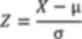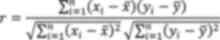Introduction
Esophageal cancer (EC) is the seventh most common
cancer globally, with ~470,000 new cases diagnosed annually; In
addition, EC ranks among the top in terms of incidence among upper
gastrointestinal tumors (1). It
has been reported that ~90% of EC cases are squamous cell
carcinoma, with most of the remaining cases being adenocarcinoma
(2). Owing to the absence of
early-stage clinical manifestations, EC is frequently diagnosed in
the advanced stages. Despite notable advancements in cancer
diagnostics and treatment, with a 5-year survival rate of <30%,
the clinical prognosis of EC remains poor (3). Therefore, the discovery of effective
targets for the treatment and early diagnosis of EC is of great
importance.
Zinc finger proteins (ZNFs), encoded by ~5% of the
human genome, form the largest family of transcription factors in
humans. Characterized by their finger-like DNA-binding motifs, ZNFs
exert important biological functions across multiple cellular
processes (4). Zinc finger motifs
are currently categorized into eight major types: C2H2, TAZ2
domain-like, zinc ribbon, treble clef, Zn2/Cys6 (5), zinc-binding loop, Gag knuckle and
metallothionein (6). A notable
body of literature has documented the biological processes
associated with ZNFs in various types of cancer. For example,
ZNF322A promotes cell proliferation, motility and invasive ability
in lung cancer through transcriptional activation of cyclin D1 and
α-adducin, coupled with inhibition of p53. Furthermore,
multivariate Cox regression analysis has revealed that ZNF322A has
a notable impact on the prognosis of lung cancer (7). Moreover, activating the PI3K/AKT
pathway induces the ZNF139-mediated promotion of bladder cancer
cell proliferation, motility and invasion (8). ZNF148 promotes breast cancer
progression by activating microRNA-335 and superoxide dismutase 2
to enhance the production of reactive oxygen species, which further
triggers pyroptotic cell death (9). To the best of our knowledge, no
studies have yet reported on the prognostic relevance of ZNF514 in
pan-cancer and EC analyses. Therefore, the impact of ZNF514 on the
development of EC warrants further investigation.
The impact of ZNF514 on the proliferation, motility
and invasiveness of EC cells was evaluated through knockdown and
overexpression experiments using Kyse-150 and Kyse-510 cells. By
using RNA sequencing (RNA-seq) analysis, followed by Gene Ontology
(GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analyses,
Gene Set Enrichment Analysis (GSEA) and Ingenuity Pathway Analysis
(IPA), the potential mechanisms underlying the functions of ZNF514
were further explored in the present study. The present study aimed
to explore the pro-tumorigenic role of ZNF514 upregulation in EC,
aiming to identify new therapeutic targets for EC and provide novel
strategies for the diagnosis and subsequent treatment of EC.
Materials and methods
Bioinformatics analysis
The Tumor Immune Estimation Resource (TIMER;
http://cistrome.shinyapps.io/timer/)
database was used to detect ZNF514 upregulation across multiple
malignancies. Data from patients with EC were obtained from TCGA
(https://gdc-portal.nci.nih.gov),
including RNA-seq data from 197 samples (184 EC tissues and 13
normal esophageal tissues). From these, 13 normal tissue samples
and their paired 13 cancer tissue samples were selected, and
Wilcoxon signed-ranks analysis were used to evaluate the
differential expression between the two groups, and data
visualization was performed using the ‘ggplot2’ (v3.5.1;
ggplot2.tidyverse.org) package in R (v4.2.2) (https://cran.r-project.org/src/base/R-4/R-4.2.2.tar.gz).
The frequency of ZNF514 gene alterations was analyzed using the
cBioPortal database (www.cbioportal.org). Data and samples were retrieved
from the esophageal carcinoma section of two TCGA-derived
databases: Firehose legacy) (gdac.broadinstitute.org/) and TCGA
(Nature 2017) (10).
Patients and specimens
Between November 2024 and January 1, 2025, six pairs
of EC samples and their corresponding adjacent non-cancerous
tissues were collected at Jinan Central Hospital (Jinan, China).
The inclusion criteria in the present study were as follows: i)
Pathological biopsy confirms esophageal cancer); ii) patients had
complete pathological data; iii) patients provided written informed
consent; and iv) patients did not receive any radiotherapy,
immunotherapy or chemotherapy before surgery. The six pairs of
cancerous samples and their corresponding adjacent non-cancerous
tissues were immediately frozen to preserve freshness for
subsequent western blot analysis. The protocol for the present
study received approval from the Ethics Committee of Jinan Central
Hospital (Jinan, China), under ethical approval number 20241120027.
All procedures were conducted following applicable guidelines and
regulations.
Cell lines and cell culture
The Het-1a, Kyse-30, Kyse-450, Kyse-150 and Kyse-510
cells were supplied by Shandong Provincial Hospital Affiliated to
Shandong First Medical University (Jinan, China). Het-1a cells were
cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.) containing
10% FBS (Hysigen). 10% FBS was added to RPMI 1640 medium (Gibco;
Thermo Fisher Scientific, Inc.) for the remaining cell culture. All
cell cultures were maintained at 37°C in a humidified incubator
with 5% CO2.
Transfection
Kyse-150 (interference) and transfected
overexpression plasmids into the Kyse-510 (overexpression) cell
line, which were pre-cultured in 6-well plates with suitable growth
medium. ZNF514 small interfering (si)RNA (Beijing Tsingke Biotech
Co., Ltd.) or overexpression plasmids (Guangzhou RiboBio Co., Ltd.)
were transfected using Lipofectamine 3000 (Invitrogen; Thermo
Fisher Scientific, Inc.) and p3000 (Guangzhou RiboBio Co., Ltd.).
siRNA was transfected using Lipofectamine 3000 alone, while
plasmids were transfected using both Lipofectamine 3000 and p3000.
A total of 2.5 µg siRNA was mixed with 5 µl Lipofectamine 3000 to
form a Lipofectamine-siRNA complex. Alternatively, 2.5 µg of
plasmid was mixed with 5 µl of Lipofectamine 3000 and 5 µl P3000 to
prepare a Lipofectamine-P3000-ZNF514 plasmid complex. After
incubating the complexes at room temperature for 15 min, they were
added to the cells. Following transfection at 37°C for 8 h, the
medium was replaced. The cells were then further incubated at 37°C
for 24–48 h before RNA or protein extraction was performed. siRNA
sequences targeting ZNF514 were as follows: si-ZNF514-1: Forward
5′-CCCUUAGCAGAGAUUAUAA-3′ and Reverse 5′-UUAUAAUCUCUGCUAAGGG-3′;
si-ZNF514-2: Forward 5′-GGUCACACUUCAUCCCUUA-3′ and Reverse
5′-UAAGGGAUGAAGUGUGACC-3′ and si-ZNF514-3: Forward
5′-GGGCCUUCUAGUAUCCAAA-3′ and Reverse 5′-UUUGGAUACUAGAAGGCCC-3′.
The negative control sequences were: Forward
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse
5′-ACGUGACACGUUCGGAGAATT-3′. The negative control for siRNA
transfection is non-targeting siRNA. In the overexpression
experiment, both the ZNF514 overexpression plasmid and the control
plasmid (empty pcDNA3.1 plasmid) used pcDNA3.1 as the vector
backbone.
RNA extraction and reverse
transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted from Het-1a, Kyse-30,
Kyse-450, Kyse-150 and Kyse-510 cells using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). RT of 1 µg
mRNA into cDNA was performed using the Evo M-MLVRT Mix Kit Ver.2
(Hunan Accurate Biotechnology Co., Ltd.), according to the
manufacturer's instructions. Gene expression was quantified with
SYBR Green PCR (Hunan Accurate Biotechnology Co., Ltd.) using 10 ng
cDNA as the template. Thermocycling conditions were as follows:
Initial pre-denaturation at 95°C for 30 sec, followed by 40 cycles
of a denaturation step at 95°C for 30 sec, an annealing step at
55°C for 30 sec, and extension step at 72°C for 30 sec. The primers
used were as follows: ZNF514, forward 5′-ACAAATCTGCCACCACCCTTA-3′,
reverse 5′-TGTTTCCCCCTAAAGTCTGCC-3′; and GAPDH, forward
5′-GCACCGTCAAGGCTGAGAAC-3′ and reverse 5′-TGGTGAAGACGCCAGTGGA-3′
(Azenta US, Inc.). Expression levels were normalized to GAPDH and
analyzed using the 2−ΔΔCq method (11).
Western blotting
Het-1a, Kyse-30, Kyse-450, Kyse-150 and Kyse-510
cells were lysed in RIPA buffer (Beijing Solarbio Science &
Technology Co., Ltd.) containing protease/phosphatase inhibitors.
Protein concentration was determined using a BCA kit (Beyotime
Institute of Biotechnology) to ensure equal loading and the protein
samples were mixed with sample buffer and heated at 98°C for 5 min.
25 µg quantified protein lysate per lane was separated by 10%
SDS-PAGE gel electrophoresis and transferred to PVDF membranes. The
membranes were blocked in 0.1% TBST containing 5% non-fat milk for
1 h at room temperature, then incubated overnight at 4°C with
primary antibodies diluted in 0.1% TBST buffer. The primary
antibodies used included rabbit polyclonal anti-ZNF514 antibody
(1:2,000; cat. no. YT6669; ImmunoWay Biotechnology Company) and
rabbit polyclonal anti-GAPDH antibody (1:10,000; cat. no. bs-2188R;
Beijing Biosynthesis Biotechnology Co., Ltd.). Subsequently, the
membranes were incubated with HRP-conjugated goat anti-rabbit
secondary antibody (1:10,000; cat. no. bs-0295G-HRP Beijing
Biosynthesis Biotechnology Co., Ltd.) at room temperature for 1 h
and detected using enhanced chemiluminescence reagents
(MilliporeSigma). Immunoreactive bands were visualized using a
Tanon 5200 imaging system (Molecular Devices, LLC) and analyzed
with ImageJ software (version 1.8.0; National Institutes of
Health).
Immunohistochemistry (IHC)
Esophageal cancer tissue samples collected were
fixed in 4% paraformaldehyde at room temperature for 24 h, then
embedded in paraffin and cut into 3-µm-thick paraffin sections. The
sections were deparaffinized with xylene, followed by rehydration
in a graded ethanol series. Subsequently, the sections were
immersed in 1× citrate buffer (pH 6.0), heated to boiling using a
pressure cooker, and then cooled to room temperature. Next, the
sections were washed 3 times with PBS, 5 min each time. They were
then placed in 3% hydrogen peroxide solution and incubated at room
temperature for 25 min in the dark, followed by 3 washes with PBS
(5 min per wash). 3% BSA (cat. no. GC305010; Servicebio) was added
dropwise to cover the tissue evenly, and the sections were blocked
at room temperature for 30 minutes. The sections were incubated
overnight at 4°C with rabbit polyclonal anti-ZNF514 antibody
(1:200; cat. no. YT6669; ImmunoWay Biotechnology Company). After
that, HRP-conjugated goat anti-rabbit secondary antibody (1:200;
cat. no. GB23303; Servicebio) was added at room temperature for 50
min. Thereafter, the slides were stained with DAB (cat. no. G1212;
Servicebio) and counterstained with hematoxylin at room temperature
for 3 min. The sections were dehydrated, cleared, and mounted with
neutral mounting medium. Finally, observations were made under a
light microscope.
