Introduction
Metabolic-associated fatty liver disease (MAFLD) is
newly approved nomenclature intended to expand the diagnostic
criteria and avoid the stigma associated with the condition
previously known as non-alcoholic fatty liver disease (NAFLD). This
condition includes a range of liver conditions, from simple fatty
liver (hepatic steatosis), which can be detected using imaging or
histological methods, to metabolic-associated steatohepatitis
(MASH), which is associated with inflammation and can result in
more severe liver damage (1–3). The
development of MAFLD is related to lipid accumulation,
lipotoxicity, oxidative stress and endoplasmic reticulum stress
(ERS) (4), and the mechanism is
complex. The US Food and Drug Administration approved resmetirom as
a therapeutic drug for MASH in March 2024 (5), marking a major breakthrough in the
treatment of MASH. However, the mechanism of MAFLD needs to be
further explored to lay the foundation for developing more
effective treatment strategies and preventive measures for
MAFLD.
The excessive accumulation of lipid droplets (LDs)
is a specific characteristic of MAFLD. Triglycerides (TGs)
accumulate in hepatic LDs, which regulate intracellular fatty acid
flux by storing and releasing fatty acids for oxidation or
re-esterification into TGs or other complex lipids, thus preventing
cytotoxic events (6,7). Perilipin (PLIN)5 is a member of the
PLIN protein family, which has been reported to be positively
associated with TG storage, fatty acid oxidation, lipotoxicity and
metabolic dysfunction (8). PLIN5
is predominantly expressed in tissues with high oxidative
metabolism, such as the liver, heart, skeletal muscle and brown
adipose tissue (BAT) (9). The
absence of PLIN5 can protect against liver injury in MAFLD by
reducing inflammasome activation (10). Furthermore, our previous studies
have shown that knockout of PLIN5 can attenuate high-fat
diet (HFD)-induced MAFLD in mice (11,12);
however, its underlying molecular mechanism remains unclear.
Ferroptosis is a form of iron-dependent,
non-apoptotic cell death characterized by lipid peroxidation. This
unique mode of cell death is regulated by several cellular
metabolic pathways, including iron metabolism and ERS, as well as
various disease-related signaling pathways, such as
glucose-regulated AMPK signaling (13). Notably, ferroptosis has gained
attention in the field of liver disease as the liver is susceptible
to oxidative damage, and excessive iron accumulation is a major
feature in most primary liver diseases (14). Iron overload accelerates the
dysfunction of lipid metabolism and liver injury in MAFLD, and
inhibition of ferroptosis can be used to prevent and protect
against various chronic liver injuries caused by iron deposition
and abnormal lipid metabolism (15). MAFLD alters iron metabolism in the
body and MAFLD progression is accompanied by extensive lipid
accumulation (16–18). Therefore, ferroptosis is considered
an emerging strategy in MAFLD therapeutics.
Activating transcription factor 3 (ATF3) belongs to
the ATF/cAMP response element-binding (CREB) transcription factor
protein family, which controls the expression of target genes by
attaching to the specific DNA sequence TGACGTCA (19). ATF3 sensitizes gastric cancer cells
to cisplatin through the induction of ferroptosis by blocking
Nrf2/Keap1/xCT signaling (19). In
addition, it has been shown that ATF3 has the ability to suppress
the transcriptional activity of SLC7A11, a crucial gene
involved in ferroptosis, thereby triggering the process of
ferroptosis (20). Furthermore,
ATF3-dependent induction of RIPK3 causes a modal shift in
hepatocellular death from apoptosis to necroptosis, and has an
important role in MASH (21).
Therefore, it could be hypothesized that ATF3 may participate in
the regulation of ferroptosis in MAFLD.
This present study aimed to investigate the
involvement of PLIN5 in MAFLD progression and to determine if its
role is related to ferroptosis. Comprehensive experiments were
conducted to elucidate the underlying molecular mechanisms using
both cellular and animal models.
Materials and methods
Animal models
A total of 20 C57BL/6J mice were obtained from
Beijing Huafukang Biotechnology Co., Ltd. In addition, 20
PLIN5−/− mice were generated at the Nanjing
Biomedical Research Institute of Nanjing University (Nanjing,
China) and were identified via gene expression.
PLIN5−/− mice were generated using a CRISPR/Cas9
system in the C57BL/6 background. In vitro transcribed guide
RNA (gRNA) was co-injected with Cas9 protein into mouse fertilized
eggs. Guided by the gRNA, the Cas9 protein binds to the target
genomic site and induces double-strand breaks. These breaks are
subsequently repaired via non-homologous end joining, leading to
deletion mutations and ultimately resulting in gene knockout. Four
single gRNAs (sgRNA1, sgRNA2, sgRNA3 and sgRNA4, listed in Table SI) targeting PLIN5 exon 4
were designed using an online CRISPR design tool (http://chopchop.cbu.uib.no/). The genotypes of the
PLIN5−/− mice were amplified by PCR analysis with
the knockout (PLIN5−/−) and wild-type (WT) primer
pairs (Table SII). The PCR was
performed using the system and program detailed in Tables SIII and SIV by the Nanjing Biomedical Research
Institute of Nanjing University (data not shown). Mice were housed
in a specific pathogen-free-grade rodent facility, with the
temperature maintained at 20–26°C, relative humidity controlled at
40–70%, and under a 12-h light/dark cycle. Specific
pathogen-free-grade C57BL/6J male mice and
PLIN5−/− male mice (age, 6–7-weeks; weight, ~20
g) were randomly divided into groups fed a normal diet ad
libitum (ND) or a HFD (n=10/group; n=40 mice total). Mice in
the ND group were given a normal chow diet and ultrapure water,
whereas those in the HFD group were given a diet high in fat with a
45% fat-supplied ratio (cat. no. H10045; Beijing HFK Bioscience
Co., Ltd.) and sugar water for 20 weeks (14). All mice were housed at the Animal
Center of Zhengzhou University (Zhengzhou, China) under suitable
conditions. For the investigations, the mice were anesthetized via
inhalation of isoflurane, employing an initial induction
concentration of 4% followed by a maintenance concentration of
1.5%. Once the mice had been fed for 20 weeks, liver specimens were
collected and the mice were immediately sacrificed by cervical
dislocation under deep anesthesia. Death was confirmed by the
cessation of vital signs, including the absence of a palpable
heartbeat, fixed and dilated pupils, and unresponsiveness to
mechanical stimuli. The Animal Ethics Committee of Zhengzhou
University approved the protocols for the animal experiments
(approval no. KY2023006). The animals received humane care
according to the criteria outlined in the Guide for the Care and
Use of Laboratory Animals prepared by the National Academy of
Sciences and published by the National Institutes of Health
(22).
Cell culture
The murine hepatocyte AML12 cell line was purchased
from CHI Scientific, Inc., and was cultured in DMEM/F-12 (1:1)
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (cat. no. F8318; MilliporeSigma), 40 ng/ml
dexamethasone (to maintain the mature phenotype and specific
function of liver cells; cat. no. D4902-25MG; MilliporeSigma) and
100 U/ml penicillin/streptomycin (cat. no. 15140122; Gibco; Thermo
Fisher Scientific, Inc.), and incubated at 37°C with a constant
supply of 5% CO2. To establish a hepatic steatosis model
in vitro, AML12 cells were stimulated with palmitic acid
(PA; 0.5 mM; cat. no. KC002) and oleic acid (OA; 1.0 mM; cat. no.
KC006) (both from Kun Chuang Biotechnology) [dissolved in 0.5%
fatty acid-free bovine serum albumin (BSA); cat. no. KC002; Kun
Chuang Biotechnology] at the indicated concentrations for 48 h. The
control group was treated with 0.5% fatty acid-free BSA (21).
Biochemical analysis
To assess the antioxidant capacity of the mouse
liver samples from five mice per group and AML12 cells, commercial
assay kits (Wuhan Elabscience Biotechnology Co., Ltd.) were used to
measure the levels of glutathione (GSH; cat. no. E-BC-K030-M) and
malondialdehyde (MDA; cat. no. E-BC-K025-M), according to the
manufacturer's instructions. To measure the levels of ferroptosis
in mice, the Cell Ferrous Iron (Fe2+) Fluorometric Assay Kit (cat.
no. E-BC-F101; Wuhan Elabscience Biotechnology Co., Ltd.) was used
to detect levels of Fe2+, according to the
manufacturer's instructions.
RNA sequencing (RNA-seq) and
bioinformatics analysis
When the AML12 cell density reached ~70% confluence,
the cells were stimulate with PA (0.5 mM) and OA (1.0 mM) at 37°C
for 48 h, and continuously supply 5% CO2. Following
treatment, the cells were lysed with TRIzol® reagent
(cat. no. 15596026; Invitrogen; Thermo Fisher Scientific, Inc.) and
homogenized via repetitive pipetting. Total RNA was isolated using
chloroform phase separation followed by isopropanol precipitation.
RNA integrity was verified using an Agilent Bioanalyzer 2100 system
(RNA integrity number >8.0; Agilent Technologies, Inc.), and
purity was confirmed using a NanoDrop spectrophotometer (A260/A280
ratio ≥1.9; NanoDrop; Thermo Fisher Scientific, Inc.). RNA-seq
library preparation and paired-end sequencing (150 bp) were
performed by Shanghai OE Biotech Co., Ltd., using an Illumina
NovaSeq 6000 platform (Illumina, Inc.) with the NovaSeq 6000 S4
Reagent Kit (300 cycles; cat. no. 20027466; Illumina, Inc.), The
loading concentration of the final library was adjusted to 10–20
pM, according to standardized protocols. The raw RNA-seq data
generated in the current study have been deposited in the NCBI
Sequence Read Archive under accession number PRJNA1295086
(https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1295086/).
Using the GeneCards database (https://www.genecards.org/), an association between
PLIN5 and ferroptosis was searched for using the key words ‘PLIN5’
and ‘ferroptosis’. Gene set enrichment analysis was performed using
the Gene Ontology (GO; http://geneontology.org/) and Kyoto Encyclopedia of
Genes and Genomes (KEGG; http://www.kegg.jp//) databases. Protein-protein
interaction (PPI) network analysis was carried out using the STRING
database (https://string-db.org/).
Single-cell RNA-seq
Once the mice had been fed for 20 weeks, liver
tissues from five randomly selected mice in each group were used
for single-cell RNA-seq. Livers were perfused with collagenase
IV/DNase I and incubated at 37°C for 15–30 min. Cells were then
filtered through a 40-µm strainer and viable cells were enriched
via flow cytometry (CytoFLEX S V4-B2-Y4-RO; Beckman Coulter, Inc.).
