Introduction
Preeclampsia (PE) is a pregnancy-specific
multisystem disorder that typically arises after the 20th week of
gestation. It is primarily defined by new-onset hypertension
(systolic blood pressure ≥140 mmHg or a diastolic blood pressure of
≥90 mmHg) and proteinuria (urinary protein excretion of ≥0.3 g/24
h) (1). Hypertension, a hallmark
clinical feature of PE, may develop gradually or abruptly and is
often accompanied by symptoms such as headaches and blurred vision
(2). Proteinuria reflects renal
impairment and its progression may lead to generalized edema,
extending from the lower limbs to the entire body (3). PE can also result in hepatic and
renal dysfunction, coagulation abnormalities and intrauterine
growth restriction. Globally, the prevalence of PE ranges from 2 to
8% (4), with increased rates
observed in developing countries (5). High-risk factors include nulliparity,
maternal age extremes (<18 or >35 years), multiple
gestations, family history of PE (6), chronic hypertension, diabetes,
obesity (7), genetic
predisposition, environmental influences, immune dysregulation
(8) and paternal contributions,
such as paternal history of PE in previous pregnancies (doubling
the risk in subsequent partners), partner change leading to loss of
maternal immune tolerance, inadequate maternal exposure to paternal
seminal fluid (associated with shorter cohabitation duration) and
genetic contributions via paternal human leukocyte antigens that
disrupt maternal-fetal immune crosstalk (9). The pathogenesis of PE is
multifactorial, involving abnormal placentation, immune dysfunction
and endothelial injury. Inadequate trophoblast invasion results in
shallow placental implantation and defective remodeling of the
uterine spiral arteries, leading to reduced placental perfusion
(10). This hypoperfusion induces
placental hypoxia and oxidative stress, contributing to cellular
damage via lipid peroxidation, DNA oxidation and protein
modification (11,12). These changes culminate in
widespread cellular and tissue dysfunction, a key feature of PE.
Furthermore, an aberrant maternal immune response to the
feto-placental unit triggers excessive production of inflammatory
mediators, disrupting the immune balance required for a healthy
pregnancy (13).
Post-translational protein modifications (PTMs) of proteins carry
out a central role in these processes by regulating protein
structure, function, localization and stability. Key PTMs include,
phosphorylation, acetylation and particularly ubiquitination (due
to its involvement in cell signaling, gene regulation) and cell
cycle control, all of which are associated with the onset and
progression of PE (14–16).
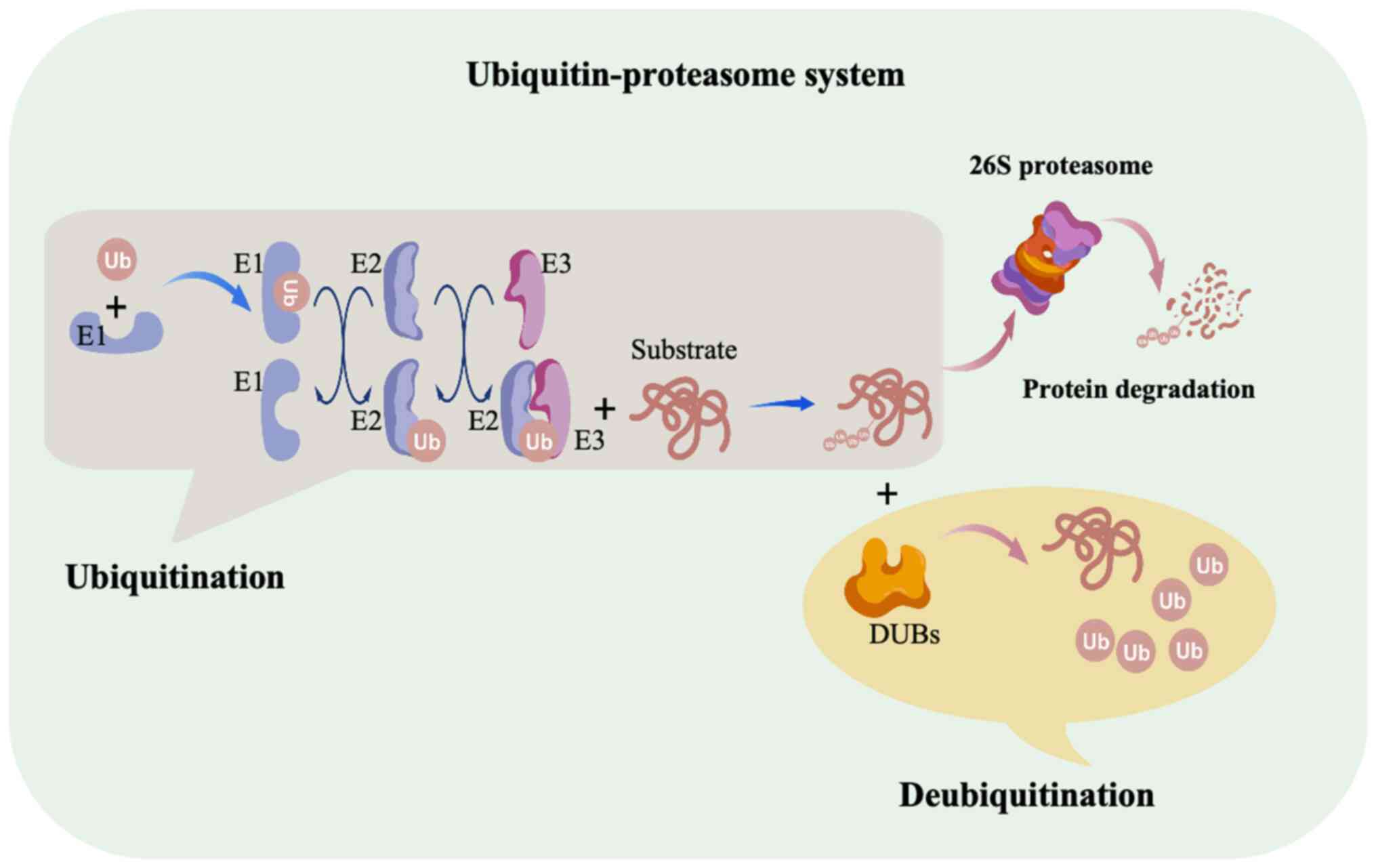
Ubiquitin-proteasome system (UPS) regulates cellular
homeostasis through ubiquitination and deubiquitination (Fig. 1) (17). Ubiquitination is a PTM process that
involves the attachment of ubiquitin molecules to target proteins.
This process requires three main types of enzymes:
Ubiquitin-activating enzymes (E1), ubiquitin-conjugating enzymes
(E2) and ubiquitin ligases (E3). First, the E1 enzyme activates
ubiquitin, which is then transferred to the E2 enzyme. The E3
ligase subsequently facilitates the transfer of ubiquitin to
specific substrate proteins (18).
Ubiquitination can occur as monoubiquitination or
polyubiquitination, with different ubiquitin chain types regulating
protein stability, activity and subcellular localization. These
modifications are integral to diverse cellular processes, including
cell cycle progression, signal transduction and gene expression
(19). Conversely,
deubiquitination, which is the removal of ubiquitin moieties, is
mediated by deubiquitinating enzymes (DUBs), which counteract
ubiquitination and fine-tune protein turnover and function. To
date, >100 DUBs have been identified and are classified into ≥9
families based on conserved domains and sequence homology. These
include ubiquitin-specific proteases, ubiquitin C-terminal
hydrolases, Machado-Joseph disease protein domain proteases,
ovarian tumor proteases, motif-interacting with
ubiquitin-containing novel DUB family, JAB1/MPN/MOV34
metalloenzymes, permuted papain fold peptidases, zinc
finger-containing ubiquitin peptidase 1 and monocyte
chemoattractant protein-induced protein-1 (20). The balance between ubiquitination
and deubiquitination is essential for cellular integrity and
function.
