Introduction
As the core regulatory mechanism of immune
homeostasis, immune checkpoints prevent autoimmune damage by
regulating T-cell activity (1).
However, in the tumor microenvironment, tumor cells evade immune
surveillance by overexpressing PD-L1, which engages PD-1 on
activated T cells, leading to functional suppression and T cell
exhaustion (2); this discovery has
driven the clinical translation of immune checkpoint inhibitors
(ICIs). As monoclonal antibody drugs, ICIs reactivate T
cell-mediated antitumor effects by blocking inhibitory signaling
pathways (3). Since the approval
of the first CTLA-4 inhibitor by the US Food and Drug
Administration (FDA) in 2011, PD-1/PD-L1 inhibitors have expanded
to the treatment of over a dozen solid tumors, including melanoma
and non-small cell lung cancer, markedly improving patient survival
rates (4). Notably, the 5-year
survival rate for advanced melanoma increased from 15% with
traditional therapies to 50% with the use of ipilimumab monotherapy
or combination regimens involving PD-1 inhibitors such as nivolumab
(5). However, alongside the
therapeutic breakthroughs, immune-related adverse events (irAEs)
have emerged as a new clinical challenge. Among these AEs,
cardiovascular toxicity, particularly ICI-related myocarditis, has
a high mortality rate of 46%, although it has an incidence of
<2% (6). The use of combination
therapy further increases the risk (3–5);
therefore, the early diagnosis and management of ICI-related
myocarditis have become critical issues in the field of cancer
immunotherapy (7–9).
Notably, the heart is considered a specific target
of immune attack (10); however,
the underlying reasons for the heightened susceptibility in
specific patient populations and the pathological mechanisms
driving ICI-related myocarditis remain incompletely elucidated. The
present review aimed to explore the immunopathological mechanisms
of ICI-related myocarditis. By summarizing the existing literature,
the review aimed to identify critical knowledge gaps in current
research and propose future directions, including the development
of novel therapeutic strategies and molecular targets. Ultimately,
this work aims to provide clinicians with actionable insights to
optimize the safety and efficacy of immunotherapy while advancing
prognostic outcomes for ICI-related myocarditis.
Epidemiological features of ICI-related
myocarditis
The development of ICI-related myocarditis is
closely associated with immune hyperactivation and therapeutic
mechanisms, with combination therapies posing a particularly
elevated risk. Epidemiological data indicate that monotherapy with
ICIs results in myocarditis incidence rates of 0.5, 2.4 and 3.3%
for anti-PD-1, anti-PD-L1 and anti-CTLA-4 inhibitors, respectively.
However, combining PD-1 and CTLA-4 inhibitors increases the
incidence to 2.4% (11), with
mortality rising sharply from 36% (monotherapy) to 67% (7,12).
Chinese cohort studies report a domestic ICI-related myocarditis
incidence of 1.05–1.06%, consistent with global epidemiological
trends (13,14). Identified high-risk factors include
diabetes mellitus, heart failure, a history of acute coronary
syndrome and advanced age (>80 years) (11,15).
Although the overall incidence remains <2%, the high fatality
rate renders ICI-related myocarditis a critical clinical challenge
in cancer immunotherapy.
ICI-related myocarditis typically occurs within
weeks of initiation, with a median onset of 34 days (IQR: 21–75
days), with timing and risk varying by cancer type (7,11).
Patients with melanoma, especially those receiving anti-PD-1/CTLA-4
combination therapy, predominantly develop early-onset myocarditis
(≤2 weeks) (11). Conversely,
patients with thymic epithelial tumors (TETs) more frequently
experience delayed-onset myocarditis (≥6 weeks), likely due to
thymic dysfunction and impaired central immune tolerance
predisposing to T cell-mediated autoimmunity (16). Patients with TETs also demonstrate
a higher incidence of severe irAEs, contributing to their high
myocarditis risk (17). Beyond
tumor-specific factors, host characteristics also modulate risk,
including advanced age (>64 years), elevated body mass index
(>28 kg/m2) and prior cardiovascular medication use
(15,18). These findings underscore the need
for tailored surveillance strategies integrating both
patient-specific and cancer-specific factors to enable early
detection and timely intervention.
Clinical manifestations, diagnosis and
differential diagnosis of ICI-related myocarditis
Clinical manifestations
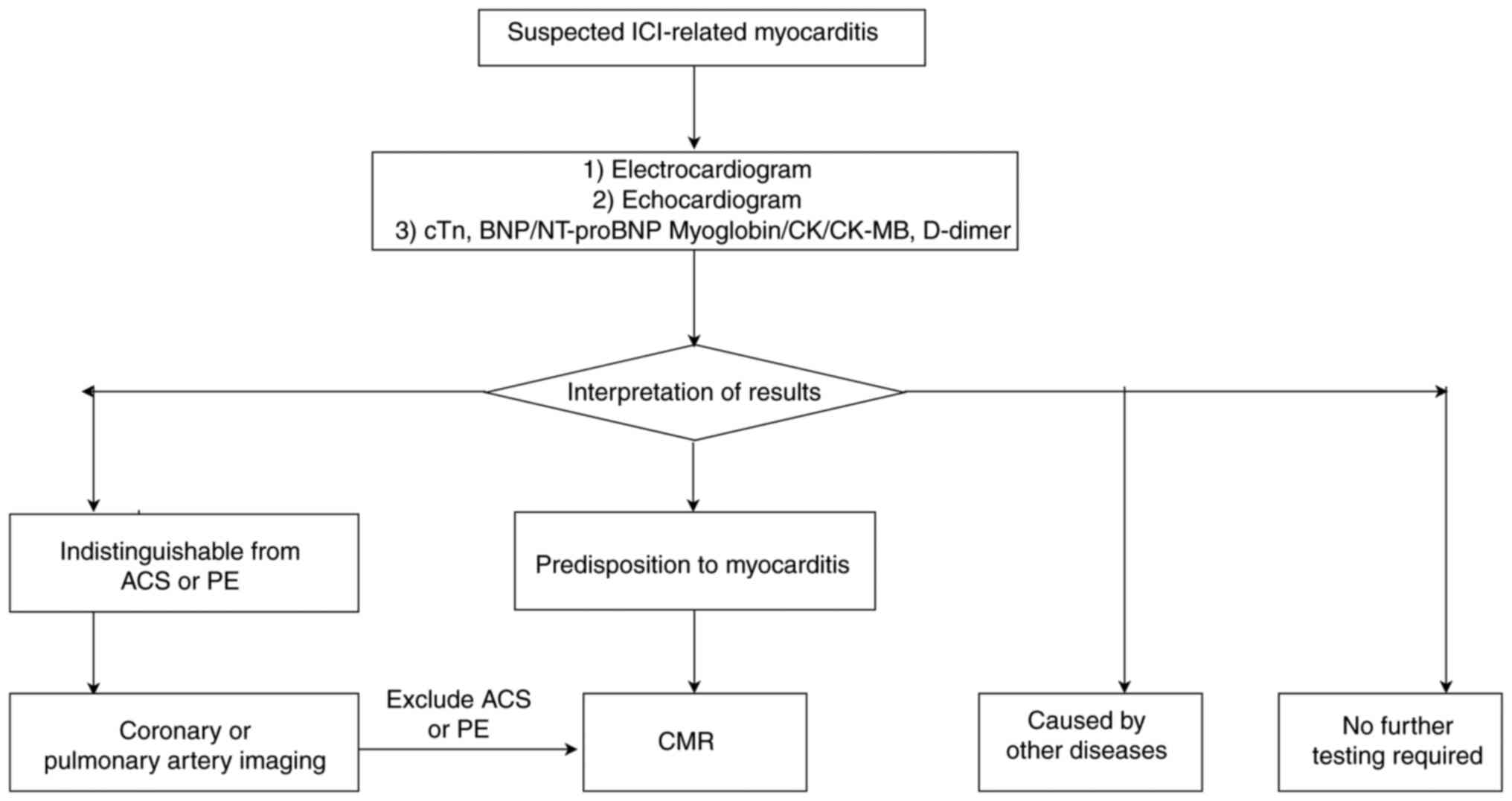
The diagnosis of ICI-related myocarditis requires
the integration of clinical manifestations, laboratory markers,
electrocardiogram findings and imaging features. Patients typically
present with chest pain, dyspnea, palpitations and fatigue. In
severe cases, the condition may progress to heart failure,
cardiogenic shock or cardiac arrest (19). Physical examination may reveal
hypotension, tachyarrhythmias, jugular vein distension or other
signs (cold, clammy skin, prolonged capillary refill and oliguria)
of hemodynamic compromise (9,19,20)
(Fig. 1).
Diagnosis
Laboratory and imaging profiles in ICI-related
myocarditis
The laboratory profile of ICI-related myocarditis is
typically characterized by elevated myocardial injury and cardiac
stress biomarkers. Multiple studies have demonstrated abnormal
elevation of cardiac troponin (cTn)T and cTnI in 46–94% of cases
(21,22). This also discovered a critical
threshold of cTnT ≥32 times the upper reference limit, which was
significantly associated with adverse myocardial toxicity events
(high-risk cohort), whereas levels below this threshold indicated a
better prognosis (low-risk group). However, it was revealed that
some patients may present without cTnI or creatine kinase (CK)
elevation (22). The prevalence of
elevated natriuretic peptides [B-type natriuretic peptide (BNP) and
N-terminal pro-BNP (NT-proBNP)] exhibits interstudy variability: A
large-scale study reported NT-proBNP elevation in 77.5% (n=185) of
cases (23), contrasting with 100%
positivity in a 14-patient cohort (22), suggesting that sensitivity may be
sample size-dependent. While myoglobin, CK-MB, aspartate
aminotransferase and lactate dehydrogenase often show nonspecific
elevation (24,25), cTns demonstrate superior
specificity over natriuretic peptides, which may be confounded by
non-inflammatory left ventricular dysfunction or acute cardiac
stress. This may lead to increased wall tension and neurohormonal
activation, elevating BNP/NT-proBNP levels even in the absence of
myocardial inflammation (23,26).
Although individual biomarkers lack diagnostic specificity, their
combined interpretation provides critical value in the evaluation
of ICI-related myocarditis.
Electrocardiogram (ECG) abnormalities are essential
diagnostic elements, detecting atrial fibrillation, ventricular
arrhythmias and conduction disturbances. Inflammatory involvement
of the cardiac conduction system may manifest as intraventricular
conduction delay, PR prolongation or complete heart block (27). Cardiac magnetic resonance (CMR)
notably enhances diagnostic accuracy through structural and
functional assessment, with particular sensitivity in quantifying
myocardial involvement (28).
Prospective studies have validated the T1 mapping technology of CMR
as achieving 92% sensitivity for myocardial edema detection via
extracellular volume and T1 value quantification, enabling early
identification of myocarditis (28,29).
Existing diagnostic criteria and
procedures
Diagnostic confirmation of ICI-related myocarditis
requires multimodal integration of clinical presentation, biomarker
profiles and imaging findings. Current international consensus
advocates the Bonaca diagnostic framework comprising four pillars
(30): i) Characteristic symptoms
(including chest pain and heart failure); ii) ECG abnormalities
(ST-T changes and conduction blocks); iii) elevated myocardial
injury markers (cTnT ≥14 ng/l or CK-MB ≥5 µg/l); iv) CMR hallmarks
(T2-weighted edema and late gadolinium enhancement). Salem et
al (31) refined this protocol
through a multidisciplinary team (MDT) model: Primary screening via
ECG with high-sensitivity cTn, followed by CMR evaluation for
suspected cases, culminating in MDT interpretation. While
endomyocardial biopsy remains the diagnostic gold standard, its
invasive nature and sampling variability restrict application to
diagnostically ambiguous cases (20).
Differential diagnosis
Core challenges in differential diagnosis
In clinical practice, establishing differential
diagnosis for ICI-related myocarditis presents multifaceted
challenges. First, the clinical manifestations demonstrate marked
heterogeneity and nonspecificity, ranging from asymptomatic
troponin elevation to fulminant heart failure or life-threatening
arrhythmia. These presentations show substantial overlap with viral
myocarditis and ischemic myocardial injury, while requiring
differentiation from irAEs, particularly neuromuscular
complications such as myositis and myasthenia gravis (32). Second, the coexistence of
preexisting cardiovascular comorbidities and treatment-related
complications in patients with cancer adds to the diagnostic
complexity (33).
