Introduction
Sepsis is characterized by life-threatening organ
dysfunction resulting from an aberrant host response to infection
(1). Gastrointestinal injury (GI)
is prevalent among individuals with sepsis and has been linked to
increased mortality rates (2). In
2017 alone, an estimated global sepsis incidence was 48.9 million
cases (3). GI complications can
stem from either underlying causes of sepsis, such as peritonitis
originating from the abdominal cavity, or systemic pro-inflammatory
responses seen in cases of sepsis and septic shock (4). A potential explanation for GI disease
in sepsis could be attributed to disruptions in bowel peristalsis
due to extensive use of sedatives and prolonged mechanical
ventilation. The pathology of sepsis involves a complex interplay
between different biological systems that results in severe
dysregulation of the inflammatory network (5). Despite advances in understanding
sepsis, the mechanisms underlying sepsis-induced GI remain complex
and poorly understood, highlighting the need for further research
(6).
Mitochondria are critical organelles for energy
production via oxidative phosphorylation, a reaction conjugated
with producing reactive oxygen species (ROS). Tadokoro et al
(7) showed that doxorubicin
inhibited mitochondrial phospholipid hydroperoxide glutathione
peroxidase (GPX4) expression, resulting in mitochondrial-dependent
ferroptosis. Ferroptosis is an iron-dependent cell death that
differs from apoptosis and necrosis due to excessive accumulation
of peroxidized polyunsaturated fatty acids, which are principally
oxidized polyunsaturated fatty acids by ROS (8). Ferroptosis occurs in various cells
and is vital in sepsis-induced multiple organ injury (9,10).
It is particularly crucial to inhibit ferroptosis caused by
oxidative damage.
In the repair process of acute GI, diverse molecular
signals are involved in regulating mitochondrial malfunction.
Sirtuin 3 (SIRT3), a mitochondrial deacetylase, mitigates
mitochondrial oxidative damage and apoptosis by peroxiredoxin-3
(PRDX3). SIRT3 is known to regulate the acetylation status of
several mitochondrial proteins, thereby influencing their activity
and stability (11). PRDX3
functions as an efficient scavenger of hydrogen peroxide
(H2O2) to safeguard cells against oxidative
damage, especially in intestinal ischemia/reperfusion (I/R) injury
(11). PRDX3 functions by
oxidizing and forming its inactive dimer form to clear
H2O2 and previous studies have demonstrated
its effective inhibition of oxidative stress, apoptosis, and
mitigation of cellular damage (11,12).
Transgenic mice overexpressing PRDX3 exhibit reduced mitochondrial
H2O2 production and oxidative damage compared
with control mice (13).
Atractylodin is a bioactive compound derived from
Atractylodes lancea (Thunb.) DC., which has been widely used
to treat various gastrointestinal diseases, including dyspepsia,
flatulence, nausea and diarrhea (14). Atractylodin has been reported to
exert anti-inflammatory effects in various inflammatory diseases.
Lipopolysaccharide-induced acute lung injury was ameliorated by
atractylodin inhibiting the nucleotide-binding oligomerization
domain-like receptor protein 3 inflammasome and Toll-like receptor
4 pathways (15).
Lipopolysaccharide- and D-galactosamine-induced acute liver failure
was also attenuated by atractylodin via suppressing inflammation
and oxidative stress (13). While
investigating the effects of atractylodin on the gastrointestinal
tract, a study demonstrated the ameliorative effects of
atractylodin on intestinal dysmotility, constipation, and diarrhea
in an experimental rat model (15). Studies have highlighted its
potential therapeutic effects, particularly in the context of
immune modulation and anti-inflammatory activities (16,17).
Despite the obvious anti-inflammatory effects of atractylodin, few
studies have been conducted on its molecular targets.
Based on the aforementioned backgrounds, it was
hypothesized that atractylodin might attenuate mitochondrial
dysfunction in sepsis-induced GI by activating SIRT3 to deacetylate
PRDX3. Thus, the present study aimed to perform molecular
biological experiments to verify the potential of atractylodin in
GI. It is hoped that the present study will provide a new strategy
for alleviating GI.
Materials and methods
Antibodies and reagents
Isoflurane was obtained from RWD Life Science.
Anhydrous ethanol analytical reagent (AR), xylene (AR), paraffin
(56–58°C), water-soluble eosin Y staining solution, Tween 20,
sodium chloride (AR), trichloromethane (AR) and 3%
H2O2 were purchased from Sinopharm Chemical
Reagent Co., Ltd. Hematoxylin was obtained from MilliporeSigma.
Dimethyl sulfoxide (DMSO), 10 mM of phosphate-buffered saline
buffer (PBS), loading buffer, 4′,6-diamidino-2-phenylindole (DAPI)
solution, anti-fluorescence attenuation sealant, IL-4, IL-6, IL-10,
IL-1β and IL-1α, enzyme-linked immunosorbent assay (ELISA) kits and
H2O2, malondialdehyde, Fe2+ and
mitochondrial respiratory chain complex activity assay kits were
purchased from Beijing Solarbio Science & Technology Co., Ltd.
GSH/GSSG detection kit was from Nanjing Jiancheng Bioengineering
Institute. Protease phosphatase inhibitor mixture, RIPA lysate
(strong), BCA protein assay kit, JC-1 mitochondrial membrane
potential assay kit and dihydroethidium (DHE) assay kit were
purchased from Shanghai Biyuntian Biotechnology Co., Ltd. The
details of the antibodies used are listed in Table SI.
Animal treatment
Male C57 BL/6 mice aged 6–8 weeks, weighing 22–25 g,
free from specific pathogens (n=90), were purchased from Jiangsu
Huachuang Xinnuo Pharmaceutical Technology Co., Ltd. The mice were
housed at 23°C and 50% relative humidity on a 12/12-h light/dark
cycle. Basic feed was processed by Beijing Huanyu Zhongke
Biotechnology according to the national standard (GB 14924.3-2010).
The model of sepsis was induced by performing cecal ligation and
perforation. Briefly, the mice were anesthetized using 4%
isoflurane, followed by a sterile midline laparotomy of ~2 cm to
expose the cecum. Half of the distal end of the cecum was ligated
at its center, and then a 21-gauge needle was inserted between the
ligature site and the end of the cecum to extrude a small amount of
cecal contents. The cecum was gently reduced and the laparotomy
site was sutured. In the Sham group, animals underwent laparotomy
and intestinal manipulation to expose the cecum without ligation or
puncture. Monitoring was performed twice a day (once in the morning
and once in the evening) and recorded every 4 h for 48 h after
surgery. All mice received resuscitation through subcutaneous
injection of normal saline at a 24 ml/kg body weight dose.
Subsequently, all mice were anesthetized with pentobarbital sodium
(40 mg/kg) and sacrificed for further analysis, including
collection of serum as well as stomach and colon tissue. The animal
experiments in the present study was conducted in the Liyang
Hospital of Chinese Medicine and approved by the Experimental
Animal Ethics Committee of Liyang Hospital of Chinese Medicine
(Jiangsu, China; approval no. 2024LY-02-02-03).
The solvent DMSO was employed to dissolve
atractylodin and prepare a storage solution. Prior to
administration, the solution was diluted to the desired
concentration using saline, ensuring that the final concentration
of DMSO did not exceed 0.1%. The mice were randomly allocated into
five groups: Sham operation group (Sham group), the cecal ligation
perforation sepsis group (Model group), and the atractylodin
treatment group (low, medium, and high doses), with 12 mice in each
group.
One hour before surgery, the atractylodin treatment
group received intraperitoneal injections of atractylodin at a
concentration of 10 mg/kg/d for the low-dose group, a concentration
of 20 mg/kg/d for medium-dose group, and a high-dose concentration
of 40 mg/kg/d for high dose group. Post-surgery, mice received
once-daily intraperitoneal injection doses of the designated
atractylodin dese for 7 days. The dosage of the drug was obtained
from the previous studies (18,19).
Clinical evaluation was performed with no pulsation
on palpation of the carotid artery, detection of absence of corneal
reflex, and secondary confirmation. In addition, the animals that
died naturally were subjected to pathological analysis to exclude
experimental interference factors. The present study strictly
adhered to the following criteria to determine the timing of
sacrifice through daily weight monitoring, a behavioral scoring
system, and veterinary assessment, ensuring that animals did not
suffer avoidable pain. Sacrifice were performed when one of the
following situations occurred: i) Weight loss: A rapid weight loss
of 15–20%; ii) weakness and loss of mobility: Unable to stand for
more than 24 h, loss of appetite, dehydration; iii) infection and
wound problems: A board-like abdomen upon abdominal palpation
(indicating diffuse peritonitis); bloody discharge around the anus
(indicating intestinal ischemic necrosis); iv) abnormal body
temperature: A deviation of 4°C from the normal body temperature
for more than 24 h; v) pain and behavioral abnormalities: Obvious
signs of pain (such as aggression, aimless running), neurological
symptoms (convulsions, paralysis).
A total of 90 C57BL/6 mice were used in the present
study, of which six died naturally (autopsy showed sepsis), and the
remaining mice were sacrificed at the end of the experiment.
Isolfurane 4% (oxygen flow 2 l/min) was used for induction, and the
oxygen flow rate was adjusted to 1.8% during the maintenance phase.
The depth of anesthesia was verified by blood gas analysis after
operation. Animals were sacrificed with an overdose of sodium
pentobarbital (150 mg/kilogram) by intraperitoneal injection.
Clinical evaluation was performed with no pulsation on palpation of
the carotid artery, detection of absence of corneal reflex, and
secondary confirmation. In addition, the animals that died
naturally were subjected to pathological analysis to exclude
experimental interference factors.
Histological examination
The stomach and colon tissue of the mice was fixed
for 12–24 h at room temperature in 4% paraformaldehydeTissue
embedded in paraffin was cut into 3 µm thick sections. The tissue
sections were deparaffinized with xylene (twice, each for 5 min),
followed by hydration with decreasing concentrations of ethanol.
The sections were stained with hematoxylin for 15 min, then rinsed
with tap water. The sections were then incubated in acid-alcohol
for 30 sec, immersed in tap water for 15 min, and stained with
eosin for 5 min. The sections were dehydrated with a gradient of
ethanol (95, 95, 100, 100%, each for 2 sec) and cleared with xylene
twice (each for 1 min). Finally, the sections were cleared with
xylene, mounted with neutral resin, and observed under an inverted
microscope.
Western blotting
After extracting total protein from the tissue using
RIPA lysis buffer (Beyotime Institute of Biotechnology; cat. no.