Single-cell capture, cDNA library construction and sequencing were
performed by Shanghai Bohao Biotechnology Co., Ltd., using the 10×
Genomics Chromium platform (targeting 5,000-10,000 cells/sample;
10× Genomics, Inc.) with the 10× Genomics Chromium Next GEM Single
Cell 3′Reagent Kit v3.1 (cat. no. 1000268; 10× Genomics, Inc.),
according to standardized protocols. An Agilent Bioanalyzer 2100
System (Agilent Technologies, Inc.) was used to detect sample
integrity. The final loading concentration of the library was ≥10
pM, and the sequencing was performed using a paired-end strategy
with a read length of 150 bp. Raw sequencing data were deposited in
the China National Center for Bioinformation under accession number
CRA018673 (https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA018673).
The DESeq2 package (1.34.0; http://bioconductor.org/packages/release/bioc/html/DESeq2.html)
was used to identify differentially expressed genes (DEGs); genes
with a P-value <0.05 and |log2 fold-change (FC)|>1
were defined as DEGs. A volcano map of the DEGs was generated using
the ggplot2 (https://ggplot2.tidyverse.org/) in R. DEGs and
ferroptosis-related genes from the FerrDb database (http://www.zhounan.org/ferrdb) were collated into
Venn diagrams; overlapping DEGs were selected for subsequent
analyses.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA extraction from AML12 cells and mouse liver
tissue from five mice per group was performed using a Cell/Tissue
Total RNA Rapid Extraction Kit (Novogene Co., Ltd.), according to
the manufacturer's instructions. The extracted RNA was then
reverse-transcribed into cDNA using an RT kit (Toyobo Co., Ltd.)
according to the manufacturer's protocol. Subsequently, cDNA,
primers and SYBR Green Master Mix (Toyobo Co., Ltd.) were combined
in the appropriate proportions, and PCR amplification was carried
out using the LightCycler 480 system (Roche Diagnostics). qPCR was
performed using a three-step protocol, as follows: Initial
denaturation at 95°C for 10 min for 1 cycle; 35 cycles of
denaturation at 95°C for 15 sec, annealing at 60°C for 15 sec and
extension at 72°C for 45 sec; and a final extension step at 72°C
for 7 min for 1 cycle. The relative abundance of mRNA was
normalized to GAPDH mRNA. Gene expression levels were
calculated using the 2−ΔΔCq method. The data are
expressed as the ratio of the mean of three independent repeats,
and each experiment was repeated at least three times. The primers
used for qPCR are listed in Table
SV.
Western blotting (WB)
Total protein was extracted from AML12 cells and
mouse liver tissues from three mice per group using high-efficiency
radioimmunoprecipitation assay lysis buffer containing protease
inhibitors (Beijing Solarbio Science & Technology Co., Ltd.).
Protein concentrations were measured using a BCA Protein Assay Kit
(Beijing Solarbio Science & Technology Co., Ltd.).
Subsequently, proteins (20 µg/lane) were separated by SDS-PAGE on
10% gels and were transferred to nitrocellulose membranes (Pall
Life Sciences), which were blocked with 8% skim milk in
Tris-buffered saline containing 0.1% Tween-20 (TBST) for 2 h at
room temperature. The membranes were then incubated overnight at
4°C with primary antibodies, and after washing with TBST, the
membranes were incubated with HRP-conjugated AffiniPure Goat
Anti-rabbit IgG (H + L) secondary antibodies at room temperature
for 2 h, before being washed in TBST. The target proteins were
detected using enhanced chemiluminescence (ECL) detection reagent
(Epizyme; Ipsen Pharma) and signal intensities were captured using
an ECL western-blotting analysis system (Tanon Science and
Technology Co., Ltd.). Semi-quantitative analysis was performed
using ImageJ version 1.8.0t software (National Institutes of
Health). All primary and secondary antibodies used are listed in
Table SVI.
Plasmid transfection
PLIN5 overexpression and ATF3
knockdown plasmids, along with their corresponding negative
controls, were commercially obtained from Suzhou GenePharma Co.,
Ltd. The PLIN5 overexpression plasmid was constructed using
the pcDNA3.1(+) backbone, while the ATF3 short hairpin
(sh)RNA and its scrambled negative control were cloned into the
pGPU6/GFP/Neo plasmid. For transient transfection, AML12 cells were
seeded at a density of 2.5×105 cells/well in 6-well
plates and cultured for 24 h to reach 70–80% confluence.
Transfection was performed using Lipofectamine® 3000
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's instructions. Specifically, 2.5 µg plasmid DNA
and 5 µl Lipofectamine 3000 reagent were diluted separately in
Opti-MEM (cat. no. 31985070; Gibco; Thermo Fisher Scientific,
Inc.), then combined and incubated for 15 min at room temperature
to form transfection complexes. The complexes were added dropwise
to the cells, which were then incubated at 37°C in a 5%
CO2 environment. The culture medium was replaced with
fresh medium at 6 h, and after 48 h of total incubation, the cells
were harvested for subsequent experiments. The sequences of shRNAs
are listed in Table SVII. The
negative control used for overexpression consisted of an empty
pcDNA3.1(+) vector, whereas a non-targeting scrambled shRNA was
used as the control for knockdown experiments. Following
transfection, the cells were treated with PAOA for 48 h. In
addition, in the pcDNA3.1 PLIN5 + ferrostatin-1 (FER-1)
group, the cells were transfected with pcDNA3.1 PLIN5,
pretreated with 2 µM FER-1 (cat. no. HY-100579; MedChemExpress) at
37°C in a 5% CO2 cell culture incubator for 1 h, and
then treated with PAOA for an additional 48 h.
Oil red O (ORO) staining
AML12 cells were cultured in a 6-well plate and then
fixed with 4% paraformaldehyde at room temperature for 30 min,
followed by staining with ORO staining solution for 30 min at room
temperature (Nanjing Jiancheng Bioengineering Institute), according
to the manufacturer's instructions. Subsequently, the cells were
thoroughly rinsed with distilled water to remove unbound dye and
images were acquired using a light microscope (Nikon Corporation).
Quantification of the staining results was conducted using ImageJ
(version 1.8.0).
FerroOrange staining
AML12 cells were fixed with 4% paraformaldehyde at
room temperature for 15 min. After washing, the nuclei were stained
with the nuclear dye green cyanine SYTO (Jiangsu Kaiji
Biotechnology) for 10 min at room temperature. Subsequently,
Fe2+ was detected by incubating the cells with
FerroOrange dye (Tocris Bioscience) for 30 min at 37°C in the dark.
Cellular nuclei and Fe2+ signals were visualized using a
confocal microscope (Zeiss LSM 900; Carl Zeiss AG). All
experimental procedures were carried out according to the
instructions provided with the reagents.
H&E staining
Histological analysis was performed on liver tissues
from three mice per group fixed in 4% paraformaldehyde for 24 h at
room temperature, followed by paraffin embedding and slicing into
5-µm sections. The sections were then deparaffinized in xylene and
rehydrated through a graded ethanol series (100, 95, 85 and 70%) to
distilled water. Subsequently, nuclei were stained with Harris
hematoxylin for 5 min at room temperature, differentiated in 1%
acid alcohol and blued in Scott's tap water. The cytoplasm and
extracellular matrix were counterstained with eosin Y for 2 min at
room temperature. Finally, sections were dehydrated through a
graded ethanol series, cleared in xylene and mounted with neutral
balsam. Histopathological examination was conducted using a light
microscope (Nikon Corporation).
Masson's trichrome staining
Histological analysis was performed on liver tissues
from three mice per group fixed in 4% paraformaldehyde for 24 h at
room temperature, followed by paraffin embedding and slicing into
5-µm sections. All staining procedures were carried out at room
temperature. Sections were stained with Weigert's hematoxylin (5–10
min), 1% eosin ethanol (5 min) and Masson composite solution (5
min), followed by differentiation in 1% hydrochloric acid ethanol
(30 sec) and 2% acetic acid (30 sec). Dehydration was performed
using graded ethanol series (70, 85, 95 and 100%). Finally,
histopathological images were acquired using a light microscope
(Nikon Corporation).
TG analysis
The concentration of TGs of mouse liver tissues from
five mice per group was quantified using a commercial TG assay kit
(cat. no. BC0625; Beijing Solarbio Science & Technology Co.,
Ltd.) in accordance with the manufacturer's protocol. TG was
extracted with isopropanol and subsequently saponified using KOH to
release glycerol and free fatty acids. The liberated glycerol was
oxidized by iodine to generate formaldehyde, which then underwent
condensation with acetylacetone in the presence of chloride ions to
yield a yellow-colored product. This chromophore displays a
distinct absorption peak at 420 nm, and the absorbance intensity is
proportional to the original TG concentration. This
well-established method allows accurate and reproducible
determination of TG levels.
Total cholesterol (TC) analysis
The concentration of TC in mouse liver tissues from
five mice per group was determined using a commercial TC assay kit
(cat. no. BC1985; Beijing Solarbio Science & Technology Co.,
Ltd.) according to the manufacturer's instructions. The extracted
cholesterol esters were first hydrolyzed to free cholesterol by
cholesterol esterase. The free cholesterol was then oxidized by
cholesterol oxidase to generate hydrogen peroxide. In the presence
of peroxidase, hydrogen peroxide reacted with 4-aminoantipyrine and
phenol to form a red quinoneimine dye. The absorbance of this
colored product was measured at 500 nm, and its intensity is
proportional to TC concentration.
Alanine aminotransferase (ALT)
analysis
The concentration of ALT was determined using a
commercial ALT assay kit (cat. no. BC1555; Beijing Solarbio Science
& Technology Co., Ltd.) according to the manufacturer's
instructions. After mouse anesthesia, as aforementioned, whole
blood (~500 µl/mouse) was collected from the retro-orbital plexus
of the 40 mice using sterile glass capillary tubes (cat. no.
71900-10; KIMBLE®; DWK Life Sciences) and immediately
transferred to 1.5-ml microcentrifuge tubes. The mice were
subsequently sacrificed by cervical dislocation under deep
anesthesia. The blood was allowed to clot at room temperature for
30 min and the clots were then pelleted by centrifugation at 2,000
× g for 15 min at 4°C. Subsequently, the supernatant (serum) was
carefully aspirated, aliquoted and stored at −80°C until further
analysis.
The serum samples from five mice per group were
diluted with physiological saline at a ratio of 1:10. A total of 5
µl serum was mixed with 290 µl working reagent containing Tris
buffer (pH 7.8), L-alanine, α-ketoglutarate, NADH and lactate
dehydrogenase. The samples were incubated at 37°C for 30 min to
allow the enzyme to fully react with the substrate. After the
reaction was complete, 50 µl NADH chromogenic reagent was added to
each well, and the OD value change was measured at a wavelength of
505 nm. ALT activity in the sample was calculated based on the
standard curve.
Statistical analysis
Data are presented as the mean ± standard error of
mean of at least three independent experiments. All statistical
analyses were performed using GraphPad Prism software version 9.5.1
(Dotmatics), and data were analyzed using one-way analysis of
variance followed by Bonferroni's post hoc test. P<0.05 was
considered to indicate a statistically significant difference.
