Introduction
Acute carbon monoxide poisoning (ACOP) is one of the
most common acute poisoning incidents globally. In the United
States, ACOP is associated with 50,000-100,000 emergency visits and
1,500-2,000 fatalities each year (1). The pathophysiological mechanism of
ACOP is complex, and can eventually lead to the dysfunction of
multiple organs, which is occasionally life-threatening in severe
cases. The kidneys are a highly metabolic and oxygen-dependent
organ, rendering them sensitive to hypoxia and oxidative stress.
Therefore, acute renal injury (AKI) is a notable complication of
ACOP (2). It has been reported
that ~18% of patients with ACOP experience AKI, which not only
markedly increases short-term mortality, but is also associated
with the development of chronic renal disease (CKD) (3). Therefore, investigating the causes of
ACOP could promote the development of targeted therapies to prevent
or alleviate AKI, thus improving patient outcomes and reducing the
risk of progression to CKD.
Evidence has suggested that the mechanisms of
ACOP-induced AKI (ACOP-AKI) involve several interrelated factors,
such as decreased oxygen transport, mitochondrial respiratory
inhibition, reperfusion injury, oxidative stress and the activation
of abnormal inflammatory signal pathways (4–7). CO
exposure may also trigger autophagy by inducing the formation of
mitochondrial reactive oxygen species (8). These findings underscore the
complexity of the molecular pathways involved in ACOP-AKI and
emphasize the need for further studies to uncover these
mechanisms.
Previous advances in RNA sequencing (RNA-seq)
technology have enabled comprehensive analysis of transcriptional
alterations in complex tissues (9–12).
This technique is applied to identify changes in gene expression
and their functional significance, thus offering a novel approach
for understanding the molecular mechanisms underlying the
development of complicated diseases. However, to date,
transcriptional modifications associated with ACOP-AKI remain yet
to be fully elucidated.
The present study aimed to assess the molecular
mechanisms underlying ACOP-AKI using RNA-seq technology. The
present study hypothesized that ACOP-AKI was associated with the
abnormal activation of important factors and signaling pathways. To
assess this hypothesis, an ACOP mouse model was established, and
RNA-seq was employed to comprehensively analyze gene expression
profiles in renal tissues following exposure to CO, mainly focusing
on important components and signaling pathways associated with
inflammation and apoptosis. The primary objective of the present
study was to elucidate the molecular mechanisms underlying ACOP-AKI
and establish a theoretical basis for the development of targeted
therapeutic interventions.
Materials and methods
Animals
A total of 60 adultC57BL/6 mice (male; age, 8–12
weeks; weight, 22–26 g) were obtained from the Animal Experimental
Center of Zunyi Medical University. All experimental protocols were
approved by the Animal Care Committee of Zunyi Medical University
(approval no. zyfy-an-2024-0710/0358) and supervised by the Ethics
Committee of the Affiliated Hospital of Zunyi Medical University.
Mice were maintained under controlled environmental conditions
(temperature, 22–25°C; humidity, 55–60%; 12-h light/dark cycle)
with free access to food and water. All procedures adhered to
China's guidelines for animal research, with efforts made to
minimize animal use and alleviate potential distress, thus
complying with ethical standards.
Animal models
The ACOP mouse model was established as previously
described (13). Briefly, mice
were placed in a transparent plastic chamber at 22–24°C. To monitor
CO levels, two CO detectors were positioned, one at the center and
another near the edge of the chamber. The chamber was then
gradually filled with CO gas. The exposure protocol involved an
initial phase of 1,000 ppm CO for 40 min, followed by a higher
concentration of 3,000 ppm CO for 20 min, or until the mice
displayed signs of unconsciousness. Following exposure to CO, the
animals were promptly transferred to fresh ambient air and observed
until they fully regained consciousness. Mice were anesthetized
with 2% isoflurane in air (2–3 l/min) on days 1, 3 and 7.
Subsequently, ~0.5–1 ml of whole blood was collected via the
retro-orbital venous plexus equally divided into anticoagulant and
plain tubes for the preparation of plasma and serum. Both plasma
and serum were isolated following centrifugation at 1,000 × g for
10 min at 4°C. Following blood collection, deep anesthesia was
maintained with 2% isoflurane, and the mice were sacrificed
directly. Different euthanasia methods were selected according to
different experimental purposes: In the experiments exploring the
time course of ACOP-induced renal injury and conducting
transcriptome analysis, to minimize animal suffering and preserve
the overall morphological structure of tissues, the experimental
animals were sacrificed by cervical dislocation under isoflurane
anesthesia. The collected renal tissues were mainly used for
phenotypic observation studies. In the inhibitor intervention
experiments, under the same isoflurane anesthesia, sacrifice was
performed by transcardiac perfusion with ice-cold 0.1 M
phosphate-buffered saline (PBS) to harvest renal tissues, which was
mainly to meet the higher tissue quality requirements for mechanism
research.
Experimental groups and process
design
To explore the temporal changes in kidney injury
subsequent to ACOP, a total of 24 mice were randomly divided into
four groups (n=6): The control, ACOP 1-day, ACOP 3-day and ACOP
7-day groups. The degree of kidney injury was evaluated at each
time point by histopathological and biochemical analyses. Follow-up
experiments were performed at the time point exhibiting the most
severe injury (determined by the highest tubular injury score).
Total RNA was extracted from the renal tissue of both the control
and ACOP model groups for RNA sequencing (RNA-seq) analysis. Raw
reads were quality-checked and adapter-trimmed using Fastp
(v0.23.2, github.com/OpenGene/fastp). The resulting high-quality
clean reads were then aligned to the reference genome using HISAT2
(v2.2.1, http://daehwankimlab.github.io/hisat2/). Gene-level
read counts were generated from the alignment files using
FeatureCounts (v2.0.3, http://subread.sourceforge.net/). Finally,
differentially expressed genes (DEGs) between the control and model
groups were identified using the DESeq2 (v1.40.2, http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
package, with a significance threshold set at an absolute fold
change ≥2 and an adjusted P-value <0.05.
Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathway enrichment analysis was then performed to identify key
signaling pathways involved in kidney injury. Based on the KEGG
results, representative upregulated genes were selected as
candidate targets and validated using western blotting. An in
vivo intervention utilizing the PI3K inhibitor LY294002 was
performed to assess the functional role of specific signaling
pathways in ACOP-AKI. A total of 36 mice were randomly allocated
into four groups (n=9) in the inhibitor experiments. The control
group consisted of mice housed in identical transparent plastic
cages as those in the experimental groups without CO treatment or
injection. The ACOP group consisted of mice exposed to CO to
establish an ACOP model.
Subsequently, mice were subject to intraperitoneal
injections with varying components depending on their group. For
the ACOP + LY294002 group, LY294002 was initially dissolved in DMSO
at a concentration of <0.1% and was subsequently diluted in 0.9%
NaCl, resulting in a final injection dose of 10 mg/kg body weight.
Mice in the ACOP group were treated with an intraperitoneal
injection of an equivalent volume of 0.9% NaCl solution without
DMSO, matching the injection volume of LY294002 in the ACOP +
LY294002 group. In the ACOP + vehicle group, mice were also exposed
to CO and then received an intraperitoneal injection of a 0.9% NaCl
solution containing 0.1% DMSO. The first injection was administered
within 2 h post-modeling via CO exposure, followed by daily
injections until the designated experimental endpoint. Mice were
then sacrificed for subsequent analysis.
Measurement of blood CO
concentration
The concentration of CO in blood was measured using
CO assay kit (cat. no. A101-3-1; Nanjing Jiancheng Bioengineering
Institute), according to the manufacturer's instructions. Total
hemoglobin (Hb) concentration was determined. The optical density
(OD) was measured at a wavelength of 540 nm (A540) using a
microplate reader (Varioskan Flash; Thermo Fisher Scientific,
Inc.). The Hb concentration was calculated as follows:
Hb=A540×73.54. Subsequently, the percentage of carboxyhemoglobin
(COHb) was determined. The ODs of test and control samples were
measured at 568 nm (A568) and 581 nm (A581). The ΔOD value was
calculated using the following formula: ΔOD=ΔOD of test sample
(A568-A581)-ΔOD of control sample (A568-A581). The COHb was then
calculated using the following equation: COHb (%)=(0.822× ΔOD +
0.001) ×100. The final blood CO concentration was calculated as
follows: CO concentration=[COHb (%) × Hb (g/l)
×106x4]/64456.
Kidney function assessment
Blood urea nitrogen (BUN) and serum creatinine (SCr)
were measured with commercially available assay kits (C013-2-1 and
C011-2-1; both Nanjing Jiancheng Bioengineer Institute, China),
according to the manufacturer's protocols. The OD was measured at a
wavelength of 450 nm, and the concentrations of BUN and SCr were
calculated based on a standard curve.
