Introduction
The liver has a strong regenerative capacity;
however, chronic injury can lead to extracellular matrix
accumulation, fibrosis and eventually liver failure, representing a
major global health burden (1). In
2022, liver diseases such as cirrhosis, viral hepatitis and
hepatocellular carcinoma (HCC) accounted for 2 million deaths
worldwide (2). In China, the
increasing prevalence of liver diseases highlights the requirement
for more effective therapies (3).
Conventional treatments offer limited efficacy in conditions such
as non-alcoholic steatohepatitis (NASH), alcoholic liver disease
(ALD) and HCC (4–7). Therefore, it is important to identify
shared molecular mechanisms to improve the diagnosis of liver
diseases and identify more effective treatment strategies.
Poly(ADP-ribose) polymerase-1 (PARP-1), a key enzyme
in the DNA damage response (DDR), also regulates cellular
metabolism by modulating oxidized nicotinamide adenine dinucleotide
(NAD+) levels and sirtuin 1 (SIRT1) activity (8–10).
PARP-1 expression is upregulated in both primary and recurrent HCC
and has been implicated in chemoresistance (11). The involvement of PARP-1 in the
progression of various liver diseases, including non-alcoholic
fatty liver disease (NAFLD), ALD and HCC, suggests its potential as
a therapeutic target (12–14).
Unlike previous studies focusing on single disease
types, the present review comprehensively analyzes the role of
PARP-1 in multiple liver pathologies, including NASH, ALD, fibrosis
and HCC, revealing shared molecular mechanisms. The dual role of
PARP-1 in liver diseases is systematically summarized, and its
functions in liver regeneration and pathological fibrosis are
integrated to provide a comprehensive understanding of its
regulatory mechanisms. In addition to summarizing established
evidence, novel mechanistic insights into the regulation of tumor
metabolism, transcriptional programming and immune microenvironment
remodeling by PARP-1 in HCC are discussed, and the therapeutic
synergy between PARP inhibitors and immune checkpoint blockade is
highlighted. Furthermore, the translational potential of PARP-1
inhibitors for autoimmune liver diseases (AILDs) is proposed based
on preclinical evidence, addressing a critical research gap.
Overall, the present review synthesizes current knowledge on the
expression, regulatory networks and mechanisms of PARP-1 in various
liver diseases, ultimately evaluating its clinical diagnostic and
therapeutic value.
Overview of liver disease
In 2022, liver disease resulted in 2 million
fatalities worldwide, or 4% of total deaths globally, ranking it as
the 11th largest cause of death (2). The main causes of mortality are
complications arising from cirrhosis and HCC, while acute hepatitis
accounts for a lower proportion of fatalities. The primary global
causes of cirrhosis include viral hepatitis, alcohol use and NAFLD,
which lead to a progressive deterioration of hepatic function.
Other factors also contribute to liver disease, including age, sex,
body composition, diet, physical activity, microbiome, and history
of alcohol and tobacco use (15).
The liver performs essential biological tasks such
as protein synthesis, glucose and lipid metabolism, detoxification
and bile production. It exhibits marked immunological activity and
functions as a systemic barrier against intestinal infections and
toxins by maintaining a localized immune-regulating milieu and
promoting tolerance to prevalent antigens (16). Optimal liver function is essential
for the body to effectively combat bacteria and viruses; therefore,
individuals with liver dysfunction are particularly susceptible to
infections (17). The immune
response generated by the liver relies upon its distinctive
architecture, the presence of essential immune cells such as
Kupffer cells (KCs), continuous immunological surveillance and
rapid recruitment of immune cells (16,18).
Chronic alcohol use increases the permeability of the gut to
bacterial lipopolysaccharide (LPS) (19), elevates endotoxin levels in the
hepatic portal vein, and activates KCs via interactions with
Toll-like receptor-4 (14).
The liver has strong regenerative capacity and can
recover considerably after resection. However, conditions such as
ALD and chronic hepatitis B (CHB) may overly activate this
regenerative response, resulting in the excessive accumulation of
extracellular matrix and collagen, leading to liver fibrosis.
Furthermore, decompensated liver fibrosis leads to the formation of
hepatic scar tissue, which is known as cirrhosis. This
significantly reduces liver function, and may progress to liver
failure and death (1).
PARP-1, a DNA repair enzyme, plays a key role in
hepatocyte apoptosis by promoting cell death and increasing the
synthesis of pro-inflammatory mediators when overactivated by ROS
and reactive nitrogen species, thereby influencing the self-healing
capacity of the liver (10). NAFLD
is a major contributor to chronic liver disease (CLD), with a
spectrum ranging from moderate steatosis to advanced NASH. NASH is
characterized by inflammation and progressive hepatocyte injury,
which can lead to cirrhosis (20)
and subsequently increase the risk of HCC.
Overview of PARP-1
PARPs are ADP-ribosyltransferases that catalyze
ADP-ribosylation, a post-translational protein modification.
ADP-ribosylation involves the cleavage of NAD+ to
generate single ADP-ribose units, oligomers or polymers that are
covalently linked to serine, glutamate, aspartate, arginine and
lysine residues in target proteins (21). The PARP family comprises 17
proteins, with PARP-1 accounting for 85–90% of total PARP activity
(22). PARP-1 contains several
conserved and functionally distinct domains, including an
auto-modification domain, a 55-kDa catalytic domain, and two zinc
finger domains responsible for DNA binding (23).
PARP-1 is present in all human tissues, with the
highest concentrations in the lymph nodes, appendix, brain,
placenta, prostate, spleen and testes. It is an essential element
in the DNA base excision repair mechanism (24,25)
and participates in the repair of single-strand and double-strand
breaks (DSBs) (26). Human PARPs,
namely PARP-1, −2 and −3, are categorized as DDR proteins because
of their involvement in DNA repair and reliance on DNA damage for
activation (22). Among these,
PARP-1 is the predominant enzyme, characterized by its abundance
and strong enzymatic activity. It serves as the principal signaling
factor in DDR, where its rapid activation is the most critical
stage in the DDR process (8).
PARP-1 serves as a primary sensor of DNA damage, capable of
detecting DNA breaks within 3 sec of their occurrence, and triggers
the recruitment of repair complexes via the ADP-ribosylation of
proteins near the damage site. Its activity is further influenced
by co-factors, such as histone PARylation factor 1 (HPF1), which
enhances the catalytic activity of PARP-1 and modifies substrate
selectivity. HPF1 interacts with the PARP-1 catalytic domain and
colocalizes with it at sites of DNA damage (8).
A study evaluating the hepatotoxicity of columbin
(CLB) demonstrated that CLB induces DNA damage both in vitro
and in vivo, which is associated with the upregulation of
PARP-1 (27). PARP-1 also
interacts with SIRTs. SIRT1-7 are a family of
NAD+-dependent protein deacetylases with extensive roles
in metabolism and aging. In the absence of PARP-1, NAD+
levels rise, potentially increasing SIRT1 activity (22,28).
In addition, PARP-1 and AMP-activated protein kinase (AMPK)
mutually reinforce each other: The activation of PARP-1 stimulates
AMPK, which in turn phosphorylates and activates PARP-1. Within the
reactive oxygen species (ROS)-PARP-1-AMPK pathway, the interaction
between PARP-1 and AMPK promotes the nuclear export of AMPK, which
induces autophagic flux and cellular apoptosis (22,29).
PARP-1 plays an essential role in DNA repair and
cellular stress responses in the liver. During oxidative stress,
such as exposure to hydrogen peroxide, PARP-1 activation preserves
hepatocyte survival, while PARP-1 inhibition or genetic ablation in
primary hepatocytes diminishes oxidation-induced necrotic cell
death, suggesting a preventive mechanism against oxidative injury
(10).
A pilot study revealed that PARP-1 knockout mice
exhibit impaired liver regeneration and reduced hepatocyte
proliferation. It also showed that PARP-1 is required for both the
early liver response and late tissue repair during liver
regeneration, and that PARP-1 inhibitors can reduce hepatocyte
proliferation (30). These results
indicate that PARP-1 and its catalytic activity are essential for
hepatocyte proliferation. This effect appears to be associated with
Yes-associated protein (YAP), a key driver of hepatocyte
proliferation (31). In PARP-1
knockout mice, YAP activity and the expression of cell
cycle-related proteins in liver tissue are suppressed, resulting in
reduced hepatocyte proliferation and impaired liver repair during
liver regeneration (30). However,
another study showed that chronic or excessive activation, such as
that observed in severe or long-standing liver disease, depletes
NAD+ and promotes fibrosis, ultimately impairing liver
regeneration (32). In view of
this duality, the inhibition of pathological activation while
retaining the beneficial functions of PARP-1 is a therapeutic
challenge.
Chromatin relaxation is essential for efficient DNA
repair, and PARP-1 functions as a crucial mediator of this process.
The inhibition of PARP-1 activity directly affects chromatin
structure. Through poly(ADP-ribosyl)ation, PARP-1 binds to and
modifies chromatin remodelers, thereby altering chromatin structure
and facilitating the ensuing DDR (33).
PARP-1 also plays a pivotal role in inflammation. It
activates nuclear factor κB (NF-κB), nuclear factor of activated T
cells and activator protein 1, leading to the production of
inflammatory cytokines, including tumor necrosis factor-α (TNF-α)
and interleukin (IL)-1β, and effector T cell cytokines, including
IL-4 and IL-5. Excessive activation of PARP-1 promotes cell death
and tissue damage, thereby exacerbating the inflammatory response
(34). These functions are
particularly relevant to chronic inflammatory liver conditions,
including viral hepatitis, ALD and drug-induced liver injury
(DILI).
PARP-1 is essential for sustaining proper immune
function, and its dysregulation contributes to immune-mediated
disorders. Under physiological conditions, PARP-1 expression is
typically low but becomes significantly upregulated during early
liver injury, chronic liver fibrosis and HCC (10). Consequently, aberrant PARP-1
activity is associated with the progression of several liver
disorders.
Function of PARP-1 in liver disease
Hepatitis
Viral hepatitis
Viral infection is a major cause of acute hepatitis,
which leads to elevated liver transaminase levels and impaired
liver function (35). China is
among the top 20 nations with the highest viral hepatitis burdens.
Between 2016 and 2022, the proportion of people living with viral
hepatitis confirmed hepatitis B cases in China increased from 19 to
24%, while that of hepatitis C cases increased from 22 to 33%
(36). CHB infection is the major
cause of HCC. Transforming viral proteins such as hepatitis B virus
(HBV) regulatory protein X (HBx) are key oncogenic factors
(37). HBx induces the degradation
of the structural maintenance of chromosome 5/6 complex, a process
implicated in HBV-related HCC. This impairs homologous
recombination (HR)-mediated DNA repair, thereby contributing to
carcinogenesis. The accumulation of double-stranded DNA (dsDNA)
breaks in cancer cells with HR deficit (HRD) can be lethal. PARP-1
inhibition prevents the repair of single-strand DNA breaks, which
subsequently exacerbates the accumulation of dsDNA breaks (38,39).