Results
PLIN5 deficiency inhibits ferroptosis
in lipotoxic hepatocytes in vitro and in vivo
First, an in vitro PAOA-induced hepatocyte
steatosis model was constructed by treating AML12 cells with PAOA
for 48 h. RNA was collected for transcriptome sequencing, and gene
set enrichment analysis with GO and KEGG was performed to analyze
the cell signaling pathways involved in lipid accumulation and
inflammation. The results revealed that PAOA treatment activated
signaling pathways associated with lipid accumulation and
inflammation, such as ‘fatty acid metabolic process’, ‘response to
lipopolysaccharide’, ‘response to fatty acid’, ‘immune response’,
‘inflammatory response’, ‘innate immune response’, ‘IL-17 signaling
pathway, ‘NF-kappa B signaling pathway’ and ‘TNF signaling pathway’
(Fig. S1B and D). In addition,
using ORO staining to assess LD content, followed by statistical
analysis with ImageJ, the LD content was markedly increased in
PAOA-treated AML12 cells compared with that in the BSA control
group (Fig. S1A). These data
indicated that the lipotoxic cell model was successfully
constructed. Furthermore, the expression levels of PLIN5 were
increased in AML12 cells treated with PAOA compared with those in
the BSA group (Fig. S1E and
F).
To investigate the impact of PLIN5 on ferroptosis
under lipotoxic conditions, the key words ‘PLIN5’ and ‘ferroptosis’
were searched in the GeneCards database to determine their
association. A total of 113 genes were identified to be associated
with both PLIN5 and ferroptosis. According to GO enrichment
analysis, the significantly regulated terms were primarily related
to ferroptosis and lipid metabolism signaling, such as
‘ferroptosis’, ‘PPAR signaling pathway’ and ‘fatty acid
biosynthesis’, indicating that these genes were not only associated
with ferroptosis, but were also closely related to biological
processes such as lipid metabolism (Fig. S1C). The primary changes in
biochemical characteristics associated with ferroptosis are iron
overload and decreased activity of the ferroptosis marker gene GSH
peroxidase 4 (GPX4) in MAFLD development (23,24).
In the present study, PPI analysis revealed an indirect association
between PLIN5 and GPX4 (Fig.
S1G).
The present study revealed that PAOA-treated AML12
cells exhibited elevated Fe2+ and significantly reduced
GPX4 expression under lipotoxic conditions compared with those in
the BSA group (Fig. 1A and B),
indicating that PAOA treatment induced ferroptosis in vitro.
Notably, Fe2+ levels were significantly decreased in the
livers of mice in the PLIN5−/− HFD group compared
with those in the WT HFD group (Fig.
1C). The successful knockout of PLIN5 is shown in
Fig. S2A and B. It has previously
been reported that patients with MAFLD have elevated levels of the
lipid peroxidation marker MDA (25). The current study also measured the
redox markers MDA and GSH in the liver tissues from WT and
PLIN5−/− mice fed a ND or HFD for 20 weeks. After
HFD feeding, MDA was increased and GSH was significantly decreased
in mice from both groups; by contrast, MDA levels were
significantly decreased and GSH levels were significantly increased
in the PLIN5−/− HFD group compared with those in
the WT HFD group (Fig. 1D and
E).
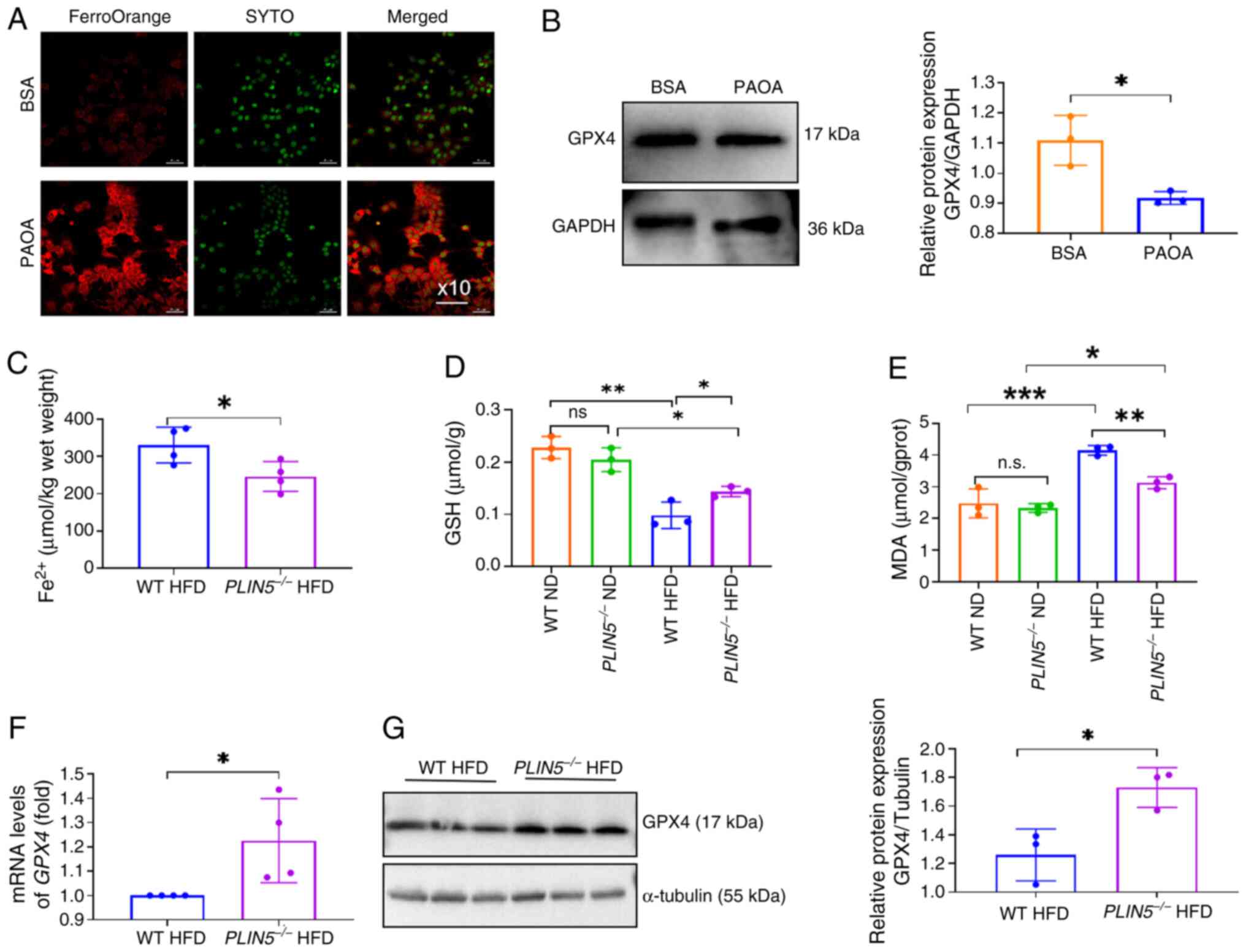 | Figure 1.PLIN5 deficiency inhibits
ferroptosis in lipotoxic hepatocytes in vitro and in
vivo. (A) Fe2+ fluorescence intensity in AML12 cells
observed using a fluorescence microscope (magnification, ×10), with
red indicating Fe2+ fluorescence and green indicating
nuclei. (B) Western blot analysis of GPX4 protein expression in
BSA- or PAOA-treated cells was performed, with GAPDH used as an
internal control. (C) Fe2+ content in the livers of mice
in the WT HFD group and PLIN5−/− HFD group was
measured (n=5 mice/group). (D) Liver GSH content of mice in each
group was detected (n=5 mice/group). (E) Liver MDA content of mice
in each group was detected (n=5 mice/group). (F) mRNA levels of
GPX4 in the liver tissues of mice in the WT HFD and
PLIN5−/− HFD groups were analyzed by quantitative
polymerase chain reaction (n=5 mice/group). (G) Western blot
analysis of GPX4 protein expression in the liver tissues of mice
from the WT HFD and PLIN5−/− HFD groups was
performed, with tubulin used as an internal control (n=3
mice/group). All experiments were repeated at least three times
(n=3). *P<0.05, **P<0.01, ***P<0.001, n.s., not
significant. Fe2+, ferrous ion; GSH, glutathione; GPX4,
GSH peroxidase 4; HFD, high-fat diet; MDA, malondialdehyde; PAOA,
palmitic acid and oleic acid; PLIN5, perilipin 5; WT,
wild-type. |
Notably, GPX4 expression levels were significantly
higher in the liver tissues of the PLIN5−/− HFD
group compared with those in the WT HFD group (Fig. 1F and G). Consistent with our
previous reports (10,11). H&E staining revealed a marked
reduction in lipid droplets in the PLIN5−/− group
compared with those in the WT group (Fig. S2C). Furthermore, Masson's
trichrome staining indicated a decrease in liver fibrosis in
PLIN5−/− mice (Fig.
S2D), and TG and TC levels were significantly reduced compared
with those in the WT group (Fig. S2E
and F). Additionally, serum ALT levels were lower in the
PLIN5−/− group compared with those in the WT
group (Fig. S2G). These findings
indicated that PLIN5 knockout could attenuate hepatic
steatosis and injury, and suggested that PLIN5 deficiency
may delay the progression of MAFLD in mice by inhibiting
ferroptosis.
Overexpression of PLIN5 promotes lipid
accumulation and ferroptosis in hepatocytes
To further explore how PLIN5 affects ferroptosis, a
PLIN5 overexpression cell model was established (Fig. 2A and B).
PLIN5-overexpressing cells were pretreated with FER-1, a
selectively effective inhibitor of ferroptosis, and all cell groups
were treated with PAOA. The results showed that LD accumulation was
significantly increased in the pcDNA3.1-PLIN5 group compared
with that in the pcDNA3.1 group, which was reversed by the addition
of FER-1 (Fig. 2C). Furthermore,
Fe2+ fluorescence intensity was significantly enhanced
in the pcDNA3. 1-PLIN5 group treated with PAOA, which was
reversed by the addition of FER-1 (Fig. 2D). Furthermore, the MDA content was
increased and GSH levels were significantly decreased in
pcDNA3.1-PLIN5 cells treated with PAOA, which was reversed
by treatment with FER-1 (Fig. 2E and
F). These findings revealed that PLIN5 overexpression
could promote lipid accumulation and ferroptosis in PAOA-treated
hepatocytes, and that the ferroptosis inhibitor FER-1 could
partially alleviate lipid accumulation, oxidative damage and
FE2+ content caused by PLIN5 overexpression.
