Introduction
Keloids are a common, chronic and inflammatory skin
disorder, characterized by benign fibroproliferative overgrowth of
the skin (1). Keloids grow
continuously and aggressively beyond the original wound boundaries,
resulting from cutaneous injury or abnormal wound healing
associated with inflammatory and fibrotic conditions (2–4).
Although it is not a life-threatening condition, the incidence of
keloids is relatively high, thus reducing the quality of life of
patients due to pain, pruritus and cosmetic reasons (5,6).
Various modalities, including surgery, radiation therapy and
intralesional steroid injections, have been used for the treatment
of keloids; however, they commonly relapse (7–9). In
addition, the underlying molecular mechanisms of keloid formation
remain largely unclear, thus restricting the development of
therapeutic strategies.
Numerous studies have investigated genes and
pathways associated with the pathophysiological processes involved
in the development of keloids, thus providing novel insights into
their onset (10–13). Notably, emerging evidence has
suggested that inflammatory etiologies, particularly chronic
inflammation, may be involved in the pathogenesis of keloids
(14,15). Furthermore, previous studies have
indicated that various pro-inflammatory cytokines/chemokines and
their receptors could be notably upregulated in keloids and closely
associated with keloid inflammation (16–18).
For example, IL-17 can enhance the expression of C-X-C motif
chemokine ligand (CXCL)12 in keloids, which in turn promotes the
infiltration of C-X-C motif chemokine receptor (CXCR)4+
cells in keloid scars (18–20).
In addition, existing data have suggested that T helper 2
(Th2)-related cytokines, such as IL-4 and IL-13, may act as crucial
mediators of keloid inflammation (21). Mechanistically, IL-4 and IL-13
could be involved into skin fibrosis via excessive fibroblast
proliferation and collagen deposition in keloids (21).
Previous cDNA microarray analyses have been
performed to compare the gene expression profiles of fibroblasts
between normal skin and keloid tissues. These analyses have
identified several gene and pathway alterations, such as those in
hypoxia-inducible factor 1α (HIF-1α) (11), the fibrosis and Wnt signaling
pathway (22) and the
extracellular matrix (23), all of
which are closely associated with the pathogenesis of keloids.
Although gene chips could identify dysregulated
genes in keloid tissues, the small sample size and considerable
heterogeneity may affect data reproducibility. Therefore, the
present study retrieved several microarray datasets from the Gene
Expression Omnibus (GEO) database, with the aim of identifying
common differentially expressed genes (DEGs) between keloid
fibroblasts (KFs) and normal fibroblasts (NFs). Then, gene
knockdown assays and RNA sequencing were performed to investigate
the expression profiles, functions and potential pathogenic
mechanisms of DEGs in keloids.
Materials and methods
Microarray data processing and
screening of target DEGs
Three independent keloid datasets, namely GSE145725
(11), GSE7890 (22) and GSE44270 (23), were obtained from the GEO
(https://www.ncbi.nlm.nih.gov/geo/). A
total of 19 samples from GSE145725 (10 NF and 9 KF samples), 10
samples from GSE7890 (5 NF and 5 KF samples) and 12 samples from
GSE44270 (3 NF and 9 KF samples) were selected for further analysis
(details are shown in Table I).
The original expression matrix was normalized and analyzed using R
package (version 4.2.1) (24).
DEGs were screened using the Limma package (25). Genes with P<0.05 and |log2 fold
change (FC)|>1 were defined as DEGs in GSE145725, GSE7890 and
GSE44270. TBtools was used to construct a Venn diagram to identify
common DEGs from the three datasets (26).
 | Table I.Details of datasets used in the
present study. |
Table I.
Details of datasets used in the
present study.
| First author,
year | GEO accession
no. | Number of KF
samples | Number of NF
samples | KF samples | NF samples | Experiment
type/platform | (Refs.) |
|---|
| Smith et
al, | GSE7890 | 5 | 5 | GSM194109; | GSM194118; | Expression | (22) |
| 2008 |
|
|
| GSM194110; | GSM194119; | profiling by |
|
|
|
|
|
| GSM194111; | GSM194120; | array/ |
|
|
|
|
|
| GSM194112; | GSM194121; | GPL570 |
|
|
|
|
|
| GSM194113 | GSM194122 |
|
|
| Hahn et
al, | GSE44270 | 9 | 3 | GSM1081582; | GSM1081608; | Expression | (23) |
| 2013 |
|
|
| GSM1081583; | GSM1081609; | profiling by |
|
|
|
|
|
| GSM1081584; | GSM1081610 | array/ |
|
|
|
|
|
| GSM1081585; |
| GPL6244 |
|
|
|
|
|
| GSM1081586; |
|
|
|
|
|
|
|
| GSM1081587; |
|
|
|
|
|
|
|
| GSM1081588; |
|
|
|
|
|
|
|
| GSM1081589; |
|
|
|
|
|
|
|
| GSM1081590 |
|
|
|
| Kang et
al, | GSE145725 | 9 | 10 | GSM4331585; | GSM4331594; | Expression | (11) |
| 2020 |
|
|
| GSM4331586; | GSM4331595; | profiling by |
|
|
|
|
|
| GSM4331587; | GSM4331596; | array/ |
|
|
|
|
|
| GSM4331588; | GSM4331597; | GPL6244 |
|
|
|
|
|
| GSM4331589; | GSM4331598; |
|
|
|
|
|
|
| GSM4331590; | GSM4331599; |
|
|
|
|
|
|
| GSM4331591; | GSM4331600; |
|
|
|
|
|
|
| GSM4331592; | GSM4331601; |
|
|
|
|
|
|
| GSM4331593 | GSM4331602, |
|
|
|
|
|
|
|
| GSM4331603 |
|
|
Patients and specimen collection
All subjects (three patients with keloids and three
healthy controls) were recruited from February to May 2023 from the
Department of Dermatology, Peking University Shenzhen Hospital
(Shenzhen, China). The average age of patients with keloids was
28.3 years (range, 25–30 years), while the average age of healthy
individuals was 28.7 years (range, 23–37 years). A total of two men
and one woman were diagnosed with keloids, and sex-matched healthy
controls (two men and one woman) were included in the present
study. All of the patients with keloids enrolled in the current
study did not receive any topical therapy or systemic treatment for
≥1 month before skin biopsy. Biopsies from the non-lesional skin of
healthy volunteers were used as controls. Specimens were
immediately placed in ice-cold 1X PBS pre-mixed with 1X penicillin
and 1X streptomycin. The present study was approved by the Ethics
Committee of Peking University Shenzhen Hospital and was conducted
in accordance with The Declaration of Helsinki. Written informed
consent was obtained from all participants.
Cell culture
Primary human dermal fibroblasts were isolated from
skin tissues obtained from the three patients with keloids and the
three healthy controls as previously described (27). Firstly, the epidermis and adipose
tissues were removed from the specimens obtained from the healthy
donors/patients with keloids. Subsequently, the dermis was cut into
0.25 cm2 sections and allowed to attach to a 10-cm
culture dish. Fibroblasts began to migrate out and proliferate from
the edges of the dermis onto the culture dish. This process yields
a pure population of fibroblasts for subsequent studies. The
fibroblasts obtained from keloid patients were designated KF#1,
KF#2 and KF#3, while those derived from healthy donors were labeled
NF#1, NF#2 and NF#3. The fibroblast cells were cultured in DMEM
supplemented with 10% fetal bovine serum, 100 U/ml penicillin and
100 µg/ml streptomycin (all Gibco; Thermo Fisher Scientific, Inc.)
in a CO2 incubator with 5% CO2 at 37°C.
Reagents, small interfering (si)RNAs
and antibodies
siRNAs targeting POSTN and IL-4R, as well as the
controls (si-CTL, forward, 5′-CCUAAGGUUAAGUCGCCCU-3′ and reverse,
5′-AGGGCGACUUAACCUUAGG-3′) were from Guangzhou RiboBio Co., Ltd.
The siRNA sequences targeting the POSTN gene were as follows:
si-POSTN#1, forward, 5′-GUGACAGUAUAACAGUAAA-3′ and reverse,
5′-UUUACUGUUAUACUGUCAC-3′; si-POSTN#2, forward,
5′-GGAUCAGCGCCUCCUUAAA-3′ and reverse, 5′-UUUAAGGAGGCGCUGAUCC-3′.
The siRNA sequences targeting the IL-4R gene were as follows:
si-IL-4R#1, forward, 5′-GUCAACAUUUGGAGUGAAA-3′ and reverse,
5′-UUUCACUCCAAAUGUUGAC-3′; si-IL-4R#2, forward,
5′-CUUCCACCUUCGGGAAGUA-3′ and reverse, 5′-UACUUCCCGAAGGUGGAAG-3′.
Antibodies against POSTN (cat. no. ab14041) and IL-4R (cat. no.
ab203398) were from Abcam, antibodies against phosphorylated
(p)-STAT1 (Tyr701) (cat. no. 9167), p-STAT1 (Ser727) (cat. no.
8826) and STAT1 (cat. no. 14994) were from Cell Signaling
Technology, Inc., whereas the antibody against β-actin (cat. no.
AF5003) was purchased from Beyotime Institute of Biotechnology. The
SuperSignal West Femto Chemiluminescent Substrate Kit used for
signal detection in western blotting was obtained from Thermo
Fisher Scientific, Inc. (cat. no. 34095). All other reagents or
chemicals were purchased from Thermo Fisher Scientific, Inc. unless
otherwise mentioned.
Western blot analysis
Western blotting was performed as described
previously (28). Briefly, protein
samples from fibroblasts were harvested using ice-cold RIPA buffer
containing 10 mM Tris pH 7.4, 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1
mM NaF, 20 mM Na4P2O7, 1% Triton
X-100, 10% glycerol, 0.1% SDS and 0.5% deoxycholate, and protease
and phosphatase inhibitor cocktails. The protein concentration was
determined using the BCA method (cat. no. 5000002; Bio-Rad
Laboratories, Inc.). A total of 10 µg/lane protein was separated
using a 10% SDS-PAGE gel, after which they were transferred onto
PVDF membranes. The membranes were blocked with blocking buffer
(cat. no. ST023; Beyotime Institute of Biotechnology) for 1 h at
room temperature, and incubated with primary antibodies (1:1,000)
as follows: phosphorylated (p)-STAT1 (Tyr701) (cat. no. 9167),
p-STAT1 (Ser727) (cat. no. 8826) and STAT1 (cat. no. 14994; all
Cell Signaling Technology, Inc.; β-actin (cat. no. AF5003) was from
Beyotime Institute of Biotechnology. The membranes were incubated
with primary antibodies overnight at 4°C. Subsequently, the
corresponding secondary antibodies, goat anti-rabbit IgG H&L
(HRP) (cat. no. ab205718; 1:10,000; Abcam) for p-STAT1 (Tyr701),
p-STAT1 (Ser727) and STAT1 and m-IgGκ BP-HRP (cat. no. sc-516102;
dilution: 1:10,000; Santa Cruz Biotechnology, Inc.) for β-actin,
were incubated for 2 h at room temperature. The signals were
detected using the SuperSignal West Femto Chemiluminescent
Substrate Kit (cat. no. 34095; Thermo Fisher Scientific, Inc.). The
images were analyzed using ImageJ software (version 1.54; National
Institutes of Health) and the relative content of the target
protein was expressed as the gray value of the target
protein/β-actin gray value. A total of three biological replicates
was performed for each treatment group.