In addition to altering protein function, HBV infection regulates
the expression of certain long non-coding RNAs (lncRNAs) in
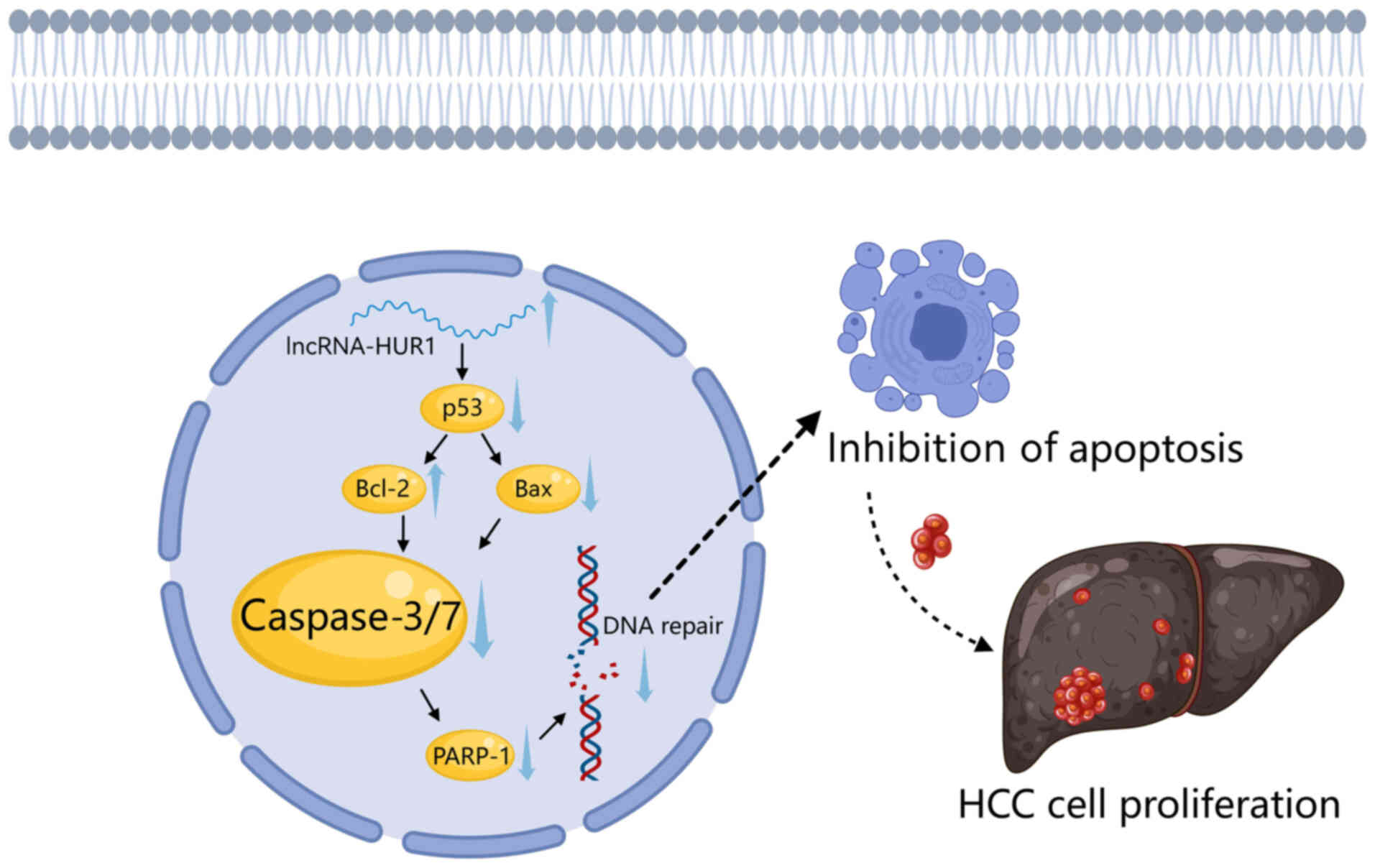
infected cells. For example, HBx inhibits p53 activity, leading to
the upregulation of lncRNA-HUR1 expression, which promotes cell
proliferation and cancer progression (40). The overexpression of lncRNA-HUR1
reduces caspase-3/7 activity and PARP-1 cleavage, while lncRNA-HUR1
knockdown increases caspase-3/7 activity and promotes PARP-1
cleavage (41). Together, these
findings underscore the crucial role of PARP-1 in HBV-induced
HCC.
In addition to HBV, other viral hepatitis subtypes
may also interact with DNA damage and repair pathways. Although
PARP-1 has not been extensively studied in these contexts, existing
data suggest a potential involvement that is worthy of further
exploration. Hepatitis C virus (HCV) is a leading cause of CLD and
HCC that induces persistent oxidative stress and DNA damage in
infected hepatocytes. Viral proteins, including core protein and
the non-structural (NS) proteins NS3 and NS5A disrupt host DNA
repair pathways by targeting key components, such as the ataxia
telangiectasia-mutated (ATM)-nibrin-meiotic recombination 11
complex, thereby promoting chromosomal instability and malignant
transformation (42,43). Although direct studies of PARP-1 in
HCV-infected liver tissue are limited, the well-established roles
of this enzyme in DDR and inflammation suggest that PARP-1 may be
activated under conditions of HCV-induced genomic stress,
potentially contributing to fibrosis and cancer progression.
Hepatitis D virus (HDV), which often co-occurs with HBV, is
associated with more severe disease and heightened oxidative stress
(44), factors that may also
induce PARP-1 activation. However, direct evidence associating
PARP-1 with HDV-induced liver injury is lacking.
Collectively, these observations highlight the
importance of investigating the role of PARP-1 in diverse forms of
viral hepatitis, as its regulatory role in DNA repair, cell death
and the inflammatory response may have major implications for the
diagnosis and treatment of various liver infections.
ALD
Liver damage, inflammation, fibrosis, cirrhosis and
cancer resulting from prolonged or excessive alcohol consumption
are hallmarks of ALD. With 3.3 million associated fatalities, or 6%
of all deaths globally, alcohol-related damage is one of the most
prevalent preventable causes of mortality. Liver damage occurs in
20–30% of individuals who misuse alcohol (45). A key pathogenic characteristic of
ALD is hepatocyte apoptosis mediated by PARP-1. Under normal
physiological conditions, alcohol dehydrogenase is the primary
enzyme that metabolizes ethanol in the liver. However, chronic
alcohol intake stimulates the expression of cytochrome P450 2E1,
which metabolizes ethanol to acetaldehyde and produces considerable
amounts of ROS as byproducts (46). Acetaldehyde is a toxic metabolite
that forms adducts with cellular macromolecules and further
amplifies oxidative stress. ROS can induce oxidative DNA damage,
which is sensed by PARP-1 and activates the TGF-β1-Smad pathway. In
the early stages of DNA damage, PARP-1 binds to sites of DNA damage
and uses NAD+ as a substrate to synthesize PARPs
including PARP-1 (47). However,
persistent or severe DNA damage leads to the overactivation of
PARP-1, which consumes large amounts of NAD+ for
poly(ADP-ribosyl)ation, leading to NAD+ and ATP
depletion along with impaired mitochondrial function and energy
metabolism, ultimately triggering hepatocyte necrosis or monocytic
cell death (48,49).
Preventing and treating ALD requires the reversal of
this imbalance caused by PARP-1 overactivation that leads to
hepatocyte necrosis or monocyte death (50). In ALD, hepatocytes and liver
macrophages exhibit upregulated C-C chemokine ligand type 2 (CCL2)
levels. A pharmacological study of mice revealed that
alcohol-induced liver damage can be prevented or reversed by
blocking the activation of C-C chemokine receptor type 2 and 5
(51). Alcohol-induced increases
in PARP-1 activity have also been shown to mediate hepatocyte
apoptosis (14,52). In Raw264.7 macrophages, PARP-1
activation promotes the nuclear translocation of LPS-induced NF-kB,
and increased KC activation exacerbates alcohol-induced liver
steatosis by increasing hepatic M1 macrophage marker gene
expression. Consistent with this, treatment of mice with the PARP-1
inhibitor PJ34 downregulates hepatic M1 marker gene expression
(14). This suggests that limiting
KC activation helps to restore hepatic lipid balance, an important
protective mechanism of PARP-1 inhibition in alcohol-induced
hepatic steatosis. Certain medications, such as cenicriviroc, have
been demonstrated to reduce hepatocyte apoptosis and alleviate
CCL2-induced hepatic steatosis (52).
NAFLD
Based on histological features, NAFLD can be
classified as NASH, which comprises <20% of cases, and
non-alcoholic fatty liver (NAFL), which comprises the remaining
>80% (53,54). NAFL is defined as hepatic steatosis
without substantial hepatocyte injury, or minor lobular
inflammation is present, while NASH is characterized by hepatocyte
damage in addition to steatosis (54). With an estimated global incidence
of 25%, NAFLD is a growing public health concern. In the United
States, severe NASH is now the second most frequent indication for
liver transplantation in men and the most common indication in
women (55). Nearly 75% of
individuals in the United States with risk factors such as diabetes
and obesity develop NAFLD (56).
In early NAFLD, the gut vascular barrier is
disrupted, which allows intestinal bacteria to proliferate, leading
to increased translocation of LPS to the liver, thereby triggering
pyroptosis. LPS stimulation also activates PARP-1, which increases
signaling via NF-κB, a crucial transcription factor that regulates
the production of pro-inflammatory cytokines. Specifically, PARP-1
interacts with transcription factors and co-activators, including
p300, to increase the transcriptional activity of NF-κB. This then
influences the expression of cytokines and other pro-inflammatory
mediators, including inducible nitric oxide synthase and
cyclooxygenase-2, thereby contributing to the inflammatory response
in hepatic tissue (34). In
high-fat diet (HFD)-induced mouse models, DNA damage activates
PARP-1, which stimulates the activation of NLR family pyrin domain
containing 3 (NLRP3) and its downstream signaling proteins
(57). This is associated with
increased levels of cleaved PARP-1 together with elevated blood
levels of IL-1β and C-reactive protein (58). Beyond inflammation, the development
of NAFLD is strongly influenced by the SIRT1/AMPK pathway and
NAD+ homeostasis. PARP-1 contributes to this
dysregulation by depleting NAD+ and interfering with
SIRT1 activity, which is essential for AMPK activation (59). SIRT1 modulates hepatic lipid
metabolism by regulating the activity of peroxisome
proliferator-activated receptors (PPARs) (60). PARP-1 activity also disrupts
hepatic lipid homeostasis by modulating the SIRT1/PPARα axis. It
depletes intracellular NAD+, directly
poly(ADP-ribosyl)ates PPARα, and impairs both SIRT1 activity and
the PPARα-mediated transcription of β-oxidation genes, leading to
suppressed mitochondrial function and fatty acid oxidation
(61,62). This promotes lipid accumulation and
the progression of steatosis. By contrast, the activation of SIRT1
enhances hepatic lipid catabolism and mitochondrial efficiency,
serving as a critical counterbalance in the maintenance of
metabolic adaptation during NAFLD progression (63). Collectively, these insights
highlight the pivotal regulatory function of the PARP-1/SIRT1/PPARα
axis in the pathophysiology of NAFLD.
DILI
Unanticipated liver damage caused by drugs or other
xenobiotics, known as DILI, is a major concern in clinical trials
and drug development (64). A wide
range of agents, including medications, natural products and
supplements, can cause DILI (65).
DILI is challenging to diagnose, requires the rigorous elimination
of other causes, and is underreported to pharmacovigilance
authorities; therefore, its prevalence is challenging to determine
(66,67). While traditional and nutritional
supplements are the primary causes of DILI in Asia, drug responses
to conventional pharmaceuticals are most prevalent in the United
States and Europe (67). With an
estimated 23.8 cases per 100,000 individuals, the frequency of DILI
is high in China (68). However,
the actual incidence could be much higher than this.
The mechanisms underlying liver injury vary
depending on the causative agents. Experimental models hased on
DILI principles have been developed to assess the efficacy of liver
protective and anti-fibrosis interventions. For example, carbon
tetrachloride can induce liver fibrosis and injury, processes that
are often accompanied by DNA damage and repair processes, with
PARP-1 playing a key role in the DDR (49). The overactivation of PARP-1
promotes the binding of transcription factor activator protein 1 to
the TGF-β1 gene, leading to chronic chemical damage in human and
animal liver cells (69). Another
example of a liver injury-inducing agent is CLB, which increases
ROS production and depletes glutathione. The resulting oxidative
stress promotes DNA breaks and upregulates PARP-1 expression
(27).
PARP-1 is activated during acetaminophen overdose,
resulting in poly(ADP-ribosyl)ation of the pregnane X receptor.
This post-translational modification augments the transcription of
the cytochrome P450, family 3, subfamily A, polypeptide 11 enzyme,
thereby increasing the production of toxic acetaminophen
metabolites (70). Furthermore,
anti-tuberculosis medications, including isoniazid and rifampicin,
can induce marked hepatic damage by inducing an inflammatory
response and oxidative stress, subsequently activating the NLRP3
inflammasome. This induces PARP-1 activation, which contributes to
hepatic damage (71).
AILD
There are three principal types of AILD, a chronic
immune-mediated illness that affects hepatic and bile duct cells,
which are autoimmune hepatitis (AIH), primary biliary cholangitis
(PBC) and primary sclerosing cholangitis (PSC) (72,73).