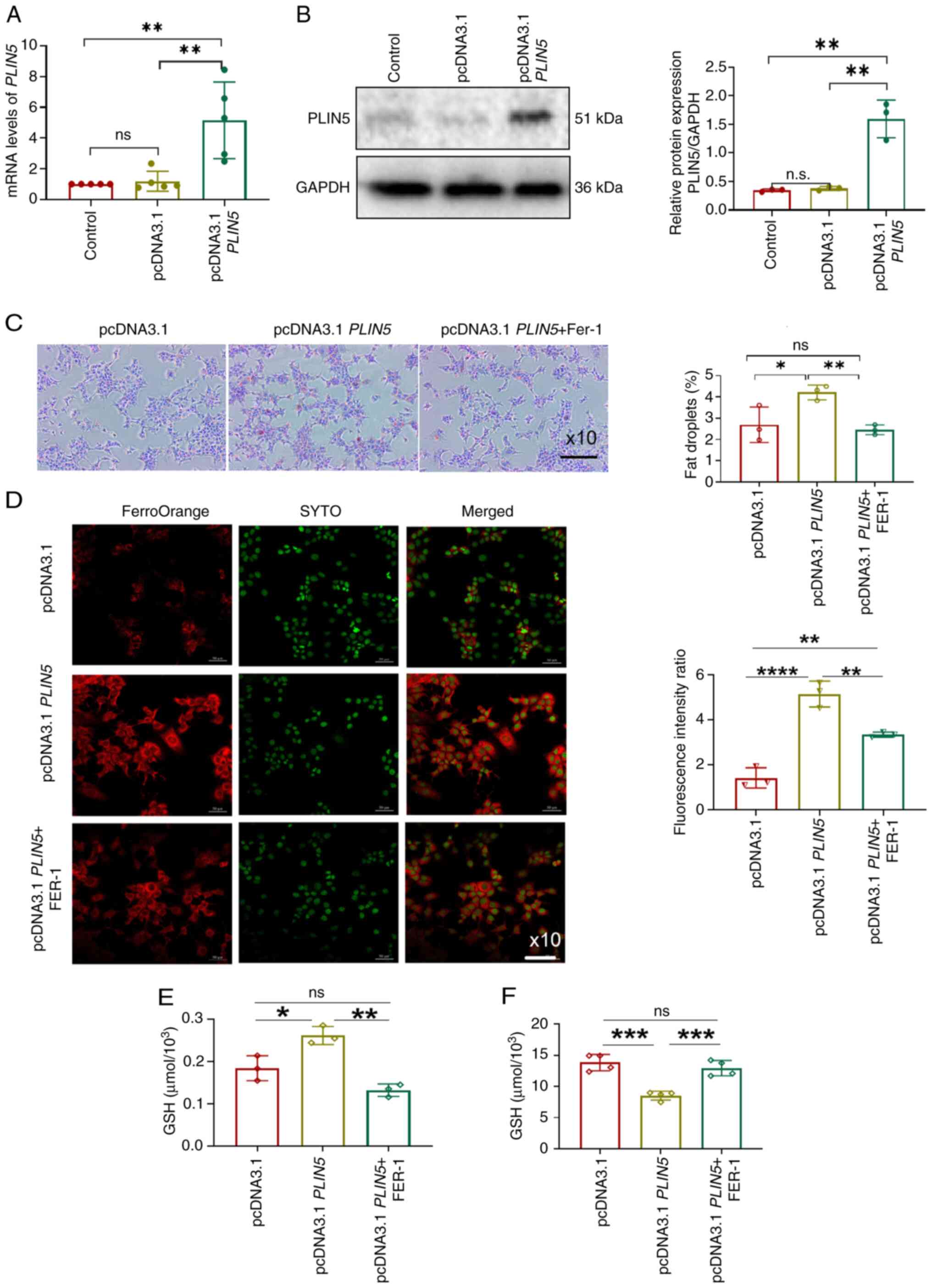 | Figure 2.Overexpression of PLIN5
promotes lipid accumulation and ferroptosis in hepatocytes. (A)
mRNA levels of PLIN5 levels were detected via quantitative
polymerase chain reaction. (B) PLIN5 protein expression was
detected via western blotting. (C) LD changes in cells were
observed using a microscope (magnification, ×10), with nuclei
stained blue and LDs stained red. Quantitative analysis was
performed using ImageJ. (D) Fe2+ fluorescence intensity
in cells was observed using a fluorescence microscope
(magnification, ×10), with nuclei stained green and Fe2+
fluorescence stained red, followed by quantitative analysis. (E)
MDA levels were detected in the three cell groups. (F) GSH levels
were detected in the three groups of cells. *P<0.05,
**P<0.01, ***P<0.001, ****P<0.0001, n.s., not significant.
Fe2+, ferrous ion; GSH, glutathione; LD, lipid droplet;
MDA, malondialdehyde; PLIN5, perilipin 5; FER-1, ferrostatin-1. |
PLIN5-induced ferroptosis is
associated with ATF3
As aforementioned, the present study initially
discovered that PLIN5 may modulate ferroptosis both in vivo
and in vitro. To further elucidate how PLIN5 affects
ferroptosis signaling, an in-depth analysis was subsequently
performed by mining single-cell RNA-seq data. This strategy enabled
identification of cell type-specific ferroptosis regulators
downstream of PLIN5. The current study analyzed single-cell RNA-seq
data obtained from the liver tissues from 5 mice from the
PLIN5−/− HFD group and 5 mice from the WT HFD
group to elucidate the functional genes involved in regulating the
effects of PLIN5 knockout in MAFLD. The results revealed
that there were 235 DEGs (log2 FC>1, P<0.05)
between the PLIN5−/− HFD and WT HFD groups, with
110 upregulated and 125 downregulated genes (Fig. 3A). Notably, these genes were
intersected with the FerrDb database and seven ferroptosis-related
DEGs were identified, including two upregulated genes: Fascin
actin-bundling protein 1 (FSCN1) and MYC, and five
downregulated genes: ATF3, glycerophosphodiester
phosphodiesterase domain containing 5 (GDPD5),
lactotransferrin (LTF), PLIN2 and protein regulator
of cytokinesis 1 (PRC1) (Fig.
3B). GO and KEGG analyses were performed on the aforementioned
genes (Fig. 3C). The DEGs
associated with ferroptosis were involved in various biological
processes, including ‘carboxylic acid transport’, ‘organic acid
transport’ and ‘monocarboxylic acid transport’. The genes were
enriched in molecular functions related to ‘repressing
transcription factor binding’, ‘mRNA binding’ and ‘ubiquitin
protein ligase binding’. In addition, these DEGs were enriched in
the following cellular components: ‘Myelin sheath’, ‘contractile
ring’ and ‘invadopodium’. KEGG analysis revealed associations with
‘acute myeloid leukemia’, as well as the ‘PPAR signaling pathway’
and ‘Wnt signaling pathway’. The results of RT-qPCR showed that,
compared with those in the WT HFD group, the mRNA expression levels
of FSCN1 (P<0.001) and MYC (P<0.05) were
significantly higher in the PLIN5−/− HFD group,
whereas the mRNA levels of ATF3, GDPD5 (P<0.05), LTF,
PLIN2 and PRC1 were significantly lower; among these,
the most significant difference was observed in ATF3)
(Fig. 3D). Therefore, PLIN5
knockout-induced inhibition of ferroptosis in MAFLD may be
associated with ATF3.
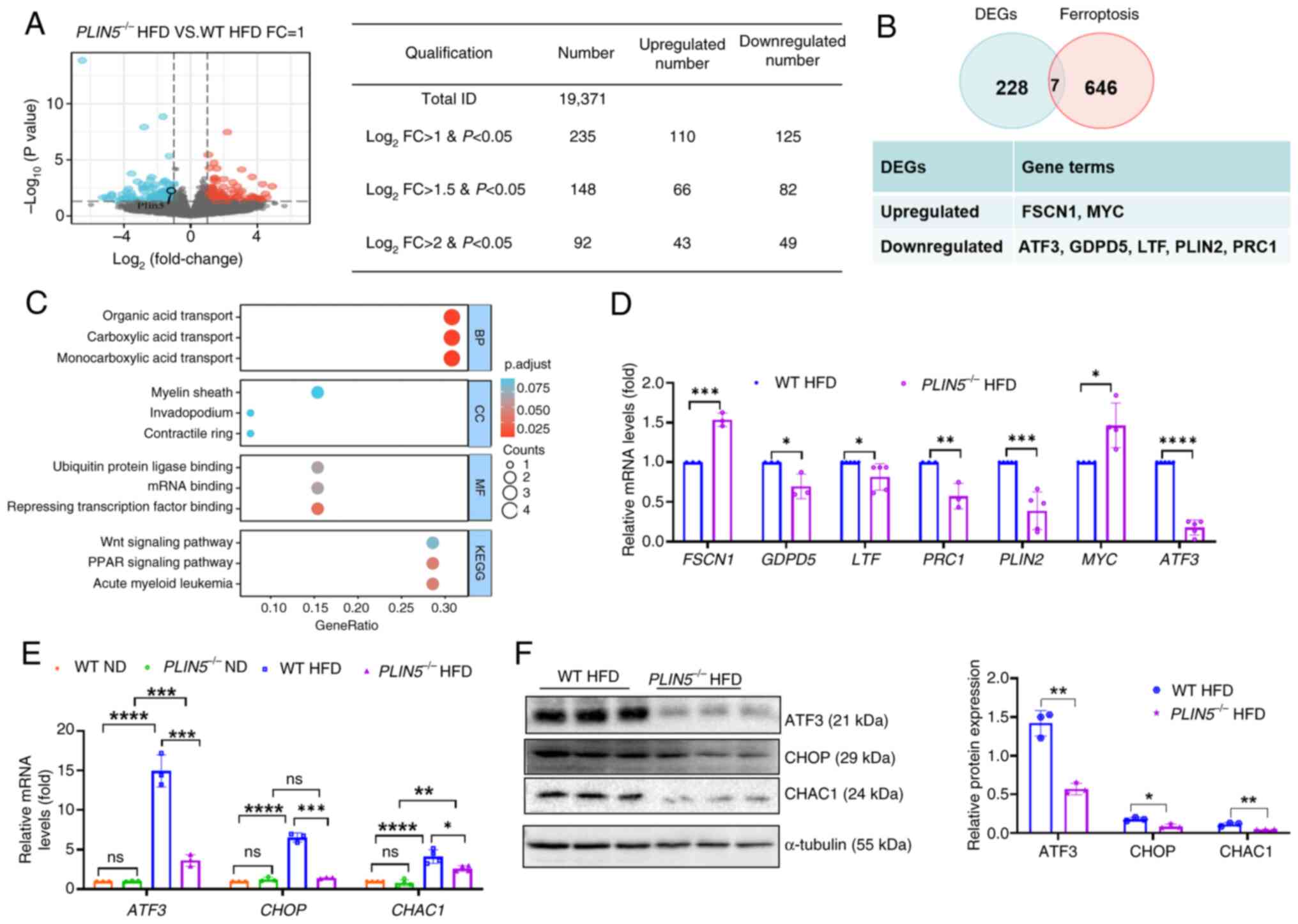 | Figure 3.PLIN5 affects ferroptosis
through ATF3/CHOP/CHAC1 signaling. (A) DEGs in mouse liver tissues
from the PLIN5−/− HFD group are shown compared to
those in the WT HFD group (red dots represent upregulated genes,
whereas blue dots represent downregulated genes). (B) DEGs
associated with ferroptosis in the PLIN5−/− HFD
and WT HFD groups were analyzed (log2 FC>1,
P<0.05). (C) KEGG pathway and GO term enrichment analyses of
different genes were performed. (D) mRNA levels of PRC1, LTF,
GDPD5, FSCN1, MYC, ATF3 and PLIN2 in the liver tissues
of WT HFD and PLIN5−/− HFD mice were measured
(n=5 mice/group). (E) mRNA levels of ATF3, CHOP and
CHAC1 in the liver tissues of mice in each group were
detected (n=5 mice/group). (F) Western blot analysis of ATF3, CHOP
and CHAC1 protein expression in the liver tissues of mice from the
WT HFD and PLIN5−/− HFD groups was performed,
with tubulin as the internal reference. Semi-quantitative analysis
performed using ImageJ (n=3 mice/group). All experiments were
repeated at least three times (n=3). *P<0.05, **P<0.01,
***P<0.001, ****P<0.0001, n.s., not significant. ATF3,
activating transcription factor 3; CHAC1, cation transport
regulator-like protein 1; CHOP, C/EBP homologous protein; DEGs,
differentially expressed genes; FC, fold-change; FSCN1,
fascin actin-bundling protein 1; GDPD5,
glycerophosphodiester phosphodiesterase domain containing 5;
GPX4, glutathione peroxidase 4; HFD, high-fat diet; KEGG,
Kyoto Encyclopedia of Genes and Genomes; ND, normal diet;
LTF, lactotransferrin; PLIN, perilipin; PRC1,
protein regulator of cytokinesis 1; WT, wild-type; BP, biological
process; CC, cellular component; MF, molecular function. |
ATF3 is a member of the ATF/CREB transcription
factor family and is upregulated in mouse models of non-alcoholic
steatohepatitis (NASH) (26). The
C/EBP homologous protein (CHOP) signaling pathway is involved in
the synergistic interaction between ferroptosis and apoptosis
(27). ATF3 and CHOP regulate the
transcription of cation transport regulator-like protein 1 (CHAC1)
(28), and elevated CHAC1 is an
important marker of ferroptosis (29,30).