Immunofluorescence staining
POSTN levels in KFs were detected using
immunofluorescence staining as described previously (29). Briefly, cells were fixed using 4%
paraformaldehyde at room temperature for 15 min, followed by
permeabilization for 10 min at room temperature using 0.1% Triton
X-100 (cat. no. P0096; Beyotime Institute of Biotechnology). Cells
were blocked with blocking solution (cat. no. P0102; Beyotime
Institute of Biotechnology) for 1 h at room temperature, followed
by incubation with the primary antibody against POSTN (cat. no.
ab14041; dilution: 1:100; Abcam) at 4°C overnight. After washing
off the primary antibody, an Alexa Fluor555-labeled donkey
anti-rabbit IgG (H+L) secondary antibody (cat. no. A0453; 1:200;
Beyotime Institute of Biotechnology) was applied for 30 min at room
temperature. For immunofluorescence staining, cells were
counterstained with DAPI (cat. no. ab104139; Abcam) at room
temperature for 15 min, and fluorescence microscopy (Nikon Inc.,
Melville, NY) was used for observation.
RNA isolation and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted with TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). Subsequently,
RT-qPCR was performed as described previously (29). Briefly, total RNA was
reverse-transcribed into cDNA using ReverTra Ace qPCR RT kit (cat.
no. FSQ-201; Toyobo Life Science) with the following temperature
protocol: 37°C for 15 min, 50°C for 5 min, and 98°C for 5 min. qPCR
was performed using iTaq™ Universal SYBR® Green Supermix
(cat. no. 1725121; Bio-Rad Laboratories, Inc.) with an initial
denaturation at 95°C for 30 sec and the following thermocycling
conditions: 95°C for 10 sec, 59°C for 25 sec and 72°C for 30 sec
for 40 cycles. The relative mRNA expression levels of target genes
were normalized to those of GAPDH using the 2−ΔΔCq
method (30). All of the primers
used in the present study were designed using the online software
PrimerBank (https://pga.mgh.harvard.edu/primerbank/index.html).
The primer sequences were as follows: GAPDH-forward (F),
5′-ACAACTTTGGTATCGTGGAAGG-3′, GAPDH-reverse (R),
5′-GCCATCACGCCACAGTTTC-3′; POSTN-F, 5′-CTCATAGTCGTATCAGGGGTCG-3′,
POSTN-R, 5′-ACACAGTCGTTTTCTGTCCAC-3′; IL-4R-F,
5′-CGTGGTCAGTGCGGATAACTA-3′, IL-4R-R, 5′-TGGTGTGAACTGTCAGGTTTC-3′.
CCNA1-F, 5′-GAGGTCCCGATGCTTGTCAG-3′, CCNA1-R,
5′-GTTAGCAGCCCTAGCACTGTC-3′; CCNA2-F, 5′-CGCTGGCGGTACTGAAGTC-3′,
CCNA2-R, 5′-GAGGAACGGTGACATGCTCAT-3′; CCNB1 F,
5′-AATAAGGCGAAGATCAACATGGC-3′, CCNB1-R,
5′-TTTGTTACCAATGTCCCCAAGAG-3′; CCNB2 F, 5′-CCGACGGTGTCCAGTGATTT-3′,
CCNB2-R, 5′-TGTTGTTTTGGTGGGTTGAACT-3′; CCNC-F,
5′-CCTTGCATGGAGGATAGTGAATG-3′, CCNC-R,
5′-AAGGAGGATACAGTAGGCAAAGA-3′; CCND3-F, 5′-TACCCGCCATCCATGATCG-3′,
CCND3-R, 5′-AGGCAGTCCACTTCAGTGC-3′; CCNE2-F
5′-TCAAGACGAAGTAGCCGTTTAC-3′, CCNE2-R,
5′-TGACATCCTGGGTAGTTTTCCTC-3′; CDK1-F 5′-AAACTACAGGTCAAGTGGTAGCC-3′
and R, 5′-TCCTGCATAAGCACATCCTGA-3′; CDK2-F,
5′-CCAGGAGTTACTTCTATGCCTGA-3′, CDK2-R, 5′-TTCATCCAGGGGAGGTACAAC-3′;
CDK3-F, 5′-GAAGGTAGAGAAGATCGGAGAGG-3′, CDK3-R,
5′-GTCCAGCAGTCGGACGATG-3′; CDK4-F, 5′-ATGGCTACCTCTCGATATGAGC-3′,
CDK4-R, 5′-CATTGGGGACTCTCACACTCT-3′; CDK5-F,
5′-GGAAGGCACCTACGGAACTG-3′, CDK5-R, 5′-GGCACACCCTCATCATCGT-3′;
JAK1-F, 5′-CTTTGCCCTGTATGACGAGAAC-3′, JAK1-R,
5′-ACCTCATCCGGTAGTGGAGC-3′; JAK2-F, 5′-TCTGGGGAGTATGTTGCAGAA-3′,
JAK2-R, 5′-AGACATGGTTGGGTGGATACC-3′; STAT1-F,
5′-CGGCTGAATTTCGGCACCT-3′, STAT1-R, 5′-CAGTAACGATGAGAGGACCCT-3′;
STAT2-F, 5′-CCAGCTTTACTCGCACAGC-3′, STAT2-R,
5′-AGCCTTGGAATCATCACTCCC-3′; IFNAR2-F,
5′-TCATGGTGTATATCAGCCTCGT-3′, IFNAR2-R,
5′-AGTTGGTACAATGGAGTGGTTTT-3′; IFNGR1-F,
5′-TCTTTGGGTCAGAGTTAAAGCCA-3′, IFNGR1-R,
5′-TTCCATCTCGGCATACAGCAA-3′; CXCL1-F, 5′-GCCAGTGCTTGCAGACCCT-3′,
CXCL1-R, 5′-GGCTATGACTTCGGTTTGGG-3′; CXCL2-F,
5′-CAAACCGAAGTCATAGCCAC-3′ and R, 5′-TCTGGTCAGTTGGATTTGCC-3′;
CXCL5-F, 5′-AGCTGCGTTGCGTTTGTTTAC-3′, CXCL5-R,
5′-TGGCGAACACTTGCAGATTAC-3′; CXCL8-F, 5′-TCTGTCTGGACCCCAAGGAA-3′,
CXCL8-R, 5′-GCATCTGGCAACCCTACAACA-3′; CXCL12-F,
5′-ATTCTCAACACTCCAAACTGTGC-3′, CXCL12-R,
5′-ACTTTAGCTTCGGGTCAATGC-3′; CCL2-F, 5′-CAGCCAGATGCAATCAATGCC-3′,
CCL2-R, 5′-TGGAATCCTGAACCCACTTCT-3′; CCL28-F,
5′-TGCACGGAGGTTTCACATCAT-3′, CCL28-R, 5′-TTGGCAGCTTGCACTTTCATC-3′;
IL33-F, 5′-GTGACGGTGTTGATGGTAAGAT-3′ and R,
5′-AGCTCCACAGAGTGTTCCTTG-3′; IL34-F, 5′-CCTGGCTGCGCTATCTTGG-3′,
IL34-R, 5′-AGTGTTTCATGTACTGAAGTCGG-3′; LIF-F,
5′-CCAACGTGACGGACTTCCC-3′, LIF-R, 5′-TACACGACTATGCGGTACAGC-3′;
LIFR-F, 5′-TGGAACGACAGGGGTTCAGT-3′, LIFR-R,
5′-GAGTTGTGTTGTGGGTCACTAA-3′; TSLP-F,
5′-ATGTTCGCCATGAAAACTAAGGC-3′, TSLP-R, 5′-GCGACGCCACAATCCTTGTA-3′;
GDF5-F, 5′-GCTGGGAGGTGTTCGACATC-3′, GDF5-R,
5′-CACGGTCTTATCGTCCTGGC-3′; FAS-F, 5′-AGATTGTGTGATGAAGGACATGG-3′,
FAS-R, 5′-TGTTGCTGGTGAGTGTGCATT-3′; OSMR-F,
5′-AATGTCAGTGAAGGCATGAAAGG-3′, OSMR-R, 5′-GAAGGTTGTTTAGACCACCCC-3′;
COL4A5-F, 5′-TGGACAGGATGGATTGCCAG-3′, COL4A5-R,
5′-GGGGACCTCTTTCACCCTTAAAA-3′; COL6A6-F,
5′-TTCAACATTGCTCCCCATAAGG-3′ and R, 5′-GTGTTTGTATTCCCACCCATCT-3′;
DST-F, 5′-CTACCAGCACTCGAACCAGTC-3′, DST-R,
5′-GCCGAAGCTAATGCAAGAGTTG-3′; MMP3-F, 5′-CTGGACTCCGACACTCTGGA-3′,
MMP3-R, 5′-CAGGAAAGGTTCTGAAGTGACC-3′.
Cell transfection, IFNα, IFNβ, IL-4
and/or IL-13 treatment, cell counting Kit-8 (CCK-8) assay, EdU Cell
Proliferation assay and cell cycle assay
Cells were cultured until they reached 70%
confluence and were then transfected with 100 nM POSTN or IL-4R
siRNAs at 37°C for 24 h using Lipofectamine® RNAiMAX
Transfection Reagent (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Cells were
transfected with siRNAs for 24 h prior to subsequent experiments.
Cells were treated with recombinant human IFNs or IL-4/IL-13. For
IFNα and IFNβ treatment, cells were incubated with recombinant
human IFNα (cat. no. 300-02AA; 50 ng/ml) and IFNβ (cat. no.