Histopathological analysis
Renal tissue samples were fixed in 4%
phosphate-buffered formaldehyde for 24–48 h at room temperature and
subsequently embedded in paraffin. The embedded tissues were sliced
into 5 µm-thick sections and were then subjected to hematoxylin and
eosin (H&E) as well as periodic acid-Schiff (PAS) staining.
Specifically, hematoxylin staining was performed for 5–8 min at
room temperature, followed by eosin staining for 1–2 min under the
same conditions. For the PAS staining, sections were oxidized in
periodic acid solution for 5–10 min at room temperature and then
stained with Schiff's reagent for 15–30 min at room temperature.
After staining, the sections were incubated with diaminobenzidine
for 5 min at room temperature, counterstained with hematoxylin for
1–2 min at room temperature, and observed under a light microscope.
Tubular injury was assessed according to a five-tier grading system
that evaluated the extent of brush border loss, tubular dilation,
cast formation, and cellular necrosis, as referenced in (14). The specific grading scheme was
defined as: 0, no pathological changes; 1, less than 10%
involvement; 2, 10–25% involvement; 3, 26–50% involvement; 4,
51–75% involvement; and 5, >75% involvement.
Immunohistochemical staining
Renal tissue samples were fixed in 4%
phosphate-buffered formaldehyde for 24–48 h at room temperature.
Paraffin-embedded tissue sections (thickness, 5 µm) were
deparaffinized and rehydrated through a descending series of
ethanol to water. Antigen retrieval was performed using citrate
buffer (pH 6.0) at 95°C for 20 min. Endogenous peroxidase activity
was blocked following tissue incubation with 3% hydrogen peroxide
for 10 min at room temperature. Subsequently, sections were blocked
with 10% normal goat serum (cat. no. AR0009, Boster Biological
Technology) for 1 h at room temperature. The sections were then
incubated at 4°C overnight with a primary antibody against
neutrophil gelatinase-associated lipocalin (NGAL; rabbit; dilution,
1:200; cat. no. ab125075; Abcam). Sections were washed three times
with phosphate-buffered saline (PBS; pH 7.4) and incubated with an
HRP-conjugated goat anti-rabbit IgG secondary antibody (dilution
1:500; cat. no. ab6721; Abcam) for 30 min at room temperature.
Following secondary antibody incubation, the sections were washed
again with PBS. The tissues were then counterstained with
hematoxylin for 1–2 min at room temperature. Images were captured
under an optical microscope and analyzed with ImageJ software
(v1.8. National Institutes of Health).
TUNEL assay
The kidney tissue samples were fixed in 4%
paraformaldehyde at room temperature for 24–48 h prior to paraffin
embedding. Paraffin-embedded sections (5 µm) were deparaffinized
and rehydrated as aforementioned, followed by antigen retrieval
with citrate buffer (pH 6.0) at 95°C for 20 min. The tissue
sections were then incubated with a TUNEL reaction mixture (One
Step TUNEL Apoptosis Assay Kit, Beyotime, C1089) for 1 h at 37°C.
Subsequently, the sections were counterstaining with hematoxylin
(0.1% w/v, Sigma-Aldrich, H9627) for 1–2 min at room temperature,
and mounted with antifade mounting medium (Beyotime, P0126).
Finally, to assess cell apoptosis, images from five randomly
selected fields of view/section were captured under a light
microscope.
RNA-seq analysis
Total RNA was extracted using a TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc., cat. no.
15596026). RNA integrity and concentration were determined using
the Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.).
Subsequently, for library preparation, high-quality RNA samples
(RNA integrity number ≥7.0) were further analyzed using the NEBNext
Ultra RNA Library Prep Kit (New England BioLabs, Inc., cat. no.
E7530L). The final libraries were quantified using the
Qubit™ dsDNA HS Assay kit (Invitrogen; Thermo Fisher
Scientific, Inc., cat. no. Q32851) and diluted to a loading
concentration of 1.8 nM. Paired-end sequencing (2×150 bp) was
performed on the Illumina NovaSeq 6000 platform with the NovaSeq
6000 S4 Reagent kit (300 cycles; Illumina, Inc., cat. no.
20028312). Then the data were processed and analyzed using Fastp
(v0.23.2, github.com/OpenGene/fastp). High-quality reads were then
aligned to the reference genome using HISAT2 (v2.2.1, http://daehwankimlab.github.io/hisat2/),
followed by gene read counting using FeatureCounts (v2.0.3,
http://subread.sourceforge.net/).
Differential expression analysis was ultimately performed using the
DESeq2 (v1.40.2, http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
software.
Candidate gene selection
A panel of candidate genes, including cytokine
signaling 6 (SOCS6), mesothelin (MSLN), glutathione-specific
γ-glutamylcyclotransferase 1 (CHAC1) and C-C motif chemokine ligand
4 (CCL4), was preselected for initial screening based on their
previously reported associations with drug-induced organ injury,
cell stress response, inflammation or immune regulation pathways
(14–17). CCL4 was prioritized for detailed
reporting in the present study because it demonstrated the most
consistent and significant dysregulation trends in our preliminary
assessments (data not shown).
ELISA
The serum levels of TNF-α, IL-6 and IL-1β were
quantified according to the manufacturer's instructions using Mouse
TNF-α ELISA Kit (Order NO. D721217, Sangon Biotech, Shanghai,
China), Mouse IL-6 ELISA Kit (Order NO. D721022, Sangon Biotech,
Shanghai), and Mouse IL-1β ELISA kit (Order NO. D721017, Sangon
Biotech, Shanghai, China).
Western blot analysis
Total proteins were extracted from kidney tissue
using RIPA lysis buffer (Beyotime Institute of Biotechnology),
supplemented with protease and phosphatase inhibitors. Protein
concentration was measured using a BCA protein assay kit (Thermo
Fisher Scientific, Inc.). Equal amounts of protein extracts (50 µg
per lane) were separated by 10% SDS-PAGE, depending on the
molecular weight of each target protein, and were then transferred
onto PVDF membranes (MilliporeSigma). Following blocking with 5%
non-fat milk in TBS-Tween-20 for 1 h at room temperature, the
membranes were incubated at 4°C overnight with primary antibodies
against NGAL (rabbit; dilution, 1:5,000; cat. no. ab125075; Abcam),
C-C motif chemokine ligand 4 (CCL4; rabbit; dilution, 1:800; cat.
no. 26614–1-AP; Proteintech Group, Inc.), phosphorylated
(p)-protein kinase B (p-Akt; rabbit; dilution, 1:3,000; cat. no.
28731-1-AP; Proteintech Group, Inc.), Akt (rabbit; dilution,
1:5,000; cat no. 10176-2-AP; Proteintech Group, Inc.), NF-κB p65
(Ser536) (rabbit; dilution, 1:5,000; cat. no. 80379-2-RR;
Proteintech Group, Inc.), NF-κB p65 (rabbit; dilution, 1:3,000; cat
no. 10745-1-AP; Proteintech Group, Inc.), IL-6 (mouse; dilution,
1:3,000; cat. no. 66146-1-Ig; Proteintech Group, Inc.), IL-1β
(rabbit; dilution, 1:1,000; cat. no. 26048-1-AP; Proteintech Group,
Inc.), TNF-α (rabbit; dilution, 1:1,000; cat. no. 17590-1-AP;
Proteintech Group, Inc.), Bax (rabbit; dilution, 1:8,000; cat no.
50599-2-Ig; Proteintech Group, Inc.) and Bcl-2 (mouse; dilution,
1:1,000; cat. no. 26593-1-AP; Proteintech Group, Inc.). Membranes
were washed three times with TBST (0.05% Tween-20), and incubated
with HRP-conjugated goat anti-rabbit (cat. no. SA00001-2) or goat
anti-mouse (1:5,000; cat. no. SA00001-1; Proteintech) secondary
antibodies (dilution, 1:5,000; Proteintech Group, Inc.) for 1 h at
room temperature. GAPDH (rabbit; dilution 1:20,000; cat. no.
10494-1-AP; Proteintech Group, Inc.) and β-actin (rabbit; dilution,
1:20,000; cat. no. 66009-1-Ig; Proteintech Group, Inc.) served as
internal loading controls. The proteins were visualized using the
WesternBright® ECL detection kit (Advansta Inc.)
combined with the ChemiDoc XRS+ imaging system (Bio-Rad
Laboratories, Inc.). The signal intensity of each band was
subsequently quantified using ImageLab software v6.1 (Bio-Rad
Laboratories, Inc.).
Statistical analysis
All data are expressed as the mean ± SD. All
statistical analyses were performed using SPSS 22.0 (IBM Corp.).