P0013), the BCA method is used to detect protein concentration. The
protein extraction of stomach and colon tissues was boiled with
gel-loading buffer for 10 min at 100°C, 50 µg protein was loaded
per lane, and resolved by 12% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis, and transferred to nitrocellulose membranes.
The membrane was blocked with 5% BSA in 1X PBST buffer (0.01%
Tween-20) for 90 min at room temperature and was probed with GAPDH)
antibody (1:5,000; Proteintech Group, Inc., 10494-1-AP) or GPX4
antibody (1:1,000; Abcam, ab125066), Mitochondrial import receptor
subunit TOM20 homolog (1:1,000; Abcam, ab186735), transferrin
receptor protein 1 (TFR1) antibody (1:1,000; Abcam, ab214039),
PRDX3 antibody (1:1,000; Abcam, ab73349), SIRT3 antibody (1:1,000;
Proteintech Group, Inc., 10099-1-AP), SLC7A11 antibody (1:1,000;
Proteintech Group, Inc., 32384-1-AP), occludin antibody (1:1,000;
Abcam, ab216327), and zona occludens protein 1 (ZO-1) antibody
(1:1,000; Proteintech Group, Inc.; cat. no. 21773-1-AP), for 2 h at
room temperature. The membrane was washed three times (10 min each
in 1X PBST) and incubated with secondary anti-rabbit IgG (H+L)
antibody, (horseradish peroxidase conjugate) (1:5,000; Cell
Signaling Technology, Inc., #7074), for 90 min at room temperature.
After being washed three times (15 min each in 1X PBST), the
membrane was incubated in a 2.0 mM DAB solution prepared in PBS for
5 min. The image of the immunoblot was digitalized with the GelDoc
XR+ system and saved in TIF format. The protein levels were
quantified by ImageJ software (National Institutes of Health,
version 1.8.0) and normalized to GAPDH as an internal control.
Co-immunoprecipitation (Co-IP)
For immunoprecipitation assays,
Co-Immunoprecipitation kit (Beyotime Institute of Biotechnology;
cat. no. P2175) was used following manufacturer instructions.
Briefly, the tissue was collected and rinsed with PBS.
Subsequently, tissue was lysed in the EBC buffer supplemented with
protease inhibitors [50 mM tris (pH 7.5), 120 mM NaCl, and 0.5%
NP-40]. Following ultrasonic lysis (power: 60%, ultrasonic
intermittent time: 1 min, ultrasonic frequency: three times,
ultrasonic exposure 10 s/time, 4°C), Protein A beads conjugated
with anti-PRDX3 antibody (1:500, Abcam, cat. no. ab222807) were
added to the lysate at a ratio of 20 µl bead suspension per 500 µl
protein sample, followed by overnight immunoprecipitation at 4°C.
The next day, the immunoprecipitate was pelleted by centrifugation
at 500 × g for 3 min at 4°C. Then, the resulting product was washed
in the lysis buffer. The immunoprecipitate was denatured at 100°C
for 15 min with 50 µl 2X SDS protein loading buffer. The
immunoprecipitate, input samples, and other lysates (10 µl) were
separated by 10% SDS-PAGE and transferred to a PVDF) membrane for
subsequent Western blotting. To detect acetylation of PRDX3,
anti-Peroxiredoxin 3/PRDX3 antibody (Abcam; cat. no. ab222807) to
precipitate PRDX3 from the samples. Subsequently, the
Acetylated-lysine Antibody (Cell Signaling Technology, Inc.; cat.
no. 9441) was used to assess the acetylated levels of PRDX3 through
immunoimprinting.
Measurement of ROS levels
DHE, a ROS-level indicative fluorescence probe
(λex=535 nm, λem=610 nm), was used to detect intracellular
superoxide anions. Fresh tissues are frozen (−20°C) and then cut
into 10 µm thick sections using a DAKEWE 6250 cryostat microtome.
The probe of DHE was incubated with tissue slices and the intensity
of red fluorescence could reflect the level of ROS under a
fluorescence microscope.
TdT-mediated dUTP Nick-End Labeling
(TUNEL) staining
One Step TUNEL Apoptosis Assay Kit (Beyotime
Institute of Biotechnology; cat. no. C1090) was used to detect
apoptotic cells. After incubating with the TUNEL regent in the dark
for 1 h at 37°C, cells were stained with DAPI (10 min at room
temperature). Apoptotic cells showed red fluorescence. Cells were
counted in three randomly selected fields of view using an inverted
fluorescence microscope (20X magnification, Olympus IX83, Olympus
Corporation).
Determination of
H2O2 content
H2O2 content was determined by
H2O2 Content Detection Kit (Beijing Solarbio
Science & Technology Co., Ltd.). The H2O2
detection reagent was melted in an ice bath. Next, the sample
extraction solution or standard was added to the detection well,
followed by the H2O2 detection reagent. This
was gently mixed and allowed to rest at room temperature (15–30°C)
for 5 min. A volume of 200 µl was aliquoted into a 96-well plate to
measure the absorbance at 415 nm. Subsequently, the concentration
of H2O2 in the sample was determined using a
standard curve. Absorbance values were measured at 415 nm using a
96-well plate according to the manufacturer's instructions.
Detection of markers of oxidative
stress
To clarify the effect of atractylodin on
sepsis-induced mitochondrial oxidative damage, we detected the
activity of catalase (CAT) in tissues using the Catalase (CAT)
Activity Assay Kit (Solarbio, BC0205), determined the activity of
superoxide dismutase 2 (SOD2) in tissues with the Superoxide
Dismutase (SOD) Isoenzyme Activity Assay Kit (Solarbio, BC5255),
and measured the content of glutathione/oxidized glutathione
(GSH/GSSG) in tissues by means of the Total Glutathione
(T-GSH)/Oxidized Glutathione (GSSG) Assay Kit (Nanjing Jiancheng
Bioengineering Institute, A061-1-2), all in accordance with the
instructions provided by the kit manufacturers. In addition, the
contents of hydrogen peroxide (H2O2) and
malondialdehyde (MDA) in tissues were detected using the Hydrogen
Peroxide (H2O2) Content Assay Kit (Solarbio,
BC3595) and the Malondialdehyde (MDA) Content Assay Kit (Solarbio,
BC025), respectively.
Measurement of mitochondrial membrane
potential
The mitochondrial membrane potential was assessed
utilizing an advanced mitochondrial membrane potential assay kit
incorporating JC-1 (cat. no. C2003S; Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. In
summary, mitochondria were isolated using a tissue mitochondria
isolation kit in accordance with the manufacturer's protocol. The
isolated mitochondria were subsequently incubated with 0.5 ml of
JC-1 fluorescent dye for 30 min at 4°C in the absence of light,
followed by analysis via flow cytometry (BECKMANCOULTER, CytoFLEX)
using FlowJo software (v10.6.2, FlowJo, FlowJo Enterprise).
Measurement of mitochondrial complexes
activity
The activity of complex I–IV in mitochondrial was
determined with the micro mitochondrial respiratory chain complex
I, II, III or IV activity assay kit (Beijing Solarbio Science &
Technology Co., Ltd.) according to the manufacturer's instructions.
Briefly, stomach and colon tissues isolated from two respective
mice in the same group were pooled and suspended in the
mitochondrial complex extraction buffer, followed by gentle
homogenization. The homogenate was centrifuged at 600 × g for 10
min at 4°C to remove cell debris and nuclei, and the supernatant
was centrifuged again at 11,000 × g for 15 min at 4°C. The
resultant pellet was resuspended in the extraction buffer and
crushed by ultrasonication (power: 60%; intermittent time: 1 min;
frequency: three times; 10 s/time, 4°C). The complex activity of
mitochondrial homogenates in the respective reaction buffer was
then measured spectrophotometrically at 340 nm (complex I), 605 nm
(complex II), and 550 nm (complex III and IV), respectively.
Immunofluorescence
Immunofluorescence staining was performed on
paraffin-embedded tissue sections. Following dewaxing and
hydration, antigen retrieval was performed using citrate buffer (pH
6.0), microwave treatment for 3 min. The sections are blocked with
1% goat serum (Beijing Solarbio Science & Technology Co., Ltd.;
cat. no. SL038) in PBS at room temperature for 1 h and incubated
with the primary antibody overnight at 4°C (Table SI. Subsequently, the sections are
washed with PBST, incubated with goat anti-rabbit Alexa Fluor 647
secondary antibody (Thermo Fisher Scientific, Inc.) at a 1:400
dilution at room temperature for 1 h, followed by DAPI (10 µg/ml)
staining for 10 min at room temperature, and mounting. Finally, the
sections are observed under a fluorescence microscope (20X,
Olympus, Tokyo, Japan) and quantified using. ImageJ software
(National Institutes of Health, version 1.8.0).
Transmission electron Microscopy
(TEM)
TEM analyses were accomplished using HT7700 Hitachi
Transmission Electron Microscope (Hitachi High-Technologies
Corporation). TEM was used to observe the mitochondrial state of
tissues. Freshly stomach and colon tissues were quickly cut into 1
mm cubes fixed overnight in 2.5% glutaraldehyde (4°C) and then
post-fixed with 1% osmium tetroxide (4°C, 2 h), dehydrated through
a graded ethanol series. Embedding was performed in a 1:1 resin
(EMBed-812) and propylene oxide (Electron Microscopy Services) mix
for 1 h at room temperature, followed by a 2:1 resin:propylene
oxide mixture overnight at room temperature. Tissue were then
placed in 100% resin for 3 h at room temperature. Ultrathin
sections (70 nm) were collected and double-stained with uranyl
acetate and lead citrate (room temperature for 15 min). Finally,
the sections were visualized with a JEM1200ex Electron Microscope
(JEOL).
Adeno-associated virus (AAV) vector
experimental protocols
AAV serotype 9 (AAV9)-short hairpin (sh)SIRT3 and
AAV9-shNC were synthesized by Jikai Gene Chemical Technology Co.,
Ltd. Male WT C57Bl/6J mice (8-week-old) were injected in the tail
vein with 2.5×1011 viral genome particles of AAV 9.
AAV9-mediated gene transfer and expression were allowed for 2 weeks
before subsequent experiment.
Statistical analysis
Data are expressed as mean ± standard deviation.
Comparisons among groups were tested with a one-way analysis of
variance followed by the Tukey test. An unpaired t-test is used
between the two groups (GraphPad Prism version 5; Dotmatics).
P<0.05 was considered to indicate a statistically significant
difference.
Results
In vivo safety
The present study initiated dose safety studies in
mice to determine safety of atractylodin. C57BL/6 mice were treated
with atractylodin (10, 20, 40 mg/kg/day) for 2 weeks to simulate
long-term administration. Following common discovery-stage
practices, multiple indices of liver function and renal function
were examined. As shown in Fig.