A clinical phenotype known as overlap syndrome occurs when symptoms
of multiple categories co-exist within the spectrum of AILD;
however, the majority of patients fit the criteria of one specific
type (74). AILD has a
male-to-female ratio of 1:5, and an annual yearly incidence
worldwide of 1.37 cases per 100,000 individuals. PBC mostly affects
women, and has a prevalence of 39.2 per 100,000 in the United
States and 4.75 per 100,000 in Korea (75).
Studies have shown that the pathophysiology of
autoimmune disorders is associated with increased PARP-1 activation
(76,77). Trichloroethene exposure induces
autoimmune reactions, leading to increased apoptosis, immune
activation and the upregulation of PARP-1 expression following the
development of anti-single-stranded DNA antibodies (78). Concanavalin A (ConA) causes liver
damage via a mechanism comparable to the clinical alterations and
immunological responses observed in patients with AIH (79). In AIH model mice, the liver
exhibits widespread necrosis, marked macrophage infiltration, and
elevated levels of the pro-inflammatory cytokine TNF-α (80). Following ConA induction, PARP-1
levels and TNF-α production increase, which contributes to PARP-1
activation (80).
Although PARP-1 has been implicated in the
regulation of inflammation and T cell-mediated immune responses,
direct evidence for the use of PARP inhibitors in animal models of
AILD, including AIH, PBC and PSC, is currently lacking. The
ConA-induced hepatitis model has been widely employed to
investigate the immunopathogenesis of AIH, owing to its ability to
simulate CD4+ T cell-mediated hepatic injury (81). Notably, genetic ablation of PARP-2,
but not PARP-1, confers significant protection against ConA-induced
liver damage, suggesting isoform-specific roles of PARP enzymes in
immune liver injury (82).
However, the role of PARP-1 remains incompletely understood in this
context. Given the established involvement of PARP-1 in the
amplification of NF-κB-mediated proinflammatory cytokine release
and the modulation of CD4+ T cell polarization (83), it is plausible that PARP-1
inhibition may mitigate immune-driven hepatic inflammation in AIH
(84). Accordingly, future studies
utilizing pharmacological PARP-1 inhibitors such as PJ34 or
olaparib, or gene-silencing approaches in AIH models, such as the
ConA-induced hepatitis model, are warranted to determine whether
PARP-1 is a viable therapeutic target in AILD.
Liver fibrosis
Persistent liver injury, such as hepatitis, can lead
to liver fibrosis. This condition is characterized by the buildup
of extracellular matrix proteins, primarily cross-linked type I and
III collagen, which replace damaged normal tissue following
cholestatic and hepatotoxic injury. The resulting fibrous scarring
impairs the structural integrity of the liver tissue and disrupts
the homeostatic function of hepatocytes (85). Liver fibrosis may eventually
develop into cirrhosis, which is associated with high morbidity and
mortality, and increases the risk of HCC and chronic liver failure
(86).
Hepatic stellate cells (HSCs) are non-parenchymal
cells located in the subendothelial region of the liver and are the
major mediators of liver fibrosis. A promising strategy to mitigate
the development of liver fibrosis is the induction of senescence in
activated HSCs (87). HSC
apoptosis can be accurately predicted based on changes in PARP-1
levels (88). Studies have shown
that autophagy exacerbates liver fibrosis by breaking down lipid
droplets in HSCs, thereby activating the HSCs. Conversely, the
suppression of autophagy promotes HSC death and reduces
proliferation. Elevated levels of cleaved PARP are associated with
HSC apoptosis, which can be induced by pharmacological agents such
as berberine (BBR) and carvedilol (89,90).
Specifically, BBR blocks autophagy in HSCs and downregulates
autophagy protein 5 expression, which leads to HSC death (90). In addition, PARP-1 activation is
associated with increased TGF-β expression. In activated KCs,
upregulated PARP-1 expression promotes TGF-β production, further
stimulating HSC activation (10).
HCC
Liver cancer ranks fourth in cancer-related
mortality and is the sixth most prevalent cancer worldwide, with
HCC accounting for >80% of cases (91,92).
With a yearly incidence rate of 1–6%, cirrhosis of any etiology is
the biggest risk factor for HCC. Indeed, HCC is the primary cause
of mortality in individuals with cirrhosis (93). Compared with nearby normal tissues,
HCC exhibits numerous chromosomal aberrations and elevated PARP
expression, which accumulates during DNA replication (94,95).
PARP-1 inhibition has been suggested to be an effective treatment
for HCC, particularly in BRCA-deficient tumors (94).
Emerging evidence indicates that PARP-1 enhances
glycolysis and metabolic reprogramming in tumor cells by physically
interacting with hypoxia inducible factor-1α, stabilizing it, and
acting as a transcriptional coactivator (96). This promotes the expression of
hexokinase 2 and other glycolytic enzymes, thereby reinforcing the
Warburg effect in HCC and facilitating tumor proliferation
(97). Beyond its role in DNA
repair, PARP-1 also functions as a transcriptional coregulator,
interacting with NF-κB and STAT5 to facilitate the expression of
pro-inflammatory and tumorigenic genes (98), which promotes chronic inflammation,
cell proliferation and angiogenesis in HCC. PARP-1 also modulates
the tumor immune microenvironment through multiple mechanisms. PARP
inhibition leads to the accumulation of cytosolic DNA, activation
of the cyclic GMP-AMP synthase-stimulator of IFN genes-type I IFN
pathway and upregulation of programmed death ligand-1 (PD-L1),
thereby reshaping immune cell infiltration and enhancing antitumor
immunity (99). A preclinical
study of HCC demonstrated that olaparib upregulates PD-L1 via the
suppression of microRNA-513, and its combination with
anti-programmed cell death protein-1 (PD-1) therapy significantly
enhances CD8+T-cell infiltration and antitumor efficacy
(100). Broader oncology studies
indicate that PARP inhibitor plus immune checkpoint inhibitor
combinations are well-tolerated and exhibit synergistic antitumor
effects, which justifies their translational exploration in HCC
(101–103).
Caspase-3 is an effector protein in cancer cell
apoptosis, which promotes apoptosis by cleaving PARP-1 (104). Notably, HCC progression is
associated with decreased levels of both PARP-1 and cleaved
caspase-3. DNA damage repair fails when PARP-1 is cleaved, as the
cleaved form of PARP-1 can no longer repair DNA (104–106). Peptides derived from Laminaria
japonica promote apoptosis by increasing cleaved caspase-9 and
caspase-3 levels and activating PARP, potentially through upstream
apoptotic signal-regulating kinase 1 (ASK1) phosphorylation and
subsequent p38 MAPK activation (107). Factors upstream of PARP-1, such
as JNK1/2 and ERK1/2, may also regulate cell death following PARP-1
inhibition (108). This mechanism
can kill cancer cells and provides a potential therapeutic avenue
for HCC. However, the upregulation of MET expression in HCC
presents a challenge: Oxidative DNA damage activates MET, which
interacts with and phosphorylates PARP-1 at tyrosine 907, thereby
limiting the effectiveness of PARP-1 inhibitors in HCC (109). Future studies on the use of PARP
inhibitors for the treatment HCC should account for this
mechanism.
A summary of the targets and associated molecules or
complexes of PARP-1 in liver diseases is presented in Table I.
 | Table I.Targets, associated molecules and
complexes of poly(ADP-ribose) polymerase-1 in liver diseases. |
Table I.
Targets, associated molecules and
complexes of poly(ADP-ribose) polymerase-1 in liver diseases.
| First author/s,
year | Liver disease
type | Target, associated
molecule or complex | (Refs.) |
|---|
| Mukhopadhyay et
al, 2014 | Liver fibrosis | TGF-β | (10) |
| Zhang et al,
2016 | ALD | NF-κB | (14) |
| Liu et al,
2018 | Viral
hepatitis | lncRNA-HUR1 | (40) |
| Yin et al,
2022 | ALD | C-C chemokine
ligand type 2 | (51) |
| Ye et al,
2022 | NAFLD | NLRP3 | (57) |
| Salomone et
al, 2017 | NAFLD | SIRT1, AMPK | (60) |
| Gallyas and Sumegi,
2020 | Drug-induced liver
injury | Activator protein
1, TGF-β1 | (69) |
| Liu et al,
2023 | Autoimmune liver
disease | TNF-α | (80) |
| Ishteyaque et
al, 2024 | Hepatocellular
carcinoma | Caspase-3 | (104) |
PARP-1-mediated cellular signaling pathways
in liver disease
The onset and progression of liver diseases involve
intricate molecular pathways that are poorly understood and
associated with a poor prognosis. PARP-1 activity is both directly
or indirectly regulated by a variety of signals, rendering it a
potentially useful biomarker for detecting the onset of liver
disorders and monitoring their course. This section summarizes the
key cellular signaling pathways in which PARP-1 participates and
their contributions to the pathogenesis of various liver
disorders.
The regulation of tumor cell growth and death is
strongly influenced by lncRNAs. Numerous lncRNAs that promote the
proliferation of tumor cells also decrease their apoptosis
(110). For example, Chen et
al (41) reported that
lncRNA-HUR1 increases Bcl-2 expression and reduces Bax expression
by suppressing p53 activity. This inhibits caspase-3/7 activation
via the intrinsic apoptosis pathway, thereby preventing PARP-1
cleavage and cell death. Conversely, the study demonstrated that in
cell lines with stable lncRNA-HUR1 knockdown, Bcl-2 expression is
reduced, Bax expression is increased and caspase-3/7 activation is
promoted, which in turn accelerates PARP-1 cleavage and cell death
(41). Although few lncRNAs have
been thoroughly investigated in the context HCC, lncRNAs in general
have been demonstrated to be essential for the development and
progression of HCC (111),
indicating their potential as both diagnostic and therapeutic
targets (Fig. 1).
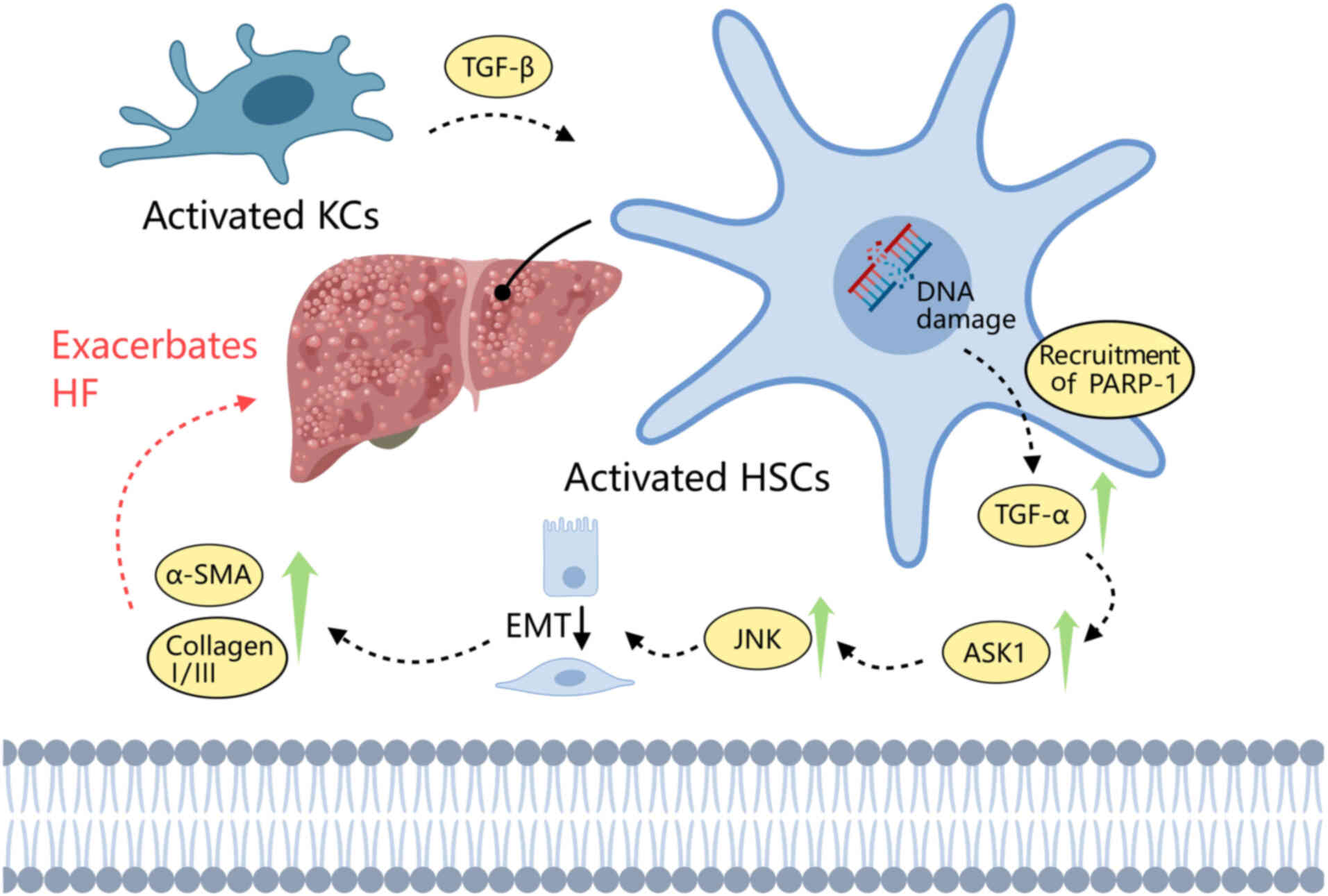
PARP-1 can sense DNA damage and trigger the
TGF-β1-Smad signaling pathway. TGF-β1 is produced by endothelial,
hematopoietic and connective tissue cells, and has been associated
with heart failure in several studies (49,112,113). A main cause of hepatic fibrosis
(HF) is the activation of HSCs. When DNA damage occurs, PARP-1 is
recruited and activates TGF-β1-Smad pathway by upregulating TNF-α
in HSCs, which increases ASK1-JNK phosphorylation. This exacerbates
HF by promoting epithelial-to-mesenchymal transition and the
deposition of α-smooth muscle actin (α-SMA) and collagen I/III.