300-02BC; 50 ng/ml) (both from PeproTech; Thermo Fisher Scientific,
Inc.) at 37°C for 15 min when cells reached 90% confluence, control
cells were treated with PBS under the same conditions. For IL-4
and/or IL-13 treatment, cells were incubated with recombinant human
IL-4 (cat. no. 200-04; 50 ng/ml) and/or IL-13 (cat. no. 200-13; 50
ng/ml) (both from PeproTech; Thermo Fisher Scientific, Inc.) at
37°C for 24 h when cells reaching 90% confluent, control cells were
treated with PBS under the same conditions. Following the
manufacturer's instructions, the Cell Counting Kit-8 (CCK-8) assay
(cat. no. A311-01; Vazyme) was performed to evaluate cell
proliferation rates. Cells were seeded on a 96-well plates with a
density of 1×104 KFs/well for the POSTN knockdown
experiment. After siRNA transfection for 24 h at 37°C, CCK-8
reagent was added to the KFs and incubated for 2 h at 37°C.
Finally, a microplate reader (Infinite 200PRO; Tecan Group, Ltd.)
was used to measure the absorbance at 450 nm. EdU Cell
Proliferation assay was performed using an EdU Cell Proliferation
Kit with Alexa Fluor 555 according to the manufacturer's
instruction (cat. no. MA0425-1; Dalian Meilun Biology Technology
Co., Ltd.). For nuclear staining, cells were counterstained with
Hoechst at room temperature for 10 min and confocal microscopy was
used to acquire images. The cell cycle assay was performed as
described previously (31).
Briefly, trypsinised cells were fixed in pre-cooled 70% ethanol at
4°C for 30 min. Following incubation in 50 µg/ml propidium iodide
solution in the presence of RNase for 30 min at room temperature,
the cells were analyzed using a flow cytometry instrument (FACS
Calibur; Becton Dickinson, Franklin, NJ), and the data were
processed with the MODFIT software (Verity Software House, Topsham,
ME). At least three independent experiments were carried out for
each treatment.
RNA sequencing (RNA-seq) and data
analysis
Total RNA was extracted from si-POSTN- and
si-CTL-transfected KFs using TRI Reagent® (cat. no.
T9424; MilliporeSigma). Purified RNA was quantified and qualified,
before library quality was assessed on an Agilent 2100 Bioanalyzer
(Agilent Technologies, Inc.). The cDNA library was constructed
using a Fast RNA-seq Lib Prep kit V2 (cat. no. RK20306; ABclonal)
and the library was sequenced using Illumina Novaseq 6000 platform
(Illumina, Inc.) with the Novaseq 6000 S4 reagent Kit (cat. no.
20027466; Illumina, Inc.) by Novogene Co., Ltd. Sequencing was
carried out on the Illumina platform using paired-end sequencing
with a read length of 150 bp. The final library was quantified
using a Qubit 4.0 Fluorometer (Thermo Fisher Scientific, Inc.) and
the final concentration was adjusted to 1.5 nM. The original
expression matrix was normalized and analyzed using HiSaT2 (v2.0.5;
daehwankimlab.github.io/hisat2/) and featureCounts (v1.5.0-p3;
subread.sourceforge.net/). DEseq2 software (v1.20.0; http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
was used to detect the DEGs between the two groups; DEGs with
|log2FC|>1 and FDR<0.05 were considered significant.
Visualization of a boxplot, heatmap and volcano plot, as well as
gene set enrichment analysis (GSEA) and Reactome analysis were
performed using R software (v4.3.2; http://www.r-project.org/).
Statistical analysis
All experiments were repeated at least three times
unless otherwise stated. Data are presented as the mean ± SEM.
Statistical analysis was conducted using GraphPad Prism 9.0
software (Dotmatics). For two-group comparisons, unpaired Student's
t-test was applied, while one-way ANOVA followed by Tukey's post
hoc test was employed for multiple group comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
POSTN is upregulated in KFs
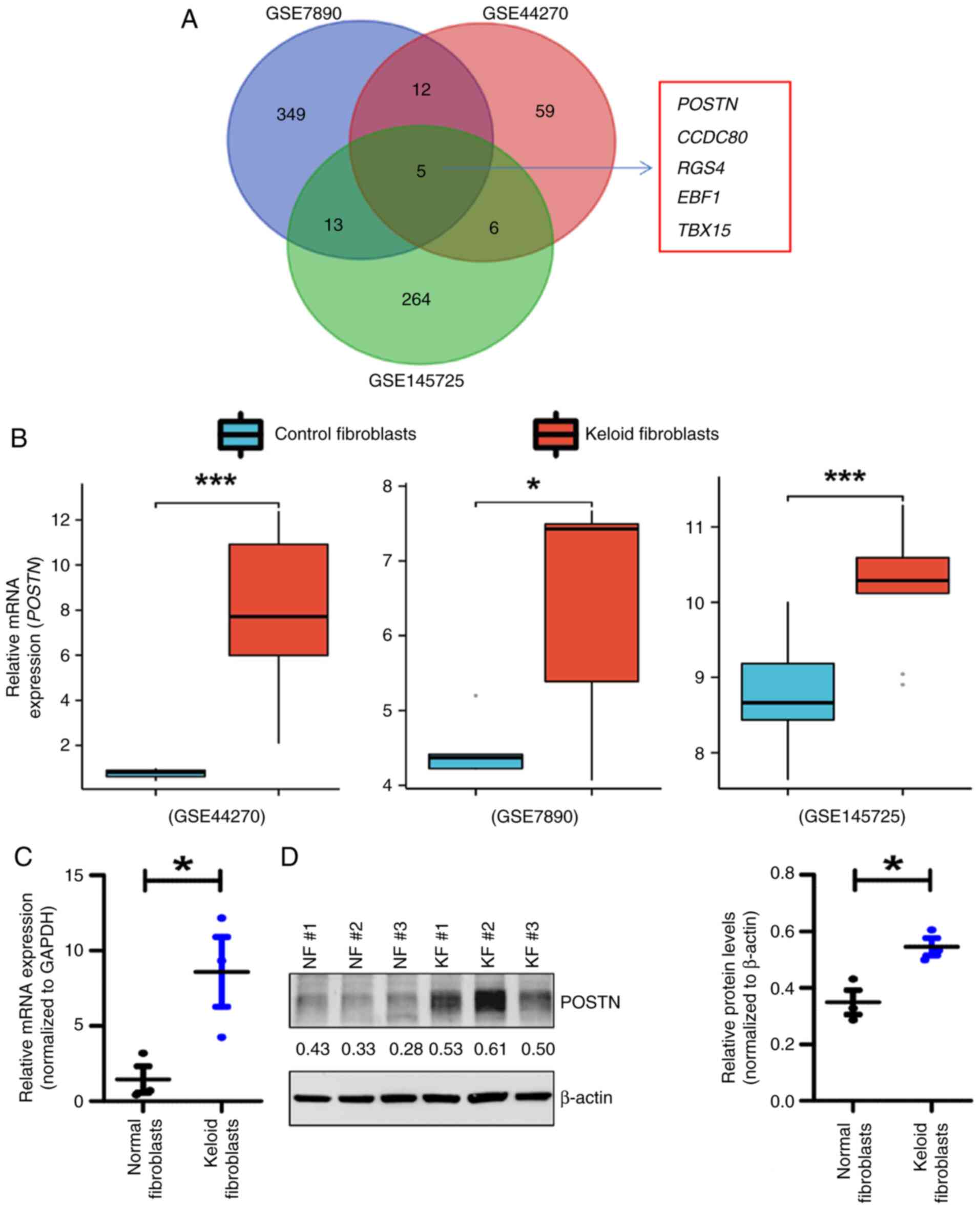
GSE7890, GSE44270 and GSE145725 datasets,
encompassing the expression profiles of genes in control
fibroblasts from healthy individuals and KFs from patients, were
downloaded from the GEO database. DEGs were defined as those with
significant differential expression between KFs and NFs, with
P<0.05 and |log2FC|>1. Upregulated DEGs were considered those
which were highly expressed in KFs compared with NFs. Notably, a
total of 82,379 and 288 upregulated DEGs were identified in the
GSE44270, GSE7890 and GSE145725 datasets, respectively (Fig. 1A). Additionally, a Venn diagram
revealed that five upregulated DEGs overlapped among the
aforementioned three datasets. Specifically, POSTN, CCDC80, RGS4,
EBF1 and TBX15 were upregulated in KFs compared with NFs. Among
these five genes, POSTN was more significantly upregulated in KFs
(Fig. 1B). Previous studies have
indicated that POSTN is markedly dysregulated in keloids, thus
supporting its potential role in the pathogenesis of keloid
(32,33). Therefore, POSTN was identified as a
gene that could be potentially associated with keloid development.
Subsequently, to detect the expression profile of POSTN in KFs and
NFs derived from patients recruited for the present study, RT-qPCR,
western blotting and immunofluorescence assays were performed.
Consistent with the results of bioinformatics analysis, the mRNA
expression levels of POSTN were significantly higher in KFs
compared with those in NFs (Fig.
1C). Furthermore, western blot analysis (Fig. 1D) and immunofluorescence staining
assays (Fig. S1) demonstrated
that the protein expression levels of POSTN were markedly increased
in KFs compared with those in NFs. Based on these results, it was
hypothesized that POSTN could represent a potential modulator of
keloid pathogenesis.