In the context of PE, dysregulation of this balance
may impair trophoblast functions such as proliferation, invasion
and migration, and fusion. It may also disrupt apoptosis, provoke
endothelial cell stress responses and exacerbate endoplasmic
reticulum (ER) and oxidative stress in trophoblasts. Understanding
the roles of ubiquitination and deubiquitination in PE pathogenesis
is therefore key for identifying novel molecular targets and
developing effective therapeutic interventions.
The role of ubiquitination in PE
Ubiquitin ligase constitutively
photomorphogenic 1 (COP1)
Serine/threonine kinase 40 (STK40), a Ser
(serine)/threonine protein kinase, interacts with cyclin-dependent
kinase inhibitor 1C (P57Kip2), a cyclin-dependent kinase inhibitor
essential for trophoblast cell fusion (21,22).
During this fusion process, STK40 downregulates P57Kip2 protein
levels by binding to its CDK/cyclin domains, thereby promoting
COP1-mediated ubiquitination. This process reduces P57Kip2 protein
levels without affecting its mRNA expression (23), highlighting a post-translational
regulatory mechanism key for trophoblast fusion. Both STK40-P57Kip2
interaction and COP1-mediated ubiquitination are essential for this
regulatory pathway (21). In
hypertensive disorders of pregnancy, such as PE, STK40 is
abnormally upregulated, potentially impairing trophoblast fusion
(21), by accelerating the
degradation of P57Kip2. This disruption may contribute to the
development of PE. Furthermore, STK40 overexpression or knockdown
modulates the expression of genes associated with P57Kip2,
components of the E3 ubiquitin ligase complex and trophoblast cell
markers, thereby affecting overall trophoblast function. In
summary, STK40, through its interaction with COP1 and regulation of
P57Kip2 ubiquitination, may carry out a pivotal role in the
pathogenesis of PE by disrupting trophoblast fusion and cell cycle
regulation when upregulated.
Ubiquitin ligase ankyrin repeat and
SOCS box containing protein 4 (ASB4)
ASB4 is an E3 ubiquitin ligase that targets specific
proteins for proteasomal degradation (24,25).
It is involved in pregnancy and has been implicated in the
development of PE (25).
ASB4-knockout mice exhibit hallmark symptoms of PE, including
hypertension, proteinuria and reduced offspring numbers (25,26).
These abnormalities are exacerbated in mice subjected to a high-fat
diet, consistent with a study that associate the ASB4 gene
polymorphisms to obesity-related PE in humans (27). Mechanistically, ASB4 mediates the
ubiquitination and subsequent degradation of inhibitor of DNA
binding 2 (ID2), an inhibitor of DNA-binding proteins (25). This degradation relieves the
suppression of basic helix-loop-helix transcription factors, which
are essential for cellular differentiation (28). The function of ASB4 is thus key for
trophoblast differentiation, implantation and placental development
(25). Additionally, ASB4
deficiency impairs angiogenesis, with reduced vascular endothelial
growth factor (VEGF) expression levels observed in the blood
vessels of Asb4-deficient mice, although the regulatory
mechanisms remain unclear (27).
For instance, while reduced ID2 expression levels promote VEGF
secretion in hepatocellular carcinoma, the impact of elevated ID2
levels on VEGF production in trophoblasts is not yet understood
(29,30). In summary, ASB4 regulates protein
turnover during pregnancy and is important for trophoblast
differentiation and placental function. Its dysfunction may be a
critical factor in the pathogenesis of PE.
Ubiquitin ligase neural precursor cell
expressed developmentally down-regulated protein 4 (NEDD4)
NEDD4, an E3 ubiquitin ligase, contributes to the
pathogenesis of PE by mediating K48-linked ubiquitination, leading
to proteasomal degradation of its substrates (31). Thrombospondin 1 (THBS1), which is
downregulated in PE placental tissues and inversely correlates with
blood pressure levels (32),
inhibits the interaction between NEDD4 and TGF-β-activated kinase 1
(TAK1) (33). Loss of THBS1
enhances NEDD4-mediated ubiquitination and degradation of TAK1,
thereby impairing trophoblast fusion and function (32). Moreover, THBS1 deficiency induces
necroptosis in trophoblast cells, a form of programmed cell death
accompanied by the release of damage-associated molecular patterns,
which can activate inflammatory responses and disrupt cellular
homeostasis (34). Notably, this
necroptosis can be reversed by specific necroptosis inhibitors such
as Necrostatin-1 and GSK'872, while the pan-caspase inhibitor
Z-VAD-FMK is ineffective in this context (4). Furthermore, THBS1 silencing
inhibits trophoblast cell proliferation, migration and invasion,
while promoting cell cycle arrest and apoptosis (4). In summary, THBS1 carries out a
protective role in maintaining trophoblast cell function by
preventing NEDD4-mediated TAK1 ubiquitination and degradation. In
PE, downregulation of THBS1 activates this ubiquitination pathway,
triggering necroptosis and inflammatory responses that contribute
to disease progression. These findings underscore the critical role
of THBS1 in trophoblast regulation and suggest potential
therapeutic targets for PE prevention and treatment.
Ubiquitin ligase neural precursor cell
expressed developmentally downregulated gene 4-like (NEDD4L)
NEDD4L, a ubiquitin ligase of the NEDD4 family,
mediates the addition of ubiquitin to target proteins, such as
epithelial sodium channel (ENaC), promoting their degradation or
altering their function (35).
ENaC is a key transporter that regulates sodium reabsorption in the
distal convoluted tubule and collecting duct of the kidney
(36). NEDD4L regulates ENaC
activity through the ubiquitination pathway. In patients with PE,
although NEDD4L expression does not change considerably, ENaC
levels in urinary extracellular vesicles are notably upregulated,
which may contribute to sodium retention and hypertension symptoms
in patients with PE (37). These
findings suggest that regulatory mechanisms other than NEDD4L may
influence the expression and activity of ENaC in PE. Overall, these
findings offer valuable insights into the pathophysiology of PE and
highlight novel therapeutic targets to regulate sodium reabsorption
and blood pressure in patients with PE.
Ubiquitin ligase ubiquitin protein
ligase E3A (UBE3A)
A molecular regulatory axis involving microRNA
(miR)-218-5p, UBE3A and special AT-rich sequence binding-protein 1
(SATB1) has been identified in the context of PE pathogenesis
(38). UBE3A, an E3 ubiquitin
ligase, mediates their ubiquitination of target proteins, marking
them for proteasomal degradation (39). Among its substrates, UBE3A
specifically targets the SATB1 protein, a key regulator of
trophoblast cell function. Reduced SATB1 expression levels are
associated with impaired trophoblast migration and invasion and
have been implicated in the development of PE (40,41).
miR-218-5p negatively regulates UBE3A expression levels, thereby
attenuating UBE3A-mediated ubiquitination and degradation of SATB1
(38). This inhibition helps
maintain SATB1 protein stability, promoting trophoblast cell
infiltration and mitigating ER and oxidative stress. In animal
models of PE, administration of miR-218-5p agomir was revealed to
alleviate pathological features, enhance trophoblast invasion and
reduce ER and oxidative stress (38). These findings suggest that
miR-218-5p may have therapeutic potential in PE by stabilizing
SATB1 through the suppression of UBE3A. Moreover, the expression
levels of miR-218-5p, UBE3A and SATB1 could serve as predictive or
prognostic biomarkers for PE. In summary, the
miR-218-5p/UBE3A/SATB1 axis carries out a key role in maintaining
trophoblast cell function, and its modulation offers a promising
molecular target for PE prevention and treatment.