Laboratory assessments are confounded by limited
specificity of conventional biomarkers (such as cTnT and CK), since
nonspecific factors including skeletal muscle injury and systemic
infections may alter their values. Furthermore, imaging modalities
currently lack sufficient discriminatory power to reliably
distinguish ICI-related myocarditis from other myocardial
pathologies (34,35). Collectively, these diagnostic
ambiguities underscore the critical need for developing novel
biomarker panels and advanced imaging techniques to improve
diagnostic precision.
Need for novel diagnostic tools and
technologies
Given the limitations of current diagnostic
methodologies, emerging technologies have expanded possibilities to
address these challenges. Liquid biopsy demonstrates distinct
advantages in early detection and disease stratification through
the capture of circulating biomarkers, including circulating tumor
cells and exosomes. These circulating components exhibit tissue
specificity while dynamically reflecting alterations in the cardiac
immune microenvironment, thereby providing molecular evidence for
differential diagnosis and therapeutic monitoring (36).
In artificial intelligence applications, machine
learning-based risk prediction models integrating clinical
characteristics, laboratory parameters and imaging biomarkers can
effectively identify populations at high risk of adverse cardiac
events. These models demonstrate superior predictive performance
compared with conventional scoring systems, offering clinicians a
robust foundation for early intervention strategies (37).
The synergistic application of multi-omics
technologies (including genomics, proteomics and metabolomics) has
further advanced the diagnostic paradigm for ICI-related
myocarditis. Single-cell sequencing of epicardial adipose tissue
has revealed that a CD8+ T cell to regulatory T cell
(Treg) ratio exceeding 2.5 serves as an early indicator of severe
myocarditis, enabling precision-guided immunosuppressive therapy
(38).
Mechanism
Physiological functions of immune
checkpoints and myocardial protection
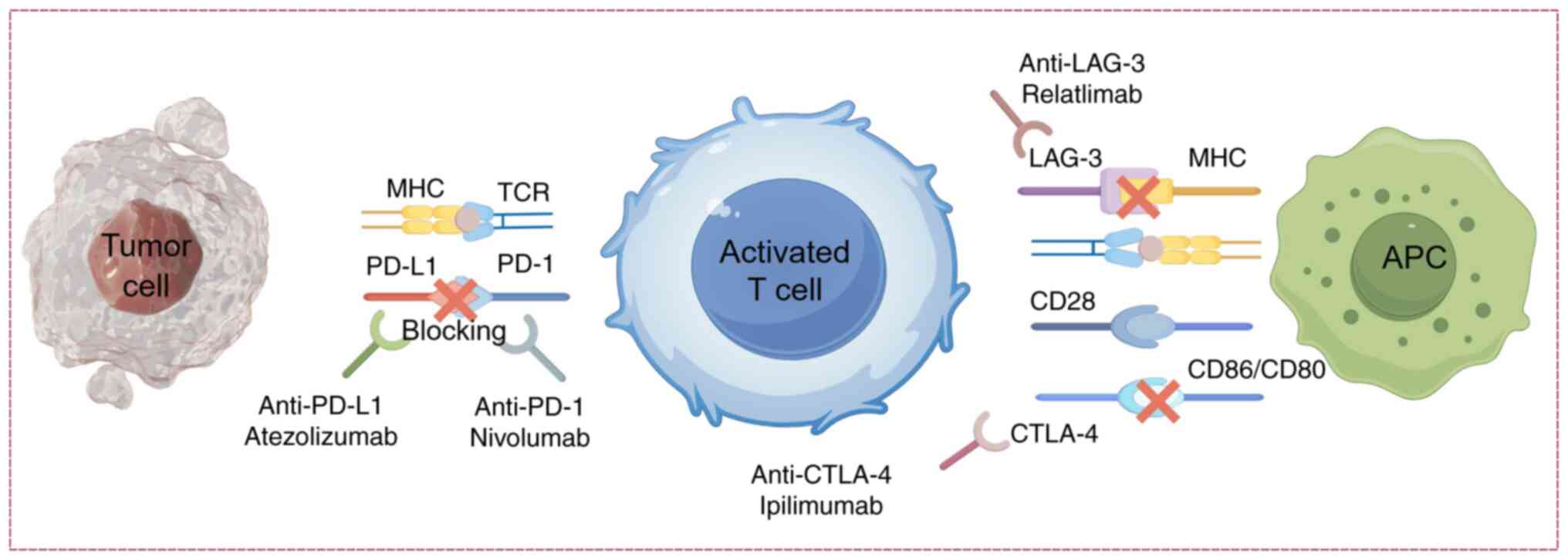
PD-1/PD-L1 signaling pathway
Immune checkpoints serve key roles in maintaining
immune homeostasis and regulating immune responses. Key
immunosuppressive receptors, including CTLA-4, PD-1/PD-L1 and
LAG-3, prevent immune system overactivation by modulating T-cell
activation thresholds (39). The
PD-1/PD-L1 axis exerts immunoregulatory functions through
ligand-receptor interactions. As a CD28 superfamily member, PD-1 is
ubiquitously expressed on activated T cells, B lymphocytes,
monocytes, natural killer (NK) cells and dendritic cells (DCs).
Upon PD-L1 engagement, PD-1 recruits phosphatases SHP-1/2 to
inhibit T-cell receptor (TCR) signaling, thereby maintaining
peripheral immune tolerance (40).
Constitutive PD-L1 expression on cardiomyocytes and endothelial
cells provides local protection against autoimmune damage by
simultaneously suppressing regional T-cell responses (41).
ICIs restore T cell-mediated antitumor activity
through blockade of these inhibitory signals, potentially
disrupting immune equilibrium. Anti-PD-1/PD-L1 monoclonal
antibodies competitively inhibit PD-1/PD-L1 interactions, reversing
T-cell functional suppression and activating downstream signaling
pathways (such as PI3K/AKT and RAS/MAPK) to amplify immune
responses (42,43). This mechanistic foundation has
enabled PD-1 inhibitors, such as pembrolizumab, to achieve
therapeutic breakthroughs across multiple solid malignancies,
including tumors with specific genomic profiles (such as non-small
cell lung cancer or black chromocele) (44).
However, immune checkpoint blockade may reduce
T-cell activation thresholds, permitting aberrant activation of
autoreactive T cells that mount immune attacks against cardiac
tissue, a central pathogenic mechanism underlying ICI-related
myocarditis (45). The dual nature
of therapeutic benefits and risks emphasizes the critical need for
deeper understanding of immune checkpoint physiology and pathology
(Fig. 2).
Immunoregulatory function of
CTLA-4
CTLA-4, as a key immune checkpoint molecule, serves
a critical role early in T-cell activation by competitively binding
to CD80/CD86 molecules on the surface of antigen-presenting cells
(APCs), antagonizing the CD28 co-stimulatory signal, and thus
inhibiting T-cell proliferation and activation to maintain immune
homeostasis (46,47) (Fig.
2).
The cardioprotective role of CTLA-4 is particularly
notable: CTLA-4-deficient murine models develop lethal myocarditis
and pancreatitis, demonstrating its essential function in cardiac
autoimmunity (48). A previous
investigative study from Harbin Medical University revealed that
CTLA-4 m2a antibodies exacerbate myocardial damage in experimental
autoimmune myocarditis through CCL5+ neutrophil subset
infiltration, establishing a theoretical framework for
preventing/treating CTLA-4-associated myocarditis (49). Concurrently, anti-CTLA-4 antibodies
activate T helper (Th)17 cells and aggravate pressure
overload-induced heart failure, elucidating the immunological
pathway through which CTLA-4 signaling dysregulation precipitates
cardiac injury (50). In addition,
CTLA-4 has a central role in Treg-mediated immune suppression by
regulating cytokine secretion (such as by inhibiting the production
of IFN-γ), T-cell differentiation and proliferation, and
contact-dependent signaling between cells, thereby balancing the
intensity of the adaptive immune response (51).
In oncological therapeutics, anti-CTLA-4 antibodies
enhance T-cell activation via ligand binding blockade, thereby
exerting antitumor effects (52).
However, CTLA-4 inhibitors may induce cardiotoxicity, including
de novo onset or exacerbation of heart failure. These
findings underscore the necessity for systematic evaluation of the
dynamic equilibrium between immune activation and cardiac function
during treatment, mandating rigorous assessment of potential
cardiovascular risks
Co-inhibitory role of LAG-3
LAG-3, a type I transmembrane protein predominantly
expressed on Tregs and exhausted T cells, exerts immunosuppressive
effects on the tumor microenvironment through binding to MHC class
II molecules on APCs, thereby inhibiting T-cell activation and
proliferation (53).
Mechanistically, LAG-3 demonstrates synergistic inhibitory effects
with PD-1 in CD8+ T cells. Their co-expression
potentiates T-cell exhaustion and suppresses IFN-γ-mediated
antitumor immunity (54). This
functional interplay has been substantiated in murine models of
melanoma, where PD-1/LAG-3 double-deficient CD8+ T cells
have been reported to exhibit enhanced tumor clearance and
survival, while dual-knockout mice developed spontaneous T
cell-mediated myocarditis, confirming their joint role in
maintaining immune homeostasis and restraining autoimmune responses
(Fig. 2) (55).
The clinical translation of this synergistic
mechanism has yielded notable breakthroughs. Combination therapy
employing anti-LAG-3 (such as relatlimab) and anti-PD-1 (for
example, nivolumab) antibodies enhances T cell-mediated tumor
cytotoxicity and promotes effector cytokine secretion, including
TNF-α, IFN-γ and IL-2 (56). The
landmark RELATIVITY-047 phase III trial demonstrated superior
clinical outcomes in patients with advanced melanoma receiving
combination therapy vs. monotherapy, leading to its US FDA approval
in 2022 (marketed as Opdualag) (57). This therapeutic milestone
underscores the pivotal role of co-inhibitory receptor networks in
advancing cancer immunotherapy paradigms.
Pathophysiological mechanisms of
ICI-related myocarditis
To maintain immune homeostasis, the immune system
must accurately distinguish between self and non-self antigens
during the process of eliminating pathogens and malignant cells.
This immunological balance is essential for mounting effective
immune responses while simultaneously preventing autoimmunity. To
achieve this, the host relies on two complementary tolerance
mechanisms-central tolerance and peripheral tolerance. Disruption
of these mechanisms, particularly through immune checkpoint
inhibition, may lead to loss of self-tolerance and the development
of immune-related adverse events, such as immune checkpoint
inhibitor-related myocarditis (58).
Central tolerance primarily involves thymic negative
selection, where self-reactive T lymphocytes are eliminated during
development. Peripheral tolerance encompasses multifaceted
regulatory processes: i) Functional suppression by Tregs and ii)
dynamic modulation of T-cell activation thresholds through immune
checkpoint signaling. These mechanisms collectively establish
immunological safeguards, including the maintenance of immune
privileged sites that protect vital organs from autoimmune
attack.
The synergistic operation of central and peripheral
tolerance systems enables accurate identification of pathogenic
threats while preserving tissue integrity. Disruption of this
equilibrium underlies the pathogenesis of ICI-related myocarditis,
where checkpoint inhibition may override protective mechanisms,
leading to aberrant immune targeting of cardiac tissue (59).
Destruction of immune tolerance
mechanisms
Defects in central tolerance
Central tolerance establishment relies on a biphasic
thymic selection process encompassing positive and negative
selection (60). During positive
selection, immature T-cell survival is determined by
moderate-affinity interactions between TCRs and self-peptide-MHC
complexes presented by thymic epithelial cells. T cells exhibiting
optimal affinity thresholds differentiate into CD4+
helper or CD8+ cytotoxic T-cell lineages, whereas those
with insufficient affinity undergo apoptosis due to failed survival
signaling (61). Negative
selection subsequently eliminates high-affinity autoreactive T
cells through self-antigen presentation by medullary thymic
epithelial cells (mTECs) and DCs (62). This dual filtration mechanism
ensures exclusive export of foreign antigen-responsive mature T
cells to secondary lymphoid organs, thereby maintaining
immunological homeostasis. However, this system demonstrates
critical deficiencies in purging tissue-specific antigens,
particularly evident in cardiac autoimmunity.
