Introduction
Atrial fibrillation (AF), characterized by irregular
and rapid heartbeat originating in the atria, poses a considerable
global health concern (1).
Currently, AF affects >33.5 million people worldwide, including
2.7–6.1 million people in the United States, and the prevalence of
AF is influenced by predisposing factors, including age,
hypertension, diabetes and structural heart disease (2). Furthermore, lifestyle factors such as
obesity and physical inactivity also markedly contribute to the
onset of AF, highlighting the potential for preventive strategies
(3). The complex interplay of
these risk factors contributes to the initiation and perpetuation
of AF, creating a challenging clinical landscape. Given its
association with cardiovascular complications, such as stroke and
heart failure, understanding the epidemiology of AF is key for
developing targeted interventions (4). Therefore, current research focuses on
elucidating the complex mechanisms of AF pathogenesis, developing
novel therapeutic interventions and refining prognostic assessment
tools (5,6).
The peroxisome proliferator-activated receptor
(PPAR) signaling pathway, a nuclear receptor pathway regulating
various physiological processes such as energy metabolism, lipid
metabolism, inflammation, includes the ligand-activated
transcription factors PPARα, PPARβ/δ and PPARγ (7,8).
Activated by ligands such as fatty acids, PPARs form heterodimers
with retinoid X receptors and regulate target gene transcription
through PPAR response elements in DNA, impacting lipid metabolism,
glucose homeostasis, inflammation and cell differentiation
(9,10). Essential in metabolic regulation,
the PPAR pathway is a therapeutic target for diabetes,
cardiovascular disease and metabolic disorders (11). Zhu et al (12) demonstrated the role of the PPAR
pathway in AF: PPARα and PPARγ attenuate angiotensin (Ang)
II-induced AF and fibrosis in mice, demonstrated by the protective
effects of angiopoietin-like 4 treatment. Zheng et al
(13) further established a
connection between the PPAR signaling pathway and AF, identifying
mannose receptor C-type 2 as a key inhibitory gene that suppresses
AF progression, offering potential diagnostic and therapeutic
avenues. Xu et al (14)
highlighted the protective role of the PPAR-γ activator
pioglitazone in mitigating age-associated vulnerability to AF
through enhanced antioxidant potential and suppression of apoptosis
via mitochondrial signaling pathways in a rat model. The
aforementioned studies highlight the potential of the PPAR
signaling pathway as an important research area for understanding
and managing AF, providing potential therapeutic strategies.
Heat shock protein D 1 (HSPD1) encodes the
mitochondrial chaperonin protein, HSP60 (15). It is key for maintaining
mitochondrial protein folding, ensuring proper protein transport
within the mitochondria and contributing to cellular homeostasis
(16). Dysregulation of HSPD1 is
associated with a range of diseases, including neurodegenerative
disorder and cardiovascular diseases, as its expression can be
induced in response to cellular stress to protect cells from damage
(17). van Marion et al
(18) demonstrated that elevated
levels of HSPD1 in atrial tissue are associated with persistent
stages of AF. Altered levels of HSPA5 in the right atrial appendage
and HSPA1 in the left atrial appendage are associated with the
development of postoperative AF and AF recurrence following
arrhythmia surgery. Oc et al (19) investigated the association between
pre- and postoperative circulating HSP70 levels and the development
of AF following coronary artery bypass surgery; HSP70 may have a
protective effect only when localized in cells, but it loses this
protection when released into the blood. Meijering et al
(20) demonstrated the protective
role of HSPs, including HSPD1, in preventing electrical,
contractile and structural remodeling of cardiomyocytes. This
underscores the potential for upstream therapy to prevent AF
progression and recurrence by upregulating the heat shock response
system.
Given the importance of AF in cardiac etiology and
its complexity, the present study aimed to clarify the causes of AF
development, with a focus on the HSPD1 gene. Peptide hormone Ang II
stimulates the release of aldosterone and constriction of blood
vessels, which are key mechanisms in the regulation of blood
pressure and fluid balance (21).
Therefore, the present study aimed to evaluate the effect of HSPD1
knockdown on angiotensin II (Ang II)-induced fibrotic response of
cardiac fibroblasts (CF), including evaluation of cell viability,
expression of inflammatory and AF-related markers. In addition, the
potential regulatory association between HSPD1 and PPAR signaling
pathways in AF was explored. The present study aimed to enhance the
understanding of the pathogenesis of AF and suggest potential
therapeutic targets.
Materials and methods
AF-related datasets
A total of two gene expression datasets associated
with AF were retrieved from the Gene Expression Omnibus
(ncbi.nlm.nih.gov/gds/; accession nos. GSE31821 and GSE14975). The
GSE31821 dataset contains four AF samples (three from the auricle
of a patient with AF and one from the pulmonary vein of a patient
with AF) and two control samples. From this dataset, three AF
samples (auricles from patients with AF) and two control samples
were selected for analysis. The GSE14975 dataset includes 5 AF and
five samples of patients with sinus rhythm (as normal controls).
The data samples were all downloaded in MINiML format.
Screening and enrichment analysis of
differentially expressed genes (DEGs) in the GSE31821 dataset
Differential expression analysis was performed on
five samples in the GSE31821 dataset using the Limma package
(version 3.52.4) in R (version 4.2.2;
bioconductor.org/packages/limma/). The threshold for fold change
was >1.3 or <0.77 with a significance level of P<0.05 to
identify DEGs. Subsequently, the Database for Annotation,
Visualization and Integrated Discovery (DAVID) database
(david.ncifcrf.gov/) was used to perform Gene Ontology (GO) and
Kyoto Encyclopedia of Genes and Genomes (KEGG) (22) enrichment analysis. GO includes
biological process (BP), cellular component (CC) and molecular
function (MF).
Candidate gene identification and
expression analysis in AF
To explore the interactions between DEGs,
protein-protein interaction (PPI) network analysis was performed
using the Search Tool for Retrieval of Interacting Genes (STRING)
database (string-db.org/). The resulting network was visualized
with Cytoscape (version 3.8.0; cytoscape.org/), using the maximum
correlation criterion (MCC), maximum neighborhood component (MNC)
and degree algorithms to identify key nodes. To determine the
commonalities between the top 10 genes identified by MCC, MNC and
degree algorithms, the Venn diagram tool from the Bioinformatics
& Evolutionary Genomics platform
(bioinformatics.psb.ugent.be/webtools/Venn/) was employed for
intersection analysis. Using the GSE31821 and GSE14975 datasets,
the expression patterns of overlapping genes were investigated in
normal and AF samples. The results were visualized with the R tool
ggplot2 (version 3.4.0; ggplot2.tidyverse.org/).
Cell lines and culture
Rat CFs were obtained from Stemrecell (Shanghai)
Biotechnology Co., Ltd. (cat. no. STM-CE-3303). The rat CFs were
all cells that had been passaged ≤3 times. They were cultured in
Dulbecco's Modified Eagle Medium (Thermo Fisher Scientific)
containing 1% penicillin-streptomycin and 10% fetal bovine serum
(FBS; Gibco, Thermo Fisher Scientific, Waltham, MA, USA) in 5%
CO2 at 37°C.
Cell treatment and transfection
To elucidate its effects, Ang II was administered to
CFs at 1, 10, 100 nM and 1 and 10 µM for 6, 12, 24 and 48 h at
37°C. To activate the PPAR signaling pathway, CFs were exposed to
10 µM thiazolidinedione (TZD; Sigma-Aldrich; Merck KGaA) for 48 h
at 37°C. CFs were cultivated in six-well plates; 2×105
cells/well were seeded in 24-well plates for transfection. Specific
small interfering RNA (siRNA) targeting HSPD1 and negative control
siRNA (both purchased from GenePharma, Shanghai, China) was
transfected into CFs to achieve knockdown of HSPD1 expression. To
enable effective knockdown of HSPD1, cells were cultured for 48 h
after transfection to allow for effective knockdown of HSPD1. siRNA
sequences for si-HSPD1 and si-negative control (NC) were used at a
final concentration of 80 µM, with sequences as follows:
si-HSPD1-1: forward: 5′-GGGCCAAAGGGAAGAACAGUGAUUA-3′ and reverse:
5′-UAAUCACUGUUCUUCCCUUUGGCCC-3′; si-HSPD1-2: Forward:
5′-GGAGAGGUGUGAUGUUGGCUGUUGA-3′ and reverse:
5′-UCAACAGCCAACAUCACACCUCUCC-3′ and si-NC: forward:
5′-GGGAAGAGGAAAGCAGAGUACCUUA-3′ and reverse:
5′-UAAGGUACUCUGCUUUCCUCUUCCC-3′. Following the manufacturer's
instructions, cells were transfected using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.), and subsequent experiments were conducted 48 h
post-transfection at 37°C'.
Reverse transcription-quantitative
(RT-q)PCR
Total RNA was extracted from 1×106 CFs
using TRIzol® (Thermo Fisher Scientific, Inc.) according
to the manufacturer's protocol. For cDNA synthesis, a PrimeScript
RT kit (Takara Bio, Inc.) was used at 42°C for 30 min, followed by
85°C for 5 min to inactivate the enzyme qPCR was performed with the
StepOnePlus Real-Time PCR System and SYBR Green PCR Master Mix
(both Applied Biosystems; Thermo Fisher Scientific, Inc.) according
to the manufacturer's instructions. Gene expression levels were
quantified and normalized to GAPDH. All target expression levels
were computed with the 2−ΔΔCq method. Thermocycling
conditions were as follows: initial denaturation at 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec and 60°C for 30 sec.
