 cells treated with
rupatadine showed increased pro-apoptotic markers and decreased
anti-apoptotic markers, similar to AsPC-1 cells, indicating
rupatadine's consistent pro-apoptotic effect (P<0.05). Relative
densities of individual markers had been quantified using Image J
software and then were normalized to that of β-actin in each group.
Values are presented as mean ± SD of three independent experiments.
*P<0.05 vs. the control after 24 h; +P<0.05 vs.
the control after 48 h. HPSC, human pancreatic stellate cells;
Mcl-1, Myeloid cell leukemia-1; c-, cleaved; cas3, caspase 3. |
Following the treatment with rupatadine, a
concentration-dependent (0–25 µM) increase in the pro-apoptotic
markers, PARP and cleaved caspase-3, and a decrease in the
anti-apoptotic marker, Mcl-1, were observed in AsPC-1 pancreatic
cancer cells, as evidenced by Western blot analysis (P<0.05)
(Fig. 1C). A similar trend was
noted in MIA PaCa-2 cells, demonstrating the compound's consistent
apoptotic induction across different cell lines (P<0.05)
(Fig. 1D). Data are presented as
mean ± SD from three independent experiments. Additionally, Western
blot analysis was employed to assess the impact of rupatadine on
TGF-β signaling and EMT markers. The results indicate that
Rupatadine significantly suppresses the expression of TGFβRI and
diminishes the phosphorylated to total SMAD2/3 ratio in a
dose-dependent manner in AsPC-1 cells (P<0.05) (Fig. 2A), implicating the inhibition of
the TGFβ1 signaling pathway at the receptor level. The analysis
further revealed that Rupatadine upregulated the epithelial marker
E-cadherin while downregulating mesenchymal markers N-cadherin,
Vimentin, and Snail, highlighting its role in thwarting EMT
(P<0.05). Parallel results in MIA PaCa-2 cells reinforced
rupatadine's efficacy in targeting both the TGFβ signaling pathway
and EMT (Fig. 2B).
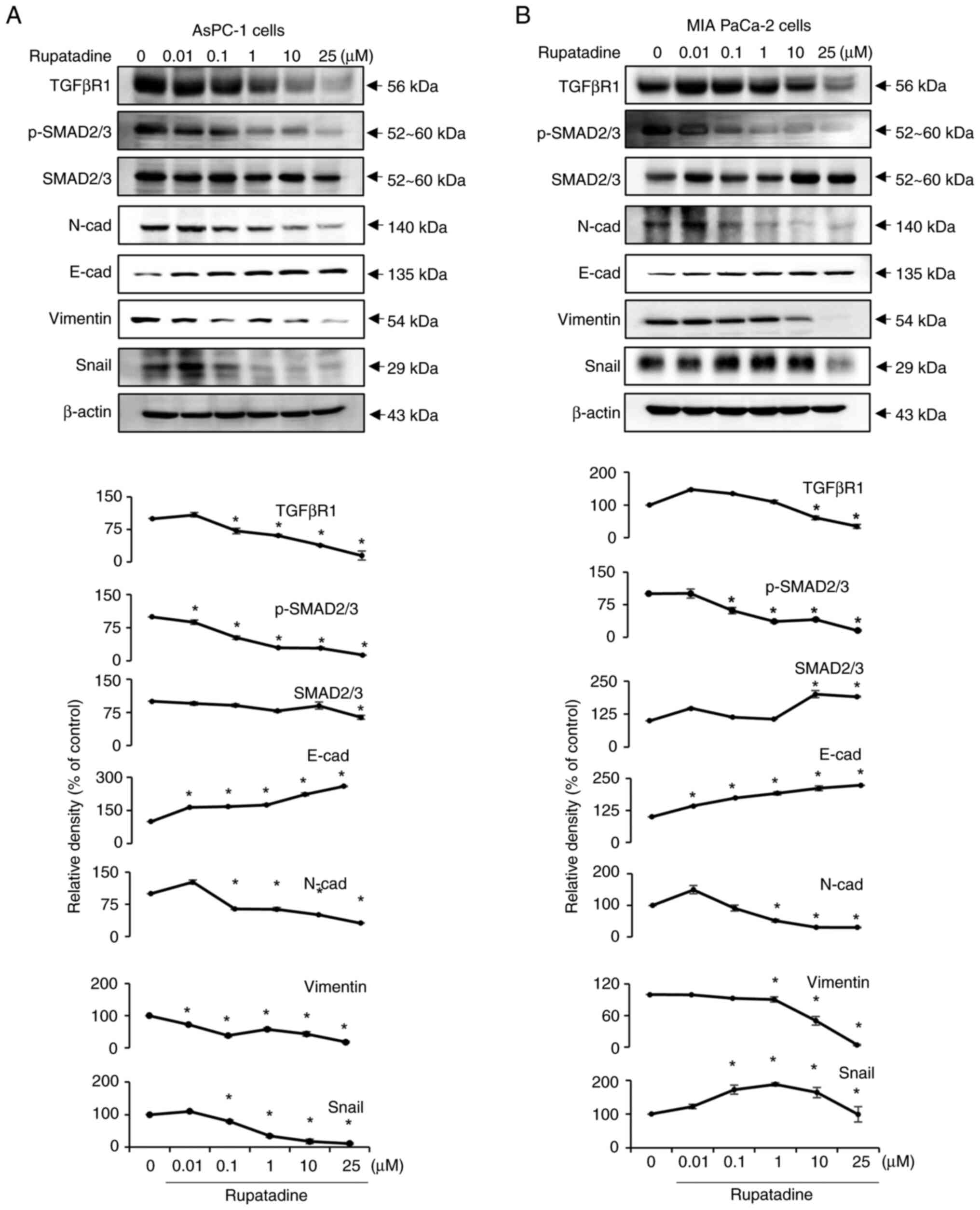 | Figure 2.Effects of rupatadine on TGF-β
signaling and EMT marker regulation. (A) In AsPC-1 pancreatic
cancer cells, rupatadine exhibits a dose-dependent decrease in
TGF-βRI expression and reduces the ratio of p-SMAD2/3 to total
SMAD2/3, suggesting an inhibition of the TGFβ1 pathway. (B) In MIA
PaCa-2 cells, analogous dose-dependent effects of rupatadine are
observed, consistent with findings in AsPC-1 cells, further
validating the drug's role in modulating TGF-β signaling and EMT.