Notably, the present results showed that the expression levels of
ATF3, CHOP and CHAC1 were significantly lower in the
PLIN5−/− HFD group compared with those in the WT
HFD group, and compared with in the ND group, their expression
levels were significantly increased in the HFD group (Fig. 3E and F). Moreover, single-cell
transcriptome data were analyzed and it was revealed that knocking
out PLIN5 significantly reduced the expression of
ACSL4 in hepatocytes in PLIN5−/− HFD mice
compared with that in the WT HFD group; however, there was no
difference in the expression of SLC7A11 (Fig. S3). These findings suggested that
knocking out PLIN5 can inhibit ferroptosis. Notably,
overexpression of PLIN5 could promote the expression of,
ATF3, CHOP and CHAC1 with PAOA treatment compared
with that in the pcDNA3.1 group, which was reversed by FER-1
treatment (Fig. 4B-E). By
contrast, compared with those in the pcDNA3.1 group, GPX4
expression levels were significantly decreased by PLIN5
overexpression; however, GPX4 was significantly enhanced in the
pcDNA3.1 PLIN5 + FER-1 group (Fig. 4A and E). Taken together, these
findings suggested that ATF3 may be involved in the
PLIN5-associated regulation of HFD-induced hepatocyte
ferroptosis.
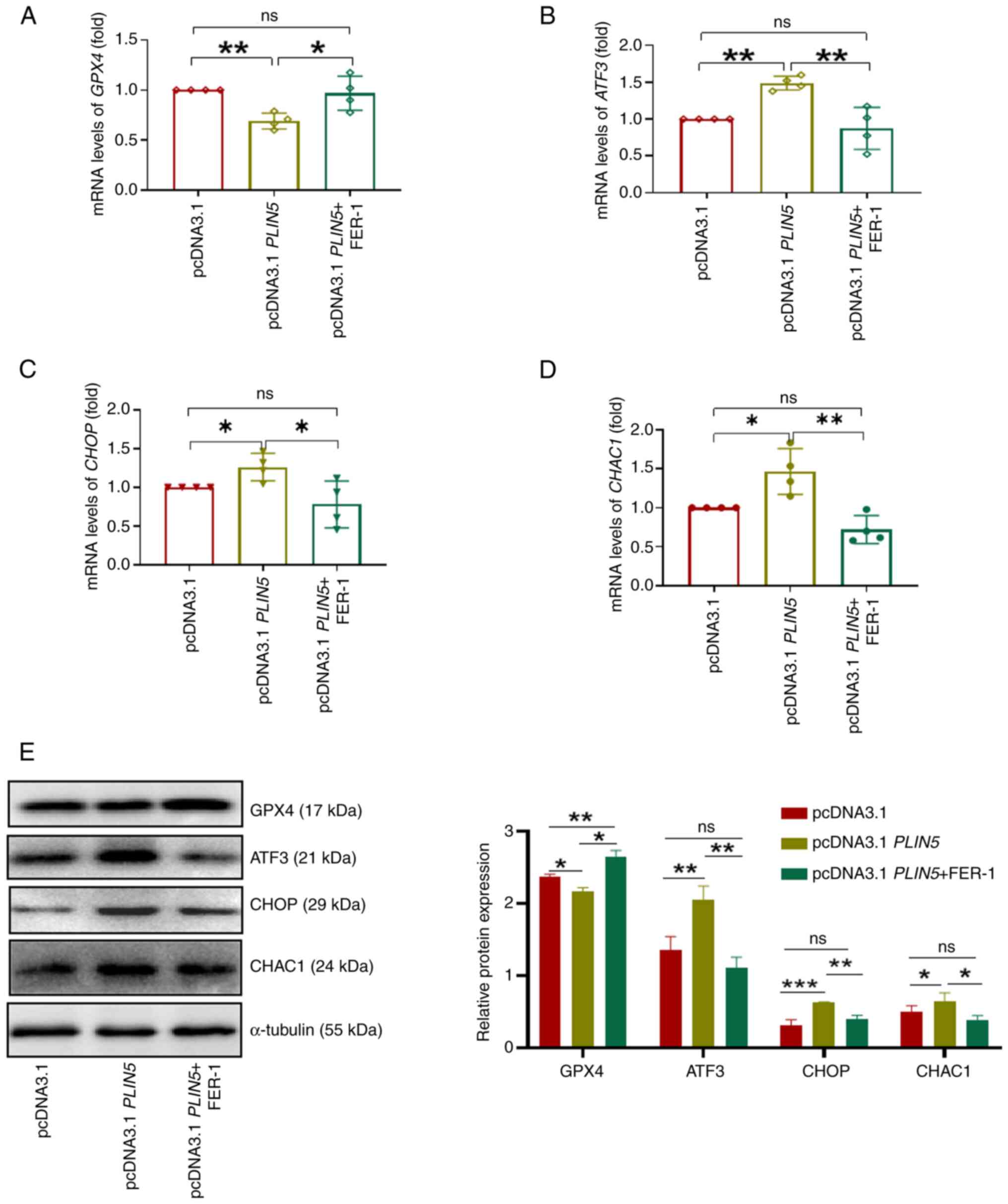 | Figure 4.PLIN5 affects ferroptosis via
ATF3/CHOP/CHAC1 signaling. The mRNA expression levels of (A)
GPX4, (B) ATF3, (C) CHOP and (D) CHAC1
in cells from each group were measured. (E) Western blot analysis
of GPX4, ATF3, CHOP and CHAC1 protein expression in cells from
different groups was performed, normalized to tubulin as an
internal control. Semi-quantitative analysis was performed using
ImageJ. All experiments were repeated at least three times.
*P<0.05, **P<0.01, ***P<0.001, n.s., not significant.
ATF3, activating transcription factor 3; CHAC1, cation transport
regulator-like protein 1; CHOP, C/EBP homologous protein; FER-1,
ferrostatin-1; GPX4, glutathione peroxidase 4; PAOA,
palmitic acid and oleic acid; PLIN5, perilipin 5. |
Knockdown of ATF3 alleviates lipid
accumulation and ferroptosis induced by overexpression of
PLIN5
To confirm that ATF3 is involved in the process of
PLIN5-regulated ferroptosis, ATF3 expression was knocked down in
the cells (Fig. 5A and B). PAOA
stimulation was used to establish cell models, and then ORO
staining was performed. The results showed that ATF3
knockdown significantly reduced PAOA-induced lipid accumulation in
hepatocytes and also partially reversed lipid accumulation induced
by PLIN5 overexpression (Fig.
5C). In addition, ATF3 knockdown partially reversed the
Fe2+ fluorescence intensity induced by PLIN5
overexpression, and PAOA-induced oxidative damage was also
alleviated, as determined by MDA and GSH detection (Fig. 5D-F). Collectively, these results
suggested that as a key regulator, PLIN5 could enhance lipid
peroxidation and ferroptosis in hepatocytes.