The primer sequences used are listed in Table I. ACTA2 is the gene encoding
α-smooth muscle actin (α-SMA). COL1A1 is the gene encoding Collagen
I. COL3A1 is the gene encoding COL3A1.
 | Table I.Primer sequences for reverse
transcription-quantitative PCR. |
Table I.
Primer sequences for reverse
transcription-quantitative PCR.
| Gene | Direction | Sequence,
5′à3′ |
|---|
| HSPD1 |
|
|
|
| Forward |
CCAGCCTTGGATTCATT |
|
| Reverse |
CAAGCATGGCATCATAGCC |
| Col1a1 |
|
|
|
| Forward |
TCCTGCCGATGTCGCTATCC |
|
| Reverse |
TCGTGCAGCCATCCACAA |
| Col3a1 |
|
|
|
| Forward |
GCCTTCTACACCTGCTCCTG |
|
| Reverse |
AGCCACCCATTCCTCCGACT |
| ACTA2 | Forward |
GAGCGTGGCTATTCCTTCGTG |
|
| Reverse |
CAGTGGCCATCTCATTTTCAA |
|
|
| AGT |
| GAPDH |
|
|
|
| Forward |
CAATCCTGGGCGGTACAACT |
|
| Reverse |
TACGGCCAAATCCGTTCACA |
Western blotting (WB)
WB was performed using protein lysates extracted
from 1×106 CFs with RIPA lysis buffer (Thermo Fisher
Scientific, Inc.) supplemented with protease and phosphatase
inhibitors. The concentration of protein was quantified with the
BCA protein assay kit. Subsequently, 30 µg/lane total protein was
loaded onto a 10% SDS-PAGE gel and the separated proteins were
transferred onto PVDF membranes (MilliporeSigma). Blocking of
membranes was performed out with 5% skimmed milk at room
temperature for 1 h. Subsequently, membranes were incubated
overnight at 4 °C with primary antibodies against HSPD1 (Abcam,
ab190828), Collagen I (Abcam, ab138492), Collagen III (Abcam,
ab184993), α-SMA (Cell Signaling Technology, #14968), atrial
natriuretic peptide (ANP) (Abcam, ab225844), MMP-2 (Abcam,
ab92536), PPARα (Abcam, ab314112), PPARγ (Abcam, ab272718),
carnitine palmitoyltransferase I (CPT-1; Cell Signaling Technology,
#41803), sirtuin (SIRT) 3 (Abcam, ab246522) and β-major
histocompatibility complex (MHC) (Cell Signaling Technology, Inc.;
cat. no. #64038) (all 1:1,000), along with the respective
horseradish peroxidase-conjugated goat anti-rabbit IgG H&L
secondary antibody (1:2,000; Abcam, ab6721) at room temperature for
1 h. GAPDH (Abcam, 1:5,000) served as an internal loading control.
Visualization of protein bands was performed using an enhanced
chemiluminescence kit (BOSTER, Wuhan, China), and ChemiDoc imaging
system (Bio-Rad Laboratories, Inc.). Densitometric analysis of the
bands was performed using ImageJ software (version 1.8.0; National
Institutes of Health; http://imagej.nih.gov/).
Cell counting kit-8 (CCK-8) assay
Viability of CFs was assessed using the CCK-8 assay
(Dojindo Europe GmbH). CFs were cultivated at a density of
5×103 cells/well in 96-well plates 37°C in a humidified
atmosphere containing 5% CO2 for 24 h prior to
treatment. Following the aforementioned treatments, 10 µl CCK-8
solution was added for 2 h at 37°C in a 5% CO2
atmosphere to facilitate the reaction. The absorbance was measured
at 450 nm using a microplate reader (Thermo Fisher Scientific,
Inc.).
Cell migration assay
Cell migration was evaluated using Transwell assay.
Transfected CF cells (5×104 cells/well) were suspended
in serum-free DMEM (Thermo Fisher Scientific, Waltham, MA, USA) in
the upper chamber of Transwell plates. The lower chamber contained
medium with 10% FBS. After incubation at 37°C for 24 h, migrated
cells were fixed with 4% paraformaldehyde at room temperature for
30 min and stained with DAPI (1 µg/ml) for 10 min at room
temperature. The number of migrated cells was determined by
counting the nuclei in five randomly selected fields of view under
an inverted fluorescence microscope (magnification, ×200).
ELISA
Cell culture supernatant were collected by
centrifugation at 1,000 × g for 10 min at 4°C to remove cell
debris, and were subsequently diluted 1:2 with the sample diluent
provided in the ELISA kit. TNF-α, IL-1β and IL-8 levels were
determined using ELISA kits (Cusabio, Technology, LLC), according
to the manufacturers' instructions: TNF-α (CSB-E11987r, Cusabio
Technology LLC, Wuhan, China), IL-1β (CSB-E08055r, Cusabio
Technology LLC, Wuhan, China), and IL-8 (MBS8579416, MyBiosource,
USA). The enzyme-linked secondary antibody (Abcam, ab6721, 1:5,000)
was added and incubated at room temperature for 1 h. After three
wash steps with 1×PBS containing 0.05% Tween-20 (each for 5 min at
room temperature), the wells were incubated with TMB
(3,3′,5,5′-tetramethylbenzidine) substrate solution for 15 min at
room temperature. A stop solution was added to and then absorbance
at the 450 nm was measured using a microplate reader. The
concentrations of TNF-α, IL-1β, and IL-8 were determined using a
standard curve generated using standards of known
concentration.
Statistical analysis
Statistical analysis was performed using R software
(version 4.2.2; r-project.org/). Data are presented as the mean ±
SD of triplicate experiments. Data were analyzed by one-way ANOVA
followed by Tukey's post hoc test. P<0.05 was considered to
indicate a statistically significant difference.
Results
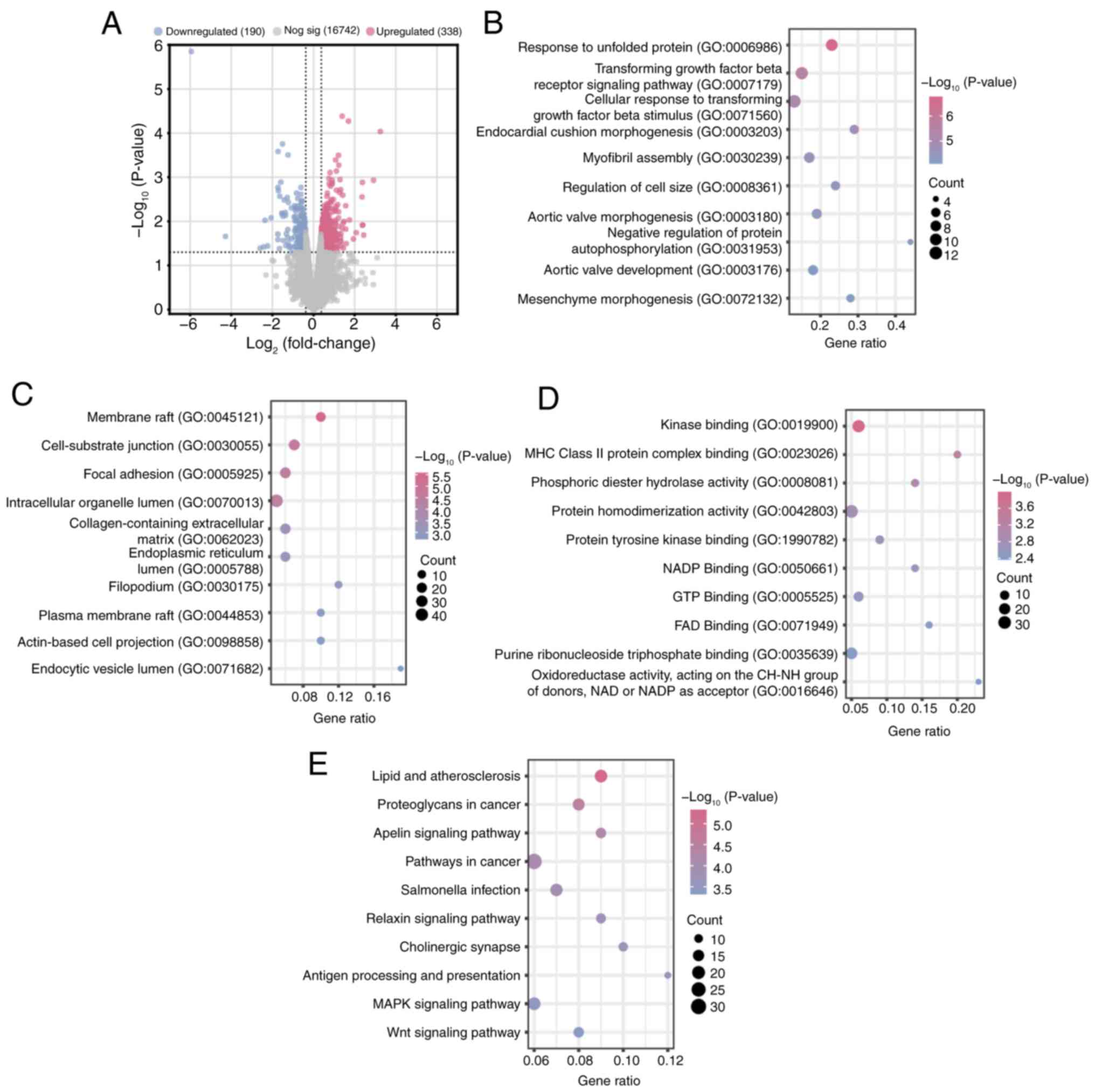
DEGs in the GSE318221 dataset are
enriched in inflammation-and remodeling-related pathways in AF
Using the R programming package, 338 up- and 190
downregulated DEGs were identified from cases and normal samples
within the GSE318221 dataset (Fig.