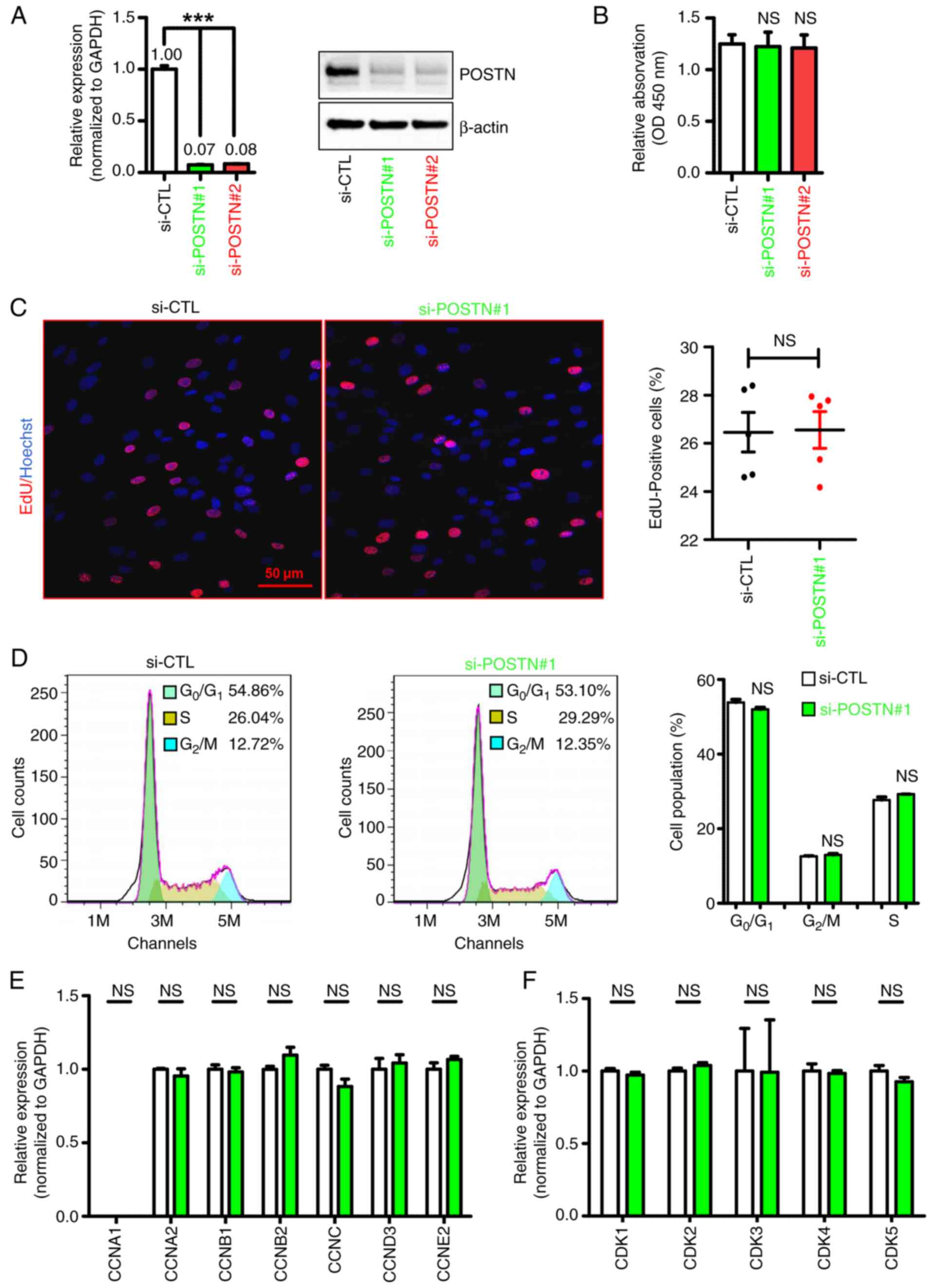
POSTN knockdown exerts limited effects
on the proliferation of KFs
To assess the regulatory role of POSTN in the
pathogenesis of keloids, the endogenous expression of POSTN was
silenced in KFs using siRNA (si-POSTN) technology. As shown in
Fig. 2A, POSTN expression was
significantly decreased in KFs transfected with si-POSTN compared
with that in the control cells; the knockdown efficiencies of
si-POSTN#1 and si-POSTN#2 were 93 and 92%, respectively (Fig. 2A). Since the knockdown efficiency
of si-POSTN#1 is higher than that of si-POSTN#2, we selected
si-POSTN#1 for subsequent experiments unless otherwise noted. The
CCK-8 assay demonstrated that POSTN silencing exerted no effect on
the proliferation of KFs (Fig.
2B). Additionally, the EdU incorporation assay revealed that
cell proliferation was comparable between KFs transfected with
si-POSTN and si-CTL (Fig. 2C),
thus suggesting that POSTN silencing had no notable effect on
fibroblast proliferation. Furthermore, flow cytometry was utilized
to examine whether POSTN knockdown could affect cell cycle
distribution in KFs. As shown in Fig.
2D, no changes were detected in the proportion of cells in the
G0/G1 or S phases of the cell cycle between
the si-POSTN and si-CTL groups, thus indicating that POSTN
knockdown did not affect the cell cycle distribution of KFs. To
further investigate the effect of POSTN on KF proliferation, the
expression levels of cell proliferation- and cell cycle-related
genes were detected in si-POSTN- and si-CTL-transfected KFs. In
line with the aforementioned results, the RT-qPCR analysis results
showed that POSTN knockdown did not alter the expression levels of
the majority of cell proliferation- and cell cycle-related genes in
KFs, since the expression levels of CCNA1/A2/B1/B2/C/D3/E2 and
CDK1/2/3/4/5 were comparable between the si-POSTN and si-CTL groups
(Fig. 2E and F). Collectively,
these findings suggested that POSTN knockdown exerted limited
effects on the proliferation of KFs.
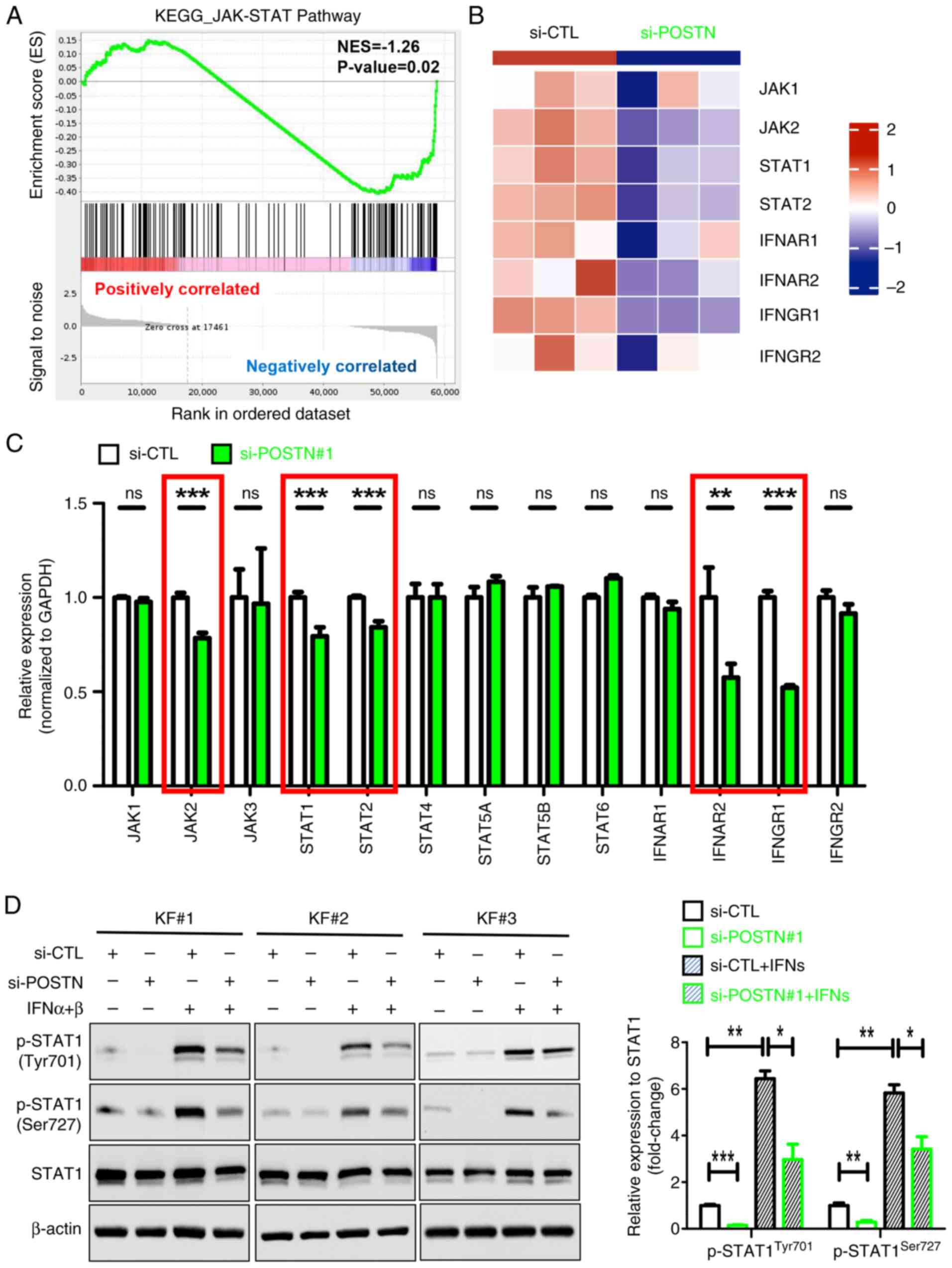
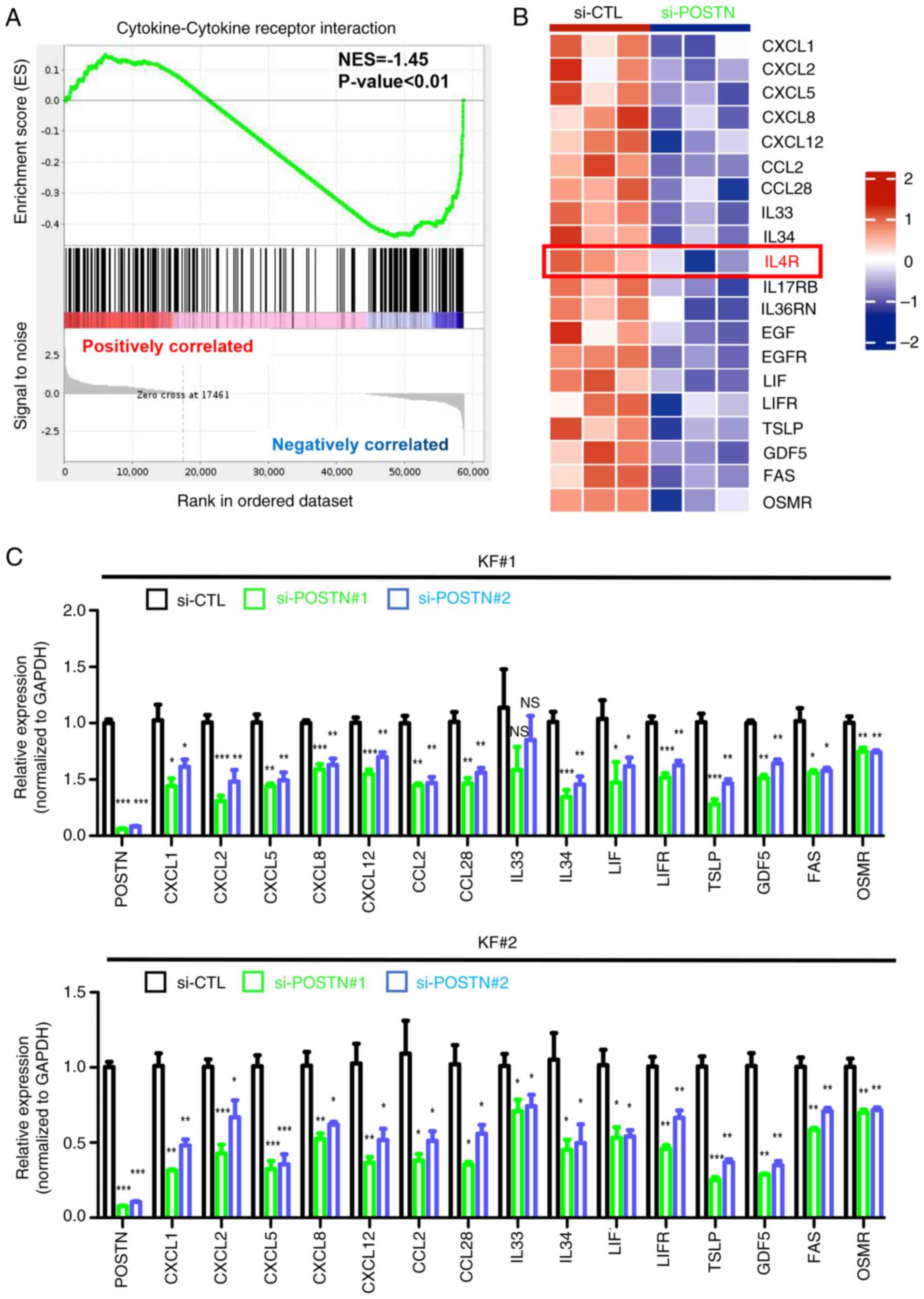
POSTN knockdown inhibits the JAK/STAT
signaling and the expression of proinflammatory factors in KFs
To further explore the role of POSTN in KFs, cells
transfected with si-POSTN and si-CTL were subjected to RNA-seq.