Relative densities of individual markers had been quantified using
Image J software and then were normalized to that of β-actin in
each group. Values are presented as mean ± SD of three independent
experiments. *P<0.05 vs. the control. EMT,
epithelial-mesenchymal transition; TGF-βRI, transforming growth
factor β receptor type I; p-, phosphorylated; N-cad, N-cadherin;
E-cad, E-cadherin. |
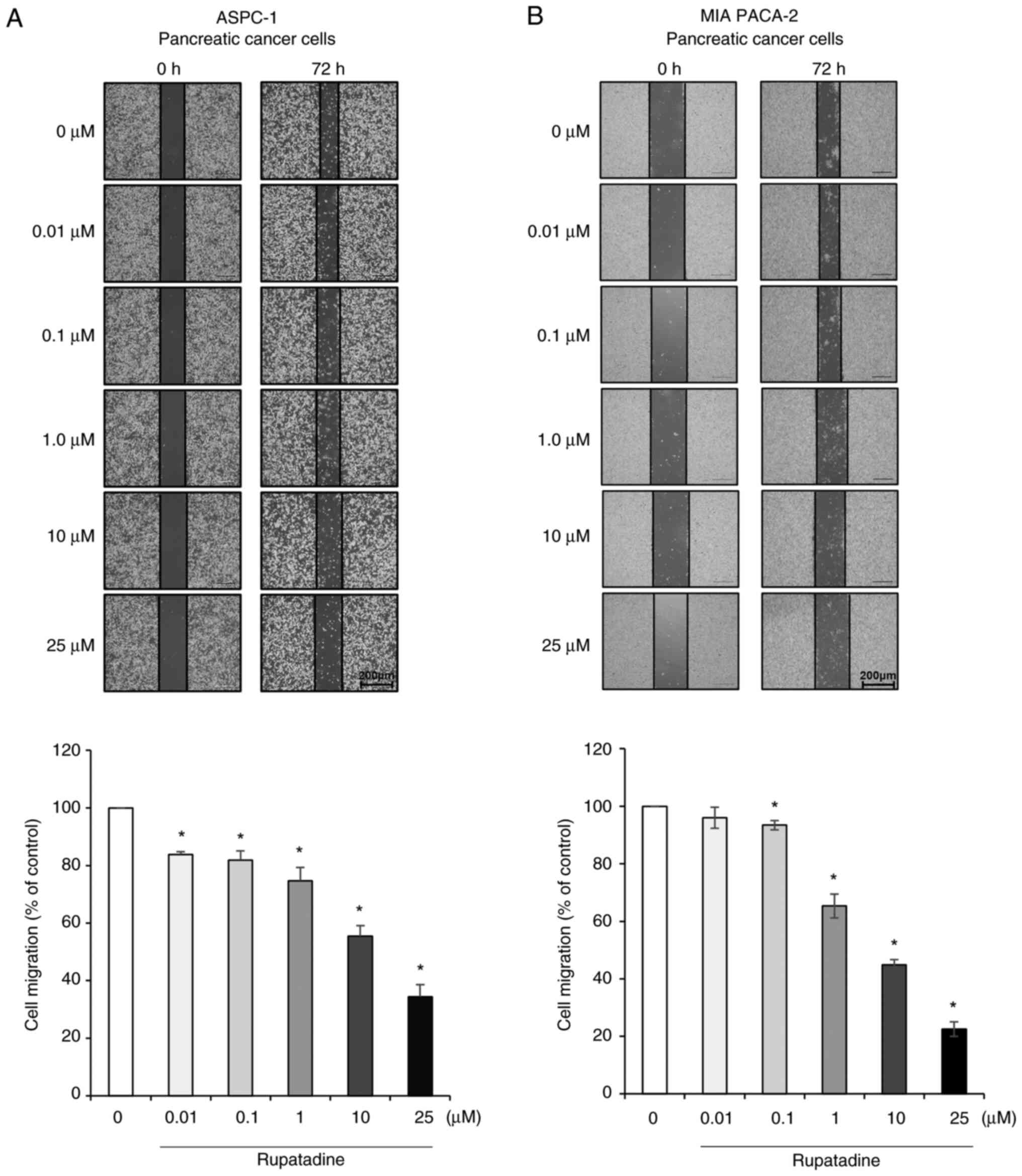
Effects of rupatadine on migration of
pancreatic cancer cells
The impact of rupatadine on the migration of
pancreatic cancer cells was evaluated using a wound healing assay.