EdU assay
Kyse-150 and Kyse-510 cells were plated into 96-well
plates at a density of 2×104 cells/well and incubated
for 24 h. Cell proliferation was evaluated using the EdU assay kit
(cat. no. C0071S; Beyotime Institute of Biotechnology) according to
the manufacturer's instructions. Briefly, cells were treated with
10 µM EdU at 37°C for 3 h, fixed in 4% paraformaldehyde at room
temperature for 10 min, permeabilized with 0.3% Triton X-100 at
room temperature for 10 min, and sequentially stained with Azide
488 for 30 min and Hoechst 33342 for 10 min at room temperature.
Fluorescence imaging was performed using a fluorescence microscope
(Olympus Corporation) to visualize EdU-positive (proliferating)
cells and total nuclei.
Cell Counting Kit (CCK)-8 cell
proliferation assay
Kyse-150 and Kyse-510 cells in the exponential
growth phase were seeded into 96-well plates at a density of 2,000
cells per well. At 24, 48, 72, and 96 h after cell adhesion, 10 µl
of CCK-8 reagent (cat. no. BA00208; Beijing Biosynthesis
Biotechnology Co., Ltd.) was added to each well, respectively.
After incubation at 37°C for 2 h, the optical density (OD) values
were measured at a wavelength of 450 nm using a microplate reader
(Model Spectra Max i3×; Molecular Devices).
Wound healing assay
Kyse-150 and Kyse-510 cells were seeded into 6-well
plates and cultured in medium containing 10% FBS until reaching 95%
confluence. Uniform scratches were created using sterile pipette
tips, after which the cells were incubated in serum-free medium at
37°C. Images were captured via phase-contrast microscope at 0 and
24 h to monitor scratch healing. Quantitative analysis of scratch
healing was performed using ImageJ software [(version 1.8.0); NIH,
USA], and the cell migration rate (%) was calculated using the
formula: [(initial scratch width-final scratch width)/initial
scratch width] ×100. Finally, observations were made under a light
microscope.
Colony formation assay
Kyse-150 and Kyse-510 cells were plated in 6-well
plates at a density of 2×103 cells/well. The culture
medium was replaced every 3 days; after 14 days of incubation, the
colonies were fixed with 4% paraformaldehyde for 30 min at room
temperature, stained with hematoxylin (Beijing Solarbio Science
& Technology Co., Ltd.) for 20 min at room temperature, scanned
and counted manually. Clones were defined as colony composed of
>50 cells.
Transwell assays
Transwell assays were performed in 24-well plates.
For the migration assay, 2×104 Kyse-150 or Kyse-510
cells were resuspended in serum-free medium and seeded into
uncoated Transwell chambers (pore size, 8 µm; Corning, Inc.), with
the lower chamber containing medium supplemented with 10% FBS. In
the invasion assay, Transwell chambers were pre-coated with
Matrigel and incubated at 37°C for 1 h. Subsequently,
2×104 cells were seeded into the upper chamber, while
the lower chamber was filled with medium supplemented with 10% FBS.
After incubation at 37°C for 24–48 h, non-migrated/non-invaded
cells on the upper surface of the membrane were removed. Cells that
had invaded to the lower surface were fixed with 4%
paraformaldehyde and stained with 0.1% crystal violet at room
temperature for 15 min. Finally, observations were made under a
light microscope.
RNA-seq
Total RNA was extracted from Kyse-150 cells using
the MJzol Animal RNA Extraction kit (cat. no.T102096; MagBeads) in
accordance with the manufacturer's instructions. RNA integrity was
assessed by determining the RNA Integrity Number using an Agilent
4200 TapeStation (Agilent Technologies, Santa Clara, CA, USA).
Qualified total RNA was purified using the RNAClean XP Kit (cat.
no. A63987, Beckman Coulter, Inc.) and the RNase-Free DNase Set
(cat. no. 79254, QIAGEN, GmbH). Subsequently, the purified total
RNA underwent mRNA isolation, fragmentation, first-strand cDNA
synthesis, second-strand cDNA synthesis, end repair, 3′-end
adenylation, adapter ligation, and enrichment steps, following the
protocol provided with the Dual-mode mRNA Library Prep Kit (cat.
no. 12310ES96, YEASen). The resulting products were purified using
HieffNGS DNA Selection Beads (cat. no. 12601ES56, YEASen). Library
concentration was quantified using the ExKubit dsDNA HS Assay kit
(cat. no. NGS00-3012, Excell) and a Qubit® 2.0
Fluorometer, while library size was determined using an Agilent
4200 TapeStation (Agilent Technologies) and diluted to 2 nM loading
concentration. Sequencing was performed on an Illumina NovaSeq 6000
platform using a paired-end 150 bp mode.
Data analysis for gene expression
Raw sequencing reads underwent preprocessing to
remove ribosomal RNA reads, sequencing adapters, short-fragment
sequences and other low-quality reads. The cleaned reads were then
mapped to the human hg38 reference genome using HISAT2 (version
2.0.4; http://daehwankimlab.github.io/hisat2/), allowing for
up to two mismatches. Following genome alignment, StringTie
(version 1.3.0; http://ccb.jhu.edu/software/stringtie/) was employed
with reference annotation to calculate fragment per kilobase of
transcript per million mapped reads (FPKM) values for known gene
models. Differential gene analysis between samples was performed
using edgeR (https://bioconductor.org/packages/release/bioc/html/edgeR.html)
(12). After obtaining P-values,
multiple hypothesis testing correction was conducted. The threshold
for P-values was determined by controlling the False Discovery Rate
(FDR) (13), and the corrected
P-values were referred to as q-values, with fold-changes estimated
based on FPKM values across samples. Significant DEGs were
identified as those with a FDR value above the threshold
(Q<0.05) (When the total number of DEGs in all comparisons is
less than 50, the threshold will be automatically adjusted to a
P-value ≤0.05) and fold-change >2 (or
|log2(fold-change)|>1). DEGs with a
|log2(Fold-change)|>2 and P<0.05 were uploaded to
the STRING database (string-db.org/) to construct protein-protein
interaction (PPI) networks. A high-confidence threshold, indicated
by an interaction score of >0.9, was applied to filter
significant protein interactions and generate the PPI network. To
identify the enriched biological processes, molecular functions,
and cellular components, Gene Ontology analysis (GO; http://www.geneontology.org/) was conducted. Kyoto
Encyclopedia of Genes and Genomes analysis (KEGG; clusterProfiler
3.10.1; https://biocon
ductor.org/packages/release/bioc/html/clusterProfiler.html) was
applied to detect cellular biochemical processes closely associated
with the DEGs. To identify signaling pathways significantly
associated with ZNF514 expression levels, we performed Gene Set
Enrichment Analysis (GSEA; http://www.gsea-msigdb.org/gsea/index.jsp).
IPA
Canonical pathway analysis and upstream regulator
analysis for various DEGs were performed using IPA. (QIAGEN Inc.;
http://digitalinsights.qiagen.com/IPA). In enrichment
analysis, we use the z-score to quantify the degree of deviation in
the overall activation/inhibition trend of a pathway. The
formula:
where X represents the raw data, µ denotes the mean,
and σ is standard deviation. The direction of the z-score indicates
the activation or inhibition trend of the pathway, while the
absolute value of the z-score reflects the significance level.
Additionally, we use -log(P-value) to enhance the visual
distinguishability of ‘significant differences’. Specifically, a
larger -log(P-value) corresponds to a smaller P-value, which in
turn means the statistics are more reliable.
Statistical analysis
Data are presented as the mean ± standard deviation
of at least three independent experiments. A paired-samples t-test
was used to compare differences between two paired groups, while an
unpaired t-test was applied to analyze differences between two
independent samples. For comparisons involving more than two
groups, a one-way analysis of variance was employed, followed by
Dunnett's post-hoc test. P<0.05 was considered to indicate a
statistically significant difference. All analyses were performed
using Prism v8.0 software (Dotmatics). To quantify the linear
association between two continuous variables, the Pearson
correlation coefficient was calculated following standard
statistical protocols. For two variables X and Y with
a total sample size of n, the Pearson correlation
coefficient was computed using the formula:
Results
ZNF514 is upregulated in EC
The TIMER database was used to investigate ZNF514
expression across different tumor types (Fig. 1A) and it was revealed that ZNF514
was commonly upregulated in various types of cancer, including EC,
bladder urothelial carcinoma, cholangiocarcinoma and colon
adenocarcinoma. Additionally, a quantitative comparison of ZNF514
expression using Log2(TPM+1) normalization across EC
samples and matched healthy tissue revealed that ZNF514 was
upregulated in most EC samples vs. healthy tissues (Fig. 1B). Additionally, the cBioPortal
database was utilized to analyze the alteration types and
frequencies of ZNF514 in EC from two different data sources from
TCGA database (Nature 2017/Firehose Legacy). The analysis revealed
that, among the 541 patients in the Nature 2017 data source, 8
patients had mutations (1.48%), 4 patients had amplifications
(0.74%) and the gene alteration frequency was 2.22% (Fig. 1C). Among 185 patients in the
Firehose legacy dataset, 0 patients had mutations (0%), while two
patients had amplifications (1.08%), and the gene alteration
frequency was 1.08% (Fig. 1C).
Subsequently, RT-qPCR and western blotting were employed to examine
the mRNA and protein expression levels of ZNF514 in Het-1a and
multiple EC cell lines (Kyse-30, Kyse-150, Kyse-450 and Kyse-510).
The results demonstrated that, with the exception of Kyse-450, both
the protein (Fig. 1D and F) and
mRNA expression levels (Fig. 1H)
of ZNF514 were significantly elevated in these EC cell lines
compared with those in the Het-1a cell line. To further assess the
expression of ZNF514 in tissues, surgically resected EC tissues and
adjacent healthy tissues were collected and analyzed through
western blotting (Fig. 1E and G)
and IHC (Fig. 1I). The results
showed that ZNF514 expression levels were higher in EC tissues than
those in adjacent healthy tissues. These findings suggested that
ZNF514 was upregulated in EC tissues and cells, indicating a close
relationship between ZNF514 and the development and progression of
EC.