S1 atractylodin treatment did not markedly impair liver or
renal function. The results showed that there was no significant
difference in body weight, which proved the safety of the three
groups of doses.
Atractylodin mitigates sepsis-induced
GI
To evaluate the effects of atractylodin on acute GI,
the present study created a model for sepsis-induced GI. The model
of sepsis was induced by performing cecal ligation and perforation.
Then, the corresponding treatments were performed in each group.
The stomach and colon tissue morphology of each group were observed
by H&E staining. The Sham group exhibited no evidence of
intestinal mucosal injury in the pathological examination results.
However, in the model group, there was an increase in epithelial
space, a decrease in the number of villous epithelial cells and
crypt cells, a disordered arrangement of villous epithelium, and
some overflow of villous tips. Atractylodin treatment markedly
attenuated sepsis-induced acute GI (Fig. 1A).
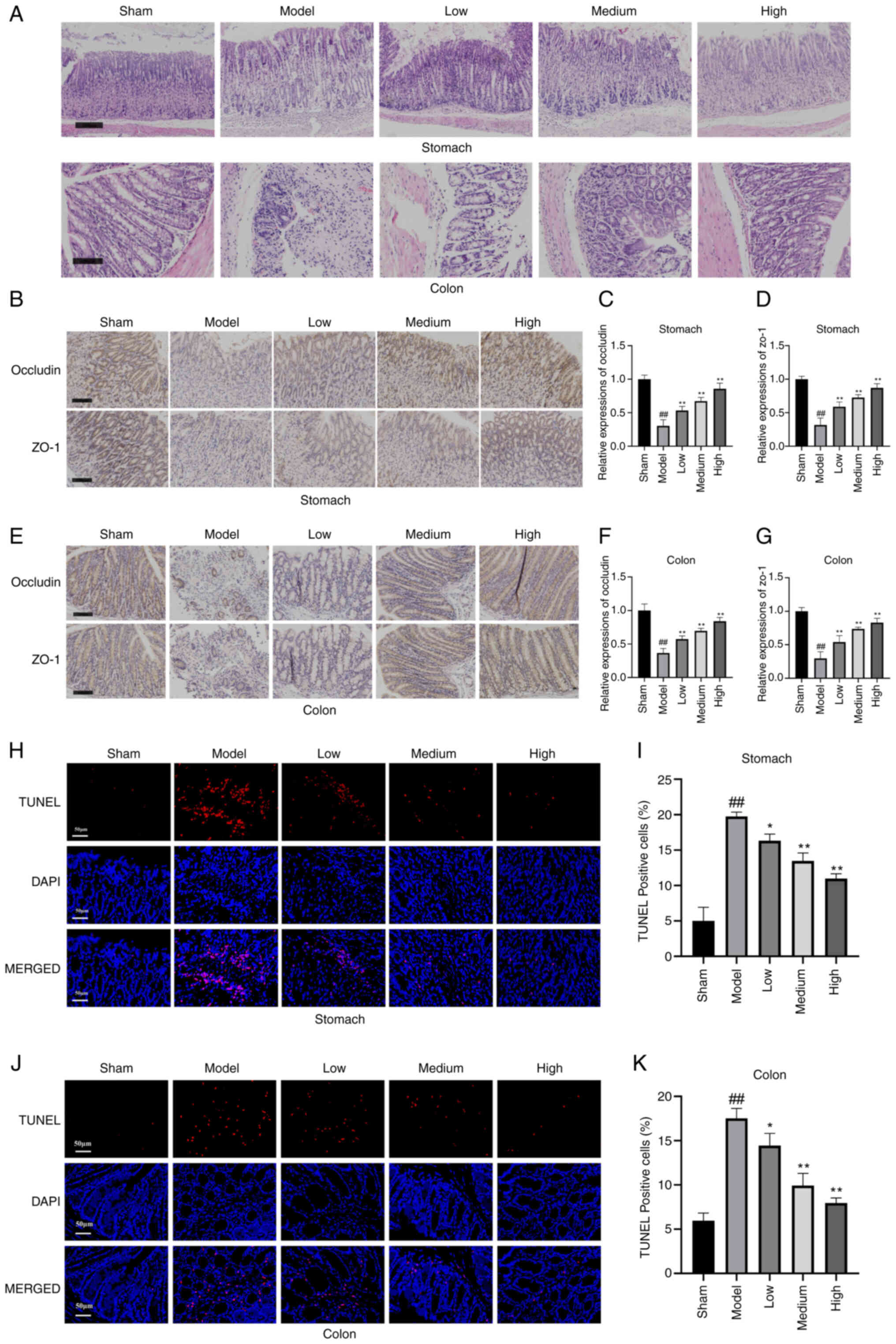 | Figure 1.Atractylodin mitigates sepsis-induced
GI. (A) H&E staining of mousee stomach (scale bar, 200 µm) and
colon tissue. (B) The immunohistochemical detection of tissue
damage in the stomach; scale bar, 100 µm. (C) Relative expression
of Occludin in the stomach. (D) Relative expression of ZO-1 in the
stomach. (E) The immunohistochemical detection of tissue damage in
the colon, scale bar 100 µm. (F) Relative expression of occludin in
the colon. (G) Relative expression of ZO-1 in the colon. The (H)
stomach tissue and (J) colon tissue cell apoptosis were analyzed
via TUNEL assay. Blue is nuclei stained with DAPI, while red is
TUNEL-positive cells stained with TUNEL; scale bar 50 µm. The
percentage of TUNEL-positive cells in (I) stomach tissues and (K)
colon tissues. TUNEL-positive cells (%) were calculated by the
number of positive cells divided by the total number of cells. Data
are combined from n=3 biological repeats. *P<0.05, **P<0.01
vs. the model group, ##P<0.01 vs. the Sham group. GI,
gastrointestinal injury; H&E, hematoxylin-eosin; TUNEL,
terminal dUTP nick end labeling; ZO-1, Zona occludens protein
1. |
ZO-1 and Occludin are essential intestinal
epithelial tight junction proteins. The present study investigated
occludin and ZO-1 expression in the stomach and colon tissue with
immunohistochemistry (Fig. 1B and
E). The groups treated with atractylodin in low, medium and
high doses appeared to relieve the pathological injuries to
different degrees compared with the model group. The results
demonstrated that the levels of ZO-1 and occludin in the model
group were markedly lower than those in the Sham group, indicating
impaired colon barrier function. Compared with the model group, the
expression levels of ZO-1 and Occludin were markedly higher in all
atractylodin treated groups (Fig. 1C,
D, F and G).
The present study then assessed the role of
atractylodin in modulating apoptosis of stomach and colon tissues
in the mice using TUNEL staining. The number of TUNEL-positive
cells was increased in mice's stomach and colon tissues in the
model group, whereas atractylodin blocked this elevation
(P<0.01; Fig. 1H-K). these
results demonstrated that atractylodin represses the apoptosis of
stomach and colon tissues in mice.
To further evaluate the effects of atractylodin on
sepsis-induced GI mice, the level of pro-inflammatory cytokines
were determined in the serum and tissues. The cytokines of IL-1β,
IL-6, TNF-α, IL-4, and IL-10 were evaluated in the blood, stomach
tissue, and colon tissue using ELISA (Fig. S2). Atractylodin effectively
prevented the increase in pro-inflammatory cytokines, while
increasing levels of anti-inflammatory cytokines IL-4, and
IL-10.
Atractylodin improves mitochondrial
dysfunction
Fig. 2A and B
illustrates changes in antioxidant defense system indicators such
as SOD2 and CAT levels in experimental mice. In the treatment
group, there was a significant increase in CAT, SOD 2, and GSH/GSSG
levels when compared with the model group in stomach and colon
tissues. As shown in Fig. 2A-B,
atractylodin treatment markedly decreased levels of
H2O2 and MDA in a dose-dependent manner. In
addition, mitochondrial morphology changes and function were
evaluated by TEM. TEM revealed more disorganized, swollen, and
damaged mitochondria in the model group. This mitochondrial damage
was mitigated in the atractylodin-treated group in stomach and
colon tissues (Fig. 2C).
Semiquantitative western blotting analysis showed a trend to
increase in TOM20/GAPDH ratio, which suggests an increase in
mitochondrial number or mitochondrial dimensions in
atractylodin-treated group compared with the model group (Fig. 2D-G).
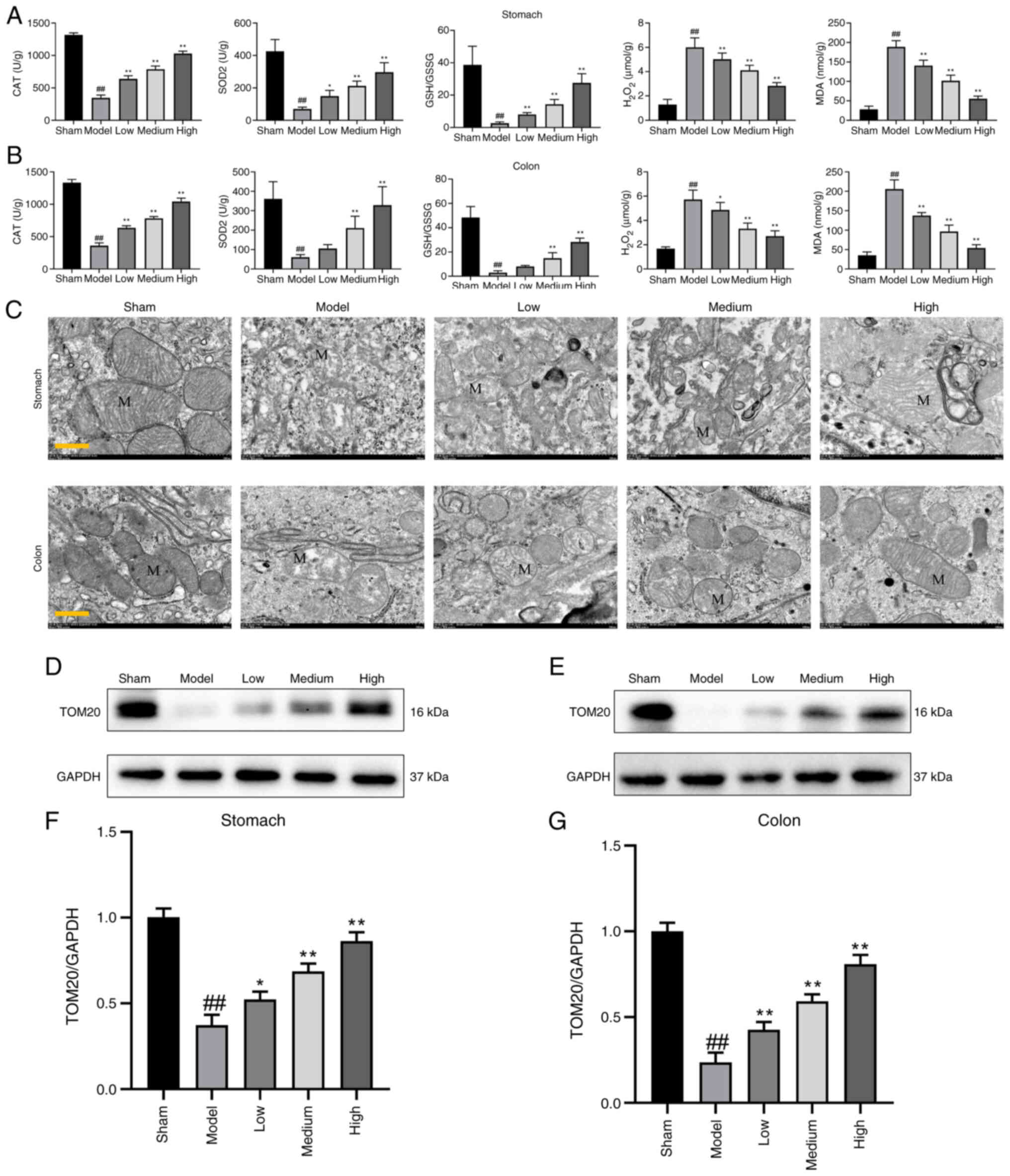 | Figure 2.Effect of atractylodin on regulating
mitochondrial dysfunction. (A) Regulation of mitochondrial
oxidative damage markers: CAT, SOD 2, GSH/GSSG,
H2O2, MDA in the stomach, and (B) colon
tissue. (C) TEM images of mitochondrial morphology of each group.