The differences between two groups were compared by unpaired
Student's t-test, while those among multiple groups were compared
by one-way ANOVA. For multiple comparisons following ANOVA,
Bonferroni correction was applied. P<0.05 was considered to
indicate a statistically significant difference.
Results
ACOP induces AKI in mice
To evaluate whether ACOP treatment induces kidney
injury in mice, a mouse model of ACOP exposure was established, and
blood samples were collected at 1, 3 and 7 days following CO
exposure to assess blood CO levels, renal function parameters and
renal histopathology (Fig. 1A).
The results demonstrated that blood CO levels were markedly
elevated in ACOP-treated mice compared with control mice, thus
verifying the successful establishment of the mouse model (Fig. 1B). In addition, SCr and BUN levels
were markedly increased at all time points following ACOP treatment
compared with the control group, thus indicating substantial renal
dysfunction (Fig. 1C and D).
Histological analysis, using H&E and PAS staining, revealed
that the glomerular structure remained intact, although mild
mesangial matrix expansion and prominent tubular injury were
observed in the ACOP group (Fig.
1E). On day 1 post-ACOP exposure, renal tubular epithelial
cells displayed marked swelling, peritubular capillary congestion,
cytoplasmic dissolution and nuclear loss (red arrows). On day 3,
severe vacuolar degeneration, tubular dilation, brush border
disruption and extensive nuclear detachment were evident (yellow
arrows). However, on day 7, partial tubular regeneration was
observed, although vacuolar changes, tubular dilation and brush
border damage persisted (blue arrows). To assess tubular injury,
the tubular injury scoring method was applied (18), which showed a significant increase
in injury scores in ACOP treated mice compared with control mice,
with the highest injury scores observed on day 3. This finding was
consistent with the morphological one (Fig. 1F). In addition, the expression
levels of NGAL, an early biomarker of AKI, were detected.
Immunohistochemical staining demonstrated markedly widespread NGAL
expression in renal tubules in mice in the ACOP group compared with
the control group (Fig. 1E and G).
Western blot analysis further verified that NGAL was markedly
upregulated in the ACOP group compared with the control group; this
was most noticeable on days 1 and 3, followed by a decrease on day
7 (Fig. 1H and I). Collectively,
the aforementioned results suggested that exposure of mice to ACOP
could successfully induce AKI.
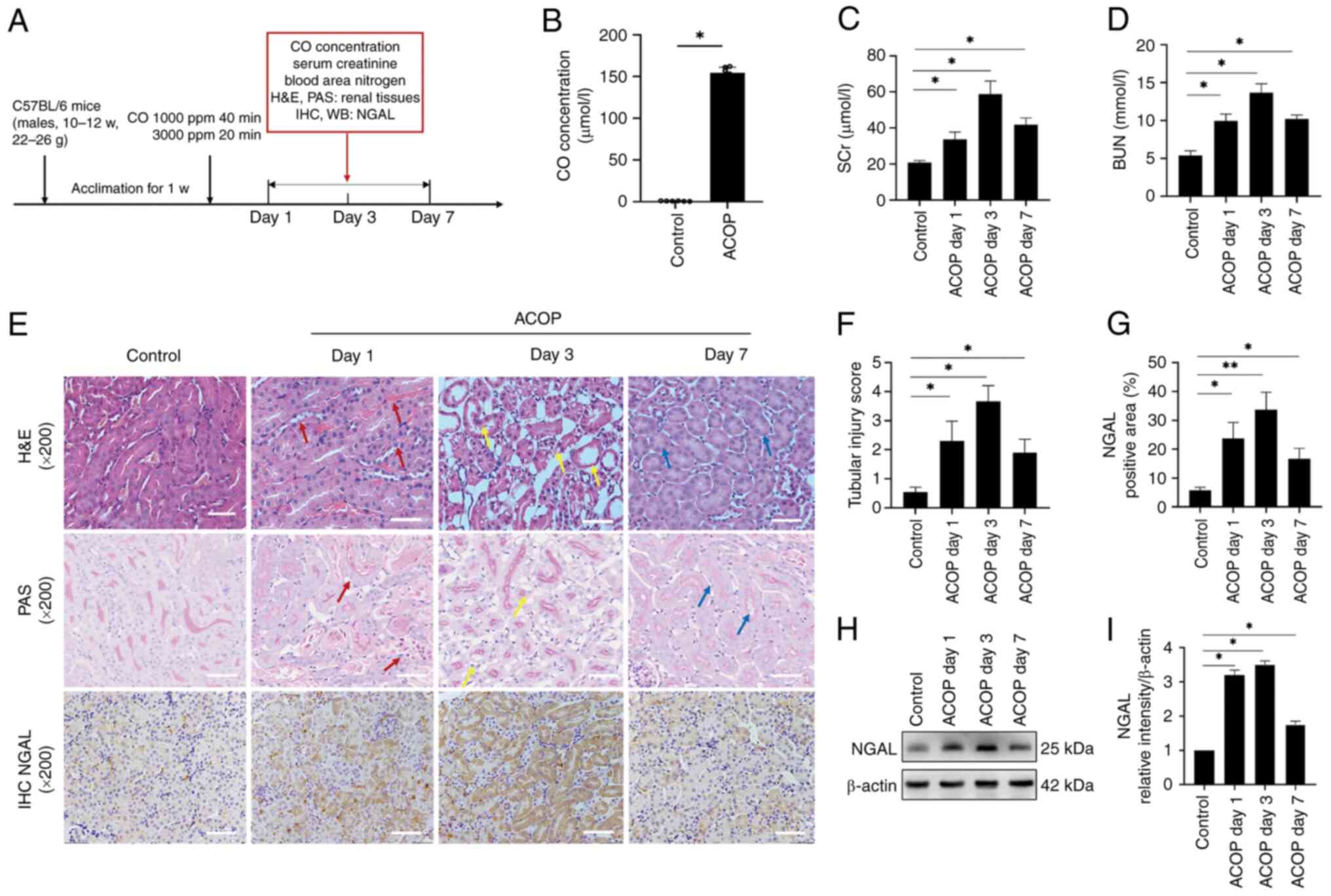 | Figure 1.ACOP induces acute kidney injury in
mice. (A) Schematic illustration of the ACOP mouse model and
experimental design. (B) Bar graph showing CO concentrations in the
blood of mice in the control and ACOP groups. (C) SCr and (D) BUN
levels at different time points after ACOP exposure are shown. (E)
Representative H&E, PAS and NGAL immunohistochemical staining
of kidney tissues at different time points after mice exposure to
ACOP exposure. On day 1, tubular epithelial cells displayed marked
swelling, peritubular capillary congestion and cytoplasmic
dissolution with nuclear loss, as indicated by the red arrows. On
day 3, extensive vacuolar degeneration, tubular dilation, brush
border disruption and nuclear shedding were observed (yellow
arrows). By day 7, signs of tubular regeneration, with persisted
vacuolar degeneration, tubular dilation and brush border damage
(blue arrows), were observed. Scale bar, 100 µm. (F) Quantification
of tubular injury scoring. (G) Quantification of NGAL protein
expression in renal tissue detected by immunohistochemistry. (H)
Expression of NGAL based on immunoblot bands. (I) Semi-quantitation
of NGAL expression. Data are expressed as the mean ± SD (n=6).
*P<0.05 and **P<0.01 vs. control. ACOP, acute carbon monoxide
poisoning; NGAL, neutrophil gelatinase-associated lipocalin; SCr,
serum creatinine; BUN, blood urea nitrogen; CO, carbon monoxide; w,
week; ppm, parts per million; H&E, Hematoxylin and Eosin; PAS,
periodic acid-Schiff; IHC, immunohistochemistry; WB, western blot
analysis. |
DEGs and signaling pathways in
ACOP-AKI
To further explore the molecular mechanism of
ACOP-AKI, RNA-seq analysis was carried out on the kidney tissue
samples from the ACOP model and control groups on day 3, identified
as the peak of kidney injury. Based on the analysis of differential
gene expression, the top 20 DEGs with the highest average
expression levels were presented in Table SI, including log2(fold
change) and P-value. The volcano plot in Fig. 2A depicts the distribution of DEGs,
revealing significant up- and downregulation trends, whereas the
heatmap in Fig. 2B focuses on
genes functionally linked to the PI3K/Akt pathway. A KEGG pathway
enrichment analysis of DEGs revealed that the ‘PI3K/Akt signaling
pathway’ was markedly enriched in the ACOP group (Fig. 2C).
