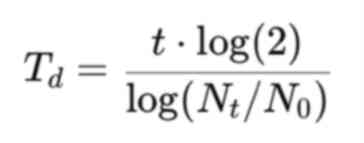Introduction
Cell adhesion molecules fulfill critical roles in
embryonic development, tissue repair and intercellular
communication (1,2). In cancer, dynamic regulation of
adhesion and detachment underlies tumor invasion and metastasis
(3). The progression and
metastasis of cancer are coordinated by intricate molecular
mechanisms that transcend mere tumor cell proliferation,
encompassing dynamic interactions with the surrounding tumor
microenvironment. Among these mechanisms, cell adhesion molecules
(CAMs) have emerged as pivotal regulators of malignant behaviors,
including cancer cell adhesion, migration and invasion and also
interaction with the vascular endothelium, thereby facilitating
metastatic dissemination and the acquisition of aggressive
phenotypes (4,5). Major CAM families, including
cadherins, integrins, selectins and members of the immunoglobulin
superfamily, contribute to tumor progression through distinct, yet
often interrelated, signaling pathways. For example, the loss of
E-cadherin expression is a well-established trigger of
epithelial-mesenchymal transition, promoting the enhanced motility,
invasiveness and metastatic potential of cancer cells (6). Integrins, which mediate
cell-extracellular matrix interactions, exert critical roles in
cellular migration and angiogenesis. Alterations in the expression
of specific integrin subunits have previously been associated with
unfavorable clinical outcomes in various malignancies (7). Moreover, accumulating evidence has
shown that CAMs function not only as structural mediators, but also
as active transducers of intracellular signaling cascades that
regulate tumor cell survival, therapeutic resistance and immune
evasion (8). In particular,
studies have highlighted that alterations in the expression and
function of CAMs are closely associated with the progression of
gastric cancer (9–14). Dysregulated CAM expression, such as
reduced E-cadherin or aberrant integrin profiles, has been linked
to enhanced tumor invasiveness, lymph node metastasis and poor
clinical prognosis in patients with gastric carcinoma (2,3,6–8,11).
These findings suggest that CAMs play a critical role not only in
early tumor development but also in the acquisition of metastatic
traits in gastric cancer. The present study focused on a specific
CAM, gicerin, that has been implicated in gastric cancer
progression. Through an analysis of its expression profile and
functional roles, the aim of this study was to elucidate its
potential as a novel therapeutic target in gastric cancer
treatment.
Gicerin, originally identified from chicken gizzard
smooth muscle, shares high homology with mammalian CD146/MelCAM
(15–19). As described in more detail below,
it has five immunoglobulin-like domains in its extracellular region
and two major isoforms [long (L)-gicerin and short (S)-gicerin)]
have been shown to be generated by alternative splicing (19–22).
Gicerin is a single-pass transmembrane glycoprotein localized to
the cell membrane and comprises a large extracellular domain, a
single transmembrane segment and a short cytoplasmic domain. The
extracellular region contains five immunoglobulin-like loop
structures, two of the V-set type and three of the C2-set type, as
well as several potential N-linked glycosylation sites. The
cytoplasmic domain is predicted to contain four serine/threonine
phosphorylation sites, suggesting that gicerin may be involved in
cell adhesion and intracellular signal transduction.
Gicerin has been shown to engage in both homophilic
and heterophilic interactions, including those with specific
laminins, influencing processes such as cell migration and neurite
extension (19,20). Although it is widely expressed
during embryogenesis, gicerin is limited in normal adult tissues to
sites such as vascular endothelium and muscle (19,20).
However, re-expression of gicerin in malignant tumors has been
shown to be associated with enhanced invasiveness and metastatic
potential (15,19,20).
Gastric cancer is a common gastrointestinal
malignancy, frequently metastasizing to the liver and peritoneum,
with occasional, but significant, pulmonary metastases (23–25).
Studies have indicated that various cell adhesion molecules are
critical regulators of metastasis in gastric cancer, highlighting
them as potential therapeutic targets (9–14,26,27).
Cell adhesion molecules, such as E-cadherin, claudin, contactin 1,
mucin 1 and L1 cell adhesion molecule, are known to facilitate or
inhibit tumor progression and gicerin is likely to have a
comparable role in gastric cancer.
NUGC-4, a poorly differentiated gastric
adenocarcinoma cell line, is commonly used for in vivo
modeling of tumor growth and metastasis (28). In the present study, it was shown
that NUGC-4 cells expressed gicerin and the study also evaluated
how anti-gicerin antibodies affect their subcutaneous
tumorigenicity and lung metastatic potential in nude mice.
Materials and methods
Immunization and purification of
anti-gicerin antibodies
To generate polyclonal antibodies against gicerin,
10 female BALB/c mice (6–8 weeks old) were immunized with
recombinant gicerin protein. The primary immunization was performed
subcutaneously with 50 µg of recombinant rat gicerin protein (aa
1–545; accession number Q9EPF2-1) (29), which exhibits ~70% peptide-sequence
homology to the human gicerin ortholog, emulsified in an equal
volume of Complete Freund's Adjuvant (MilliporeSigma). Booster
injections were administered on days 14 and 28 using an identical
dose of antigen emulsified in Incomplete Freund's Adjuvant
(MilliporeSigma). On day 42, mice were deeply anesthetized using
isoflurane (induction at 3–5% and maintenance at 2–3% in oxygen)
and the absence of reflex responses (pedal withdrawal and palpebral
reflexes) was confirmed before proceeding. For terminal blood
collection, cardiac puncture was performed under deep anesthesia
using a 1 ml syringe with a 23-gauge needle. Blood (~0.5–1.0 ml)
was withdrawn from the left ventricle. Following the procedure,
sacrifice of the mice was performed by cervical dislocation while
under deep anesthesia, or alternatively by thoracotomy to confirm
the cessation of cardiac activity. Finally, mortality was verified
by the absence of a heartbeat and fixed, dilated pupils. All animal
procedures were conducted in accordance with institutional
guidelines and were approved by the Animal Care and Use Committee
of Kyoto Prefecture University (approval no. KPU240401). A total of
55 mice, including those reserved as backups, were used for
antibody production and tumor cell transplantation in the present
study.
Collected blood samples were allowed to clot at room
temperature for 1 h prior to centrifugation at 1,500 × g for 10 min
at 4°C to isolate serum. The sera from all mice were pooled and
total IgG was purified using a Protein G Sepharose column (GE
Healthcare) following the manufacturer's protocol. Bound IgG was
eluted with 0.1 M glycine-HCl buffer (pH 2.7) and immediately
neutralized with 1 M Tris-HCl (pH 9.0). The eluates were
subsequently dialyzed against phosphate-buffered saline (PBS) (pH
7.4) and the IgG concentrations were determined
spectrophotometrically. Similarly, serum was collected from
non-immunized normal mice and IgG was purified using protein G
affinity chromatography. The resulting antibodies were used as
pre-immune IgG.
For the isolation of gicerin-specific antibodies,
affinity purification was performed using a custom-made column
prepared by covalently coupling recombinant gicerin protein to
CNBr-activated Sepharose 4B beads (Cytiva). The purified IgG was
subsequently loaded onto the affinity column equilibrated with PBS.
After extensive washing, bound antibodies were eluted with 0.1 M
glycine-HCl buffer (pH 2.7) and immediately neutralized, as
aforementioned. The eluted antibodies were then dialyzed against
PBS and protein concentrations were quantified using a BCA Protein
Assay Kit (Thermo Fisher Scientific, Inc.). The final purified
anti-gicerin antibodies were subsequently adjusted to a
concentration of 1 mg/ml in PBS and stored at −80°C until use. The
overview of antibody production and purification is shown in
Fig. 1A.
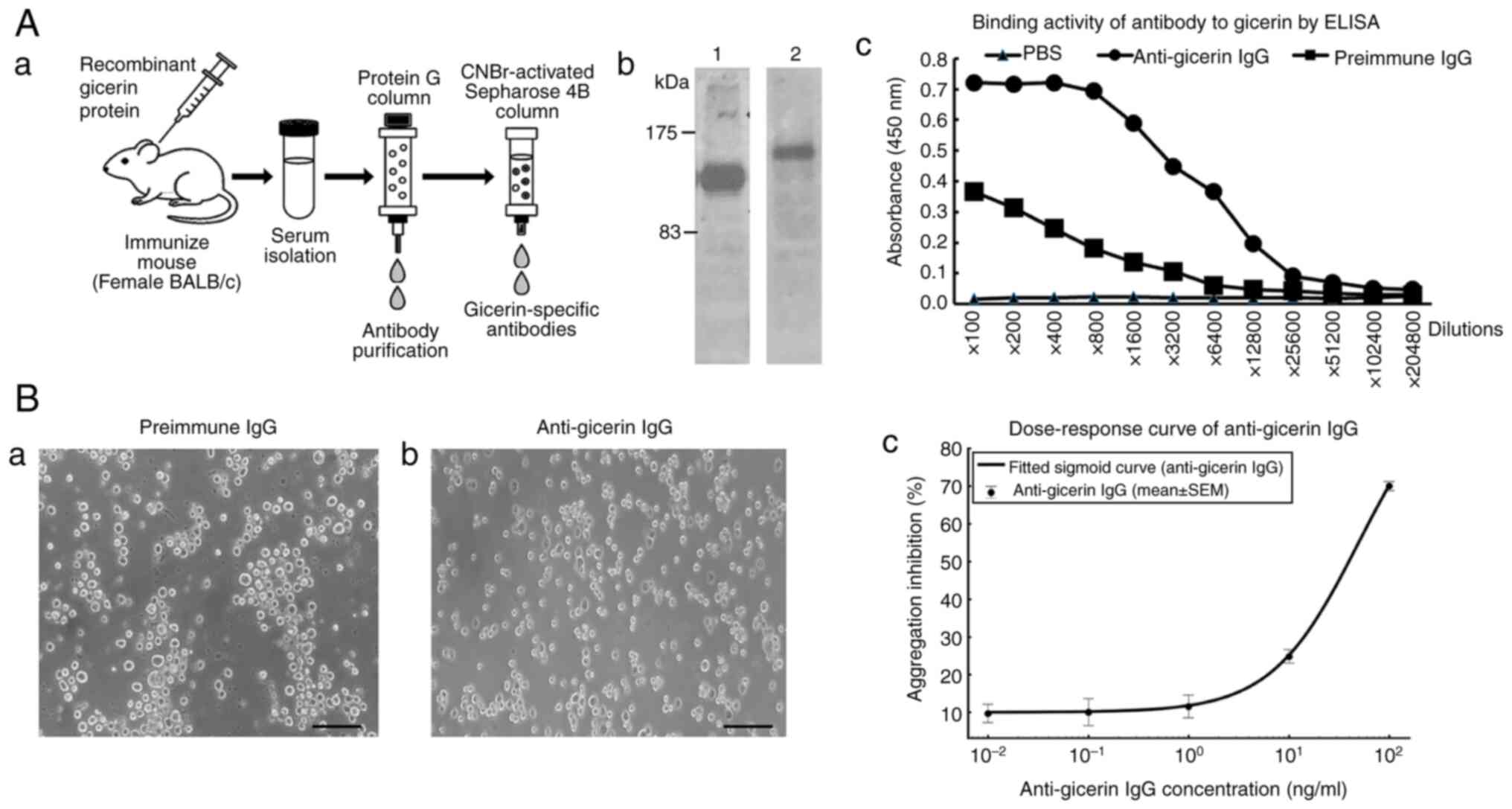 | Figure 1.Generation of mouse anti-gicerin
antibody and validation of its binding activity. (A) The procedure
for generating and purifying the murine anti-gicerin antibody is
summarized. (a) BALB/c mice were immunized with recombinant gicerin
protein, followed by IgG purification from serum. The IgG fraction
was subsequently affinity-purified using a Sepharose column
conjugated with gicerin protein. (b) As shown by SDS-PAGE, the
gicerin recombinant protein immunized as the antigen for antibody
production (lane 1) appeared as a single band at ~120 kDa, while
the final purified mouse IgG (lane 2) exhibited a single band at
~150 kDa. Binding activity of mouse IgG to gicerin proteins by
ELISA. (c) Recombinant proteins of gicerin were bound to plate at 2
µg/ml overnight at 4°C. Samples were blocked with 1% blocking
buffer (Block Ace) in distilled water for 2 h. Serial dilutions of
pre-immune or anti-gicerin IgG were incubated at room temperature
for 2 h. After washing with PBS, HRP-conjugated polyclonal antibody
against mouse IgGs were reacted for 1 h at room temperature. Then,
3,3′,5,5′-tetramethylbenzidine was added to each well and plates
were read at 450 nm utilizing a Multiscan JX (Thermo Fisher
Scientific, Inc.). ELISA demonstrated that the purified antibody
exhibited strong binding activity to gicerin protein, with an ELISA
titer of 12,800 when compared to pre-immune IgG. (B) Neutralizing
activity of anti-gicerin IgG in cell adhesion. (a) In the cell
aggregation assay, L-929 cells expressing gicerin formed
multicellular aggregates upon the addition of pre-immune IgG,
indicating cell-cell adhesion. (b) When the anti-gicerin IgG was
added to the suspension cultures, a marked inhibition of cell-cell
aggregation was observed. Scale bars, 100 µm; magnification, ×200.