Western blot analysis of mitochondrial membrane TOM20 markers in
(D) stomach and (E) colon tissue. Scale bar 500 nm. Relative
quantitation of the intensity of TOM20 in the (F) stomach and (G)
colon tissue. Values are means ± SD of n=6 (A, B, F and G) or n=3
(D and E) experiments. *P<0.05, **P<0.01 vs. the model group,
##P<0.01 vs. the Sham group. CAT, catalase; SOD 2,
superoxide dismutase; GSH/GSSG, glutathione/glutathione (oxidized);
MDA, malondialdehyde TEM, transmission electron microscopy; TOM20,
mitochondrial import receptor subunit TOM20 homolog; MDA,
malondialdehyde. |
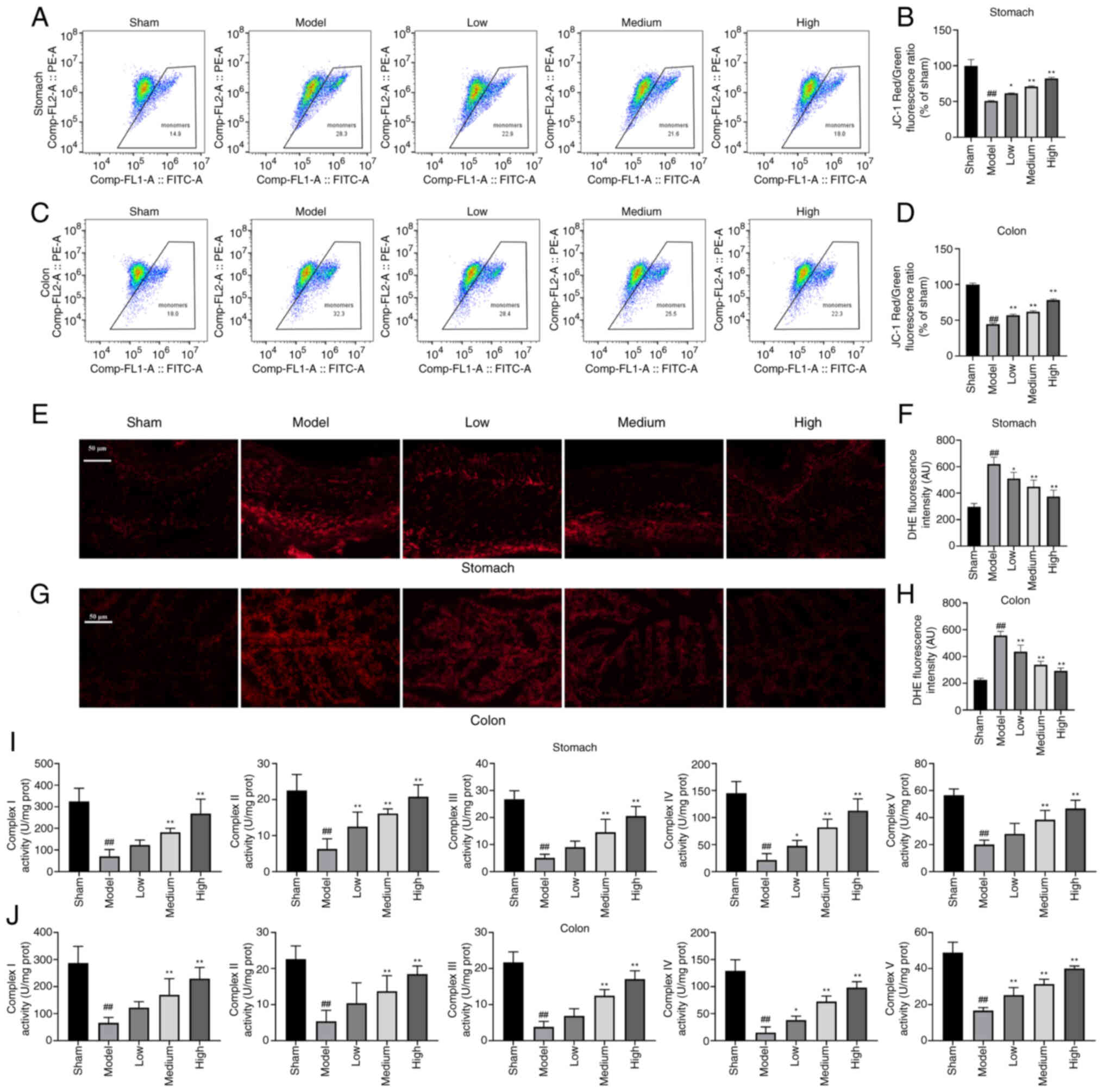
The mitochondrial function was further examined. To
assess the transmembrane potential, a flow cytometric-based assay
was performed to measure transmembrane potential in each group in
the stomach and colon tissue. Cells of the gastric and colon tissue
of mice in each group were isolated and the mitochondrial membrane
potential was detected using a mitochondrial membrane potential
detection kit. We found that the potential for the dissipation of
mitochondrial transmembrane could be attenuated by atractylodin in
each group of stomach and colon tissue (Fig. 3A-D). To examine the ROS levels,
stomach and colon tissue were stained with DHE, a superoxide
indicator. The intensity of DHE fluorescence was markedly decreased
in all atractylodin concentration groups compared with the model
group (Fig. 3E-H). Mitochondrial
complexes I, II, III, IV, and V activities were also markedly
enhanced in each atractylodin concentration group compared with
those in the model group (Fig. 3I and
J).
Atractylodin inhibits ferroptosis in
mice with sepsis-induced GI
To address the role of atractylodin in ferroptosis
of sepsis-induced GI, the Fe2+ levels in stomach and
colon tissues were determined. The results showed that the level of
Fe2+ in stomach and colon tissues of mice was markedly
decreased in the atractylodin-treated group (Fig. 4A and F). Based on the
aforementioned results, the essential proteins related to
ferroptosis were detected in each group. Western blotting showed
that the levels of GPX4 and Solute carrier family 7 member 11
(SLC7A11) in the atractylodin-treated group were markedly higher
compared with those of the model group. At the same time, TFR1 was
decreased in both stomach tissues (Fig. 5B-E) and colon tissues (Fig. 5G-J). these results showed that
atractylodin inhibited ferroptosis in mice with sepsis-induced
GI.
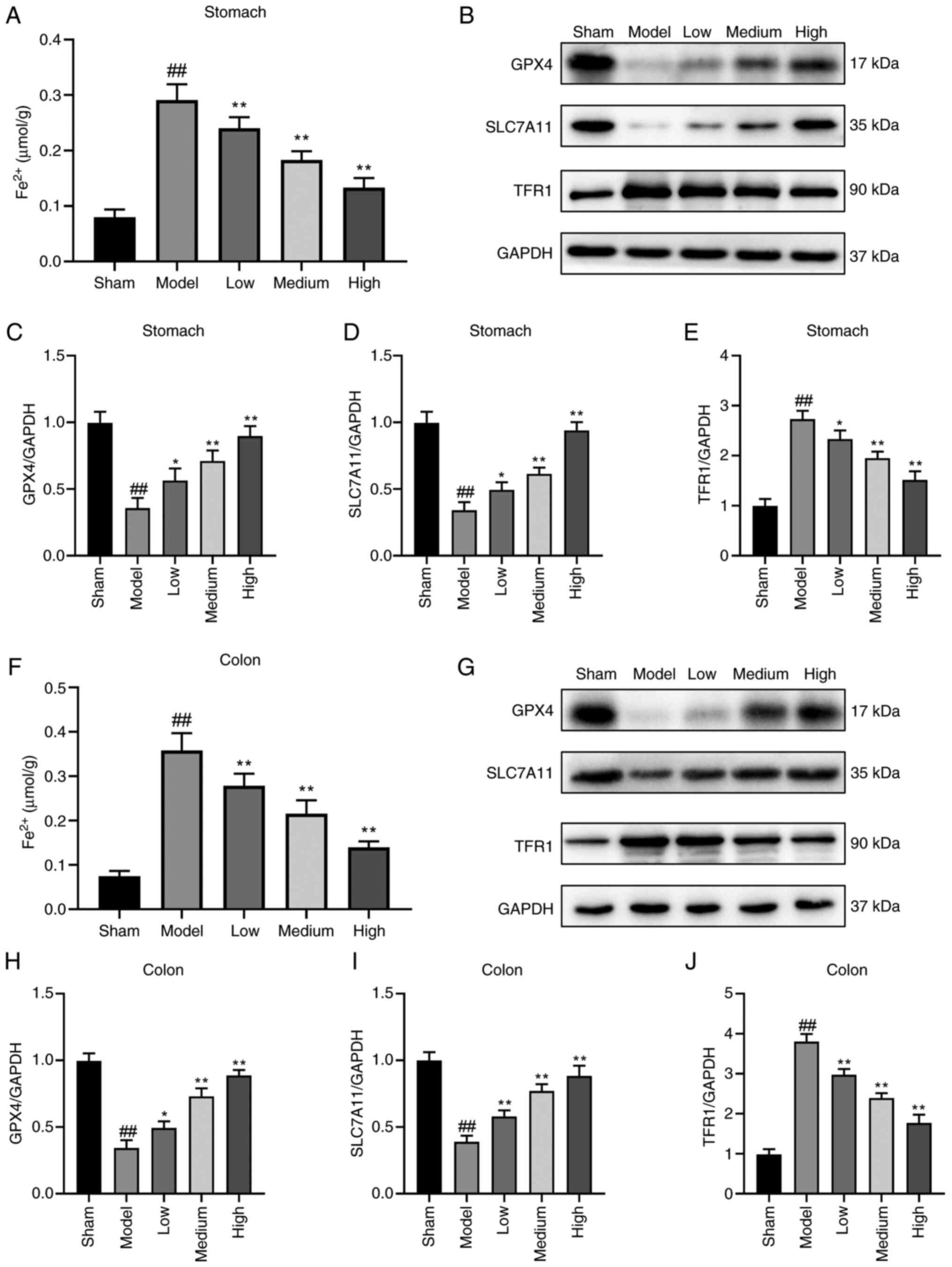 | Figure 4.Atractylodin treatment improves the
prognosis of sepsis-induced ferroptosis in the stomach and colon
tissues. (A) Determination of the expression levels of
Fe2+ in stomach tissues. (B) Protein expression levels
of GPX4, SLC7A11, TFR1 and GAPDH in stomach tissue in each group.