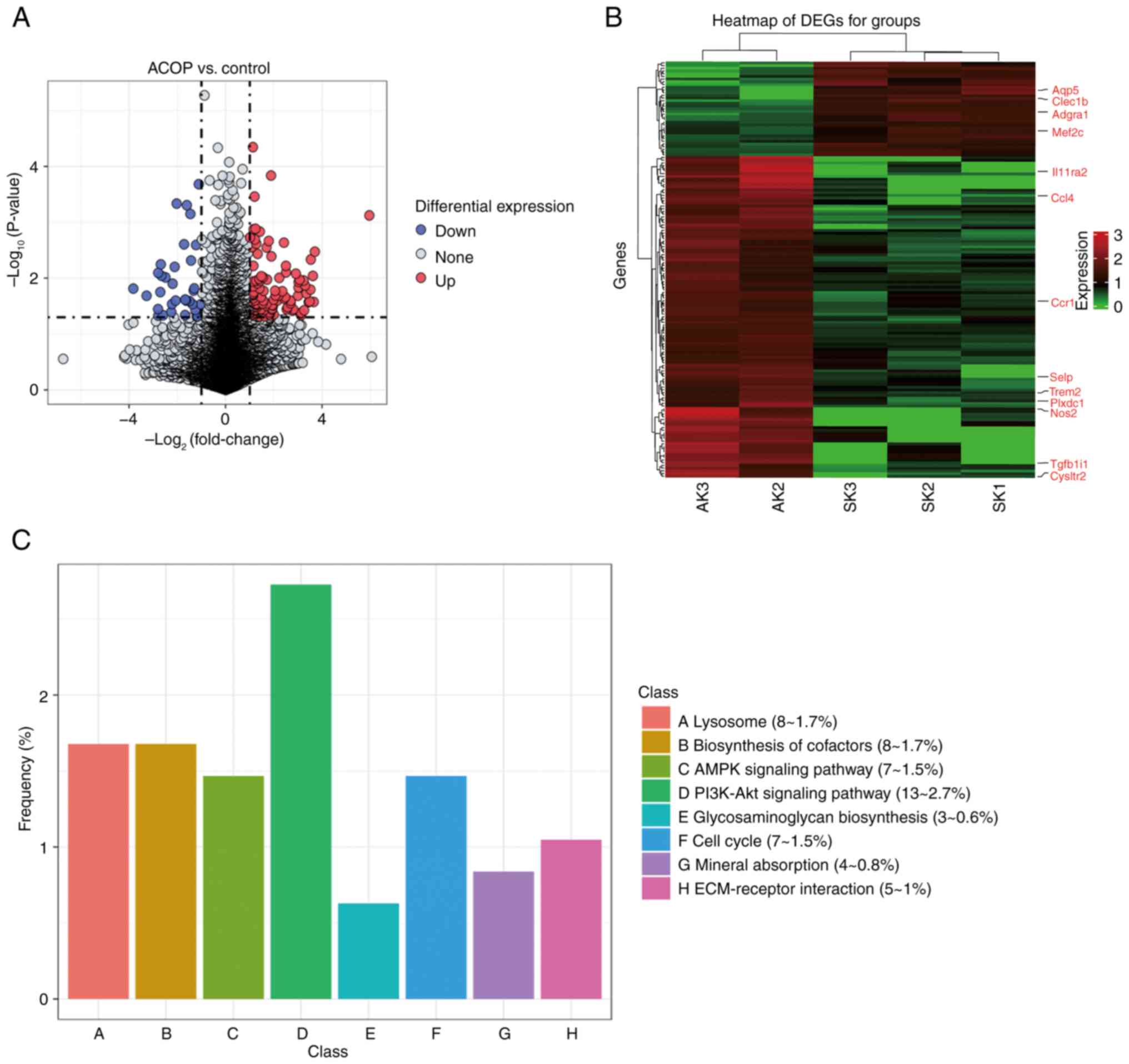 | Figure 2.C-C motif chemokine ligand 4 is
upregulated in ACOP-treated mice via the PI3K/Akt signaling
pathway. (A) Volcano plots showing identified DEGs. Red and blue
dots indicate markedly upregulated and downregulated genes,
respectively, while gray dots represent genes that do not meet the
differential expression criteria. (B) Heatmap of DEGs. Red and
green indicate the upregulated and downregulated genes,
respectively. (C) Kyoto Encyclopedia of Genes and Genomes pathway
enrichment analysis of DEGs. DEG, differentially expressed genes;
ACOP, acute carbon monoxide poisoning; AMPK, AMP-activated protein
kinase; ECM, extracellular matrix; AK, acute kidney; SK, sham
kidney Aqp5, aquaporin 5; Clec1b, C-type lectin domain family 1
member B; Adgra1, adhesion G protein-coupled receptor A1; Mef2c,
myocyte enhancer factor 2C; Il11ra2, interleukin 11 receptor
subunit α2; Ccl4, C-C motif chemokine ligand 4; Ccr1, C-C motif
chemokine receptor 1; Selp, selectin P; Trem2, triggering receptor
expressed on myeloid cells 2; Plxdc1, plexin domain containing 1;
Nos2, nitric oxide synthase 2; Tgfb1i1, transforming growth factor
β1 induced transcript 1; Cysltr2, cysteinyl leukotriene receptor
2. |
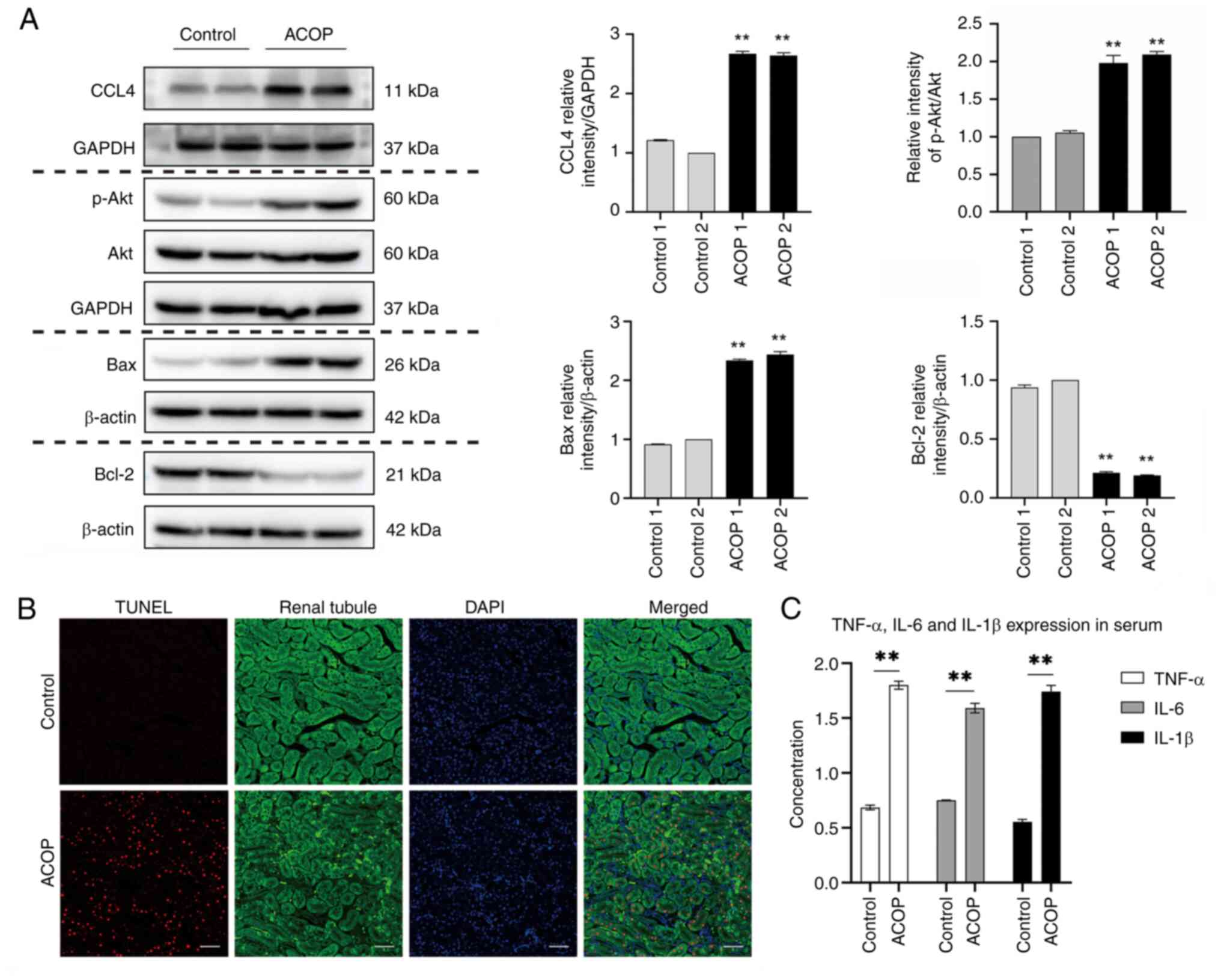
ACOP upregulates CCL4 expression in
renal tissue, activates the PI3K/Akt pathway and elevates renal
cell apoptosis and systemic inflammatory cytokine levels
Through candidate gene screening and validation,
CCL4 exhibited consistent patterns at both the transcriptional and
protein levels in the present model (data not shown), indicating
its potential role in the mechanisms underlying ACOP-AKI. In
addition, the present study observed that in ACOP-exposed mice, the
p-Akt/Akt ratio and Bax protein levels were markedly increased,
whereas Bcl-2 expression was markedly decreased when compared with
control mice (Fig. 3A). TUNEL
staining assays further revealed that the number of TUNEL-positive
cells was notably increased in renal tissues of mice in the ACOP
group compared with the control group. Merged images demonstrated
co-localization of TUNEL signals with DAPI-stained nuclei,
confirming the induction of cellular apoptosis in the renal tissue
following ACOP injury. Furthermore, the apoptosis appeared to be
predominantly localized within the renal tubule (Fig. 3B). Furthermore, the ELISA results
demonstrated markedly elevated serum levels of the systemic
pro-inflammatory cytokines TNF-α, IL-6 and IL-1β in the ACOP group
compared with the control group (Fig.