(c) Dose-response curves were generated by plotting the aggregation
index against the logarithmic antibody concentrations. Nonlinear
regression analysis determined that the IC50 of
anti-gicerin IgG was 49.5 ng/ml, indicating its potent neutralizing
effect on cell adhesion. IC50, half-maximal inhibitory
concentration. |
Sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and Coomassie Brilliant Blue (CBB)
Staining
Recombinant gicerin protein and mouse antibodies
were separated by SDS-PAGE on 8% gels. Protein samples were mixed
with Laemmli sample buffer, heated at 95°C for 5 min and loaded
onto an 8% polyacrylamide gel. Electrophoresis was carried out
under reducing conditions using a Mini-PROTEAN Tetra System
(Bio-Rad Laboratories, Inc.) at a constant voltage of 100 V. After
electrophoresis, the gel was briefly rinsed with distilled water
and stained using Coomassie Brilliant Blue R-250 staining solution
(Bio-Rad Laboratories, Inc.) for 1 h at room temperature. The gel
was subsequently destained with a solution containing 40% methanol
and 10% acetic acid until protein bands became clearly visible.
Enzyme linked immunosorbent assay
(ELISA)
Each well of the polystyrene ELISA plates (Sumitomo
Bakelite Co., Ltd.) was coated with 0.2 µg of recombinant gicerin
proteins in PBS and the plate was incubated over night at 4°C. Each
of the following incubation steps were preceded by washing the
wells twice with PBS containing 0.05% Tween 20. The wells were
blocked for nonspecific binding by the addition of a commercial
blocking buffer (DS Pharma Biomedical Co., Ltd.) and were incubated
at 37°C for 2 h. The serial dilutions of pre-immune IgG and
anti-gicerin IgG were added vertically to the wells and kept for
incubation at 37°C for 1 h. The HRP-conjugated rabbit IgG diluted
(1:5,000) in PBS was dispensed into each well. The plate was
incubated for 1 h at 37°C. Later, a substrate buffer containing TMB
(Sumitomo Bakelite Co., Ltd.) was added to each well and kept for
incubation at 37°C for 15 min. The reaction was terminated by the
addition of a stopping reagent (1.25M sulfuric acid). The
absorbance was recorded at 450 nm using the ELISA plate reader (DS
Pharma Biomedical Co., Ltd.). The ELISA titer was defined as the
highest dilution factor of anti-gicerin IgG that yielded an
absorbance value at least three times higher than that obtained
with pre-immune IgG.
Neutralization activity of
anti-gicerin IgG on cell-cell interactions
Reaggregation assay and determination of
IC50 for anti-gicerin antibody: A reaggregation assay
was performed to evaluate the inhibitory effect of anti-gicerin
mouse IgG on cell-cell adhesion in gicerin transfected L929 cells
(RIKEN BioResource Research Center; cat. no. RCB2619) (30). The cells were cultured in
Dulbecco's Modified Eagle Medium (DMEM; high glucose) supplemented
with L-glutamine, sodium pyruvate, 10% fetal bovine serum (FBS) and
antibiotics (Nacalai Tesque, Inc.) and maintained at 37°C in a
humidified atmosphere containing 5% CO2. Then, the cells
were detached using 0.02% EDTA in calcium- and magnesium-free PBS
to obtain a single-cell suspension. The cells were then washed
twice with calcium- and magnesium-containing Hank's Balanced Salt
Solution (HBSS) and resuspended at a concentration of
1×106 cells/ml. Serial dilutions of purified
anti-gicerin mouse IgG (0, 0.01, 0.1, 1, 10 and 100 ng/ml) or
control non-immune mouse IgG were prepared in HBSS. Equal volumes
of antibody solution and cell suspension were mixed and transferred
to 96-well U-bottom low-adhesion plates (100 µl/well). The mixtures
were incubated at 37°C for 30 min to allow antibody binding.
Subsequently, the plate was gently agitated on an orbital shaker
(50 rpm) and incubated for an additional 45 min at 37°C to promote
cell-cell reaggregation. Following incubation, the degree of cell
aggregation in each well was assessed by phase-contrast microscopy.
A cell aggregate composed of at least four cells was counted as a
single unit for analysis (21).
Images of representative fields (five per well) were captured at
×200 magnification. Aggregation was quantified by calculating the
inhibition of cell aggregation (%), defined as follows: Inhibition
of cell aggregation (%)=100 × (1-number of single cells/total
number of cells).
Cell line and culture conditions
NUGC-4 cells (JCRB Cell Bank; cat no. JCRB0834),
HUVEC cells [JCRB Cell Bank; cat. no. IFO50271; F10 (passage 10)]
and murine melanoma B16 cells (RIKEN BioResource Research Center;
cat. no. RCB1283) were cultured in DMEM (high glucose) supplemented
with L-glutamine, sodium pyruvate, 10% FBS and antibiotics (Nacalai
Tesque, Inc.) and maintained at 37°C in a humidified atmosphere
containing 5% CO2.
Confirmation of gicerin expression on
NUGC-4 cells and HUVECs
Monolayers of NUGC-4 and HUVEC cells were fixed
using Zamboni's fixative and incubated with a murine polyclonal
anti-gicerin antibody (1:500; generated as aforementioned) at 37°C,
followed by an FITC-conjugated goat anti-mouse IgG (1:100;
MilliporeSigma; cat. no. AP124F) at 37°C. In addition, tumor
tissues harvested from mice were fixed in Zamboni's solution and
frozen sections (20 µm thickness) were subjected to
immunofluorescence staining using the anti-gicerin primary antibody
followed by the FITC-conjugated secondary antibody as
aforementioned. Fluorescence microscopy revealed the distinct
membrane-localized expression of gicerin on cells. A commercially
available mounting medium containing DAPI was employed following
the manufacturer's instructions. While nuclear staining was
achieved, the combined display of phase-contrast, FITC (gicerin)
and DAPI slightly reduced the clarity of cell contours in our
setup; therefore, the final images are presented without DAPI to
best illustrate the relationship between cell morphology and
gicerin expression in the same field of view.
Protein extraction and western blot
analysis
Cultured NUGC-4, HUVEC and B16 cells were rinsed
twice with calcium- and magnesium-free Hanks' balanced salt
solution and collected using a cell scraper. The harvested
monolayer cells were subsequently homogenized in PBS using a
Polytron homogenizer. The homogenates were centrifuged at 1,000 × g
for 10 min at 4°C to remove cellular debris, followed by
centrifugation of the supernatants at 18,000 × g for 30 min at 4°C.
The resulting pellets were resuspended in 10 mM Tris-acetate buffer
(pH 8.0) containing 1 mM EDTA and 0.5% Nonidet P-40 and incubated
at 4°C for 1 h with continuous rotation. The suspensions were
subsequently centrifuged at 40,000 × g for 90 min at 4°C and the
final supernatants were collected for western blot analysis, as
described by Taniura et al (31). Protein concentrations were
quantified using BCA Protein Assay Reagent (Pierce; Thermo Fisher
Scientific, Inc.) at an absorbance of 562 nm, with bovine serum
albumin as a standard. Equal amounts of total protein (20 µg)
extracted from HUVEC, NUGC-4 and B16 cells, along with 10 ng
recombinant gicerin protein and mouse IgG, were separated by
SDS-PAGE using 7.5% polyacrylamide gels and electrotransferred on
to polyvinylidene difluoride (PVDF) membranes (Bio-Rad
Laboratories, Inc.). The membranes were blocked with Bullet
Blocking One reagent (Nacalai Tesque, Inc.; cat. no. 1379) and
subsequently incubated for 1 h at room temperature with primary
anti-gicerin antibodies which were generated as aforementioned)
diluted 1:1,000 in 2% skimmed milk in Tris-buffered saline (TBS)
buffer (150 mM NaCl/10 mM Tris-HCl, pH 8.0). After six washes with
TBST buffer (TBS buffer containing 0.05% Tween-20), the membranes
were incubated for 1 h at room temperature with horseradish
peroxidase-conjugated goat anti-rabbit IgG (Nacalai Tesque, Inc.;
cat. no. 21860-61l) diluted 1:1,000 in TBS buffer containing 2%
skimmed milk. Following four washes with TBST and two final washes
with TBS, immunoreactive bands were visualized using 0.05%
diaminobenzidine (MilliporeSigma) in 50 mM Tris-HCl (pH 7.6)
containing 0.03% hydrogen peroxide for 5 min at room temperature.