Quantification of relative protein expression of (C) GPX4, (D)
SLC7A11 and (E) TFR1in the stomach tissue. (F) Determination of the
expression levels of Fe2+ in colon tissues. (G) Protein
expression levels of GPX4, SLC7A11, TFR1 and GAPDH in colon tissue
in each group. (H-I) Quantifying relative protein expression of (H)
GPX4, (I) SLC7A11 and (J) TFR1 in the colon tissue. Values are
means ± SD of n=6 (A and F) or n=3 (B-E and G-J) experiments.
*P<0.05, **P<0.01 vs. the model group, ##P<0.01
vs. the Sham group. GPX4, Phospholipid hydroperoxide glutathione
peroxidase; SLC7A11, Solute carrier family 7 member 11; TFR1,
transferrin receptor protein 1; GAPDH, glyceraldehyde-3-phosphate
dehydrogenase. |
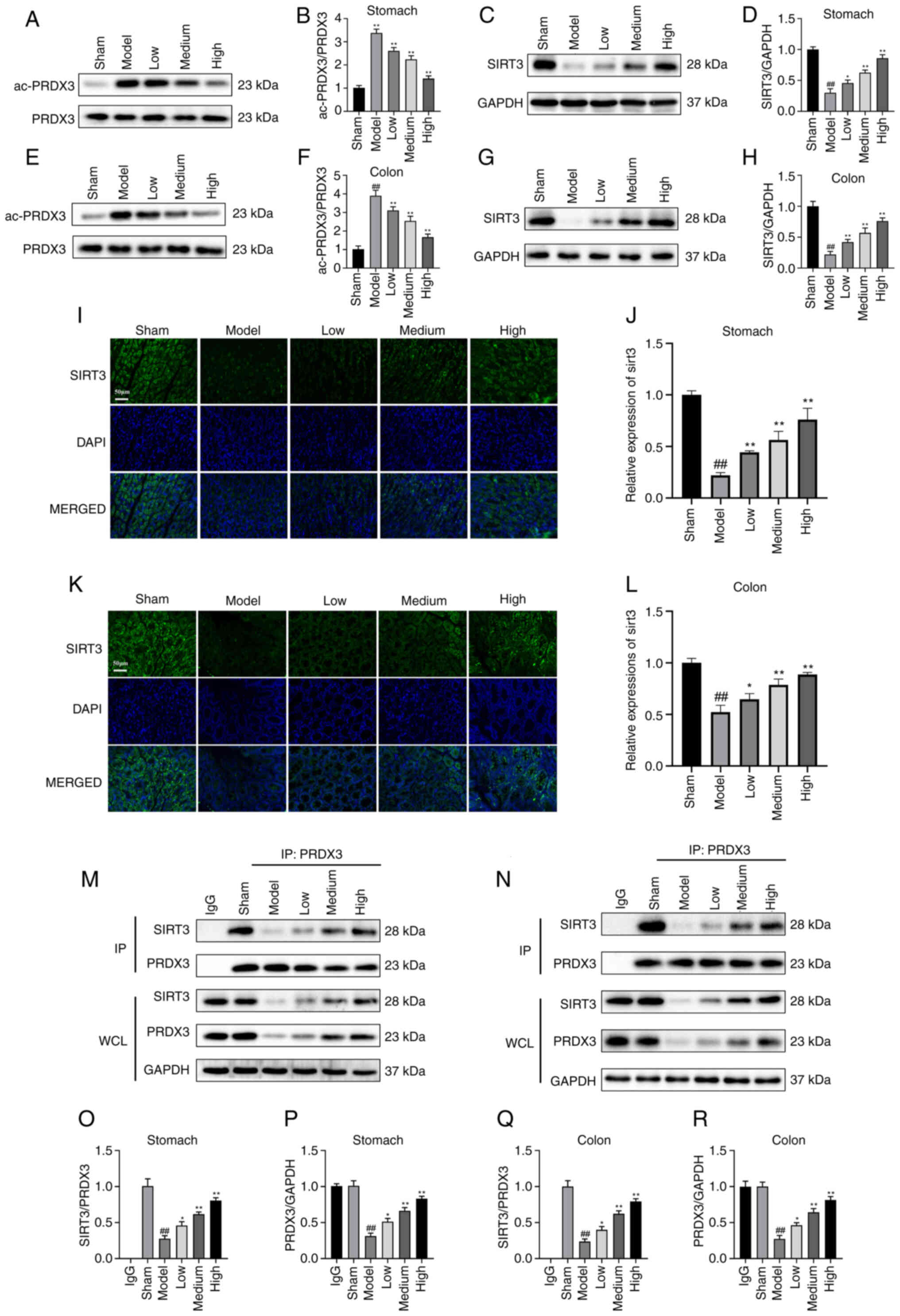
Atractylodin upregulate SIRT3 while
suppress acetylation of PRDX3
It has been suggested that SIRT3-mediated
deacetylation of PRDX3 could alleviate mitochondrial oxidative
injury (11). The present study
examined the expression of Ac-PRDX3 and SIRT3 in the stomach and
colon tissues of mice. The expression of Ac-PRDX3/PRDX3 in the
stomach tissues of mice in the atractylodin-treated group was lower
than that in the model group. The high-dose atractylodin-treated
group showed the lowest Ac-PRDX3/PRDX3 levels in stomach tissues
(Fig. 5A and B). As shown in
Fig. 5C and D, the SIRT3
expression markedly increased in the high-medium- and low-dose
groups compared with the model group. The same trend was observed
in the colon tissues (Fig. 5E-H).
The expression of SIRT3 increased in the stomach and colon tissues
of mice in the atractylodin-treated group compared with the model
group (Fig. 5C, D, G and H). To
explore how atractylodin regulates SIRT3 expression in cells, SIRT3
was colocalized with the nuclei marker DAPI using
immunofluorescence in the stomach and colon tissues. As shown in
Fig 5I and K, The increased SIRT3
expression with the atractylodin treatment groups was shown by
immunofluorescence. The expression of SIRT3 increased with an
increased dose of atractylodin in the stomach and colon tissues of
mice in atractylodin treated group compared with the model group
(Fig. 5J and L).
To assess whether atractylodin promotes the binding
of SIRT3 to PRDX3, a co-immunoprecipitation assay was performed.
The results showed that the binding of SIRT3 with PRDX3 was
increased in all atractylodin treatment groups (Fig. 5M and N). The high doses of the
atractylodin group showed increased SIRT3/PRDX3 and PRDX3/GAPDH in
the stomach and colon tissues (Fig. 5N
and O).
Atractylodin abolishes sepsis-induced
mitochondrial dysfunction in GI through mediation of SIRT3/PRDX3
signaling
The downstream regulatory mechanism of the SIRT3 and
PRDX3 was explored in sepsis-induced GI. To confirm the involvement
of SIRT3 in the reparative effect on sepsis-induced GI, shSIRT3 was
intravenously injected into the mice through the tail vein to knock
down SIRT3. Fig. 6A and B showed
that the protein levels of SIRT3 were markedly downregulated by 68%
following injection with shSIRT3, as compared with the mice
injected with shNC. Mice were divided into five groups: sham group,
model group, model + atractylodin group, AAV-shSIRT3 group, and
AAV-shSIRT3 + atractylodin group. The expression of GPX4 in the
stomach and colon tissue was measured by eastern blotting. The
results demonstrated that in the stomach and colon tissue, the GPX4
expression in the model group was markedly reduced compared with
the sham group (Fig. 6C and E).
However, it was obviously upregulated in the model mice treated
with high-atractylodin. Compared with the model combined with
atractylodin group, the GPX4 level of both the AAV-shSIRT3 and the
AAV-shSIRT3 + atractylodin group was markedly decreased (Fig. 6D and F). These results suggested
that SIRT3 plays a key role in ferroptosis-mediated GI. Western
blotting also demonstrated that the protein expression of TOM20,
ZO-1 and occludin was markedly decreased in the AAV-shSIRT3 +
atractylodin treated group compared with the model combined with
atractylodin group in the stomach (Fig. 6G-J) and colon tissues (Fig. 6K-N). These results showed that
atractylodin inhibited mitochondrial oxidative stress by increasing
SIRT3 expression. Moreover, mitochondrial complexes I, II/III, IV
and V activities were also markedly attenuated in AAV-shSIRT3+
atractylodin-treated mice, as compared with model combined with
atractylodin mice in the stomach (Fig.
6O-S) and colon tissues (Fig.
6T-X).