1A). DEGs were subjected to GO and KEGG enrichment analysis
using the DAVID database (Fig.
1B-E). The enriched terms included ‘myofibril assembly’ and
‘regulation of cell size’ (BP), ‘membrane raft’ and ‘focal
adhesion’ (CC), ‘NADP binding’ and ‘kinase binding’ (MF).
Additionally, the DEGs were significantly enriched in key pathways
such as ‘lipid and atherosclerosis’, ‘MAPK signaling pathway’ and
‘Wnt signaling pathway’, highlighting their potential roles in the
pathogenesis of AF.
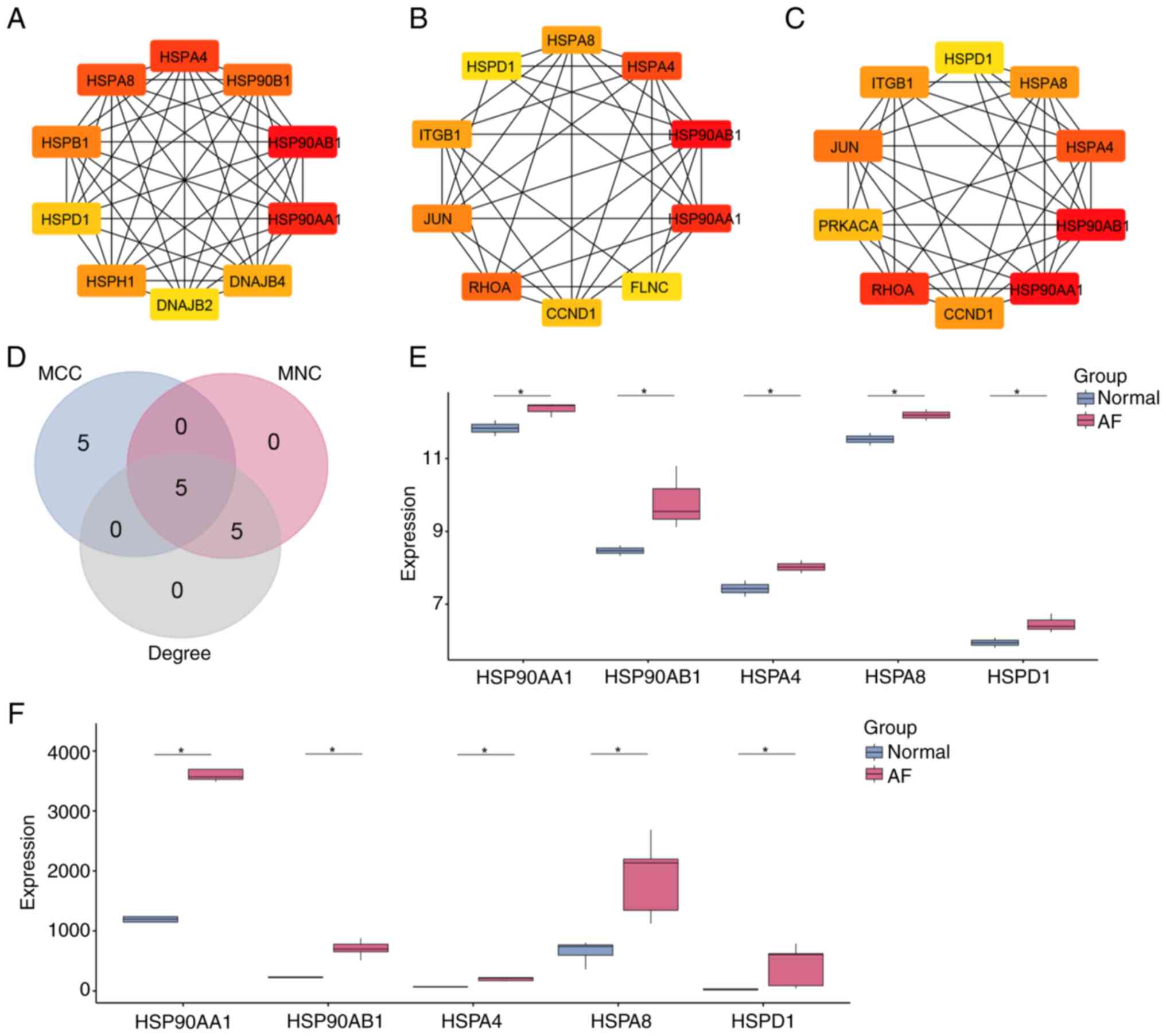
HSPD1 is a hub gene in AF
PPI network analysis was performed using the STRING
database on GSE31821 DEGs, and Cytoscape software visualized the
top 10 genes detected by MCC, MNC and degree algorithms (Fig. 2A-C). Specifically, the MCC network
encompassed 10 nodes and 44 edges, the MNC network comprised 10
nodes and 36 edges and the degree network included 10 nodes and 38
edges. Intersection analysis revealed five overlapping genes among
the top 10 genes identified in the MCC, MNC and degree networks
(Fig. 2D). Further investigation
of expression patterns within AF samples in the GSE31821 and
GSE14975 datasets revealed that five genes (HSP90AA1, HSP90AB1,
HSPA4, HSPA8 and HSPD1) were significantly upregulated (Fig. 2E and F), suggesting their potential
as targets for therapeutic intervention in AF. Among the five
overlapping genes, the roles of HSP90AA1, HSP90AB1, HSPA4 and HSPA8
in AF have been well-documented in previous studies (23–26).
In contrast, the function of HSPD1 in AF remains largely
unexplored. Therefore, HSPD1 was selected as the hub gene for
further investigation in this study.
Ang II enhances CF activation and
fibrosis marker expression
Notably, a significant increase in CF viability was
observed 24 h after exposure to Ang II at concentrations greater
than 1 nM (Fig. 3A). Cell
viability continued to increase over time and reached its highest
recorded level at 48 h following induction with 1 µM Ang II
(Fig. 3B). The migratory capacity
of CFs was assessed using Transwell assay, demonstrating a notable
rise in cell migration following induction with 1 µM Ang II for 48
h (Fig. 3C). RT-qPCR and WB data
revealed a substantial upregulation of HSPD1 expression in CFs 48 h
post-induction with 1 µM Ang II (Fig.
3D and E). Additionally, collagen I and III and α-SMA
expression levels were significantly increased in CFs following 48
h induction with 1 µM Ang II, as demonstrated by both RT-qPCR and
WB (Fig. 3F-H). These findings
underscore the key role of Ang II in regulating CF viability,
migration and expression of fibrosis-related markers, elucidating
its mechanism of action in cardiac fibrosis.
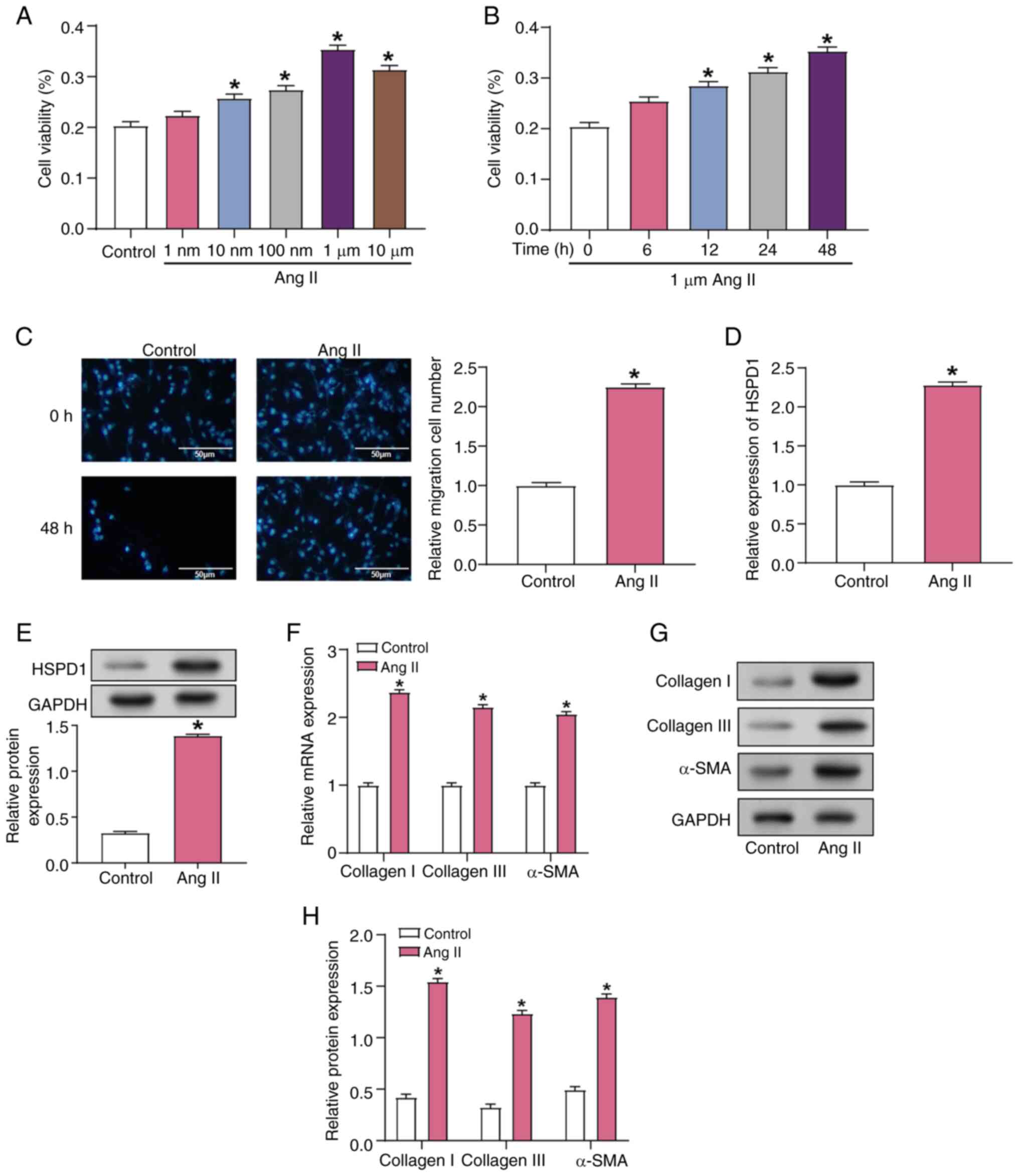 | Figure 3.Effects of Ang II induction on
fibrosis in CFs. (A) Viability of CFs treated with different
concentrations of Ang II for 24 h, measured using the Cell Counting
Kit-8 assay. *P<0.05 vs. the Control group. (B) Cell viability
of CFs treated with 1 µM Ang II for 0, 6, 12, 24, and 48 h.