Notably, a total of 3,893 DEGs were identified between the two
groups, including 1,856 upregulated and 2,037 downregulated genes
(Fig. S2). Subsequently, GSEA was
performed to investigate the potential biological functions of
these DEGs, which could be involved in keloid pathogenesis. The
results showed that the majority of genes were enriched in
‘JAK/STAT signaling pathway’, ‘cytokine-cytokine receptor
interaction’, ‘NF-kappa B’, ‘natural killer cell-mediated
cytotoxicity’, ‘antigen processing’ and ‘TNF’ signaling pathways
(Fig. S3). Emerging evidence has
suggested that the JAK/STAT signaling pathway and proinflammatory
cytokines, such as CXCL12, C-C motif chemokine ligand (CCL)2 and
thymic stromal lymphopoietin (TSLP), serve crucial roles in the
pathogenesis of keloids (19,20,34).
Therefore, the current study mainly focused on the regulatory
effect of POSTN on the ‘JAK/STAT signaling pathway’ (Fig. 3A) and ‘cytokine-cytokine receptor
interaction’ (Fig. 4A) in KFs.
Heatmap analysis showed that the expression of the JAK/STAT
pathway-related DEGs was evidently altered in si-POSTN-transfected
KFs compared with in si-CTL-transfected KFs (Fig. 3B). In addition, the expression
levels of JAK/STAT pathway-related genes, including JAK2, STAT1/2,
IFNAR2 and IFNGR1, were significantly decreased in POSTN-depleted
KFs (Fig. 3C, marked in red
boxes). Furthermore, POSTN silencing markedly reduced the protein
expression levels of p-STAT1 (Tyr701) and p-STAT1 (Ser727) in KFs,
regardless of IFN-α/β treatment (Fig.
3D). Collectively, these results suggested that POSTN may be
involved in the activation of JAK/STAT signaling in KFs.
The expression pattern of DEGs associated with
‘cytokine-cytokine receptor interaction’ is presented in a heatmap
in Fig. 4B. To further validate
the significance of POSTN expression in the production of
proinflammatory cytokines/chemokines, POSTN was silenced in KFs
derived from two different patients with keloids using two sets of
specific si-POSTN constructs. RT-qPCR indicated mRNA expression
levels of keloid inflammation-related DEGs, including
CXCL1/CXCL2/CXCL5/CXCL8/CXCL12, CCL2/CCL28, IL33/IL34, LIF/LIFR,
TSLP, GDF5, FAS and OSMR, were similar between the two patients
(Fig. 4C). Notably, the majority
of DEGs were downregulated in POSTN-depleted KFs compared with in
control cells, thus indicating that POSTN could modulate the
expression of inflammatory cytokines in KFs. Taken together, the
aforementioned findings suggested that POSTN could serve a key role
in maintaining keloid inflammation.
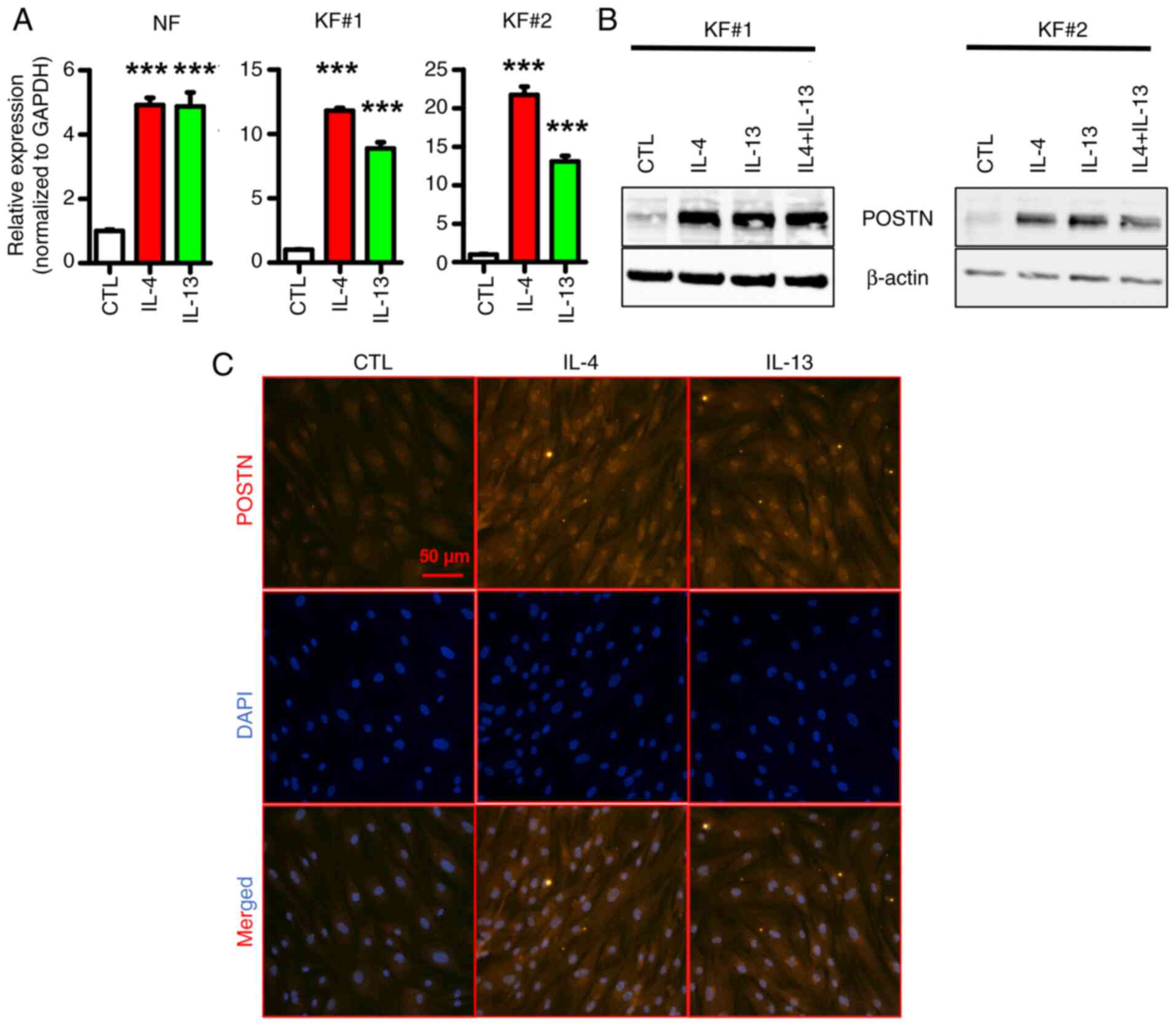
IL-4 and/or IL-13 upregulates POSTN in
KFs
Previous studies have demonstrated that IL-4 and
IL-13, two signature type 2 cytokines, can induce the expression of
POSTN in inflammation-related diseases such as atopic dermatitis
and bronchial asthma (35–37). Notably, another study showed that
Th2 markers are significantly upregulated in keloid lesions
compared with in normal skin, thus suggesting that keloid
pathogenesis could be triggered by Th2 cytokines (32). Therefore, the present study
explored whether IL-4 and/or IL-13 could induce POSTN expression in
KFs. As shown in Fig. 5A and B,
treatment of primary fibroblasts with IL-4 and/or IL-13 notably
induced POSTN expression, at both the mRNA (Fig. 5A) and protein levels (Fig. 5B). Additionally, the
immunofluorescence staining results further verified that IL-4 and
IL-13 could induce the protein expression of POSTN in KFs (Fig. 5C). These results suggested that
IL-4 and/or IL-13 could upregulate POSTN in KFs.