The reaction was stopped by rinsing in distilled water. Images of
the membranes were captured using a ChemiDoc imaging system
(Bio-Rad Laboratories, Inc.) as previously described (19,20).
Subcutaneous tumor transplantation
model
Subcutaneous implantation of tumor cells into the
cervical region of nude mice offers several advantages (32). The cervical area provides a soft
and accessible site that facilitates precise and reproducible
injection of the cell suspension, minimizing procedural
variability. Tumors formed in this region tend to grow outward
beneath the skin, allowing for easy visual assessment and accurate
measurement of tumor size using calipers. Additionally, the
relatively stable blood supply in the cervical skin reduces the
risk of ulceration, thereby enabling long-term observation and
reliable evaluation of therapeutic efficacy. Since the tumor
remains localized in the cervical area, vital organs such as those
of the gastrointestinal and urinary systems are less likely to be
affected, contributing to the maintenance of the general condition
of the animals and allowing for consistent assessment of drug
effects and toxicity. Moreover, because cervical tumors rarely
interfere with the mobility or feeding behavior of the animals,
this model supports the ethical conduct of experiments while
minimizing distress to the animals. After harvesting the NUGC-4
cells with trypsin-EDTA (Nacalai Tesque, Inc.), the cells were
split into two groups. One group was incubated with anti-gicerin
IgG (10 µg/ml), whereas the other was incubated with pre-immune IgG
(10 µg/ml) for 1 h at 37°C. Cells were washed and resuspended in
serum-free DMEM (3×107 cells/ml) and 0.2 ml cell
suspension was injected subcutaneously into the dorsal cervical
region of nude mice (9-week-old female BALB/c Slc-nu/nu mice; n=5
per group). Tumor volume was calculated every other day starting at
1-week post-inoculation using the formula (AxB2)/2,
where A and B represent the largest and smallest tumor diameters,
respectively.
After the final measurement, mice were deeply
anesthetized using isoflurane (induction at 3–5% and maintenance at
2–3% in oxygen) and the absence of reflex responses (pedal
withdrawal and palpebral reflexes) was confirmed before proceeding
with the experiment. Sacrifice was performed by cervical
dislocation while the mice were in a state of deep anesthesia, or
alternatively by thoracotomy to confirm the cessation of cardiac
activity. Mortality was verified by the absence of a heartbeat and
fixed, dilated pupils. In accordance with institutional animal care
guidelines, the maximum allowable tumor volume was limited to 1.5
cm3. Mice were sacrificed immediately upon reaching this
limit or upon exhibiting signs of pain, ulceration or impaired
mobility; however, no animals exhibited such conditions during the
course of the experiment.
Pathologically, the tumors were excised, 10%
buffered-formalin-fixed for 48 h at room temperature,
paraffin-embedded and stained with hematoxylin for 5 min at room
temperature (22–25°C), followed by eosin staining for 3 min at room
temperature [hematoxylin and eosin (H&E) staining] for a
histopathological assessment of local invasion. For
immunofluorescence staining, tumor tissues were fixed in Zamboni's
fixative, followed by cryoprotection in 20% sucrose solution for 24
h at 4°C. The samples were subsequently frozen at −30°C and 20-µm
frozen sections were prepared using a cryostat. The frozen sections
were blocked with 1% non-fat dry milk in PBS containing 0.1%
Tween-20 for 1 h at room temperature, and were subsequently
incubated with a murine polyclonal anti-gicerin antibody (1:500;
generated as aforementioned) at 37°C, followed by an
FITC-conjugated goat anti-mouse IgG (1:100; MilliporeSigma; cat.
no. AP124F) at 37°C as aforementioned. Fluorescence microscopy
revealed the distinct membrane-localized expression of gicerin on
cells.
Hematogenous pulmonary metastasis
model
NUGC-4 cells (2.5×106 cells/ml) were
pretreated with anti-gicerin antibodies or pre-immune IgG under the
same conditions. Each mouse (n=5 per group) received a 0.1 ml
tail-vein injection of either of the treated cell suspensions.
After 1 week, mice were sacrificed and lungs were collected,
formalin-fixed, paraffin-embedded and stained with H&E, as
aforementioned. The number of metastatic foci was counted in all
lung lobes and expressed as nodules per cm2. In these
experiments, mice were deeply anesthetized using isoflurane
(induction at 3–5% and maintenance at 2–3% in oxygen) and the
absence of reflex responses (pedal withdrawal and palpebral
reflexes) was confirmed prior to the performance of any procedure.
Sacrifice was performed as aforementioned and mortality was
verified by the absence of a heartbeat and the presence of fixed,
dilated pupils. In cases where mice exhibited clinical signs such
as respiratory distress or lethargy, sacrifice was to have been
performed immediately to minimize suffering; however, no animals
exhibited such symptoms during the course of the experiment.
In vitro cell adhesion of NUGC-4 cells to gicerin
proteins: Cell adhesion experiments were carried out as
reported previously (21).
Aliquots (1 ml) of PBS containing 10ng/ml of recombinant gicerin
proteins were spotted on a 35 mm culture dish and incubated for 1 h
in a humidified 5% CO2 in incubator at 37°C. Then the
dishes were washed with DMEM three times and preincubated with DMEM
containing 10% FBS for 1 h to block nonspecific binding. Aliquots
(2 ml) of the cell suspension (1×106 cells/ml) were
plated on the test dishes. To assess the inhibition of adhesion
activity by anti-gicerin antibody, the cells were preincubated for
30 min at 37°C with the pre-immune IgG or anti-gicerin IgG at a
final concentration of 100 ng/ml before plating on the dishes. The
test dishes were incubated for 1 h at 37°C, washed twice with DMEM
and the adhesion cells were observed by phase-contrast
microscopy.
In vitro cell adhesion of NUGC-4 cells
to vascular endothelial cells
Endogenous gicerin-positive HUVEC cells, a vascular
endothelial cell line, were cultured on dishes with DMEM containing
10% FBS at 37°C and used as feeder layers. The NUGC-4 cell
suspensions were preincubated with anti-gicerin polyclonal
antibodies or pre-immune IgG for 1 h at 37°C. After five washing in
PBS and centrifugation at 800 × g for 10 min at 4°C, the NUGC-4
cells were resuspended in DMEM and seeded on to the HUVEC
monolayers. Following a 30 min incubation at 37°C, the cultures
were gently washed with DMEM and removed and the NUGC-4 cells
adhering to the feeder layers were observed under a light
microscope (Nikon Corporation). The cell number per area was
determined for five areas and the average scores and standard
deviation (SD) were calculated.
Measurement of doubling time of NUGC-4
cells with antibodies
NUGC-4 cells were seeded into 6-well tissue culture
plates at a density of 2×104 cells per well in complete
DMEM supplemented with 10% FBS. The cells were incubated at 37°C in
a humidified atmosphere containing 5% CO2. After 4 h to
allow for cell attachment, 10 µl of phosphate-buffered saline
(PBS), 10 µl of pre-immune IgG, or 10 µl (10 ng/ml) of anti-gicerin
IgG (10 ng/ml) was added to each well, respectively. For each
treatment group, a total of four independent plates (n=4) were
prepared and analyzed. At 0, 24, 48 and 72 h after seeding, one
well from each plate was harvested. Cells were gently washed with
PBS, detached using 0.25% trypsin-EDTA and resuspended in culture
medium. Cell viability was assessed using trypan blue exclusion and
viable cells were counted with a hemocytometer. Only unstained
(viable) cells were included in the count.
Doubling time (Td) was calculated using
the following formula:
where t is the elapsed time (in h),
N0 is the number of viable cells at time 0 and
Nt is the number of viable cells at time t. The mean
doubling time were calculated for each treatment group. Statistical
comparison was performed between the pre-immune IgG group and
anti-gicerin IgG using Student's t-test.
In vitro cell migration assay
NUGC-4 cells on culture dishes coated with gicerin
protein (as aforementioned) were cultured in DMEM supplemented with
10% FBS at 37°C in a humidified atmosphere containing 5%
CO2 until they reached confluence. A straight scratch
was made across the center of each well using a sterile yellow
200-µl micropipette tip to detach cells along the line. The medium
was carefully aspirated to remove the detached cells and replaced
with fresh serum-free DMEM. The cells were then treated with either
pre-immune IgG (100 ng/ml) or anti-gicerin IgG (100 ng/ml) and
incubated under the same conditions. Cell migration into the
scratched area was monitored daily under a light microscope for up
to 96 h.
Complement-dependent cytotoxicity
(CDC) assay
NUGC-4 cells were cultured in DMEM supplemented with
10% FBS and seeded into 24-well culture plates (Corning, Inc.).
Cells were subsequently incubated at 37°C in a humidified
atmosphere containing 5% CO2 until they reached
semi-confluence. After removal of the culture medium, cells were
gently rinsed with PBS. Subsequently, serum-free DMEM containing
either pre-immune IgG or anti-gicerin polyclonal antibody was added
to each well. The final antibody concentration was adjusted to 10
µg/ml. Cells were incubated at 37°C for 1 h. Following antibody
treatment, cells were washed gently two or three times with PBS to
remove any unbound antibodies. Fresh mouse serum
(complement-active) or heat-inactivated mouse serum (treated at
56°C for 30 min) was subsequently diluted in serum-free DMEM to a
final concentration of 20% and added to the cells. After a 1 h
incubation at 37°C, the supernatant containing non-adherent cells
was collected and temporarily maintained at 37°C, while the
adherent cells were detached using trypsin-EDTA. The cell
suspension and supernatant were mixed in the tubes and subsequently
centrifuged at 300 × g for 5 min. The pellet was then resuspended
in either PBS or serum-free DMEM. Cell viability was subsequently
assessed using the trypan blue exclusion method. Equal volumes of
cell suspension and 0.4% trypan blue solution were mixed and the
mixture was loaded onto a hemocytometer. Viable (unstained) and
non-viable (blue-stained) cells were counted under a light
microscope and the cell death rate (as a percentage) was calculated
using the following formula: Cell death rate (%)=number of
blue-stained cells/total number of cells ×100. At least five
independent wells were analyzed per treatment group.
Statistical analysis
All quantitative data are presented as mean ±
standard deviation (SD). For in vivo experiments, n=5 mice
per group; for in vitro assays, at least n=4 independent
replicates per group unless otherwise indicated. Two-tailed,
unpaired Student's t-tests were used for single pairwise
comparisons and one-way analysis of variance (ANOVA) followed by
Tukey's honestly significant difference (HSD) test was used for
≥3-group comparisons. Exact P-values are reported on the graphs
and/or in the figure legends; where thresholds are shown,
significance is denoted as *P<0.05, **P<0.01 and *P<0.001;
‘ns’ indicates P≥0.05. Error bars on all graphs represent SD and
all ‘±’ values denote SD. P<0.05 was considered to indicate a
statistically significant difference.