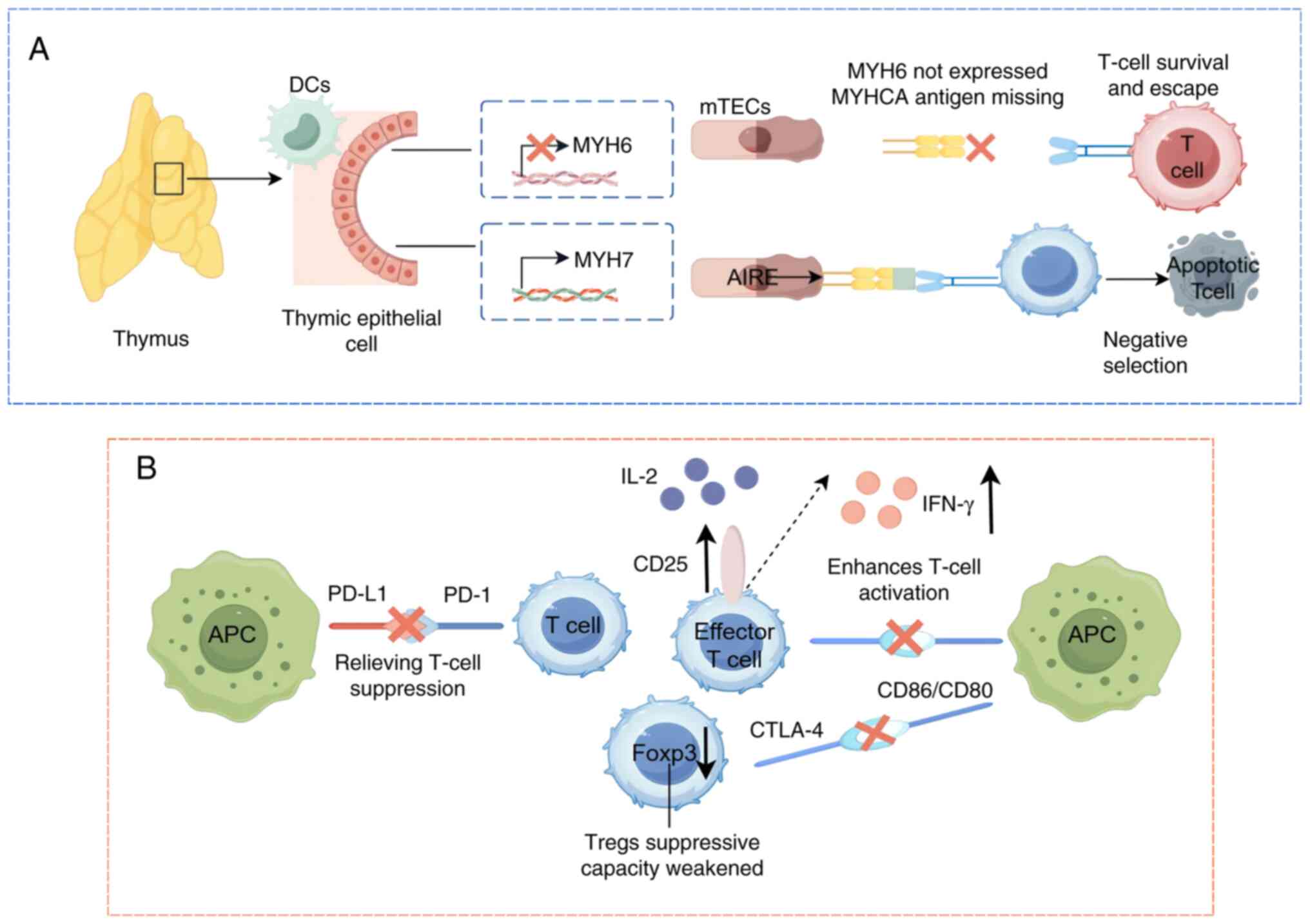
The inefficacy of negative selection represents a
key limitation in central tolerance. Thymic deletion fails to
eliminate T cells targeting myosin heavy chain-α (MYHCA), a
cardiac-specific protein encoded by the MYH6 gene and predominantly
expressed in the atrioventricular myocardium (63,64).
This defect arises from the absence of MYH6 expression in the
thymus. While mTECs mediate negative selection by regulating
ectopic tissue antigen expression via the autoimmune regulator
(63), impaired MYH6 transcription
in mTECs allows MYHCA-reactive T cells to evade thymic deletion
during development (65). These T
cells therefore escape to peripheral tissues, where they may be
activated under specific conditions, potentially triggering an
autoimmune response. As a result, central tolerance is not entirely
effective and relies on other mechanisms, such as peripheral
tolerance, to compensate for its deficiencies, thus maintaining
immune system homeostasis and preventing the onset of autoimmune
diseases (Fig. 3A).
Disruption of peripheral
tolerance
Peripheral tolerance maintains immune homeostasis by
regulating the function of mature T cells. Its core mechanisms
include the immunosuppressive function of Tregs and immune
checkpoint signaling regulation. Tregs, as a subset of
CD4+ T cells (accounting for 5–7%) (66), suppress the function of effector T
cells through various mechanisms, including: Sequestration of the
pro-survival signal molecule IL-2, secretion of anti-inflammatory
cytokines such as TGFβ, IL-10 and IL-35, direct lysis of APCs and
effector T cells, mediating trogocytosis and inducing the
generation of other immunosuppressive T cell subsets within the
local microenvironment (67).
Additionally, insufficient co-stimulatory signals during T-cell
activation (such as low expression of CD80/CD86 on APCs) can lead
to T-cell anergy or apoptosis, which is further suppressed by Tregs
(68).
The immune checkpoint mechanism in peripheral
tolerance mainly involves co-inhibitory receptors on the surface of
T cells, such as CTLA-4, PD-1/PD-L1 and LAG-3 (69). CTLA-4 inhibits co-stimulatory
signals by competitively binding to CD80/CD86, serving a negative
regulatory role in the early stages of T-cell activation (4). PD-1 antagonizes the TCR/CD28
signaling pathway by recruiting tyrosine phosphatases, inhibiting
the activation of effector T cells (70,71).
In addition, CTLA-4 and PD-1 can enhance the immunosuppressive
function of Tregs (72). LAG-3
interferes with the interaction between T cells and APCs by binding
to MHC class II molecules (73).
These molecules together constitute a dynamic regulatory network of
peripheral tolerance.
The core mechanism by which ICIs induce cardiac
autoimmunity lies in the dual disruption of peripheral tolerance.
On one hand, ICIs block the CTLA-4 or PD-1/PD-L1 signaling axis,
thereby relieving physiological suppression of effector T cells.
This leads to their overactivation and subsequent attack on
myocardial tissue (74).
Experimental evidence has demonstrated that in PD-L1-deficient
murine models of myocarditis, the inflammatory response is
exacerbated, confirming the direct protective role of PD-1/PD-L1 in
maintaining cardiac immune homeostasis (75). On the other hand, ICIs impair Treg
function. For example, CTLA-4 deficiency induces fatal
lymphoproliferative disorders, and anti-CTLA-4 therapy not only
diminishes Treg-mediated suppression but also dysregulates PD-1
signaling, resulting in compounded tolerance defects (76). Additionally, ICI therapy disrupts
endogenous cardiac protective mechanisms (such as myocardial PD-L1
expression), which potentiates CD8+ T cell cytotoxicity
against cardiac tissue (77).
Concurrently, LAG-3 inhibition exacerbates inflammatory
cardiomyopathy (78). The systemic
collapse of peripheral tolerance networks ultimately culminates in
cardiac-specific autoimmune pathology (Fig. 3B).
Cross-reactivity of cardiac
antigens
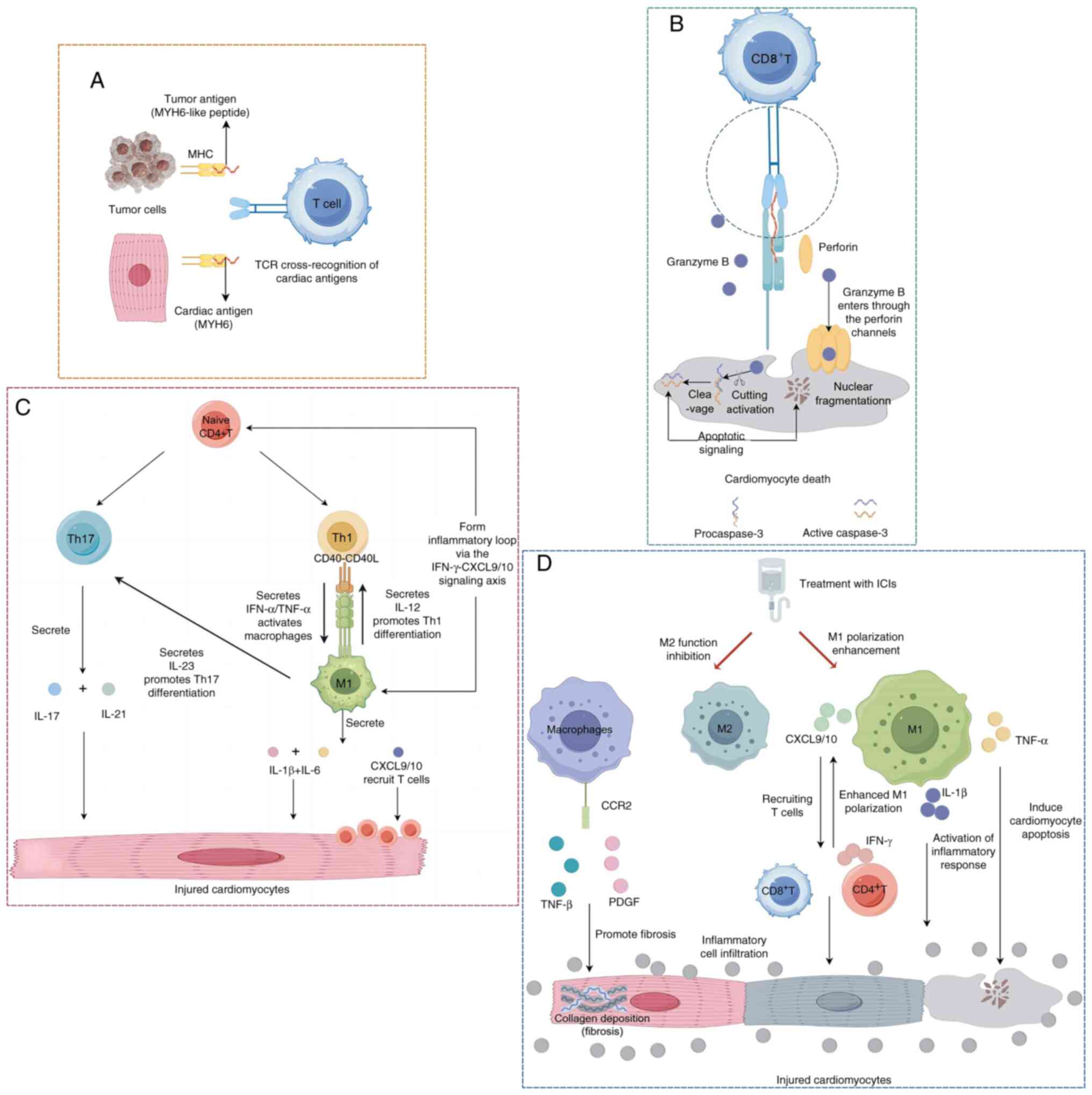
The pathogenesis of ICI-related myocarditis may
predominantly involve tumor-cardiac cross-antigen reactivity.
Substantial evidence has indicated that tumor antigens (such as
MYH6) share homologous epitopes with myocardial structural proteins
(including troponin), which can activate cross-reactive T cells
capable of simultaneously targeting both neoplastic and cardiac
tissues (79). Experimental
validation using immunocompetent A/J murine models has demonstrated
that repeated administration of anti-PD-1 antibodies induces
myocarditis, with myocardial infiltration of α-myosin-specific
CD4+ and CD8+ T cells identified through TCR
analysis. Notably, these cross-reactive T-cell populations have
also been detected in the peripheral blood, spleen, mediastinal
lymph nodes and cardiac tissues of healthy control mice, suggesting
pre-existing autoreactive lymphocytes that become pathogenic upon
immune checkpoint inhibition (80). Johnson et al (81) revealed that TCR-β clonotypic
sequences were highly overlapped between the myocardium, tumors and
skeletal muscles of patients with myocarditis, indicating that
cross-reactive T cells can mediate multi-organ damage. Clinically,
the frequent co-occurrence of myositis or myasthenia gravis in
patients with ICI-related myocarditis aligns with the shared
antigenic epitope theory. Particularly, MYHC antigens expressed in
both cardiac and skeletal muscles may serve as critical targets for
these cross-reactive immune responses (82). Collectively, these findings
establish that cross-antigen-driven immune mechanisms underlie the
simultaneous multi-organ damage observed in this clinical syndrome
(Fig. 4A).