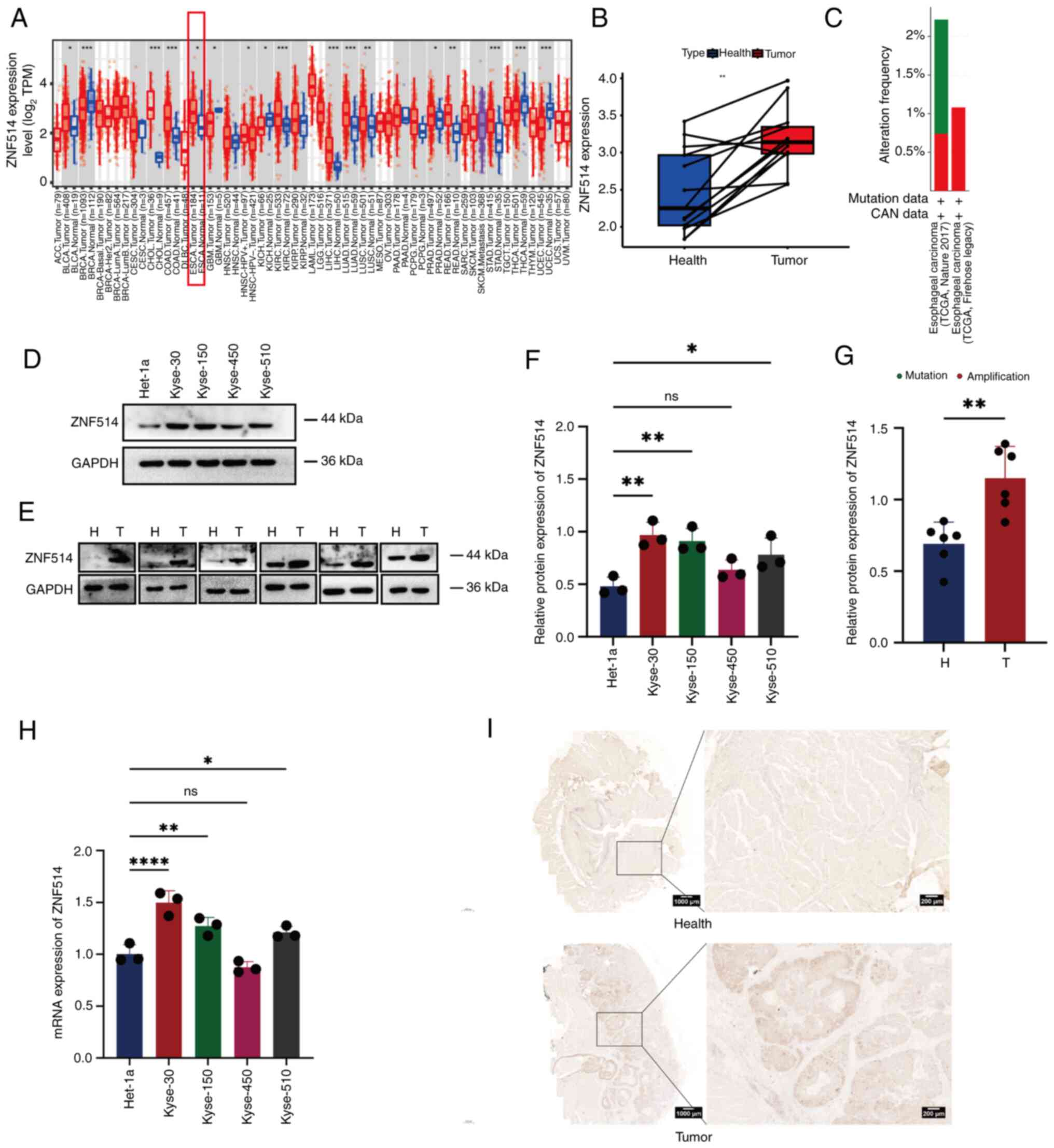 | Figure 1.ZNF514 is significantly upregulated
in EC tissues and cells. (A) Analysis of RNA expression differences
of ZNF514 in normal and cancer tissues across various types of
cancer using the Tumor Immune Estimation Resource database. (B) RNA
expression differences between H and paired cancer tissues in
patients with EC. (C) Gene alteration frequency of ZNF514 in EC.
(D) Western blot analysis of ZNF514 protein expression differences
in Het-1a and EC cell lines. (E) Western blot analysis of ZNF514
protein expression differences in the tumors and adjacent tissues
of patients with EC. Semi-quantitative analyses of ZNF514 protein
expression in (F) Het-1a and EC cell lines, and (G) tumor and
adjacent tissues of patients with EC normalized to GAPDH. (H)
Reverse transcription-quantitative PCR analysis of ZNF514
expression levels in Het-1a and EC cell lines. (I) Representative
images of immunohistochemical staining for ZNF514 in the tumors and
adjacent tissues of patients with EC. Scale bar, 1000 µm.
*P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001. H,
Healthy; T, tumor; ZNF514, zinc finger protein 514; EC, esophageal
cancer; ns, not significant; CNA, copy number alteration; TCGA, The
Cancer Genome Atlas. |
Silencing ZNF514 leads to suppression
of EC cell migration, invasion and proliferation
To investigate the functional role of ZNF514 in EC
progression, phenotypic assays were performed following ZNF514
knockdown in Kyse-150 cells. First, the transfection efficiency of
three ZNF514-specific siRNAs (siZNF514-1, siZNF514-2 and
siZNF514-3) was validated using RT-qPCR (Fig. 2A) and western blotting (Fig. 2B and C). Based on these results,
siZNF514-2 and siZNF514-3 were selected for subsequent functional
analyses. Transwell migration and invasion assays (Fig. 2D-F), wound healing assay (Fig. 2G and H), colony formation assay
(Fig. 2I and J), CCK-8
proliferation assay (Fig. 2K) and
EdU assay (Fig. 2L and M)
collectively showed that compared with the si-NT group, ZNF514
knockdown significantly inhibited migration, invasion and
proliferation in EC cells. These findings indicated that silencing
ZNF514 exerted a tumor-suppressive effect by restricting the
migratory, invasive and proliferative capacities of EC cells.
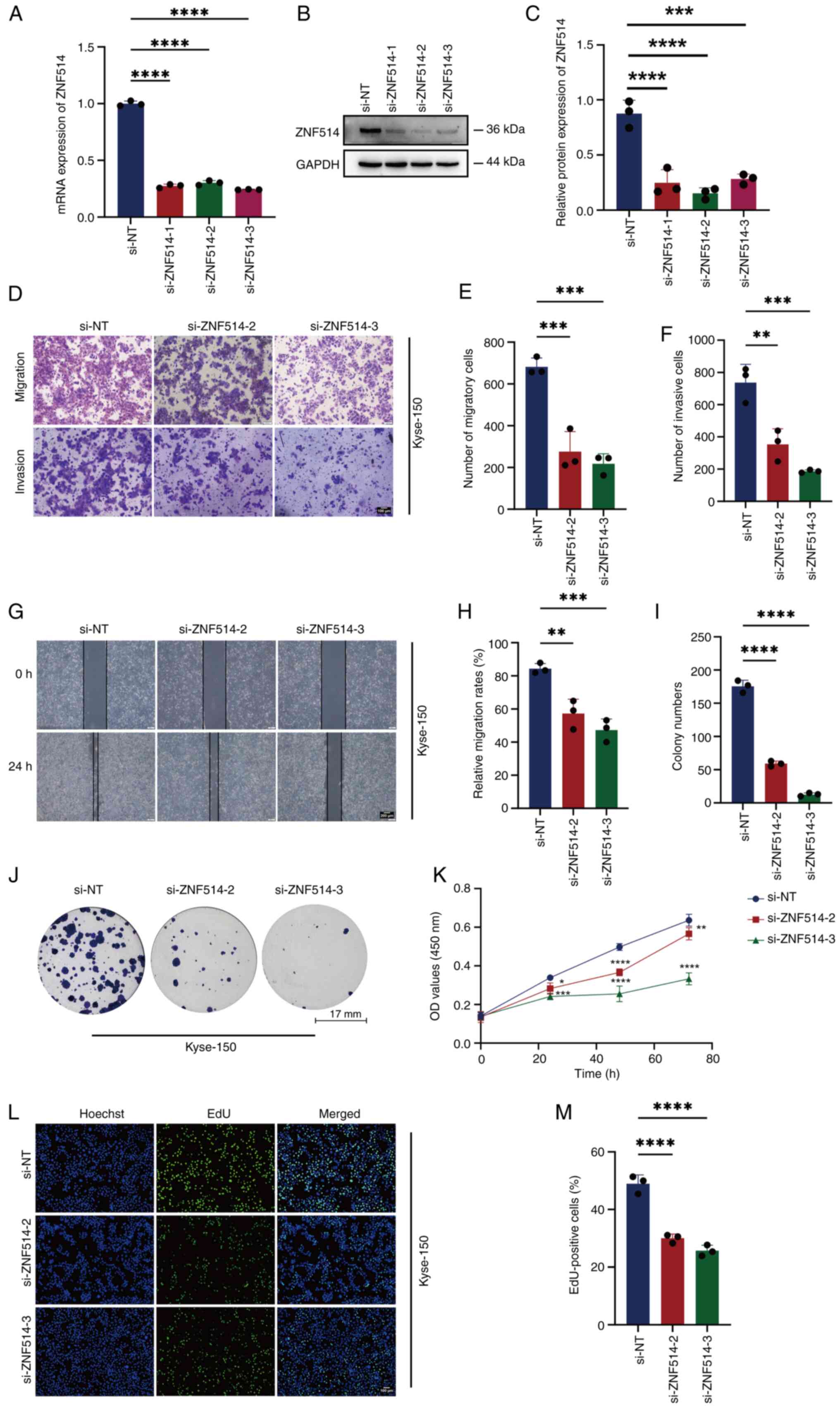 | Figure 2.Downregulation of ZNF514 inhibits
proliferation, migration and invasion of Kyse-150 cells. ZNF514
mRNA and protein expression levels in Kyse-150 cells transfected
with targeted siRNAs were analyzed using (A) reverse
transcription-quantitative PCR and (B) western blot analysis,
alongside (C) semi-quantitative analysis of ZNF514 protein levels.
(D) Effects of ZNF514 knockdown on the migration and invasion of
Kyse-150 cells was assessed using Transwell assays; (E)
quantitative analysis of the migration assay, (F) quantitative
analysis of the invasion assay. Scale bar, 100 µm. (G) Wound
healing assays were conducted to evaluate the effect of ZNF514
knockdown on the migration of Kyse-150 cells; (H) quantitative
analysis of cell migration rate. Scale bar, 200 µm. (I)
Quantitative analysis of colony formation, (J) colony formation,
(K) Cell Counting Kit −8, (L) EdU assays, and (M) quantitative
analysis of the EdU assay were utilized to evaluate the effects of
ZNF514 knockdown on the proliferation of Kyse-150 cells. Scale bar,
100 µm. *P<0.05, **P<0.01, ***P<0.001 and ****P<0.0001
vs. si-NT. NT, non-targeting; ZNF514, zinc finger protein 514;
CCK-8, Cell Counting Kit-8; si, small interfering; EdU,
5-ethynyl-2′-deoxyuridine. |
Overexpression of ZNF514 increases
migration, invasion and proliferation of EC cells
To explore the oncogenic role of ZNF514 in EC,
phenotypic analyses were conducted following ZNF514 overexpression
in Kyse-510 cells. First, the transfection efficiency of the ZNF514
overexpression plasmid was validated using RT-qPCR (Fig. 3A) and western blotting (Fig. 3B and C). Cells with stable
overexpression then underwent functional assays. Transwell
migration and invasion assays (Fig.