3C). The aforementioned data indicated that ACOP may have
upregulated the expression of CCL4, accompanied by activation of
the PI3K/Akt signaling pathway. Furthermore, ACOP induced
significant inflammatory responses and notable apoptosis.
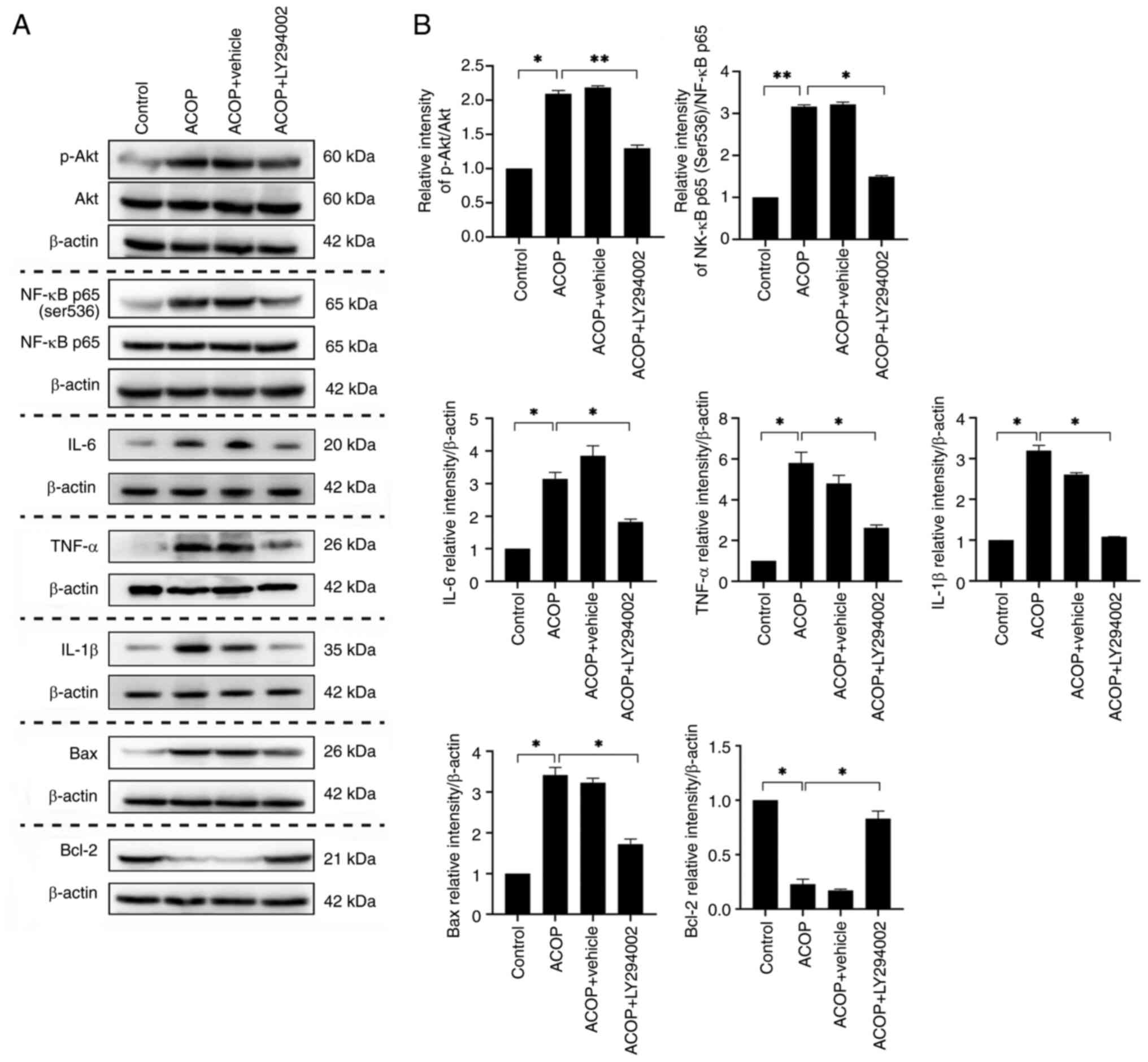
LY294002 attenuates ACOP-AKI
To investigate the role of the PI3K/Akt signaling
pathway in ACOP-AKI, mice were treated with the PI3K inhibitor
LY294002, and renal function and histopathological changes were
evaluated on day 3 (Fig. 4A).
Biochemical analysis showed that mice in the ACOP group exhibited
markedly elevated serum levels of SCr and BUN compared with the
control group. In addition, LY294002 treatment partially yet
markedly ameliorated these increased serum levels (Fig. 4B and C). Morphological assessment
revealed pronounced tubular damage in the kidneys of ACOP-treated
mice, which was markedly alleviated in the ACOP + LY294002 group
(Fig. 4D). Consistently, the
tubular injury score was markedly lower in the ACOP + LY294002
group compared with the ACOP group (Fig. 4E). Immunohistochemical analysis
demonstrated a significant increase in NGAL expression in the ACOP
group compared with the control group, which was markedly reduced
following treatment with LY294002 (Fig. 4F). Finally, western blot analysis
was performed to assess the NGAL protein expression levels
(Fig. 4G), and densitometric
quantification verified that LY294002 markedly suppressed NGAL
expression in the kidneys of ACOP-treated mice (Fig. 4H). These findings suggested that
LY294002 could effectively mitigate ACOP-AKI.