Cellular and molecular mechanisms
T cell-mediated direct injury
T cells serve as central effector cells in
ICI-related myocarditis. These cells recognize cardiac antigens and
directly attack cardiomyocytes, resulting in tissue damage
(83,84). This process is initiated by the
binding of TCRs to MHC-antigen complexes presented on the surface
of cardiomyocytes, thereby triggering cytotoxic responses.
Role of CD8+ T cells
CD8+ cytotoxic T lymphocytes serve a
pivotal role in immune-mediated myocarditis. They induce
cardiomyocyte death through the release of perforin and granzyme B.
Perforin facilitates transmembrane pore formation, enabling
granzyme B influx and subsequent activation of the
caspase-dependent apoptotic cascade, ultimately leading to
cardiomyocyte death (85–87). Pathological analyses of myocardial
biopsies have revealed notable CD8+ T-cell infiltration
in focal necrotic regions, with infiltration levels positively
associated with the degree of T-cell clonal expansion (79). Mechanistically, CD8+ T
cells interact with cardiomyocytes via MHC class I molecules and
release cytotoxic mediators; especially when receiving anti-CTLA-4
combined with anti-PD-1 treatment, T-cell activation and
proliferation are enhanced, further aggravating myocardial damage
(88). Animal models have
demonstrated that CD8+ T-cell depletion markedly
attenuates myocardial inflammation and fibrosis (77). In addition, peripheral blood
analyses of patients with ICI-related myocarditis have detected
clonally expanded activated cytotoxic CD8+ T-cell
subsets. A prominent increase is observed in terminally
differentiated effector memory CD8+ T cells, whereas
naïve T cells, central memory T cells and other effector memory
subsets remain unaltered (89).
This distinct immunophenotypic shift underscores the critical
involvement of CD8+ T cells in the pathogenesis of
ICI-related myocarditis, with their activation status closely
associated with disease severity (Fig.
4B).
Role of CD4+ T cells
CD4+ T cells drive the inflammatory
cascade through cytokine networks in ICI-related myocarditis.
Although the number of infiltrating myocardial CD4+ T
cells is limited, the pro-inflammatory cytokines they secrete, such
as IFN-γ and TNF-α, synergistically promote the recruitment and
activation of monocytes/macrophages while directly activating
apoptotic pathways in myocardial cells, thereby exacerbating tissue
damage (90). Clinical evidence
has demonstrated a significant positive association between the
density of myocardial CD4+ T-cell infiltration and the
severity of the inflammatory response, indicating that their
regulatory activity is closely associated with the degree of
disease progression. In a
Ctla-4+/−Pdcd1−/−mouse model, CD4+
T cells and macrophages form a self-amplifying inflammatory loop
via the IFN-γ-CXCL9/10 signaling axis, leading to persistent
deterioration of the inflammatory microenvironment (91). Furthermore, within the inflammatory
milieu, cytokines drive the differentiation of CD4+ T
cells into Th1/Th17 subsets. These subsets recruit neutrophils and
activate cytotoxic T cells, thereby contributing to
multidimensional mechanisms of myocardial injury (65) (Fig.
4C).
T-cell hyperactivation and autoimmune
reactivity
The CTLA-4 protein exhibits superior binding
affinity for B7 molecules (CD80/CD86) on APCs compared with CD28.
This competitive interaction suppresses CD28-mediated T-cell
activation signals, serving as a critical negative regulatory
mechanism (52). Anti-CTLA-4
antibodies (such as ipilimumab) disrupt CTLA-4/B7 binding, thereby
restoring CD28 co-stimulatory signals and promoting T-cell
proliferation, differentiation and cytokine production. The
PD-1/PD-L1 pathway, on the other hand, inhibits T-cell function
through phosphorylation of the intracellular immune receptor
tyrosine-based inhibitory motif, which recruits phosphatases such
as SHP-2 to antagonize TCR and CD28 downstream signaling (40). PD-1 inhibitors (such as nivolumab
and pembrolizumab) block ligand binding, consequently alleviating
T-cell activity suppression and inducing aberrant activation of
effector T cells (42,43). Histopathological studies have
demonstrated T-cell infiltration within the myocardial tissues of
patients of ICI-related myocarditis, with clonal T cell populations
showing homology to tumor-infiltrating effector T cells (35,92).
Clinical evidence indicates that ICI-induced T-cell clonal
expansion drives myocardial tissue targeting, ultimately
precipitating myocarditis (81).
Mechanistically, ICIs interfere with signaling pathways such as
CTLA-4 and PD-1/PD-L1, lowering the T-cell activation threshold and
disrupting peripheral immune tolerance, which leads to abnormal
T-cell activation and inappropriate immune attacks on myocardial
tissue.
Macrophages and inflammatory
amplification
Infiltration and pro-inflammatory mediator
release
The pathogenesis of ICI-related myocarditis is
characterized by prominent macrophage infiltration, as evidenced by
histopathological analyses (93).
Clinical investigations have demonstrated substantial
co-infiltration of CD68+ macrophages with
CD4+/CD8+ T lymphocytes within myocardial
tissues, suggesting their synergistic role in propagating
inflammatory responses (94).
Activated M1-type macrophages further amplify the inflammatory
response by secreting pro-inflammatory cytokines (TNF-α, IL-1β and
IL-6) and chemokines (CXCL9 and CXCL10), which recruit T cells and
other immune cells to infiltrate the myocardial tissue, thereby
forming an inflammatory cascade amplification (77). Experimental models utilizing
CTLA-4/PD-1-deficient mice recapitulate these findings, showing
concurrent infiltration of CD68+ macrophages and T cells
in myocardial lesions. These macrophages exacerbate tissue damage
through sustained release of IL-1β and other inflammatory mediators
(95). Compared with other types
of myocarditis (such as dilated or lymphocytic myocarditis), the
myocardial tissue of patients with ICI-related myocarditis shows a
notable enrichment of CXCL9+/CXCL10+
macrophage populations (92).
Antigen presentation and T-cell
interactions
Macrophages simultaneously have antigen-presenting
function in ICI-related myocarditis. They potentially present
cardiac autoantigens (such as α-myosin) via MHC molecules,
consequently activating autoreactive T cells and establishing a
pathogenic positive feedback loop (96). Experimental models have
demonstrated that monocyte-derived CCR2+ macrophages
within cardiac tissue secrete CXCL9/CXCL10. These chemokines engage
CXCR3 receptors on T cells, driving T-cell activation and
sustaining a pro-inflammatory microenvironment (91). Furthermore, IFN-γ secreted by
activated T cells induces macrophage activation of the JAK/STAT
signaling pathway. This reciprocal interaction forms a
bidirectional regulatory network that amplifies inflammatory
responses (97). These experiments
collectively suggest that macrophages and T cells interact and
jointly promote the inflammatory response.
Macrophage phenotypic plasticity
Under physiological conditions, M2 macrophages
facilitate tissue repair through apoptotic cell clearance and
secretion of anti-inflammatory cytokines (such as IL-10) (98). In ICI-related myocarditis, however,
this homeostatic balance shifts toward dominance of the
pro-inflammatory M1 phenotype (99). Animal model studies have shown that
inhibiting macrophage chemotaxis (such as by blocking MCP-1
signaling) or targeting the IFN-γ pathway can reduce the
infiltration of pro-inflammatory macrophages and improve myocardial
damage (90,100). Single-cell sequencing has further
revealed the heterogeneity of macrophage subsets and reported that
the CCR2+ subset exacerbates pathological myocardial
remodeling by activating pro-fibrotic pathways (such as TGF-β
signaling), providing a theoretical basis for its potential as a
therapeutic target (101). In
summary, macrophages in ICI-related myocarditis act both as
inflammatory effector cells and as key nodes in immune regulation.
Future research should further explore the specific functions of
these subsets and the mechanisms of phenotypic conversion to
provide a theoretical basis for immune interventions (Fig. 4D).
Other immune cells
Role of B cells
Current evidence does not establish a direct
pathogenic role of B cells in ICI-related myocarditis. While murine
models demonstrate detectable IgG-secreting plasma cells and
anti-myosin autoantibodies, these antibodies likely represent
secondary responses to antigen exposure following cardiomyocyte
necrosis, given the rare observation of B-cell infiltration in
clinical myocardial biopsies (102). Nevertheless, extrapolating from
B-cell functions in other subtypes of myocarditis and immune
checkpoint regulation of B-cell activity, it may be proposed that B
cells could contribute indirectly through antibody-mediated
immunity or T-cell collaboration. Studies have shown that in
PD-1-deficient mice, there is overactivation of B cells and
hypergammaglobulinemia, while PD-1 helps maintain immune
homeostasis by inhibiting B-cell proliferation and the secretion of
pro-inflammatory factors such as IL-6 (103,104). These findings suggest that this
indirect regulatory mechanism may explain some of the clinical
manifestations of humoral immune abnormalities in patients with
ICI-related myocarditis.
Role of NK cells
The pathological role of NK cells in ICI-related
myocarditis remains incompletely defined. Studies have indicated
that NK cells express immune checkpoint molecules, including PD-1
and CTLA-4, and their cytotoxic activity may be modulated by
checkpoint signaling pathways. Prolonged antigenic stimulation can
upregulate PD-1 expression on NK cells, leading to impaired
cytotoxic function (105,106). Consequently, ICI therapy may
reverse this inhibitory state, potentiating NK cell cytotoxicity
and the release of pro-inflammatory mediators such as IFN-γ,
thereby contributing to the pathogenesis of myocarditis (107). Nevertheless, existing evidence
has not confirmed direct infiltration of NK cells into myocardial
tissue or their role in mediating cardiomyocyte injury.
Genetic and microenvironmental
determinants
Genetic predisposition
The HLA-DQ8 allele, encoding a specific MHC class II
heterodimer, exhibits strong associations with autoimmune
pathologies including type 1 diabetes mellitus and celiac disease.
Its molecular structure markedly increases the risk of ICI-related
myocarditis in carriers by enhancing autoantigen presentation and
T-cell activation (108). This
histocompatibility complex potentiates autoimmune responses through
structural facilitation of self-antigen presentation and subsequent
CD4+ T-cell activation, conferring elevated risk for
ICI-related myocarditis in genetically predisposed individuals
(109). Mechanistic validation
comes from murine models engineered with humanized HLA-DQ8
expression on an MHC class II-deficient background. These
preclinical models develop severe concurrent myocarditis and
myositis following anti-PD-1 therapy, recapitulating the human
disease phenotype (110). This
evidence suggests that specific genetic backgrounds, such as
HLA-DQ8, may significantly increase the susceptibility to
ICI-related myocarditis (111).
Thymic dysfunction
Patients with TETs exhibit a markedly elevated
incidence of myocarditis following ICI therapy. These individuals
frequently develop severe arrhythmia and myositis, complications
that may progress to respiratory failure or fatal outcomes
(111–113). The elevated levels of serum
anti-acetylcholine receptor antibodies in this population suggest
that thymus-related autoimmune disorders are one of the causes of
the disease (114).
Mechanistically, the thymus regulates T-cell
development and central tolerance via CTLA-4 and PD-1 signaling. In
patients with TETs, thymic structural disruption creates dual
pathological consequences: i) Absence of cardiac-specific antigens
(such as MYHCA) in the thymic medulla permits autoreactive T cells
to evade negative selection (16);
ii) the tumor microenvironment may induce the formation of T-cell
subsets with potential self-targeting tendencies. After ICI
treatment relieves immune inhibitory signals, these T-cell subsets
become activated and breach immune tolerance, leading to an attack
on cardiac tissue (115). This
thymic dysfunction, in combination with the effects of ICI therapy,
ultimately results in a markedly increased risk of developing
myocarditis.
Gut microbiome
The molecular mimicry between peptides derived from
gut commensal microbiota and cardiac myosin (such as MYH6) may
activate cardiac-specific Th17 cells, thereby triggering autoimmune
responses against myocardial tissue. Studies have suggested that
such cross-reactive peptides drive ICI-related myocarditis by
promoting Th17 cell activation and cross-recognition of
tumor/microbial antigens. Furthermore, variations in gut microbiota
composition among patients may modulate these immune responses,
thereby influencing disease susceptibility (89,116).
Other aspects
Myocardial electrophysiology and conduction
system injury
ICI-related cardiac electrophysiological
abnormalities can induce arrhythmias through multiple pathways.