After creating a scratch in a confluent monolayer of AsPC-1 cells,
the closure of the wound was monitored in response to increasing
concentrations of rupatadine (0.01–25 µM). Increased concentrations
of rupatadine, specifically at 25 µM, were correlated with a
significant expansion of the scratch area, illustrating a
dose-dependent suppression of cell migration in AsPC-1 cells
(Fig. 3A). An analogous effect was
recorded in MIA PaCa-2 cells at the 25 µM concentration of
rupatadine (Fig. 3B). Data are
presented as mean ± SD from three independent experiments.
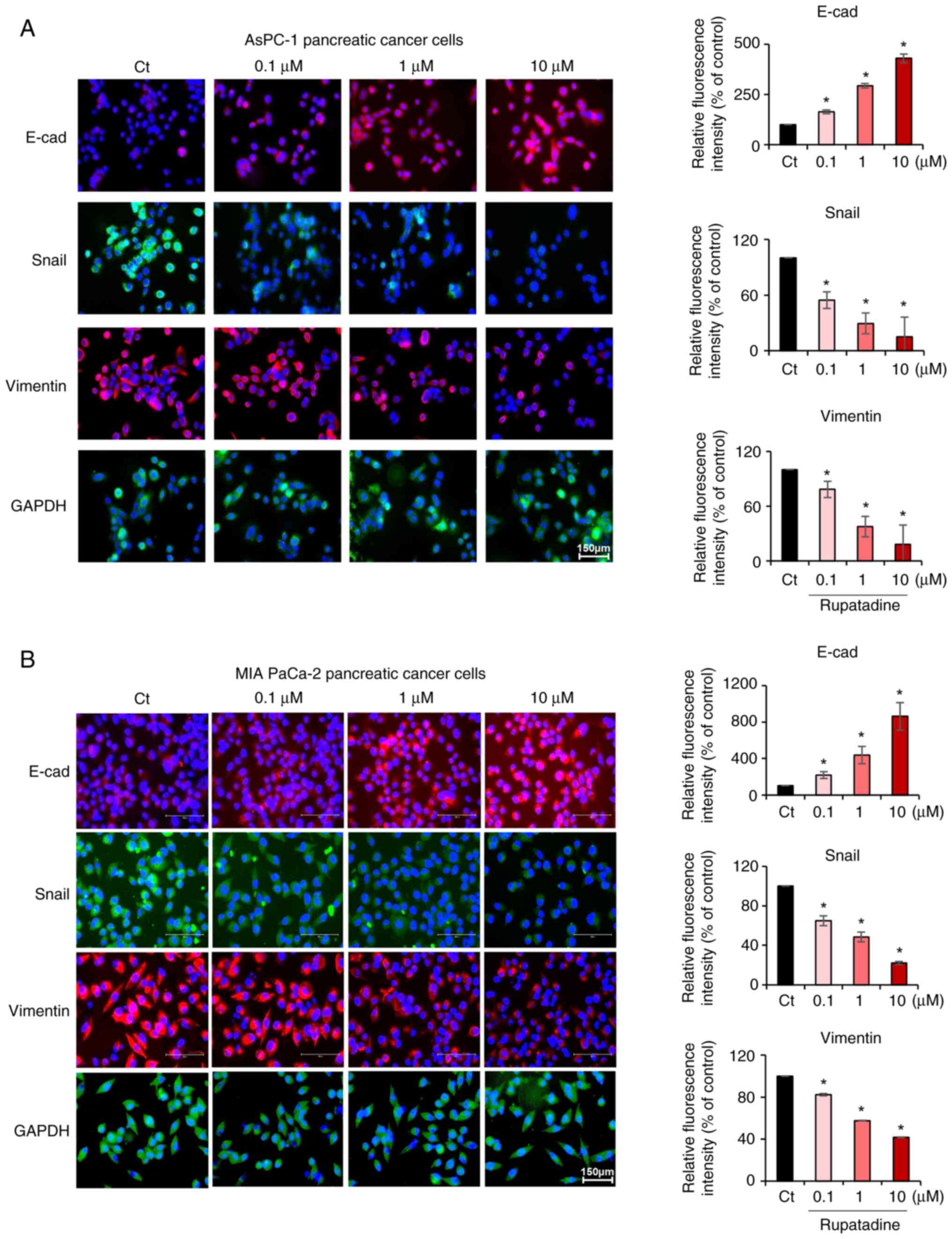
Effects of rupatadine on EMT of
pancreatic cancer cells
The influence of rupatadine on EMT in AsPC-1
pancreatic cancer cells was investigated through immunofluorescence
analysis of EMT markers following treatment with rupatadine.