Ubiquitin ligase casitas B-lineage
lymphoma (Cbl)
Cbl-mediated ubiquitination of Met disrupts
hepatocyte growth factor (HGF) signaling in early-onset PE (E-PE)
(42), which is defined as PE
diagnosed before 34 weeks of gestation (43). HGF regulates trophoblast
migration/invasion via Met receptor activation-activating the
MEK/Erk, PI3K and STAT3 pathways (44), which are essential for placental
nutrient exchange. These processes are essential for the role of
the placenta in mediating nutrient exchange between the mother and
fetus. In mice, the absence of HGF or Met results in embryonic
lethality and placental dysfunction, highlighting the importance of
the HGF/Met pathway (45,46). Furthermore, in E-PE, the
suppression of this signaling pathway associates with enhanced
internalization and subsequent ubiquitination of the Met receptor.
Normally, the Met receptor undergoes CAV-1-mediated internalization
and Cbl-mediated ubiquitination after activation to maintain
signaling homeostasis (47).
However, under hypoxic stress in E-PE, CAV-1 binding to Met
increases, leading to more frequent Met internalization. Meanwhile,
Cbl expression levels are reduced, impairing the ubiquitination and
degradation of Met (47). This
leads to Met accumulation in trophoblast cells, forming aggregates
that disrupt HGF/Met signaling, ultimately diminishing the
invasiveness of trophoblast cells (47). Consequently, this disruption is
closely associated with PE development, affecting placental
formation and function, which can compromise fetal blood and
nutrient supply. The hypoxic environment and disrupted Met
signaling may create a cycle that worsens trophoblast cell
dysfunction and contributes to PE pathogenesis. The effect of
hypoxia on the internalization of membrane receptors, as observed
in receptors such as glutamate receptor 1 (48) and the transferrin receptor
(49), may similarly apply to
preeclamptic placentas.
These findings suggest that dysregulated
ubiquitin-mediated degradation, particularly involving the Met
receptor, is central to the pathogenesis of E-PE. In conclusion,
aberrant Met receptor internalization and ubiquitination, along
with suppression of the HGF/Met signaling pathway, likely carry out
a key role in the early stages of PE. Understanding this mechanism
provides valuable insight into the molecular basis of PE and
highlights potential therapeutic targets for its prevention and
treatment.
Ubiquitin ligase tripartite motif
containing 72 (TRIM72)
Cytotrophoblast-derived mesenchymal stem cells
(CVMSCs) have emerged as a promising therapeutic approach for PE
due to their robust self-renewal capacity and low immunogenicity
(50). Recent studies highlight
the role of CVMSC-derived exosomes in modulating trophoblast
behavior through the ubiquitination pathway (50–52),
particularly by regulating p53 protein levels. As a tumor
suppressor, p53 governs key cellular processes, including
proliferation, apoptosis and genomic stability (53). Li et al (52) demonstrated that CVMSC-derived
exosomes upregulate the expression of TRIM72, an E3 ubiquitin
ligase that directly interacts with p53, promoting its
ubiquitination and subsequent proteasomal degradation. This
downregulation of p53 reduces trophoblast apoptosis while enhancing
their proliferation and migration, which are essential functions
for proper placental development and function. The ubiquitination
cascade involves the sequential action of ubiquitin-activating,
-conjugating and -ligating enzymes, with E3 ligases such as TRIM72
conferring substrate specificity. The upregulation of TRIM72 and
the consequent suppression of p53 and its downstream signaling
highlight a key mechanism by which CVMSC-derived exosomes support
trophoblast viability and function (52). By attenuating p53-mediated
apoptosis, these exosomes may help restore placental function and
mitigate PE progression. Therefore, CVMSC-derived exosomes regulate
trophoblast apoptosis, proliferation and migration via
TRIM72-mediated p53 ubiquitination, offering a novel therapeutic
strategy for PE.
Ubiquitin ligase ring finger protein
123 (RNF123)
PE, a hypertensive disorder of pregnancy, is
characterized by impaired trophoblast cell invasiveness, often
associated with the upregulation of Cyclin G2 (CCNG2) (54). Elevated CCNG2 expression in the
placentas of patients with PE associates with diminished
trophoblast migration, invasion and endothelial-like network
formation, key processes for normal placental development (54). CCNG2 inhibits the JNK-dependent
Wnt/PCP signaling pathway by promoting the ubiquitination and
proteasomal degradation of the Dvl2 protein, thereby reducing the
expression of downstream epithelial-mesenchymal transition (EMT)
markers and MMPs (54–57). While CCNG2 does not alter Dvl2 mRNA
levels, it decreases Dvl2 protein stability via the
ubiquitin-proteasome pathway (54,57).
Recent studies have identified RNF123, an E3 ubiquitin ligase, as a
downstream effector of CCNG2 (54). RNF123 interacts with Dvl2,
facilitating its ubiquitination and degradation, thereby impairing
trophoblast cell function. By enhancing RNF123 expression and its
interaction with Dvl2, CCNG2 suppresses the Wnt/PCP-JNK signaling
cascade, ultimately limiting trophoblast migration, invasion and
network formation (54). These
findings provide mechanistic insights into the pathogenesis of PE
and highlight potential molecular targets for therapeutic
intervention.
Ubiquitin ligase β-transducin
repeat-containing protein (β-TrCP)
Severe PE is also associated with reduced
trophoblast invasiveness and increased expression of β-TrCP, a
component of the Skp1-cullin1-F-box E3 ubiquitin ligase complex.
β-TrCP mediates the ubiquitination and degradation of several key
regulatory proteins (58). β-TrCP
inhibits the expression of Snail, a zinc finger transcription
factor that promotes EMT by repressing E-cadherin (59). Overexpression of β-TrCP suppresses
trophoblast migration and invasion, whereas its knockdown enhances
these functions (60). β-TrCP also
regulates VEGFR2 protein levels, a key factor in angiogenesis,
through the ubiquitin-proteasome pathway (60). Research reveals that silencing
β-TrCP prolongs the half-life of Snail protein and the inhibition
of trophoblast migration and invasion by β-TrCP can be partially
reversed with the proteasome inhibitor MG132 (59–61).
Notably, PE placental tissues exhibit considerably
reduced expression of miR-135a-5p, which negatively associates with
β-TrCP levels (62). Functional
studies demonstrate that miR-135a-5p enhances trophoblast migration
and invasion in vitro (62)
and directly targets β-TrCP in trophoblast cells (62). Overexpression of β-TrCP counteracts
the promotive effects of miR-135a-5p on trophoblast invasion and
migration (62). At the molecular
level, miR-135a-5p increases N-cadherin, vimentin and β-catenin
expression while suppressing E-cadherin levels, effects that are
attenuated by β-TrCP overexpression (62). These findings suggest that β-TrCP
contributes to PE pathogenesis by regulating Snail through the
ubiquitin-proteasome pathway, thereby impairing EMT and trophoblast
invasiveness. As such, β-TrCP represents a promising molecular
target for the prevention and treatment of PE.
Ubiquitin ligase F-box and WD repeat
domain containing 2 (FBW2)
Glial cells missing homolog 1 (GCM1) is essential
for placental development as it activates the expression of genes
necessary for appropriate placental development by autoregulating
its promoter activity (63).