3D-F), wound healing assay (Fig.
3G and H), colony formation assay (Fig. 3I and J), CCK-8 proliferation assay
(Fig. 3K) and EdU assay (Fig. 3L and M) revealed that
ZNF514-overexpressing EC cells exhibited significantly enhanced
migration, invasion and proliferative capacity compared with
control EC cells transfected with empty vector plasmids. These
results indicated that ZNF514 may act as an oncogene to facilitate
EC progression by enhancing tumor cell motility and proliferative
potential.
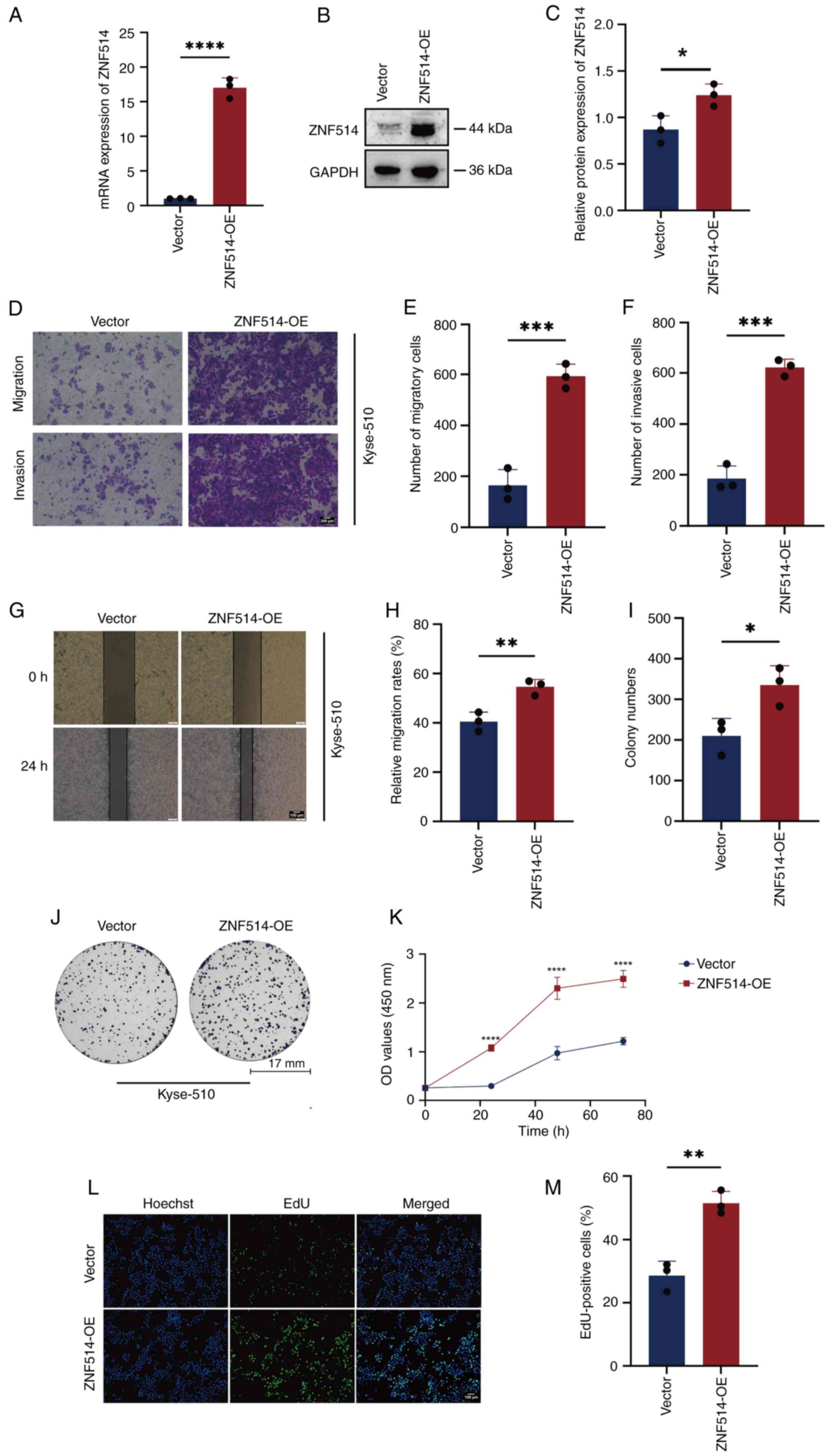 | Figure 3.OE of ZNF514 promotes proliferation,
migration and invasion of Kyse-510 cells. Analysis of ZNF514 mRNA
and protein expression in Kyse-510 cells transfected with targeted
OE plasmid using (A) reverse transcription-quantitative PCR and (B)
western blot analysis, along with (C) semi-quantification of ZNF514
protein. (D) Transwell assays were performed to evaluate the
effects of ZNF514 OE on the migration and invasion of Kyse-510
cells; (E) quantitative analysis of the migration assay, (F)
quantitative analysis of the invasion assay. Scale bar, 200 µm. (G)
Wound healing assays were conducted to assess the impact of ZNF514
OE on the migration of Kyse-510 cells. (H) quantitative analysis of
cell migration rate. Scale bar, 100 µm. (I) Quantitative analysis
of colony formation, (J) Colony formation, (K) Cell Counting Kit-8,
(L) EdU assays, and (M) quantitative analysis of the EdU assay were
utilized to evaluate the effects of ZNF514 OE on the proliferation
of Kyse-510 cells. Scale bar, 100 µm. *P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 vs. vector. ZNF514, zinc finger
protein 514; OE, overexpression; EdU,
5-ethynyl-2′-deoxyuridine. |
RNA-seq analysis following ZNF514
knockdown in Kyse-150 cells
Building on the previous findings, RNA-seq was
performed on ZNF514-knockdown Kyse-150 150 EC s to explore the
molecular mechanisms underlying ZNF514-mediated promotion of EC
tumorigenesis and progression. Genes displaying significant
expression level changes across various experimental conditions
were termed DEGs. Significant DEGs were identified using the
‘edgeR’ software, with selection criteria including a q<0.05,
fold-change >2 or |log2(fold-change)|>1). This
filtering process resulted in the identification of 1,901 DEGs,
which included 260 upregulated genes and 1,641 downregulated genes
(Fig. 4A and B). Subsequently,
Pearson correlation analysis was performed on the identified DEGs.
The Pearson correlation coefficient was 0.955, indicating an
extremely strong positive correlation between the gene expression
profiles of the si-NT group and the si-ZNF514 group. In terms of
DEGs, red dots represent up-regulated genes, blue dots represent
down-regulated genes, and gray dots represent genes with no
significant differential expression. P<2.2×10−16
(Fig. 4C). To further clarify the
functional enrichment of DEGs, GO enrichment analysis categorized
the DEGs into three primary categories: Biological processes,
cellular components and molecular functions (Fig. 4D). The analysis indicated that
these DEGs were significantly associated with molecular functions,
including ‘histone demethylase activity (H4-K20 specific)’ and
‘hedgehog receptor activity’. (Fig.
4E) Additionally, they were associated with biological
processes such as ‘histone H4-K20 demethylation’ and ‘positive
regulation of acrosome reaction’.
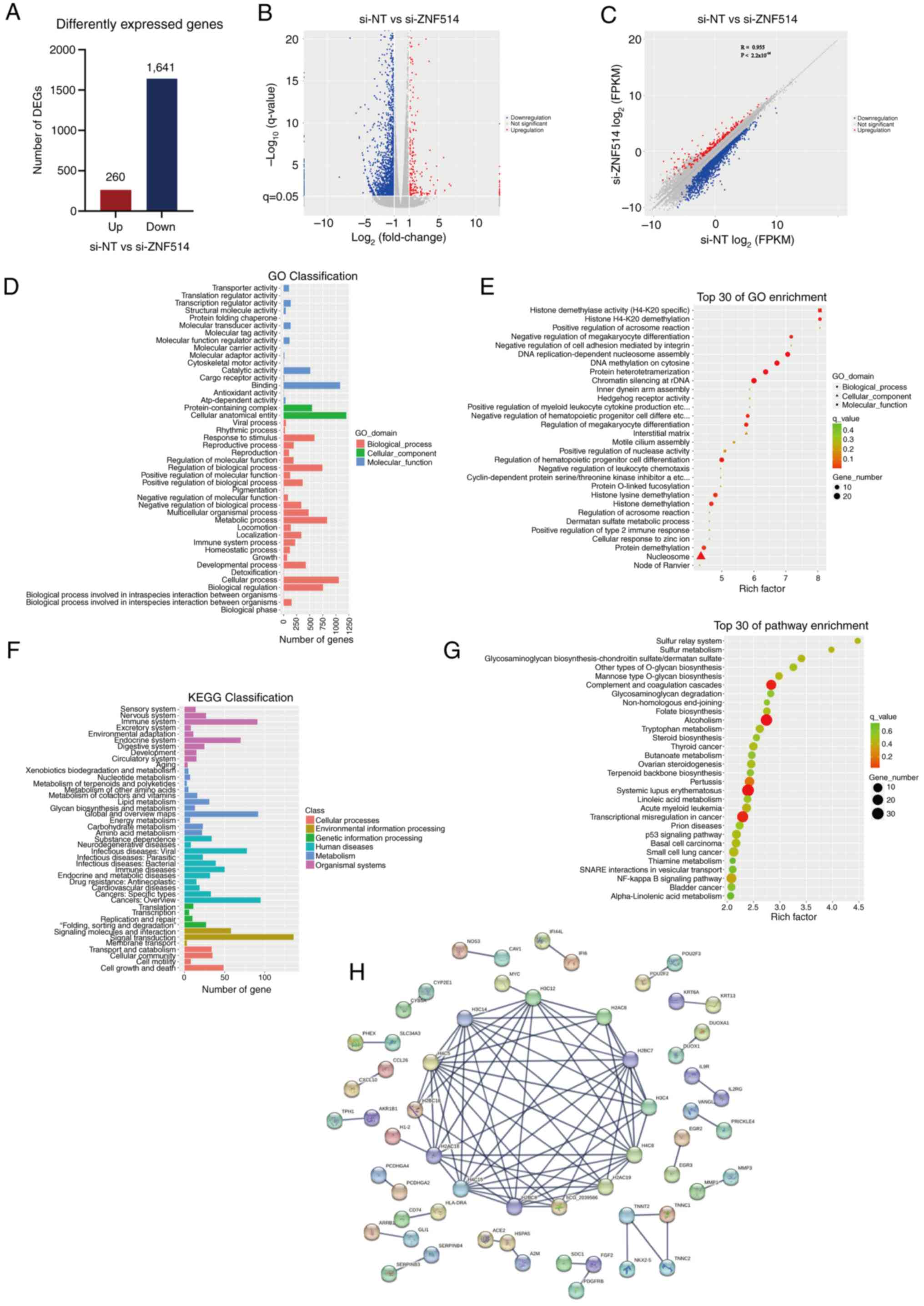 | Figure 4.RNA sequencing analysis of esophageal
cancer cells reveals the mechanisms by which downregulation of
ZNF514 contributes to cancer progression. (A) Bar graph displaying
the number of genes affected by ZNF514 knockdown, selected based on
the criteria of |log2(Fold-change)|>1 and q-value
<0.05. (B) Volcano plot illustrating the expression of DEGs,
with red indicating upregulated genes and blue signifying
downregulated genes. (C) Scatter plot showing the correlation of
DEGs, with red indicating upregulated genes and blue signifying
downregulated genes. (D) GO functional classification of DEGs, (E)
GO enrichment analysis of DEGs, (F) KEGG pathway classification of
DEGs, and (G) KEGG pathway enrichment analysis of DEGs. (H)
Protein-protein interaction analysis was conducted based on the
DEGs. DEG, differentially expressed gene; ZNF514, zinc finger
protein 514; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes
and Genomes; si-NT, siRNA-non-targeting. |
KEGG pathway analysis was performed to identify key
signaling pathways associated with the DEGs, aiming to clarify the
mechanisms through which these genes impact cancer initiation and
progression. The findings indicated that DEGs exhibited enrichment
in pathways categorized into: Cellular Processes, including
‘Cellular community’, ‘Cell growth and death’ and ‘Transport and
catabolism’; Environmental Information Processing, including
‘Signaling molecules and interaction’, ‘Membrane transport’ and
‘Signal transduction’; Genetic Information Processing including
‘Transcription’, ‘Replication and repair’, ‘folding, sorting and
degradation’ and ‘Translation’; Human Diseases, including
‘Infectious diseases: Viral’, ‘Cancers: Specific types’ and ‘Immune
diseases’; Metabolism, including ‘Energy metabolism’ and
‘Nucleotide metabolism’; and Organismal Systems, including ‘Immune
system’ and ‘Digestive system’ (Fig.