Results
Generation and purification of murine
anti-gicerin antibody
The procedure for generating and purifying the
murine anti-gicerin antibody is summarized in Fig. 1A. BALB/c mice were immunized with
recombinant gicerin protein, followed by IgG purification from
serum. The IgG fraction was subsequently affinity-purified using a
Sepharose column conjugated with gicerin protein. SDS-PAGE analysis
revealed a single band at ~150 kDa in the final purified fraction,
indicating that a highly pure IgG preparation was obtained.
Furthermore, ELISA demonstrated that the purified antibody
exhibited strong binding activity to gicerin protein, with an ELISA
titer of 12,800 when compared to pre-immune IgG.
Neutralizing activity of anti-gicerin
IgG in cell adhesion
The neutralizing activity of anti-gicerin IgG was
evaluated using a cell aggregation assay. When anti-gicerin IgG was
added to the suspension culture of gicerin-expressing L929 cells, a
marked inhibition of cell-cell aggregation was observed (Fig. 1Ba and Bb). Dose-response curves
were generated by plotting the aggregation index against the
logarithmic antibody concentrations (Fig. 1Bc). Nonlinear regression analysis
determined that the half-maximal inhibitory concentration
(IC50) of anti-gicerin IgG was 49.5 ng/ml, indicating
its potent neutralizing effect on cell adhesion.
Gicerin is expressed in NUGC-4
cells
Immunofluorescence analysis revealed the presence of
distinct membrane-associated signals in NUGC-4 cells, confirming
robust gicerin expression (Fig.
2A). As a limitation of this study, it was unable to perform
co-localization staining with another inner membrane protein. This
was due to the poor adherence of NUGC-4 cells, which made them
susceptible to detachment during the multiple steps required for
double immunofluorescence staining, such as repeated washing,
antibody incubation and prolonged handling. Western blot analysis
demonstrated that gicerin was expressed in the membrane fractions
of B16 and HUVEC cells, both of which are known to endogenously
express gicerin, similarly to the recombinant protein. In addition,
gicerin expression was also detected in NUGC-4 cells (Fig. 2B). Notably, doublet bands were
observed at ~120 kDa, indicating that NUGC-4 cells express both the
S- and the L-isoforms of gicerin (19,20).
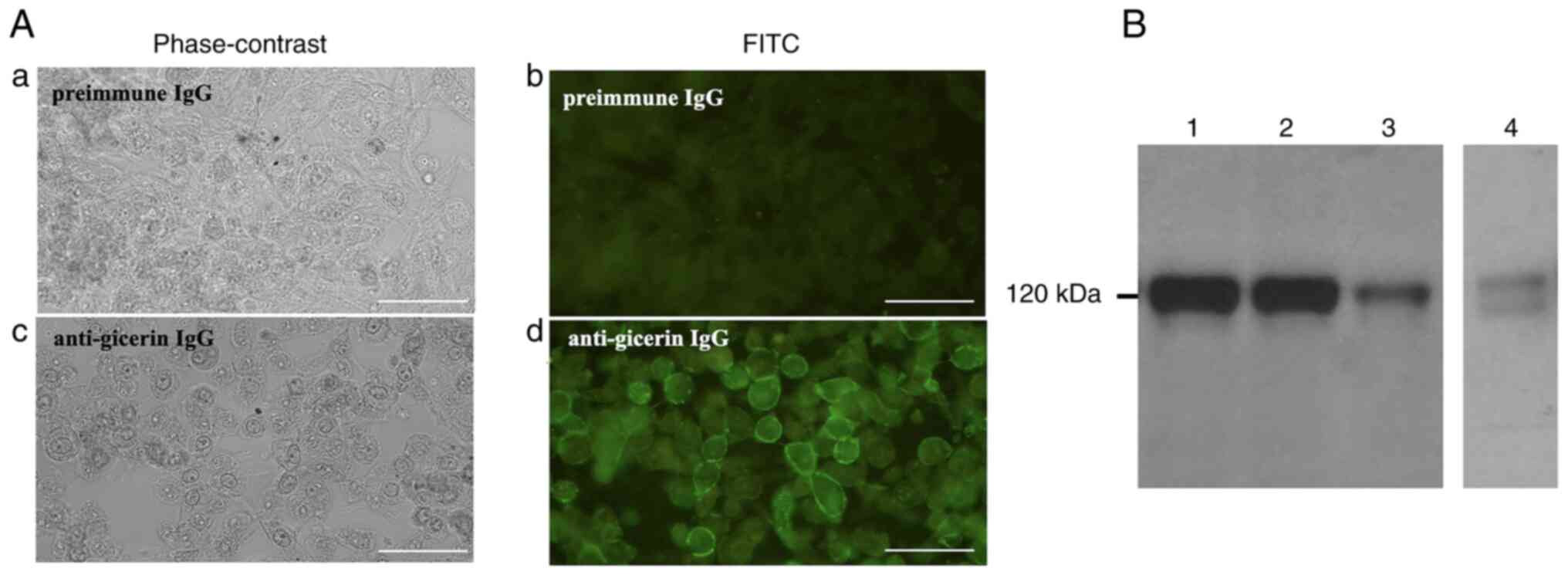 | Figure 2.Gicerin expression in NUGC-4 cells.
(A) Indirect immunofluorescence staining of NUGC-4 cells was
performed and observed under a fluorescence microscope (as shown by
the B-excitation filter). No fluorescence was detected when
pre-immune IgG was applied (a, phase contrast; b,
immunofluorescence). By contrast, strong green fluorescence was
observed on the cell surface following staining with anti-gicerin
IgG (c, phase contrast; d, immunofluorescence), indicating
membrane-associated expression of gicerin. scale bars, 50 µm;
magnification, ×400. (B) Western blot analysis of membrane
fractions from B16 (lane 2), HUVEC (lane 3) and NUGC-4 cells (lane
4) following SDS-PAGE. The recombinant gicerin protein used as the
immunogen was also subjected to electrophoresis as a positive
control and to verify the specificity and reactivity of the
antibody (lane 1). Notably, a doublet band of ~120 kDa was observed
in lane 4, consistent with the expression of both the short and
long isoforms of gicerin in NUGC-4 cells. |
Subcutaneous tumor growth and invasion
is reduced in the subcutaneous tumor model
In the subcutaneous tumor model, tumor masses
derived from cells treated with anti-gicerin antibody exhibited
markedly suppressed tumor growth compared with the control group (P
<0.01; Fig. 3A).
Immunofluorescence analysis further revealed strong gicerin
expression on the membranes of tumor cells and in the endothelial
cells of newly formed blood vessels within tumors located in the
cervicodorsal region of the implanted mice (Fig. 3B). Moreover, histopathological
analysis revealed that tumors in the control group exhibited
extensive invasion into the dermis, muscle and adipose tissue. In
addition, epithelial-like arrangements of tumor cells were observed
in the control group, thereby revealing histological features
consistent with poorly differentiated adenocarcinoma. By contrast,
tumors in the anti-gicerin antibody-treated group were more
circumscribed and exhibited markedly reduced local invasiveness.
Furthermore, frequent and prominent focal necrotic lesions were
observed in tumors from animals administered the anti-gicerin
antibody. Within these tumors, cellular degeneration, structural
disintegration, pyknosis and karyorrhexis were also frequently
noted (Fig. 3C).
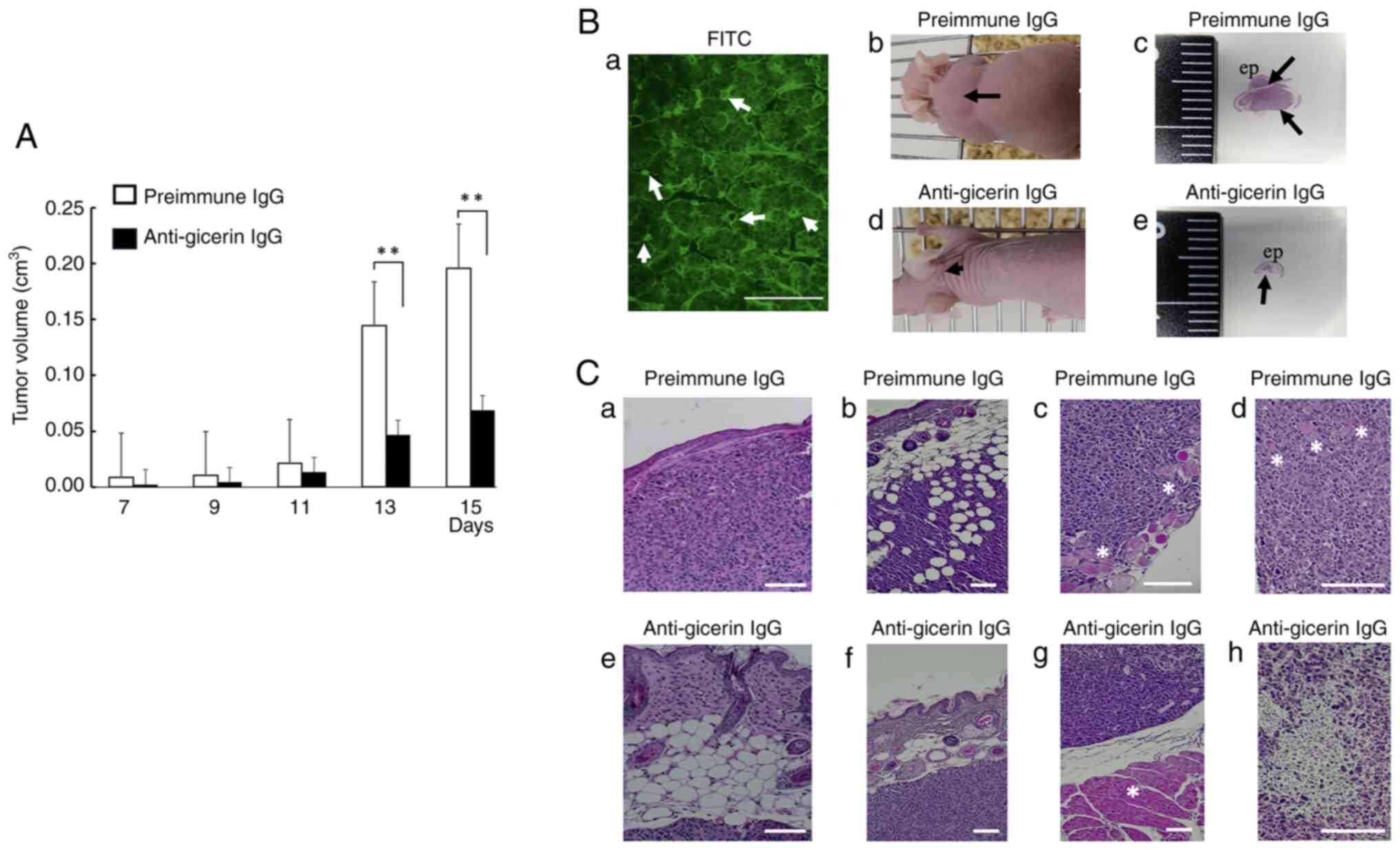 | Figure 3.Tumor growth and histopathological
features following subcutaneous injection into nude mice. (A)
NUGC-4 cells treated with either pre-immune IgG or anti-gicerin IgG
were subcutaneously injected into the dorsal neck region of nude
mice (n=5 per group). Tumor volume was measured over time using the
formula: AxB2/2, where A and B represent the maximum and
minimum tumor diameters, respectively. Data are mean ± SD (n=5 mice
per group). Two-tailed unpaired Student's t-tests were performed
between pre-immune IgG and anti-gicerin IgG at each time point.