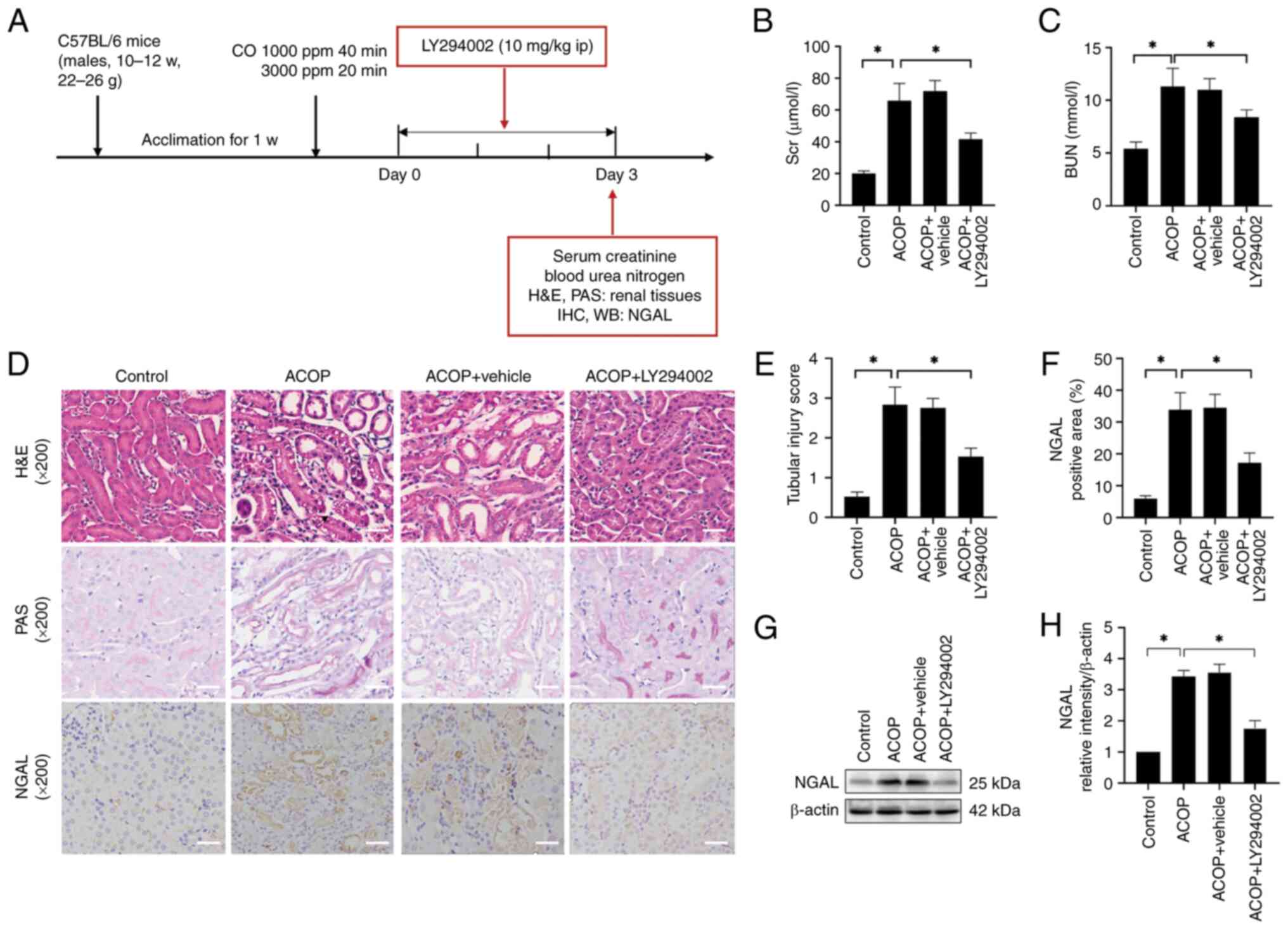 | Figure 4.Effects of LY294002 on kidney injury
in mice exposed to ACOP. (A) Schematic representation of the
establishment of the ACOP + LY294002 mouse model and experimental
design. The experiment was conducted on day 3 after ACOP exposure,
when the injury was most severe. Blood and tissue samples were
subsequently collected for serological, morphological and protein
expression analyses. (B) SCr and (C) BUN levels in control, ACOP,
vehicle-treated ACOP control and LY294002-treated ACOP groups are
shown. (D) Representative H&E, PAS and NGAL immunohistochemical
staining images of kidney tissues in different groups are shown.
Scale bar, 100 µm. (E) Tubular injury scoring is presented. (F)
Quantification of NGAL protein expression in renal tissue detected
by immunohistochemistry. (G) Expression of NGAL based on immunoblot
bands. (H) Semi-quantitation of NGAL expression. Data are expressed
as the mean ± SD (n=3). *P<0.05 vs. control. ACOP, acute carbon
monoxide poisoning; NGAL, neutrophil gelatinase-associated
lipocalin; SCr, serum creatinine; BUN, blood urea nitrogen; CO,
carbon monoxide; w, week; ppm, parts per million; PAS, periodic
acid-Schiff; IHC, immunohistochemistry; WB, western blot analysis;
ip, intraperitoneal. |
LY294002 inhibits inflammation and
apoptosis in the kidneys of ACOP mice
To further investigate the molecular mechanisms
underlying the effects of LY294002 on ACOP-AKI, western blot
analysis was employed to detect the expression levels of
inflammatory factors and apoptosis-related proteins. The results
showed that the ratio of p-Akt/Akt was markedly elevated in the
ACOP-treated group, compared with the control, accompanied by a
significant upregulation of inflammatory factors such as IL-6,
TNF-α and IL-1β. By contrast, treatment with LY294002 markedly
reduced the p-Akt/Akt ratio and the protein expression levels of
IL-6, IL-1β and TNF-α compared with the ACOP group, suggesting an
inhibitory effect of LY294002 on the expression of pro-inflammatory
cytokines.
Considering that NF-κB is a key mediator of
pro-inflammatory cytokine expression, the present study further
assessed the ratio of NF-κB p65 (Ser536)/NF-κB p65. The results
revealed that treatment with LY294002 also markedly inhibited the
elevation of this ratio in the kidneys of ACOP-treated mice.
Furthermore, ACOP treatment induced a significant increase in the
expression of the pro-apoptotic protein Bax and a significant
decrease in the expression of the anti-apoptotic protein Bcl-2 when
compared with the control group. However, LY294002 treatment
markedly suppressed ACOP-induced Bax expression and restored Bcl-2
levels (Fig. 5). In conclusion,
LY294002 effectively suppressed inflammation and apoptosis in the
kidneys of ACOP-treated mice by modulating key signaling pathways
such as PI3K/Akt and NF-κB, providing a potential pharmacological
target for the treatment of ACOP-AKI.
Discussion
ACOP is one of the most common causes of poisoning
globally and can induce AKI, potentially leading to CKD in severe
cases (19,20). In the present study, the successful
establishment of the ACOP mouse model was verified via measuring
the serum levels of CO. Renal dysfunction was observed on the first
day after exposure of mice to ACOP, which was characterized by
elevated SCr and BUN levels, accompanied by hallmark
histopathological alterations, including tubular dilatation,
epithelial cell detachment, inflammatory infiltration and aberrant
NGAL expression. A clinical study reported that the median time to
the first diagnosis of ACOP-AKI is one day after Emergency
Department (ED) admission, with the majority of patients being
diagnosed at their highest KDIGO stage within three days of ED
admission (3). Notably, renal
injury reached its peak on day 3 and exhibited partial recovery by
day 7, which was consistent with clinical observations (3). While individuals with stage I and II
AKI typically achieved a full recovery, up to 55.6% of those with
stage III AKI progressed to CKD (3,21).
The underlying mechanisms of ACOP-AKI to CKD may involve a burst of
reactive oxygen species (ROS) production and the activation of
downstream signal cascades. ROS not only directly damage cellular
components but also trigger sustained tissue injury via promoting
inflammatory responses and apoptotic signaling, potentially forming
the pathological basis for long-term sequelae of ACOP (22). Histological analyses further
revealed that metabolically active renal tubular epithelial cells
were the primary targets of injury, whereas glomerular structures
remained relatively intact. This may be attributed to the higher
oxygen demand and greater susceptibility to oxidative stress of
tubular epithelial cells (23,24).
This observation was consistent with the clinical manifestations of
patients with ACOP-AKI, who primarily present with oliguria,
proteinuria, urinary casts, and disturbances in water and
electrolyte balance (25).
The mechanisms underlying ACOP-AKI are complex,
involving CO-mediated tissue hypoxia and ischemia, mitochondrial
dysfunction, oxidative stress and inflammation. However, the
underlying molecular mechanisms remain yet to be fully elucidated
(2,23). Among the aforementioned mechanisms,
inflammation was considered to be closely related to ACOP-AKI. In
the present study, RNA-seq analysis of renal tissues from mice with
ACOP-AKI revealed a significant upregulation of multiple
inflammation-related genes. During the initial phase of the present
study, candidate genes were preselected, such as SOCS6, MSLN, CHAC1
and CCL4, for further investigation based on prior research.
Notably, CCL4 exhibited consistent trends at both the
transcriptional and protein levels in the present model, suggesting
its potential role in the mechanisms underlying ACOP-AKI. While we
are actively exploring the functional roles of other key candidate
genes, further evidence is required to substantiate these findings.
Given the novelty of CCL4 in ACOP-AKI, we prioritized reporting
these results. Furthermore, KEGG pathway enrichment analysis
indicated that the PI3K/Akt signaling pathway was markedly
activated, thus suggesting that this signaling axis could serve an
important role in the pathogenesis of ACOP-AKI.
In the present study, the RNA-seq results were
further verified at the protein and functional levels by western
blot analysis. The results demonstrated that in ACOP-treated mice,
the expression levels of CCL4 and the p-Akt/Akt ratio were markedly
increased compared with the control group. As a chemotactic factor,
CCL4 is important for immune cell recruitment and initiates local
immune responses, thus promoting inflammation (26). The PI3K/Akt signaling pathway is a
notable axis in cell growth, survival and stress responses, and its
activation can enhance cell survival signals and inhibit apoptosis.
However, chronic or excessive PI3K/Akt signaling activation can
result in an amplified inflammatory response and promote apoptosis
(27). The present study found
that renal tubular cell apoptosis was accelerated in ACOP-treated
mice, accompanied by a significant increase in Bax and decrease in
Bcl-2 expression. In addition, the serum levels of TNF-α, IL-6 and
IL-1β were also markedly elevated, thus indicating that a systemic
inflammatory response was activated. These findings supported the
involvement of CCL4 upregulation and activation of the PI3K/Akt
pathway in the process of ACOP-AKI, which may play a role in
promoting the inflammatory response and apoptosis.