Hypoxic conditions lead to GCM1 degradation and downregulation of
its target genes, which are implicated in PE pathogenesis (64,65).
Chiang et al (66)
demonstrated that under hypoxic conditions, the protein levels of
GCM1 decrease due to ubiquitin-mediated proteasomal degradation. In
this process, hypoxia inhibits the PI3K-Akt signaling pathway,
activating GSK-3β (66), which
then phosphorylates GCM1 at Ser322. This phosphorylation
facilitates F-box protein FBW2, promoting the ubiquitination and
subsequent proteasomal degradation of GCM1 (66). Notably, the phosphorylation of GCM1
at Ser322 and Ser326 is key for FBW2 recognition and is a
prerequisite for GCM1 ubiquitination and protein degradation
(66). Moreover, the use of the
GSK-3β inhibitor LiCl can prevent hypoxia-induced degradation of
GCM1, indicating a key role for GSK-3β in regulating the stability
of GCM1 (66). Given the
association of PE with hypertension, proteinuria and edema,
hypoxia-induced GCM1 degradation may lead to placental
insufficiency, contributing to PC. These findings offer valuable
insights into potential PE interventions targeting GCM1 stability
under hypoxic conditions.
Other factors influencing
ubiquitination
Long non-coding (Lnc)-double homeobox A
pseudogene 8 (DUXAP8)
DUXAP8 is a long non-coding RNA that carries out a
key role in regulating trophoblast function by modulating the
ubiquitination of poly(rC) binding protein 2 (PCBP2). DUXAP8
specifically binds to PCBP2 and inhibits its K48-linked
ubiquitination, thereby preventing its degradation via the
ubiquitin-proteasome pathway (36). While DUXAP8 overexpression
increases PCBP2 protein stability without altering its mRNA levels,
its downregulation enhances PCBP2 degradation. This regulatory
mechanism impacts the activation of the AKT/mTOR signaling pathway,
influencing trophoblast cell proliferation, migration and invasion
(67,68). Mechanistically, DUXAP8
overexpression activates the AKT/mTOR pathway, inhibits autophagy
(evidenced by decreased LC3-II and increased p62 levels), impairs
ER quality control and induces ER stress and protein aggregation
(68). In PE models, DUXAP8
overexpression is associated with a PE-like phenotype,
characterized by reduced expression of FAM134B and LC3-II and
increased protein aggregation (68). These findings underscore the role
of DUXAP8 in regulating autophagy and ER-selective autophagy in
trophoblast cells, offering new perspectives on PE pathogenesis and
potential molecular targets for its prevention and treatment.
FUNDC1
FUNDC1 is a mitochondrial autophagy receptor that
initiates mitophagy under hypoxic conditions, serving as a
protective cellular mechanism (69). Chen et al (70) demonstrated that hypoxia reduces
FUNDC1 ubiquitination in trophoblast cells, implicating this
modulation in PE pathogenesis. Using the proteasome inhibitor MG132
and the activator MF-094, they revealed that decreased FUNDC1
ubiquitination promotes mitophagy, whereas increased ubiquitination
suppresses it. Reduced ubiquitination also influences mitochondrial
membrane potential, a key apoptotic signal (70). Under hypoxia, elevated FUNDC1
levels suppress mitophagy, decrease intracellular reactive oxygen
species (ROS) and malondialdehyde, and increase antioxidant markers
glutathione and superoxide dismutase, indicating reduced oxidative
damage (70). In models of PE,
enhanced FUNDC1 ubiquitination mitigates oxidative stress-induced
damage (70). These findings
suggest that targeting FUNDC1 ubiquitination could be a novel
approach to modulate mitophagy and oxidative stress in
trophoblasts, offering potential strategies for early PE diagnosis
and treatment.
Storkhead box 1 (STOX1)
Regulation of the STOX1 protein depends largely on
Akt-mediated phosphorylation, which influences its
nucleocytoplasmic shuttling and ubiquitin-dependent degradation.
STOX1 contains regulatory motifs similar to those in the FOX gene
family, affecting its nucleocytoplasmic transport (71,72).
Two putative Akt phosphorylation sites at Ser647 and Ser944 have
been identified (70,71). Treatment with the
ubiquitin-proteasome inhibitor MG132 increases cytoplasmic STOX1
levels, confirming ubiquitin-mediated degradation (73). Mutation analyses reveal that the
S647A mutant and wild-type STOX1 localize primarily to the nucleus,
while the phosphomimetic S647E mutation prevents nuclear entry and
accelerates degradation (73).
STOX1 directly binds to the promoter of catenin α 3 (CTNNA3),
enhancing α-T-catenin expression levels, with the Y153H mutation
strengthening this interaction and increasing mRNA expression
levels (73). Matrigel invasion
assays reveal that downregulation of STOX1 or α-T-catenin markedly
enhances trophoblast invasion, while their overexpression inhibits
it (73). Silencing STOX1 or
CTNNA3 in trophoblast cells induces a shift toward a
non-proliferative cell type (73).
In vitro, trophoblasts with the STOX1 HH genotype exhibit
reduced proliferation and increased CTNNA3 expression levels upon
STOX1 upregulation (73). These
findings indicate that Akt-mediated phosphorylation controls
nucleocytoplasmic transport and ubiquitin-dependent degradation of
STOX1, thereby regulating CTNNA3 transcription. Variations in the
STOX1 genotype associate with trophoblast invasive and
proliferative capacities, suggesting that STOX1 carries out a key
role in the pathogenesis PE. Since PE is characterized by impaired
trophoblast invasion leading to placental insufficiency and
maternal complications, STOX1 regulation may be a considerable
factor in disease development.
In summary, ubiquitination is a key PTM in PE, a
pregnancy disorder marked by reduced trophoblast invasiveness. It
regulates protein homeostasis, cellular signaling and membrane
dynamics through enzymes such as STK40, ASB4, NEDD4, NEDD4L, UBE3A,
Cb1, TRIM72, RNF123, β-TrCP and FBW2, which modulate trophoblast
function via ubiquitination and degradation of key proteins
(Fig. 2 and Table I). These ubiquitin ligases
represent promising candidates for elucidating the pathogenesis of
PE.