Significance indicators: ns (P≥0.05), *P<0.05, **P<0.01. (B)
On day 15 post-injection, tumor tissues were examined both
histologically and using immunofluorescence analysis. (a) Gicerin
expression was detected in the tumor cell membranes, especially in
epithelial-like structures, as well as in neovascular structures
(denoted by the arrows) scale bars, 100 µm; magnification, ×200.
Compared with mice injected with pre-immune IgG (b and c), those
injected with anti-gicerin IgG displayed smaller tumor masses and
reduced tumor areas histologically with hematoxylin and eosin
staining (arrows) (d and e). (C) In the pre-immune IgG group,
histological evidence of tumor invasion into the dermis (a),
adipose tissue (b), subcutaneous muscle (c) and undifferentiated
adenocarcinoma (d) regions was noted (arrows indicate invasion into
the muscle). (e-g) By contrast, mice treated with anti-gicerin IgG
showed markedly reduced tumor invasiveness. (h) Furthermore, signs
of cellular degeneration, structural disintegration, pyknosis and
karyorrhexis were frequently observed within tumors treated with
anti-gicerin antibody. Scale bars, 100 µm; magnification: a and e:
×100; b, f and g: ×50; and c, d and h, ×200. |
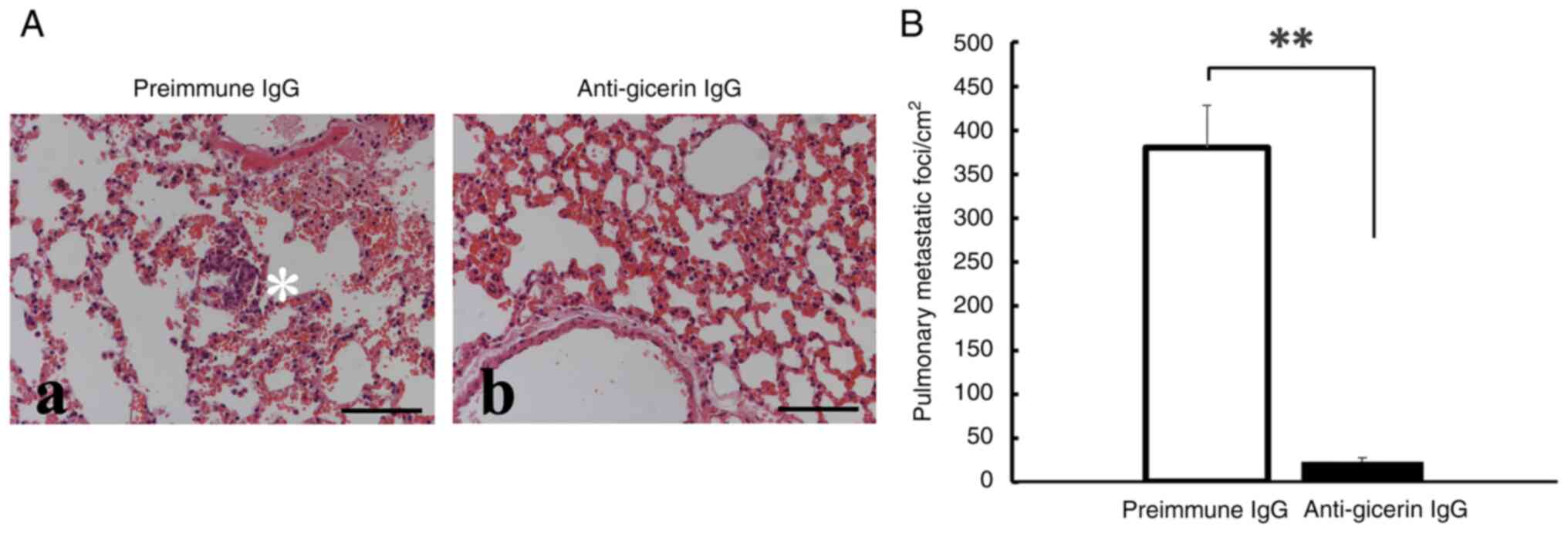
Pulmonary metastasis is suppressed in
the tail-vein injection model
In the tail-vein injection model, mice that received
pre-immune IgG-treated cells were found to develop multiple tumor
nodules in the lung parenchyma, often accompanied by hemorrhagic
changes (Fig. 4). By contrast,
mice injected with anti-gicerin antibody-treated cells exhibited
markedly fewer metastatic lesions, indicating substantial
inhibition of hematogenous spread.
Inhibition of NUGC-4 cell adhesion to
gicerin-coated surfaces by anti-gicerin antibody
To evaluate the role of gicerin in mediating cell
adhesion, culture dishes were coated with recombinant gicerin
protein and seeded with NUGC-4 cells. A substantial number of cells
adhered to the gicerin-coated surface under control conditions.
However, in the presence of anti-gicerin antibody, cell adhesion to
the gicerin-coated areas was markedly inhibited, as evidenced by a
significant reduction in the number of adherent cells (Fig. 5A). These findings demonstrated that
the anti-gicerin antibody effectively neutralizes the homophilic
binding activity of gicerin, thereby suppressing gicerin-mediated
adhesion of NUGC-4 cells.
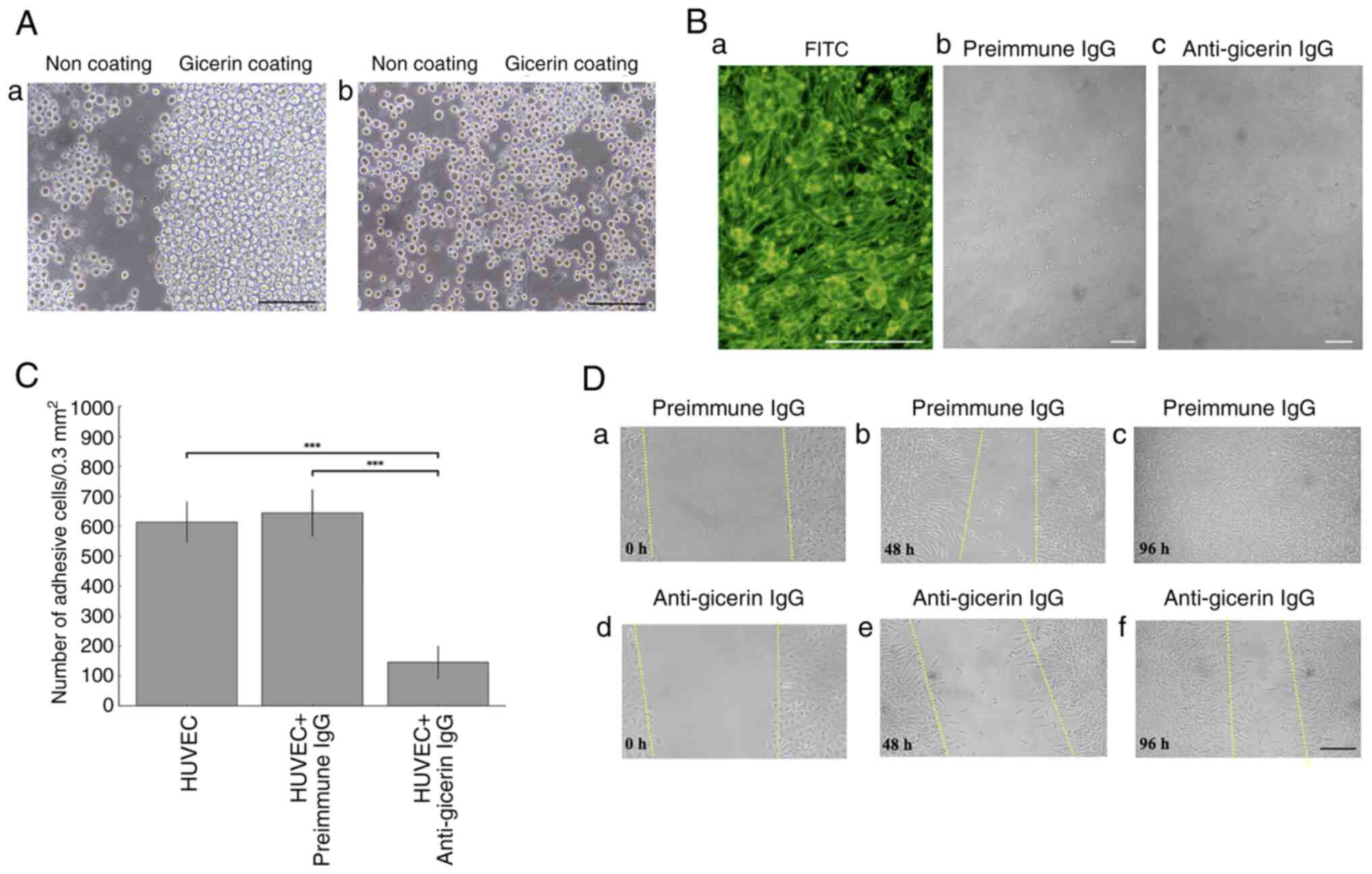 | Figure 5.(A) Inhibition of NUGC-4 cell
adhesion to gicerin protein by anti-gicerin antibody. Only the
right half of each culture dish was coated with recombinant gicerin
protein. NUGC-4 cell adhesion was assessed under a phase-contrast
microscope. (a) The cells predominantly adhered to the
gicerin-coated region, indicating that NUGC-4 cells exhibit
homophilic binding through gicerin molecules expressed on their
surface. Adhesion was not inhibited by pre-immune IgG. (b) By
contrast, treatment with anti-gicerin IgG markedly suppressed the
attachment of NUGC-4 cells to the gicerin-coated area. These
findings demonstrate that anti-gicerin IgG effectively blocks the
homophilic adhesion ability of NUGC-4 cells via gicerin. Scale
bars, 100 µm; magnification ×200. (B) Adhesion of NUGC-4 cells to
HUVEC monolayers. (a) Immunofluorescence staining revealed gicerin
expression on both adherent NUGC-4 cells and HUVEC feeder cells.
NUGC-4 cells treated with either (b) pre-immune IgG or (c)
anti-gicerin IgG were seeded on to confluent HUVEC monolayers.