To further verify the functional involvement of the
PI3K/Akt pathway, mice in the ACOP group were treated with the
LY294002 PI3K inhibitor (28). The
results demonstrated that LY294002 markedly improved renal
dysfunction, as evidenced by the reduced SCr and BUN levels, and
improved the morphological integrity of the renal tubular
structures. Immunohistochemical and western blot analyses further
revealed a significant decrease in the expression levels of NGAL in
the LY294002-treated group compared with the ACOP model group, thus
providing additional evidence for the nephroprotective effects of
PI3K inhibition.
As a key regulatory factor in inflammatory
responses, PI3K has emerged as a potential treatment target for
inflammatory diseases (29).
Previous studies showed that LY294002 could inhibit the PI3K/Akt
pathway, eventually downregulating NF-κB and pro-inflammatory
factors, such as TNF-α and IL-1β, in the spinal cords of bone
cancer pain-model rats (30).
NF-κB is a key transcription factor, which is notably involved in
several inflammatory reactions. The NF-κB-induced release of
downstream inflammatory mediators not only enhances the recruitment
of immune cells but can also lead to cell damage and apoptosis
through various mechanisms, such as generating excessive oxidative
stress or directly activating apoptotic pathways (31,32).
In particular, TNF-α and IL-1β are considered significant
pro-apoptotic factors.
In the present study, the activation of the PI3K/Akt
pathway was associated with the phosphorylation of NF-κB p65 (Ser
536), which may enhance its transcriptional activity and promote
the expression of pro-inflammatory mediators, including TNF-α, IL-6
and IL-1β. These inflammatory mediators further promoted the
expression of the pro-apoptotic protein Bax, while reducing the
expression levels of the anti-apoptotic protein Bcl-2, eventually
aggravating renal cell apoptosis. This mechanism was reversed by
LY294002 treatment. The aforementioned mechanism was consistent
with that reported in a previous study, suggesting that
NF-κB-mediated inflammation could also be involved in CO
poisoning-induced neurological injury (33). However, PI3K/Akt may also have
affected apoptosis via additional downstream targets, such as
mammalian target of rapamycin or forkhead box O. Nevertheless,
further studies are needed to verify the involvement of these
pathways.
However, the present study had some limitations.
Previous studies have demonstrated that CCL4 can activate a variety
of downstream signaling pathways, including the PI3K/Akt pathway
(34), and induce the
phosphorylation of NF-κB p65 via its receptor C-C chemokine
receptor type (CCR)5 (35). This
process promotes cell senescence and dysfunction via increasing ROS
production and activating inflammatory responses (17). Although the results of RNA-seq
showed that CCL4 was markedly upregulated at the transcription
level and was closely associated with the activation of the
PI3K/Akt pathway, the particular role of CCL4 in ACOP-AKI and its
causal association with the PI3K/Akt pathway should be further
verified. Functional experiments, such as gene knockout or
antibody-mediated neutralization are necessary to validate this
association. In addition, other differentially expressed
inflammation-related genes identified through RNA-seq analysis,
such as MSLN, SOCS6, CHAC1, CCR1, nitric oxide synthase 2 and
P-selectin, could also interact with the PI3K/Akt axis, warranting
further exploration into their molecular interaction mechanisms.
Furthermore, clinical data correlating the CCL4/Akt axis with the
severity of AKI in patients with ACOP remains lacking. Therefore,
future studies exploring this potential association should be
performed for improved understanding of its clinical relevance.
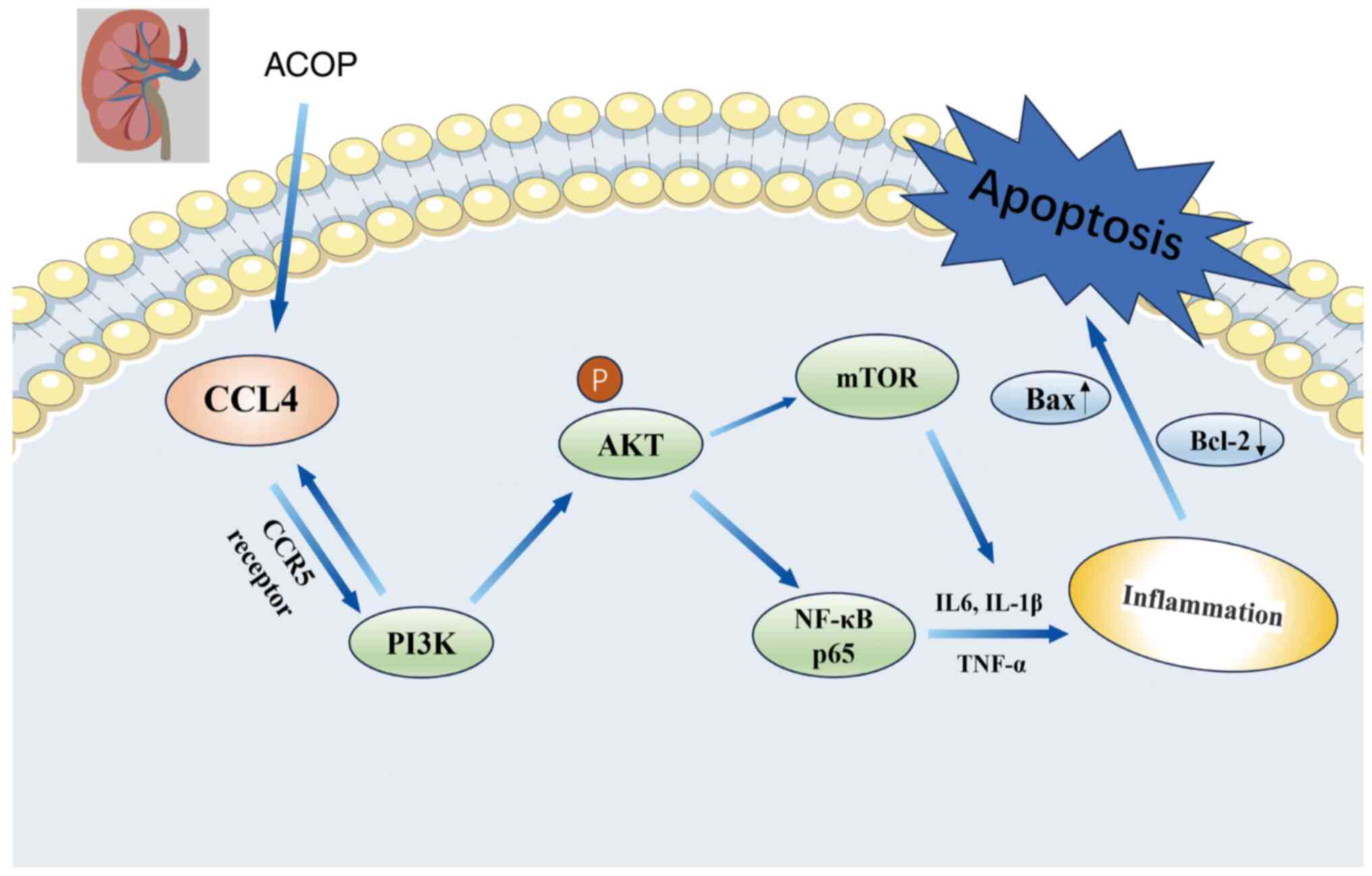
In conclusion, the present study demonstrated that
during ACOP, CCL4 expression and the PI3K/Akt pathway were
upregulated, resulting in renal dysfunction and promoting
inflammatory responses and apoptosis. Additionally,
LY294002-induced inhibition of the PI3K/Akt pathway attenuated
NF-κB-mediated inflammatory signaling, thus alleviating AKI in ACOP
mice via suppressing inflammatory responses and cell apoptosis
(Fig. 6). To the best of our
knowledge, the present study was the first study to utilize RNA-seq
technology to identify abnormal gene expression and changes in
signaling pathways in the renal tissues of ACOP-exposed mice. These
findings could provide important experimental evidence and
theoretical insights for further elucidating the molecular
mechanisms underlying ACOP-AKI.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by the National Natural Science
Foundation of China (grant no. 82260254), Medical Research Union
Fund for High-quality health development of Guizhou Province (grant
no. 2024GZYXKYJJXM0081), Guizhou Province Basic Research Program
[grant no. (2023)568], Guizhou Province Basic Research
Program-[grant no. (2025)387] and Science and Technology Fund
Project of Guizhou Provincial Health Commission (grant no.
2024GZWJKJXM1070).
Availability of data and materials
The data generated in the present study may be found
in the Sequence Read Archive under accession number PRJNA1234475 or
at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1234475.