Increasing the concentration of rupatadine (0.1–10 µM) resulted in
a concentration-dependent increase in the immunofluorescence of the
epithelial marker E-cadherin, and a decrease in the
immunofluorescence of mesenchymal markers Snail and Vimentin
(P<0.05) (Fig. 4A). Similar
effects were observed in MIA PaCa-2 pancreatic cancer cells, with
rupatadine treatment leading to an increased immunofluorescence in
E-cadherin and a decreased immunofluorescence in Snail and Vimentin
(P<0.05) (Fig. 4B). Data are
presented as mean ± SD from three independent experiments.
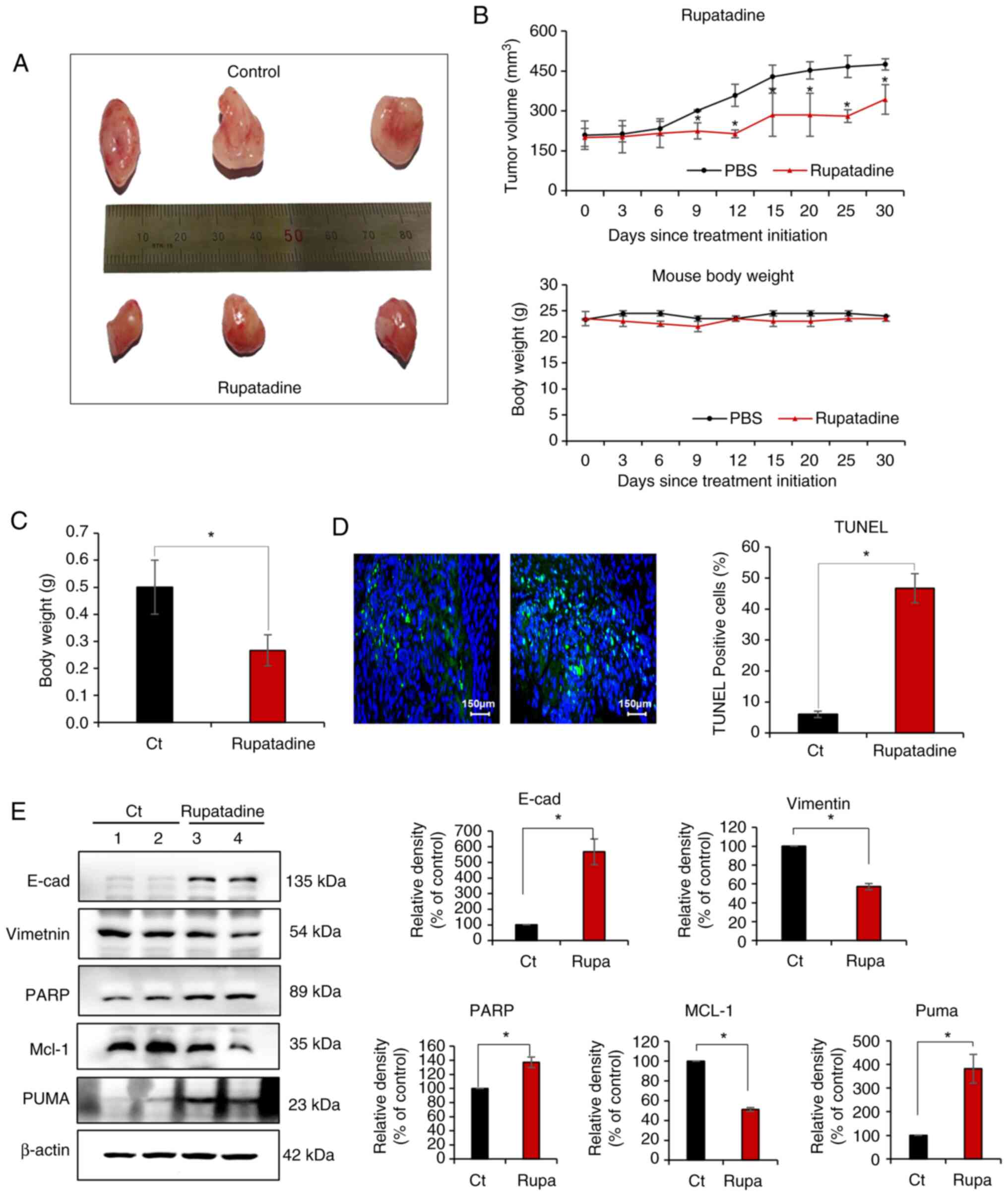
Anticancer efficacy of rupatadine in a
pancreatic cancer xenograft mouse model
The in vivo xenograft mouse model of
pancreatic cancer was established by direct injection of AsPC-1
cells (1×106 cells) into the pancreas of BALB/c nude
mice after laparotomy under anesthesia. Two weeks later, the mice
were treated with rupatadine (3 mg/kg) administered
intraperitoneally, three times a week for three weeks. On 21 days
after initial treatment, the animals were euthanized, and the
tumors were surgically removed for subsequent analysis. Fig. 5A presents a representative
illustration depicting the difference in tumor size between the
control group and the rupatadine-treated group on 21 days after
initial treatment. Tumors in the control group weighed between 0.4
and 0.6 g, whereas those treated with Rupatadine weighed between
0.2 and 0.3 g. To assess the effects of rupatadine on tumor
progression over time, tumor volume was measured at multiple time
points following treatment initiation. The rupatadine-treated group
exhibited a significantly slower tumor growth rate compared to the
control group (Fig. 5B top). After
21 days of treatment, the average tumor volume in the control group
continued to increase, whereas the tumor volume in the
rupatadine-treated group showed a marked reduction (Fig. 5B top). In contrast, no significant
differences in body weight were observed between the two groups
throughout the experimental period, indicating that rupatadine
treatment did not cause notable systemic toxicity (Fig. 5B bottom). Consistently, tumor
weight analysis performed on day 21 after the initial treatment
revealed a significant reduction in the rupatadine-treated group
compared to the control group (P<0.05), further supporting its
tumor-suppressive effects (Fig.