Dihydrokaempferol has been shown to decrease TNF-α transcription
and inhibit the PARP-1-regulated TGF-β1 pathway, thereby reducing
the levels of phosphorylated (p-)Smad 2/3 and p-ERK 1/2 (MAPK1).
This is associated with suppression of the ability of HSCs to
produce α-SMA and collagen I/III, contributing to the mitigation of
HF, potentially by inhibition of the downstream NF-κB and ASK1-JNK
signaling pathways (49) (Fig. 2).
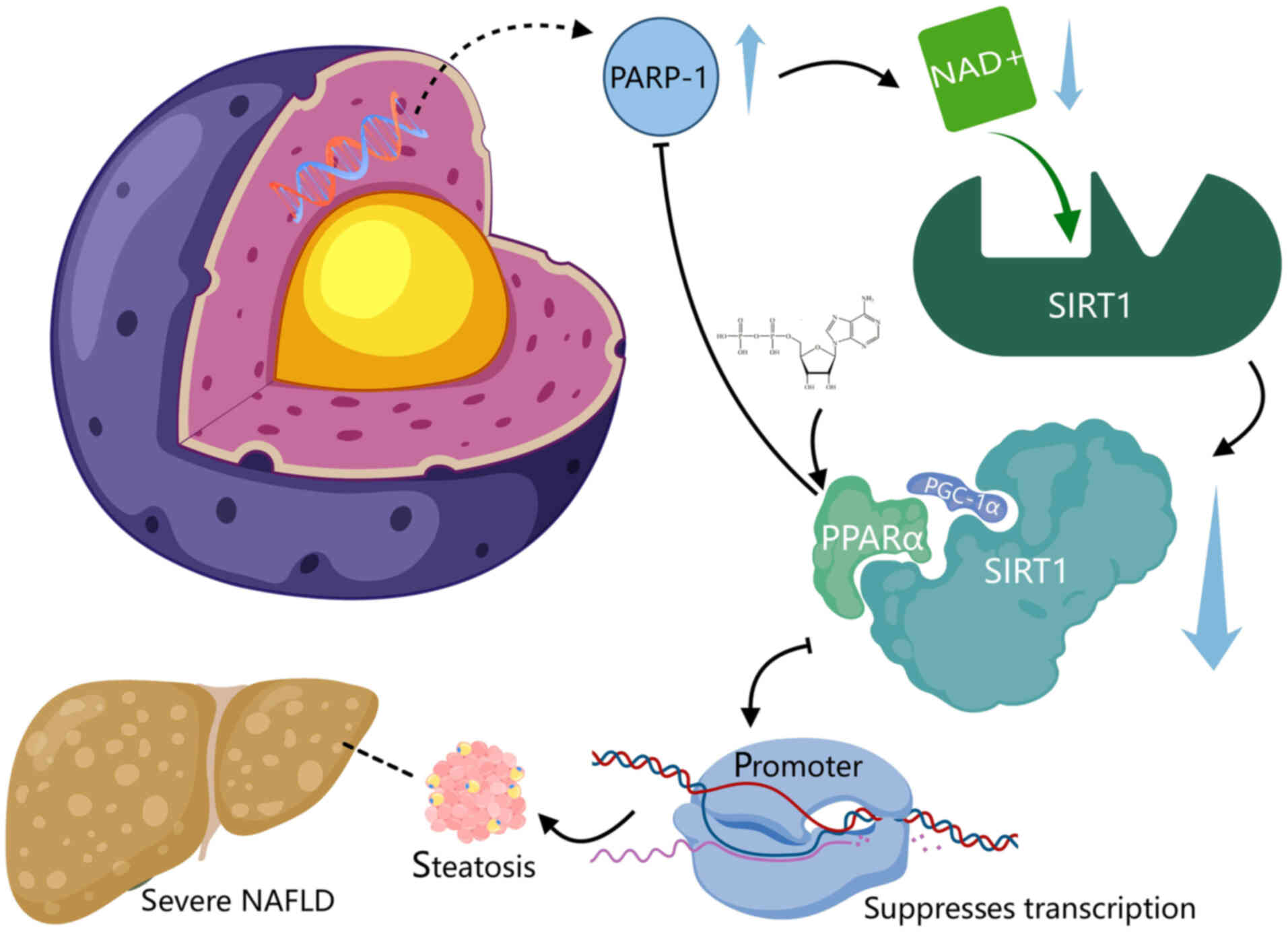
SIRT1 is an NAD+-dependent deacetylase
that plays a key role in the regulation of PPARα activity. Its
expression and activity are regulated by NAD+, which
serves as its substrate (114,115). Consequently, intracellular
NAD+ levels can directly influence PPARα signaling. In
the liver, PPARα signaling is markedly inhibited in the absence of
SIRT1. The inhibition of PARP-1 can raise intracellular
NAD+ levels, thereby increasing SIRT1 activity, and
upregulating PPARα signaling and mitochondrial oxidation. The
PARP-1-mediated poly(ADP-ribosyl)ation of PPARα suppresses its
transcriptional activity by preventing PPARα from binding to SIRT1
and from recruiting the PPARα/SIRT1/PPARg coactivator-1α complex to
target promoters (115). Notably,
the liver PARP activity of patients with severe NAFLD is much
greater than that of patients with simple steatosis, suggesting
that aberrant PARP-1 activation may contribute to the
downregulation of liver PPARα expression in these individuals
(115). These findings suggest
that inhibition of PARP-1 may have therapeutic potential in the
prevention and treatment of NAFLD (Fig. 3).
The accumulation of β-catenin has been observed in a
mouse model of liver cancer (116). Valanejad et al (117) identified a subset of 250-bp human
chromosomal domains, known as aggressive liver cancer domains
(ALCDs), that are located close to genes associated with various
cancer pathways and are regulated by PARP-1. Hepatoblastoma (HBL)
is a type of pediatric liver cancer that primarily affects children
<3 years old (118). Notably,
the PARP-1-ALCD axis modulates β-catenin, a potent promoter of
aggressive liver cancer, which is markedly increased in aggressive
HBL (117). PARP-1 activation can
contribute to the development of HBL by activating the
Wnt/β-catenin pathway and inducing post-translational changes in
tumor suppressor genes. Conversely, PARP-1 inhibition can decrease
cell proliferation, block Wnt/β-catenin signaling, and restore the
expression of tumor suppressor genes (119). These findings suggest that
targeting proteins associated with the Wnt/β-catenin pathway may
represent a potential therapeutic approach for liver cancer.
The PI3K/Akt/mTOR signaling pathway inhibits
apoptosis while enhancing cell survival and proliferation, and is
associated with angiogenesis, carcinogenesis, invasion, and
metastasis in several cancers, including colorectal cancer and
non-small cell lung cancer (120,121). Numerous studies have examined the
effects of natural substances on HCC, some of which target this
pathway. For example, in one study, the treatment of HepG2 cells
with 13-acetoxysarcocrassolide reduced p-PI3K, p-AKT and p-mTOR
levels, indicating suppression of the PI3K/Akt/mTOR signaling
pathway. This inhibition decreased the mTOR-mediated
phosphorylation of p70S6K and its downstream effectors S6 and
eukaryotic translation initiation factor 4B, and also increased
cleaved PARP-1 levels, indicating that the induced apoptosis was
facilitated by mitochondrial dysfunction. These findings suggest
that PARP-1 activity may be associated with the phosphorylation of
AKT, which influences downstream mTOR signaling and promotes tumor
cell proliferation and apoptotic resistance (122).
Mechanistically, PARP-1 further promotes HCC growth
by forming a positive feedback loop with the PI3K/AKT/mTOR
signaling axis. PARP-1 activation increases the phosphorylation of
AKT at serine 473, which activates mTOR complex 1 (mTORC1) targets
such as ribosomal protein S6 kinase 1 and eukaroytic translation
initiator factor 4E binding protein 1, thereby driving protein
synthesis, cell proliferation and survival while inhibiting
apoptosis (123–125). Conversely, PARP-1 inhibition
downregulates AKT activity via upregulation of PH domain
leucine-rich repeat protein phosphatase 1, highlighting the direct
role of PARP-1 in the modulation of AKT/mTOR signaling (126). The activation of AKT/mTOR
signaling has been demonstrated to support the stability and
activity of PARP-1, with PARP-1 activation modulating mTORC1
through AMPK-dependent mechanisms, and AKT activation enhancing DDR
components, including PARP-1 itself (124,127). This mutual reinforcement
establishes a negative feedback loop of pro-survival signaling and
genomic maintenance that promotes HCC progression and therapy
resistance. Preclinical studies indicate that disrupting either
PARP-1 or the PI3K/AKT/mTOR pathway can disrupt this feedback loop,
reduce tumor viability, and sensitize cells to treatment (125,126,128).
These findings indicate that PARP-1 regulates
several cellular signaling pathways and is crucial for the onset,
progression and clinical signs of several liver diseases. This
suggests the potential of PARP-1 as a prognostic indicator and
possible target for liver disease therapy.
Potential clinical value of PARP-1 in liver
disease
In emerging nations, particularly in the
Asia-Pacific region, liver diseases are becoming widely
acknowledged as major health concerns. While viral hepatitis
remains the primary cause of morbidity and death, ALD and NAFLD are
contributing to the liver disease burden at an accelerating rate
(129).
PARP-1 expression in upregulated in numerous
cancers, and prompt evaluation of its expression is important when
devising a treatment plan and monitoring therapeutic effectiveness.
Positron emission tomography (PET) imaging is an efficient,
noninvasive, real-time technique that can be used to assess PARP-1
activity throughout the body. In 2016, researchers conducted
preclinical evaluations and the first-in-human study of PARP
activity in cancer. Notably, the study observed increased uptake of
18F fluorthanatrace (18F-FTT) in a patient
with HCC (130). However,
investigations of PARP-targeted imaging in liver diseases,
including NASH, ALD and HCC, remain at the preclinical stage
(131), including a study of
PARP-targeted PET imaging in NASH animal models (132). Clinical trials of PARP-1 PET
probes such as 18F-FTT have so far been limited to other
tumor types, including breast, ovarian and prostate cancer
(133), with no direct
applications in liver pathology. This highlights a critical gap and
opportunity for the future clinical translation of PARP-1 in liver
diseases.
A number of tracers targeting PARP-1 have been
developed, including 18F-FTT and the olaparib analog
18F-PARPi. Patients with elevated PARP-1 expression can
be identified by PET imaging using 18F-FTT (134). A recent study designed a
high-affinity PARP-1 molecule, 18F-BIBD-300, by chemical
modification of the main pharmacophore of 18F-FTT. Since
18F-FTT has been used to diagnose liver
cancer,18F-BIBD-300 has been suggested to have potential
for this purpose (135).