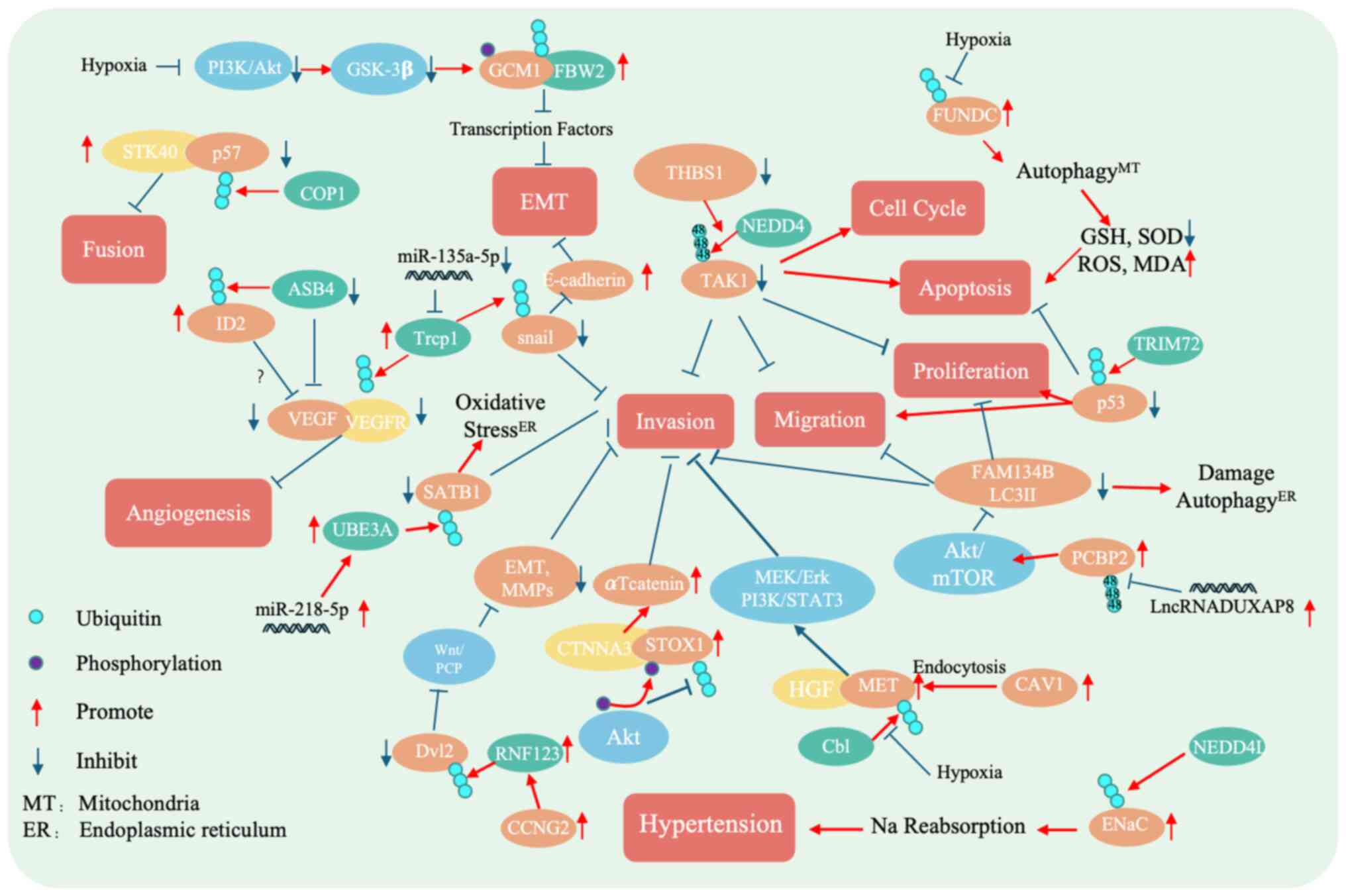 | Figure 2.Mechanistic diagram of ubiquitinating
enzymes in preeclampsia. COP1 promotes the degradation of p57,
thereby impairing trophoblast fusion. The ASB4 targets ID2 for
degradation, inhibiting angiogenesis. Silencing THBS1 enhances the
NEDD4-mediated ubiquitination and degradation of TAK1, which
suppresses trophoblast fusion, proliferation, migration and
invasion, while increasing the cell cycle arrest and apoptosis.
NEDD4L adds ubiquitin to ENaC, promoting its degradation or
altering its function. miRNA-218-5p upregulates the UBE3A
expression, which facilitates the degradation of SATB1, thereby
inhibiting trophoblast migration, invasion and ER/oxidative stress.
Cb1 promotes MET degradation, reducing trophoblast invasion. TRIM72
facilitates p53 degradation, suppressing apoptosis and enhancing
proliferation and migration. The upregulation of RNF by CCNG2
promotes Dvl12 degradation, suppressing EMT markers and MMP
expression via the Wnt/PCP pathway, thereby reducing cell invasion.
β-TrCP1 degrades Snail and VEGFRs, blocking EMT and angiogenesis.
FBW2 degrades GCM1, disrupting the EMT processes. The
downregulation of LncRNA-DUXAP8 enhances PCBP2 ubiquitination and
degradation, which activates the Akt/mTOR signaling pathway. This
activation decreases FAM134B, a known inhibitor of cell
proliferation, invasion, migration and ER stress. Hypoxia reduces
FUNDC1 ubiquitination, which promotes trophoblast autophagy. Akt
phosphorylates STOX1, influencing its ubiquitination and stability,
and modulating trophoblast invasion and proliferation. miRNA,
microRNA; ER, endoplasmic reticulum; GCM1, glial cells missing
transcription factor 1; FBW2, F-box and wd repeat domain containing
2; STK40, serine/threonine kinase 40; COP1, constitutively
photomorphogenic 1; ASB4, ankyrin repeat and SOCS box containing 4;
ID2, inhibitor of DNA binding 2; Trcp1, transient receptor
potential cation channel subfamily C member 1; SATB1, special
AT-rich sequence binding-protein 1; UBE3A, ubiquitin-protein ligase
E3A; THSB1, thrombospondin 1; NEDD4, neural precursor cell
expressed developmentally downregulated 4; TAK1, TGF-β-activated
kinase 1; FUNDC1, FUN14 domain-containing protein 1; GSH,
glutathione; SOD, superoxide dismutase; ROS, reactive oxygen
species; MDA, malondialdehyde; TRIM72, tripartite motif containing
72; FAM124B, family with sequence similarity 124 member B; LC2II,
microtubule-associated protein 1 light chain 3-II; MEK,
mitogen-activated protein kinase kinase; HGF, hepatocyte growth
factor; MET, mesenchymal-epithelial transition factor; Cbl, casitas
B-lineage lymphoma; RNF123, ring finger protein 123; Dvl2,
dishevelled segment polarity protein 2; PCP, planar cell polarity;
CCNG2, cyclin G2; ENaC, epithelial sodium channel; CAV1, caveolin
1; PCBP2, poly (rC)-binding protein 2; MT, mitochondria. |
 | Table I.List of ubiquitinating enzymes, their
substrates and functions associated with PE. |
Table I.
List of ubiquitinating enzymes, their
substrates and functions associated with PE.
| First author/s,
year | Ubiquitinating
enzymes | Substrates | Associated
proteins | Functions | Expression in
PE | (Refs.) |
|---|
| Zhang et al,
2024 | COP1 | P57Kip2 | STK40 | Promotes
ubiquitination and degradation of p57Kip2, affecting trophoblast
cell fusion | Up | (18) |
| Li et al,
2024; Ferguson et al, 2007 | ASB4 | ID2 | VEGF | Mediates ID2
ubiquitination and proteasomal degradation, and affects trophoblast
cell differentiation | Down | (21,24) |
| Ling et al,
2014; Lasorella et al, 2005 | NEDD4 | TAK1 | THBS1 | Promotes TAK1
ubiquitination and degradation, and affects trophoblast
proliferation, migration, invasion, cell cycle and apoptosis | - | (28,29) |
| Murao et al,
2021; Manning and Kumar, 2018; Busst et al, 2013 | NEDD4L | ENaC | - | Mediates ENaC
ubiquitination and degradation, and affects sodium retention | No change | (34–36) |
| Leung et al;
2023; Wang et al, 2019; Rao et al, 2018 | UBE3A | SATB1 | miR-218-5p, | Promotes SATB1
ubiquitination and degradation, and affects trophoblast cell
invasion, migration and endoplasmic reticulum/oxidative stress | Down | (37,39,40) |
| Uehara et
al, 1995 | Cb1 | MET | CAV1, HGF, MEK/ERK,
PI3K/STAT3 | Promotes MET
ubiquitination and degradation, and affects trophoblast
invasion | Down | (45) |
| Uder et al,
2018 | TRIM72 | p53 | - | Promotes p53
ubiquitination and degradation, and affects trophoblast apoptosis,
proliferation and migration | Up | (50) |
| Li et al,
2021 | RNF123 | Dvl2 | CCNG2, Wnt/PCP,
EMT, MMPs | Promotes Dvl2
ubiquitination and degradation, and affects trophoblast
invasion | UP | (52) |
| van Amerongen and
Nuss, 2009; Lerner and Ohlsson, 2015; Orian et al, 2000 | β-TrCP | Snail | E-cadherin, VEGFR,
miR-135-5p | Promotes Snail
ubiquitination and degradation, and affects trophoblast invasion,
EMT and angiogenesis | UP | (56–58) |
| Yu et al,
2002 | FBW2 | GCM1 | PI3K/Ak,
GSK-3β | Promotes GCM1
ubiquitination and degradation, and affects trophoblast EMT | - | (64) |
Identifying their substrates could enable targeted
regulation using Proteolysis-Targeting Chimeras (PROTACs), an
innovative drug development strategy. PROTACs consist of three
components: A ligand that binds the target protein, a linker and a
ligand recruiting an E3 ubiquitin ligase. By bringing the target
protein into proximity with the E3 ligase, PROTACs facilitate
ubiquitination and subsequent proteasomal degradation, enabling
precise protein control (74).