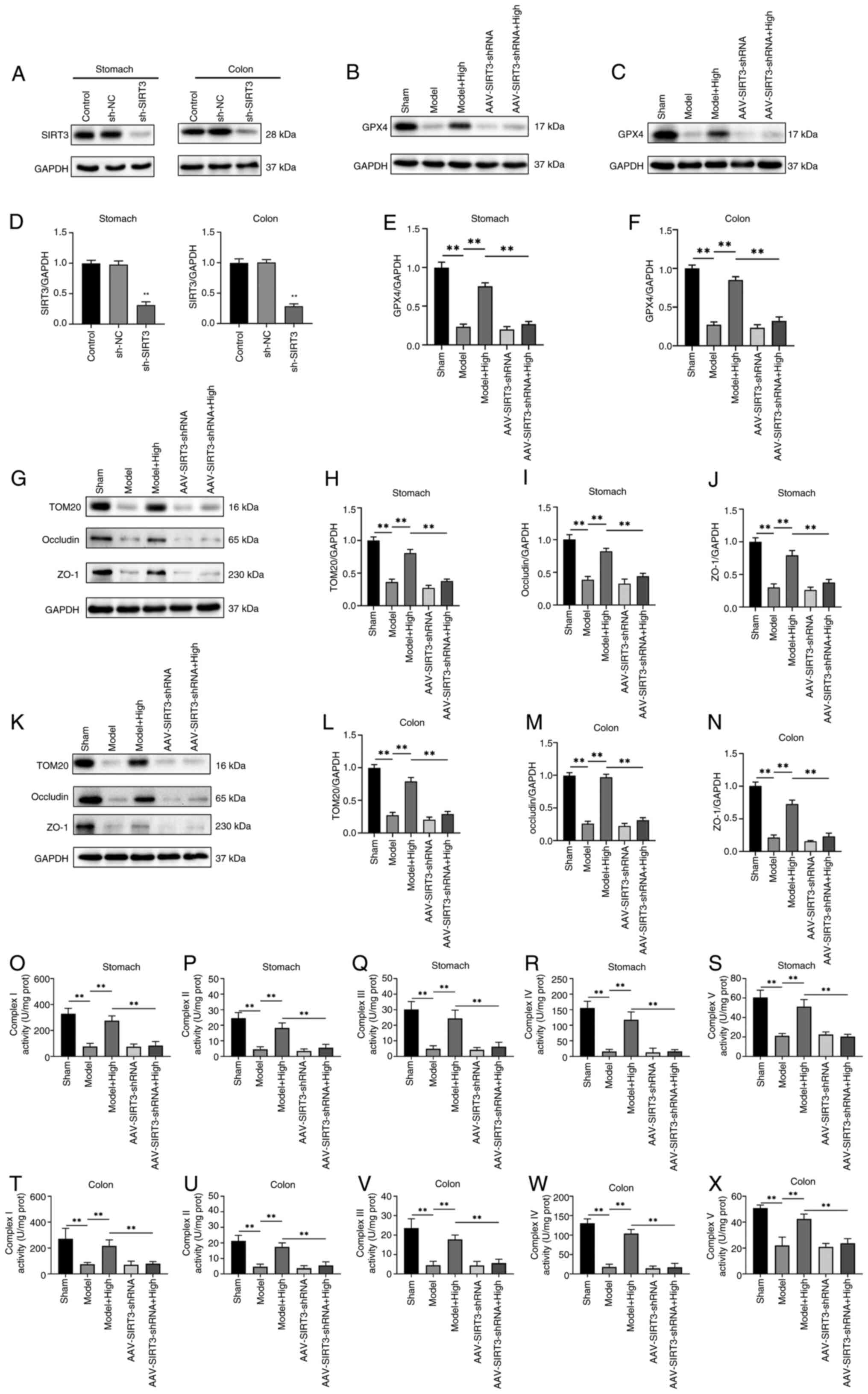 | Figure 6.Regulatory mechanism of SIRT3 and
PRDX3 in sepsis-induced ferroptosis-mediated mitochondrial
oxidative damage. (A) Western blot analysis of SIRT3 protein level
in the stomach and colon tissue. Mice were injected with sh-SIRT3
or control shRNA (shRNA-NC). (B) Western blot analysis of GPX4
protein level in stomach and (C) Western blot analysis of GPX4
protein level in colon tissue. (D) Relative SIRT3/GAPDH expression
obtained using ImageJ software. Relative quantitative evaluation of
the GPX4/GAPDH expression in (E) stomach and (F) colon tissue of
each group. (G) Western blot analysis of TOM20, occludin, and ZO-1
protein level in stomach tissue of each group. Relative
quantitative evaluation of (H) TOM20, (I) occludin and (J) ZO-1
protein expression level in stomach tissue in each group. (K)
Western blot analysis of TOM20, occludin, and ZO-1 protein level in
stomach and colon tissue of each group. Relative quantitative
evaluation of (L) TOM20, (M) occludin and (N) ZO-1 protein
expression level in colon tissue. Activity of mitochondrial
respiratory chain complex (O) I, mitochondrial respiratory chain
complex II (P), mitochondrial respiratory chain complex III (Q),
mitochondrial respiratory chain complex IV (R), mitochondrial
respiratory chain complex V (S) in the stomach tissue. (T-X) The
activity of mitochondrial respiratory chain complex I (T),
mitochondrial respiratory chain complex II (U), mitochondrial
respiratory chain complex III (V), mitochondrial respiratory chain
complex IV (W), mitochondrial respiratory chain complex V (X) in
the colon tissue. Values are means ± SD of n=3 (A-N) or n=6 (O-X)
experiments. **P<0.01. SIRT3, NAD-dependent protein deacetylase
sirtuin-3, mitochondrial; PRDX3, peroxiredoxin-3; sh, short
hairpin; GPX4, Phospholipid hydroperoxide glutathione peroxidase;
TOM20, mitochondrial import receptor subunit TOM20 homolog; ZO-1,
zona occludens protein 1. |
Discussion
Sepsis is a clinical syndrome characterized by an
aberrant inflammatory response to infection, leading to organ
dysfunction. Target organ dysfunction caused by sepsis and multiple
organ dysfunction syndrome are the primary causes of patient
mortality, with acute GI being particularly prevalent (3). The gastrointestinal tract is the
target for inflammatory mediators in sepsis and is a significant
source of these mediators (20).
Studies have reported varying degrees of GI in patients with sepsis
(21), making it the most common
complication associated with this condition. Impaired
gastrointestinal function results in bacterial translocation and
toxin transfer into the bloodstream, exacerbating inflammation and
impairing multiple organ function. Consequently, sepsis and
gastrointestinal dysfunction mutually reinforce each other,
creating a vicious cycle. Furthermore, since the gastrointestinal
tract is often the initial site affected by multiple organ
dysfunction syndrome resulting from sepsis, actively improving
gastrointestinal function of patients is significant in enhancing
the success rate of sepsis treatment.
Atractylodin is classified as an acetylene compound,
and modern pharmacological studies have demonstrated its
anti-inflammatory and antioxidant properties (22). The present study constructed a
cecum ligation perforation model to investigate how atractylodin
alleviates sepsis-associated gastrointestinal damage. ZO-1 and
occludin are crucial components of tight junctions, and their
downregulated expression or reduced activity can impair the
formation of tight junctions between cells, compromising the vital
defense barrier function of the intestinal mucosa and increasing
the risk of enteroborne infections caused by harmful bacteria and
toxins penetrating the bloodstream (23). The present study indicated that the
administration of atractylodin upregulated the expression of ZO-1
and occludin in gastrointestinal tissue, suggesting its potential
to enhance gastrointestinal tissue barrier function and reduce
inflammation occurrence. Histological examination using H&E and
immunohistochemical staining also revealed improved intestinal
damage repair and enhanced barrier function in both stomach and
colon tissues among mice in the all-administration group.
The concept of ferroptosis was initially proposed as
a form of iron-dependent programmed cell death, distinct from
apoptosis, cell necrosis, and autophagy (24). Various cellular metabolic events
including redox homeostasis, iron load, mitochondrial function and
lipid metabolism regulate ferroptosis. Mitochondria play crucial
regulatory roles in the process of iron death and are essential for
cellular resistance against it (25). Maintaining mitochondrial integrity
is an effective strategy for preventing iron death across different
cell types. The present study used electron microscopy to
demonstrate structural disorder, swelling and damage in the model
group mice; however, treatment with atractylodin reduced
mitochondrial damage while increasing the number and size of
mitochondria in the administration group. Upregulation of CAT, SOD2
and GSH/GSSG proteins indicated that atractylodin could mitigate
cell damage; meanwhile, downregulation of
H2O2 and MDA proteins suggested an
improvement in mitochondrial oxidative stress.
Mitochondria are the primary sites for ATP
generation in animal and plant cells. During respiratory oxidation,
asymmetric protons and other ions are distributed on both sides of
the inner membrane of mitochondria, resulting in mitochondrial
membrane potential (MMP) (26).
Maintaining a normal MMP is essential for sustaining mitochondrial
oxidative phosphorylation and ATP production, which contributes to
maintaining cellular physiological functions (27). The present study employed flow
cytometry and mitochondrial membrane potential detection kits to
investigate the effects of atractylodin on mitochondrial
transmembrane energy consumption and membrane potential in stomach
and colon tissues. The results revealed that atractylodin can
decrease mitochondrial transmembrane energy consumption while
enhancing the membrane potential.
The mitochondrial respiratory chain is a key
component of cellular energy metabolism, functioning as a
continuous reaction system that consists of a series of hydrogen
transfer reactions and electron transfer reactions in a specific
sequence, commonly referred to as the electron transport chain
(28). This respiratory chain
reaction efficiently synthesizes abundant ATP molecules while
simultaneously removing hydrogen atoms from metabolites to generate
water (29). Assessing the
activity of mitochondrial complexes can provide insights into the
effectiveness of electron transport in redox processes involved in
oxidative phosphorylation and cell death. The present study
demonstrated significant enhancements in mitochondrial complex I,
II, III, IV and V activities among mice exposed to different
concentrations of atractylodin. Furthermore, elevated levels of
mitochondrial ROS induce ferritin autophagy and increase
intracellular iron content, ultimately leading to ferroptosis
(30). Gastric and colon tissues
were stained with DHE, a superoxide indicator. Compared with the
model group, each concentration group treated with atractylodin
exhibited markedly reduced fluorescence intensity of DHE,
indicating that atractylodin effectively mitigates mitochondrial
ROS levels.
The accumulation of large quantities of free
Fe2+ can also induce ferroptosis (31) due to its role as a cofactor for
various metabolic enzymes, such as lipid oxygenase, thereby
enhancing their activity and promoting the production of lipid
peroxides. Fe2+ level catalyzed by the Fenton reaction
leads to the generation of peroxy and hydroxyl radicals, which
further react with lipid peroxides, resulting in the substantial
production of lipid ROS. Ultimately, these processes culminate in
the induction of ferroptosis (31). The present study assessed the
Fe2+ level and observed a significant reduction in the
atractylodin-administration group. The SLC7A11-GPX4 axis is the
pivotal system involved in resistance against ferroptosis (31). GPX4 is a glutathione peroxidase
that utilizes glutathione to detoxify lipid peroxides and inhibit
ferroptosis. TFR1 is a membrane protein widely expressed across
various cell and tissue types within the human body. Western
blotting analysis revealed markedly higher levels of GPX4 and
SLC7A11 in the administration group treated with atractylodin
compared with those in the model group. It also downregulated TFR1
expression.