18F-PARPi and 18F-FTT both undergo
hepatobiliary clearance, which may limit their effectiveness in the
imaging of abdominal lesions, such as liver metastases (136). 18F-FPyPARP, a
6-fluoronicotinoyl analog of 18F-PARPi, has been found
to be an excellent radioactive tracer for PARP expression that is
simpler to manufacture and less lipophilic than
18F-PARPi (136).
PARP inhibitors have emerged as leading cancer
treatments for BRCA-deficient patients with ovarian and breast
cancers (137). Notably, although
all PARP-1 drugs are classified as PARP inhibitors, not all PARP
inhibitors are selective for PARP-1. Thus, PARP-1 inhibitors
constitute a distinct subgroup within the broader category of PARP
inhibitors. The clinical utility of PARP inhibitors may be
attributed to their ability to efficiently kill cells with HRD in
BRCA1/2-deficient cancers by driving the accumulation of unrepaired
DNA damage and inducing synthetic lethality (138).
Common adverse reactions reported in cancer trials
of various PARP inhibitors, including olaparib and niraparib,
include anemia, leukopenia, thrombocytopenia, nausea, vomiting and
fatigue (139,140). While these adverse reactions have
been observed in patients with cancer, patients with liver disease
often have cytopenias, such as thrombocytopenia due to cirrhosis
(141); therefore, PARP
inhibitors may exacerbate hematological complications that are
already present. Nausea and vomiting are also common symptoms of
PARP inhibitors, occurring in >20% of patients, which may
exacerbate malnutrition in patients with advanced liver disease
(142). In addition, niraparib
has been associated with nephrotoxicity; impaired hepatic albumin
synthesis, such as that occurring in cirrhosis, may increase free
drug concentrations, thereby exacerbating nephrotoxicity (143,144).
Evidence for the efficacy of PARP-1 inhibitors in
liver diseases, including NAFLD/NASH, ALD, liver fibrosis/cirrhosis
and HCC, comes primarily from cell and animal models. For example,
one study demonstrated that PARP-1 inhibition contributed to the
antifibrotic effect of puerarin in mice (145), suggesting that targeting PARP-1
has therapeutic potential for liver fibrosis. Another study used
mouse models of NASH to verify that PARP inhibition and PARP-1
deletion reduce hepatic triglyceride accumulation, metabolic
disorders, inflammation and/or fibrosis (146), indicating that PARP inhibition
may be a promising treatment strategy for fatty hepatitis. Limited
data from patients are also available: A pilot study using liver
samples from patients with NAFLD confirmed that PARP-1 is activated
in human fatty liver, as well as in a HFD-induced mouse model
(61). In addition, a study at the
University of Minnesota analyzed liver tissue samples from patients
with alcoholic and HBV-related cirrhosis (stage 3–4 fibrosis). The
results of the study demonstrated the key pathogenic role of PARP-1
in liver fibrosis and the potential of PARP-1 inhibitors to restore
liver function after fibrosis (10). Collectively, these studies suggest
that repurposing PARP-1 inhibitors to treat liver diseases
associated with injury, inflammation and fibrosis may have
potential clinical applicability.
PARP-1 inhibitor monotherapy appears to be largely
ineffective in HCC, likely due to the BRCA nature of this cancer
type. However, one study investigated non-BRCA1/2 gene
abnormalities, and found that they confer sensitivities to PARP
inhibition comparable to those observed with BRCA1/2 mutations
(147). In addition, marked
suppression of HCC cell growth was observed both in vitro
and in vivo following treatment with a combination of
wild-type p53-induced protein phosphatase 1 and PARP inhibitors
(138). Synergistic strategies
comprising PARP inhibitors and radiotherapy have also been shown to
improve HCC management, with olaparib increasing the
radiosensitivity of HCC cells by promoting extensive DSBs (148). In addition, dual inhibition of
PARP-1/2 and tankyrase 1/2 exhibited potent anticancer activity
with strong synergy, suggesting an expanded therapeutic scope for
PARP-1 inhibitors (149).
Likewise, both ALD and NAFLD may be prevented by
blocking PARP-1. Since NAD+ plays a crucial role in
acute alcoholic liver injury, preserving NAD+ levels by
blocking PARP may help to maintain liver homeostasis (12). In addition, the pharmacological
inhibition or genetic deletion of PARP-1 has been shown to protect
against ethanol-induced hepatocyte damage and increased fatty acid
oxidation, thereby preventing fatty liver development in a rat
model of NAFLD (19).
Despite the encouraging results obtained for PARP-1
inhibitors in preclinical studies, several obstacles remain in the
translation of these discoveries into successful clinical
applications. A major issue is the intrinsic disparity between
animal or in vitro systems and the intricate pathophysiology
of human disease. Preclinical models cannot accurately replicate
the tumor microenvironment, genetic diversity or immune responses
observed in patients. In addition, drug metabolism and
pharmacokinetics differ significantly across species, resulting in
variations in efficacy and toxicity profiles. Another major barrier
is the development of resistance mechanisms in patients, which are
not always predictable from preclinical data (150). Collectively, these factors
contribute to the frequent failure of preclinically promising drugs
in clinical trials and highlight the requirement for improved
prediction models and biomarkers. Nevertheless, PARP-1 inhibition
remains a promising treatment strategy for various liver diseases,
if these translational challenges can be addressed.
Future expectations
As research has progressed, understanding of the
role of PARP-1 in liver disorders has advanced. Studies have shown
that conditions such as ALD (52),
liver cirrhosis (88) and HCC
(94,95) are associated with elevated PARP-1
levels, and that reducing PARP-1 expression and/or activity in
hepatocytes can alleviate these disorders. This section of the
review examines future directions for PARP-1 research by
integrating the latest findings from both medical and and
pharmaceutical perspectives.
PARP-1 inhibitors are commonly used to treat
malignancies, such as breast and ovarian cancers, with BRCA1/2
mutations (151). However, these
inhibitors have limited effectiveness in individuals who develop
resistance to PARP-1 inhibitors and in other malignancies, such as
HCC (152). Therefore, clinical
development has focused on overcoming PARP-1 inhibitor
resistance.
First, PARP-1 inhibitors can be combined with other
DNA damage repair inhibitors. Resistance to PARP inhibitors is
often not due to target protein mutations, but arises from the
ability of tumor cells to change their DNA damage repair pathways
(153). In addition to PARP-1,
several other proteins participate in DNA damage repair. For
example, DSBs recruit ATM proteins, which mediate checkpoint
signaling and DNA repair (154).
Replication stress activates ATM and Rad3-related (ATR), which
stabilize and restart replication forks, with checkpoint kinases 1
and 2 acting downstream of ATR and ATM (155). The effectiveness of DDR
inhibitors as monotherapies depends on their specific biological
activity, and combining inhibitors may be a beneficial therapeutic
approach when complementary pathways are targeted (156).
Secondly, PARP-1 inhibitors can be combined with
immune checkpoint inhibitors. PD-1, PD-L1 and cytotoxic
T-lymphocyte-associated antigen-4 are the most studied
immunological checkpoints. Immune checkpoint inhibitors targeting
these molecules exert antitumor effects by stimulating the tumor
immune response (157). Treatment
with a combination of PARP and immune checkpoint inhibitors has
been shown to exhibit higher antitumor efficacy than monotherapy.
Immune checkpoint inhibitors are able to sensitize tumor cells to
PARP inhibitors, which represents a promising therapeutic approach
(158).
A recent study identified numerous novel liver
cancer susceptibility genes and characterized the landscape of rare
genetic variants of liver cancer in a Chinese population. These
variants were found to be strongly associated with the risk of
liver cancer. Notably, the nuclear RNAi-defective 2 (NRDE2) gene
was shown to enhance the assembly and activity of the casein kinase
2 complex, thereby promoting the phosphorylation of mediator of DNA
damage checkpoint protein 1, initiating the HR repair pathway for
DNA DSB repair, and ultimately inhibiting the development of liver
cancer. NRDE2 may be a potential synthetic lethal target as rare
genetic mutations that impair its HR repair function markedly
increase the susceptibility of HCC to PARP-1 inhibitors. Overall,
the study demonstrated that NRDE2 is a regulatory component that
suppresses HCC and facilitates DNA damage repair, and that its
deficiency in HCC cells results in greater susceptibility to PARP-1
inhibitors (159). This study
identified NRDE2 as a potential biomarker for the treatment of HCC
with PARP-1 inhibitors. However, further studies are necessary to
determine whether NRDE2 deficiency could be used as a biomarker for
PARP-1 inhibitor response in other cancer types or if the loss of
other genes may have an impact comparable to that of NRDE2
(158).
Conclusions
PARP-1 plays a crucial role in liver disease by
managing responses to DNA damage, inflammation, apoptosis and
metabolic balance. When PARP-1 is dysregulated, it significantly
contributes to the progression of conditions such as NAFLD, ALD,
viral hepatitis, autoimmune liver diseases, hepatic fibrosis and
the serious development of HCC. Research has shown that inhibiting
PARP-1 can help reduce liver cell injury, decrease inflammation and
fibrosis, and enhance antitumor immunity, highlighting its
potential as a therapeutic target. Additionally, combining PARP-1
inhibitors with immune checkpoint blockers or agents that target
DNA repair may improve treatment effectiveness. Future research
should focus on the specific roles of different PARP-1 isoforms,
the mechanisms behind treatment resistance and how to apply these
findings in clinical settings. Overall, PARP-1 represents a
promising target for both treatment and diagnosis, with the
potential to improve outcomes for various liver diseases.
Acknowledgements
Not applicable.
Funding
This study was supported by the Natural Science Research Project
of Anhui Educational Committee (grant no. 2024AH050702) and the
Anhui Traditional Chinese Medicine Inheritance and Innovation
Research Project (grant no. 2024CCCX288).
Availability of data and materials
Not applicable.
Authors' contributions
KH, HT and SC wrote the manuscript. YL and RW
created the figures and table. YJ and BC supervised the research,
revised the manuscript, obtained financial support, conceptualized
the review and performed the literature search. All authors read
and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Neshat SY, Quiroz VM, Wang Y, Tamayo S and
Doloff JC: Liver Disease: induction, progression, immunological
mechanisms, and therapeutic interventions. Int J Mol Sci.
22:67772021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Devarbhavi H, Asrani SK, Arab JP, Nartey
YA, Pose E and Kamath PS: Global burden of liver disease: 2023
Update. J Hepatol. 79:516–537. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xiao J, Wang F, Wong NK, He J, Zhang R,
Sun R, Xu Y, Liu Y, Li W, Koike K, et al: Global liver disease
burdens and research trends: Analysis from a Chinese perspective. J
Hepatol. 71:212–221. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Younossi ZM, Loomba R, Rinella ME,
Bugianesi E, Marchesini G, Neuschwander-Tetri BA, Serfaty L, Negro
F, Caldwell SH, Ratziu V, et al: Current and future therapeutic
regimens for nonalcoholic fatty liver disease and nonalcoholic
steatohepatitis. Hepatology. 68:361–371. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Alvarado-Tapias E, Pose E, Gratacós-Ginès
J, Clemente-Sánchez A, López-Pelayo H and Bataller R:
Alcohol-associated liver disease: Natural history, management and
novel targeted therapies. Clin Mol Hepatol. 31 (Suppl 1):S112–S133.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Llovet JM, De Baere T, Kulik L, Haber PK,
Greten TF, Meyer T and Lencioni R: Locoregional therapies in the
era of molecular and immune treatments for hepatocellular
carcinoma. Nat Rev Gastroenterol Hepatol. 18:293–313. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ishikawa T: Efficacy and features of
balloon-occluded transarterial chemoembolization for hepatocellular
carcinoma: A narrative review. Transl Gastroenterol Hepatol.