Conventional small molecules typically inhibit protein function by
binding active sites, which is ineffective against undruggable
targets lacking accessible pockets (75). PROTAC technology overcomes this
limitation by inducing degradation independent of active-site
binding, thus expanding the druggable proteome (76). In complex diseases such as PE,
characterized by dysregulated protein networks (77), PROTACs offer a promising approach
for precise therapeutic intervention (78). By selectively targeting aberrant
proteins involved in ubiquitination, PROTACs could provide more
effective treatments (78). This
novel strategy not only addresses current challenges in PE drug
development but also holds considerable potential for advancing
targeted therapies, offering new hope for improved management of
this complex condition.
The role of deubiquitination in PE
Ubiquitin-specific protease (USP)22:
Regulating protein stability
USP22 is a regulator of trophoblast function. The
interaction between USP22 and a disintegrin and metalloproteinase 9
(ADAM9) carries out a key role in modulating trophoblast cell
function during the pathogenesis of PE (79). ADAM9, a metalloproteinase involved
in various physiological processes, is regulated by the
deubiquitinating activity of USP22, which removes ubiquitin
moieties from ADAM9, thereby enhancing its stability and
maintaining its activity (79,80).
Experimental data reveal that USP22 overexpression notably
stabilizes ADAM9 in trophoblast cells. This stabilization was
confirmed through co-immunoprecipitation assays, demonstrating a
direct interaction between USP22 and ADAM9 (79). However, the increased stability of
ADAM9 exerts detrimental effects on trophoblast function. USP22
overexpression suppresses trophoblast proliferation, migration and
invasion while promoting apoptosis (79). Moreover, USP22 inhibits EMT, a key
process required for trophoblast invasiveness (79). Silencing ADAM9 reverses these
effects, indicating that USP22 modulates trophoblast behavior
primarily through ADAM9 stabilization (79). Additionally, the USP22-ADAM9
interaction influences the Wnt/β-catenin signaling pathway, which
governs cellular proliferation, migration and EMT (79). Elevated levels of USP22 and ADAM9
in placental tissues from patients with PE further support their
involvement in the development of PE.
In summary, USP22 contributes to PE pathogenesis by
stabilizing ADAM9 through deubiquitination, thereby inhibiting
trophoblast proliferation, migration, invasion and EMT. This
regulation is partly mediated via the Wnt/β-catenin signaling
pathway. Understanding the USP22-ADAM9 axis offers novel insights
into the molecular mechanisms underlying PE and highlights
potential therapeutic targets for its prevention and treatment.
COP9 signalosome (CSN): Modulating
protein stability
In the context of PE, the CSN complex, particularly
its CSN1 and CSN5 subunits, carry out a key role in regulating
protein stability (81). The CSN
complex modulates the UPS by removing Nedd8 from Cullin-RING
ubiquitin ligases, a process primarily mediated by the
deubiquitinating activity of the Jab1/CSN5 subunit (82,83).
In PE placentas, the expression of CSN1 and CSN5 is markedly
upregulated and predominantly localized in vascular endothelial
cells, syncytiotrophoblasts, stromal cells and Hofbauer cells
(84). CSN1 contributes to the
regulation of transcription factor stability and influences lipid
metabolism and insulin-mediated gluconeogenesis. Meanwhile,
Jab1/CSN5 is directly involved in protein degradation through its
DUB activity (85–89). The elevated levels of CSN1 and CSN5
observed in PE placentas are associated with key pathological
changes characteristic of the disorder (84). Notably, the accumulation of
hypoxia-inducible factors (HIFs), HIF-1α and HIF-2α in PE placentas
may result from impaired proteasome activity. Jab1/CSN5 interacts
with HIF-1α and promotes its stabilization, offering a potential
explanation for this phenomenon (84). This aberrant stabilization may
impair normal trophoblast function and contribute to defective
placental development. These findings provide new insights into the
molecular mechanisms of PE and may contribute to the development of
new therapeutic strategies. Additionally, understanding the
regulatory role of the CSN complex may offer potential targets for
the treatment of PE, helping to improve placental function and
prevent the onset of the disease.
USP14: Linking inflammation and
hormone regulation
Upregulation of USP14 is associated with the
exacerbation of inflammatory responses in PE. USP14 promotes the
activation of the NF-κB signaling pathway through its
deubiquitinating activity, leading to the increased production of
proinflammatory cytokines such as TNF-α, IL-6 and IL-1β (89). This inflammatory effect has been
reported in trophoblast cell models exposed to
hypoxia/reoxygenation, mimicking PE conditions. Increased USP14
expression in these models associates with NF-κB activation and
higher levels of inflammatory cytokines. Notably, treatment with
the USP14 inhibitor IU1 inhibits the hypoxia/reoxygenation-induced
activation of NF-κB and MAPK, effectively reducing proinflammatory
cytokine production (89). These
findings suggest that USP14 carries out a key role in driving
inflammation in PE, and its inhibition may represent a promising
therapeutic strategy to mitigate inflammatory responses in
trophoblast cells, potentially improving outcomes in PE.
In addition to its role in inflammation, USP14 also
participates in hormone regulation by modulating aromatase
expression in placental trophoblast cells. This regulation involves
the transcription factor HAND1, whose stability is controlled by
the regulator of G protein signaling 2 (RGS2). RGS2 promotes the
ubiquitin-mediated degradation of HAND1, whereas USP14 counteracts
this process by removing ubiquitin moieties, thereby stabilizing
HAND1 (90). Stabilized HAND1
enhances repression of the aromatase gene, ultimately reducing
aromatase expression. This dual functionality of USP14 (in both
promoting inflammation and regulating hormone synthesis) highlights
its central role in the pathogenesis of PE.
In placental tissues of patients with PE, the
expression of RGS2 and aromatase is reduced, while HAND1 expression
is increased (90,91). These findings suggest that
dysregulation of the RGS2-HAND1 pathway may impair aromatase
expression and consequently disrupt estrogen synthesis, a process
essential for pregnancy maintenance. Clinical analyses reveal a
positive correlation between RGS2 and aromatase expression in both
normal and PE placental tissues, underscoring the importance of the
RGS2-aromatase axis in estrogen regulation and its potential
utility as a biomarker for PE and associated obstetric conditions.