4F). The bubble plot illustrated that DEGs were significantly
enriched in pathways related to ‘Complement and coagulation
cascades’, ‘Alcoholism’, ‘Pertussis’, ‘Systemic lupus
erythematosus’ and ‘Transcriptional misregulation in cancer’
(Fig. 4G). The STRING database was
used for PPI analysis and to build a PPI network of DEGs. The
resulting comprehensive overview revealed that the ZNF514-related
protein network primarily consisted of histone families, which were
structural constituents of chromatin, and associated troponins of
the troponin complex, including histones H2A, H2B, H3 and the
troponin proteins TNNC1, TNNC2, TNNT2 and NKX2-5 (Fig. 4H).
GSEA of the RNA-seq
GSEA was carried out on the DEGs obtained through
RNA-seq analysis. The enrichment score (ES) was calculated and
normalized to produce the normalized ES (NES), thereby
standardizing the gene set data. By calculating the ES and
evaluating its significance, enrichment plots for various pathways
were generated. In the analysis results, the criteria of P<0.05,
|NES|>1 and q-value <0.25 were regarded as significantly
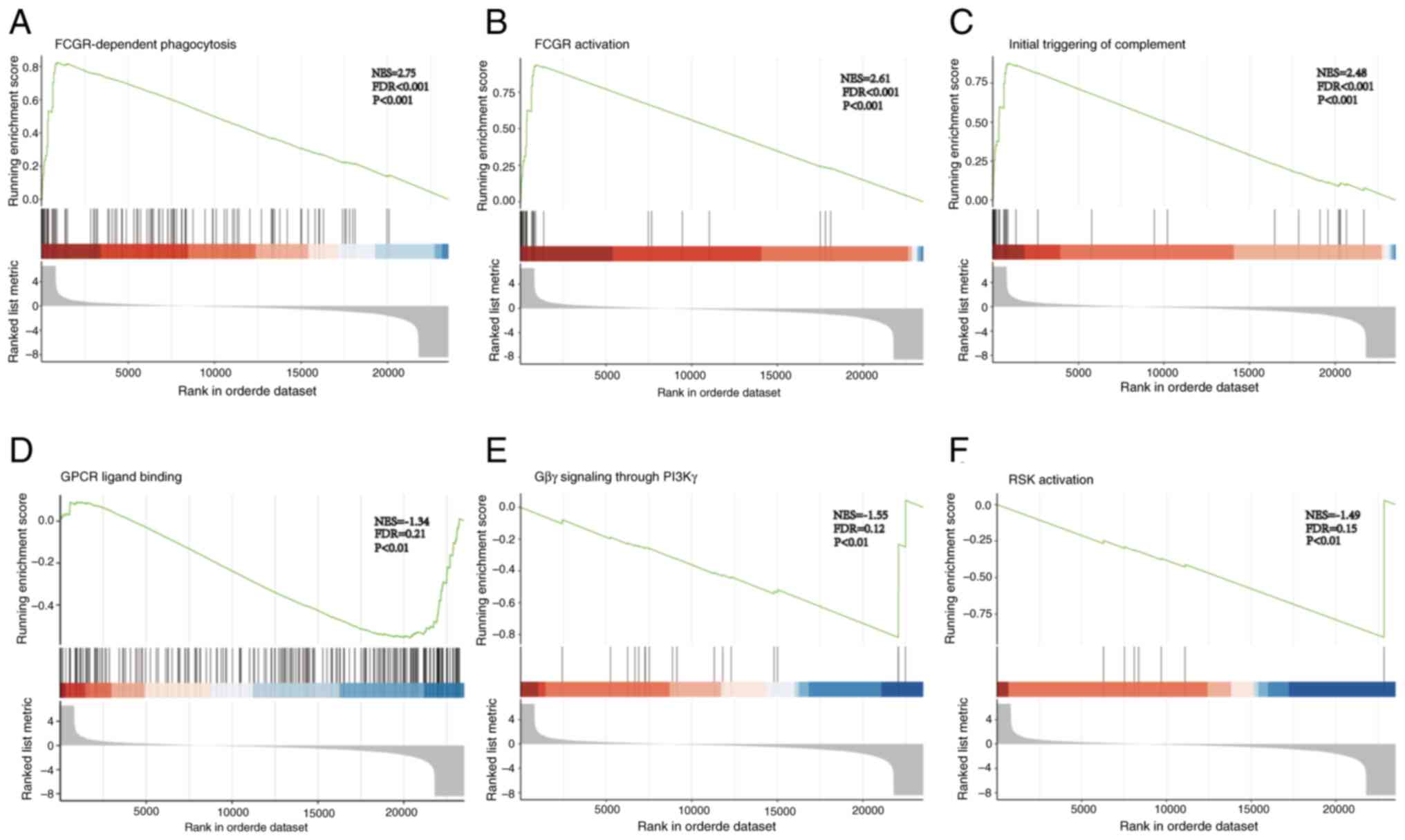
enriched. GSEA revealed that the Fcγ receptor (FCGR)-dependent
phagocytosis (Fig. 5A), FCGR
activation (Fig. 5B) and the
initial triggering of complement (Fig.
5C) were activated. Additionally, G-protein coupled receptor
(GPCR) ligand binding (Fig. 5D),
G-protein βγ (Gβγ) signaling through PI3Kγ (Fig. 5E) and ribosomal S6 kinase (RSK)
activation (Fig. 5F) were
inhibited. These results suggested that ZNF514 regulation may
affect EC immunosurveillance via FCGR-dependent phagocytosis, FCGR
activation, and initial complement triggering; it may also
influence EC occurrence and development by regulating oncogenic
signaling cascades through GPCR ligand binding, Gβγ signaling via
PI3Kγ, and RSK activation.
IPA of the DEGs identified by RNA-seq
analysis uncovers their potential mechanisms of influence on EC
progression
To investigate the mechanism by which ZNF514
influences EC progression, IPA of the DEGs was performed. After
ZNF514 knockdown in Kyse-150 cells, significantly upregulated genes
included A2M, CTC_435M103, IGHG1, IGHA1, RP11_140H171, TGFB2-OT1,
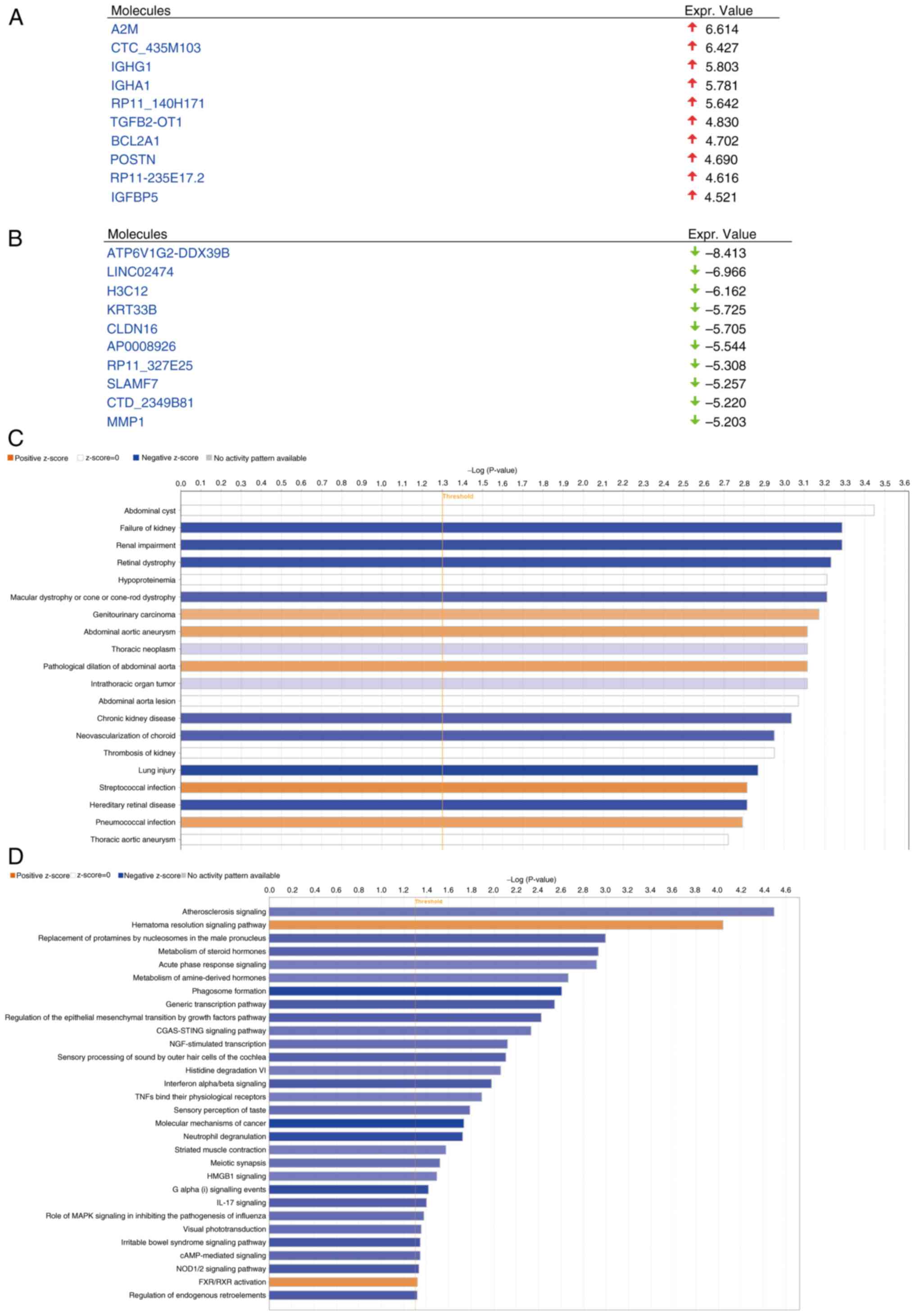
BCL2A1, POSTN, RP11-235E17.2 and IGFBP5 (Fig. 6A). Conversely, the significantly
downregulated genes included ATP6V1G2-DDX39B, LINC02474, H3C12,
KRT33B, CLDN16, AP0008926, RP11_327E25, SLAMF7, CTD_2349B81 and
MMP1 (Fig. 6B). Enrichment
analysis indicated that these genes were significantly associated
with multiple pathologies, such as ‘Failure of kidney’, ‘Renal
impairment’, ‘Retinal dystrophy’, ‘Macular dystrophy or cone-rod
dystrophy’, ‘Genitourinary carcinoma’ and ‘Abdominal aortic
aneurysm’. (Fig. 6C).