5C). Data are presented as mean ± SD from three independent
experiments.
Subsequently, a TUNEL assay was conducted to
quantify apoptosis in the excised tumors from the
rupatadine-treated groups. The results of the TUNEL assay revealed
a significantly higher apoptosis index in the tumors treated with
rupatadine compared to those in the control groups (P<0.05)
(Fig. 5D). Western blot analysis
was utilized to assess the differential expression of markers
associated with EMT and apoptosis in the excised tumor tissues
(Fig. 5E). In the group treated
with rupatadine, a notable upregulation of the epithelial marker
E-cadherin was observed, in contrast to the control group.
Conversely, the mesenchymal marker vimentin was significantly
downregulated (P<0.05) in the rupatadine-treated tumors.
Furthermore, the rupatadine-treated tumors demonstrated a
significant elevation in the levels of pro-apoptotic markers, PARP
and PUMA, while the expression of the anti-apoptotic marker Mcl-1
was markedly reduced (P<0.05). Data are presented as mean ± SD
from three independent experiments.
Effects of rupatadine on histological
changes, apoptosis, EMT, and pharmacokinetics in a pancreatic
cancer xenograft mouse model
Tumor tissues derived from a mouse xenograft model
of pancreatic cancer underwent Hematoxylin and Eosin (H&E)
staining for comparative histological examination. In the
rupatadine-treated group, a notable reduction in tumor cell density
was observed (Fig. 6A). This was
followed by immunohistochemical staining of the excised tumor
tissues from each experimental group. Analysis of the stained
tissues revealed that, in the rupatadine-treated group, there was a
significant increase in the percentage of immunoreactive areas for
a pro-apoptotic marker, BIM, compared to the control group.
Conversely, the expression of Mcl-1, an anti-apoptotic marker, was
significantly decreased (P<0.05) (Fig. 6B top). Furthermore, the
rupatadine-treated group exhibited a significant increase in the
percentage of immunoreactive areas for the epithelial marker
E-cadherin, alongside a significant decrease in Snail, a
mesenchymal marker (P<0.05) (Fig.
6B bottom). Overall, these findings suggest that rupatadine
treatment can attenuate the EMT process and enhance apoptosis in
pancreatic cancer tissues within this mouse xenograft model.
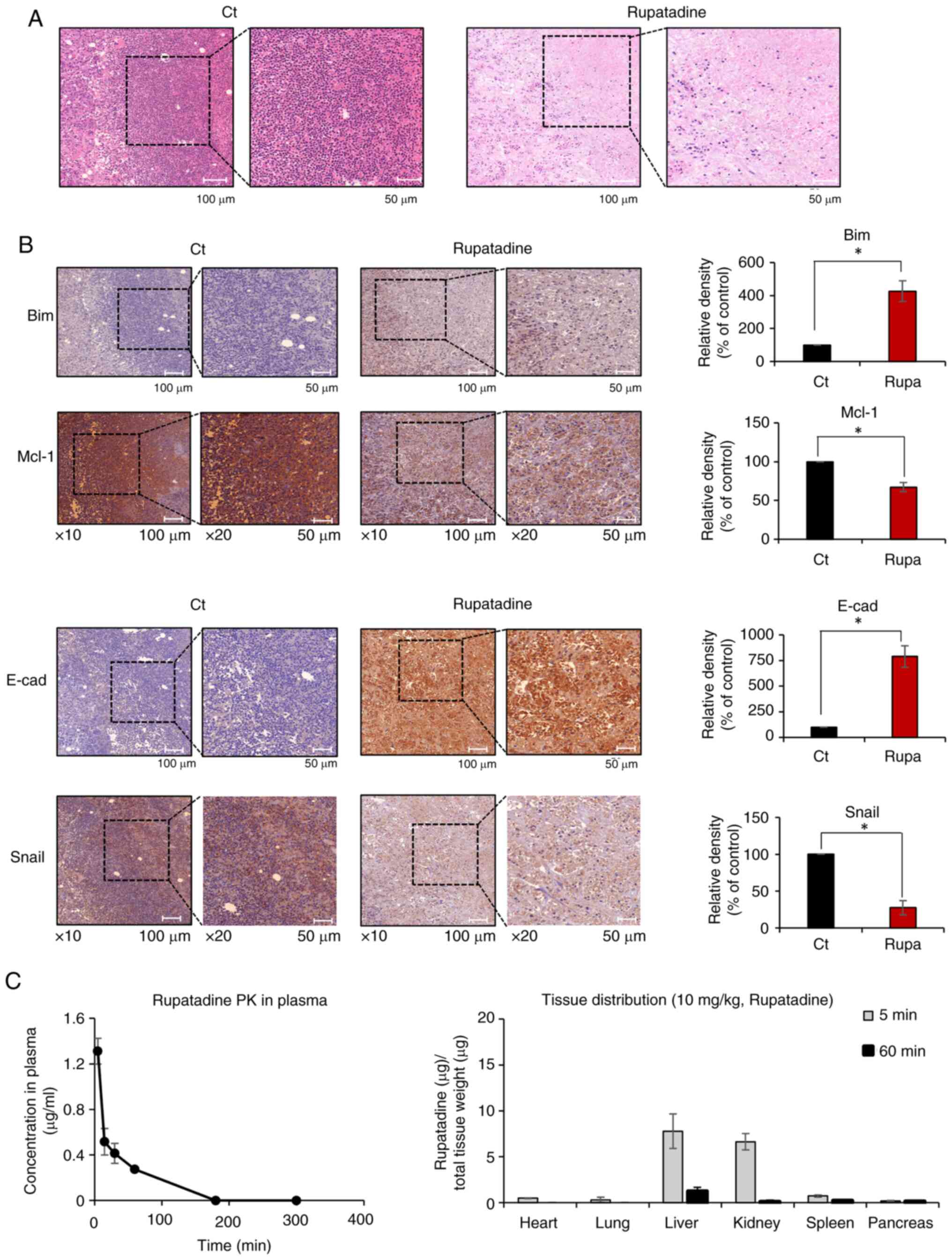 | Figure 6.Impact of rupatadine on histological
changes in a pancreatic cancer xenograft mouse model. (A) H&E
staining of excised tumor tissues, demonstrating a notable
reduction in tumor cell density in the rupatadine-treated group.
(B) Immunohistochemical analysis for the markers of apoptosis and
EMT. Rupatadine-treated group exhibited increased immunoreactivity