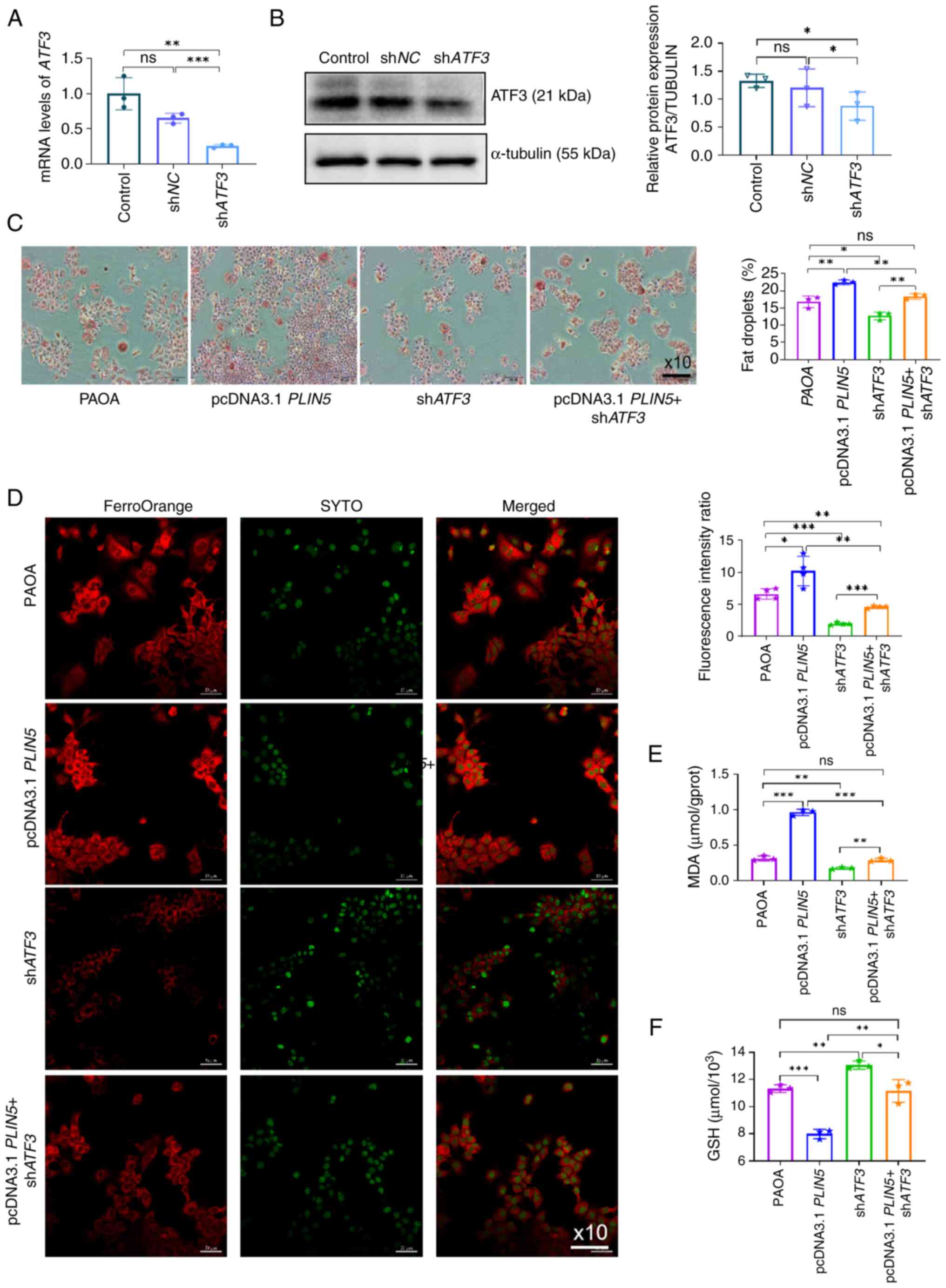 | Figure 5.Knockdown of ATF3 alleviates
lipid accumulation and ferroptosis induced by overexpression of
PLIN5. (A) ATF3 mRNA expression in AML12 cells was
analyzed by quantitative polymerase chain reaction 48 h after
transfection with shATF3 plasmid. (B) Semi-quantitative analysis of
ATF3 protein expression in AML12 cells 48 h after transfection with
shATF3 plasmid was performed, normalized to tubulin as an internal
control, using ImageJ. (C) LDs were observed in cells from
different intervention groups using a microscope (magnification,
×10), with nuclei stained blue and LDs stained red and quantitative
analysis was performed using ImageJ. (D) Fe2+
fluorescence intensity in cells from different intervention groups
was observed using a fluorescence microscope, with nuclei stained
green and Fe2+ fluorescence stained red, followed by
quantitative analysis. (E) MDA content was measured in cells from
different intervention groups. (F) GSH content was measured in
cells from different intervention groups. All experiments were
repeated at least three times. *P<0.05, **P<0.01,
***P<0.001, n.s., not significant. ATF3, activating
transcription factor 3; Fe2+, ferrous ion; GSH,
glutathione; LD, lipid droplet; MDA, malondialdehyde; NC, negative
control; PAOA, palmitic acid and oleic acid; PLIN5,
perilipin 5; sh, short hairpin. |
Discussion
Over time, the increasing incidence of MAFLD and
associated complications leading to mortality have become of
increasing concern (31). The
development of MAFLD is associated with lipid accumulation,
oxidative stress, ERS and lipotoxicity (4). The pathogenesis of MAFLD is complex,
and there are currently no specific drugs that can be used to
reverse MAFLD. Notably, different types of hepatocellular death in
MAFLD, including apoptosis, necrosis, necroptosis and pyroptosis,
have been studied extensively (32,33).
Ferroptosis is a form of regulated cell death that is
iron-dependent and non-apoptotic, induced by lipid peroxidation and
controlled by the integration of oxidative and antioxidant systems
(34). Key steps in the formation
of ferroptosis include Fe2+ accumulation and lipid
peroxidation, and ACSL4 and GPX4 positively and negatively regulate
ferroptosis, respectively (24,25).
Previous studies have shown that ferroptosis is
exacerbated in MAFLD, that it serves an important role as a trigger
for the initiation of inflammation in steatohepatitis and that it
affects the progression of NASH (18,35,36).
Given that ferroptosis is critical in regulating the progression of
MAFLD, understanding the molecular mechanisms underlying
ferroptosis and identifying novel targets to inhibit its occurrence
may be an effective approach in the treatment of abnormal
ferroptosis-related MAFLD. The present study provides compelling
evidence demonstrating that the downregulation of PLIN5 can
inhibit the occurrence of ferroptosis. Mechanistically, PLIN5 may
influence the progression of MAFLD and the occurrence of
ferroptosis by regulating ATF3 activation. Knockout of PLIN5
was shown to inhibit expression of the ferroptosis-related genes
ATF3, CHOP and CHAC1; in addition, knockdown of
ATF3 could alleviate PLIN5 overexpression-induced
ferroptosis and lipid accumulation in cells. Notably, it has been
reported that ATF1, ATF3 and ATF4 can bind to the
PLIN5 promoter and induce its expression in hepatocytes
(37). It may thus be speculated
that a positive feedback loop between PLIN5 and ATF3
could contribute to the pathogenesis of MAFLD. Moreover, emerging
evidence has indicated that phosphorylation of PLIN5 by protein
kinase A triggers PLIN5 nuclear translocation and regulates the
transcriptional expression of PPARγ coactivator 1-α (38). However, current evidence
demonstrates that PLIN5 primarily functions as a transcriptional
co-activator or auxiliary factor. It therefore could be
hypothesized that PLIN5 may participate in regulating ATF3
transcriptional expression; however, this requires further
validation in luciferase reporter assays and chromatin
immunoprecipitation experiments. In addition, a previous study
showed that JNK phosphorylation was significantly reduced in a
moues model of MAFLD after loss of PLIN5 (39). In another experiment, the JNK
inhibitor JM-2 was reported to significantly protect mouse livers
from HFD-induced lipid accumulation and apoptosis (40). Therefore, other cell death pathways
regulated by PLIN5 may also contribute to the development of
MAFLD.
PLINs have been recognized as key proteins involved
in lipid accumulation; in particular, the role of PLIN5 in the
liver has been extensively studied (9–12).
Different models of MAFLD have been established in
PLIN5−/− mice and have yielded differing results.
These conflicting findings may be due to differences in the
MAFLD/MASH model and stages of the disease studied. For example,
Mass-Sanchez et al (41)
demonstrated that loss of PLIN5 protects against worsening
of MAFLD by regulating inflammatory signaling, mitochondrial
function and lipid metabolism, while in a mouse model of
MAFLD-induced hepatocellular carcinoma, PLIN5 knockout was
found to suppress phosphorylated-STAT3 and attenuate the
inflammatory response, thereby mitigating severe liver injury.
However, in various NASH models, PLIN5 knockout has been
shown to worsen NASH-associated characteristics in mice given a
high-fat/high-cholesterol (HFHC) diet, such as lipid accumulation,
inflammation and hepatic fibrosis; by contrast, overexpression of
PLIN5 has been shown to ameliorate methionine and
choline-deficient (MCD) diet-induced NASH and ferroptosis (42). In a previous study, key points
reported regarding the major models were reviewed, as well as the
feeding conditions evaluated in each of these models, underpinning
the notion that the role of PLIN5 in metabolism appears to be
tissue-specific (43). In the
current study, PLIN5 was highly expressed in the MAFLD model, and
knockout of PLIN5 could reverse MAFLD. Notably, it was
demonstrated that the loss of PLIN5 could reduce ferroptosis
and upregulate the expression of GPX4; however, these
findings differ from those reported in other studies (42,44),
where knockout of PLIN5 diminished HFHC diet-induced
ferroptosis and overexpression of PLIN5 ameliorated MCD
diet-induced NASH and ferroptosis (42). Furthermore, PLIN5
overexpression has been suggested to ameliorate ferroptosis via the
PIR/NF-κB axis in PA-stimulated HL-1 cells (44). In summary, the regulation of
ferroptosis by PLIN5 varies in different disease models.
A previous study demonstrated that knocking out
PLIN5 enhances insulin resistance in skeletal muscles by
reducing the storage of TGs (45).
Moreover, PLIN5 deletion protects against MAFLD and
hepatocellular carcinoma by modulating lipid metabolism and
inflammatory responses (41).
Gallardo-Montejano et al (46) demonstrated that promoting PLIN5
function in BAT was associated with healthy remodeling of
subcutaneous white adipose tissue, and an improvement in systemic
glucose tolerance, as well as diet-induced hepatic steatosis. These
conflicting results may be attributed to the methods used in
previous PLIN5-related research related to MAFLD, all of which have
focused on systemic rather than local knockout of PLIN5.
Therefore, crosstalk in the biological effects of PLIN5 in
different tissues is possible. Furthermore, systemic knockout may
have off-target metabolic effects. Given that PLIN5 exhibits
distinct biological functions across different tissues and cell
types, liver-specific knockout mice will be essential to further
elucidate the role of PLIN5 in liver metabolism. Furthermore, ex
vivo liver organoid models, with their operational simplicity,
reproducibility and technical accessibility (47,48),
may serve as an ideal platform for elucidating how
microenvironmental factors mediate the regulatory role of PLIN5 in
MAFLD pathogenesis.
ATF3 is a member of the ATF/CREB family of
transcription factors (20).
Recent research has demonstrated that liver macrophage ATF3
simultaneously inhibits hepatocyte lipogenesis and hepatic stellate
cell activation in mice (49).
Basak et al (50) showed
that ATF3 can form a complex with RGS7, which is required for
PA-dependent oxidative stress and cell death. ATF3 has also
been identified as a key gene in ferroptosis following spinal cord
injury (51), and the soy-derived
compound 6j can inhibit liver cancer cell proliferation via
ATF3-mediated ferroptosis (52).
In addition, evidence has suggested that ATF3 is involved in
erastin-induced ferroptosis (53).
Therefore, ATF3 may have a critical role in regulating ferroptosis
during MASH progression. The present study identified a set of
genes associated with ferroptosis, and demonstrated that ATF3
expression was significantly lower in the
PLIN5−/− HFD group compared with that in the WT
HFD group.
The CHOP signaling pathway is also involved in the
synergistic interaction between ferroptosis and apoptosis (28), and ATF3/CHOP/BCL2 signaling can
control ERS-associated apoptosis (54). CHAC1 is the downstream target of
the ATF4/CHOP axis; it possesses γ-glutamyl cyclotransferase
activity and also degrades GSH. Notably, depletion of GSH is a
crucial factor in apoptosis initiation and execution (55). Endoplasmic reticulum-mediated
apoptosis is also activated via the ATF4/CHOP/CHAC1 cascade
(56). Given these findings, it
may be hypothesized that ATF3 is the interacting target of PLIN5,
which affects ferroptosis, oxidative damage and lipid accumulation.