Compared with the pre-immune IgG group, the number of NUGC-4 cells
attached to HUVEC layers was noticeably reduced in the anti-gicerin
IgG group. Scale bar, 100 µm. Magnification: a, ×200; and b and c,
×50. (C) The number of NUGC-4 cells adhering to HUVECs was markedly
lower in the anti-gicerin IgG group compared with the pre-immune
control. Adherent cells were counted in randomly selected 0.3
mm2 areas under light microscopy. Data are mean ± SD
(n=5 independent replicates per group). Group differences were
analyzed by one-way ANOVA followed by Tukey's HSD test across PBS,
pre-immune IgG and anti-gicerin IgG conditions. Anti-gicerin IgG
differed markedly from the other groups (***P<0.001), whereas
other pairwise comparisons were not significant unless indicated.
(D) Inhibition of NUGC-4 cell migration by anti-gicerin antibody.
NUGC-4 cells were seeded on culture dishes pre-coated with gicerin
protein and incubated until they reached confluence. (a) A linear
scratch was made across the cell monolayer using the tip of a
yellow pipette to create a cell-free area. (b) Culture media
containing either pre-immune IgG or anti-gicerin IgG was then added
and cell migration into the scratched area was monitored over time
using phase-contrast microscopy. (b) In the presence of pre-immune
IgG, numerous cells migrated into the scratch area by 48 h and (c)
the wound was completely closed by 96 h. (d) By contrast, dishes
treated with anti-gicerin IgG showed (e) markedly reduced cell
migration at 48 h and (f) a large cell-free area remained even
after 96 h. These findings indicate that anti-gicerin IgG inhibits
gicerin-mediated cell motility. The dashed straight line indicates
the edge of the wound at each time point after the initial scratch
procedure. Scale bar, 100 µm; magnification: ×100. |
In vitro cell adhesion of the NUGC-4
cells to vascular endothelial cells
When HUVECs were used as a feeder layer and NUGC-4
cells were seeded, a substantial number of NUGC-4 cells adhered to
the surface of HUVECs. Immunofluorescence staining subsequently
revealed that gicerin was positively expressed in the monolayer of
HUVEC cells, as well as in the NUGC-4 cells adhering to them
(Fig. 5B). In this experiment,
two-color staining was attempted to visualize HUVECs, which served
as the feeder layer and the NUGC-4 cells adhered to them. However,
successful fluorescence-based visualization could not be achieved.
The reason for this was that the adhesion of NUGC-4 cells to HUVECs
was weak and during the double-staining procedure, where gicerin on
HUVECs was labeled red with rhodamine and gicerin on NUGC-4 cells
was labeled green using a FITC-conjugated antibody, the NUGC-4
cells detached. Therefore, it was not possible to perform dual
staining with different dyes. In this condition, NUGC-4 cells
adhered to the surface of HUVECs with a small, rounded morphology.
Based on this morphology and the observed fluorescence, both NUGC-4
cells and HUVECs were confirmed to express gicerin, as evidenced by
the green fluorescence emitted by both cell types. Notably, the
fluorescence intensity was stronger in NUGC-4 cells. These findings
clearly demonstrate gicerin positivity in both cell types through
their morphological and fluorescence characteristics.
The addition of an anti-gicerin antibody markedly
decreased the number of NUGC-4 cells that adhered to HUVECs
compared with the addition of pre-immune IgG, demonstrating that
the anti-gicerin antibody could inhibit the adhesion of NUGC-4
cells to vascular endothelial cells (Fig. 5C).
In vitro cell migration assay
To examine the involvement of gicerin in the
migratory potential of NUGC-4 cells, an in vitro scratch
assay were performed. Notably, NUGC-4 cells treated with pre-immune
IgG exhibited markedly greater migration into the scratched area
compared with those treated with anti-gicerin IgG (Fig. 5D). By day 4, the scratched area was
completely closed in the pre-immune IgG group, whereas cell
migration was markedly delayed in the group treated with
anti-gicerin antibody. These findings suggested that gicerin
expression facilitates the migration of NUGC-4 cells.
Measurement of cell death by CDC
assay
When pre-immune IgG was used, the addition of fresh
serum did not induce cell death. By contrast, significant levels of
cell death were observed when fresh murine serum was added to
NUGC-4 cells preincubated with anti-gicerin IgG; however, no cell
death occurred when heat-inactivated serum was used (Fig. 6A). Taken together, these results
suggested that complement activity in the serum, engaged by the
anti-gicerin IgG bound to NUGC-4 cells, is responsible for inducing
cell death (Fig. 6B). The doubling
time of NUGC-4 cells was 23.4 h. When pre-immune IgG was added, the
doubling time was 24.7 h and when anti-gicerin IgG was added, it
was 22.5 h (Fig. 6C). These
findings suggest that the addition of anti-gicerin IgG does not
affect the proliferation of gastric cancer cells: no significant
difference between pre-immune IgG and anti-gicerin IgG groups;
(P<0.05).
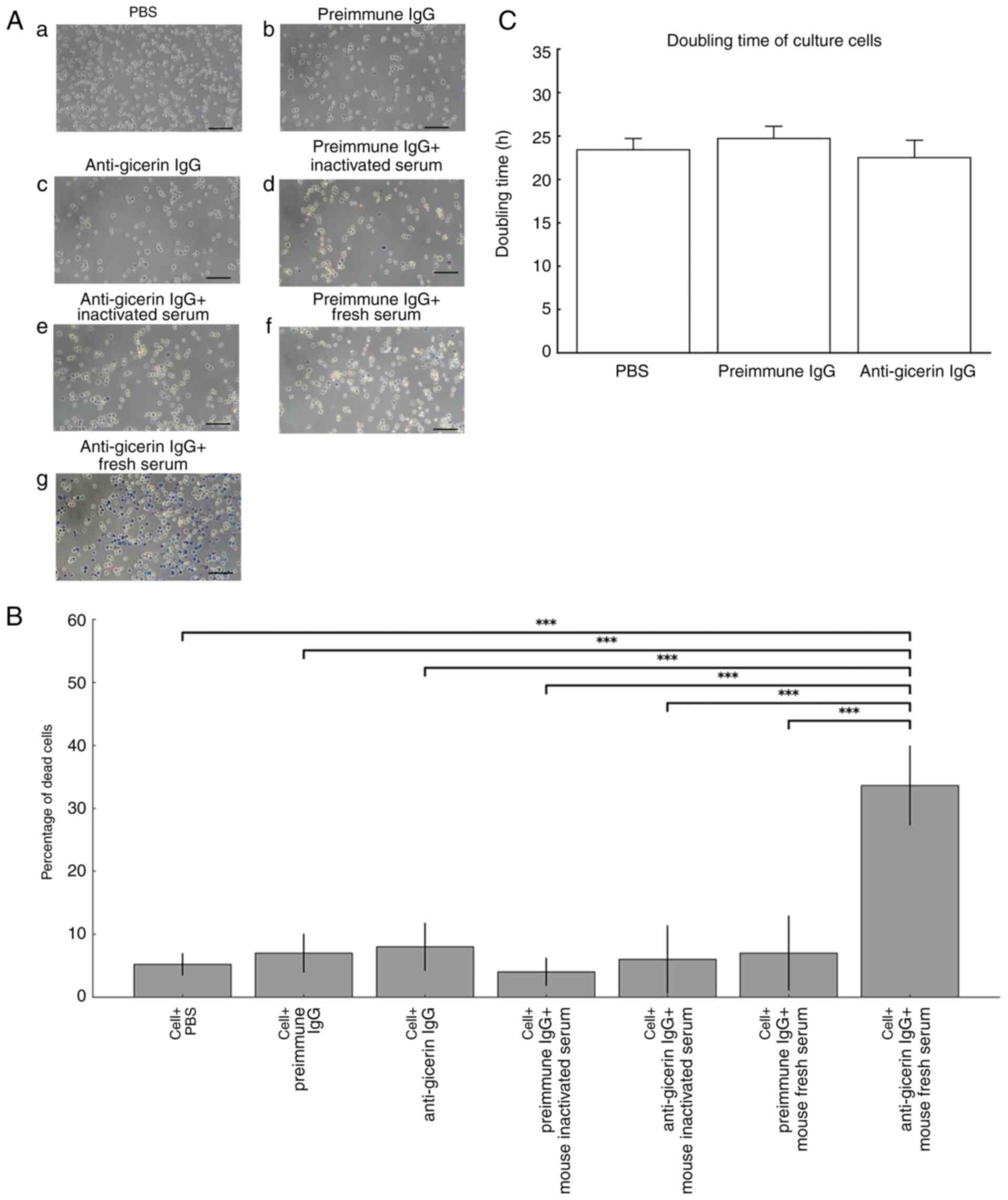 | Figure 6.Complement-dependent cytotoxicity
assay. (A) NUGC-4 cells were incubated with either pre-immune IgG
or anti-gicerin IgG, followed by treatment with either fresh mouse
serum (complement-active) or heat-inactivated serum (heated at 56°C
for 30 min). After a 1 h incubation at 37°C, non-adherent cells
were removed and adherent cells were detached by treating the cells
with trypsin-EDTA. Cell viability was assessed using the trypan
blue exclusion method under light microscopy (a, with PBS; b, with
pre-immune IgG, c, with anti-gicerin IgG; d, with pre-immune IgG
and inactivated serum; e, with anti-gicerin IgG and inactivated
serum; f, pre-immune IgG and fresh serum; g, with anti-gicerin IgG
and fresh serum. (B) Viable (unstained) and nonviable
(blue-stained) cells were counted and the percentages of dead cells
were calculated. Data are mean ± SD (n=5 independent replicates per
group). Seven groups were compared by one-way ANOVA with Tukey's
HSD; only the cell + anti-gicerin IgG + fresh mouse serum group
differed markedly from all other groups (***P<0.001). (C)
Doubling times of NUGC-4 cells with anti-gicerin antibody. The
cells were cultured with PBS, pre-immune IgG and anti-gicerin IgG
and doubling times in each condition were calculated. As shown in
the graph, anti-gicerin antibody did not affect the proliferation
of NUGC-4 cells under in vitro conditions, similar to
pre-immune IgG. Data are presented as mean ± SD (n=4 independent
replicates per group). Differences among the three groups (PBS,
pre-immune IgG and anti-gicerin IgG) were evaluated by one-way
ANOVA followed by Tukey's HSD post hoc test; no statistically
significant differences were detected. |
Discussion
The present study demonstrated that gicerin was
highly expressed in NUGC-4 gastric cancer cells and that the
specific binding of these cells to anti-gicerin antibodies markedly
suppressed subcutaneous tumor growth, local invasion and pulmonary
metastasis in an in vivo model. These observations are all
consistent with previous studies, which suggest that gicerin
fulfills a critical role in tumor metastasis (16,19,20).