IL-4R is essential for
IL-4/IL-13-induced POSTN upregulation in KFs
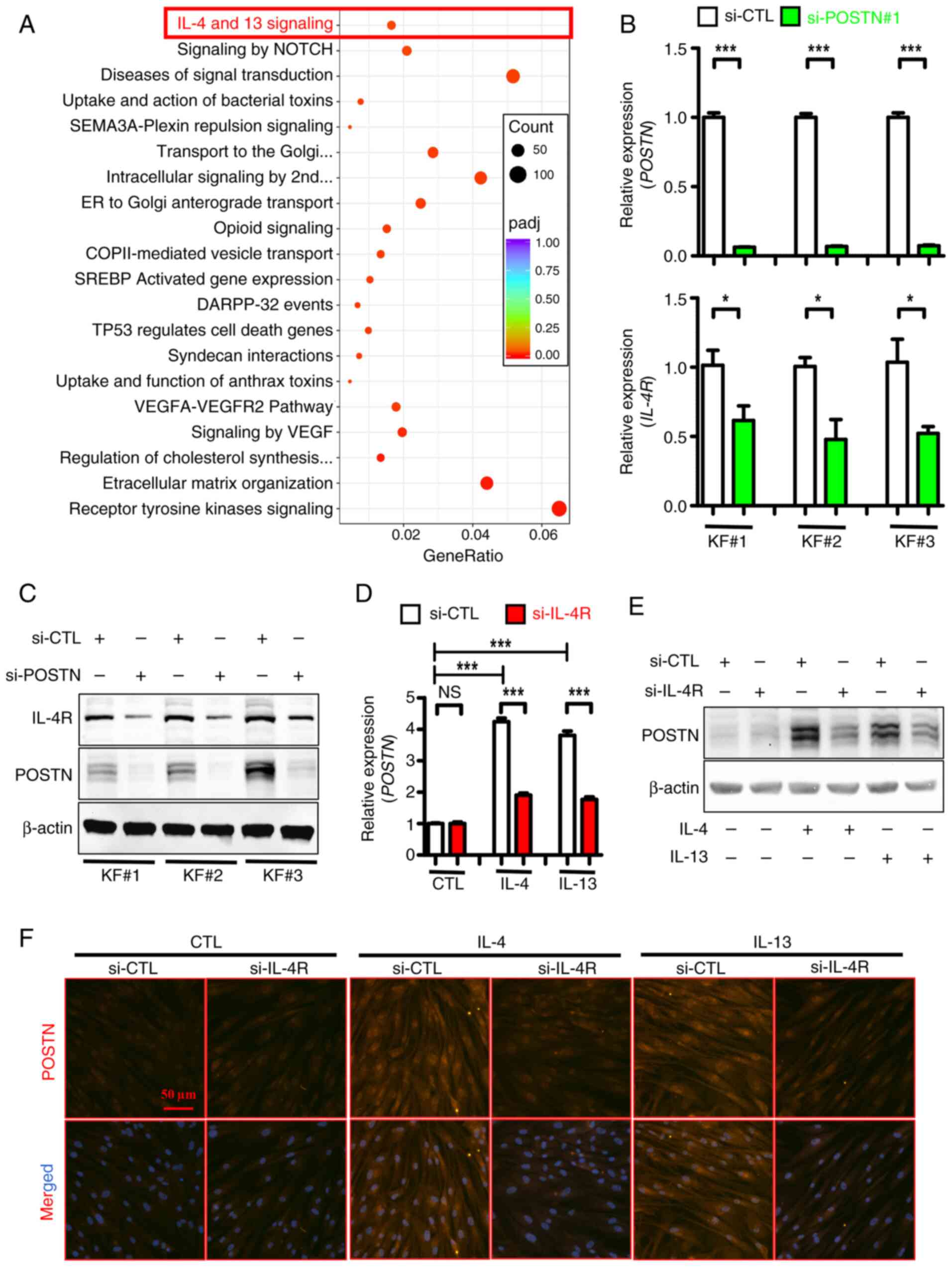
Notably, Reactome enrichment analysis revealed that
the ‘IL-4 and IL-13 signaling’ pathway was significantly enriched
in DEGs between POSTN-depleted and control KFs (Fig. 6A, highlighted in red). This finding
was consistent with previous reports suggesting that POSTN is
strongly involved in Th2 cell-mediated inflammation (38,39).
Notably, the mRNA (Fig. 6B) and
protein (Fig. 6C) expression
levels of IL-4R, a cytokine receptor for both IL-4 and IL-13, were
notably inhibited in POSTN-depleted KFs derived from three patients
with keloids. Additionally, protein expression levels of IL-4R and
POSTN were higher in KFs compared with NFs, suggesting a positive
association of the expression between these two proteins in primary
fibroblasts (Fig. S4A). The
aforementioned finding indicated that POSTN could positively
regulate IL-4R expression in KFs. Subsequently, to verify the role
of Th2 signaling in inducing POSTN expression, IL-4R was silenced
in KFs using specific siRNAs (Fig.
S4B) and POSTN expression was then detected in the presence or
absence of IL-4/IL-13 treatment. As shown in Fig. 6D, treatment of KFs with human
recombinant IL-4/IL-13 upregulated POSTN, while its mRNA levels
were notably reduced in IL-4R-depleted KFs induced by IL-4/IL-13.
In line with this finding, western blot analysis showed that
stimulation of KFs with IL-4/IL-13 markedly increased the protein
levels of POSTN, which were reduced following IL-4R knockdown
(Fig. 6E). Subsequent
immunofluorescence analysis further verified that the protein
levels of POSTN were reduced in IL-4R-depleted KFs compared with
control KFs in the presence of IL-4/IL-13 stimulation (Fig. 6F). Collectively, these results
indicated that IL-4R, which could be positively regulated by POSTN,
was necessary for the IL-4/IL-13-induced upregulation of POSTN in
KFs, thus supporting the presence of a positive feedback loop
between POSTN and Th2 signaling.
POSTN knockdown inhibits the
expression of genes associated with extracellular matrix formation
in KFs
Reactome enrichment analysis also revealed
significant enrichment of extracellular matrix organization-related
signaling in DEGs between si-POSTN- and si-CTL-transfected KFs
(Fig. 6A). This was further
supported by a heatmap analysis (Fig.
S5A), which highlighted the significance of POSTN in regulating
extracellular matrix remodeling. Additionally, RT-qPCR analysis
verified that the mRNA expression levels of extracellular
matrix-related DEGs, including COL4A5, COL6A6, DST, and MMP3, were
significantly decreased in POSTN-depleted KFs (Fig. S5B). Overall, POSTN could
positively regulate extracellular matrix formation in KFs, a
process involved in the pathogenesis of keloids.
Discussion
Keloids are a chronic and inflammatory skin
condition, characterized by fibroproliferative overgrowth of the
skin (1). Keloids are considered
to result from abnormal wound healing in susceptible individuals
following trauma, inflammation, surgery and burns (2). Keloids are not life-threatening;
however, they can affect the quality of life of patients due to
pain and itching, and they can easily relapse even if surgically
removed (5,6,40).
The precise mechanism underlying keloid pathogenesis remains
unknown; therefore, identifying the pathological mechanisms of
keloids is of importance for the development of novel and efficient
therapeutic strategies.
Previous microarray analyses have identified several
genetic alterations associated with keloid pathogenesis, such as
HIF-1α (11), Wnt signaling
(22) and extracellular matrix
(23). However, the sample sizes
in the aforementioned studies were relatively small and the
heterogeneity was high, possibly resulting in biased results. To
accurately screen genes involved in keloid formation, three
datasets, namely GSE7890, GSE44270 and GSE145725, from the GEO
database were compared in the current study to identify common DEGs
between KFs and NFs. By comprehensively analyzing the gene
expression profiles of these three GEO datasets, a total of five
common DEGs overlapping between the three datasets were identified,
namely POSTN, CCDC80, RGS4, EBF1 and TBX15, which were all
upregulated in KFs compared with NFs. Among these five genes, it
has been reported that POSTN is markedly dysregulated in keloids,
thus indicating its possible role in keloid pathogenesis (32,33).
Consistently, the mRNA and protein levels of POSTN were also
elevated in KFs isolated from patients with keloids. To the best of
our knowledge, only a few studies have systematically investigated
the role of POSTN in keloid formation (32,33,41).
Therefore, POSTN, a notable candidate gene associated with keloid
development, was selected for further investigation.
POSTN, a matricellular protein that belongs to the
fasciclin family, serves key roles in skin development, skin
remodeling/repair and skin-related diseases (42–44).
A previous study demonstrated that POSTN is substantially expressed
in the dermis of patients with psoriasis, eventually leading to
epidermal hyperplasia and the pathogenesis of psoriasis (36). Another study on atopic dermatitis
also suggested that POSTN may be involved in the pathogenesis of
allergic skin inflammation by inducing TSLP production and
disrupting barrier function (37).
However, the role of POSTN in keloids has not been systematically
investigated. In the present study, RNA interference technology was
applied to evaluate the role of POSTN in KFs, which are the most
common cells involved in keloid formation. Notably, POSTN knockdown
had limited effects on the proliferation of KFs, as evidenced by
the comparable cell counts, EdU incorporation rates and cell cycle
distribution between KFs transfected with si-POSTN and those
transfected with si-CTL. Keloid is featured by unrestricted
fibroproliferative overgrowths of the skin (45). The current study revealed that
POSTN silencing could not affect the growth and proliferation of
KFs; this could be due to the limited sensitivity of the examined
fibroblasts to growth inhibition. Additionally, certain types of
fibroblasts may exert distinct biological functions. It would
therefore be of interest to investigate other functions of POSTN,
such as its involvement in fibrosis, extracellular matrix
production and inflammation, within these particular types of
KFs.
To further explore the role of POSTN in keloid
pathogenesis, KFs transfected with si-POSTN or si-CTL were
subjected to RNA-seq. The results demonstrated that the JAK/STAT
pathway, which has previously been involved in the pathogenesis of
keloids (46), was markedly
affected by POSTN knockdown in KFs. Additionally, GSEA further
revealed that the cytokine-cytokine receptor interaction pathway
was significantly altered by POSTN silencing. These findings were
further validated using two sets of si-POSTN constructs in KFs
derived from two different patients with keloids. Specifically, the
expression levels of genes associated with keloid inflammation,
including CXCL1/CXCL2/CXCL5/CXCL8/CXCL12, CCL2/CCL28, IL33/IL34,
TSLP, GDF5 and OSMR, were markedly decreased in POSTN-deficient
KFs, when compared with control cells. Based on these observations,
it was hypothesized that POSTN could be involved in the
pathogenesis of keloids, at least in part, by regulating the
inflammatory response within KFs.
It has been well established that chronic
inflammation and immune responses serve key roles in keloid
formation (14–16). Several studies have reported the
upregulation of proinflammatory cytokines in keloid lesions,
including TSLP, IL-4, IL-13, CXCL8, CXCL12 and CCL2 (4–7,19,34,47,48).
Furthermore, Wu et al (32)
reported that keloid tissues are characterized by an inflammatory
microenvironment, including activated Th2 (IL-4R and TSLP), Th1
(CXCL10/11) and Th17/Th22 (CCL20) pathways, underscoring their
roles in keloid pathogenesis. Among the aforementioned cytokines,
CXCL12 is considered a critical mediator, and a previous study
showed that CXCL12 is upregulated in keloid scars compared with
normal skin (19). However, the
underlying mechanism remains unclear. Herein, the current study
demonstrated that POSTN knockdown could downregulate CXCL12 in KFs,
thus providing a potential explanation for its dysregulation in
keloids. Consequently, CXCL12 accumulation could be involved in
keloid pathology via the CXCL12/CXCR4 axis, thus promoting cell
chemotaxis and the recruitment of inflammatory cells into keloid
tissues, eventually sustaining chronic inflammation (20). Notably, TSLP, which was also shown
to be positively regulated by POSTN in KFs in the present study, is
known to induce the infiltration of CXCR4+ fibrocytes by
upregulating CXCL12 in keloid scars (19). A previous study also revealed the
hyperactivation of MCP-1 (CCL2) in keloid lesions (49), which is in consistent with the
findings of the current study. In addition, elevated CCL2 release
in keloid tissues could augment fibroblast proliferation, thus
initiating keloid development (49). The aforementioned findings
collectively support the role of inflammatory cytokines and
chemokines in promoting the pathogenesis of keloids.