In addition, the human-specific lncRNA UCA1 has been identified as
a key regulator in the pathogenesis of PE through its interaction
with profilin 1 (PFN1) in placental tissues and maternal serum. RNA
pulldown, mass spectrometry and RNA immunoprecipitation assays have
confirmed a direct interaction between UCA1 and PFN1. Functioning
as a molecular scaffold, UCA1 recruits the DUB USP14 to form a
UCA1-USP14-PFN1 complex (92).
This complex modulates the ubiquitination and stability of PFN1
(92). Protein half-life assays
demonstrate that UCA1 overexpression extends the half-life of PFN1
by reducing its ubiquitination and preventing proteasomal
degradation. Conversely, treatment with the USP14 inhibitor b-AP15
increases PFN1 ubiquitination and promotes its degradation,
confirming the role of USP14 in stabilizing PFN1 (92). Importantly, in the context of PE,
UCA1-mediated stabilization of PFN1 activates the RhoA/ROCK
signaling pathway, resulting in excessive production of ROS in
vascular endothelial cells. This oxidative stress contributes to
endothelial injury, a hallmark of PE, and is associated with
maternal vascular dysfunction and hypertension (92).
In summary, USP14 carries out a multifaceted role in
the pathogenesis of PE through three key mechanisms: First,
promoting inflammatory responses via NF-κB signaling; second,
modulating aromatase expression and estrogen synthesis by
regulating the RGS2-HAND1 axis; and third, stabilizing PFN1 to
activate the RhoA/ROCK pathway and induce oxidative stress in
vascular endothelial cells. These findings highlight USP14 as a
central mediator in PE and a potential therapeutic target for its
prevention and treatment.
USP8: Restoring trophoblast
function
USP8 has been identified as a key regulator of
trophoblast cell function in the context of PE. It enhances the
stability and membrane localization of ENaC by removing their
ubiquitin tags, thereby preventing proteasomal degradation. This
stabilization promotes trophoblast proliferation, migration and
invasion, which are key processes for normal placental development
(93). In PE placentas, USP8
expression is considerably reduced, correlating with impaired
trophoblast function. The downregulation of USP8 disrupts ENaC
stability and surface expression, contributing to the functional
defects observed in trophoblast cells during PE (93). Additionally, microRNA miR-874-3p
negatively regulates USP8 by binding to its 3′ untranslated region,
further suppressing its expression. This inhibition reduces ENaC
levels on the trophoblast membrane and exacerbates dysfunction
(93). However, overexpression of
USP8 can counteract the effects of miR-874-3p, restoring ENaC
expression and rescuing trophoblast proliferation, migration and
invasion (93). These findings
highlight the key role of the USP8/ENaC axis and its regulation by
miR-874-3p in PE pathogenesis. USP8 represents a promising
therapeutic target, with potential interventions aimed at enhancing
its activity or expression to restore normal trophoblast function
and improve placental development. Targeting this pathway may offer
innovative strategies for the prevention and treatment of PE.
USP17: Modulating inflammatory
pathways
USP17, a DUB, has emerged as a potential regulator
in the pathogenesis of PE due to its markedly reduced expression in
the placental tissues of patients with PE (16). USP17 maintains protein homeostasis
by removing ubiquitin tags, thereby stabilizing key regulatory
proteins. One such target is histone deacetylase 2 (HDAC2), whose
stability is enhanced by USP17-mediated deubiquitination. This
stabilization promotes trophoblast proliferation, migration and
invasion, which are processes essential for normal placental
development (16,94). HDAC2 also interacts with STAT1, and
through deacetylation, enhances STAT1 activity (16). Activated STAT1, in turn, suppresses
the NF-κB-signaling pathway, a key mediator of inflammatory
responses that is associated with the development of PE (16). Thus, USP17 may influence PE
progression through the HDAC2/STAT1/NF-κB signaling axis,
associating protein stability to inflammatory regulation. These
findings underscore the therapeutic potential of USP17 in
modulating inflammation and improving trophoblast function in
PE.
DUBs are integral to the pathogenesis of PE, a
pregnancy disorder characterized by impaired trophoblast function.
USP22 stabilizes ADAM9, influencing trophoblast behavior via the
Wnt/β-catenin signaling pathway. The COP9 signalosome modulates
protein stability through the UPS, potentially affecting
trophoblast activity. USP14 promotes NF-κB-mediated inflammation,
regulates the RGS2-HAND1-aromatase axis affecting estrogen
synthesis and stabilizes PFN1 to preserve vascular integrity. USP8
stabilizes ENaC, enhancing trophoblast function and is negatively
regulated by miR-874-3p. Lastly, USP17 stabilizes HDAC2, thereby
influencing STAT1 and suppressing NF-κB activation, the key to
controlling inflammation in PE (Fig.
3 and Table II). Despite
these promising targets, developing therapies that modulate protein
regulation remains challenging. Currently, >85% of proteins are
considered ‘undruggable’, as traditional small-molecule drugs often
fail to bind and modulate them effectively (95). Targeted protein degradation
technologies offer a potential solution, yet diseases such as PE,
driven by dysregulated ubiquitination, require even more precise
strategies. DUBs carry out key roles in the pathogenesis of PE by
regulating trophoblast function, inflammatory responses and hormone
signaling pathways. Dysregulation of specific DUBs can lead to
trophoblast dysfunction, compromised placental development and the
onset of clinical manifestations associated with PE. Traditional
drug development strategies often lack the specificity required to
modulate DUB activity effectively. Broad-spectrum inhibitors may
induce off-target effects, disrupting essential molecular pathways
and complicating therapeutic outcomes. In this context,
deubiquitinase-targeting chimeras (DUBTACs) represent a novel and
promising therapeutic approach for PE. DUBTACs are
heterobifunctional molecules designed to recruit DUBs to specific
target proteins. Each molecule comprises a ligand that binds to the
target protein, a chemical linker and a DUB-recruiting moiety. By
facilitating this proximity, DUBTACs promote the removal of
polyubiquitin chains from the target protein, thereby preventing
its proteasomal degradation and enhancing its stability and
function (96). Therefore, DUBTAC
technology represents a promising avenue for developing targeted
therapies to address the complex molecular mechanisms underlying
PE, offering a new avenue for effective disease management.