9:482024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pandey N and Black BE: Rapid detection and
signaling of DNA damage by PARP-1. Trends Biochem Sci. 46:744–757.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Galindo-Campos MA, Bedora-Faure M, Farrés
J, Lescale C, Moreno-Lama L, Martínez C, Martín-Caballero J,
Ampurdanés C, Aparicio P, Dantzer F, et al: Coordinated signals
from the DNA repair enzymes PARP-1 and PARP-2 promotes B-cell
development and function. Cell Death Differ. 26:2667–2681. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mukhopadhyay P, Rajesh M, Cao Z, Horváth
B, Park O, Wang H, Erdelyi K, Holovac E, Wang Y, Liaudet L, et al:
Poly (ADP-ribose) polymerase-1 is a key mediator of liver
inflammation and fibrosis. Hepatology. 59:1998–2009. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu XL, Xing BC, Han HB, Zhao W, Hu MH, Xu
ZL, Li JY, Xie Y, Gu J, Wang Y and Zhang ZQ: The properties of
tumor-initiating cells from a hepatocellular carcinoma patient's
primary and recurrent tumor. Carcinogenesis. 31:167–174. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gariani K, Ryu D, Menzies KJ, Yi HS, Stein
S, Zhang H, Perino A, Lemos V, Katsyuba E, Jha P, et al: Inhibiting
poly ADP-ribosylation increases fatty acid oxidation and protects
against fatty liver disease. J Hepatol. 66:132–141. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Guillot C, Hall J, Herceg Z, Merle P and
Chemin I: Update on hepatocellular carcinoma breakthroughs:
Poly(ADP-ribose) polymerase inhibitors as a promising therapeutic
strategy. Clin Res Hepatol Gastroenterol. 38:137–142. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Wang C, Tian Y, Zhang F, Xu W, Li
X, Shu Z, Wang Y, Huang K and Huang D: Inhibition of
poly(ADP-ribose) polymerase-1 protects chronic alcoholic liver
injury. Am J Pathol. 186:3117–3130. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hardy T and Mann DA: Epigenetics in liver
disease: From biology to therapeutics. Gut. 65:1895–1905. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng ML, Nakib D, Perciani CT and
MacParland SA: The immune niche of the liver. Clin Sci (Lond).
135:2445–2466. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Piano S, Bunchorntavakul C, Marciano S and
Rajender Reddy K: Infections in cirrhosis. Lancet Gastroenterol
Hepatol. 9:745–757. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubes P and Jenne C: Immune responses in
the liver. Annu Rev Immunol. 36:247–277. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mukhopadhyay P, Horváth B, Rajesh M, Varga
ZV, Gariani K, Ryu D, Cao Z, Holovac E, Park O, Zhou Z, et al: PARP
inhibition protects against alcoholic and non-alcoholic
steatohepatitis. J Hepatol. 66:589–600. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
De Siervi S, Cannito S and Turato C:
Chronic liver disease: latest research in pathogenesis, detection
and treatment. Int J Mol Sci. 24:106332023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Santinelli-Pestana DV, Aikawa E, Singh SA
and Aikawa M: PARPs and ADP-ribosylation in chronic inflammation: A
focus on macrophages. Pathogens. 12:9642023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Vida A, Márton J, Mikó E and Bai P:
Metabolic roles of poly(ADP-ribose) polymerases. Semin Cell Dev
Biol. 63:135–143. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li WH, Wang F, Song GY, Yu QH, Du RP and
Xu P: PARP-1: A critical regulator in radioprotection and
radiotherapy-mechanisms, challenges, and therapeutic opportunities.
Front Pharmacol. 14:11989482023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Szántó M, Gupte R, Kraus WL, Pacher P and
Bai P: PARPs in lipid metabolism and related diseases. Prog Lipid
Res. 84:1011172021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Demin AA, Hirota K, Tsuda M, Adamowicz M,
Hailstone R, Brazina J, Gittens W, Kalasova I, Shao Z, Zha S, et
al: XRCC1 prevents toxic PARP1 trapping during DNA base excision
repair. Mol Cell. 81:3018–3030.e5. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ogawa K, Masutani M, Kato K, Tang M,
Kamada N, Suzuki H, Nakagama H, Sugimura T and Shirai T: Parp-1
deficiency does not enhance liver carcinogenesis induced by
2-amino-3-methylimidazo[4,5-f]quinoline in mice. Cancer Lett.
236:32–38. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liao Y, Wang X, Ran G, Zhang S, Wu C, Tan
R, Liu Y, He Y, Liu T, Wu Z, et al: DNA damage and up-regulation of
PARP-1 induced by columbin in vitro and in vivo. Toxicol Lett.
379:20–34. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Cantó C, Houtkooper RH, Pirinen E, Youn
DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux
PA, Cettour-Rose P, et al: The NAD(+) precursor nicotinamide
riboside enhances oxidative metabolism and protects against
high-fat diet-induced obesity. Cell Metab. 15:838–847. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu AY, Sekar P, Huang DY, Hsu SH, Chan CM
and Lin WW: Spatiotemporal roles of AMPK in PARP-1- and
autophagy-dependent retinal pigment epithelial cell death caused by
UVA. J Biomed Sci. 30:912023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ju C, Liu C, Yan S, Wang Y, Mao X, Liang M
and Huang K: Poly(ADP-ribose) Polymerase-1 is required for
hepatocyte proliferation and liver regeneration in mice. Biochem
Biophys Res Commun. 511:531–535. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yimlamai D, Christodoulou C, Galli GG,
Yanger K, Pepe-Mooney B, Gurung B, Shrestha K, Cahan P, Stanger BZ
and Camargo FD: Hippo pathway activity influences liver cell fate.
Cell. 157:1324–1338. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cover C, Fickert P, Knight TR,
Fuchsbichler A, Farhood A, Trauner M and Jaeschke H:
Pathophysiological role of poly(ADP-ribose) polymerase (PARP)
activation during acetaminophen-induced liver cell necrosis in
mice. Toxicol Sci. 84:201–208. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Andronikou C and Rottenberg S: Studying
PAR-dependent chromatin remodeling to tackle PARPi resistance.
Trends Mol Med. 27:630–642. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pazzaglia S and Pioli C: Multifaceted role
of PARP-1 in DNA repair and inflammation: Pathological and
therapeutic implications in cancer and non-cancer diseases. Cells.
9:412019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Llaneras J, Riveiro-Barciela M,
Rando-Segura A, Marcos-Fosch C, Roade L, Velázquez F,
Rodríguez-Frías F, Esteban R and Buti M: Etiologies and features of
acute viral hepatitis in Spain. Clin Gastroenterol Hepatol.
19:1030–1037. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cooke GS, Flower B, Cunningham E, Marshall
AD, Lazarus JV, Palayew A, Jia J, Aggarwal R, Al-Mahtab M, Tanaka
Y, et al: Progress towards elimination of viral hepatitis: A lancet
gastroenterology and hepatology commission update. Lancet
Gastroenterol Hepatol. 9:346–365. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yeh SH, Li CL, Lin YY, Ho MC, Wang YC,
Tseng ST and Chen PJ: Hepatitis B virus DNA integration drives
carcinogenesis and provides a new biomarker for HBV-related HCC.
Cell Mol Gastroenterol Hepatol. 15:921–929. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Funato K, Otsuka M, Sekiba K, Miyakawa Y,
Seimiya T, Shibata C, Kishikawa T and Fujishiro M: Hepatitis B
virus-associated hepatocellular carcinoma with Smc5/6 complex
deficiency is susceptible to PARP inhibitors. Biochem Biophys Res
Commun. 607:89–95. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou L, Liu CH, Lv D, Sample KM, Rojas Á,
Zhang Y, Qiu H, He L, Zheng L, Chen L, et al: Halting
hepatocellular carcinoma: Identifying intercellular crosstalk in
HBV-driven disease. Cell Rep. 44:1154572025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu N, Liu Q, Yang X, Zhang F, Li X, Ma Y,
Guan F, Zhao X, Li Z, Zhang L and Ye X: Hepatitis B
virus-upregulated LNC-HUR1 promotes cell proliferation and
tumorigenesis by blocking p53 activity. Hepatology. 68:2130–2144.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Y, Wen J, Qi D, Tong X, Liu N and Ye
X: HBV-upregulated Lnc-HUR1 inhibits the apoptosis of liver cancer
cells. Sheng Wu Gong Cheng Xue Bao. 38:3501–3514. 2022.(In
Chinese). PubMed/NCBI
|
|
42
|
Machida K, Tsukamoto H, Mkrtchyan H, Duan
L, Dynnyk A, Liu HM, Asahina K, Govindarajan S, Ray R, Ou JH, et
al: Toll-like receptor 4 mediates synergism between alcohol and HCV
in hepatic oncogenesis involving stem cell marker Nanog. Proc Natl
Acad Sci USA. 106:1548–1553. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pal S, Polyak SJ, Bano N, Qiu WC,
Carithers RL, Shuhart M, Gretch DR and Das A: Hepatitis C virus
induces oxidative stress, DNA damage and modulates the DNA repair
enzyme NEIL1. J Gastroenterol Hepatol. 25:627–634. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Smirnova OA, Ivanova ON, Mukhtarov F,
Valuev-Elliston VT, Fedulov AP, Rubtsov PM, Zakirova NF, Kochetkov
SN, Bartosch B and Ivanov AV: Hepatitis Delta virus antigens
trigger oxidative stress, activate antioxidant Nrf2/ARE pathway,
and induce unfolded protein response. Antioxidants (Basel).
12:9742023. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bajaj JS: Alcohol, liver disease and the
gut microbiota. Nat Rev Gastroenterol Hepatol. 16:235–246. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Contreras-Zentella ML, Villalobos-García D
and Hernández-Muñoz R: Ethanol metabolism in the liver, the
induction of oxidant stress, and the antioxidant defense system.
Antioxidants (Basel). 11:12582022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang Y, Luo W and Wang Y: PARP-1 and its
associated nucleases in DNA damage response. DNA Repair (Amst).
81:1026512019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Langelier MF and Pascal JM: PARP-1
mechanism for coupling DNA damage detection to poly(ADP-ribose)
synthesis. Curr Opin Struct Biol. 23:134–143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Huang H, Wei S, Wu X, Zhang M, Zhou B,
Huang D and Dong W: Dihydrokaempferol attenuates
CCl4-induced hepatic fibrosis by inhibiting PARP-1 to
affect multiple downstream pathways and cytokines. Toxicol Appl
Pharmacol. 464:1164382023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ma XY, Zhang M, Fang G, Cheng CJ, Wang MK,
Han YM, Hou XT, Hao EW, Hou YY and Bai G: Ursolic acid reduces
hepatocellular apoptosis and alleviates alcohol-induced liver
injury via irreversible inhibition of CASP3 in vivo. Acta Pharmacol
Sin. 42:1101–1110. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Yin F, Wu MM, Wei XL, Ren RX, Liu MH, Chen
CQ, Yang L, Xie RQ, Jiang SY, Wang XF and Wang H: Hepatic NCoR1
deletion exacerbates alcohol-induced liver injury in mice by
promoting CCL2-mediated monocyte-derived macrophage infiltration.
Acta Pharmacol Sin. 43:2351–2361. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Ambade A, Lowe P, Kodys K, Catalano D,
Gyongyosi B, Cho Y, Iracheta-Vellve A, Adejumo A, Saha B, Calenda
C, et al: Pharmacological inhibition of CCR2/5 signaling prevents
and reverses alcohol-induced liver damage, steatosis, and
inflammation in mice. Hepatology. 69:1105–1121. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang XJ and Malhi H: Nonalcoholic fatty
liver disease. Ann Intern Med. 169:ITC65–ITC80. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Guo X, Yin X, Liu Z and Wang J:
Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural
products for prevention and treatment. Int J Mol Sci. 23:154892022.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Harrison SA, Allen AM, Dubourg J,
Noureddin M and Alkhouri N: Challenges and opportunities in NASH
drug development. Nat Med. 29:562–573. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Saiman Y, Duarte-Rojo A and Rinella ME:
Fatty liver disease: Diagnosis and stratification. Annu Rev Med.
73:529–544. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Ye H, Ma S, Qiu Z, Huang S, Deng G, Li Y,
Xu S, Yang M, Shi H, Wu C, et al: Poria cocos polysaccharides
rescue pyroptosis-driven gut vascular barrier disruption in order
to alleviates non-alcoholic steatohepatitis. J Ethnopharmacol.