Primarily mediated by inflammatory responses (myocarditis), these
abnormalities trigger myocardial fibrosis and localized scar
formation. Such anatomical remodeling establishes a pathological
substrate for reentrant arrhythmias (such as atrial fibrillation
and ventricular fibrillation) through the creation of ectopic
electrical foci or conduction block zones (117,118). The electrophysiological
regulation of the cardiac conduction system is intrinsically linked
to macrophage functionality through electrical coupling mediated by
connexin 43 (Cx43) with cardiomyocytes (119). Hulsmans et al (119) demonstrated that
macrophage-specific Cx43 ablation causes atrioventricular
conduction delays, whereas CD11b deficiency induces progressive
atrioventricular blocks, implicating macrophage-dependent Cx43
signaling in both physiological conduction and arrhythmogenesis.
These synergistic mechanisms collectively amplify arrhythmia
susceptibility during ICI therapy.
Protective effects of estrogen
The sex disparity (the incidence or severity is
higher in women) observed in ICI-related myocarditis incidence may
be attributed to the estrogen-estrogen receptor (ER)
β-mesencephalic astrocyte-derived neurotrophic factor (MANF)
signaling axis. Mechanistic studies have demonstrated that estrogen
activates ERβ signaling, upregulating MANF. This neurotrophic
factor confers cardioprotection through two key mechanisms:
Suppressing myocardial T-cell infiltration and inhibiting
pro-inflammatory cytokine release (including IFN-γ and TNF-α)
(120,121).
Experimental data from female murine models have
revealed that ICI treatment induces 17-β-estradiol depletion,
concomitant with reduced cardiac MANF expression, which exacerbates
myocarditis progression. Pharmacological intervention with
exogenous estrogen or ERβ-selective agonists restores MANF levels,
markedly attenuating myocardial inflammation and fibrotic
remodeling (122). These findings
elucidate the molecular basis of estrogen-mediated protection and
provide a rationale for sex-specific therapeutic strategies in
ICI-associated cardiac toxicity.
Future directions
Biomarker development and diagnostic
technologies
Advancements have notably improved early diagnostic
strategies for ICI-related myocarditis. Myocardium-specific
autoantibodies (such as anti-myosin antibodies) and TCR repertoire
analysis have the potential for early diagnosis of ICI-related
myocarditis. Notably, T-cell infiltration and upregulation of PD-L1
expression has been detected in the myocardial tissue of patients
with ICI-related myocarditis, suggesting a T cell-mediated immune
injury mechanism (95).
Additionally, the combined detection of myocardial-specific
microRNAs (miRs), such as miR-208a, and cTnI enhances diagnostic
sensitivity, as miR-208a is released earlier than cTnI alone
(123). Furthermore, optimization
of imaging diagnostics and artificial intelligence-assisted systems
may address current diagnostic limitations (124). Future efforts should integrate
multi-omics biomarker datasets with dynamic monitoring platforms to
establish non-invasive, high-sensitivity diagnostic frameworks.
Innovative targeted therapeutic
strategies
The standard first-line treatment for ICI-related
myocarditis involves high-dose corticosteroids (such as
methylprednisolone, 1,000 mg/day for 3 days), followed by a
tapering regimen over several weeks. For steroid-refractory cases,
immunosuppressants such as abatacept, mycophenolate mofetil or
infliximab may be employed. Early treatment within 72 h is critical
for improved outcomes (125).
Advances in targeted therapeutic strategies for
ICI-related myocarditis have emerged as a critical research focus.
Current efforts have concentrated on three principal domains: i)
Immune checkpoint regulation: By antagonizing CTLA-4 or modulating
downstream PD-1 molecules (such as PI3K/AKT/mTOR), these strategies
aim to balance immune activation and myocardial protection
(126). ii) Cytokine-targeted
intervention: Implementation of IL-6 receptor antagonists
(tocilizumab) and TNF-α inhibitors may alleviate the onset of
inflammation. Clinical evidence has indicated that survival
outcomes are enhanced in critically ill patients when combined with
extracorporeal membrane oxygenation support (127). iii) Development of specific
monoclonal antibodies: Antibody drugs targeting myocardial-specific
antigens, such as cTns, may reduce nonspecific attacks, although
this technology is still in the preclinical research phase
(89). Additionally, innovative
combination therapies have garnered attention. For example,
low-dose corticosteroids combined with JAK/STAT pathway inhibitors
(such as baricitinib) can synergistically suppress hyperactivated
immune responses while reducing the toxicity associated with
conventional treatments (128).
Nevertheless, current evidence remains predominantly derived from
limited clinical cohorts, underscoring the imperative for
large-scale randomized controlled trials to validate safety and
efficacy profiles. Future investigations should prioritize
multi-omics-driven target discovery and advanced drug delivery
platforms to optimize myocardial-specific immunomodulatory
precision.
Multidisciplinary collaboration
Rechallenge with ICIs after immune-related
myocarditis remains a controversial clinical issue. Rechallenge may
be considered under stringent conditions: Complete resolution of
myocarditis symptoms, normalization of troponin and NT-proBNP,
resolution of imaging findings and consensus by an MDT. Emerging
guidelines suggest cautious use and rechallenge is typically
avoided unless no alternatives exist (125).
The management of ICI-related myocarditis
necessitates MDT collaboration involving cardiology, oncology and
immunology to optimize early detection and personalized
interventions. Real-time coordination between oncologists and
cardiologists enables balanced therapeutic decision-making,
including ICI dose adjustments or co-administration of
immunomodulatory agents to mitigate cardiotoxicity without
compromising antitumor efficacy (Table
I) (129–131). In the future, standardized
collaborative processes need to be established to improve
prognosis.
 | Table I.Graded treatment strategies for
ICI-related myocarditis. |
Table I.
Graded treatment strategies for
ICI-related myocarditis.
| Severity | First-line
treatment | Second-line
treatment | Monitoring and key
actions |
|---|
| Mild (Grade 1):
Suspected | 1) Hospitalization
for | If symptoms develop
or | Serial
monitoring: |
| or confirmed,
asymptomatic | monitoring. | biomarkers
worsen, | -Troponin |
| with cardiac
biomarker | 2) Initiation of
cortico- | escalation to
moderate/ | -ECG |
| elevation or ECG
changes | steroids:
Prednisone | severe treatment
protocol. | -Echocardiography
(LVEF) |
|
| 1-2 mg/kg/day
orally |
| Decision to resume
ICI is |
|
| (or
methylprednisolone |
| highly
individualized if |
|
| IV
equivalent). |
| symptoms resolve
and |
|
| 3) Discontinuation
of ICI therapy. |
| biomarkers
normalize. |
| Moderate to
severe | 1) Immediate
hospitali- | If no clinical
improvement | Intensive
monitoring: |
| (Grade 2–3): | zation (often to
cardiac | within 24–72 h of
high-dose | -Frequent troponin,
ECG |
| Significant
symptoms | unit). | steroids: |
-Echocardiography |
| (e.g., chest pain,
dyspnea), | 2) High-dose
cortico | -Add second-line
immuno- | -Assess for
multi-organ ir |
| biomarker
elevation, LVEF | steroids:
Methylpredni- | suppressants: | AEs (e.g.,
myositis, MG). |
| dysfunction or
arrhythmias | solone 1–2
mg/kg/day IV. | • Mycophenolate
mofetil | -Manage heart
failure/ |
|
| 3) Permanent
disconti- | • Antithymocyte
globulin | arrhythmias as
per |
|
| nuation of
ICI. | (ATG) | standard
guidelines. |
|
|
| • Abatacept
(especially for fulminant cases) |
|
|
|
| -CONTRAINDICATED:
Infliximab (associated with increased cardiovascular mortality and
heart failure). |
|
|
Life-threatening | 1) Admit to
ICU. | Initiate urgently
(concurrently | ICU-level
monitoring: |
| (Grade 4):
Hemodynamic | 2) Pulse-dose | or if no immediate
response): | -Hemodynamics
(e.g., |
| instability,
cardiogenic |
corticosteroids: | -Second-line
agents: ATG or | Swan-Ganz
catheter) |
| shock, lethal
arrhythmias |
Methylprednisolone | Abatacept. | -Continuous
telemetry |
|
| 1,000 mg/day
IV. | -Mechanical
circulatory | -Multi-organ
function |
|
| 3) Add IV
Immuno-globulin (IVIG). | support: Consider
VA-ECMO for cardiogenic shock. | support. |
|
| 4) Permanently
discontinue ICI. | -Temporary pacing
for high-grade heart block. |
|
|
|
| -Heart
failure/arrhythmia support. |
|
Conclusion
ICI-related myocarditis represents a critical
challenge in cancer immunotherapy. While ICIs have revolutionized
survival outcomes in malignancies, their cardiovascular toxicity
substantially limits their clinical utility. Current diagnostic
strategies integrating biomarker profiling, cardiac imaging and
histopathological confirmation remain constrained by delayed
detection and suboptimal specificity. Current research is focused
on three core issues: i) The pathophysiological mechanisms,
particularly the role of T-cell clonal expansion and the
cross-reactivity between myocardial antigen epitopes, which remain
to be fully elucidated; ii) the absence of validated predictive
biomarkers and risk stratification tools; iii) existing stratified
treatment strategies lack evidence-based support. Future research
is needed in the following areas: Firstly, systematic biology
methods should be employed to analyze dynamic changes in the immune
microenvironment, and single-cell sequencing techniques should be
used to reveal the clonotype characteristics of myocardium-specific
TCRs. Secondly, multi-omics predictive models integrating genomics,
proteomics and metabolomics should be developed. Additionally,
interdisciplinary research platforms spanning oncology, cardiology
and immunology should be established. Particular emphasis should be
placed on cohort studies and biobank development, as these will
provide crucial support for optimizing diagnostic and therapeutic
pathways. With the ongoing expansion of indications for
immunotherapy, further elucidating the molecular mechanisms of
ICI-related myocarditis and establishing prevention and treatment
strategies will become key directions for future research. Current
research must prioritize elucidating risk stratification mechanisms
to achieve a precise equilibrium between therapeutic efficacy and
cardiotoxicity.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
LD was the primary contributor to the writing of
the manuscript. QX and YD proposed the topic, oversaw the
literature selection and synthesis, and critically revised the
manuscript for accuracy and clarity; these two authors contributed
equally to this article. Data authentication is not applicable. All
authors read and approved the final manuscript, were responsible
for all aspects of the work and approved its submission.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liu X, Hogg GD and DeNardo DG: Rethinking
immune checkpoint blockade: ‘Beyond the T cell’. J Immunother
Cancer. 9:e0014602021. View Article : Google Scholar
|
|
2
|
Blank C, Gajewski TF and Mackensen A:
Interaction of PD-L1 on tumor cells with PD-1 on tumor-specific T
cells as a mechanism of immune evasion: Implications for tumor
immunotherapy. Cancer Immunol Immunother. 54:307–314. 2005.
View Article : Google Scholar
|
|
3
|
Naimi A, Mohammed RN, Raji A, Chupradit S,
Yumashev AV, Suksatan W, Shalaby MN, Thangavelu L, Kamrava S,
Shomali N, et al: Tumor immunotherapies by immune checkpoint
inhibitors (ICIs); the pros and cons. Cell Commun Signal.
20:442022. View Article : Google Scholar
|
|
4
|
Korman AJ, Garrett-Thomson SC and Lonberg
N: The foundations of immune checkpoint blockade and the ipilimumab
approval decennial. Nat Rev Drug Discov. 21:509–528. 2022.
View Article : Google Scholar
|
|
5
|
Wei J, Li W, Zhang P, Guo F and Liu M:
Current trends in sensitizing immune checkpoint inhibitors for
cancer treatment. Mol Cancer. 23:2792024. View Article : Google Scholar
|
|
6
|
Cone EB, Haeuser L and Reese SW: Immune
checkpoint inhibitor monotherapy is associated with less cardiac
toxicity than combination therapy. PLoS One. 17:e02720222022.
View Article : Google Scholar
|
|
7
|
Salem JE, Manouchehri A, Moey M,
Lebrun-Vignes B, Bastarache L, Pariente A, Gobert A, Spano JP,
Balko JM, Bonaca MP, et al: Cardiovascular toxicities associated
with immune checkpoint inhibitors: An observational, retrospective,
pharmacovigilance study. Lancet Oncol. 19:1579–1589. 2018.
View Article : Google Scholar
|
|
8
|
Gougis P, Jochum F, Abbar B, Dumas E,
Bihan K, Lebrun-Vignes B, Moslehi J, Spano J, Laas E, Hotton J, et
al: Clinical spectrum and evolution of immune-checkpoint inhibitors
toxicities over a decade-a worldwide perspective.