Pathway enrichment analysis identified several
significantly affected pathways (negative Z-score indicates that
the pathway is significantly inhibited, while a positive Z-score
indicates the pathway is significantly activated.), encompassing
the ‘Atherosclerosis Signaling’ pathway, ‘Hematoma Resolution
Signaling Pathway’, ‘Regulation of the Epithelial Mesenchymal
Transition by Growth Factors Pathway’, ‘NOD1/2 Signaling Pathway’,
‘Molecular Mechanisms of Cancer’, ‘IL-17 Signaling’ pathway and
‘CGAS-STING Signaling Pathway’. Based on the Z-value results,
including the following pathways were significantly inhibited:
‘Molecular Mechanisms of Cancer’; ‘Regulation of the Epithelial
Mesenchymal Transition by Growth Factors Pathway’; ‘NOD1/2
Signaling Pathway’ and ‘IL-17 Signaling Pathway’ (Fig. 6D). The present study found that
‘Molecular Mechanisms of Cancer’ was the pathway with the furthest
Z-score from 0 (−4.323), indicating that the pathway was subjected
to the most significant inhibition. Subsequent analysis of the
pathways within ‘Molecular Mechanisms of Cancer’ revealed that the
PI3K/AKT pathway was widely inhibited (Fig. 7A). Therefore, it was highly
plausible that the low expression of ZNF514 exerted its effects on
cellular biological functions by suppressing the upstream PI3K/AKT
pathway, which in turn would lead to the inhibition of multiple
downstream pathways. The ‘Regulation of the Epithelial Mesenchymal
Transition by Growth Factors Pathway’ was implicated in
epithelial-mesenchymal transition (EMT) regulation, with multiple
pathways, such as the Ras/MEK and PI3K/AKT pathways, found to be
inhibited following ZNF514 knockdown. Fig. S1 illustrates the key molecules
involved in these pathways. Furthermore, the related molecules in
the ‘STAT3 pathway’ (Fig. S2) and
‘NOD1/2 Signaling Pathway’ (Fig.
S3) were significantly inhibited. Additionally, the related
molecules of NF-κB in the ‘Molecular Mechanisms of Cancer’ were
significantly inhibited. The upstream factors of ZNF514 were also
identified, with TBX3 (Fig. 7B),
and TLR5 (Fig. 7C) being
recognized as significant upstream regulators. Notably, TBX3 was
significantly activated, while TLR5 was significantly
inhibited.
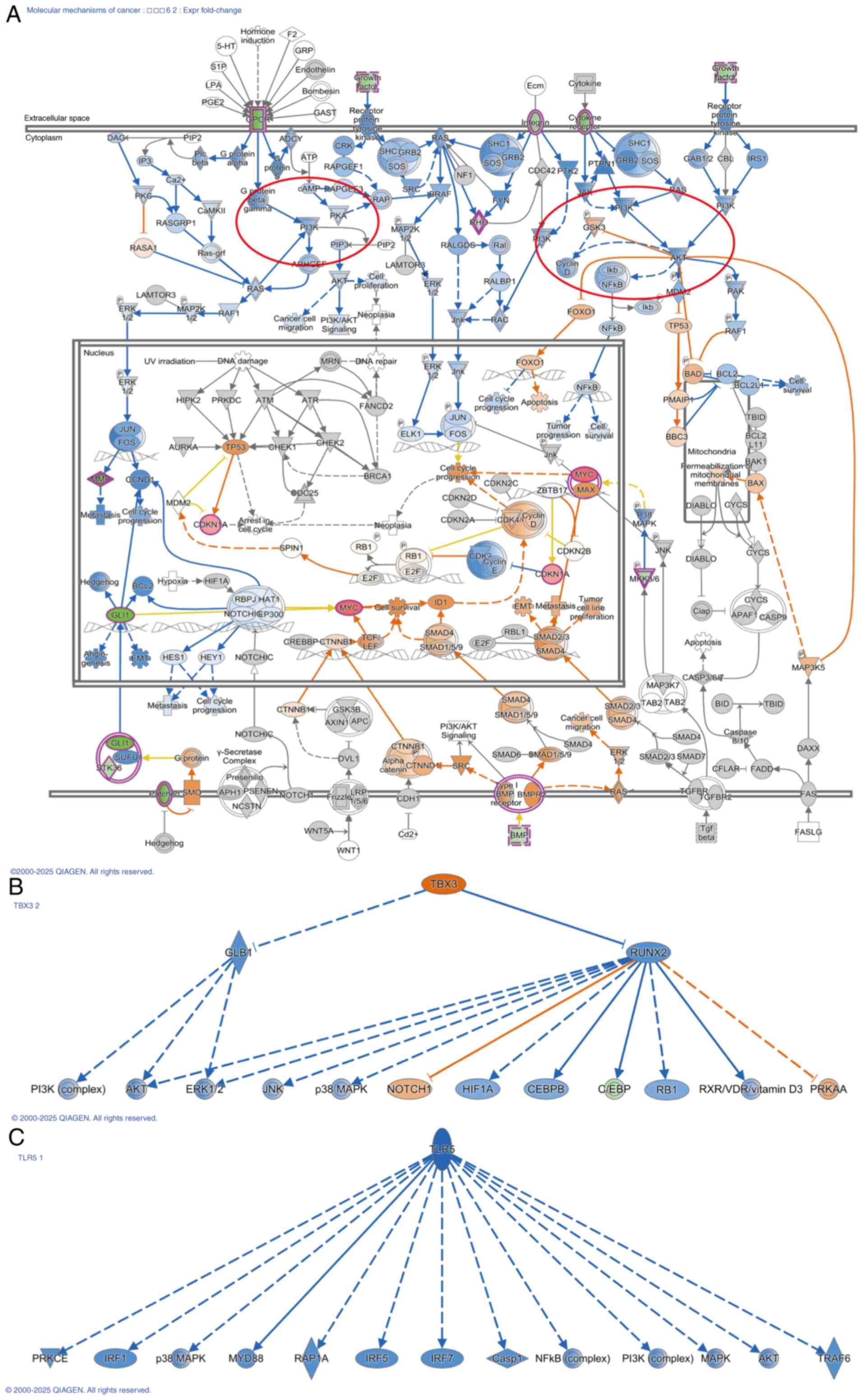 | Figure 7.IPA shows that ZNF514 knockdown
affects multiple cancer-related pathways. (A) The affected PI3K/AKT
pathway, and its upstream and downstream relationships in the
Molecular Mechanisms in Cancer pathway. (B) Activation of TBX3, and
(C) inhibition of TLR5, with the downstream molecules influenced by
these factors shown. Solid line, direct interaction; dashed line,
indirect interaction. Arrow, activation; Blocked arrow, inhibition.
Blue, leads to inhibition; Orange, leads to activation; Yellow,
findings inconsistent with state of downstream molecule; Gray,
effect not predicted. |
In conclusion, the present findings suggested that
the antitumor mechanisms of ZNF514 downregulation might be linked
to the suppression of the PI3K/AKT and Ras/MEK pathways, as well as
the STAT3 pathway, nucleotide oligomerization domain receptor
(NOD)1/2 pathway and NF-κB pathway. Therefore, low expression of
ZNF514 may suppress EC progression by inhibiting these classical
oncogenic pathways, making ZNF514 a notable target for EC
treatment.
Discussion
ZNFs, defined by their conserved zinc finger motifs,
represent one of the largest families of transcription factors in
the human genome and are important in a number of diverse
biological processes, such as differentiation, cell development,
metabolic regulation and apoptosis (14). The C2H2 is the largest class among
all zinc finger types, comprising the sequence CX2CX3FX5LX2HX3H.
Upon interaction with zinc ions, the two cysteine and two histidine
residues fold into a finger-like structure comprising an α-helix
and a double-stranded anti-parallel β-sheet (5). Each C2H2-type ZNF typically contains
KRAB, BTB, SCAN and SET domains, and multiple zinc finger motifs
capable of binding to DNA sequences. ZNF514, a member of the
KRAB-type C2H2 ZNF family, encompasses two core domains: The
N-terminal KRAB domain (residues 74–111), which acts as a key
transcriptional repressor by recruiting corepressors such as
KRAB-associated protein 1 to modulate chromatin states and
facilitate epigenetic regulation; and seven C2H2 zinc finger
domains at the C-terminus (residues 307–400), each featuring a
conserved Cys2/His2 motif that enables sequence-specific DNA
binding and thereby mediates the regulation of gene promoter
regions, underpinning its function as a transcription factor
(15–17).
Studies have shown that ZNF family can perform
distinct functions with various partners, potentially leading to
antagonistic interactions with different partners. For example,
zinc finger E-box-binding homeobox 1 is a transcriptional repressor
closely associated with cell differentiation genes, which can
interact with yes-associated protein 1 to promote the invasive
phenotype of cancer (18). ZNF
family exhibit diverse regulatory mechanisms for various downstream
genes by recruiting different chromatin modifiers. For example,
ZNF217 modifies downstream genes by interacting with REST
corepressor 1, lysine demethylase 1, histone deacetylase 2 and
C-terminal binding protein, thereby influencing their expression
(19).
Additionally, post-translational modifications of
ZNF family provide another layer of regulation. Phosphorylation of
serine or threonine residues on ZNF-linked peptides has previously
been reported. During mitosis, ZNFs such as Ikaros, Sp1 and YY1
exhibit high levels of phosphorylation on threonine/serine residues
in the linked peptides of ZNF family, leading to a loss of
DNA-binding ability (5).