The current study showed that POSTN expression was
increased in KFs, while the expression of inflammatory factors was
reduced in POSTN-depleted KFs, thus indicating a positive
association between POSTN and inflammatory signaling in keloid
pathogenesis. Therefore, targeting the inflammatory
microenvironment by reducing the expression of these cytokines
and/or inhibiting POSTN, could reverse the inflammatory process and
offer a potential treatment strategy for keloid therapy. However,
due to the lack of a robust in vivo model for keloids, it
remains challenging to definitively conclude that POSTN could be
directly involved in keloid formation through regulating the
inflammatory responses. Therefore, the establishment of an
effective animal model for keloids could help in addressing this
limitation. It has been reported that IL-4 and IL-13 generation is
enhanced in keloid lesions compared with in normal skin (50). However, herein, abnormal expression
of IL-4/IL-13 in POSTN-deficient KFs was not observed (data not
shown), possibly due to the fact that IL-4/IL-13 are primarily
secreted by immune cells rather than fibroblasts. In terms of scar
formation, the primary cellular sources of IL-4 and IL-13 are
CD4+ Th2 cells, basophils, mast cells, eosinophils,
natural killer T cells and type 2 innate lymphoid cells (51,52).
These cytokines can promote collagen deposition, thus contributing
to the pathogenesis of scars. Additionally, IL-4 and IL-13 can
enhance POSTN expression in fibroblasts, thus facilitating scar
formation and progression (53).
Consistent with this, Nangole and Agak (54) showed that the plasma levels of IL-4
and IL-13 are significantly elevated in patients with keloids
compared with in healthy subjects, supporting the notion that
IL-4/IL-13 signaling and immune responses could serve key roles in
keloid formation.
Emerging evidence has suggested that IL-4 and/or
IL-13, two signatures of type 2 cytokines, can induce the
expression of POSTN in allergic diseases (35–37).
The present study demonstrated that IL-4 and/or IL-13 upregulated
POSTN in KFs, thus indicating that Th2 signals could upregulate
POSTN in keloids. In line with this result, a previous study showed
that POSTN is expressed in Th2-associated fibroblasts in atopic
dermatitis (55), further
confirming a crosstalk between Th2 immunity and keloid
pathogenesis. Notably, the IL-4 and IL-13 signaling pathway was
significantly enriched in POSTN-related DEGs in KFs in the present
study. Additionally, POSTN knockdown could inhibit the expression
of IL-4R, a cytokine receptor for both IL-4 and IL-13, thus
suggesting that POSTN may positively regulate IL-4R expression in
KFs. Notably, IL-4R was necessary for IL-4/IL-13-induced POSTN
upregulation, underlying a positive feedback loop between POSTN and
Th2 signals in keloids. Previous studies have also revealed that
targeting IL-4Ra using dupilumab results in both atopic dermatitis
improvement and shrinkage of keloids (50,56).
Moreover, IL-4/IL-13 signaling is activated and the mRNA levels of
Th2-related factors have been shown to be increased in keloid
lesions (21,56). In addition, it has been reported
that keloids and other Th2-skewed diseases, including atopic
dermatitis (57) and asthma
(58), share several overlapped
clinical features. RNA-seq data have also linked keloids to a
Th2-skewed immune profile, thus indicating a close association
between keloid pathogenesis and Th2-related gene expression
signatures (32). Collectively,
IL-4 and/or IL-13 could induce POSTN expression, while, in turn,
the elevated POSTN expression could promote IL-4R expression, thus
suggesting a positive feedback loop and a novel mechanism that
could be involved in the pathogenesis of keloids. Due to the lack
of an effective mouse model for keloids, the regulatory effect of
IL-4/IL-13 on POSTN expression could not be investigated in
vivo. This represents a limitation of the present study.
Nonetheless, an in vivo model for keloid research should be
established in the future, potentially through an organoid-based
approach, to further explore the interaction between POSTN and
IL-4/IL-13 signaling in vivo.
In conclusion, the results of the present study
demonstrated that POSTN was upregulated by IL-4 and/or IL-13 in
KFs. Functionally, POSTN could promote inflammation in KFs without
affecting their proliferation. Furthermore, POSTN could positively
regulate IL-4R expression, which may be essential for
IL-4/IL-13-induced POSTN upregulation in KFs, thus suggesting that
a positive feedback loop between POSTN and Th2 signaling could be
involved in keloid development. However, further studies are needed
to verify the clinical relevance of POSTN in keloid pathogenesis,
and to assess the therapeutic potential of targeting POSTN and/or
Th2 signaling in keloid treatment.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by grants from the National
Natural Science Foundation of China (grant nos. 82103726 and
82203900), the Guangdong Basic and Applied Basic Research
Foundation (grant nos. 2023A1515010575 and 2025A1515010947), the
Shenzhen Science and Technology Program (grant nos.
JCYJ20210324110008023 and JCYJ20230807095809019), the Shenzhen
Sanming Project (grant no. SZSM202311029), the Shenzhen Key Medical
Discipline Construction Fund (grant no. SZXK040) and the Shenzhen
High-level Hospital Construction Fund and Peking University
Shenzhen Hospital Scientific Research Fund (grant no.
KYQD2024378).
Availability of data and materials
The sequencing data generated in the present study
may be found in the NCBI Gene Expression Omnibus under accession
number GSE307210 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE307210.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
BJ, FZ and CH contributed to the experimental
design, performance, data collection/analysis and manuscript
preparation. XL, KZ, JG and JW helped to perform in vitro
experiments. WZ, YZ and BY contributed to data analysis and
discussed the results. BY and CH supervised the study. WZ, BY and
CH contributed to funding acquisition. FZ and CH confirm the
authenticity of all the raw data. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The studies involving human participants were
reviewed and approved by the Ethics Committee from the Peking
University Shenzhen Hospital (protocol code 2022-070). Written
informed consent to participate in the study was provided by the
participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jiang Z, Chen Z, Xu Y, Li H, Li Y, Peng L,
Shan H, Liu X, Wu H, Wu L, et al: Low-frequency ultrasound
sensitive piezo1 channels regulate Keloid-related characteristics
of fibroblasts. Adv Sci (Weinh). 11:e23054892024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cohen AJ, Nikbakht N and Uitto J: Keloid
disorder: Genetic basis, gene expression profiles, and
immunological modulation of the fibrotic processes in the skin.
Cold Spring Harb Perspect Biol. 15:a0412452023. View Article : Google Scholar
|
|
3
|
Wang ZC, Zhao WY, Cao Y, Liu YQ, Sun Q,
Shi P, Cai JQ, Shen XZ and Tan WQ: The roles of inflammation in
keloid and hypertrophic scars. Front Immunol. 11:6031872020.
View Article : Google Scholar
|
|
4
|
Zhang M, Chen H, Qian H and Wang C:
Characterization of the skin keloid microenvironment. Cell Commun
Signal. 21:2072023. View Article : Google Scholar
|
|
5
|
Delaleu J, Charvet E and Petit A: Keloid
disease: Review with clinical atlas. Part I: Definitions, history,
epidemiology, clinics and diagnosis. Ann Dermatol Venereol.
150:3–15. 2023. View Article : Google Scholar
|
|
6
|
Hawash AA, Ingrasci G, Nouri K and
Yosipovitch G: Pruritus in keloid scars: Mechanisms and treatments.
Acta Derm Venereol. 101:adv005822021. View Article : Google Scholar
|
|
7
|
Walsh LA, Wu E, Pontes D, Kwan KR, Poondru
S, Miller CH and Kundu RV: Keloid treatments: An evidence-based
systematic review of recent advances. Syst Rev. 12:422023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Naik PP: Novel targets and therapies for
keloid. Clin Exp Dermatol. 47:507–515. 2022. View Article : Google Scholar
|
|
9
|
Ekstein SF, Wyles SP, Moran SL and Meves
A: Keloids: A review of therapeutic management. Int J Dermatol.
60:661–671. 2021. View Article : Google Scholar
|
|
10
|
Kim J, Kim B, Kim SM, Yang CE, Song SY,
Lee WJ and Lee JH: Hypoxia-induced Epithelial-To-Mesenchymal
transition mediates fibroblast abnormalities via ERK activation in
cutaneous wound healing. Int J Mol Sci. 20:25462019. View Article : Google Scholar
|
|
11
|
Kang Y, Roh MR, Rajadurai S, Rajadurai A,
Kumar R, Njauw CN, Zheng Z and Tsao H: Hypoxia and HIF-1α regulate
collagen production in keloids. J Invest Dermatol. 140:2157–2165.
2020. View Article : Google Scholar
|
|
12
|
Guo C, Liang L, Zheng J, Xie Y, Qiu X, Tan
G, Huang J and Wang L: UCHL1 aggravates skin fibrosis through an
IGF-1-induced Akt/mTOR/HIF-1α pathway in keloid. FASEB J.
37:e230152023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gu JJ, Deng CC, Feng QL, Liu J, Zhu DH,
Cheng Q, Rong Z and Yang B: Relief of extracellular matrix
deposition repression by downregulation of IRF1-mediated TWEAK/Fn14
signaling in keloids. J Invest Dermatol. 143:1208–1219. 2023.
View Article : Google Scholar
|
|
14
|
Wang Y, Chen Y, Wu J and Shi X: BMP1
Promotes keloid by inducing fibroblast inflammation and
fibrogenesis. J Cell Biochem. 125:e306092024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mao J, Chen L, Qian S, Wang Y, Zhao B,
Zhao Q, Lu B, Mao X, Zhai P, Zhang Y, et al: Transcriptome network
analysis of inflammation and fibrosis in keloids. J Dermatol Sci.
113:62–73. 2024. View Article : Google Scholar
|
|
16
|
Wang X, Wang X, Liu Z, Liu L, Zhang J,
Jiang D and Huang G: Identification of inflammation-related
biomarkers in keloids. Front Immunol. 15:13515132024. View Article : Google Scholar
|
|
17
|
Lee SY, Lee AR, Choi JW, Lee CR, Cho KH,
Lee JH and Cho ML: IL-17 induces autophagy dysfunction to promote
inflammatory cell death and fibrosis in keloid fibroblasts via the
STAT3 and HIF-1α dependent signaling pathways. Front Immunol.
13:8887192022. View Article : Google Scholar
|
|
18
|
Lee SY, Kim EK, Seo HB, Choi JW, Yoo JH,
Jung KA, Kim DS, Yang SC, Moon SJ, Lee JH and Cho ML: IL-17 induced
stromal Cell-derived Factor-1 and profibrotic factor in
Keloid-derived skin fibroblasts via the STAT3 pathway.