EClinicalMedicine. 70:1025362024. View Article : Google Scholar
|
|
9
|
Hu JR, Florido R, Lipson EJ, Naidoo J,
Ardehali R, Tocchetti CG, Lyon AR, Padera RF, Johnson DB and
Moslehi J: Cardiovascular toxicities associated with immune
checkpoint inhibitors. Cardiovasc Res. 115:854–868. 2019.
View Article : Google Scholar
|
|
10
|
Yousif LI, Screever EM, Versluis D,
Aboumsallem JP, Nierkens S, Manintveld OC, de Boer RA and Meijers
WC: Risk factors for immune checkpoint inhibitor-mediated
cardiovascular toxicities. Curr Oncol Rep. 25:753–763. 2023.
View Article : Google Scholar
|
|
11
|
Mahmood SS, Fradley MG, Cohen JV, Nohria
A, Reynolds KL, Heinzerling LM, Sullivan RJ, Damrongwatanasuk R,
Chen CL, Gupta D, et al: Myocarditis in patients treated with
immune checkpoint inhibitors. J Am Coll Cardiol. 71:1755–1764.
2018. View Article : Google Scholar
|
|
12
|
Moslehi JJ, Salem JE, Sosman JA,
Lebrun-Vignes B and Johnson DB: Increased reporting of fatal immune
checkpoint Inhibitor-associated myocarditis. Lancet. 391:9332018.
View Article : Google Scholar
|
|
13
|
Wang F, Sun X, Qin S, Hua H, Liu X, Yang L
and Yang M: A retrospective study of immune checkpoint
inhibitor-associated myocarditis in a single center in China.
Cancer Commun. 9:162020.
|
|
14
|
Wang F, Qin S, Lou F, Chen FX, Shi M,
Liang X, Jiang H, Jiang Y, Chen Y, Du Y, et al: Retrospective
analysis of immune checkpoint Inhibitor-associated myocarditis from
12 cancer centers in China. J Clin Oncol. 38:e151302020. View Article : Google Scholar
|
|
15
|
Oren O, Yang EH, Molina JR, Bailey KR,
Blumenthal RS and Kopecky SL: Cardiovascular health and outcomes in
cancer patients receiving immune checkpoint inhibitors. Am J
Cardiol. 125:1920–1926. 2020. View Article : Google Scholar
|
|
16
|
Fenioux C, Abbar B, Boussouar S, Bretagne
M, Power JR, Moslehi JJ, Gougis P, Amelin D, Dechartres A, Lehmann
LH, et al: Thymus alterations and susceptibility to immune
checkpoint inhibitor myocarditis. Nat Med. 29:3100–3110. 2023.
View Article : Google Scholar
|
|
17
|
Shelly S, Agmon-Levin N, Altman A and
Shoenfeld Y: Thymoma and autoimmunity. Cell Mol Immunol. 8:199–202.
2011. View Article : Google Scholar
|
|
18
|
Zamami Y, Niimura T, Okada N, Koyama T,
Fukushima K, Izawa-Ishizawa Y and Ishizawa K: Factors associated
with immune checkpoint inhibitor-related myocarditis. JAMA Oncol.
5:1635–1637. 2019. View Article : Google Scholar
|
|
19
|
Puzanov I, Subramanian P, Yatsynovich YV,
Jacobs DM, Chilbert MR, Sharma UC, Ito F, Feuerstein SG, Stefanovic
F, Hicar MD, et al: Clinical characteristics, time course,
treatment and outcomes of patients with immune checkpoint
inhibitor-associated myocarditis. J Immunother Cancer.
9:e0025532021. View Article : Google Scholar
|
|
20
|
Bonaca MP, Olenchock BA, Salem JE, Wiviott
SD, Ederhy S, Cohen A, Stewart GC, Choueiri TK, Di Carli M,
Kumbhani DJ, et al: Myocarditis in the setting of cancer
therapeutics: Proposed case definitions for emerging clinical
syndromes in Cardio-oncology. Circulation. 140:80–91. 2019.
View Article : Google Scholar
|
|
21
|
Lehmann LH, Heckmann MB, Bailly G, Finke
D, Procureur A, Power JR, Stein F, Bretagne M, Ederhy S, Moslehi J,
et al: Cardiomuscular biomarkers in the diagnosis and
prognostication of immune checkpoint inhibitor myocarditis.
Circulation. 148:473–486. 2023. View Article : Google Scholar
|
|
22
|
Barac A, Wadlow RC, Deeken JF and
deFilippi C: Cardiac troponin I and T in ICI myocarditis screening,
diagnosis, and prognosis. J Am Coll Cardiol. 6:804–807. 2024.
|
|
23
|
Zotova L: Immune checkpoint
Inhibitors-related myocarditis: A review of reported clinical
cases. Diagnostics. 13:12432023. View Article : Google Scholar
|
|
24
|
Vasbinder A, Chen Y, Procureur A, Gradone
A, Azam TU, Perry D, Shadid H, Anderson E, Catalan T, Blakely P, et
al: Biomarker trends, incidence, and outcomes of immune checkpoint
Inhibitor-induced myocarditis. JACC CardioOncol. 4:689–700. 2022.
View Article : Google Scholar
|
|
25
|
Caio G: Myocarditis with immune checkpoint
blockade. N Engl J Med. 376:291–292. 2017.
|
|
26
|
Semeraro GC, Cipolla CM and Cardinale DM:
Role of cardiac biomarkers in cancer patients. Cancers (Basel).
13:54262021. View Article : Google Scholar
|
|
27
|
Song W, Zheng Y, Dong M, Zhong L, Bazoukis
G, Perone F, Li G, Ng CF, Baranchuk A, Tse G and Liu T:
Electrocardiographic features of immune checkpoint
inhibitor-associated myocarditis. Curr Probl Cardiol.
48:1014782023. View Article : Google Scholar
|
|
28
|
Faron A, Isaak A, Mesropyan N, Reinert M,
Schwab K, Sirokay J, Sprinkart AM, Bauernfeind FG, Dabir D, Pieper
CC, et al: Cardiac MRI depicts immune checkpoint Inhibitor-induced
myocarditis: A prospective study. Radiology. 301:602–609. 2021.
View Article : Google Scholar
|
|
29
|
Correction to. Routine application of
cardiac magnetic resonance imaging in patients with suspected
myocarditis from immune checkpoint inhibitor therapy. Eur Heart J.
46:3042025. View Article : Google Scholar
|
|
30
|
Pereyra M, Farina J, Mahmoud AK, Scalia I,
Tagle-Cornell MC, Kenyon C, Abbas MT, Baba N, Herrmann J, Arsanjani
R and Ayoub C: The prognostic value of criteria for diagnosis of
Immune Checkpoint Inhibitor Related Myocarditis: A comparison of
the Bonaca et al. Criteria and European Society of Cardiology
(ESC)-International Cardio-Oncology Society (ICOS) guidelines.
Circulation. 150 (Suppl 1):A41420442024. View Article : Google Scholar
|
|
31
|
Salem JE, Bretagne M, Abbar B,
Leonard-Louis S, Ederhy S, Redheuil A, Boussouar S, Nguyen LS,
Procureur A, Stein F, et al: Abatacept/Ruxolitinib and screening
for concomitant respiratory muscle failure to mitigate fatality of
Immune-checkpoint inhibitor myocarditis. Cancer Discov.
13:1100–1115. 2023. View Article : Google Scholar
|
|
32
|
Thibault C, Vano Y, Soulat G and Mirabel
M: Immune checkpoint inhibitors myocarditis: Not all cases are
clinically patent. Eur Heart J. 39:35532018.
|
|
33
|
Domen H, Kaga K, Hida Y, Honma N, Kubota
R, Yagi Y and Matsui Y: Investigation of lung cancer patients with
cardiovascular disease. Kyobu Geka. 68:266–70. 2015.(In
Japanese).
|
|
34
|
Murtagh G, deFilippi C, Zhao Q and Barac
A: Circulating biomarkers in the diagnosis and prognosis of immune
checkpoint inhibitor-related myocarditis: Time for a risk-based
approach. Front Cardiovasc Med. 11:13505852024. View Article : Google Scholar
|
|
35
|
Palaskas N, Lopez-Mattei J, Durand JB,
Iliescu C and Deswal A: Immune checkpoint inhibitor myocarditis:
Pathophysiological characteristics, diagnosis, and treatment. J Am
Heart Assoc. 9:e0137572020. View Article : Google Scholar
|
|
36
|
Pi JK, Chen XT, Zhang YJ, Chen XM, Wang
YC, Xu JY, Zhou JH, Yu SS and Wu SS: Insight of immune checkpoint
inhibitor related myocarditis. Int Immunopharmacol. 143:1135592024.
View Article : Google Scholar
|
|
37
|
Heilbroner SP, Few R, Mueller J, Chalwa J,
Charest F, Suryadevara S, Kratt C, Gomez-Caminero A and Dreyfus B:
Predicting cardiac adverse events in patients receiving immune
checkpoint inhibitors: A machine learning approach. J Immunother
Cancer. 9:e0025452021. View Article : Google Scholar
|
|
38
|
Zhang C, Bockman A and DuPage M: Breaking
up the CD8+ T cell: Treg pas de deux. Cell Mol Immunol. 42:941–942.
2024.
|
|
39
|
Yu L, Sun M, Zhang Q, Zhou Q and Wang Y:
Harnessing the immune system by targeting immune checkpoints:
Providing new hope for oncotherapy. Front Immunol. 13:9820262022.
View Article : Google Scholar
|
|
40
|
Keir ME, Butte MJ, Freeman GJ and Sharpe
AH: PD-1 and its ligands in tolerance and immunity. Annu Rev
Immunol. 26:677–704. 2008. View Article : Google Scholar
|
|
41
|
Boussiotis VA: Molecular and biochemical
aspects of the PD-1 checkpoint pathway. N Engl J Med.
375:1767–1778. 2016. View Article : Google Scholar
|
|
42
|
Yi M, Zheng X, Niu M, Zhu S, Ge H and Wu
K: Combination strategies with PD-1/PD-L1 blockade: Current
advances and future directions. Mol Cancer. 21:282022. View Article : Google Scholar
|
|
43
|
Gaikwad S, Agrawal MY, Kaushik I,
Ramachandran S and Srivastava SK: Immune checkpoint proteins:
Signaling mechanisms and molecular interactions in cancer
immunotherapy. Semin Cancer Biol. 86:137–150. 2022. View Article : Google Scholar
|
|
44
|
Malmberg R, Zietse M, Dumoulin DW,
Hendrikx JJMA, Aerts JGJVA, van der Veldt AAM, Koch BCP, Sleijfer
S, van Leeuwen RWF, Koch BCP, et al: Alternative dosing strategies
for immune checkpoint inhibitors to improve cost-effectiveness: A
special focus on nivolumab and pembrolizumab. Lancet Oncol.
23:e552–e561. 2022. View Article : Google Scholar
|
|
45
|
Wolchok JD, Chiarion-Sileni V, Gonzalez R,
Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D,
Ferrucci PF, et al: Overall survival with combined nivolumab and
ipilimumab in advanced melanoma. N Engl J Med. 377:1345–1356. 2017.
View Article : Google Scholar
|
|
46
|
Xu D, Wang H, Bao Q, Jin K, Liu M, Liu W,
Yan X, Wang L, Zhang Y, Wang G, et al: The anti-PD-L1/CTLA-4
bispecific antibody KN046 plus lenvatinib in advanced unresectable
or metastatic hepatocellular carcinoma: A phase II trial. Nat
Commun. 16:14432025. View Article : Google Scholar
|
|
47
|
Schaub J and Tang SC: Beyond checkpoint
inhibitors: The three generations of immunotherapy. Clin Exp Med.
25:432025. View Article : Google Scholar
|
|
48
|
Buehning F, Lerchner T, Vogel J,
Hendgen-Cotta UB, Totzeck M, Rassaf T and Michel L: Preclinical
models of cardiotoxicity from immune checkpoint inhibitor therapy.
Basic Res Cardiol. 120:171–185. 2025. View Article : Google Scholar
|
|
49
|
Wu MM, Yang YC, Cai YX, Jiang S, Xiao H,
Miao C, Jin XY, Sun Y, Bi X, Hong Z, et al: Anti-CTLA-4 m2a
antibody exacerbates cardiac injury in experimental autoimmune
myocarditis mice by promoting Ccl5-Neutrophil infiltration. Adv
Sci. 11:e24004862024. View Article : Google Scholar
|
|
50
|
Shang AQ, Yu CJ, Bi X, Jiang WW, Zhao ML,
Sun Y, Guan H and Zhang ZR: Blocking CTLA-4 promotes pressure
Overload-induced heart failure via activating Th17 cells. FASEB J.