Furthermore, research has indicated that ZNFs can affect tumor cell
migration and invasion by regulating the EMT process. The A20
protein belongs to the Cys2/Cys2 ZNF family (20), where it induces the
monoubiquitination of Snail1 at three lysine residues, thereby
decreasing its phosphorylation. Consequently, the monoubiquitinated
Snail1 is stabilized in the nucleus and promotes EMT and basal-like
breast cancer metastasis induced by transforming growth factor-β1
(21). Multiple ZNFs have emerged
as potential biomarkers and effective therapeutic targets in cancer
(22–24). However, research on ZNF514 in EC is
limited and the mechanisms by which it functions in EC remain to be
elucidated.
In the present study, ZNF514 exhibited significant
upregulation in EC tissues. Furthermore, functional assays, such as
siRNA-mediated knockdown experiments, supported the role of ZNF514
in promoting EC cell migration, invasion and proliferation.
Mechanistically, ZNF514 inhibition may suppress tumorigenesis by
disrupting multiple oncogenic pathways, including PI3K/AKT,
Ras/MEK, STAT3, NOD1/2 and NF-κB signaling cascades. Collectively,
the findings of the present study established ZNF514 as a central
regulator of EC progression, highlighting its potential as a
therapeutic target and prognostic biomarker.
In the present study, RNA-seq analysis of Kyse-150
cells after ZNF514 knockdown, followed by GO and KEGG analyses,
GSEA and IPA of the transcriptomics data, indicated that ZNF514
modulated multiple signaling pathways, functioning as an upstream
regulator and consequently impacting EC progression. GSEA further
revealed that ZNF514 knockdown in Kyse-150 cells activated
FCGR-dependent phagocytosis, increased FCGR activation and promoted
early complement triggering compared with normal Kyse-150 cells.
Additionally, GPCR ligand binding, Gβγ signaling via PI3Kγ and RSK
activation were significantly inhibited. Meanwhile, IPA
corroborated the reliability of the GSEA findings, showing that the
Ras/MEK, PI3K/AKT, STAT3, NOD1/2 and NF-κB pathways were inhibited
in the transcriptome following ZNF514 knockdown.
FCGRs are a family of transmembrane glycoproteins
that exert anti-pathogen effects by specifically binding to the Fc
region of IgG antibodies. These receptors are expressed across
diverse immune cell types, establishing a robust defense mechanism
to eliminate invading pathogens (25). Studies have shown that the
bispecific antibody MDX-H210 can target FCGR1 (FcγRI) on cytotoxic
effector cells with upregulated FcγRI expression, thereby
activating these effector cells to target HER2/neu-overexpressing
malignancies (26).
The complement system is part of the innate immune
system and protects the host against pathogen invasion after
activation via the classical, alternative and lectin pathways
(27). The complement system
exerts dual effects on cancer progression. Furthermore, the
interactions of complement components are specific to each cancer
type, and are regulated by tumor cell characteristics and the tumor
microenvironment (28). The
effects of the complement system on malignancies are determined by
a combination of complement-mediated antitumor and cytotoxicity,
along with C5a/C5aR1 axis-mediated protumor chronic inflammation
that hinders antitumor T-cell responses (29). Although the mechanisms by which
complement system activation via ZNF514 downregulation affects
tumor cells are yet to be elucidated, the increase in cytotoxic
effects resulting from FCGR activation suggests that the activation
of the complement system in the present study may have enhanced
immune cell toxicity, thereby inhibiting tumor progression.
Therefore, ZNF514 downregulation may have promoted the activation
of FCGRs and the complement system, thereby enhancing cytotoxic
effects to kill tumor cells.
GPCRs, which constitute ~4% of the human genome,
represent the largest family of cell surface receptors. These
receptors mediate cell communication by transducing external
signals into internal cellular responses (30). By binding to ligands, they activate
various Gα proteins, including Gαs, Gαi, Gαq and Gα12/13,
subsequently triggering downstream signaling cascades. These
signaling cascades regulate cell migration, transcription,
proliferation and survival via kinases and phosphatases, including
MAPK, AKT and mTOR (31). This
indicates that GPCR activation is closely associated with cancer
initiation and progression.
An important step in neutrophil chemotaxis involves
the transformation of PIP2 into PIP3 by PI3Kγ, which is also
important for the metastasis of multiple cancer types (32). The Gβγ heterodimer can recruit
PI3Kγ to the cell membrane and activate the kinase activity of
PI3Kγ via allosteric regulation, enabling PI3Kγ to convert PIP2
into PIP3. This activates the PI3K signaling pathway and
subsequently triggers pro-tumorigenic signaling cascades (32). RSK is a signaling molecule located
downstream of the Ras/MAPK signaling pathway and its activation
promotes cellular biological functions such as proliferation,
growth, motility and survival (33). Thus, downregulation of ‘Gβγ
signaling through the PI3Kγ’ and RSK pathways may inhibit tumor
progression, a finding that the present study emphasizes.
Furthermore, transcriptomic IPA revealed that the
tumor-suppressive mechanisms associated with ZNF514 downregulation
may involve inhibition of the Ras/MEK, PI3K/AKT, STAT3, NOD1/2 and
NF-κB pathways. EGF is important in governing cell growth,
proliferation and differentiation. Additionally, it is linked to
cancer stemness and EMT (34),
with the Ras/Raf/MAPK pathway considered a traditional downstream
signaling pathway of EGF/EGF receptor interactions. The Ras-ERK
signaling cascade is activated by growth factors that bind to
receptor tyrosine kinases (35,36),
leading to the activation of the small G-protein Ras. Subsequently,
MEK, RAF and ERK are activated sequentially in a
phosphorylation-dependent cascade, with ERK1/2 ultimately
phosphorylating downstream effectors which regulate numerous
physiological processes, including proliferation, transcription,
survival, cell adhesion, growth and differentiation (35,37,38).
Aberrant activation of the PI3K/AKT/mTOR pathway markedly
contributes to carcinogenesis by driving cancer initiation and
progression via its impact on cell proliferation (39,40),
autophagy (41,42), apoptosis (43), angiogenesis (44,45)
and EMT (46,47). Activation of STAT3 is closely
associated with various epithelial (48), mesenchymal (49) and hematological tumors (50), and STAT3 activation is key for the
malignant phenotype (51).
Research indicates that STAT3 binds to the TWIST promoter to
mediate its transcriptional upregulation, consequently triggering
EMT (52).
NOD1 primarily participates in innate inflammatory
immune responses by mediating the NF-κB, MAPK and autophagy-related
pathways. Uncontrolled apoptosis regulated by NOD1 may induce an
immunosuppressive microenvironment that facilitates tumor
progression (53). By contrast,
NOD2 is associated with chronic inflammation and cancer development
(54). The NF-κB pathway has long
been recognized as a prototypical pro-inflammatory signaling
pathway that is frequently overactivated in tumors (55). Additionally, NF-κB activation is
observed in cancer cells and the tumor microenvironment of most
solid tumors and hematological malignancies. This activation
contributes to genetic and epigenetic alterations, metabolic
reprogramming, the acquisition of cancer stem cell traits, EMT,
invasion, angiogenesis and metastasis (55).
Through bioinformatics analyses and in vitro
cell experiments, the present study revealed the important role of
ZNF514 in the progression of EC. ZNF514 may serve a notable role in
regulating cell motility and the invasive potential of EC cells.
The present study aimed to explore the role of high ZNF514
expression in the mechanism of EC progression using EC cell models
and did not perform in vivo experiments. In future research,
it is necessary to establish EC animal models, such as
immunodeficient mouse xenograft models, and combine techniques,
such as in vivo imaging and histopathological analysis, to
further validate the pro-cancer effects and molecular pathways of
ZNF514 in the in vivo environment, and to elaborate its role
in driving EC initiation and progression. Meanwhile, the small
sample size of the present study also constitutes a limitation. In
the future, we aim to expand the sample size, conduct more
comprehensive clinical validations, and further explore the
relationship between ZNF514 expression and the prognosis of
patients with EC. In addition, the lack of validation experiments
based on RNA-seq and pathway analysis was also a limitation of the
present study. The study of the mechanism of ZNF514 in EC remains
in its initial stage and further experimental verification is
needed. This validation will not only establish a solid foundation
for the present conclusions but also provide a scientific rationale
for developing ZNF514-targeted small-molecule compounds and their
clinical translation.
The present study represents, to the best of our
knowledge, the first report on the role of ZNF514 in EC,
demonstrating that ZNF514 may be highly expressed in patients with
EC. Its overexpression may drive the proliferation, motility and
invasiveness of EC cells. The present study also revealed that
ZNF514 can modulate the biological behavior of EC cells by engaging
multiple signaling pathways. Its mechanism may predominantly
regulate EC progression via the PI3K/AKT pathway, underscoring its
multifunctional role. The findings of the present study merit
further investigation into the specific molecular mechanisms
underlying the diverse regulatory functions of ZNF514. Furthermore,
the present study established a robust theoretical framework for
exploring the specific mechanisms underlying the multifaceted
function of ZNF514. This not only facilitates the development of
targeted therapies across diverse cancer types, but also
accelerates the advancement of EC diagnosis and treatment in
clinical settings. Notably, the present study has laid an
experimental foundation for the molecular functions of ZNF514 in
EC.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was financially supported by the Shandong
Province Medical and Health Development Plan (grant no.
202304020860) and the Jinan Municipal Health Commission Science and
Technology Development Plan Project (grant no. 2024302003).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author. The RNA sequencing data
generated in the present study have been deposited in the Gene
Expression Omnibus database under accession number GSE301311 or at
the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE301311.
Authors' contributions
LZ and HL conceived and designed the study. LL, GW
and CZ conducted the experiments; LL, XS and HZ performed the
bioinformatics analysis. LZ revised the manuscript. LZ and XS
confirm the authenticity of all the raw data. All authors read and
approved the manuscript.
Ethics approval and consent to
participate
The present study was approved by the institutional
review board of Jinan Central Hospital (approval no. 20241120027).
All subjects provided written informed consent form before
enrollment in the study.
Patient consent for publication
Patients provided written informed consent regarding
publishing their data.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, the AI tool
ChatGPT (https://chatgpt.com) was used to improve
the readability and language of the manuscript, and subsequently,
the authors revised and edited the content produced by the AI tools
as necessary, taking full responsibility for the ultimate content
of the present manuscript.
References
|
1
|
Deboever N, Jones CM, Yamashita K, Ajani
JA and Hofstetter WL: Advances in diagnosis and management of
cancer of the esophagus. BMJ. 385:e0749622024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Smyth EC, Lagergren J, Fitzgerald RC,
Lordick F, Shah MA, Lagergren P and Cunningham D: Oesophageal
cancer. Nat Rev Dis Primers. 3:170482017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huang FL and Yu SJ: Esophageal cancer:
Risk factors, genetic association, and treatment. Asian J Surg.
41:210–215. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lin L and Lin DC: Biological significance
of tumor heterogeneity in esophageal squamous cell carcinoma.
Cancers (Basel). 11:11562019. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jen J and Wang YC: Zinc finger proteins in
cancer progression. J Biomed Sci. 23:532016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hong K, Yang Q, Yin H, Wei N, Wang W and
Yu B: Comprehensive analysis of ZNF family genes in prognosis,
immunity, and treatment of esophageal cancer. BMC Cancer.