*P<0.05 vs. the 1 µM Ang II for 0 h group. (C) Transwell assay
of CF migration after induction with 1 µM Ang II for 48 h. Scale
bar, 50 µm. *P<0.05 vs. the Control group. (D) mRNA and (E)
protein expression of HSPD1 in CF cells induced by 1 µM Ang
II for 48 h. *P<0.05 vs. the Control group. (F) mRNA expression
of collagen I, collagen III and α-SMA in CFs following induction
with 1 µM Ang II for 48 h. *P<0.05 vs. the Control group. (G)
Representative western blotting and (H) protein expression of
collagen I, collagen III and α-SMA in CFs following induction with
1 µM Ang II for 48 h. *P<0.05 vs. Control group Ang II,
angiotensin II; CF, cardiac fibroblast; HSPD1, heat shock protein
D1. |
Downregulation of HSPD1 alleviates the
impact of Ang II induction on CF cell function
RT-qPCR and WB verified knockdown efficiency of
HSPD1, demonstrating the knockdown efficiency of si-HSPD1-2
was more pronounced (Fig. 4A and
B). HSPD1 expression in CFs was significantly decreased after
transfection with si-HSPD1-2 compared with Ang II induction alone,
as evidenced by both RT-qPCR and WB (Fig. 4C and D). Furthermore, Ang II
induction resulted in a significant increase in CF viability,
whereas HSPD1 knockdown alleviated this increase (Fig. 4E). ELISA demonstrated that the
secretion of inflammatory factors (TNF-α, IL-8 and IL-1β)
significantly increased after induction by 1 µM Ang II, while HSPD1
knockdown attenuated this increase (Fig. 4F-H). Protein expression of
AF-associated markers (ANP, β-MHC and MMP-2) was significantly
upregulated following induction with 1 µM Ang II. However, HSPD1
knockdown attenuated Ang II-induced responses and decreased ANP,
β-MHC and MMP-2 levels, as observed by RT-qPCR and WB (Fig. 4I-L). This suggested that HSPD1
served a role in regulating CF cell viability, inflammatory
responses and AF-associated markers.
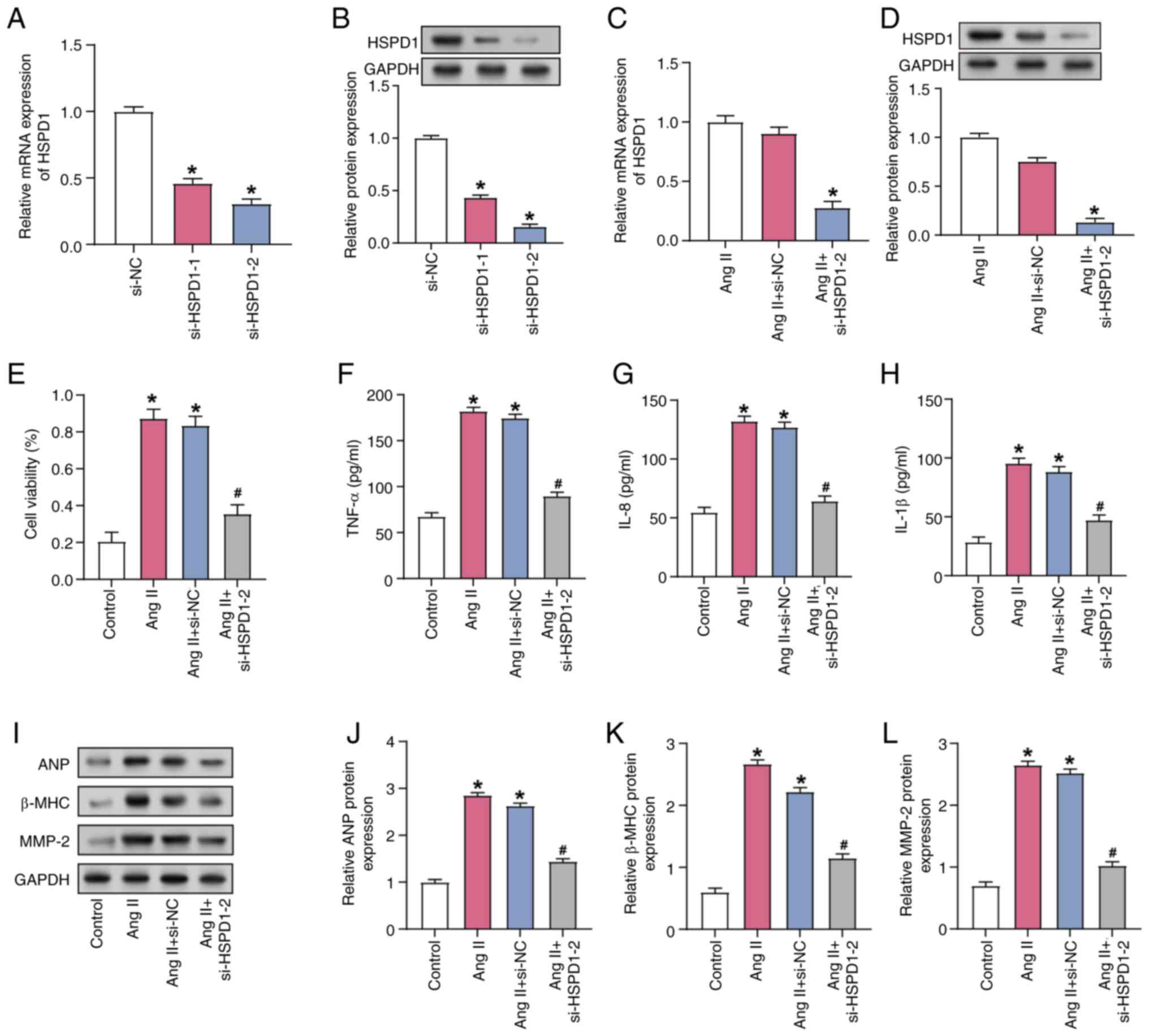 | Figure 4.Downregulation of HSPD1 improves Ang
II-induced AF. (A) mRNA and (B) protein expression of HSPD1 in
HSPD1-knockdown CFs. *P<0.05 vs. the si-NC group. (C)
mRNA and (D) protein expression of HSPD1 in CFs. *P<0.05 vs. the
Ang II+ si-NC group. (E) Cell viability of CFs was assessed by Cell
Counting Kit-8 assay. ELISA was used to detect the secretion of
inflammatory factors (F) TNF-α, (G) IL-8 and (H) IL-1β in CFs. (I)
Representative western blotting and protein expression of the
AF-associated markers (J) ANP, (K) β-MHC and (L) MMP-2 in CFs.
*P<0.05 vs. the Control group; #P<0.05 vs. the Ang
II+ si-NC group. AF, atrial fibrillation; Ang II, angiotensin II;
CF, cardiac fibroblast; si, small interfering; NC, negative
control; HSPD1, heat shock protein D1; ANP, atrial natriuretic
peptide; MHC, major histocompatibility complex. |
HSPD1 regulates Ang II-induced AF via
the PPAR signaling pathway
WB analysis revealed that HSPD1 knockdown
significantly decreased the expression of proteins involved in the
PPAR signaling pathway, including PPARα, SIRT3, CPT-1 and PPARγ, in
Ang II-induced CFs (Fig. 5A-E).
The cardioprotective effect of HSPD1 knockdown combined with
treatment with the PPAR activator TZD was investigated in Ang
II-induced CFs. Ang II-treated CFs exhibited increased viability,
which was significantly alleviated by siRNA-mediated knockdown of
HSPD1. TZD alleviated the effect of HSPD1 knockdown on Ang
II-induced CF (Fig. 5F). ELISA
demonstrated that HSPD1 knockdown led to a significant decrease in
the secretion of inflammatory factors TNF-α, IL-1β and IL-8 in Ang
II-induced CFs and addition of TZD prevented the increase of these
inflammatory factors (Fig. 5G-I).
HSPD1 knockdown significantly reduced the Ang II-induced protein
expression of AF-related markers ANP, β-MMP-2 and MHC; TZD
mitigated this reduction (Fig.
5J-M).