38:e238512024. View Article : Google Scholar
|
|
51
|
Hossen MM, Ma Y, Yin Z, Xia Y, Du J, Huang
JY, Huang JJ, Zou L, Ye Z and Huang Z: Current understanding of
CTLA-4: From mechanism to autoimmune diseases. Front Immunol.
14:11983652023. View Article : Google Scholar
|
|
52
|
Maurer MF, Lewis KE, Kuijper JL, Ardourel
D, Gudgeon CJ, Chandrasekaran S, Mudri SL, Kleist KN, Navas C,
Wolfson MF, et al: The engineered CD80 variant fusion therapeutic
davoceticept combines checkpoint antagonism with conditional CD28
costimulation for anti-tumor immunity. Nat Commun. 13:17902022.
View Article : Google Scholar
|
|
53
|
Wang J, Sanmamed MF, Datar I, Su TT, Ji L,
Sun J, Chen L, Chen Y, Zhu G, Yin W, et al: Fibrinogen-like protein
1 is a major immune inhibitory ligand of LAG-3. Cell.
176:334–347.e12. 2019. View Article : Google Scholar
|
|
54
|
Cillo AR, Cardello C, Shan F, Karapetyan
L, Kunning S, Sander C, Rush E, Karunamurthy A, Massa RC, Rohatgi
A, et al: Blockade of LAG-3 and PD-1 leads to Co-Expression of
cytotoxic and exhaustion gene modules in CD8+ T cells to promote
antitumor immunity. Cell. 187:4373–4388.e15. 2024. View Article : Google Scholar
|
|
55
|
Andrews LP, Butler SC, Cui J, Cillo AR,
Cardello C, Liu C, Brunazzi EA, Baessler A, Xie B, Kunning SR, et
al: LAG-3 and PD-1 synergize on CD8+ T cells to drive T cell
exhaustion and hinder autocrine IFN-γ-dependent anti-tumor
immunity. Cell. 187:4355–4372.e22. 2024. View Article : Google Scholar
|
|
56
|
Sordo-Bahamonde C, Lorenzo-Herrero S,
González-Rodríguez AP, Payer AR, González-García E, López-Soto A
and Gonzalez S: LAG-3 Blockade with Relatlimab (BMS-986016)
restores Anti-leukemic responses in chronic lymphocytic leukemia.
Cancers (Basel). 13:21122021. View Article : Google Scholar
|
|
57
|
Tawbi HA, Schadendorf D, Lipson EJ,
Ascierto PA, Matamala L, Gutiérrez EC, Rutkowski P, Gogas HJ, Lao
CD, De Menezes JJ, et al: Relatlimab and nivolumab versus nivolumab
in untreated advanced melanoma. N Engl J Med. 386:24–34. 2022.
View Article : Google Scholar
|
|
58
|
Schwartz RH: Historical overview of
immunological tolerance. Cold Spring Harb Perspect Biol.
4:a0069082012. View Article : Google Scholar
|
|
59
|
Chen X, Ghanizada M, Mallajosyula V, Sola
E, Capasso R, Kathuria KR and Davis MM: Differential roles of human
CD4+ and CD8+ regulatory T cells in controlling self-reactive
immune responses. Nat Immunol. 26:230–239. 2025. View Article : Google Scholar
|
|
60
|
Coutinho A, Caramalho I, Seixas E and
Demengeot J: Thymic commitment of regulatory T cells is a pathway
of TCR-dependent selection that isolates repertoires undergoing
positive or negative selection. Curr Top Microbiol Immunol.
293:43–71. 2005.
|
|
61
|
Kisielow P: How does the immune system
learn to distinguish between good and evil? The first definitive
studies of T cell central tolerance and positive selection.
Immunogenetics. 71:513–518. 2019. View Article : Google Scholar
|
|
62
|
Spetz J, Presser AG and Sarosiek KA: T
cells and regulated cell death: Kill or be killed. Int Rev Cell Mol
Biol. 342:27–71. 2019. View Article : Google Scholar
|
|
63
|
Lv H, Havari E, Pinto S, Gottumukkala
RVSRK, Cornivelli L, Raddassi K, Matsui T, Rosenzweig A, Bronson
RT, Smith R, et al: Impaired thymic tolerance to α-Myosin directs
autoimmunity to the heart in mice and humans. J Clin Invest.
121:1561–1573. 2011. View Article : Google Scholar
|
|
64
|
Sur M, Rasquinha MT, Arumugam R,
Massilamany C, Gangaplara A, Mone K, Lasrado N, Yalaka B, Doiphode
A, Gurumurthy C, et al: Transgenic mice expressing functional TCRs
specific to cardiac Myhc-α 334–352 on Both CD4 and CD8 T cells are
resistant to the development of myocarditis on C57BL/6 genetic
background. Cells. 12:23462023. View Article : Google Scholar
|
|
65
|
Nindl V, Maier R, Ratering D, De Giuli R,
Züst R, Thiel V, Scandella E, Di Padova F, Kopf M, Rudin M, et al:
Cooperation of Th1 and Th17 cells determines transition from
autoimmune myocarditis to dilated cardiomyopathy. Eur J Immunol.
42:2311–2321. 2012. View Article : Google Scholar
|
|
66
|
Raffin C, Vo LT and Bluestone JA: Treg
Cell-based therapies: Challenges and perspectives. Nat Rev Immunol.
20:158–172. 2020. View Article : Google Scholar
|
|
67
|
Cheru N, Hafler DA and Sumida TS:
Regulatory T cells in peripheral tissue tolerance and diseases.
Front Immunol. 14:11545752023. View Article : Google Scholar
|
|
68
|
ElTanbouly MA and Noelle RJ: Rethinking
peripheral T cell tolerance: Checkpoints across a T cell's journey.
Nat Rev Immunol. 21:257–267. 2021. View Article : Google Scholar
|
|
69
|
Bluestone JA and Anderson M: Tolerance in
the age of immunotherapy. N Engl J Med. 383:1156–1166. 2020.
View Article : Google Scholar
|
|
70
|
Pardoll DM: The blockade of immune
checkpoints in cancer immunotherapy. Nat Rev Cancer. 12:252–264.
2012. View Article : Google Scholar
|
|
71
|
Sharpe AH and Pauken KE: The diverse
functions of the PD1 inhibitory pathway. Nat Rev Immunol.
18:153–167. 2018. View Article : Google Scholar
|
|
72
|
Wing K, Onishi Y, Prieto-Martin P,
Yamaguchi T, Miyara M, Fehervari Z, Nomura T and Sakaguchi S:
CTLA-4 control over Foxp3+ regulatory T cell function. Science.
322:271–275. 2008. View Article : Google Scholar
|
|
73
|
Aggarwal V, Workman CJ and Vignali DAA:
LAG-3 as the third checkpoint inhibitor. Nat Immunol. 24:1415–1422.
2023. View Article : Google Scholar
|
|
74
|
Grabie N, Lichtman AH and Padera R: T cell
checkpoint regulators in the heart. Cardiovasc Res. 115:869–877.
2019. View Article : Google Scholar
|
|
75
|
Tan CL, Kuchroo JR, Sage PT, Liang D,
Francisco LM, Buck J, Thaker YR, Zhang Q, McArdel SL, Juneja VR, et
al: PD-1 restraint of regulatory T cell suppressive activity is
critical for immune tolerance. J Exp Med. 218:e201822322021.
View Article : Google Scholar
|
|
76
|
Zhang A, Ren Z, Tseng K-F, Liu X, Li H, Lu
C, Cai Y, Minna JD and Fu YX: Dual targeting of CTLA-4 and CD47 on
Treg cells promotes immunity against solid tumors. Sci Transl Med.
13:eabg86932021. View Article : Google Scholar
|
|
77
|
Guo X, Wang H, Zhou J, Li Y, Duan L, Si X
and Zhang L, Fang L and Zhang L: Clinical manifestation and
management of immune checkpoint Inhibitor-associated
cardiotoxicity. Thorac Cancer. 11:475–480. 2020. View Article : Google Scholar
|
|
78
|
Huertas RM, Serrano CS, Perna C, Gómez AF
and Gordoa TA: Cardiac toxicity of Immune-checkpoint inhibitors: A
clinical case of Nivolumab-induced myocarditis and review of the
evidence and new challenges. Cancer Manag Res. 11:4541–4548. 2019.
View Article : Google Scholar
|
|
79
|
Axelrod ML, Meijers WC, Screever EM, Qin
J, Carroll MG, Sun X, Tannous E, Zhang Y, Sugiura A, Taylor BC, et
al: T cells specific for α-Myosin drive Immunotherapy-related
myocarditis. Nature. 611:818–826. 2022. View Article : Google Scholar
|
|
80
|
Lim SY, Lee JH, Gide TN, Menzies AM,
Guminski A, Carlino MS, Breen EJ, Yang JYH, Ghazanfar S, Kefford
RF, et al: Circulating cytokines predict Immune-related toxicity in
melanoma patients receiving Anti-PD-1-based immunotherapy. Clin
Cancer Res. 25:1557–1563. 2019. View Article : Google Scholar
|
|
81
|
Johnson DB, Balko JM, Compton ML, Chalkias
S, Gorham J, Xu Y, Hicks M, Puzanov I, Alexander MR, Bloomer TL, et
al: Fulminant myocarditis with combination immune checkpoint
blockade. N Engl J Med. 375:1749–1755. 2016. View Article : Google Scholar
|
|
82
|
Wang DY, Salem JE, Cohen JV, Chandra S,
Menzer C, Ye F, Zhao S, Das S, Beckermann KE, Ha L, et al: Fatal
toxic effects associated with immune checkpoint inhibitors: A
systematic review and Meta-analysis. JAMA Oncol. 4:1721–1728. 2018.
View Article : Google Scholar
|
|
83
|
Martínez-Lostao L, Anel A and Pardo J: How
do cytotoxic lymphocytes kill cancer cells? Clin Cancer Res.
21:5047–5056. 2015. View Article : Google Scholar
|
|
84
|
Del Re DP, Amgalan D, Linkermann A, Liu Q
and Kitsis RN: Fundamental mechanisms of regulated cell death and
implications for heart disease. Physiol Rev. 99:1765–1817. 2019.
View Article : Google Scholar
|
|
85
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar
|
|
86
|
Voskoboinik I, Whisstock JC and Trapani
JA: Perforin and Granzymes: Function, dysfunction and human
pathology. Nat Rev Immunol. 15:388–400. 2015. View Article : Google Scholar
|
|
87
|
Wang W, Green M, Choi JE, Gijón M, Kennedy
PD, Johnson JK, Liao P, Lang X, Kryczek I, Sell A, et al: CD8+ T
cells regulate tumour ferroptosis during cancer immunotherapy.
Nature. 569:270–274. 2019. View Article : Google Scholar
|
|
88
|
Alshoubaki YK, Nayer B, Lu YZ, Salimova E,
Lau SN, Tan JL, Amann-Zalcenstein D, Hickey PF, Del Monte-Nieto G,
Vasanthakumar A, et al: Tregs Delivered Post-myocardial infarction
adopt an Injury-specific phenotype promoting cardiac repair via
macrophages in mice. Nat Commun. 15:64802024. View Article : Google Scholar
|
|
89
|
Zhu H, Galdos FX, Lee D, Waliany S, Huang
YV, Ryan J, Dang K, Neal JW, Wakelee HA, Reddy SA, et al:
Identification of pathogenic immune cell subsets associated with
checkpoint Inhibitor-induced myocarditis. Circulation. 146:316–335.
2022. View Article : Google Scholar
|
|
90
|
Zhang N and Bevan MJ: CD8(+) T cells: Foot
soldiers of the immune system. Immunity. 35:161–168. 2011.