296:1154572022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang JH, Byeon EH, Kang D, Hong SG, Yang
J, Kim DR, Yun SP, Park SW, Kim HJ, Huh JW, et al: Fermented
soybean paste attenuates biogenic amine-induced liver damage in
obese mice. Cells. 12:8222023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Guo R, Li Y, Song Q, Huang R, Ge X, Nieto
N, Jiang Y and Song Z: Increasing cellular NAD+ protects
hepatocytes against palmitate-induced lipotoxicity by preventing
PARP-1 inhibition and the mTORC1-p300 pathway activation. Am J
Physiol Cell Physiol. 328:C776–C790. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Salomone F, Barbagallo I, Godos J, Lembo
V, Currenti W, Cinà D, Avola R, D'Orazio N, Morisco F, Galvano F
and Li Volti G: Silibinin restores NAD+ levels and
induces the SIRT1/AMPK pathway in non-alcoholic fatty liver.
Nutrients. 9:10862017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Huang K, Du M, Tan X, Yang L, Li X, Jiang
Y, Wang C, Zhang F, Zhu F, Cheng M, et al: PARP1-mediated PPARα
poly(ADP-ribosyl)ation suppresses fatty acid oxidation in
non-alcoholic fatty liver disease. J Hepatol. 66:962–977. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Majeed Y, Halabi N, Madani AY, Engelke R,
Bhagwat AM, Abdesselem H, Agha MV, Vakayil M, Courjaret R, Goswami
N, et al: SIRT1 promotes lipid metabolism and mitochondrial
biogenesis in adipocytes and coordinates adipogenesis by targeting
key enzymatic pathways. Sci Rep. 11:81772021. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Ding RB, Bao J and Deng CX: Emerging roles
of SIRT1 in fatty liver diseases. Int J Biol Sci. 13:852–867. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Ray K: Developing a toolbox for
drug-induced liver injury. Nat Rev Gastroenterol Hepatol.
17:7142020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Kumachev A and Wu PE: Drug-induced liver
injury. CMAJ. 193:E3102021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Björnsson HK and Björnsson ES:
Drug-induced liver injury: Pathogenesis, epidemiology, clinical
features, and practical management. Eur J Intern Med. 97:26–31.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
European Association for the Study of the
Liver, . EASL clinical practice guidelines: Drug-induced liver
injury. J Hepatol. 70:1222–1261. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Li X, Tang J and Mao Y: Incidence and risk
factors of drug-induced liver injury. Liver Int. 42:1999–2014.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Gallyas F Jr and Sumegi B: Mitochondrial
protection by PARP inhibition. Int J Mol Sci. 21:27672020.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Wang C, Xu W, Zhang Y, Huang D and Huang
K: Poly(ADP-ribosyl)ated PXR is a critical regulator of
acetaminophen-induced hepatotoxicity. Cell Death Dis. 9:8192018.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Su Q, Kuang W, Hao W, Liang J, Wu L, Tang
C, Wang Y and Liu T: Antituberculosis drugs (rifampicin and
isoniazid) induce liver injury by regulating NLRP3 inflammasomes.
Mediators Inflamm. 2021:80862532021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Carbone M and Neuberger JM: Autoimmune
liver disease, autoimmunity and liver transplantation. J Hepatol.
60:210–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Trivedi PJ, Hirschfield GM, Adams DH and
Vierling JM: Immunopathogenesis of primary biliary cholangitis,
primary sclerosing cholangitis and autoimmune hepatitis: Themes and
concepts. Gastroenterology. 166:995–1019. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Yilmaz K, Haeberle S, Kim YO, Fritzler MJ,
Weng SY, Goeppert B, Raker VK, Steinbrink K, Schuppan D, Enk A and
Hadaschik EN: Regulatory T-cell deficiency leads to features of
autoimmune liver disease overlap syndrome in scurfy mice. Front
Immunol. 14:12536492023. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Heo NY and Kim H: Epidemiology and updated
management for autoimmune liver disease. Clin Mol Hepatol.
29:194–196. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang Y, Pötter S, Chen CW, Liang R, Gelse
K, Ludolph I, Horch RE, Distler O, Schett G, Distler JHW and Dees
C: Poly(ADP-ribose) polymerase-1 regulates fibroblast activation in
systemic sclerosis. Ann Rheum Dis. 77:744–751. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Su X, Ye L, Chen X, Zhang H, Zhou Y, Ding
X, Chen D, Lin Q and Chen C: MiR-199-3p promotes ERK-mediated IL-10
production by targeting poly (ADP-ribose) polymerase-1 in patients
with systemic lupus erythematosus. Chem Biol Interact. 306:110–116.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang G, Ma H, Wang J and Khan MF:
Contribution of poly(ADP-ribose)polymerase-1 activation and
apoptosis in trichloroethene-mediated autoimmunity. Toxicol Appl
Pharmacol. 362:28–34. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hao J, Sun W and Xu H: Pathogenesis of
concanavalin A induced autoimmune hepatitis in mice. Int
Immunopharmacol. 102:1084112022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Liu Q, Yang H, Kang X, Tian H, Kang Y, Li
L, Yang X, Liu H, Ren P, Kuang X, et al: A synbiotic ameliorates
con A-induced autoimmune hepatitis in mice through modulation of
gut microbiota and immune imbalance. Mol Nutr Food Res.
67:e22004282023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Liu Y, Hao H and Hou T: Concanavalin
A-induced autoimmune hepatitis model in mice: Mechanisms and future
outlook. Open Life Sci. 17:91–101. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Filliol A, Piquet-Pellorce C, Dion S,
Genet V, Lucas-Clerc C, Dantzer F and Samson M: PARP2 deficiency
affects invariant-NKT-cell maturation and protects mice from
concanavalin A-induced liver injury. Am J Physiol Gastrointest
Liver Physiol. 313:G399–G409. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Virág L and Szabó C: The therapeutic
potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev.
54:375–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Tanaka A: Emerging novel treatments for
autoimmune liver diseases. Hepatol Res. 49:489–499. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kisseleva T and Brenner D: Molecular and
cellular mechanisms of liver fibrosis and its regression. Nat Rev
Gastroenterol Hepatol. 18:151–166. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Horn P and Tacke F: Metabolic
reprogramming in liver fibrosis. Cell Metab. 36:1439–1455. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Gu L, Zhao C, Wang Y, Wang C, Yin X, Ye Q,
Liu Y, Zou X, Wang L, Zhuge Y, et al: Senescence of hepatic
stellate cells by specific delivery of manganese for limiting liver
fibrosis. Nano Lett. 24:1062–1073. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Nithyananthan S and Thirunavukkarasu C:
Arsenic trioxide, a cancer chemo drug hampers fibrotic liver
regeneration by interrupting oxidative stress rekindling and
stellate cell rejuvenation. J Cell Physiol. 235:1222–1234. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Meng D, Li Z, Wang G, Ling L, Wu Y and
Zhang C: Carvedilol attenuates liver fibrosis by suppressing
autophagy and promoting apoptosis in hepatic stellate cells. Biomed
Pharmacother. 108:1617–1627. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tan Y, Li C, Zhou J, Deng F and Liu Y:
Berberine attenuates liver fibrosis by autophagy inhibition
triggering apoptosis via the miR-30a-5p/ATG5 axis. Exp Cell Res.
427:1136002023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Singal AG, Kanwal F and Llovet JM: Global
trends in hepatocellular carcinoma epidemiology: Implications for
screening, prevention and therapy. Nat Rev Clin Oncol. 20:864–884.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Pan J, Zhang M, Dong L, Ji S, Zhang J,
Zhang S, Lin Y, Wang X, Ding Z, Qiu S, et al: Genome-Scale CRISPR
screen identifies LAPTM5 driving lenvatinib resistance in
hepatocellular carcinoma. Autophagy. 19:1184–1198. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
No authors listed. Hepatocellular
carcinoma. Nat Rev Dis Primers. 7:72021. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zai W, Chen W, Han Y, Wu Z, Fan J, Zhang
X, Luan J, Tang S, Jin X, Fu X, et al: Targeting PARP and autophagy
evoked synergistic lethality in hepatocellular carcinoma.
Carcinogenesis. 41:345–357. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Gerossier L, Dubois A, Paturel A, Fares N,
Cohen D, Merle P, Lachuer J, Wierinckx A, Saintigny P, Bancel B, et
al: PARP inhibitors and radiation potentiate liver cell death in
vitro. Do hepatocellular carcinomas have an achilles' heel? Clin
Res Hepatol Gastroenterol. 45:1015532021.PubMed/NCBI
|
|
96
|
Jeong KY and Park MH: The significance of
targeting Poly (ADP-ribose) polymerase-1 in pancreatic cancer for
providing a new therapeutic paradigm. Int J Mol Sci. 22:35092021.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Demény MA and Virág L: The PARP enzyme
family and the hallmarks of cancer part 1. Cell intrinsic
hallmarks. Cancers (Basel). 13:20422021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Zhu T, Zheng JY, Huang LL, Wang YH, Yao DF
and Dai HB: Human PARP1 substrates and regulators of its catalytic
activity: An updated overview. Front Pharmacol. 14:11371512023.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Wong CW, Evangelou C, Sefton KN, Leshem R,
Zhang W, Gopalan V, Chattrakarn S, Fernandez Carro ML, Uzuner E,
Mole H, et al: PARP14 inhibition restores PD-1 immune checkpoint
inhibitor response following IFNγ-driven acquired resistance in
preclinical cancer models. Nat Commun. 14:59832023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Sun G, Miao G, Li Z, Zheng W, Zhou C, Sun
G, Cao H, Li Z and Tang W: Inhibition of PARP potentiates immune
checkpoint therapy through miR-513/PD-L1 pathway in hepatocellular
carcinoma. J Oncol. 2022:69889232022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Konstantinopoulos PA, Waggoner S, Vidal
GA, Mita M, Moroney JW, Holloway R, Van Le L, Sachdev JC,
Chapman-Davis E, Colon-Otero G, et al: Single-arm phases 1 and 2
trial of niraparib in combination with pembrolizumab in patients
with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol.
5:1141–1149. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Yu EY, Piulats JM, Gravis G, Fong PCC,
Todenhöfer T, Laguerre B, Arranz JA, Oudard S, Massard C,
Heinzelbecker J, et al: Pembrolizumab plus olaparib in patients
with metastatic castration-resistant prostate cancer: Long-term
results from the phase 1b/2 KEYNOTE-365 cohort a study. Eur Urol.
83:15–26. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Yap TA, Bardia A, Dvorkin M, Galsky MD,
Beck JT, Wise DR, Karyakin O, Rubovszky G, Kislov N, Rohrberg K, et
al: Avelumab plus talazoparib in patients with advanced solid
tumors: The JAVELIN PARP medley nonrandomized controlled trial.
JAMA Oncol. 9:40–50. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ishteyaque S, Singh G, Yadav KS, Verma S,
Sharma RK, Sen S, Srivastava AK, Mitra K, Lahiri A, Bawankule DU,
et al: Cooperative STAT3-NFkB signaling modulates mitochondrial
dysfunction and metabolic profiling in hepatocellular carcinoma.
Metabolism. 152:1557712024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Zein N, Elewa YHA, Alruwaili MK, Dewaard
M, Alorabi M, Albogami SM, Batiha GE and Zahran MH: Barhi date
(Phoenix dactylifera) extract ameliorates hepatocellular carcinoma
in male rats. Biomed Pharmacother. 156:1139762022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Pasaol JC, Dejnaka E, Mucignat G, Bajzert
J, Henklewska M, Obmińska-Mrukowicz B, Giantin M, Pauletto M,
Zdyrski C, Dacasto M and Pawlak A: PARP inhibitor olaparib induces
DNA damage and acts as a drug sensitizer in an in vitro model of
canine hematopoietic cancer. BMC Vet Res. 21:4392025. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Wu Y, Li Y, Guo W, Liu J, Lao W, Hu P, Lin
Y and Chen H: Laminaria japonica peptides suppress liver
cancer by inducing apoptosis: Possible signaling pathways and
mechanism. Mar Drugs. 20:7042022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Luo T, Yuan Y, Yu Q, Liu G, Long M, Zhang
K, Bian J, Gu J, Zou H, Wang Y, et al: PARP-1 overexpression
contributes to Cadmium-induced death in rat proximal tubular cells
via parthanatos and the MAPK signalling pathway. Sci Rep.
7:43312017. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Dong Q, Du Y, Li H, Liu C, Wei Y, Chen MK,
Zhao X, Chu YY, Qiu Y, Qin L, et al: EGFR and c-MET cooperate to
enhance resistance to PARP inhibitors in hepatocellular carcinoma.