The present study revealed that the expression levels of CHOP and
CHAC1 were reduced in PLIN5−/− HFD mice compared
with those in WT HFD mice. Furthermore, knockdown of ATF3
alleviated lipid accumulation and ferroptosis induced by
PLIN5 overexpression. Emerging evidence has revealed that
PLIN5 not only functions as an LD-coating protein localized on LDs,
but that its phosphorylated form can also translocate into the
nucleus to participate in transcriptional regulation. This dual
functionality markedly enhances the complexity of the reciprocal
regulatory network between PLIN5 and transcription factor ATF3,
warranting further mechanistic investigation (34,57).
Owing to limitations in clinical specimen availability, the current
study was unable to examine the association between PLIN5 and
ferroptosis-related proteins (including ATF3, CHOP and CHAC1) in
human fatty liver tissues. Notably, a previous study reported that
the protein expression of PLIN5 was markedly elevated in the
severely steatotic livers of included patients (58). Moreover, ATF3 has been reported to
be highly expressed in the livers of Zucker diabetic fatty rats and
in human participants with MAFLD and/or type 2 diabetes (59).
As indicated in the current guidelines (AASLD
(60), very few drugs are
recommended for MAFLD treatment. To date, only vitamin E and the
PPARγ ligand pioglitazone have been endorsed for limited patients
by the European-and American Association for the Study of the Liver
(61). Given the central roles of
PLIN5 in LD stabilization and ATF3 in hepatic stress response, as
demonstrated by the present study, pharmacological inhibition of
these targets represents a mechanistically rational strategy for
MAFLD intervention. Patients with hypercholesterolemia routinely
receive statins, HMG-CoA reductase inhibitors that block de
novo cholesterol biosynthesis. Previous studies have
demonstrated that statins reduce PLIN5 levels in murine/human
hepatocytes, concomitant with decreased TGs and LD quantity. This
effect is mediated by an atypical sterol-regulatory sequence in the
PLIN5 promoter, where SREBP2 binding confers statin
responsiveness (62,63). Crucially, partial PLIN5
knockdown can mimic statin-induced TG reduction, while PLIN5
overexpression reverses it (62).
Thus, combining PLIN5 inhibitors with statins may yield synergistic
anti-MAFLD effects. Notably, Yu et al (64) proposed targeting ATF3 with
engineered peptide inhibitors, providing a compelling rationale for
developing ATF3-targeted therapies against MAFLD.
In conclusion, the present study identified a link
from PLIN5 to ferroptosis and MAFLD via the ATF3/CHOP/CHAC1 axis,
thereby providing insights into the mechanism of ferroptosis in
MAFLD progression. Knocking down PLIN5 may target the
ATF3/CHOP/CHAC1 signaling axis, which attenuates ferroptosis in
liver cells, eventually alleviating MAFLD. The present findings may
lay the foundation for a promising therapeutic strategy in the
treatment of MAFLD.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was supported by research grants from the
National Natural Science Foundation of China (grant no. 31471330),
the Henan Provincial Medical Science and Technology Tackling
Program Co-construction Project (grant nos. LHGJ20210483 and
LHGJ20220557), the Zhengzhou University Discipline Key Special
Project (grant no. XKZDQY202001), the Henan Province Foreign
Intelligence Introduction Program (grant no. GZS2022008), Science
and Technology Projects (grant no. 242102310228), the Zhengzhou
Science and Technology Benefit to the People Project (grant no.
2022KJHM0020) and the Key Research and Development Projects of
Henan Province (grant no. 241111210500).
Availability of data and materials
The raw RNA-seq data generated in the present study
may be found in the NCBI Sequence Read Archive under accession
number PRJNA1295086 or at the following URL: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1295086/.
The raw single-cell RNA-seq data generated in the present study may
be found in the China National Center for Bioinformation under
accession number CRA018673 or at the following URL: https://ngdc.cncb.ac.cn/gsa/search?searchTerm=CRA018673.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
Experiments were performed by YT, YL, XW and XY,
with the help of LD, WC and YX. Experiments were designed by YL and
XW, with the help of PZ and YW. LD, XZ and LL managed animals and
performed animal experiments. XW, XY and YL analyzed the data. YL
wrote the paper, with assistance from YT, XW and XY in editing and
revisions. PZ and YT supervised the work, and confirm the
authenticity of all the raw data. All authors made significant
contributions to the article, and read and approved the final
manuscript.
Ethics approval and consent to
participate
The animal study was approved by the Ethics
Committee of Zhengzhou University (approval no. KY2023006). The
study was performed in accordance with Institutional Animal Care
and Use Committee guidelines; all procedures were carried out in
accordance with institutional guidelines.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rinella ME, Neuschwander-Tetri BA,
Siddiqui MS, Abdelmalek MF, Caldwell S, Barb D, Kleiner DE and
Loomba R: AASLD Practice Guidance on the clinical assessment and
management of nonalcoholic fatty liver disease. Hepatology.
77:1797–1835. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yip TCF, Vilar-Gomez E, Petta S, Yilmaz Y,
Wong GL, Adams LA, de Lédinghen V, Sookoian S and Wong VW:
Geographical similarity and differences in the burden and genetic
predisposition of NAFLD. Hepatology. 77:1404–1427. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Syed-Abdul MM: Lipid metabolism in
Metabolic-associated steatotic liver disease (MASLD). Metabolites.
14:122024. View Article : Google Scholar
|
|
4
|
Guo X, Yin X, Liu Z and Wang J:
Non-alcoholic fatty liver disease (NAFLD) pathogenesis and natural
products for prevention and treatment. Int J Mol Sci. 23:154892022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Petta S, Targher G, Romeo S, Pajvani UB,
Zheng MH, Aghemo A and Valenti LVC: The first MASH drug therapy on
the horizon: Current perspectives of resmetirom. Liver Int.
44:1526–1536. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Herker E, Vieyres G, Beller M, Krahmer N
and Bohnert M: Lipid droplet contact sites in health and disease.
Trends Cell Biol. 31:345–358. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gluchowski NL, Becuwe M, Walther TC and
Farese RV Jr: Lipid droplets and liver disease: From basic biology
to clinical implications. Nat Rev Gastroenterol Hepatol.
14:343–355. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Najt CP, Khan SA, Heden TD, Witthuhn BA,
Perez M, Heier JL, Mead LE, Franklin MP, Karanja KK, Graham MJ, et
al: Lipid Droplet-derived monounsaturated fatty acids traffic via
PLIN5 to allosterically activate SIRT1. Mol Cell. 77:810–824.e8.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mason RR and Watt MJ: Unraveling the roles
of PLIN5: Linking cell biology to physiology. Trends Endocrinol
Metab. 26:144–152. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Asimakopoulou A, Engel KM, Gassler N,
Bracht T, Sitek B, Buhl EM, Kalampoka S, Pinoé-Schmidt M, van
Helden J, Schiller J and Weiskirchen R: Deletion of perilipin 5
protects against hepatic injury in nonalcoholic fatty liver disease
via missing inflammasome activation. Cells. 9:13462020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ma Y, Yin X, Qin Z, Ke X, Mi Y, Zheng P
and Tang Y: Role of Plin5 deficiency in progression of
Non-alcoholic fatty liver disease induced by a High-fat diet in
mice. J Comp Pathol. 189:88–97. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin X, Dong L, Wang X, Qin Z, Ma Y, Ke X,
Li Y, Wang Q, Mi Y, Lyu Q, et al: Perilipin 5 regulates hepatic
stellate cell activation and high-fat diet-induced non-alcoholic
fatty liver disease. Animal Model Exp Med. 7:166–178. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wu J, Wang Y, Jiang R, Xue R, Yin X, Wu M
and Meng Q: Ferroptosis in liver disease: New insights into disease
mechanisms. Cell Death Discov. 7:2762021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou X, Fu Y, Liu W, Mu Y, Zhang H, Chen J
and Liu P: Ferroptosis in chronic liver diseases: Opportunities and
challenges. Front Mol Biosci. 9:9283212022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang S, Liu Z, Geng J, Li L and Feng X: An
overview of ferroptosis in Non-alcoholic fatty liver disease.
Biomed Pharmacother. 153:1133742022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang H, Axinbai M, Zhao Y, Wei J, Qu T,
Kong J, He Y and Zhang L: Bioinformatics analysis of
Ferroptosis-related genes and immune cell infiltration in
non-alcoholic fatty liver disease. Eur J Med Res. 28:6052023.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yin X, Mi Y, Wang X, Li Y, Zhu X, Bukhari
I, Wang Q, Zheng P, Xue X and Tang Y: Exploration and validation of
Ferroptosis-associated genes in ADAR1 Deletion-induced NAFLD
through RNA-seq analysis. Int Immunopharmacol. 134:1121772024.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fu D, Wang C, Yu L and Yu R: Induction of
ferroptosis by ATF3 elevation alleviates cisplatin resistance in
gastric cancer by restraining Nrf2/Keap1/xCT signaling. Cell Mol
Biol Lett. 26:262021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhao X, Wang Z, Wu G, Yin L, Xu L, Wang N
and Peng J: Apigenin-7-glucoside-loaded nanoparticle alleviates
intestinal ischemia-reperfusion by ATF3/SLC7A11-mediated
ferroptosis. J Control Release. 366:182–193. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Inaba Y, Hashiuchi E, Watanabe H, Kimura
K, Oshima Y, Tsuchiya K, Murai S, Takahashi C, Matsumoto M,
Kitajima S, et al: The transcription factor ATF3 switches cell
death from apoptosis to necroptosis in hepatic steatosis in male
mice. Nat Commun. 14:1672023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
National Research Council (US) Institute
for Laboratory Animal Research, . The Development of Science-based
Guidelines for Laboratory Animal Care: Proceedings of the November
2003 International Workshop. National Academies Press; Washington
DC: 2004
|
|
23
|
Hu Y, He W, Huang Y, Xiang H, Guo J, Che
Y, Cheng X, Hu F, Hu M, Ma T, et al: Fatty acid synthase-suppressor
screening identifies sorting Nexin 8 as a therapeutic target for
NAFLD. Hepatology. 74:2508–2525. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ren Y, Mao X, Xu H, Dang Q, Weng S, Zhang
Y, Chen S, Liu S, Ba Y, Zhou Z, et al: Ferroptosis and EMT: Key
targets for combating cancer progression and therapy resistance.