PRDX is a potent thiol peroxidase family that
comprises at least six subtypes in mammalian cells (32). Isoforms PRDX1, PRDX2 and PRDX6 are
localized in the cytoplasm, while PRDX4 is found in the endoplasmic
reticulum. PRDX5 is located in both the peroxisome and
mitochondria, whereas PRDX3 is the dominant species within the
mitochondria (33). As PRDX3 is
the most abundant and effective enzyme for
H2O2 elimination in mitochondria, it serves
as an important mitochondrial antioxidant protein and acts as a
target for nearly 90% of HO produced in the matrix (34). The clearance of HO by PRDX3 occurs
through its oxidation to an inactive dimer form (35), with previous studies reporting its
ability to effectively inhibit oxidative stress, and apoptosis
(11–13,36),
and decrease cell damage. Transgenic mice overexpressing PRDX3 have
shown reduced production of H2O2 and
decreased oxidative damage within their mitochondria compared with
control mice (37). SIRT3, a
highly conserved nicotinamide adenine dinucleotide (NAD)-dependent
deacetylase primarily expressed in mitochondria, regulates several
mitochondrial proteins involved in fatty acid oxidation, oxidative
phosphorylation, and antioxidant reaction systems (38). The immediate clearance of ROS by
SIRT3 is not feasible, while PRDX3 undergoes reversible acetylation
(39). Therefore, it was
hypothesized that SIRT3 plays a role in sepsis combined with GI
through deacetylation of PRDX3. The present study revealed the
downregulation of PRDX3 expression along with upregulation of SIRT3
expression in gastric tissues treated with atractylodin. To
investigate how atractylodin regulates SIRT3 expression within
cells, immunofluorescence was employed to co-localize SIRT3 with
nuclear marker DAPI within gastric and colon tissues.
Immunofluorescence analysis demonstrated increased SIRT3 expression
upon treatment with atractylodin. Co-IP revealed an enhanced
interaction between SIRT3 and PRDX3 in the group treated with
atractylodin. Moreover, when SIRT3 was downregulated using
AAV-shSIRT3, the regulatory effect of atractylodin on mitochondrial
and cellular ferroptosis was attenuated. These findings suggested
that SITR3 mediated the inhibitory effects of atractylodin on cell
ferroptosis in sepsis-induced GI.
Research indicates that SIRT3 within mitochondria
undergoes SUMOylation under physiological conditions, markedly
inhibiting its deacetylase activity (40). Under conditions of metabolic
stress, the desumoylation enzyme SENP1 translocates to
mitochondria, restoring the deacetylation activity of SIRT3 by
removing its SUMOylation modification (41). This process regulates the
acetylation levels of mitochondrial proteins, thereby influencing
metabolic functions (41).
Notably, resveratrol, a natural activator of SIRT3, has been shown
to upregulate the expression of SOD2 and catalase via the
SIRT3/FoxO3a signaling axis, ROS and lipid peroxide levels, and
inhibit ferroptosis by enhancing the GSH/GPX4 pathway activity.
This activity effectively mitigates intestinal I/R injury (42). These findings demonstrate the
regulatory mechanisms of the dynamic post-translational
modifications of SIRT3 and provide a theoretical foundation for the
cytoprotective effects of monomeric components of traditional
Chinese medicine by targeting the downstream effector molecules of
SIRT3. Based on these findings, it was hypothesized that
atractylotin may enhance the deacetylation of PRDX3 by SIRT3
through the desumoylation of SIRT3, which ultimately exerts its
organ-protective effect by restoring mitochondrial redox
homeostasis and inhibiting ferroptosis during sepsis-induced GI.
However, the precise molecular regulation mechanism remains to be
elucidated by future studies.
The present study acknowledges several limitations.
First, the investigation did not incorporate an analysis of the
effect of interval dosing on efficacy, as comparisons between short
and long treatment courses were not conducted. As a mechanistic
study exploring the role of atractylodin in sepsis treatment, the
primary objective was to verify its fundamental efficacy and
dose-response relationship. To optimize dosing frequency and
duration, it is essential to integrate pharmacokinetic data.
Secondly, further research is required to elucidate the precise
molecular regulatory mechanisms involved in downstream
processes.
The present basic science study offers essential
preclinical evidence supporting the potential therapeutic efficacy
of atractylodin in the treatment of sepsis and establishes a
mechanistic basis that underscores the need for further
translational research. Prior to initiating clinical evaluation, it
is imperative to conduct comprehensive preclinical safety and
toxicology studies in relevant animal models to evaluate potential
systemic toxicity, organ-specific adverse effects and establish
safe dosage ranges for initial human trials. Subsequent rigorous
investigations into pharmacokinetics and pharmacodynamics (PK/PD)
are necessary to fully elucidate the absorption, distribution,
metabolism and excretion profile of atractylodin in relevant
preclinical species and to understand how drug exposure levels
correlate with the observed therapeutic outcomes and potential
toxicities. These PK/PD data are crucial for informing initial dose
selection for human studies. Following successful preclinical
validation, the potential of atractylodin would be evaluated in a
structured clinical trial program. This typically begins with Phase
1 trials in a small group of healthy volunteers or patients with
stable conditions to assess safety, tolerability and basic human
PK/PD (43). If deemed safe, Phase
2 trials would then enroll a larger cohort of sepsis patients to
investigate preliminary efficacy, explore dose-response
relationships, and further evaluate safety in the target
population. Efficacy endpoints in Phase 2 sepsis trials commonly
include changes in organ dysfunction scores, inflammatory markers,
or surrogate outcomes (44). Based
on promising Phase 2 results, Phase 3 trials would be necessary to
confirm efficacy on clinically relevant endpoints such as 28-day
mortality and time to organ failure resolution, in large,
multi-center, randomized controlled studies comparing atractylodin
to current standard of care (45).
Successfully navigating these rigorous clinical trial phases is
required to ultimately assess the safety, efficacy, and optimal
dosing of atractylodin for potential clinical use in human patients
with sepsis.
In summary, atractylodin can ameliorate
mitochondrial dysfunction in the acute gastrointestinal tract of
sepsis and mitigate ferroptosis. The underlying mechanism involves
the SIRT3/PRDX3 signaling pathway. Atractylodin holds promise as a
potential therapeutic agent for the treatment and prevention of
gastrointestinal disorders.
GI is a critical illness associated with high
morbidity and mortality. Mitochondrial oxidative stress and
ferroptosis are the key pathogenic events resulting from GI. The
present study first reported the following observations: i)
Atractylodin mitigated sepsis-induced GI. ii) PRDX3 protected
against sepsis-induced acute GI, mitochondrial oxidative damage and
ferroptosis. iii) SIRT3 deacetylates PRDX3 and can therefore
alleviate sepsis-induced acute GI mitochondrial oxidative damage
and ferroptosis.
The present study revealed that atractylodin can
alleviate mitochondrial dysfunction and decrease ferroptosis in
sepsis-induced acute GI. In addition, it was found that the
mechanisms of atractylodin are associated with SIRT3/PRDX3
signaling. Together, the findings of the present study demonstrated
that atractylodin may serve as a promising therapeutic agent for
treating and preventing GI.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 81804035), the Project of Jiangsu
Provincial Administration of Chinese Medicine (grant no.
MS2023092), the Changzhou Sci and Tech Program (grant no.
CJ20239006) and the Youth Talent Technology Project of Changzhou
Health Commission (grant no. QN202137).
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
MQC was responsible for investigation and resources.
JLW, HDZ and TTL were responsible for investigation. TW was
responsible for resources. YXH was responsible for validation,
investigation and writing the original draft. YB was responsible
for writing, reviewing and editing, resources, methodology,
investigation, funding acquisition and conceptualization. HG was
responsible for writing, reviewing and editing, validation,
resources and investigation. All authors read and approved the
final manuscript. YXH and MQI confirm the authenticity of all the
raw data.
Ethics approval and consent to
participate
The animal experiments in the present study was
conducted in the Liyang Hospital of Chinese Medicine and approved
by the Experimental Animal Ethics Committee of Liyang Hospital of
Chinese Medicine (Jiangsu, China; approval no.
2024LY-02-02-03).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Salomão R, Ferreira BL, Salomão MC, Santos
SS, Azevedo LCP and Brunialti M: Sepsis: Evolving concepts and
challenges. Braz J Med Biol Res. 52:e85952019. View Article : Google Scholar
|
|
2
|
Deitch EA: Gut-origin sepsis: Evolution of
a concept. Surgeon. 10:350–356. 2012. View Article : Google Scholar
|
|
3
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar
|
|
4
|
Clements TW, Tolonen M, Ball CG and
Kirkpatrick AW: Secondary peritonitis and intra-abdominal sepsis:
An increasingly global disease in search of better systemic
therapies. Scand J Surg. 110:139–149. 2021. View Article : Google Scholar
|
|
5
|
Rittirsch D, Flierl MA and Ward PA:
Harmful molecular mechanisms in sepsis. Nat Rev Immunol. 8:776–787.
2008. View
Article : Google Scholar
|
|
6
|
Zarjou A and Agarwal A: Sepsis and acute
kidney injury. J Am Soc Nephrol. 22:999–1006. 2011. View Article : Google Scholar
|
|
7
|
Tadokoro T, Ikeda M, Ide T, Deguchi H,
Ikeda S, Okabe K, Ishikita A, Matsushima S, Koumura T, Yamada KI,
et al: Mitochondria-dependent ferroptosis plays a pivotal role in
doxorubicin cardiotoxicity. JCI Insight. 5:e1327472020. View Article : Google Scholar
|
|
8
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar
|
|
9
|
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C,
Wu H, Deng W, Shen D and Tang Q: Ferritinophagy-mediated
ferroptosis is involved in sepsis-induced cardiac injury. Free
Radic Biol Med. 160:303–318. 2020. View Article : Google Scholar
|
|
10
|
Li J, Lu K, Sun F, Tan S, Zhang X, Sheng
W, Hao W, Liu M, Lv W and Han W: Panaxydol attenuates ferroptosis
against LPS-induced acute lung injury in mice by Keap1-Nrf2/HO-1
pathway. J Transl Med. 19:962021. View Article : Google Scholar
|
|
11
|
Wang Z, Sun R, Wang G, Chen Z, Li Y, Zhao
Y, Liu D, Zhao H, Zhang F, Yao J and Tian X: SIRT3-mediated
deacetylation of PRDX3 alleviates mitochondrial oxidative damage
and apoptosis induced by intestinal ischemia/reperfusion injury.