Notably, the cell-adhesive properties of gicerin appear to enhance
interactions between tumor cells and the surrounding tissues
(especially muscular and adipose tissues), as well as endothelial
cells, thereby promoting the processes of intravasation, embolus
formation and subsequent extravasation. Indeed, the findings in the
present study have added further support to the hypothesis that
gicerin facilitates in vitro adhesion of gastric cancer
cells to endothelial cells. L-gicerin, which possesses a longer
intracellular domain, has been shown to promote significant
proliferative activity of the cells and metastasis to multiple
organs, thereby driving tumor progression more effectively compared
with S-gicerin, which has a shorter intracellular domain (20,26).
Notably, the NUGC-4 cell line used in the present study expressed
both gicerin isoforms. Although normal muscle tissue likewise
expresses both isoforms, the biological significance of their
co-expression remains poorly understood and elucidating the
interplay between these two isoforms in NUGC-4 cells warrants
further investigation.
Furthermore, the addition of anti-gicerin IgG to the
culture medium was found not to alter the doubling time of tumor
cells in vitro, suggesting that this antibody does not
directly suppress tumor cell proliferation under these conditions.
However, in the mouse subcutaneous tumor model, the presence of
anti-gicerin IgG did markedly inhibit tumor growth, suggesting that
antibody bound to the tumor surface may interact with factors in
the tumor microenvironment, thereby contributing to tumor control
in vivo. Since tumor progression and expansion often involve
interactions between cell adhesion molecules on the tumor surface
and normal tissues, this finding highlighted the potential role of
gicerin as a molecular mediator in these processes. In addition,
the in vitro data generated in the present study
demonstrated that complement components in mouse blood can interact
with anti-gicerin IgG to induce tumor cell death; hence, complement
activation within the tumor milieu may further promote tumor cell
destruction, ultimately reducing tumor growth, local infiltration
and hematogenous metastasis to the lung. During tumorigenesis,
cancer cells not only acquire the ability to evade host regulatory
mechanisms, but also actively manipulate systemic homeostasis to
favor their own survival and proliferation (33). Tumor-derived cytokines and hormones
have been shown to influence key endocrine organs, including the
hypothalamus, pituitary, adrenal glands and thyroid, thereby
modulating central regulatory axes to establish a physiological
milieu conducive to tumor growth and progression. It has been
demonstrated that gicerin mediates homophilic interactions between
gicerin molecules themselves, which facilitate the adhesion of
tumor cells to each other, to vascular tissues and to muscular
structures (16,19,20,34–36).
In addition, heterophilic interactions between gicerin and
extracellular matrix (ECM) components have been suggested to
promote local tissue invasion by tumor cells. These findings
collectively indicate that gicerin may play a multifaceted role in
tumor progression through both structural and signaling mechanisms.
Notably, previous analyses also suggests that gicerin expression
within tumors may contribute to intratumoral angiogenesis, possibly
through factors expressed by tumor cells themselves (16,19,20,36–39).
These mechanisms are not unique to experimental tumor models but
are highly relevant to human gastric cancer as well. The current
study further expanded on this knowledge by using a mouse-derived
anti-gicerin antibody to target gicerin expressed on tumor cells
within a murine model of gastric cancer. Unlike our previous work,
which employed rabbit-derived antibodies to study gicerin function
in tumors of murine and avian origin (16,19,20,34–36),
the present study represented the first attempt, to the best of the
authors' knowledge, to directly target gicerin within a living
mouse using a homologous (murine) antibody. Strikingly, systemic
administration of the mouse-derived anti-gicerin antibody resulted
in widespread necrotic foci within the tumor tissue. This
observation suggested that the murine antibodies, by engaging with
mouse complement proteins, may have efficiently induced
complement-mediated cytotoxicity against gicerin-expressing gastric
cancer cells. This is in contrast to our in vitro findings,
where the same anti-gicerin antibody had no effect on the
proliferation rate (doubling time) of gastric cancer cells. Thus,
the induction of tumor necrosis in vivo appears to be
mediated by immune components present in the host, rather than by
direct effects of the antibody on cell proliferation. In addition
to directly inducing necrosis of the tumor itself, the
mouse-derived anti-gicerin antibody may have disrupted
gicerin-positive neovasculature within the tumor, thereby creating
a hypoxic and energy-deficient microenvironment that led to
secondary tumor cell necrosis. Furthermore, it remains an important
issue for future investigation whether the suppression of gastric
cancer progression by the antibody also involves the induction of
apoptosis in cancer cells.
Additionally, the present study demonstrated that
the antibody markedly suppressed the motility of gastric cancer
cells cultured on gicerin-coated dishes. These findings support the
hypothesis that gicerin plays a critical role in facilitating tumor
cell migration and tissue infiltration, particularly in gastric
cancer. In subsequent studies, it is planned to further dissect the
interaction between gicerin and other adhesion molecules such as
cadherins and vimentin, using in vitro chemotaxis systems
and cell motility analyses in the presence of anti-gicerin
antibodies.
The present study chose recombinant rat gicerin as
the immunogen for antibody production. This decision was based on
previous success in establishing a high-purity, large-scale
production system for recombinant rat gicerin protein, which
enabled the generation of highly sensitive neutralizing antibodies
when used to immunize rabbit (29). By contrast, our attempts to produce
recombinant human gicerin in large quantities were unsuccessful, as
the yield from cultured cells was extremely low and insufficient
for immunization. Given these technical constraints and the
sufficient cross-reactivity of rat gicerin with human gicerin
expressed on NUGC-4 gastric cancer cells, the present study opted
to use recombinant rat gicerin to immunize mice, resulting in the
successful generation of anti-gicerin IgG with potent neutralizing
activity. Inhibitory anti gicerin monoclonal antibodies (such as
AA98) bind the D4-D5 junction, stabilize the monomeric form and
thereby suppress dimerization dependent signaling and cell
migration (40). These functional
surfaces (loops on D1 and D4-D5) are structurally conserved among
mammals. The antibody in the present study was murine polyclonal,
so it probably contained multiple IgG species that collectively
recognize several conserved epitopes on human gicerin, which is
consistent with the broad neutralization observed in
adhesion/invasion/metastasis assays. The antibodies in the present
study were not raised against a short peptide. Instead, it
immunized with a purified recombinant rat gicerin soluble
extracellular domain encompassing all five Ig like domains. The
resulting product is a mouse polyclonal IgG (anti gicerin IgG)
purified to high homogeneity. As the immunogen was the full
ectodomain, the antibody preparation necessarily recognizing
multiple linear and conformational epitopes distributed across the
extracellular domains. Therefore, there is no single ‘epitope
location’ or ‘peptide sequence’ applicable to this polyclonal.
Despite overall 70–72% identity between rat and
human gicerin/CD146 in the extracellular region, functionally
important surfaces within the Ig like domains are conserved. Prior
structural work shows that inhibitory anti gicerin monoclonal
antibodies (mAbs) can bind the D4-D5 interface and attenuate
dimerization/activation. It is hypothesized that our polyclonal
anti rat gicerin IgG contains subpopulations recognizing such
conserved epitopes on human gicerin, which explained the
neutralization of adhesion/invasion/metastasis observed in NUGC 4
cells. The present study did not perform epitope mapping. As follow
up, it is planned to i) test binding to isolated human gicerin
domain fragments (D1-D5), ii) run peptide competition or Pepscan
ELISAs focusing on conserved surface loops within D1 and D4-D5 and
iii) evaluate blocking against a reference inhibitory anti
gicerin/CD146 mAb (e.g., AA98) to determine overlap. These
experiments will pinpoint the dominant neutralizing epitopes.
In the development of antibody-based therapies,
these results underscore the dual role of gicerin in gastric
cancer: not only as a structural adhesion molecule that facilitates
tumor dissemination, but also as an immunological target whose
inhibition may trigger tumor necrosis through host immune
mechanisms. These findings collectively highlighted gicerin as a
promising therapeutic target for the treatment of gastric
cancer.
Nevertheless, further studies are warranted, both to
clarify whether anti-gicerin antibodies primarily interfere with
homophilic or with heterophilic binding and to elucidate the
functions of distinct isoforms, if any should exist. Additional
investigations focusing on established tumors and the possible
synergistic effects of anti-gicerin therapy in combination with
standard chemotherapy will be essential in order to define the
clinical applicability of this strategy. Overall, these findings
have strongly supported the potential usefulness of gicerin as a
molecular target in the treatment of gastric cancer.
Gastric cancer cells actively shape their tumor
microenvironment (TME) through several mechanisms that promote
tumor survival, invasion and metastasis (41). First, gastric cancer cells secrete
a range of cytokines and growth factors, such as VEGF, IL-6 and
TGF-β, that modulate angiogenesis, stromal remodeling and immune
cell recruitment. These factors contribute to the creation of a
pro-tumorigenic environment by promoting vascularization,
suppressing immune surveillance and enabling ECM degradation.
Secondly, studies have shown that gastric cancer cells can induce
polarization of tumor-associated macrophages toward an M2-like
phenotype, which in turn supports tumor progression by secreting
anti-inflammatory cytokines and matrix-remodeling enzymes. In
addition, cancer-associated fibroblasts, often activated by
tumor-derived signals, secrete ECM components and
metalloproteinases that facilitate tumor cell migration and local
invasion (4,33,41).
Importantly, the present study suggested that the cell adhesion
molecule gicerin may also participate in regulating the TME of
gastric cancer. Gicerin-mediated interactions between tumor cells
and endothelial or stromal cells may facilitate intravasation and
immune evasion. Moreover, the observed complement-dependent
cytotoxicity induced by anti-gicerin antibodies in vivo
implied that gastric cancer cells may exploit gicerin to avoid
complement activation under normal conditions, thereby maintaining
TME homeostasis favorable to tumor growth.
However, when administering anti-gicerin antibodies
to tumor-bearing animals, it is necessary to pretreat the antibody
so as to minimize complement activation in the circulation. This
cautionary measure arises from the fact that normal vascular and
muscular tissues constitutively express gicerin; specifically,
inadvertent binding of the antibody, followed by complement
activation, could have deleterious effects on healthy tissues.
Indeed, our preliminary pilot investigations revealed that the
intravenous administration of intact anti-gicerin IgG to healthy
mice induced pulmonary edema as a serious adverse event.
Accordingly, before proceeding with in vivo therapeutic
evaluations, there is a need to optimize the antibody protein to
circumvent complement-mediated toxicity, which will thereby enhance
the safety profile of this promising therapeutic strategy.
Collectively, the observed suppression of gastric
cancer progression in vivo by the antibody may be attributed
to the complement-mediated cytotoxic effect induced by anti-gicerin
antibody in mice, as well as the inhibition of hematogenous
metastasis and local tissue invasion through suppression of cell
adhesion between gastric cancer cells and vascular endothelium.