Cell Mol Life Sci. 80:2632023. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen GH, Song CC, Pantopoulos K, Wei XL,
Zheng H and Luo Z: Mitochondrial oxidative stress mediated
Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol
Med. 180:95–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Asghari S, Hamedi-Shahraki S and Amirkhizi
F: Systemic redox imbalance in patients with nonalcoholic fatty
liver disease. Eur J Clin Invest. 50:e132112020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bathish B, Robertson H, Dillon JF,
Dinkova-Kostova AT and Hayes JD: Nonalcoholic steatohepatitis and
mechanisms by which it is ameliorated by activation of the CNC-bZIP
transcription factor Nrf2. Free Radic Biol Med. 188:221–261. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong SH, Lee DH, Lee YS, Jo MJ, Jeong YA,
Kwon WT, Choudry HA, Bartlett DL and Lee YJ: Molecular crosstalk
between ferroptosis and apoptosis: Emerging role of ER
stress-induced p53-independent PUMA expression. Oncotarget.
8:115164–115178. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mungrue IN, Pagnon J, Kohannim O,
Gargalovic PS and Lusis AJ: CHAC1/MGC4504 is a novel proapoptotic
component of the unfolded protein response, downstream of the
ATF4-ATF3-CHOP cascade. J Immunol. 182:466–476. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Stockwell BR: Ferroptosis turns 10:
Emerging mechanisms, physiological functions, and therapeutic
applications. Cell. 185:2401–2421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Golabi P, Paik JM, AlQahtani S, Younossi
Y, Tuncer G and Younossi ZM: Burden of non-alcoholic fatty liver
disease in Asia, the Middle East and North Africa: Data from global
burden of disease 2009–2019. J Hepatol. 75:795–809. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li J, Wang T, Liu P, Yang F, Wang X, Zheng
W and Sun W: Hesperetin ameliorates hepatic oxidative stress and
inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic
acid-induced HepG2 cells and a rat model of high-fat diet-induced
NAFLD. Food Funct. 12:3898–3918. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tong J, Lan X, Zhang Z, Liu Y, Sun D, Wang
X, Ou-Yang SX, Zhuang CL, Shen FM, Wang P and Li DJ: Ferroptosis
inhibitor liproxstatin-1 alleviates metabolic
dysfunction-associated fatty liver disease in mice: Potential
involvement of PANoptosis. Acta Pharmacol Sin. 44:1014–1028. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen X, Li J, Kang R, Klionsky DJ and Tang
D: Ferroptosis: Machinery and regulation. Autophagy. 17:2054–2081.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsurusaki S, Tsuchiya Y, Koumura T,
Nakasone M, Sakamoto T, Matsuoka M, Imai H, Yuet-Yin Kok C, Okochi
H, Nakano H, et al: Hepatic ferroptosis plays an important role as
the trigger for initiating inflammation in nonalcoholic
steatohepatitis. Cell Death Dis. 10:4492019. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guan Q, Wang Z, Hu K, Cao J, Dong Y and
Chen Y: Melatonin ameliorates hepatic ferroptosis in NAFLD by
inhibiting ER stress via the MT2/cAMP/PKA/IRE1 signaling pathway.
Int J Biol Sci. 19:3937–3950. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Tan Y, Jin Y, Wang Q, Huang J, Wu X and
Ren Z: Perilipin 5 Protects against cellular oxidative stress by
enhancing mitochondrial function in HepG2 cells. Cells. 8:12412019.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gallardo-Montejano VI, Saxena G, Kusminski
CM, Yang C, McAfee JL, Hahner L, Hoch K, Dubinsky W, Narkar VA and
Bickel PE: Nuclear Perilipin 5 integrates lipid droplet lipolysis
with PGC-1α/SIRT1-dependent transcriptional regulation of
mitochondrial function. Nat Commun. 7:127232016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fang Z, Xu H, Duan J, Ruan B, Liu J, Song
P, Ding J, Xu C, Li Z, Dou K and Wang L: Short-term tamoxifen
administration improves hepatic steatosis and glucose intolerance
through JNK/MAPK in mice. Signal Transduct Target Ther. 8:942023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jin L, Wang M, Yang B, Ye L, Zhu W, Zhang
Q, Lou S, Zhang Y, Luo W and Liang G: A small-molecule JNK
inhibitor JM-2 attenuates high-fat diet-induced non-alcoholic fatty
liver disease in mice. Int Immunopharmacol. 115:1095872023.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mass-Sanchez PB, Krizanac M, Štancl P,
Leopold M, Engel KM, Buhl EM, van Helden J, Gassler N, Schiller J,
Karlić R, et al: Perilipin 5 deletion protects against nonalcoholic
fatty liver disease and hepatocellular carcinoma by modulating
lipid metabolism and inflammatory responses. Cell Death Discov.
10:942024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Xu X, Qiu J, Li X, Chen J, Li Y, Huang X,
Zang S, Ma X and Liu J: Perilipin5 protects against Non-alcoholic
steatohepatitis by increasing 11-Dodecenoic acid and inhibiting the
occurrence of ferroptosis. Nutr Metab (Lond). 20:292023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mass Sanchez PB, Krizanac M, Weiskirchen R
and Asimakopoulos A: Understanding the role of perilipin 5 in
Non-alcoholic fatty liver disease and its role in hepatocellular
carcinoma: A review of novel insights. Int J Mol Sci. 22:52842021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Shen X, Zhang J, Zhou Z and Yu R: PLIN5
suppresses lipotoxicity and ferroptosis in cardiomyocyte via
modulating PIR/NF-κB Axis. Int Heart J. 65:537–547. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mason RR, Mokhtar R, Matzaris M,
Selathurai A, Kowalski GM, Mokbel N, Meikle PJ, Bruce CR and Watt
MJ: PLIN5 deletion remodels intracellular lipid composition and
causes insulin resistance in muscle. Mol Metab. 3:652–663. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gallardo-Montejano VI, Yang C, Hahner L,
McAfee JL, Johnson JA, Holland WL, Fernandez-Valdivia R and Bickel
PE: Perilipin 5 links mitochondrial uncoupled respiration in brown
fat to healthy white fat remodeling and systemic glucose tolerance.
Nat Commun. 12:33202021. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Navik U, Singh SK, Khurana A and
Weiskirchen R: Revolutionizing liver fibrosis research: The promise
of 3D organoid models in understanding and treating chronic liver
disease. Expert Rev Gastroenterol Hepatol. 19:105–110. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu Y, Gilchrist AE, Johansson PK, Guan Y,
Deras JD, Liu YC, Ceva S, Huang MS, Navarro RS, Enejder A, et al:
Engineered hydrogels for organoid models of human nonalcoholic
fatty liver disease. Adv Sci (Weinh). 12:e173322025. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu S, Li R, Gong D, Hu P, Xu J, Ai Y, Zhao
X, Hu C, Xu M, Liu C, et al: Atf3-mediated metabolic reprogramming
in hepatic macrophage orchestrates metabolic dysfunction-associated
steatohepatitis. Sci Adv. 10:eado31412024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Basak M, Das K, Mahata T, Sengar AS, Verma
SK, Biswas S, Bhadra K, Stewart A and Maity B: RGS7-ATF3-Tip60
complex promotes hepatic steatosis and fibrosis by directly
inducing TNFα. Antioxid Redox Signal. 38:137–159. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kang Y, Li Q, Zhu R, Li S, Xu X, Shi X and
Yin Z: Identification of Ferroptotic genes in spinal cord injury at
different time points: Bioinformatics and experimental validation.
Mol Neurobiol. 59:5766–5784. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tian K, Wei J, Wang R, Wei M, Hou F and Wu
L: Sophoridine derivative 6j inhibits liver cancer cell
proliferation via ATF3 mediated ferroptosis. Cell Death Discov.
9:2962023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M,
Ding HF, Zhang J, Wang H, Chen X and Yan C: ATF3 promotes
erastin-induced ferroptosis by suppressing system Xc. Cell Death
Differ. 27:662–675. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Alosaimi M, Abd-Elhakim YM, Mohamed AA,
Metwally MMM, Khamis T, Alansari WS, Eskandrani AA, Essawi WM, Awad
MM, El-Shaer RAA, et al: Green synthesized zinc oxide nanoparticles
attenuate acrylamide-induced cardiac injury via controlling
endoplasmic reticulum stress-associated apoptosis through
ATF3/CHOP/BCL2 signaling in rats. Biol Trace Elem Res.
202:2657–2671. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Crawford RR, Prescott ET, Sylvester CF,
Higdon AN, Shan J, Kilberg MS and Mungrue IN: Human CHAC1 protein
degrades glutathione, and mRNA induction is regulated by the
transcription factors ATF4 and ATF3 and a bipartite ATF/CRE
regulatory element. J Biol Chem. 290:15878–15891. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wang X, He MJ, Chen XJ, Bai YT and Zhou G:
Glaucocalyxin A impairs tumor growth via amplification of the
ATF4/CHOP/CHAC1 cascade in human oral squamous cell carcinoma. J
Ethnopharmacol. 290:1151002022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Drummer C IVth, Saaoud F, Jhala NC, Cueto
R, Sun Y, Xu K, Shao Y, Lu Y, Shen H, Yang L, et al: Caspase-11
promotes high-fat diet-induced NAFLD by increasing glycolysis,
OXPHOS, and pyroptosis in macrophages. Front Immunol.
14:11138832023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ma SY, Sun KS, Zhang M, Zhou X, Zheng XH,
Tian SY, Liu YS, Chen L, Gao X, Ye J, et al: Disruption of Plin5
degradation by CMA causes lipid homeostasis imbalance in NAFLD.
Liver Int. 40:2427–2438. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kim JY, Park KJ, Hwang JY, Kim GH, Lee D,
Lee YJ, Song EH, Yoo MG, Kim BJ, Suh YH, et al: Activating
transcription factor 3 is a target molecule linking hepatic
steatosis to impaired glucose homeostasis. J Hepatol. 67:349–359.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cusi K, Isaacs S, Barb D, Basu R, Caprio
S, Garvey WT, Kashyap S, Mechanick JI, Mouzaki M, Nadolsky K, et
al: American association of clinical endocrinology clinical
practice guideline for the diagnosis and management of nonalcoholic
fatty liver disease in primary care and endocrinology clinical
settings: Co-sponsored by the American association for the study of
liver diseases (AASLD). Endocr Pract. 28:528–562. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Paternostro R and Trauner M: Current
treatment of non-alcoholic fatty liver disease. J Intern Med.
292:190–204. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Langhi C, Marquart TJ, Allen RM and Baldán
A: Perilipin-5 is regulated by statins and controls triglyceride
contents in the hepatocyte. J Hepatol. 61:358–365. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Gao X, Nan Y, Zhao Y, Yuan Y, Ren B and
Sun C: Atorvastatin reduces lipid accumulation in the liver by
activating protein kinase A-mediated phosphorylation of perilipin
5. Biochim Biophys Acta Mol Cell Biol Lipids. 1862:1512–1519. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu M, Tang TMS, Ghamsari L, Yuen G,
Scuoppo C, Rotolo JA, Kappel BJ and Mason JM: Exponential
Combination of a and e/g intracellular peptide libraries identifies
a selective ATF3 inhibitor. ACS Chem Biol. 19:753–762. 2024.
View Article : Google Scholar : PubMed/NCBI
|