Inflammation. 43:664–672. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shin JU, Kim SH, Kim H, Noh JY, Jin S,
Park CO, Lee WJ, Lee DW, Lee JH and Lee KH: TSLP Is a potential
initiator of collagen synthesis and an activator of CXCR4/SDF-1
axis in keloid pathogenesis. J Invest Dermatol. 136:507–515. 2016.
View Article : Google Scholar
|
|
20
|
Chen Z, Gao Z, Xia L, Wang X, Lu L and Wu
X: Dysregulation of DPP4-CXCL12 balance by TGF-β1/SMAD pathway
promotes CXCR4+ inflammatory cell infiltration in keloid scars. J
Inflamm Res. 14:4169–4180. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nguyen JK, Austin E, Huang A, Mamalis A
and Jagdeo J: The IL-4/IL-13 axis in skin fibrosis and scarring:
Mechanistic concepts and therapeutic targets. Arch Dermatol Res.
312:81–92. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Smith JC, Boone BE, Opalenik SR, Williams
SM and Russell SB: Gene profiling of keloid fibroblasts shows
altered expression in multiple fibrosis-associated pathways. J
Invest Dermatol. 128:1298–1310. 2008. View Article : Google Scholar
|
|
23
|
Hahn JM, Glaser K, McFarland KL, Aronow
BJ, Boyce ST and Supp DM: Keloid-derived keratinocytes exhibit an
abnormal gene expression profile consistent with a distinct causal
role in keloid pathology. Wound Repair Regen. 21:530–544. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics and computational biology solutions
using R and Bioconductor. Springer; New York, NY: pp. 397–420.
2005, View Article : Google Scholar
|
|
25
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: Limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Chen C, Chen H, Zhang Y, Thomas HR, Frank
MH, He Y and Xia R: TBtools: An integrative toolkit developed for
interactive analyses of big biological data. Mol Plant.
13:1194–1202. 2020. View Article : Google Scholar
|
|
27
|
Williams R and Thornton MJ: Isolation of
different dermal fibroblast populations from the skin and the hair
follicle. Methods Mol Biol. 2154:13–22. 2020. View Article : Google Scholar
|
|
28
|
Huang C, Zhong W, Ren X, Huang X, Li Z,
Chen C, Jiang B, Chen Z, Jian X, Yang L, et al: MiR-193b-3p-ERBB4
axis regulates psoriasis pathogenesis via modulating cellular
proliferation and inflammatory-mediator production of
keratinocytes. Cell Death Dis. 12:9632021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Huang C, Lv X, Chen P, Liu J, He C, Chen
L, Wang H, Moness ML, Dong J, Rueda BR, et al: Human papillomavirus
targets the YAP1-LATS2 feedback loop to drive cervical cancer
development. Oncogene. 41:3761–3777. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang C, Li W, Shen C, Jiang B, Zhang K,
Li X, Zhong W, Li Z, Chen Z, Chen C, et al: YAP1 facilitates the
pathogenesis of psoriasis via modulating keratinocyte proliferation
and inflammation. Cell Death Dis. 16:1862025. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu J, Del Duca E, Espino M, Gontzes A,
Cueto I, Zhang N, Estrada YD, Pavel AB, Krueger JG and
Guttman-Yassky E: RNA sequencing keloid transcriptome associates
keloids with Th2, Th1, Th17/Th22, and JAK3-skewing. Front Immunol.
11:5977412020. View Article : Google Scholar
|
|
33
|
Xu H, Wang Z, Yang H, Zhu J and Hu Z:
Bioinformatics analysis and identification of dysregulated POSTN in
the pathogenesis of keloid. Int Wound J. 20:1700–1711. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nangole FW, Ouyang K, Anzala O, Ogengo J
and Agak GW: Multiple cytokines elevated in patients with keloids:
Is it an indication of Auto-inflammatory disease? J Inflamm Res.
14:2465–2470. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Masuoka M, Shiraishi H, Ohta S, Suzuki S,
Arima K, Aoki S, Toda S, Inagaki N, Kurihara Y, Hayashida S, et al:
Periostin promotes chronic allergic inflammation in response to Th2
cytokines. J Clin Invest. 122:2590–2600. 2012. View Article : Google Scholar
|
|
36
|
Arima K, Ohta S, Takagi A, Shiraishi H,
Masuoka M, Ontsuka K, Suto H, Suzuki S, Yamamoto K, Ogawa M, et al:
Periostin contributes to epidermal hyperplasia in psoriasis common
to atopic dermatitis. Allergol Int. 64:41–48. 2015. View Article : Google Scholar
|
|
37
|
Mitamura Y, Nunomura S, Nanri Y, Ogawa M,
Yoshihara T, Masuoka M, Tsuji G, Nakahara T, Hashimoto-Hachiya A,
Conway SJ, et al: The IL-13/periostin/IL-24 pathway causes
epidermal barrier dysfunction in allergic skin inflammation.
Allergy. 73:1881–1891. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kou K, Okawa T, Yamaguchi Y, Ono J, Inoue
Y, Kohno M, Matsukura S, Kambara T, Ohta S, Izuhara K and Aihara M:
Periostin levels correlate with disease severity and chronicity in
patients with atopic dermatitis. Br J Dermatol. 171:283–291. 2014.
View Article : Google Scholar
|
|
39
|
Akar-Ghibril N, Casale T, Custovic A and
Phipatanakul W: Allergic endotypes and phenotypes of asthma. J
Allergy Clin Immunol Pract. 8:429–440. 2020. View Article : Google Scholar
|
|
40
|
Frech FS, Hernandez L, Urbonas R, Zaken
GA, Dreyfuss I and Nouri K: Hypertrophic scars and keloids:
Advances in treatment and review of established therapies. Am J
Clin Dermatol. 24:225–245. 2023. View Article : Google Scholar
|
|
41
|
Tian J, Liu X, Zhu D and Li J: Periostin
regulates the activity of keloid fibroblasts by activating the
JAK/STAT signaling pathway. Heliyon. 10:e388212024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Yin SL, Qin ZL and Yang X: Role of
periostin in skin wound healing and pathologic scar formation. Chin
Med J (Engl). 133:2236–2238. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sonnenberg-Riethmacher E, Miehe M and
Riethmacher D: Periostin in allergy and inflammation. Front
Immunol. 12:7221702021. View Article : Google Scholar
|
|
44
|
Crawford J, Nygard K, Gan BS and O'Gorman
DB: Wound healing and fibrosis: A contrasting role for periostin in
skin and the oral mucosa. Am J Physiol Cell Physiol.
318:C1065–C1077. 2020. View Article : Google Scholar
|
|
45
|
Chen L, Su Y, Yin B, Li S, Cheng X, He Y
and Jia C: LARP6 regulates keloid fibroblast proliferation,
invasion, and ability to synthesize collagen. J Invest Dermatol.
142:2395–2405. 2022. View Article : Google Scholar
|
|
46
|
Yin Q, Wolkerstorfer A, Lapid O, Niessen
FB, Van Zuijlen PPM and Gibbs S: The JAK-STAT pathway in keloid
pathogenesis: A systematic review with qualitative synthesis. Exp
Dermatol. 32:588–598. 2023. View Article : Google Scholar
|
|
47
|
Limandjaja GC, van den Broek LJ, Waaijman
T, Breetveld M, Monstrey S, Scheper RJ, Niessen FB and Gibbs S:
Reconstructed human keloid models show heterogeneity within keloid
scars. Arch Dermatol Res. 310:815–826. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tanaka R, Umeyama Y, Hagiwara H,
Ito-Hirano R, Fujimura S, Mizuno H and Ogawa R: Keloid patients
have higher peripheral blood endothelial progenitor cell counts and
CD34+ cells with normal vasculogenic and angiogenic
function that overexpress vascular endothelial growth factor and
interleukin-8. Int J Dermatol. 58:1398–1405. 2019. View Article : Google Scholar
|
|
49
|
Liao WT, Yu HS, Arbiser JL, Hong CH,
Govindarajan B, Chai CY, Shan WJ, Lin YF, Chen GS and Lee CH:
Enhanced MCP-1 release by keloid CD14+ cells augments fibroblast
proliferation: Role of MCP-1 and Akt pathway in keloids. Exp
Dermatol. 19:e142–e150. 2010. View Article : Google Scholar
|
|
50
|
Diaz A, Tan K, He H, Xu H, Cueto I, Pavel
AB, Krueger JG and Guttman-Yassky E: Keloid lesions show increased
IL-4/IL-13 signaling and respond to Th2-targeting dupilumab
therapy. J Eur Acad Dermatol Venereol. 34:e161–e164. 2020.
View Article : Google Scholar
|
|
51
|
Lee CC, Tsai CH, Chen CH, Yeh YC, Chung WH
and Chen CB: An updated review of the immunological mechanisms of
keloid scars. Front Immunol. 14:11176302023. View Article : Google Scholar
|
|
52
|
Junttila IS: Tuning the cytokine
responses: An update on interleukin (IL)-4 and IL-13 receptor
complexes. Front Immunol. 9:8882018. View Article : Google Scholar
|
|
53
|
Zhang D, Li B and Zhao M: Therapeutic
strategies by regulating interleukin family to suppress
inflammation in hypertrophic scar and keloid. Front Pharmacol.
12:6677632021. View Article : Google Scholar
|
|
54
|
Nangole FW and Agak GW: Keloid
pathophysiology: Fibroblast or inflammatory disorders? JPRAS Open.
22:44–54. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
He H, Suryawanshi H, Morozov P,
Gay-Mimbrera J, Del Duca E, Kim HJ, Kameyama N, Estrada Y, Der E,
Krueger JG, et al: Single-cell transcriptome analysis of human skin
identifies novel fibroblast subpopulation and enrichment of immune
subsets in atopic dermatitis. J Allergy Clin Immunol.
145:1615–1628. 2020. View Article : Google Scholar
|
|
56
|
Guttman-Yassky E, Bissonnette R, Ungar B,
Suárez-Fariñas M, Ardeleanu M, Esaki H, Suprun M, Estrada Y, Xu H,
Peng X, et al: Dupilumab progressively improves systemic and
cutaneous abnormalities in patients with atopic dermatitis. J
Allergy Clin Immunol. 143:155–172. 2019. View Article : Google Scholar
|
|
57
|
Lu YY, Lu CC, Yu WW, Zhang L, Wang QR,
Zhang CL and Wu CH: Keloid risk in patients with atopic dermatitis:
A nationwide retrospective cohort study in Taiwan. BMJ Open.
8:e0228652018. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Hajdarbegovic E, Bloem A, Balak D, Thio B
and Nijsten T: The association between atopic disorders and
keloids: A Case-control study. Indian J Dermatol. 60:6352015.
View Article : Google Scholar
|