View Article : Google Scholar
|
|
91
|
Ma P, Liu J, Qin J, Lai L, Heo GS,
Luehmann H, Sultan D, Bredemeyer A, Bajapa G, Feng G, et al:
Expansion of pathogenic cardiac macrophages in immune checkpoint
inhibitor myocarditis. Circulation. 149:48–66. 2024. View Article : Google Scholar
|
|
92
|
Blum SM, Zlotoff DA, Smith NP, Kernin IJ,
Ramesh S, Zubiri L, Caplin J, Samanta N, Martin S, Wang M, et al:
Immune responses in checkpoint myocarditis across heart, blood and
tumour. Nature. 636:215–223. 2024. View Article : Google Scholar
|
|
93
|
Moslehi JJ, Brinkley DM and Meijers WC:
Fulminant myocarditis: Evolving diagnosis, Evolving biology,
evolving prognosis. J Am Coll Cardiol. 74:312–314. 2019. View Article : Google Scholar
|
|
94
|
Altan M, Toki MI, Gettinger SN,
Carvajal-Hausdorf DE, Zugazagoitia J, Sinard JH, Herbst RS and Rimm
DL: Immune checkpoint Inhibitor-associated pericarditis. J Thorac
Oncol. 14:1102–1108. 2019. View Article : Google Scholar
|
|
95
|
Salem JE, Allenbach Y, Vozy A, Brechot N,
Johnson DB, Moslehi JJ and Kerneis M: Abatacept for severe immune
checkpoint Inhibitor-associated myocarditis. N Engl J Med.
380:2377–2379. 2019. View Article : Google Scholar
|
|
96
|
Grossman Z and Paul WE: Dynamic tuning of
lymphocytes: Physiological basis, mechanisms, and function. Annu
Rev Immunol. 33:677–713. 2015. View Article : Google Scholar
|
|
97
|
Schwartz DM, Bonelli M, Gadina M and
O'Shea JJ: Type I/II Cytokines, JAKs, and new strategies for
treating autoimmune diseases. Nat Rev Rheumatol. 12:25–36. 2016.
View Article : Google Scholar
|
|
98
|
Yan L, Wang J, Cai X, Liou YC, Shen HM,
Hao J, Huang C, Luo G and He W: Macrophage plasticity: Signaling
pathways, tissue repair, and regeneration. MedComm. 5:e6582024.
View Article : Google Scholar
|
|
99
|
Bracamonte-Baran W and Čiháková D: Cardiac
autoimmunity: Myocarditis. Adv Exp Med Biol. 1003:187–221. 2017.
View Article : Google Scholar
|
|
100
|
Cao Z, Zhang Y, Jia H, Sun X, Feng Y, Wu
H, Xu B and Wei Z: Immune checkpoint inhibitors mediate myocarditis
by promoting macrophage polarization via cGAS/STING pathway.
Cytokine. 187:1568732025. View Article : Google Scholar
|
|
101
|
Chen G, Jiang H, Yao Y, Tao Z, Chen W,
Huang F and Chen X: Macrophage, a potential targeted therapeutic
immune cell for cardiomyopathy. Front Cell Dev Biol. 10:9087902022.
View Article : Google Scholar
|
|
102
|
Munir AZ, Gutierrez A, Qin J, Lichtman AH
and Moslehi JJ: Immune-checkpoint inhibitor-mediated myocarditis:
CTLA4, PD1 and LAG3 in the heart. Nat Rev Cancer. 24:540–553. 2024.
View Article : Google Scholar
|
|
103
|
Thibult ML, Mamessier E, Gertner-Dardenne
J, Pastor S, Just-Landi S, Xerri L, Chetaille B and Olive D: PD-1
is a novel regulator of Human B-cell activation. Int Immunol.
25:129–137. 2013. View Article : Google Scholar
|
|
104
|
Lu J, Wu J, Mao L, Xu H and Wang S:
Revisiting PD-1/PD-L pathway in T and B cell response: Beyond
immunosuppression. Cytokine Growth Factor Rev. 67:58–65. 2022.
View Article : Google Scholar
|
|
105
|
Esen F, Deniz G and Aktas EC: PD-1,
CTLA-4, LAG-3, and TIGIT: The roles of immune checkpoint receptors
on the regulation of human NK cell phenotype and functions. Immunol
Lett. 240:15–23. 2021. View Article : Google Scholar
|
|
106
|
Judge SJ, Dunai C, Aguilar EG, Vick SC,
Sturgill IR, Khuat LT, Stoffel KM, Van Dyke J, Longo DL, Darrow MA,
et al: Minimal PD-1 expression in mouse and human NK cells under
diverse conditions. J Clin Invest. 130:3051–3068. 2020. View Article : Google Scholar
|
|
107
|
Merino A, Zhang B, Dougherty P, Luo X,
Wang J, Blazar BR, Miller JS and Cichocki F: Chronic stimulation
drives human NK cell dysfunction and epigenetic reprograming. J
Clin Invest. 129:3770–3785. 2019. View Article : Google Scholar
|
|
108
|
Racine JJ, Bachman JF, Zhang JG, Misherghi
A, Khadour R, Kaisar S, Bedard O, Jenkins C, Abbott A, Forte E, et
al: Murine MHC-deficient nonobese diabetic mice carrying human
HLA-DQ8 develop severe myocarditis and myositis in response to
anti-PD-1 immune checkpoint inhibitor cancer therapy. J Immunol.
212:1287–1306. 2024. View Article : Google Scholar
|
|
109
|
Taneja V and David CS: Spontaneous
autoimmune myocarditis and cardiomyopathy in HLA-DQ8.NODAbo
transgenic mice. J Autoimmun. 33:260–269. 2009. View Article : Google Scholar
|
|
110
|
Taylor JA, Havari E, McInerney MF, Bronson
R, Wucherpfennig KW and Lipes MA: A spontaneous model for
autoimmune myocarditis using the human MHC molecule HLA-DQ8. J
Immunol. 172:2651–2658. 2004. View Article : Google Scholar
|
|
111
|
Konstantina T, Konstantinos R, Anastasios
K, Anastasia M, Eleni L, Ioannis S, Sofia A and Dimitris M: Fatal
adverse events in two thymoma patients treated with anti-PD-1
immune check point inhibitor and literature review. Lung Cancer.
135:29–32. 2019. View Article : Google Scholar
|
|
112
|
Chen Q, Huang DS, Zhang LW, Li YQ, Wang HW
and Liu HB: Fatal myocarditis and rhabdomyolysis induced by
nivolumab during the treatment of type B3 thymoma. Clin Toxicol
(Phila). 56:667–671. 2018. View Article : Google Scholar
|
|
113
|
Nguyen LS, Bretagne M, Arrondeau J, Zahr
N, Ederhy S, Abbar B, Pinna B, Allenbach Y, Mira JP, Moslehi J, et
al: Reversal of Immune-checkpoint inhibitor fulminant myocarditis
using Personalized-Dose-Adjusted abatacept and ruxolitinib: Proof
of concept. J Immunother Cancer. 10:e0046992022. View Article : Google Scholar
|
|
114
|
Huang GZ and Lo YL: Correlation between
acetylcholine receptor antibody levels and thymic pathology in
myasthenia gravis: A review. J Clin Neuromuscul Dis. 14:209–217.
2013. View Article : Google Scholar
|
|
115
|
Régnier P, Le Joncour A, Maciejewski-Duval
A, Darrasse-Jèze G, Dolladille C, Meijers WC, Bastarache L, Fouret
P, Bruneval P, Arbaretaz F, et al: CTLA-4 pathway is instrumental
in giant cell arteritis. Circ Res. 133:298–312. 2023. View Article : Google Scholar
|
|
116
|
Gil-Cruz C, Perez-Shibayama C, De Martin
A, Ronchi F, van der Borght K, Niederer R, Onder L, Lütge M,
Novkovic M, Nindl V, et al: Microbiota-derived peptide mimics drive
lethal inflammatory cardiomyopathy. Science. 366:881–886. 2019.
View Article : Google Scholar
|
|
117
|
Muser D, Santangeli P and Liang JJ:
Mechanisms of ventricular arrhythmias and implications for catheter
ablation. Card Electrophysiol Clin. 14:547–558. 2022. View Article : Google Scholar
|
|
118
|
Francis Stuart SD, De Jesus NM, Lindsey ML
and Ripplinger CM: The crossroads of inflammation, fibrosis, and
arrhythmia following myocardial infarction. J Mol Cell Cardiol.
91:114–122. 2016. View Article : Google Scholar
|
|
119
|
Hulsmans M, Clauss S, Xiao L, Aguirre AD,
King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, et
al: Macrophages facilitate electrical conduction in the heart.
Cell. 169:510–522.e20. 2017. View Article : Google Scholar
|
|
120
|
Zhang Y, Qin J, Amancherla K, Jing Y, Hu
Q, Liang K, Zhang Z, Ye Y, Huang LA, Nguyen TK, et al: Hormonal
therapies upregulate MANF and overcome female susceptibility to
immune checkpoint inhibitor-myocarditis. Sci Transl Med.
14:eabo19812022. View Article : Google Scholar
|
|
121
|
Feng S, Yang M, Dong P, Ding F, Hong Y,
Cai H and Liu X: Investigation of MANF regulation of glioma
stemness via STAT3/TGF-β/SMAD4/p38 pathway based on pan-cancer
analysis. Transl Oncol. 60:1024972025. View Article : Google Scholar
|
|
122
|
Zhang Y, Sun C, Li Y, Qin J, Amancherla K,
Jing Y, Hu Q, Liang K, Zhang Z, Ye Y, et al: Hormonal therapies
Up-Regulate MANF and overcome female susceptibility to immune
checkpoint inhibitor myocarditis. Sci Transl Med. 14:eabo19812022.
View Article : Google Scholar
|
|
123
|
Wang J and Han B: Dysregulated CD4+ T
cells and microRNAs in myocarditis. Front Immunol. 11:5392020.
View Article : Google Scholar
|
|
124
|
Tajczak PM and Jóźwik K: Artificial
intelligence and myocarditis-a systematic review of current
applications. Heart Fail Rev. 29:1217–1234. 2024. View Article : Google Scholar
|
|
125
|
Brahmer JR, Abu-Sbeih H, Ascierto PA,
Brufsky J, Cappelli LC, Cortazar FB, Gerber DE, Hamad L, Hansen E,
Johnson DB, et al: Society for Immunotherapy of cancer (SITC)
clinical practice guideline on immune checkpoint inhibitor-related
adverse events. J Immunother Cancer. 9:e0024352021. View Article : Google Scholar
|
|
126
|
Quan Z, Yang Y, Zheng H, Zhan Y, Luo J,
Ning Y and Fan S: Clinical Implications of the interaction between
PD-1/PD-L1 and PI3K/AKT/mTOR pathway in progression and treatment
of non-small cell lung cancer. J Cancer. 13:3434–3443. 2022.
View Article : Google Scholar
|
|
127
|
Ramayya T, Mitchell JD, Hartupee JC,
Lavine K, Ridley CH, Kotkar KD, Jimenez J, Lin CY, Alvarez-Cardona
JA, Krone RK and Campbell CM: Delayed diagnosis and recovery of
fulminant immune checkpoint Inhibitor-associated myocarditis on
VA-ECMO support. JACC CardioOncol. 4:722–726. 2022. View Article : Google Scholar
|
|
128
|
Osinga TE, Oosting SF, van der Meer P, de
Boer RA, Kuenen BC, Rutgers A, Bergmann L, Oude Munnink TH, Jalving
M and van Kruchten M: Immune checkpoint inhibitor-associated
myocarditis: Case reports and a review of the literature. Neth
Heart J. 30:295–301. 2022. View Article : Google Scholar
|
|
129
|
Lipe DN, Qdaisat A, Krishnamani PP, Nguyen
TD, Chaftari P, El Messiri N, Srinivasan A, Galvis-Carvajal E and
Reyes-Gibby CC: Myocarditis, myositis, and myasthenia gravis
overlap syndrome associated with immune checkpoint inhibitors: A
systematic review. Diagnostics (Basel). 14:17942024. View Article : Google Scholar
|
|
130
|
Schneider BJ, Naidoo J, Santomasso BD,
Lacchetti C, Adkins S, Anadkat M, Atkins MB, Brassil KJ, Caterino
JM, Chau I, et al: Management of immune-related adverse events in
patients treated with immune checkpoint inhibitor therapy: ASCO
guideline update. J Clin Oncol. 39:4073–4126. 2021. View Article : Google Scholar
|
|
131
|
Lyon AR, Lopez-Fernandez T, Couch LS,
Asteggiano R, Aznar MC, Bergler-Klein J, Boriani G, Cardinale D,
Cordoba R, Cosyns B, et al: 2022 ESC Guidelines on cardio-oncology
developed in collaboration with the European Hematology Association
(EHA), the European Society for Therapeutic Radiology and Oncology
(ESTRO) and the International Cardio-Oncology Society (IC-OS). Eur
Heart J. 43:4229–4361. 2022. View Article : Google Scholar
|