for the pro-apoptotic marker BIM and decreased expression of the
anti-apoptotic marker Mcl-1. In addition, the rupatadine-treated
group exhibited decreased immunoreactivity for the epithelial
marker E-cadherin and decreased expression of the mesenchymal
marker Snail. For 10× magnification, the scale bar is 100 µm; for
20× magnification, the scale bar is 50 µm. (C) Pharmacokinetic and
tissue distribution analysis of rupatadine in vivo. (Left)
Plasma concentration-time curve of rupatadine following intravenous
administration (10 mg/kg) in mice. Rupatadine was rapidly cleared
from plasma, with undetectable levels beyond 180 min. (Right)
Tissue distribution of rupatadine at 5 and 60 min post-injection,
showing predominant accumulation in the liver and kidneys at early
time points, with minimal retention after 60 min. Values are
presented as mean ± SD of three independent experiments.
Percentages of immunoreactive areas were measured using Image J
(National Institute of Health) and expressed as relative values to
those in control tissues. *P<0.05 vs. the control. H&E,
hematoxylin and eosin staining; EMT, epithelial-mesenchymal
transition; BIM, Bcl-2 interacting mediator of cell death; Ct,
control; Mcl-1, Myeloid cell leukemia-1; E-cad, E-cadherin; Rupa,
Rupatadine. |
To further investigate the in vivo
pharmacokinetics of rupatadine, we analyzed its plasma
concentration and tissue distribution following intravenous
injection (10 mg/kg) in mice. Plasma concentration-time profiling
demonstrated a rapid decline in rupatadine levels, with detectable
concentrations at 5, 15, 30, and 60 min, but no detectable levels
after 180 min, indicating fast clearance from circulation (Fig. 6C, left). Tissue distribution
analysis revealed that rupatadine primarily accumulated in the
liver and kidneys within 5 min post-injection but was largely
undetectable in these tissues by 60 min, suggesting rapid
metabolism and excretion (Fig. 6C,
right). These findings provide essential information regarding
rupatadine's pharmacokinetic profile and tissue clearance dynamics,
which are critical for understanding its potential as an anticancer
agent.
Discussion
Rupatadine, known for its role as a selective
histamine H1 receptor and platelet-activating factor antagonist in
treating allergic rhinitis, has gained attention for its anticancer
potential due to its capacity to induce lysosomal membrane
permeabilization. This research explores the anticancer properties
of rupatadine within in vitro and in vivo pancreatic
cancer models. Rupatadine demonstrated a concentration-dependent
reduction in the cell viability of both AsPC-1 and MIA PaCa-2
pancreatic cells, across a range of concentrations (0.001–50 µM).