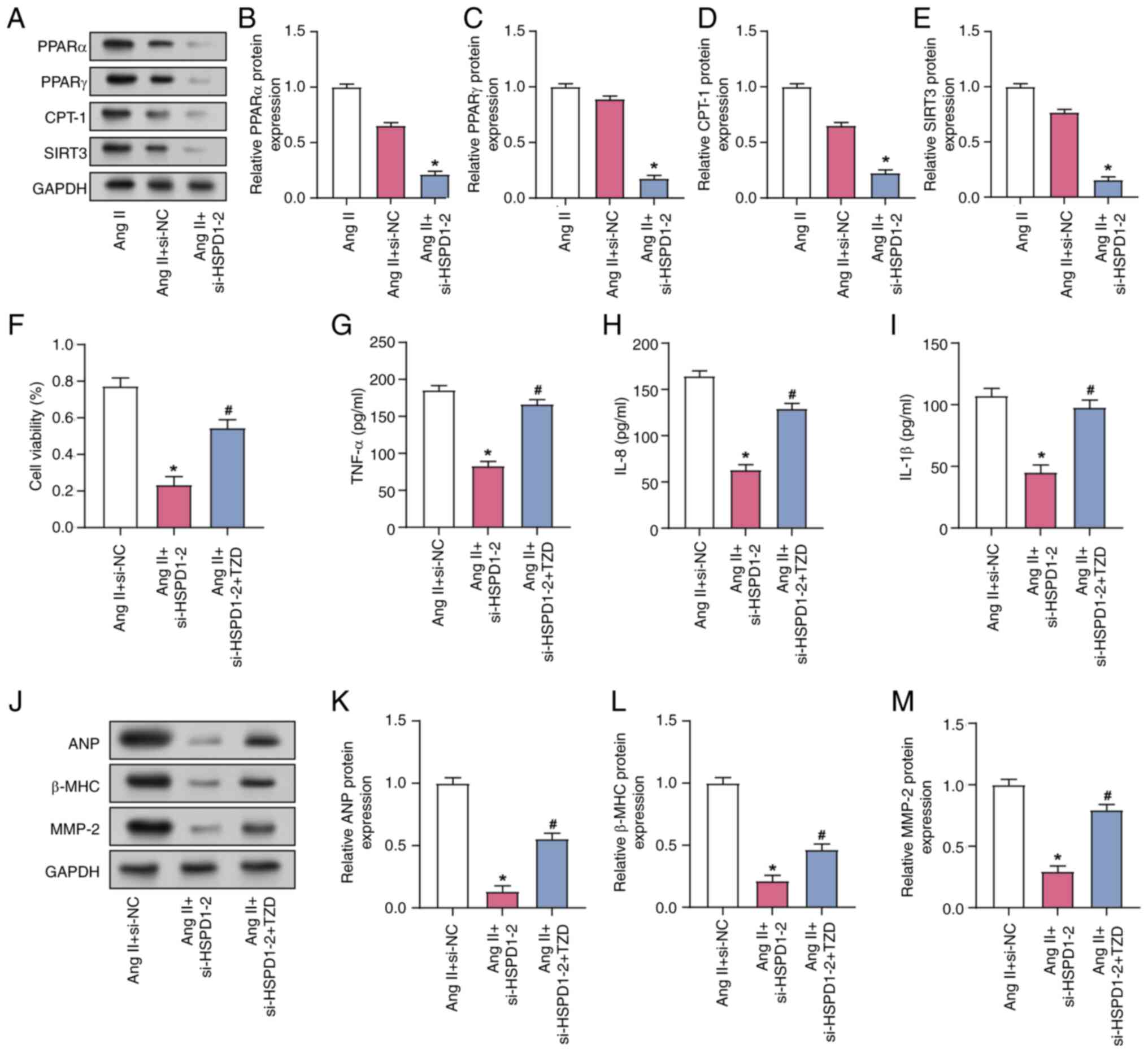 | Figure 5.Regulation of Ang II-induced AF by
HSPD1 via the PPAR signaling pathway. (A) Representative western
blotting and protein expression of PPAR signaling
pathway-associated proteins (B) PPARα, (C) PPARγ, (D) CPT-1 and (E)
SIRT3 in CFs. (F) Cell Counting Kit-8 assay of CF viability. ELISA
detection of the expression of the inflammatory factors (G) TNF-α,
(H) IL-8 and (I) IL-1β in CFs. (J) Representative western blotting
and protein expression of the AF markers (K) ANP, (L) β-MHC and (M)
MMP-2 in CFs. *P<0.05 vs. Ang II+ si-NC; #P<0.05
vs. the Ang II+si-HSPD1-2 group. AF, atrial fibrillation; TZD,
thiazolidinedione; CF, cardiac fibroblast; si, small interfering;
NC, negative control; PPAR, peroxisome proliferator-activated
receptor; CPT-1, carnitine palmitoyltransferase I; SIRT, sirtuin;
ANP, atrial natriuretic peptide; MHC, major histocompatibility
complex. |
Discussion
AF poses a challenge due to its increasing
incidence, morbidity and mortality rates (27). Traditional diagnostic methods,
including electrocardiography and echocardiography, aid in AF
diagnosis but exhibit limitations in detecting asymptomatic or
paroxysmal cases, underscoring the need for more sensitive
biomarkers (28). Treatment
strategies primarily focus on heart rate and rhythm control,
anticoagulation therapy and invasive procedures such as catheter
ablation (29). In the present
study, HSPD1 was a key target gene, belonging to the HSP60 family.
Zhou et al (30)
highlighted the role of HSPD1 in inducing regulatory T cells and
modulating immunoregulation during helminth infection. Yang et
al (31) demonstrated the role
of the HSP60 family, including HSPD1, in regulating proteostasis
and the mitochondrial unfolded protein response in hepatic
inflammation and fibrosis, influenced by microRNA (miR)-29a
therapy. Additionally, Fu et al (32) identified HSPD1 as a potential hub
gene in the PPAR signaling pathway, suggesting its involvement in
regulating myocardial changes associated with volume overload. The
aforementioned studies suggest that integrating biomarkers into
risk stratification models can enhance predictive accuracy and
guide treatment decisions, thus offering a promising avenue for
improving AF management.
Based on the bioinformatics analysis of the GSE31821
dataset and the subsequent identification of DEGs, the present
study investigated the functional enrichment of these genes and
demonstrated enrichment in pathways such as ‘MAPK signaling
pathway’ and ‘Wnt signaling pathway’. This aligns with the study by
Zheng et al (33), which
elucidated the role of sympathetic activation in
hyperthyroidism-induced AF. The aforementioned study demonstrated
that cardiomyocyte apoptosis, orchestrated via the p38 MAPK
signaling pathway, is a pivotal mechanism in this context. Wolke
et al (34) demonstrated
the centrality of the WNT signaling pathway in the intricate
landscape of AF. This pathway influences diverse facets of AF,
ranging from its developmental phases to the maintenance and
progression of arrhythmia. This understanding of the molecular
mechanisms involved in AF pathogenesis provides a foundation for
devising targeted interventions. Zhao et al (35) demonstrated the protective role of
diminished α7 nicotinic acetylcholine receptor expression in
amyloid-β-induced atrial remodeling within the context of AF and
Alzheimer's disease (AD); inhibition of mitochondrial oxidative
stress mediated by oxidation of CaMKII/mitogen-activated protein
kinase/activator protein 1 in atrial cells and mice with AD
suggests a potential therapeutic avenue for mitigating atrial
remodeling in the context of AF and neurodegenerative
disorders.
PPI network analysis of the DEGs in the GSE31821
dataset revealed five candidate genes in AF, namely HSP90AA1,
HSP90AB1, HSPA4, HSPA8 and HSPD1. Expression analysis in the
GSE31821 and GSE14975 datasets demonstrated that all five genes
were highly expressed in AF samples. In addition to HSPD1, four
other genes are associated with heart disease. Fang et al
(23) identified HSP90AA1 as a key
gene involved in the pathogenesis of AF by affecting the lipid
biosynthetic pathway. This highlights the role of HSP90AA1 in the
metabolic disorders that lead to the development of AF. Xiao et
al (24) demonstrated that
dipyridamole, a vasodilator with antiplatelet and antithrombotic
properties, can interact with HSP90AB1 and may serve as an AF
treatment target, but the specific mechanism is still unclear.
Okamoto et al (25)
demonstrated that HSPA8 acts as an accessory protein of chloride
voltage-gated channel 2 (CLCN2) in rat pulmonary vein (PV)
cardiomyocytes, altering the CLCN2 current dependence on chloride
concentration and voltage activation, which may affect the
hyperpolarization-activated chloride current associated with
spontaneous activity, leading to AF. HSPA4 serves a key role in
cardiac health by maintaining protein quality control and
preventing cardiac hypertrophic growth, and may contribute to
protection against doxorubicin-induced cardiotoxicity by enhancing
autophagy and protein stability (26). The aforementioned findings suggest
a role for HSPs in cardiac disease and are consistent with findings
of the present study, highlighting the complex molecular mechanisms
associated with AF and potential targets for therapeutic
intervention.
Ang II, a key hormone for blood pressure regulation
and cardiovascular homeostasis, serves a key role in cardiac
fibrosis. In pathological conditions such as hypertension, elevated
Ang II levels induce excessive collagen deposition and structural
alterations in atrial tissue, fostering fibrotic remodeling
associated with AF development and progression (36,37).
The activation of the renin-Ang-aldosterone system, resulting in
heightened Ang II, markedly contributes to adverse atrial
structural changes, establishing a key association between Ang II,
cardiac fibrosis and AF pathogenesis (38).
Collagen I and collagen III, as key structural
proteins, confer strength and support to connective tissue, which
is essential for tissue architecture and wound healing (39). α-SMA, involved in smooth muscle
cell and myofibroblast contraction, participates in tissue
remodeling (40). Elevated levels
of collagen I, α-SMA and collagen III signify heightened fibrotic
activity, impacting tissue structure and function. These proteins
have been studied in various pathological contexts, notably in
cardiovascular diseases such as AF, where fibrosis serves a key
role in disease progression (39,41).