Cadherins are known to strengthen intercellular adhesion and their
loss or mutation is associated with increased cancer cell invasion
and metastasis. By contrast, cell adhesion molecules belonging to
the immunoglobulin superfamily, such as gicerin, may play an
opposing role. These findings support the concept that, in
vivo, tumor progression is finely regulated by a complex
network of multiple adhesion molecules and related factors.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SU was responsible for conceptualization of the
project, performing the investigation, data analysis, writing and
reviewing the manuscript and editing. NU was responsible for
devising the methodology and validation of the data. KA performed
the animal experiments. YT was responsible for conceptualization of
the project, project administration, the acquisition of funding and
preparation of the original draft of the manuscript. SU and YT
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The use of HUVECs in this study was approved by the
institutional review committee of Kyoto Prefectural University
(Kyoto, Japan) at the time of cell purchase (approval no.
OSP20190889). All animal experiments conducted in this study were
reviewed for their scientific rationale and ethical justification
and were approved by the Animal Experiment Committee of Kyoto
Prefectural University (approval no. KPU240401). The authors
declare that the animal experiment in this study was reviewed and
approved by the institutional ethics committee without the
participation of the project leader or any of the applicants in the
review and approval process.
Patient consent for publication
Not applicable.
Competing interests
The authors declare they have no competing
interests.
References
|
1
|
Cunningham BA: Cell adhesion molecules and
signal transduction. Curr Opin Cell Biol. 7:628–633. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Albelda SM: Role of integrins and other
cell adhesion molecules in tumor progression and metastasis. Lab
Invest. 68:4–17. 1993.PubMed/NCBI
|
|
3
|
Takeichi M: Cadherins in cancer:
Implications for invasion and metastasis. Curr Opin Cell Biol.
5:806–811. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Janiszewska M, Primi MC and Izard T: Cell
adhesion in cancer: Beyond the migration of single cells. J Biol
Chem. 295:2495–2505. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Berx G and van Roy F: The
E-cadherin/catenin complex: An important gatekeeper in breast
cancer tumorigenesis and malignant progression. Breast Cancer Res.
3:289–293. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
7
|
Desgrosellier JS and Cheresh DA: Integrins
in cancer: Biological implications and therapeutic opportunities.
Nat Rev Cancer. 10:9–22. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rathinam R and Alahari SK: Important role
of integrins in the cancer biology. Cancer Metastasis Rev.
29:223–237. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jung H, Jun KH, Jung JH, Chin HM and Park
WB: The expression of claudin-1, claudin-2, claudin-3, and
claudin-4 in gastric cancer tissue. J Surg Res. 167:e185–e191.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chen DL, Zeng ZL, Yang J, Ren C, Wang DS,
Wu WJ and Xu RH: L1cam promotes tumor progression and metastasis
and is an independent unfavorable prognostic factor in gastric
cancer. J Hematol Oncol. 6:432013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lu G, Cai Z, Jiang R, Tong F, Tu J, Chen
Y, Fu Y, Sun J and Zhang T: Reduced expression of E-cadherin
correlates with poor prognosis and unfavorable clinicopathological
features in gastric carcinoma: A meta-analysis. Aging (Albany NY).
16:10271–10298. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gu L, Chen M, Guo D, Zhu H, Zhang W, Pan
J, Zhong X, Li X, Qian H and Wang X: PD-L1 and gastric cancer
prognosis: A systematic review and meta-analysis. PLoS One.
12:e01826922017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen DH, Yu JW and Jiang BJ: Contactin 1:
A potential therapeutic target and biomarker in gastric cancer.
World J Gastroenterol. 21:9707–9716. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh P, Toom S and Huang Y: Anti-claudin
18.2 antibody as new targeted therapy for advanced gastric cancer.
J Hematol Oncol. 10:1052017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsukamoto Y, Taira E, Kotani T, Yamate J,
Wada S, Takaha N, Miki N and Sakuma S: Involvement of gicerin, a
cell adhesion molecule, in tracheal development and regeneration.
Cell Growth Differ. 7:1761–1767. 1996.PubMed/NCBI
|
|
16
|
Adachi K, Hattori M, Kato H, Inai M,
Tsukamoto M, Handharyani E, Taira E and Tsukamoto Y: Involvement of
gicerin, a cell adhesion molecule, in the portal metastasis of rat
colorectal adenocarcinoma cells. Oncol Rep. 24:1427–1431.
2010.PubMed/NCBI
|
|
17
|
Tsukamoto Y, Matsumoto T, Kotani T, Taira
E, Takaha N, Miki N, Yamate J and Sakuma S: The expression of
gicerin, a cell adhesion molecule, in regenerating process of
collecting ducts and ureters of the chicken kidney after the
nephrotrophic strain of infectious bronchitis virus infection.
Avian Pathol. 26:245–255. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsukamoto Y, Taira E, Yamate J, Nakane Y,
Kajimura K, Tsudzuki M, Kiso Y, Kotani T, Miki N and Sakuma S:
Gicerin, a cell adhesion molecule, participates in the histogenesis
of retina. J Neurobiol. 33:769–780. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tsukamoto Y, Sakaiuchi T, Hiroi S, Furuya
M, Tsuchiya S, Sasaki F, Miki N and Taira E: Expression of gicerin
enhances the invasive and metastatic activities of a mouse mammary
carcinoma cell line. Int J Oncol. 23:1671–1677. 2003.PubMed/NCBI
|
|
20
|
Tsukamoto Y, Egawa M, Hiroi S, Furuya M,
Tsuchiya S, Sasaki F, Miki N and Taira E: Gicerin, an
Ig-superfamily cell adhesion molecule, promotes the invasive and
metastatic activities of a mouse fibroblast cell line. J Cell
Physiol. 197:103–109. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Taira E, Takaha N, Taniura H, Kim CH and
Miki N: Molecular cloning and functional expression of gicerin, a
novel cell adhesion molecule that binds to neurite outgrowth
factor. Neuron. 12:861–872. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Taira E, Nagino T, Taniura H, Takaha N,
Kim CH, Kuo CH, Li BS, Higuchi H and Miki N: Expression and
functional analysis of a novel isoform of gicerin, an
immunoglobulin superfamily cell adhesion molecule. J Biol Chem.
270:28681–28687. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kong JH, Lee J, Yi CA, Park SH, Park JO,
Park YS, Lim HY, Park KW and Kang WK: Lung metastases in metastatic
gastric cancer: Pattern of lung metastases and clinical outcome.
Gastric Cancer. 15:292–298. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Niimi T, Samejima J, Koike Y, Miyoshi T,
Tane K, Aokage K, Taki T, Ishii G and Tsuboi M: A case of lung
metastasis from gastric cancer presenting as ground-glass opacity
dominant nodules. J Cardiothorac Surg. 19:3652024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen ZR, Yang MF, Xie ZY, Wang PA, Zhang
L, Huang ZH and Luo Y: Risk stratification in gastric cancer lung
metastasis: Utilizing an overall survival nomogram and comparing it
with previous staging. World J Gastrointest Surg. 16:357–381. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rabben HL, Kodama Y, Nakamura M, Bones AM,
Wang TC, Chen D, Zhao CM and Øverby A: Chemopreventive effects of
dietary isothiocyanates in animal models of gastric cancer and
synergistic anticancer effects with cisplatin in human gastric
cancer cells. Front Pharmacol. 12:6134582021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li W, Zhao X, Wang H, Liu X, Zhao X, Huang
M, Qiu L, Zhang W, Chen Z, Guo W, et al: Maintenance treatment of
Uracil and Tegafur (UFT) in responders following first-line
fluorouracil-based chemotherapy in metastatic gastric cancer: A
randomized phase II study. Oncotarget. 8:37826–37834. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Akiyama S, Amo H, Watanabe T, Matsuyama M,
Sakamoto J, Imaizumi M, Ichihashi H, Kondo T and Takagi H:
Characteristics of three human gastric cancer cell lines, NU-GC-2,
NU-GC-3 and NU-GC-4. Jpn J Surg. 18:438–446. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Taira E, Kohama K, Tsukamoto Y, Okumura S
and Miki N: Characterization of Gicerin/MUC18/CD146 in the rat
nervous system. J Cell Physiol. 198:377–387. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Taira E, Nagino T, Tsukamoto Y, Okumura S,
Muraoka O, Sasaki F and Miki N: Cytoplasmic domain is not essential
for the cell adhesion activities of gicerin, an Ig-superfamily
molecule. Exp Cell Res. 253:697–703. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Taniura H, Kuo CH, Hayashi Y and Miki N:
Purification and characterization of an 82-kD membrane protein as a
neurite outgrowth factor binding protein: Possible involvement of
NOF binding protein in axonal outgrowth in developing retina. J
Cell Biol. 112:313–322. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Spyridopoulou K, Aindelis G, Lampri E,
Giorgalli M, Lamprianidou E, Kotsianidis I, Tsingotjidou A, Pappa
A, Kalogirou O and Chlichlia K: Improving the subcutaneous mouse
tumor model by effective manipulation of magnetic
nanoparticles-treated implanted cancer cells. Ann Biomed Eng.
46:1975–1987. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Slominski RM, Raman C, Chen JY and
Slominski AT: How cancer hijacks the body's homeostasis through the
neuroendocrine system. Trends Neurosci. 46:263–275. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tsukamoto Y, Taira E, Miki N and Sasaki F:
The role of gicerin, a novel cell adhesion molecule, in
development, regeneration and neoplasia. Histol Histopathol.
16:563–571. 2001.PubMed/NCBI
|
|
35
|
Tsuchiya S, Tsukamoto Y, Furuya M, Hiroi
S, Miki N, Sasaki F and Taira E: Gicerin, a cell adhesion molecule,
promotes the metastasis of lymphoma cells of the chicken. Cell
Tissue Res. 314:389–397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kirimura N, Kubota Y, Adachi K and
Tsukamoto Y: Involvement of gicerin, a cell adhesion molecule, in
the hematogenous metastatic activities of a melanoma cell line. Am
Int J Biol. 2:65–75. 2014. View Article : Google Scholar
|
|
37
|
Kohama K, Tsukamoto Y, Fruya M, Okamura K,
Tanaka H, Miki N and Taira E: Molecular cloning and analysis of the
mouse gicerin gene. Neurochem Int. 46:465–470. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Taira E, Kohama K, Tsukamoto Y, Okumura S
and Miki N: Gicerin/CD146 is involved in neurite extension of
NGF-treated PC12 cells. J Cell physiol. 204:632–637. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tsuchiya S, Tsukamoto Y, Taira E and
LaMarre J: Involvement of transforming growth factor-beta in the
expression of gicerin, a cell adhesion molecule, in the
regeneration of hepatocytes. Int J Mol Med. 19:381–386.
2007.PubMed/NCBI
|
|
40
|
Chen X, Yan H, Liu D, Xu Q, Duan H, Feng
J, Yan X and Xie C: Structure basis for AA98 inhibition on the
activation of endothelial cells mediated by CD146. iScience.
24:1024172021. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Matsuoka T and Masakazu Y: Molecular
mechanism for malignant progression of gastric cancer within the
tumor microenvironment. Int J Mol Sci. 25:117352024. View Article : Google Scholar : PubMed/NCBI
|