Cancer Res. 79:819–829. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Merdrignac A, Papoutsoglou P and Coulouarn
C: Long noncoding RNAs in cholangiocarcinoma. Hepatology.
73:1213–1226. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Huang Z, Zhou JK, Peng Y, He W and Huang
C: The role of long noncoding RNAs in hepatocellular carcinoma. Mol
Cancer. 19:772020. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen C, Chen J, Wang Y, Fang L, Guo C,
Sang T, Peng H, Zhao Q, Chen S, Lin X and Wang X: Ganoderma lucidum
polysaccharide inhibits HSC activation and liver fibrosis via
targeting inflammation, apoptosis, cell cycle, and ECM-receptor
interaction mediated by TGF-β/Smad signaling. Phytomedicine.
110:1546262023. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Ren Y, Chen Y, Tang EH, Hu Y, Niu B, Liang
H, Xi C, Zhao F and Cao Z: Arbidol attenuates liver fibrosis and
activation of hepatic stellate cells by blocking TGF-β1 signaling.
Eur J Pharmacol. 967:1763672024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang
XJ, Wang H, Du Y, Guan J, Wang X and Fu J: NAD+ improves
cognitive function and reduces neuroinflammation by ameliorating
mitochondrial damage and decreasing ROS production in chronic
cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J
Neuroinflammation. 18:2072021. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Huang K, Du M, Tan X, Yang L, Li X, Jiang
Y, Wang C, Zhang F, Zhu F, Cheng M, et al: PARP1-mediated PPARα
poly(ADP-ribosyl)ation suppresses fatty acid oxidation in
non-alcoholic fatty liver disease. J Hepatol. 66:962–977. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Nozaki T, Fujihara H, Watanabe M, Tsutsumi
M, Nakamoto K, Kusuoka O, Kamada N, Suzuki H, Nakagama H, Sugimura
T and Masutani M: Parp-1 deficiency implicated in colon and liver
tumorigenesis induced by azoxymethane. Cancer Sci. 94:497–500.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Valanejad L, Cast A, Wright M, Bissig KD,
Karns R, Weirauch MT and Timchenko N: PARP1 activation increases
expression of modified tumor suppressors and pathways underlying
development of aggressive hepatoblastoma. Commun Biol. 1:672018.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Pio L, O'Neill AF, Woodley H, Murphy AJ,
Tiao G, Franchi-Abella S, Fresneau B, Watanabe K, Alaggio R,
Lopez-Terrada D, et al: Hepatoblastoma. Nat Rev Dis Primers.
11:362025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Sha YL, Liu S, Yan WW and Dong B:
Wnt/β-catenin signaling as a useful therapeutic target in
hepatoblastoma. Biosci Rep. 39:BSR201924662019. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Maharati A and Moghbeli M: PI3K/AKT
signaling pathway as a critical regulator of epithelial-mesenchymal
transition in colorectal tumor cells. Cell Commun Signal.
21:2012023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Tan AC: Targeting the PI3K/Akt/mTOR
pathway in non-small cell lung cancer (NSCLC). Thorac Cancer.
11:511–518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Hsu CM, Lin JJ, Su JH and Liu CI:
13-Acetoxysarcocrassolide induces apoptosis in human hepatocellular
carcinoma cells through mitochondrial dysfunction and suppression
of the PI3K/AKT/mTOR/p70S6K signalling pathway. Pharm Biol.
60:2276–2285. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ethier C, Tardif M, Arul L and Poirier GG:
PARP-1 modulation of mTOR signaling in response to a DNA alkylating
agent. PLoS One. 7:e479782012. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Sun EJ, Wankell M, Palamuthusingam P,
McFarlane C and Hebbard L: Targeting the PI3K/Akt/mTOR pathway in
hepatocellular carcinoma. Biomedicines. 9:16392021. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Gallyas F Jr, Sumegi B and Szabo C: Role
of Akt activation in PARP inhibitor resistance in cancer. Cancers
(Basel). 12:5322020. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Wang S, Wang H, Davis BC, Liang J, Cui R,
Chen SJ and Xu ZX: PARP1 inhibitors attenuate AKT phosphorylation
via the upregulation of PHLPP1. Biochem Biophys Res Commun.
412:379–384. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Xu H, Ma Z, Mo X, Chen X, Xu F, Wu F, Chen
H, Zhou G, Xia H and Zhang C: Inducing synergistic DNA damage by
TRIP13 and PARP1 inhibitors provides a potential treatment for
hepatocellular carcinoma. J Cancer. 13:2226–2237. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Bhamidipati D, Haro-Silerio JI, Yap TA and
Ngoi N: PARP inhibitors: Enhancing efficacy through rational
combinations. Br J Cancer. 129:904–916. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Sarin SK, Kumar M, Eslam M, George J, Al
Mahtab M, Akbar SMF, Jia J, Tian Q, Aggarwal R, Muljono DH, et al:
Liver diseases in the Asia-Pacific region: A lancet
gastroenterology & hepatology commission. Lancet Gastroenterol
Hepatol. 5:167–228. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Michel LS, Dyroff S, Brooks FJ, Spayd KJ,
Lim S, Engle JT, Phillips S, Tan B, Wang-Gillam A, Bognar C, et al:
PET of poly (ADP-Ribose) polymerase activity in cancer: Preclinical
assessment and first in-human studies. Radiology. 282:453–463.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Destro G, Rizzo R, Rua C, Azimi RR and
Morbelli S: Exploring the radiochemistry of PARP inhibitors: A new
era in therapy and imaging. EJNMMI Radiopharm Chem. 10:372025.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Jeppesen TE, Shao T, Chen J, Patel JS,
Zhou X, Kjaer A and Liang SH: Poly(ADP-ribose) polymerase
(PARP)-targeted PET imaging in non-oncology application: A pilot
study in preclinical models of nonalcoholic steatohepatitis. Am J
Nucl Med Mol Imaging. 14:41–47. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
McDonald ES, Pantel AR, Shah PD, Farwell
MD, Clark AS, Doot RK, Pryma DA and Carlin SD: In vivo
visualization of PARP inhibitor pharmacodynamics. JCI Insight.
6:e1465922021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Lee HS, Schwarz SW, Schubert EK, Chen DL,
Doot RK, Makvandi M, Lin LL, McDonald ES, Mankoff DA and Mach RH:
The development of 18F fluorthanatrace: A PET
radiotracer for imaging poly (ADP-ribose) polymerase-1. Radiol
Imaging Cancer. 4:e2100702022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Zheng W, Huang Y, Xie Y, Yang T, Cheng X,
Chen H, Li C, Jiang Z, Yu Z, Li Z, et al: Design, synthesis, and
evaluation of [18F]BIBD-300 as a positron emission
tomography tracer for poly(ADP-ribose) polymerase-1. Mol Pharm.
21:2606–2621. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Stotz S, Kinzler J, Nies AT, Schwab M and
Maurer A: Two experts and a newbie: [18F]PARPi vs.
[18F]FTT vs. [18F]FPyPARP-a comparison of
PARP imaging agents. Eur J Nucl Med Mol Imaging. 49:834–846. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Li H, Liu ZY, Wu N, Chen YC, Cheng Q and
Wang J: PARP inhibitor resistance: The underlying mechanisms and
clinical implications. Mol Cancer. 19:1072020. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Chen M, Wang W, Hu S, Tong Y, Li Y, Wei Q,
Yu L, Zhu L, Zhu Y, Liu L, et al: Co-targeting WIP1 and PARP
induces synthetic lethality in hepatocellular carcinoma. Cell
Commun Signal. 20:392022. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Zhou Y, Zhao S, Wu T and Zhang H:
Comparison of adverse reactions caused by olaparib for different
indications. Front Pharmacol. 13:9681632022. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
LaFargue CJ, Dal Molin GZ, Sood AK and
Coleman RL: Exploring and comparing adverse events between PARP
inhibitors. Lancet Oncol. 20:e15–e28. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Peck-Radosavljevic M: Thrombocytopenia in
chronic liver disease. Liver Int. 37:778–793. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Shin S, Jun DW, Saeed WK and Koh DH: A
narrative review of malnutrition in chronic liver disease. Ann
Transl Med. 9:1722021. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Ren X, Sun P and Wang Y: PARP
inhibitor-related acute renal failure: A real-world study based on
the FDA adverse event reporting system database. Expert Opin Drug
Saf. 23:1463–1471. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Lazareth H, Delanoy N, Cohen R, Boissier
E, Ayari H, Combe P, Crespel C, Mercadier-Riaz E, Karras A,
Courbebaisse M, et al: Nephrotoxicity associated with niraparib. Am
J Kidney Dis. 76:898–900. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Wang S, Shi XL, Feng M, Wang X, Zhang ZH,
Zhao X, Han B, Ma HC, Dai B and Ding YT: Puerarin protects against
CCl4-induced liver fibrosis in mice: Possible role of PARP-1
inhibition. Int Immunopharmacol. 38:238–245. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Mukhopadhyay P, Horváth B, Rajesh M, Varga
ZV, Gariani K, Ryu D, Cao Z, Holovac E, Park O, Zhou Z, et al: PARP
inhibition protects against alcoholic and non-alcoholic
steatohepatitis. J Hepatol. 66:589–600. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Guo M and Wang SM: The BRCAness landscape
of cancer. Cells. 11:38772022. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Chen G, Zheng D, Zhou Y, Du S and Zeng Z:
Olaparib enhances radiation-induced systemic anti-tumor effects via
activating STING-chemokine signaling in hepatocellular carcinoma.
Cancer Lett. 582:2165072024. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Xu Y, Wu H, Huang L, Zhai B, Li X, Xu S,
Wu X, Zhu Q and Xu Q: Rational design, synthesis and biological
evaluation of dual PARP-1/2 and TNKS1/2 inhibitors for cancer
therapy. Eur J Med Chem. 237:1144172022. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Wang YQ, Wang PY, Wang YT, Yang GF, Zhang
A and Miao ZH: An update on poly(ADP-ribose)polymerase-1 (PARP-1)
inhibitors: Opportunities and challenges in cancer therapy. J Med
Chem. 59:9575–9598. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Chan CY, Tan KV and Cornelissen B: PARP
inhibitors in cancer diagnosis and therapy. Clin Cancer Res.
27:1585–1594. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Kim DS, Camacho CV and Kraus WL: Alternate
therapeutic pathways for PARP inhibitors and potential mechanisms
of resistance. Exp Mol Med. 53:42–51. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Mweempwa A and Wilson MK: Mechanisms of
resistance to PARP inhibitors-an evolving challenge in oncology.
Cancer Drug Resist. 2:608–617. 2019.PubMed/NCBI
|
|
154
|
Priya B, Ravi S and Kirubakaran S:
Targeting ATM and ATR for cancer therapeutics: Inhibitors in
clinic. Drug Discov Today. 28:1036622023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Wilson Z, Odedra R, Wallez Y, Wijnhoven
PWG, Hughes AM, Gerrard J, Jones GN, Bargh-Dawson H, Brown E, Young
LA, et al: ATR inhibitor AZD6738 (ceralasertib) exerts antitumor
activity as a monotherapy and in combination with chemotherapy and
the PARP inhibitor olaparib. Cancer Res. 82:1140–1152. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Jaidee R, Pocasap P, Jusakul A,
Senggunprai L, Prawan A, Hong JH, Heng HL, Kukongviriyapan V, Teh
BT and Kongpetch S: Synergistic suppression of cholangiocarcinoma
cells via DNA damage response and cell cycle arrest by dual
targeting PARP and ATM in DNA damage repair pathway. Biomed
Pharmacother. 189:1182732025. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Pandey P, Khan F, Qari HA, Upadhyay TK,
Alkhateeb AF and Oves M: Revolutionization in cancer therapeutics
via targeting major immune checkpoints PD-1, PD-L1 and CTLA-4.
Pharmaceuticals (Basel). 15:3352022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Stewart RA, Pilié PG and Yap TA:
Development of PARP and immune-checkpoint inhibitor combinations.
Cancer Res. 78:6717–6725. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Wang Y, Liu X, Zuo X, Wang C, Zhang Z,
Zhang H, Zeng T, Chen S, Liu M, Chen H, et al: NRDE2 deficiency
impairs homologous recombination repair and sensitizes
hepatocellular carcinoma to PARP inhibitors. Cell Genom.
4:1005502024. View Article : Google Scholar : PubMed/NCBI
|