Collagen I, α-SMA and collagen III serves key roles in atrial
fibrosis associated with AF, as demonstrated by Su et al
(42), who reported increased
protein levels in response to Ang II stimulation. The
aforementioned study also revealed that H2S treatment
effectively mitigates fibrosis marker protein expression, offering
a potential therapeutic avenue through miR-133a/connective tissue
growth factor axis modulation. Additionally, Li et al
(43) revealed that overexpression
of miR-10a in AF-induced rats notably increases collagen I, α-SMA
and collagen III expression, promoting cardiac fibrosis via the
TGF-β1/Smads signaling pathway, while miR-10a downregulation exerts
opposite effects. Findings of the present study demonstrated the
effect of Ang II-induced CF on cell fibrosis, including increased
production of fibrosis-related proteins (collagen I, α-SMA and
collagen III) and improved survival and migration of CFs. These
findings suggest that Ang II is a major factor in the development
of cardiac fibrotic alterations. Nakamura et al (44) reveals that cyclic compressive
loading activates the Ang II type 1 receptor and stimulates
hypertrophic differentiation of chondrocytes via a
G-protein-dependent pathway. This highlights the role of Ang II in
cartilage biology, demonstrating its ability to induce tissue
remodeling via receptor-mediated signaling. This is consistent with
the results of the present study, which demonstrated that Ang II
increases the production of fibrosis-associated proteins (collagen
I, α-SMA and collagen III), leading to enhanced fibrosis activity
and affecting tissue structure and function. The aforementioned and
present studies underscore the importance of Ang II signaling in
driving cellular responses and tissue changes, suggesting that
targeting the Ang II pathway may hold therapeutic potential in
multiple tissue contexts.
Inflammatory factors, including TNF-α, IL-8 and
IL-1β, are key mediators of the inflammatory response. IL-1β is a
proinflammatory cytokine that aids in immune responses, TNF-α is a
cytokine that causes systemic inflammation and IL-8 is a chemokine
that draws immune cells to the site of inflammation (45). These inflammatory factors are
typically associated with the pathogenesis and progression of AF.
AF biomarkers, such as ANP, β-MHC and MMP-2, provide insights into
the structural and functional changes occurring in the atria. ANP
is released in response to atrial stretch and is indicative of
atrial enlargement and pressure overload (46). β-MHC is a cardiac muscle protein
associated with cardiac remodeling; its upregulation may signify
pathological changes in the heart (47).
MMP-2, a matrix metalloproteinase, serves a role in
tissue remodeling and is implicated in cardiac fibrosis and
structural alterations associated with AF (48). Investigating these indicators aids
in comprehending the molecular processes and potential treatment
options in AF. Thijssen et al (49) revealed increased expression of
β-MHC and a downregulation of overall MHC expression during
sustained AF in goats, indicating cardiomyocyte de-differentiation
and suggesting an early response to AF may involve ischemic stress.
Yang et al (50)
demonstrated that atorvastatin treatment in a rabbit model of
pacing-induced AF effectively prevents atrial remodeling by
reducing levels of atrial myeloperoxidase, MMP-2 and MMP-9.
Additionally, the study by Xia et al (51) on hypertensive rats revealed
pronounced alterations in PV electrophysiology and histology,
characterized by decreased expression of voltage-gated sodium
channel subunit Nav1.5 and Kir (Inward rectifyimg potassium
channel) 2.1, increased interstitial fibrosis and elevated levels
of fibrosis markers (TGF-β1, MMP-2 and collagen I). These findings
underscore the potential mechanistic association between altered
gene expression, atrial remodeling and PV changes in AF. The
present study demonstrated that HSPD1 downregulation mitigates the
pathological upregulation of AF-associated biomarkers and
inflammatory mediators in Ang II-stimulated CFs, suggesting that
modulating HSPD1 expression may be a promising therapeutic strategy
to counteract the inflammatory and fibrotic processes in AF. Based
on the widespread effects of Ang II on multiple types of tissue
(21), it is plausible that HSPD1
may be involved in other types of cardiovascular tissue, such as
the vasculature, where it could contribute to vascular remodeling
and hypertension. HSPD1 has been implicated in metabolic disorders,
including obesity and diabetes, where it contributes to
mitochondrial dysfunction and insulin resistance (52,53).
These conditions are typically associated with systemic
inflammation and cardiovascular complications, suggesting that
HSPD1 may have broader systemic effects (54). Future research should explore
expression of HSPD1 and function in multiple types of tissue and
disease model to elucidate its systemic role and potential as a
therapeutic target.
A key function of the PPAR signaling pathway is in
regulating heart disease, affecting lipid metabolism, inflammation
and energy homeostasis in the heart (55). Studies have highlighted the
involvement of key proteins such as PPARα and PPARγ in regulating
cardiac function, with PPARα activation associated with improved
lipid utilization and decreased cardiac remodeling (10,56).
Conversely, the role of PPARγ in glucose metabolism and
inflammation underscores its therapeutic potential in diabetic
cardiomyopathy (57). CPT-1 is
regulated by PPARα and is key for mitochondrial fatty acid
oxidation, a key energy source for the heart (58). Furthermore, SIRT3 is affected by
PPARγ, involved in mitochondrial function and is associated with
cardiomyocyte protection against oxidative stress (59). Research on these proteins
highlights their potential as therapeutic targets for heart disease
(60,61), emphasizing the importance of the
PPAR signaling pathway in cardiovascular health. The present study
revealed that HSPD1 knockdown significantly decreased the
expression of PPAR signaling pathway-associated proteins in Ang
II-induced CFs. After HSPD1 knockdown, cell viability was
significantly decreased after Ang II treatment, whereas secretion
of inflammatory factors and levels of AF-related marker proteins
decreased. Application of the PPAR pathway activator TZD modulated
these effects, suggesting a regulatory interaction between HSPD1
and PPAR signaling pathways in mediating Ang II-induced AF. This
enhances the understanding of the role of HSPD1 in AF
pathophysiology and its potential as a target for therapeutic
intervention.
HSPD1 (also known as HSP60) is a mitochondrial
chaperonin involved in protein folding and maintaining cellular
homeostasis under stress (62).
While the present study investigated AF, the role of HSPD1 in CFs
and inflammation may also be relevant to hypertension. Hypertension
induces oxidative stress and mitochondrial dysfunction (63), which may lead to upregulated HSPD1
expression as a compensatory mechanism to protect cells.
Inflammation is a key contributor to hypertension (64). The present study demonstrated that
HSPD1 knockdown decreased the secretion of inflammatory cytokines
(for example, TNF-α, IL-8 and IL-1β) in Ang II-stimulated CFs. Ang
II induces inflammation and fibrosis, both central to
hypertension-associated complications (65). Thus, HSPD1 may promote hypertension
by regulating inflammation in cardiac and vascular tissue.
Hypertension is also associated with vascular fibrosis and
remodeling, which increase vascular stiffness and resistance
(36). The present study
demonstrated that HSPD1 upregulates fibrosis-associated markers
(for example, collagen I and III and α-SMA) in Ang II-induced CFs,
suggesting its role in hypertension through promoting fibrosis and
remodeling. Additionally, HSPD1 regulates the PPAR signaling
pathway, which is involved in lipid metabolism, inflammation and
energy homeostasis. PPAR signaling is implicated in hypertension,
with activators showing potential benefits in decreasing blood
pressure and vascular inflammation (9,58).
The downregulation of PPAR signaling proteins (PPARα, PPARγ, CPT-1
and SIRT3) following HSPD1 knockdown indicates that HSPD1 may
modulate hypertension via this pathway. In conclusion, the findings
of the present study highlight the potential role of HSPD1 in
hypertension pathogenesis. Future studies should explore HSPD1 as a
therapeutic target for hypertension by modulating mitochondrial
stress and enhancing PPAR pathway activity.
The present study identifying HSPD1 as a novel
therapeutic target for AF and potentially other types of
cardiovascular disease. By elucidating the role of HSPD1 in
mediating fibrosis, inflammation and PPAR signaling, the findings
of the present study provide novel insights into the pathogenesis
of AF and suggest that targeting HSPD1 may mitigate key
pathological processes such as fibrosis and inflammation.
Additionally, the involvement of HSPD1 in PPAR signaling highlights
its potential for broader cardiovascular applications, including
hypertension management. These results provide the groundwork for
future clinical research and development of targeted therapies
aimed at improving patient outcomes in AF and associated
conditions.
The present study has limitations. Although the
experiments reveal significant findings related to fibrosis,
inflammation and the expression of AF-associated proteins, they do
not fully capture the complexity of in vivo systems. The
absence of animal models limits the direct translation of the
findings to a physiological context. Future work should include
animal models to elucidate the role of HSPD1 in AF and to validate
the therapeutic potential of targeting the PPAR signaling
pathway.
In summary, the present study investigated the
regulatory role of HSPD1 in Ang II-induced AF and explored its
underlying mechanisms. The bioinformatics analysis revealed that
the expression of HSPD1 was significantly upregulated in AF
samples. In vitro assays demonstrated the important role of
HSPD1 in mediating the pathophysiological effects of Ang II on CFs,
emphasizing its impact on cell viability, inflammatory response and
expression of fibrosis-associated markers. Downregulation of HSPD1
attenuated Ang II-induced upregulation of key AF markers, thereby
attenuating fibrotic and inflammatory responses. Furthermore, the
present study demonstrated the regulatory role of HSPD1 on PPAR
signaling in the context of Ang II-induced AF, further elucidating
potential therapeutic avenues for AF management by integrating PPAR
signaling pathway activation via TZD. These insights contribute to
understanding the molecular complexity of AF development and
highlight HSPD1 as a potential therapeutic target.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shanghai Municipal Health
Commission (grant no. 202340055).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YZ, ZZ, WZ and NX conceived and designed the study.