In an in vivo pancreatic cancer xenograft model, intravenous
administration of rupatadine led to a significant tumor weight
reduction after 35 days of treatment. Further analysis of excised
tumor tissues from the mouse model corroborated the in vitro
findings, with rupatadine inhibiting EMT and promoting apoptosis in
tumor cells. Specifically, it increased E-cadherin levels while
decreasing Vimentin and Snail expressions, indicating a suppression
of EMT. Concurrently, it augmented the expression of the
pro-apoptotic marker PARP and reduced the anti-apoptotic marker
Mcl-1, suggesting an enhancement of apoptotic pathways.
Collectively, these findings underscore the potential of rupatadine
as a therapeutic agent in pancreatic cancer due to its ability to
concurrently inhibit EMT and activate apoptotic pathways in both
in vitro and in vivo models of pancreatic cancer.
Rupatadine, widely used as an antagonist for
histamine H1 receptors and platelet-activating factors in allergic
rhinitis treatment, has garnered attention for its potential
anticancer effects, particularly in acute myeloid leukemia (AML)
and ovarian cancer (7,9). Specifically, research has shown that
rupatadine affects AML cells at concentrations marginally higher
than those used for alleviating allergic symptoms (9). In the study, rupatadine showed no
substantial impact on the viability of normal blood cells, such as
T cells, B cells, and myeloid cells, with the exception of a noted
decrease in the myeloid cell population. Moreover, the treatment
preserved the clonogenic capacity of healthy hematopoietic
progenitor/stem cells, indicating a selective action against cancer
cells while sparing healthy cells. Additionally, in vivo
experiments demonstrated that rupatadine reduced the regeneration
potential of AML cells when transplanted into immunocompromised
mice, further supporting its potential as an anticancer agent.
These findings highlight the potential of rupatadine as a selective
treatment option for AML, revealing a new avenue for the use of
this antihistamine beyond its traditional role in allergy
management.
This study offers preliminary insights into
rupatadine's ability to inhibit EMT, an effect not widely
recognized in previous research. The inhibitory effect of
rupatadine on EMT can reasonably be ascribed to its capacity to
induce lysosomal membrane permeabilization and mitochondrial
damage. This can lead to increased cellular stress, potentially
disrupting EMT-related signaling pathways and modulating
transcription factors crucial for EMT, such as Snail, Slug, and
Twist. Additionally, rupatadine's impact on cellular stress and
mitochondrial dysfunction could reduce the metastatic potential of
cancer cells and alter autophagic flux, further contributing to the
inhibition of EMT (9). These
combined effects, stemming from rupatadine's disruption of
lysosomal and mitochondrial functions, likely contribute to its
efficacy in inhibiting EMT processes in pancreatic cancer
cells.
This study confirms that rupatadine's anticancer
efficacy is primarily derived from its dual action of inhibiting
EMT and promoting apoptosis. In addition, this study has shown that
Rupatadine can have anticancer effect by inhibiting the TGFβ1
signaling pathway which promotes cancer cell invasion,
angiogenesis, and immune evasion within the tumor microenvironment,
facilitating tumor progression and metastasis. The inhibitory
effects of Rupatadine on the TGF-β signaling pathway have been
demonstrated in numerous previous studies (10–13).
Especially, Didamoony et al (11) delineates that Rupatadine mediates
its anticancer efficacy through the inhibition of the TGF-β1
pathway, potentially by modulating associated pathways such as PAF,
NF-κB, and p65. Furthermore, Rupatadine's anticancer effects can be
partly linked to its role in causing mitochondrial and lysosomal
damage (9). As a cationic
amphiphilic drug, Rupatadine can easily enter cells and accumulate
in lysosomes, the cell's waste disposal system. This leads to the
disruption of the lysosomal membrane, releasing enzymes that
trigger cell death.
Although the AsPC-1 and MIA PaCa-2 cell lines used
in this study were obtained from an authenticated source,
additional in-house authentication and mycoplasma testing were not
performed prior to the experiments. While the passage numbers were
kept relatively low (passage 11 for AsPC-1 and passage 13 for MIA
PaCa-2), we acknowledge that prolonged culture and high passage
numbers may contribute to genetic and phenotypic variability, which
could influence experimental outcomes. Future studies should
consider regular authentication to ensure reproducibility and
minimize variability associated with cell culture conditions.
In conclusion, this study highlights rupatadine's
anticancer effects in pancreatic cancer models. The in vitro
results demonstrated rupatadine's capacity to suppress EMT and
enhance apoptosis in pancreatic cancer cells. In a pancreatic
cancer xenograft mouse model, intravenous administration of
rupatadine led to a significant tumor weight reduction. Further
analysis of the excised tumor tissue from this model reinforced the
observation that rupatadine's anticancer efficacy largely stems
from its dual action in inhibiting EMT and facilitating apoptosis.