Redox Biol. 28:1013432020. View Article : Google Scholar
|
|
12
|
Xu S, Liu Y, Yang S, Fei W, Qin J, Lu W
and Xu J: FXN targeting induces cell death in ovarian cancer
stem-like cells through PRDX3-Mediated oxidative stress. iScience.
27:1105062024. View Article : Google Scholar
|
|
13
|
Tang F, Fan K, Wang K and Bian C:
Atractylodin attenuates lipopolysaccharide-induced acute lung
injury by inhibiting NLRP3 inflammasome and TLR4 pathways. J
Pharmacol Sci. 136:203–211. 2018. View Article : Google Scholar
|
|
14
|
Koonrungsesomboon N, Na-Bangchang K and
Karbwang J: Therapeutic potential and pharmacological activities of
Atractylodes lancea (Thunb.) DC. Asian Pac J Trop Med.
7:421–428. 2014. View Article : Google Scholar
|
|
15
|
Yu C, Xiong Y, Chen D, Li Y, Xu B, Lin Y,
Tang Z, Jiang C and Wang L: Ameliorative effects of atractylodin on
intestinal inflammation and co-occurring dysmotility in both
constipation and diarrhea prominent rats. Korean J Physiol
Pharmacol. 21:1–9. 2017. View Article : Google Scholar
|
|
16
|
Xu L, Zhou Y, Xu J, Xu X, Lu G, Lv Q, Wei
L, Deng X, Shen X, Feng H and Wang J: Anti-inflammatory,
antioxidant and anti-virulence roles of atractylodin in attenuating
Listeria monocytogenes infection. Front Immunol. 13:9770512022.
View Article : Google Scholar
|
|
17
|
Lin YC, Yang CC, Lin CH, Hsia TC, Chao WC
and Lin CC: Atractylodin ameliorates ovalbumin-induced asthma in a
mouse model and exerts immunomodulatory effects on Th2 immunity and
dendritic cell function. Mol Med Rep. 22:4909–4918. 2020.
View Article : Google Scholar
|
|
18
|
Qu L, Lin X, Liu C, Ke C, Zhou Z, Xu K,
Cao G and Liu Y: Atractylodin attenuates dextran sulfate
sodium-induced colitis by alleviating gut microbiota dysbiosis and
inhibiting inflammatory response through the MAPK pathway. Front
Pharmacol. 12:6653762021. View Article : Google Scholar
|
|
19
|
Heo G, Kim Y, Kim EL, Park S, Rhee SH,
Jung JH and Im E: Atractylodin ameliorates colitis via PPARα
agonism. Int J Mol Sci. 24:8022023. View Article : Google Scholar
|
|
20
|
Vitzthum LK, Nalawade V, Riviere P, Marar
M, Furnish T, Lin LA, Thompson R and Murphy JD: Impacts of an
opioid safety initiative on US veterans undergoing cancer
treatment. J Natl Cancer Inst. 114:753–760. 2022. View Article : Google Scholar
|
|
21
|
Nullens S, Staessens M, Peleman C, Plaeke
P, Francque SM, Lammens C, Malhotra-Kumar S, De Man J and De Winter
BY: Su1190 effect of gastrointestinal barrier protection on
sepsis-induced changes of intestinal motility, inflammation and
colonic permeability. Gastroenterology. 150:S4912016. View Article : Google Scholar
|
|
22
|
Song GY, Kim SM, Back S, Yang SB and Yang
YM: Atractylodes lancea and its constituent, atractylodin,
ameliorates metabolic dysfunction-associated steatotic liver
disease via AMPK activation. Biomol Ther (Seoul). 32:778–792. 2024.
View Article : Google Scholar
|
|
23
|
Kuo WT, Odenwald MA, Turner JR and Zuo L:
Tight junction proteins occludin and ZO-1 as regulators of
epithelial proliferation and survival. Ann N Y Acad Sci.
1514:21–33. 2022. View Article : Google Scholar
|
|
24
|
Li S and Huang Y: Ferroptosis: An
iron-dependent cell death form linking metabolism, diseases, immune
cell and targeted therapy. Clin Transl Oncol. 24:1–12. 2022.
View Article : Google Scholar
|
|
25
|
Wu H, Wang F, Ta N, Zhang T and Gao W: The
multifaceted regulation of mitochondria in ferroptosis. Life
(Basel). 11:2222021.
|
|
26
|
Gu J, Liu T, Guo R, Zhang L and Yang M:
The coupling mechanism of mammalian mitochondrial complex I. Nat
Struct Mol Biol. 29:172–182. 2022. View Article : Google Scholar
|
|
27
|
Ryu KW, Fung TS, Baker DC, Saoi M, Park J,
Febres-Aldana CA, Aly RG, Cui R, Sharma A, Fu Y, et al: Cellular
ATP demand creates metabolically distinct subpopulations of
mitochondria. Nature. 635:746–754. 2024. View Article : Google Scholar
|
|
28
|
Sazanov LA: A giant molecular proton pump:
structure and mechanism of respiratory complex I. Nat Rev Mol Cell
Biol. 16:375–388. 2015. View Article : Google Scholar
|
|
29
|
Lee HJ, Svahn E, Swanson JM, Lepp H, Voth
GA, Brzezinski P and Gennis RB: Intricate role of water in proton
transport through cytochrome c oxidase. J Am Chem Soc.
132:16225–16239. 2010. View Article : Google Scholar
|
|
30
|
Chen Y, Guo X, Zeng Y, Mo X, Hong S, He H,
Li J, Fatima S and Liu Q: Oxidative stress induces mitochondrial
iron overload and ferroptotic cell death. Sci Rep. 13:155152023.
View Article : Google Scholar
|
|
31
|
Dixon SJ and Olzmann JA: The cell biology
of ferroptosis. Nat Rev Mol Cell Biol. 25:424–442. 2024. View Article : Google Scholar
|
|
32
|
Bryk R, Griffin P and Nathan C:
Peroxynitrite reductase activity of bacterial peroxiredoxins.
Nature. 407:211–215. 2000. View Article : Google Scholar
|
|
33
|
Kang SW, Chae HZ, Seo MS, Kim K, Baines IC
and Rhee SG: Mammalian peroxiredoxin isoforms can reduce hydrogen
peroxide generated in response to growth factors and tumor necrosis
factor-alpha. J Biol Chem. 273:6297–6302. 1998. View Article : Google Scholar
|
|
34
|
Cox AG, Winterbourn CC and Hampton MB:
Mitochondrial peroxiredoxin involvement in antioxidant defence and
redox signalling. Biochem J. 425:313–325. 2009. View Article : Google Scholar
|
|
35
|
Peskin AV, Low FM, Paton LN, Maghzal GJ,
Hampton MB and Winterbourn CC: The high reactivity of peroxiredoxin
2 with H(2)O(2) is not reflected in its reaction with other
oxidants and thiol reagents. J Biol Chem. 282:11885–11892. 2007.
View Article : Google Scholar
|
|
36
|
Nonn L, Berggren M and Powis G: Increased
expression of mitochondrial peroxiredoxin-3 (thioredoxin
peroxidase-2) protects cancer cells against hypoxia and
drug-induced hydrogen peroxide-dependent apoptosis. Mol Cancer Res.
1:682–689. 2003.
|
|
37
|
Lombard DB and Zwaans BM: SIRT3: As simple
as it seems? Gerontology. 60:56–64. 2014. View Article : Google Scholar
|
|
38
|
Shimazu T, Hirschey MD, Hua L,
Dittenhafer-Reed KE, Schwer B, Lombard DB, Li Y, Bunkenborg J, Alt
FW, Denu JM, et al: SIRT3 deacetylates mitochondrial
3-hydroxy-3-methylglutaryl CoA synthase 2 and regulates ketone body
production. Cell Metab. 12:654–661. 2010. View Article : Google Scholar
|
|
39
|
Hebert AS, Dittenhafer-Reed KE, Yu W,
Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ,
Higbee AJ, et al: Calorie restriction and SIRT3 trigger global
reprogramming of the mitochondrial protein acetylome. Mol Cell.
49:186–199. 2013. View Article : Google Scholar
|
|
40
|
Liang J, Zhou C, Zhang C, Liang S, Zhou Z,
Zhou Z, Wu C, Zhao H, Meng X, Zou F, et al: Nicotinamide
mononucleotide attenuates airway epithelial barrier dysfunction via
inhibiting SIRT3 SUMOylation in asthma. Int Immunopharmacol.
127:1113282024. View Article : Google Scholar
|
|
41
|
Wang T, Cao Y, Zheng Q, Tu J, Zhou W, He
J, Zhong J, Chen Y, Wang J, Cai R, et al: SENP1-Sirt3 signaling
controls mitochondrial protein acetylation and metabolism. Mol
Cell. 75:823–834.e5. 2019. View Article : Google Scholar
|
|
42
|
Wang X, Shen T, Lian J, Deng K, Qu C, Li
E, Li G, Ren Y, Wang Z, Jiang Z, et al: Resveratrol reduces
ROS-induced ferroptosis by activating SIRT3 and compensating the
GSH/GPX4 pathway. Mol Med. 29:1372023. View Article : Google Scholar
|
|
43
|
Na-Bangchang K, Kulma I, Plengsuriyakarn
T, Tharavanij T, Kotawng K, Chemung A, Muhamad N and Karbwang J:
Phase I clinical trial to evaluate the safety and pharmacokinetics
of capsule formulation of the standardized extract of
Atractylodes lancea. J Tradit Complement Med. 11:343–355.
2021. View Article : Google Scholar
|
|
44
|
Park SJ, Park J, Lee MJ, Seo JS, Ahn JY
and Cho JW: Time series analysis of delta neutrophil index as the
predictor of sepsis in patients with acute poisoning. Hum Exp
Toxicol. 39:86–94. 2020. View Article : Google Scholar
|
|
45
|
Geven C, Blet A, Kox M, Hartmann O,
Scigalla P, Zimmermann J, Marx G, Laterre PF, Mebazaa A and
Pickkers P: A double-blind, placebo-controlled, randomised,
multicentre, proof-of-concept and dose-finding phase II clinical
trial to investigate the safety, tolerability and efficacy of
adrecizumab in patients with septic shock and elevated
adrenomedullin concentration (AdrenOSS-2). BMJ Open. 9:e0244752019.
View Article : Google Scholar
|