ZZ, WZ and JW collected data and contributed to data management.
YZ, QG, ZZ, LZ, JW and NC analyzed and interpreted data. SZ
interpreted data. YZ and SZ wrote the manuscript. ZZ and JW
critically revised the manuscript for important intellectual
content. YZ and ZZ confirm the authenticity of all the raw data.
All authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Czabanski R, Horoba K, Wrobel J, Matonia
A, Martinek R, Kupka T, Jezewski M, Kahankova R, Jezewski J and
Leski JM: Detection of atrial fibrillation episodes in long-term
heart rhythm signals using a support vector machine. Sensors
(Basel). 20:7652020. View Article : Google Scholar
|
|
2
|
Bavishi A and Patel RB: Addressing
comorbidities in heart failure: Hypertension, atrial fibrillation,
and diabetes. Heart Fail Clin. 16:441–456. 2020. View Article : Google Scholar
|
|
3
|
Middeldorp ME, Ariyaratnam J, Lau D and
Sanders P: Lifestyle modifications for treatment of atrial
fibrillation. Heart. 106:325–332. 2020. View Article : Google Scholar
|
|
4
|
Carlisle MA, Fudim M, DeVore AD and
Piccini JP: Heart failure and atrial fibrillation, like fire and
fury. JACC Heart Fail. 7:447–456. 2019. View Article : Google Scholar
|
|
5
|
Karakasis P, Theofilis P, Vlachakis PK,
Korantzopoulos P, Patoulias D, Antoniadis AP and Fragakis N: Atrial
fibrosis in atrial fibrillation: Mechanistic insights, diagnostic
challenges, and emerging therapeutic targets. Int J Mol Sci.
26:2092024. View Article : Google Scholar
|
|
6
|
Huang J, Wu B, Qin P, Cheng Y, Zhang Z and
Chen Y: Research on atrial fibrillation mechanisms and prediction
of therapeutic prospects: Focus on the autonomic nervous system
upstream pathways. Front Cardiovasc Med. 10:12704522023. View Article : Google Scholar
|
|
7
|
Korbecki J, Bobiński R and Dutka M:
Self-regulation of the inflammatory response by peroxisome
proliferator-activated receptors. Inflamm Res. 68:443–458. 2019.
View Article : Google Scholar
|
|
8
|
Qiu YY, Zhang J, Zeng FY and Zhu YZ: Roles
of the peroxisome proliferator-activated receptors (PPARs) in the
pathogenesis of nonalcoholic fatty liver disease (NAFLD). Pharmacol
Res. 192:1067862023. View Article : Google Scholar
|
|
9
|
Tahri-Joutey M, Andreoletti P, Surapureddi
S, Nasser B, Cherkaoui-Malki M and Latruffe N: Mechanisms mediating
the regulation of peroxisomal fatty acid beta-oxidation by PPARα.
Int J Mol Sci. 22:89692021. View Article : Google Scholar
|
|
10
|
Montaigne D, Butruille L and Staels B:
PPAR control of metabolism and cardiovascular functions. Nat Rev
Cardiol. 18:809–823. 2021. View Article : Google Scholar
|
|
11
|
Recinella L, Orlando G, Ferrante C,
Chiavaroli A, Brunetti L and Leone S: Adipokines: New potential
therapeutic target for obesity and metabolic, rheumatic, and
cardiovascular diseases. Front Physiol. 11:5789662020. View Article : Google Scholar
|
|
12
|
Zhu X, Zhang X, Cong X, Zhu L and Ning Z:
ANGPTL4 attenuates Ang II-induced atrial fibrillation and fibrosis
in mice via PPAR pathway. Cardiol Res Pract. 2021:99353102021.
View Article : Google Scholar
|
|
13
|
Zheng P, Zhang W, Wang J, Gong Q, Xu N and
Chen N: Bioinformatics and functional experiments reveal that MRC2
inhibits atrial fibrillation via the PPAR signaling pathway. J
Thorac Dis. 15:5625–5639. 2023. View Article : Google Scholar
|
|
14
|
Xu D, Murakoshi N, Igarashi M, Hirayama A,
Ito Y, Seo Y, Tada H and Aonuma K: PPAR-γ activator pioglitazone
prevents age-related atrial fibrillation susceptibility by
improving antioxidant capacity and reducing apoptosis in a rat
model. J Cardiovasc Electrophysiol. 23:209–217. 2021. View Article : Google Scholar
|
|
15
|
Enomoto H, Mittal N, Inomata T, Arimura T,
Izumi T, Kimura A, Fukuda K and Makino S: Dilated
cardiomyopathy-linked heat shock protein family D member 1
mutations cause up-regulation of reactive oxygen species and
autophagy through mitochondrial dysfunction. Cardiovasc Res.
117:1118–1131. 2021. View Article : Google Scholar
|
|
16
|
Wachoski-Dark E, Zhao T, Khan A, Shutt TE
and Greenway SC: Mitochondrial protein homeostasis and
cardiomyopathy. Int J Mol Sci. 23:33532022. View Article : Google Scholar
|
|
17
|
Khafaga AF, Noreldin AE and Saadeldin IM:
Role of HSP in the pathogenesis of age-related inflammatory
diseases. Heat Shock Proteins in Inflammatory Diseases. Asea AAA
and Kaur P: Volume 22. Springer; Cham: pp. 341–371. 2020,
View Article : Google Scholar
|
|
18
|
van Marion DMS, Ramos KS, Lanters EAH,
Bulte LB, Bogers AJJC, de Groot NMS and Brundel BJJM: Atrial heat
shock protein levels are associated with early postoperative and
persistence of atrial fibrillation. Heart Rhythm. 18:1790–1798.
2021. View Article : Google Scholar
|
|
19
|
Oc M, Ucar HI, Pinar A, Akbulut B, Oc B,
Akyon Y, Kanbak M and Dogan R: Heat shock protein70: A new marker
for subsequent atrial fibrillation development? Artif Organs.
32:846–850. 2008. View Article : Google Scholar
|
|
20
|
Meijering RAM, Zhang D, Hoogstra-Berends
F, Henning R and Brundel BJJM: Loss of proteostatic control as a
substrate for atrial fibrillation: A novel target for upstream
therapy by heat shock proteins. Front Physiol. 3:362012. View Article : Google Scholar
|
|
21
|
Nehme A, Zouein FA, Zayeri ZD and Zibara
K: An update on the tissue renin angiotensin system and its role in
physiology and pathology. J Cardiovasc Dev Dis. 6:142019.
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Boil. 4:P32003.
View Article : Google Scholar
|
|
23
|
Fang Y, Liu L and Wang H: The four key
genes participated in and maintained atrial fibrillation process
via reprogramming lipid metabolism in AF patients. Front Genet.
13:8217542022. View Article : Google Scholar
|
|
24
|
Xiao S, Zhou Y, Liu Q, Zhang T and Pan D:
Identification of pivotal MicroRNAs and target genes associated
with persistent atrial fibrillation based on bioinformatics
analysis. Comput Math Methods Med. 2021:66802112021. View Article : Google Scholar
|
|
25
|
Okamoto Y, Nagasawa Y, Obara Y, Ishii K,
Takagi D and Ono K: Molecular identification of HSPA8 as an
accessory protein of a hyperpolarization-activated chloride channel
from rat pulmonary vein cardiomyocytes. J Biol Chem.
294:16049–16061. 2019. View Article : Google Scholar
|
|
26
|
Marques Rodrigues D: The role of heat
shock protein A4 (HSPA4) in the heart. eDiss. 2023.
|
|
27
|
Rowin EJ, Link MS, Maron MS and Maron BJ:
Evolving contemporary management of atrial fibrillation in
hypertrophic cardiomyopathy. Circulation. 148:1797–1811. 2023.
View Article : Google Scholar
|
|
28
|
Hesselkilde EZ, Carstensen H, Flethøj M,
Fenner M, Kruse DD, Sattler SM, Tfelt-Hansen J, Pehrson S,
Braunstein TH, Carlson J, et al: Longitudinal study of electrical,
functional and structural remodelling in an equine model of atrial
fibrillation. BMC Cardiovasc Disord. 19:2282019. View Article : Google Scholar
|
|
29
|
Velleca M, Costa G, Goldstein LJ, Bishara
M, Ming Boo L and Sha Q: Management of atrial fibrillation in
Europe: Current care pathways and the clinical impact of
antiarrhythmic drugs and catheter ablation. EMJ Cardiol. 7:98–109.
2019. View Article : Google Scholar
|
|
30
|
Zhou S, Jin X, Chen X, Zhu J, Xu Z, Wang
X, Liu F, Hu W, Zhou L and Su C: Heat shock protein 60 in eggs
specifically induces Tregs and reduces liver immunopathology in
mice with Schistosomiasis japonica. PLoS One. 10:e01391332015.
View Article : Google Scholar
|
|
31
|
Yang YL, Wang PW, Wang FS, Lin HY and
Huang YH: miR-29a modulates GSK3β/SIRT1-linked mitochondrial
proteostatic stress to ameliorate mouse non-alcoholic
steatohepatitis. Int J Mol Sci. 21:68842020. View Article : Google Scholar
|
|
32
|
Fu Y, Zhao D, Zhou Y, Lu J, Kang L, Jiang
X, Xu R, Ding Z and Zou Y: Identification of differential
expression genes between volume and pressure overloaded hearts
based on bioinformatics analysis. Genes (Basel). 13:12762022.