Authors' contributions
AYY and QL conceived and designed the study. YN,
XHJ, SHW, TP and TTY analyzed and interpreted data. YN, XHJ and SHW
improved the quality of the figures. XHJ, SHW, TTY and YN drafted
the manuscript. TP, AYY and QL revised the manuscript. YN and QL
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Animal Care Committee of Zunyi Medical University (approval no.
zyfy-an-2024-0710/0358) and supervised by the Ethics Committee of
the Affiliated Hospital of Zunyi Medical University. All procedures
adhered to China's guidelines for animal research, ensuring ethical
standards were met by minimizing animal usage and alleviating
potential distress.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rose JJ, Wang L, Xu Q, McTiernan CF, Shiva
S, Tejero J and Gladwin MT: Carbon monoxide poisoning:
Pathogenesis, management, and future directions of therapy. Am J
Respir Crit Care Med. 195:596–606. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Huang TL, Tung MC, Lin CL and Chang KH:
Risk of acute kidney injury among patients with carbon monoxide
poisoning. Medicine (Baltimore). 100:e272392021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim YJ, Sohn CH, Seo DW, Oh BJ, Lim KS,
Chang JW and Kim WY: Analysis of the development and progression of
carbon monoxide poisoning-related acute kidney injury according to
the kidney disease improving global outcomes (KDIGO) criteria. Clin
Toxicol (Phila). 56:759–764. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Garrabou G, Inoriza JM, Morén C, Oliu G,
Miró Ò, Martí MJ and Cardellach F: Mitochondrial injury in human
acute carbon monoxide poisoning: The effect of oxygen treatment. J
Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 29:32–51.
2011.PubMed/NCBI
|
|
5
|
Rose JJ, Bocian KA, Xu Q, Wang L,
DeMartino AW, Chen X, Corey CG, Guimarães DA, Azarov I, Huang XN,
et al: A neuroglobin-based high-affinity ligand trap reverses
carbon monoxide-induced mitochondrial poisoning. J Biol Chem.
295:6357–6371. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Teksam O, Sabuncuoğlu S, Girgin G and
Özgüneş H: Evaluation of oxidative stress and antioxidant
parameters in children with carbon monoxide poisoning. Hum Exp
Toxicol. 38:1235–1243. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thom SR, Fisher D and Manevich Y: Roles
for platelet-activating factor and *NO-derived oxidants causing
neutrophil adherence after CO poisoning. Am J Physiol Heart Circ
Physiol. 281:H923–H930. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee SJ, Ryter SW, Xu JF, Nakahira K, Kim
HP, Choi AM and Kim YS: Carbon monoxide activates autophagy via
mitochondrial reactive oxygen species formation. Am J Respir Cell
Mol Biol. 45:867–873. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Z, Gerstein M and Snyder M: RNA-Seq:
A revolutionary tool for transcriptomics. Nat Rev Genet. 10:57–63.
2009. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ozsolak F and Milos PM: RNA sequencing:
Advances, challenges and opportunities. Nat Rev Genet. 12:87–98.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stark R, Grzelak M and Hadfield J: RNA
sequencing: The teenage years. Nat Rev Genet. 20:631–656. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Conesa A, Madrigal P, Tarazona S,
Gomez-Cabrero D, Cervera A, McPherson A, Szcześniak MW, Gaffney DJ,
Elo LL, Zhang X and Mortazavi A: A survey of best practices for
RNA-seq data analysis. Genome Biol. 17:132016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Xiong B, Tian Y, Hu Q, Jiang X,
Zhang J, Chen L, Wang R, Li M, Zhou X, et al: Targeting ferroptosis
promotes functional recovery by mitigating white matter injury
following acute carbon monoxide poisoning. Mol Neurobiol.
61:1157–1174. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang P, Guan P, Ye X, Lu Y, Hang Y, Su Y
and Hu W: SOCS6 promotes mitochondrial fission and cardiomyocyte
apoptosis and is negatively regulated by quaking-mediated miR-19b.
Oxid Med Cell Longev. 2022:11213232022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Nishio T, Koyama Y, Liu X, Rosenthal SB,
Yamamoto G, Fuji H, Baglieri J, Li N, Brenner LN, Iwaisako K, et
al: Immunotherapy-based targeting of MSLN+ activated
portal fibroblasts is a strategy for treatment of cholestatic liver
fibrosis. Proc Natl Acad Sci USA. 118:e21012701182021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sun J, Ren H, Wang J, Xiao X, Zhu L, Wang
Y and Yang L: CHAC1: A master regulator of oxidative stress and
ferroptosis in human diseases and cancers. Front Cell Dev Biol.
12:14587162024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chang TT, Lin LY, Chen C and Chen JW: CCL4
contributes to aging related angiogenic insufficiency through
activating oxidative stress and endothelial inflammation.
Angiogenesis. 27:475–499. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kim MG, Yun D, Kang CL, Hong M, Hwang J,
Moon KC, Jeong CW, Kwak C, Kim DK, Oh KH, et al: Kidney VISTA
prevents IFN-γ/IL-9 axis-mediated tubulointerstitial fibrosis after
acute glomerular injury. J Clin Invest. 132:e1511892022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mattiuzzi C and Lippi G: Worldwide
epidemiology of carbon monoxide poisoning. Hum Exp Toxicol.
39:387–392. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei KY, Liao CY, Chung CH, Lin FH, Tsao
CH, Sun CA, Lu KC, Chien WC and Wu CC: Carbon monoxide poisoning
and chronic kidney disease risk: A nationwide, population-based
study. Am J Nephrol. 52:292–303. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ostermann M, Bellomo R, Burdmann EA, Doi
K, Endre ZH, Goldstein SL, Kane-Gill SL, Liu KD, Prowle JR, Shaw
AD, et al: Controversies in acute kidney injury: Conclusions from a
kidney disease: Improving global outcomes (KDIGO) conference.
Kidney Int. 98:294–309. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dent MR, Rose JJ, Tejero J and Gladwin MT:
Carbon monoxide poisoning: From microbes to therapeutics. Annu Rev
Med. 75:337–351. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Scholz H, Boivin FJ, Schmidt-Ott KM,
Bachmann S, Eckardt KU, Scholl UI and Persson PB: Kidney physiology
and susceptibility to acute kidney injury: Implications for
renoprotection. Nat Rev Nephrol. 17:335–349. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao ZB, Marschner JA, Iwakura T, Li C,
Motrapu M, Kuang M, Popper B, Linkermann A, Klocke J, Enghard P, et
al: Tubular epithelial cell HMGB1 promotes AKI-CKD transition by
sensitizing cycling tubular cells to oxidative stress: A rationale
for targeting HMGB1 during AKI recovery. J Am Soc Nephrol.
34:394–411. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Weaver LK: Carbon monoxide poisoning.
Undersea Hyperb Med. 47:151–169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ozga AJ, Chow MT and Luster AD: Chemokines
and the immune response to cancer. Immunity. 54:859–874. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Glaviano A, Foo ASC, Lam HY, Yap KCH,
Jacot W, Jones RH, Eng H, Nair MG, Makvandi P, Geoerger B, et al:
PI3K/Akt/mTOR signaling transduction pathway and targeted therapies
in cancer. Mol Cancer. 22:1382023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Vanhaesebroeck B, Perry MWD, Brown JR,
André F and Okkenhaug K: PI3K inhibitors are finally coming of age.
Nat Rev Drug Discov. 20:741–769. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Castel P, Toska E, Engelman JA and
Scaltriti M: The present and future of PI3K inhibitors for cancer
therapy. Nat Cancer. 2:587–597. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhao J, Yan Y, Zhen S, Yu L, Ding J, Tang
Q, Liu L, Zhu H and Xie M: LY294002 alleviates bone cancer pain by
reducing mitochondrial dysfunction and the inflammatory response.
Int J Mol Med. 51:422023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang H and Sun SC: NF-κB in inflammation
and renal diseases. Cell Biosci. 5:632015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Dai C, Liu D, Qin C, Fang J, Cheng G, Xu
C, Wang Q, Lu T, Guo Z, Wang J, et al: Guben Kechuan granule
attenuates bronchial asthma by inhibiting NF-κB/STAT3 signaling
pathway-mediated apoptosis. J Ethnopharmacol. 340:1191242025.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Arya AK, Sethuraman K, Waddell J, Cha YS,
Liang Y, Bhopale VM, Bhat AR, Imtiyaz Z, Dakessian A, Lee Y and
Thom SR: Inflammatory responses to acute carbon monoxide poisoning
and the role of plasma gelsolin. Sci Adv. 11:eado97512025.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Festa BP, Siddiqi FH, Jimenez-Sanchez M
and Rubinsztein DC: Microglial cytokines poison neuronal autophagy
via CCR5, a druggable target. Autophagy. 20:949–951. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Deng H, Xue P, Zhou X, Wang Y and Liu W:
CCL4/CCR5 regulates chondrocyte biology and OA progression.
Cytokine. 183:1567462024. View Article : Google Scholar : PubMed/NCBI
|