Future studies are needed to elucidate the precise mechanisms
underlying these effects. In addition, expanding the panel of
pancreatic cancer cell lines in future experiments will help
validate the generalizability of our findings and further support
the clinical relevance of rupatadine. Moreover, while the present
study focused on monotherapy, future investigations will evaluate
the combinatorial potential of rupatadine with established
chemotherapeutic agents and targeted therapies to better reflect
clinical treatment strategies. Overall, rupatadine can be
considered as a promising candidate for pancreatic cancer therapy,
offering a unique mechanism of action that could synergize
effectively with existing treatment modalities in a combination
therapy approach.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms Jeong-Yeon Seo
(Translational Research Team, Surginex Co., Ltd., Seoul, South
Korea) for manuscript processing and Ms Jennifer Lee (Translational
Research Team, Surginex Co., Ltd., Seoul, South Korea) for their
contributions to the illustrations.
Funding
This work was supported by the financial support of the Catholic
Medical Center Research Foundation made in the program year of
2021.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SJK conceptualized the study and was project
administrator. BJC analyzed and interpreted the data and wrote the
original draft. BJC, HJC, DL, JHP and THH performed the animal
experiments. HJJ, GHJ and OHK performed the in vitro
experiments. All authors have read and approved the final
manuscript. SJK and BJC confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
Our animal experiments were approved by the
Institutional Animal Care and Use Committee of the Catholic
University of Korea (approval no. CUMC-2022-0014-01) and were
conducted in accordance with institutional and national guidelines
for the care and use of laboratory animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jiang Z, Zheng X, Li M and Liu M:
Improving the prognosis of pancreatic cancer: Insights from
epidemiology, genomic alterations, and therapeutic challenges.
Front Med. 17:1135–1169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahib L, Wehner MR, Matrisian LM and Nead
KT: Estimated projection of US cancer incidence and death to 2040.
JAMA Netw Open. 4:e2147082021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Siegel RL, Miller KD, Fuchs HE and Jemal
A: Cancer statistics, 2021. CA Cancer J Clin. 71:7–33.
2021.PubMed/NCBI
|
|
4
|
Mizrahi JD, Surana R, Valle JW and Shroff
RT: Pancreatic cancer. Lancet. 395:2008–2020. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park W, Chawla A and O'Reilly EM:
Pancreatic cancer: A review. JAMA. 326:851–862. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Awais N, Satnarine T, Ahmed A, Haq A,
Patel D, Hernandez GN, Seffah KD, Zaman MA and Khan S: A systematic
review of chemotherapeutic regimens used in pancreatic cancer.
Cureus. 15:e466302023.PubMed/NCBI
|
|
7
|
Deuster E, Hysenaj I, Kahaly M, Schmoeckel
E, Mayr D, Beyer S, Kolben T, Hester A, Kraus F, Chelariu-Raicu A,
et al: The platelet-activating factor receptor's association with
the outcome of ovarian cancer patients and its experimental
inhibition by rupatadine. Cells. 10:23372021. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Faustino-Rocha AI, Ferreira R, Gama A,
Oliveira PA and Ginja M: Antihistamines as promising drugs in
cancer therapy. Life Sci. 172:27–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cornet-Masana JM, Banus-Mulet A, Carbo JM,
Torrente MA, Guijarro F, Cuesta-Casanovas L, Esteve J and Risueno
RM: Dual lysosomal-mitochondrial targeting by antihistamines to
eradicate leukaemic cells. EBioMedicine. 47:221–234. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ahmed LA, Mohamed AF, Abd El-Haleim EA and
El-Tanbouly DM: Boosting Akt pathway by rupatadine modulates
Th17/Tregs balance for attenuation of isoproterenol-induced heart
failure in rats. Front Pharmacol. 12:6511502021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Didamoony MA, Atwa AM and Ahmed LA: A
novel mechanistic approach for the anti-fibrotic potential of
rupatadine in rat liver via amendment of PAF/NF-kB p65/TGF-beta1
and hedgehog/HIF-1alpha/VEGF trajectories. Inflammopharmacology.
31:845–858. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Didamoony MA, Atwa AM and Ahmed LA:
Modulatory effect of rupatadine on mesenchymal stem cell-derived
exosomes in hepatic fibrosis in rats: A potential role for
miR-200a. Life Sci. 324:1217102023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lv XX, Wang XX, Li K, Wang ZY, Li Z, Lv Q,
Fu XM and Hu ZW: Rupatadine protects against pulmonary fibrosis by
attenuating PAF-mediated senescence in rodents. PLoS One.
8:e686312013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yuan W, Fang W, Zhang R, Lyu H, Xiao S,
Guo D, Ali DW, Michalak M, Chen XZ, Zhou C, et al: Therapeutic
strategies targeting AMPK-dependent autophagy in cancer cells.
Biochim Biophys Acta Mol Cell Res. 1870:1195372023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Paunovic V, Kosic M, Misirkic-Marjanovic
M, Trajkovic V and Harhaji-Trajkovic L: Dual targeting of tumor
cell energy metabolism and lysosomes as an anticancer strategy.
Biochim Biophys Acta Mol Cell Res. 1868:1189442021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Debnath J, Gammoh N and Ryan KM: Autophagy
and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol.
24:560–575. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bhagya N and Chandrashekar KR: Liposome
encapsulated anticancer drugs on autophagy in cancer cells -
Current and future perspective. Int J Pharm. 642:1231052023.
View Article : Google Scholar : PubMed/NCBI
|