23:3012023. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jen J, Lin LL, Lo FY, Chen HT, Liao SY,
Tang YA, Su WC, Salgia R, Hsu CL, Huang HC, et al: Oncoprotein
ZNF322A transcriptionally deregulates alpha-adducin, cyclin D1 and
p53 to promote tumor growth and metastasis in lung cancer.
Oncogene. 36:52192017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yao J, Qian K, Chen C, Liu X, Yu D, Yan X,
Liu T and Li S: Correction for: ZNF139/circZNF139 promotes
cell proliferation, migration and invasion via activation of
PI3K/AKT pathway in bladder cancer. Aging (Albany NY).
14:4927–4928. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wang Y, Gong Y, Li X, Long W, Zhang J, Wu
J and Dong Y: Targeting the ZNF-148/miR-335/SOD2 signaling cascade
triggers oxidative stress-mediated pyroptosis and suppresses breast
cancer progression. Cancer Med. 12:21308–21320. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cancer Genome Atlas Research Network;
Analysis Working Group; Asan University; BC Cancer Agency; Brigham
and Women's Hospital; Broad Institute; Brown University; Case
Western Reserve University; Dana-Farber Cancer Institute; Duke
University; Greater Poland Cancer Centre, et al, . Integrated
genomic characterization of oesophageal carcinoma. Nature.
541:169–175. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Korthauer K, Kimes PK, Duvallet C, Reyes
A, Subramanian A, Teng M, Shukla C, Alm EJ and Hicks SC: A
practical guide to methods controlling false discoveries in
computational biology. Genome Biol. 20:1182019. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ye Q, Liu J and Xie K: Zinc finger
proteins and regulation of the hallmarks of cancer. Histol
Histopathol. 34:1097–1109. 2019.PubMed/NCBI
|
|
15
|
Wang J, Chitsaz F, Derbyshire MK, Gonzales
NR, Gwadz M, Lu S, Marchler GH, Song JS, Thanki N, Yamashita RA, et
al: The conserved domain database in 2023. Nucleic Acids Res.
51:D384–D388. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lu S, Wang J, Chitsaz F, Derbyshire MK,
Geer RC, Gonzales NR, Gwadz M, Hurwitz DI, Marchler GH, Song JS, et
al: CDD/SPARCLE: The conserved domain database in 2020. Nucleic
Acids Res. 48:D265–D268. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marchler-Bauer A, Bo Y, Han L, He J,
Lanczycki CJ, Lu S, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR,
et al: CDD/SPARCLE: Functional classification of proteins via
subfamily domain architectures. Nucleic Acids Res. 45:D200–D203.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lehmann W, Mossmann D, Kleemann J, Mock K,
Meisinger C, Brummer T, Herr R, Brabletz S, Stemmler MP and
Brabletz T: ZEB1 turns into a transcriptional activator by
interacting with YAP1 in aggressive cancer types. Nat Commun.
7:104982016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qin X, Zhou K, Dong L, Yang L, Li W, Chen
Z, Shen C, Han L, Li Y, Chan AKN, et al: CRISPR screening reveals
ZNF217 as a vulnerability in high-risk B-cell acute lymphoblastic
leukemia. Theranostics. 15:3234–3256. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Enesa K and Evans P: The biology of
A20-like molecules. Adv Exp Med Biol. 809:33–48. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lee JH, Jung SM, Yang KM, Bae E, Ahn SG,
Park JS, Seo D, Kim M, Ha J, Lee J, et al: A20 promotes metastasis
of aggressive basal-like breast cancers through
multi-monoubiquitylation of Snail1. Nat Cell Biol. 19:1260–1273.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li J, Zhou Q, Zhang C, Zhu H, Yao J and
Zhang M: Development and validation of novel prognostic models for
zinc finger proteins-related genes in soft tissue sarcoma. Aging
(Albany NY). 15:3171–3190. 2023.PubMed/NCBI
|
|
23
|
Wu P, Lin Y, Dai F, Wang H, Wen H, Xu Z,
Sun G and Lyu Z: Pan-cancer analysis and experimental validation
revealed the prognostic role of ZNF83 in renal and lung cancer
cohorts. Discov Oncol. 16:13352025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kou H, Jiang S, Wu X, Jing C, Xu X, Wang
J, Zhang C, Liu W, Gao Y, Men Q, et al: ZNF655 involved in the
progression of multiple myeloma via the activation of AKT. Cell
Biol Int. 49:177–187. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gan SY, Tye GJ, Chew AL and Lai NS:
Current development of Fc gamma receptors (FcγRs) in diagnostics: A
review. Mol Biol Rep. 51:9372024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rajasekaran N, Chester C, Yonezawa A, Zhao
X and Kohrt HE: Enhancement of antibody-dependent cell mediated
cytotoxicity: A new era in cancer treatment. Immunotargets Ther.
4:91–100. 2015.PubMed/NCBI
|
|
27
|
Omori T, Machida T, Ishida Y, Sekiryu T
and Sekine H: Roles of MASP-1 and MASP-3 in the development of
retinal degeneration in a murine model of dry age-related macular
degeneration. Front Immunol. 16:15660182025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Roumenina LT, Daugan MV, Petitprez F,
Sautès-Fridman C and Fridman WH: Context-dependent roles of
complement in cancer. Nat Rev Cancer. 19:698–715. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Merle NS and Roumenina LT: The complement
system as a target in cancer immunotherapy. Eur J Immunol.
54:e23508202024. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wess J: Designer GPCRs as novel tools to
identify metabolically important signaling pathways. Front
Endocrinol (Lausanne). 12:7069572021. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Inverso D, Tacconi C, Ranucci S and De
Giovanni M: The power of many: Multilevel targeting of
representative chemokine and metabolite GPCRs in personalized
cancer therapy. Eur J Immunol. 54:e23508702024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen CL, Syahirah R, Ravala SK, Yen YC,
Klose T, Deng Q and Tesmer JJG: Molecular basis for Gβγ-mediated
activation of phosphoinositide 3-kinase γ. Nat Struct Mol Biol.
31:1198–1207. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Romeo Y and Roux PP: Paving the way for
targeting RSK in cancer. Expert Opin Ther Targets. 15:5–9. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chatterjee P, Ghosh D, Chowdhury SR and
Roy SS: ETS1 drives EGF-induced glycolytic shift and metastasis of
epithelial ovarian cancer cells. Biochim Biophys Acta Mol Cell Res.
1871:1198052024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nussinov R, Yavuz BR and Jang H: Molecular
principles underlying aggressive cancers. Signal Transduct Target
Ther. 10:422025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Du R, Shen W, Liu Y, Gao W, Zhou W, Li J,
Zhao S, Chen C, Chen Y, Liu Y, et al: TGIF2 promotes the
progression of lung adenocarcinoma by bridging EGFR/RAS/ERK
signaling to cancer cell stemness. Signal Transduct Target Ther.
4:602019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Asghar J, Latif L, Alexander SPH and
Kendall DA: Development of a novel cell-based, In-Cell Western/ERK
assay system for the high-throughput screening of agonists acting
on the delta-opioid receptor. Front Pharmacol. 13:9333562022.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Caunt CJ, Sale MJ, Smith PD and Cook SJ:
MEK1 and MEK2 inhibitors and cancer therapy: The long and winding
road. Nat Rev Cancer. 15:577–592. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tian T, Li X and Zhang J: mTOR signaling
in cancer and mTOR inhibitors in solid tumor targeting therapy. Int
J Mol Sci. 20:7552019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Mangé A, Coyaud E, Desmetz C, Laurent E,
Béganton B, Coopman P, Raught B and Solassol J: FKBP4 connects
mTORC2 and PI3K to activate the PDK1/Akt-dependent cell
proliferation signaling in breast cancer. Theranostics.
9:7003–7015. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Al-Bari MAA and Xu P: Molecular regulation
of autophagy machinery by mTOR-dependent and -independent pathways.
Ann N Y Acad Sci. 1467:3–20. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nowosad A, Jeannot P, Callot C, Creff J,
Perchey RT, Joffre C, Codogno P, Manenti S and Besson A: Publisher
correction: p27 controls ragulator and mTOR activity in amino
acid-deprived cells to regulate the autophagy-lysosomal pathway and
coordinate cell cycle and cell growth. Nat Cell Biol. 23:10482021.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
He K, Zheng X, Li M, Zhang L and Yu J:
mTOR inhibitors induce apoptosis in colon cancer cells via
CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene.
35:148–157. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Karar J and Maity A: PI3K/AKT/mTOR pathway
in angiogenesis. Front Mol Neurosci. 4:512011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim K, Kim IK, Yang JM, Lee E, Koh BI,
Song S, Park J, Lee S, Choi C, Kim JW, et al: SoxF transcription
factors are positive feedback regulators of VEGF signaling. Circ
Res. 119:839–852. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Herrerias MM and Budinger GRS: Revisiting
mTOR and epithelial-mesenchymal transition. Am J Respir Cell Mol
Biol. 62:669–670. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Karimi Roshan M, Soltani A, Soleimani A,
Rezaie Kahkhaie K, Afshari AR and Soukhtanloo M: Role of AKT and
mTOR signaling pathways in the induction of epithelial-mesenchymal
transition (EMT) process. Biochimie. 165:229–234. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Tian F, Yang X, Liu Y, Yuan X, Fan T,
Zhang F, Zhao J, Lu J, Jiang Y, Dong Z and Yang Y: Constitutive
activated STAT3 is an essential regulator and therapeutic target in
esophageal squamous cell carcinoma. Oncotarget. 8:88719–88729.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
D'Amico S, Shi J, Martin BL, Crawford HC,
Petrenko O and Reich NC: STAT3 is a master regulator of epithelial
identity and KRAS-driven tumorigenesis. Genes Dev. 32:1175–1187.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Andersson EI, Brück O, Braun T, Mannisto
S, Saikko L, Lagström S, Ellonen P, Leppä S, Herling M, Kovanen PE
and Mustjoki S: STAT3 mutation is associated with STAT3
activation in CD30+ ALK− ALCL. Cancers
(Basel). 12:7022020. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Groner B and von Manstein V: Jak Stat
signaling and cancer: Opportunities, benefits and side effects of
targeted inhibition. Mol Cell Endocrinol. 451:1–14. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Sadrkhanloo M, Entezari M, Orouei S,
Ghollasi M, Fathi N, Rezaei S, Hejazi ES, Kakavand A, Saebfar H,
Hashemi M, et al: STAT3-EMT axis in tumors: Modulation of cancer
metastasis, stemness and therapy response. Pharmacol Res.
182:1063112022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Moreno L and Gatheral T: Therapeutic
targeting of NOD1 receptors. Br J Pharmacol. 170:475–485. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang W and Wang Y: Activation of
RIPK2-mediated NOD1 signaling promotes proliferation and invasion
of ovarian cancer cells via NF-κB pathway. Histochem Cell Biol.
157:173–182. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Taniguchi K and Karin M: NF-κB,
inflammation, immunity and cancer: Coming of age. Nat Rev Immunol.
18:309–324. 2018. View Article : Google Scholar : PubMed/NCBI
|