View Article : Google Scholar
|
|
33
|
Zheng J, Zhao S, Yang Q, Wei Y, Li J and
Guo T: Sympathetic activation promotes cardiomyocyte apoptosis in a
rabbit susceptibility model of hyperthyroidism-induced atrial
fibrillation via the p38 MAPK signaling pathway. Crit Rev Eukaryot
Gene Expr. 33:17–27. 2023. View Article : Google Scholar
|
|
34
|
Wolke C, Antileo E and Lendeckel U: WNT
signaling in atrial fibrillation. Exp Biol Med (Maywood).
246:1112–1120. 2021. View Article : Google Scholar
|
|
35
|
Zhao J, Yu L, Xue X, Xu Y, Huang T, Xu D,
Wang Z, Luo L and Wang H: Diminished α7 nicotinic acetylcholine
receptor (α7nAChR) rescues amyloid-β induced atrial remodeling by
oxi-CaMKII/MAPK/AP-1 axis-mediated mitochondrial oxidative stress.
Redox Biol. 59:1025942023. View Article : Google Scholar
|
|
36
|
Zeng X and Yang Y: Molecular mechanisms
underlying vascular remodeling in hypertension. Rev Cardiovasc Med.
25:722024. View Article : Google Scholar
|
|
37
|
Sygitowicz G, Maciejak-Jastrzębska A and
Sitkiewicz D: A review of the molecular mechanisms underlying
cardiac fibrosis and atrial fibrillation. J Clin Med. 10:44302021.
View Article : Google Scholar
|
|
38
|
Mirabito Colafella KM, Bovée DM and Danser
AHJ: The renin-angiotensin-aldosterone system and its therapeutic
targets. Exp Eye Res. 186:1076802019. View Article : Google Scholar
|
|
39
|
Singh D, Rai V and Agrawal DK: Regulation
of collagen I and collagen III in tissue injury and regeneration.
Cardiol Cardiovasc Med. 7:5–16. 2023. View Article : Google Scholar
|
|
40
|
Hinz B, McCulloch CA and Coelho NM:
Mechanical regulation of myofibroblast phenoconversion and collagen
contraction. Exp Cell Res. 379:119–128. 2019. View Article : Google Scholar
|
|
41
|
Ma J, Chen Q and Ma S: Left atrial
fibrosis in atrial fibrillation: Mechanisms, clinical evaluation
and management. J Cell Mol Med. 25:2764–2775. 2021. View Article : Google Scholar
|
|
42
|
Su H, Su H, Liu CH, Hu HJ, Zhao JB, Zou T
and Tang YX: H2S inhibits atrial fibrillation-induced
atrial fibrosis through miR-133a/CTGF axis. Cytokine.
146:1555572021. View Article : Google Scholar
|
|
43
|
Li PF, He RH, Shi SB, Li R, Wang QT, Rao
GT and Yang B: Modulation of miR-10a-mediated TGF-β1/Smads
signaling affects atrial fibrillation-induced cardiac fibrosis and
cardiac fibroblast proliferation. Biosci Rep. 39:BSR201819312019.
View Article : Google Scholar
|
|
44
|
Nakamura F, Tsukamoto I, Inoue S,
Hashimoto K and Akagi M: Cyclic compressive loading activates
angiotensin II type 1 receptor in articular chondrocytes and
stimulates hypertrophic differentiation through a
G-protein-dependent pathway. FEBS Open Bio. 8:962–973. 2018.
View Article : Google Scholar
|
|
45
|
Soare AY, Durham ND, Gopal R, Tweel B,
Hoffman KW, Brown JA, O'Brien M, Bhardwaj N, Lim JK, Chen BK and
Swartz TH: P2X antagonists inhibit HIV-1 productive infection and
inflammatory cytokines interleukin-10 (IL-10) and IL-1β in a human
tonsil explant model. J Virol. 93:e01186–18. 2018.
|
|
46
|
Rao S, Pena C, Shurmur S and Nugent K:
Atrial natriuretic peptide: Structure, function, and physiological
effects: A narrative review. Curr Cardiol Rev.
17:e0511211910032021. View Article : Google Scholar
|
|
47
|
Bai L, Zhao Y, Zhao L, Zhang M, Cai Z,
Yung KKL, Dong C and Li R: Ambient air PM2.5 exposure
induces heart injury and cardiac hypertrophy in rats through
regulation of miR-208a/b, α/β-MHC, and GATA4. Environ Toxicol
Pharmacol. 85:1036532021. View Article : Google Scholar
|
|
48
|
Henriet P and Emonard H: Matrix
metalloproteinase-2: Not (just) a ‘hero’ of the past. Biochimie.
166:223–232. 2019. View Article : Google Scholar
|
|
49
|
Thijssen VLJL, van der Velden HMW, van
Ankeren EP, Ausma J, Allessie MA, Borgers M, van Eys GJJM and
Jongsma HJ: Analysis of altered gene expression during sustained
atrial fibrillation in the goat. Cardiovasc Res. 54:427–437. 2002.
View Article : Google Scholar
|
|
50
|
Yang Q, Qi X, Dang Y, Li Y, Song X and Hao
X: Effects of atorvastatin on atrial remodeling in a rabbit model
of atrial fibrillation produced by rapid atrial pacing. BMC
Cardiovasc Disord. 16:1422016. View Article : Google Scholar
|
|
51
|
Xia PP, Li LJ, Qi RD, Shi JJ, Ju WZ and
Chen ML: Electrical and histological remodeling of the pulmonary
vein in 2K1C hypertensive rats: Indication of initiation and
maintenance of atrial fibrillation. Anatol J Cardiol. 19:169–175.
2018.
|
|
52
|
Hauffe R, Rath M, Schell M, Ritter K,
Kappert K, Deubel S, Ott C, Jähnert M, Jonas W, Schürmann A and
Kleinridders A: HSP60 reduction protects against diet-induced
obesity by modulating energy metabolism in adipose tissue. Mol
Metab. 53:1012762021. View Article : Google Scholar
|
|
53
|
Kleinridders A, Lauritzen HPMM, Ussar S,
Christensen JH, Mori MA, Bross P and Kahn CR: Leptin regulation of
Hsp60 impacts hypothalamic insulin signaling. J Clin Invest.
123:4667–4680. 2013. View Article : Google Scholar
|
|
54
|
Timofeev YS, Kiselev AR, Dzhioeva ON and
Drapkina OM: Heat shock proteins (HSPs) and cardiovascular
complications of obesity: Searching for potential biomarkers. Curr
Issues Mol Biol. 45:9378–9389. 2023. View Article : Google Scholar
|
|
55
|
Bougarne N, Weyers B, Desmet SJ, Deckers
J, Ray DW, Staels B and De Bosscher K: Molecular actions of PPARα
in lipid metabolism and inflammation. Endocr Rev. 39:760–802. 2018.
View Article : Google Scholar
|
|
56
|
Wang X, Zhu XX, Jiao SY, Qi D, Yu BQ, Xie
GM, Liu Y, Song YT, Xu Q, Xu QB, et al: Cardiomyocyte peroxisome
proliferator-activated receptor α is essential for energy
metabolism and extracellular matrix homeostasis during pressure
overload-induced cardiac remodeling. Acta Pharmacol Sin.
43:1231–1242. 2022. View Article : Google Scholar
|
|
57
|
Song F, Mao YJ, Hu Y, Zhao SS, Wang R, Wu
WY, Li GR, Wang Y and Li G: Acacetin attenuates diabetes-induced
cardiomyopathy by inhibiting oxidative stress and energy metabolism
via PPAR-α/AMPK pathway. Eur J Pharmacol. 922:1749162022.
View Article : Google Scholar
|
|
58
|
Liu X, Xu X, Zhang T, Xu L, Tao H, Liu Y,
Zhang Y and Meng X: Fatty acid metabolism disorders and potential
therapeutic traditional Chinese medicines in cardiovascular
diseases. Phytother Res. 37:4976–4998. 2023. View Article : Google Scholar
|
|
59
|
Zhang J, Ren D, Fedorova J, He Z and Li J:
SIRT1/SIRT3 modulates redox homeostasis during ischemia/reperfusion
in the aging heart. Antioxidants (Basel). 9:8582020. View Article : Google Scholar
|
|
60
|
Kvandová M, Majzúnová M and Dovinová I:
The role of PPARgamma in cardiovascular diseases. Physiol Res. 65
(Suppl 3):S343–S363. 2016. View Article : Google Scholar
|
|
61
|
Metzger JM, Matsoff HN, Zinnen AD,
Fleddermann RA, Bondarenko V, Simmons HA, Mejia A, Moore CF and
Emborg ME: Post mortem evaluation of inflammation, oxidative
stress, and PPARγ activation in a nonhuman primate model of cardiac
sympathetic neurodegeneration. PLoS One. 15:e02269992020.
View Article : Google Scholar
|
|
62
|
Singh MK, Shin Y, Han S, Ha J, Tiwari PK,
Kim SS and Kang I: Molecular chaperonin HSP60: Current
understanding and future prospects. Int J Mol Sci. 25:54832024.
View Article : Google Scholar
|
|
63
|
Pokharel MD, Marciano DP, Fu P, Franco MC,
Unwalla H, Tieu K, Fineman JR, Wang T and Black SM: Metabolic
reprogramming, oxidative stress, and pulmonary hypertension. Redox
Biol. 64:1027972023. View Article : Google Scholar
|
|
64
|
Navaneethabalakrishnan S, Smith HL, Arenaz
CM, Goodlett BL, McDermott JG and Mitchell BM: Update on immune
mechanisms in hypertension. Am J Hypertens. 35:842–851. 2022.
View Article : Google Scholar
|
|
65
|
Escobales N, Nuñez RE and Javadov S:
Mitochondrial angiotensin receptors and cardioprotective pathways.
Am J Physiol Heart Circ Physiol. 316:H1426–H438. 2019. View Article : Google Scholar
|