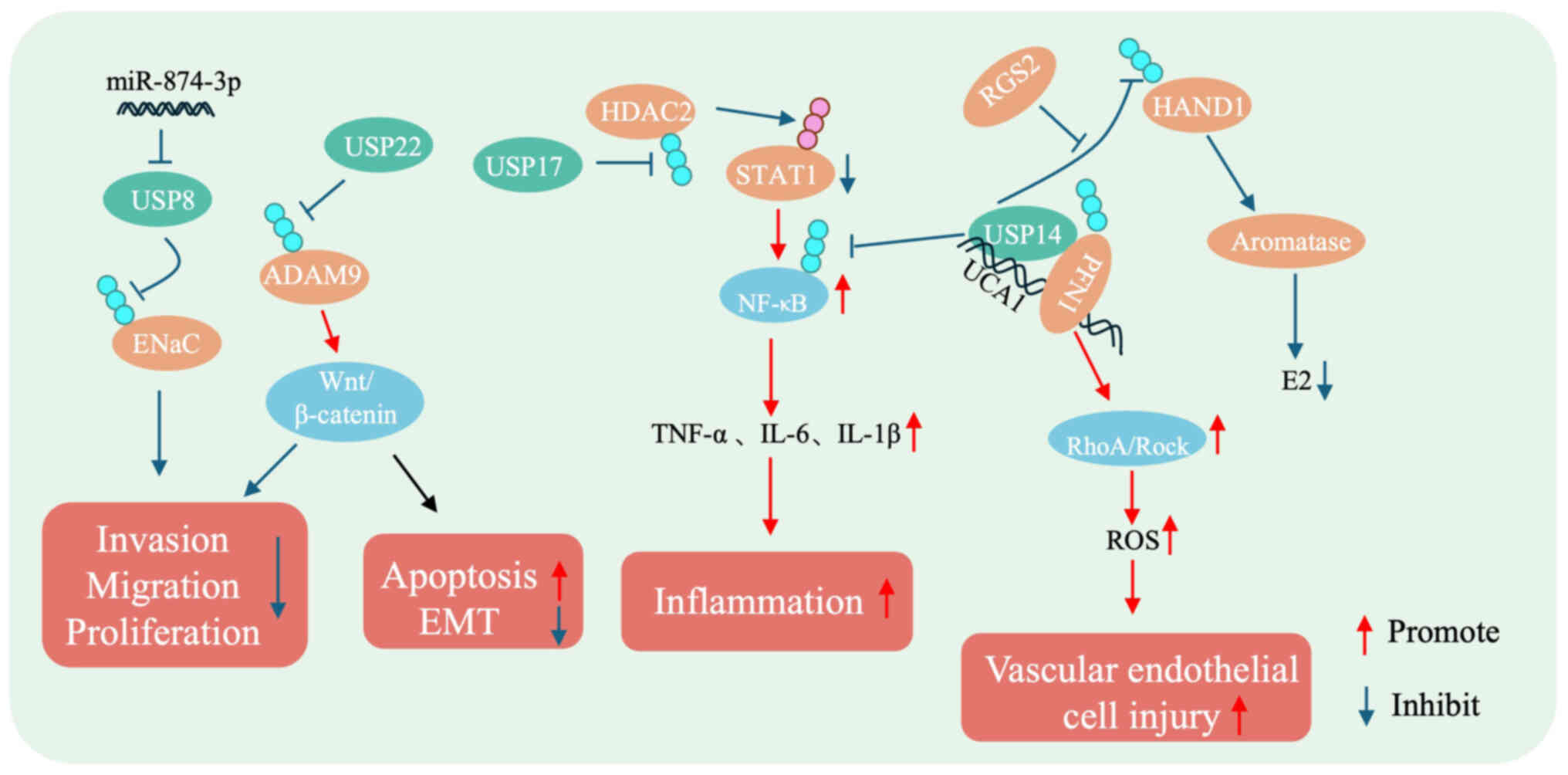 | Figure 3.Mechanistic diagram of DUBs in
preeclampsia. USP22 stabilizes ADAM9, inhibiting cell
proliferation, migration, invasion and EMT through the
Wnt/β-catenin signaling pathway. USP14 stabilizes NF-κB and HAND1,
promoting proinflammatory factors and affecting estrogen synthesis.
USP14 also stabilizes PFN1, contributing to vascular endothelial
injury via the Rho/ROCK-signaling pathway. USP8 stabilizes ENaC,
inhibiting cell invasion, migration and proliferation. USP17
stabilizes HDAC2, activating STAT1 and enhancing the secretion of
proinflammatory factors. USP, ubiquitin-specific protease; ENaC,
epithelial sodium channel; EMT, epithelial-mesenchymal transition;
ROS, reactive oxygen species; HAND1, heart and neural crest
derivatives expressed 1; PFN1, penetration 1; RGS2, regulator of
G-protein signaling 2. |
| Table II.List of deubiquitinating enzymes,
their substrates and functions associated with PE. |
Table II.
List of deubiquitinating enzymes,
their substrates and functions associated with PE.
| First author/s,
year | Deubiquitinating
enzymes | Substrates | Associated
proteins | Functions | Expression in
PE | (Refs.) |
|---|
| van Dijk et
al, 2010 | USP22 | ADAM9 | Wnt/β-catenin | Affects trophoblast
cell invasion, migration, proliferation, |
|
|
|
|
|
|
| apoptosis and
EMT | Up | (71) |
| Wei and Deng,
2003 | COP9 | HIF-1α | - | Affects trophoblast
functions | Up | (81) |
| Dentin et
al, 2007; | USP14 | NF-κB, | TNF-α, IL-6, | Affects
inflammatory responses, vascular endothelial | Up | (87–90) |
| Kato and
Yoneda-Kato, |
| HAND1, | IL-1β, RGS2, | cell injury and E2
secretion |
|
|
| 2009; Zhao et
al, 2021; |
| PFN1 | aromatase, |
|
|
|
| Tang et al,
2023 |
|
| RhoA/Rock, |
|
|
|
|
|
|
| ROS |
|
|
|
| Perschbacher et
al, 2020 | USP8 | ENaC | - | Affects trophoblast
proliferation, migration and invasion | Down | (91) |
| Wu et al,
2022; | USP17 | HDAC2 | STAT1, NF-κB | Affects trophoblast
proliferation, migration, invasion and | Down | (92,93) |
| Zhang et al,
2023 |
|
|
| inflammatory
responses |
|
|
Summary and future perspectives
The present review highlights the key roles of E3
ubiquitin ligases and DUBs in the pathogenesis of PE, emphasizing
their regulatory functions in trophoblast behavior, inflammatory
responses, protein stability and hormonal signaling.
Ubiquitination, mediated by E3 ligases such as STK40 (21), ASB4 (27) and NEDD4 (4), influences key processes including
trophoblast fusion, differentiation, invasion and migration, which
are essential functions for proper placental development and are
implicated in the onset of PE (47). Conversely, deubiquitination,
carried out by DUBs such as USP22 (79), the COP9 signalosome (84) and USP14 (89), also carry out a pivotal role by
maintaining protein homeostasis and modulating inflammation
(89) and vascular integrity
(84). As research into
ubiquitination and deubiquitination mechanisms in PE advances,
future directions are increasingly centered on translational
applications. Emerging therapeutic technologies, including siRNA
(97), PROTAC, DUBTAC (98) and therapeutic antibodies (99), offer promising strategies for
targeting dysregulated E3 ligases and DUBs. siRNA technology
enables selective silencing of mRNA transcripts encoding specific
E3 ligases or DUBs, allowing for precise modulation of pathogenic
pathways in PE (100). Compared
with conventional small-molecule drugs, siRNA therapy offers higher
specificity and reduced off-target effects. PROTAC technology
represents an innovative approach for targeting previously
‘undruggable’ proteins by co-opting E3 ligases to tag
disease-related proteins for proteasomal degradation (76). Tailoring PROTACs to target proteins
implicated in PE could enable highly specific regulation of their
abundance and activity (101). As
a revolutionary approach, DUBTACs function by recruiting DUBs to
specific target proteins, stabilizing them through the removal of
polyubiquitin chains and preventing their degradation. Given the
central roles of DUBs in trophoblast function, immune regulation
and hormone signaling (79),
DUBTACs present a promising avenue for therapeutic intervention in
PE (102). By specifically
binding to E3 ligases or DUBs, therapeutic antibodies can block
their interactions with other proteins, offering an alternative
strategy for targeted PE therapy.
Future research should also focus on elucidating the
intricate interactions between E3 ubiquitin ligases, DUBs and other
signaling pathways involved in the pathogenesis of PE. A more
comprehensive understanding of these complex regulatory networks is
essential for the development of effective and multifaceted
therapeutic strategies. In addition, exploring the role of
epigenetic modifications in regulating the expression and activity
of E3 ligases and DUBs may uncover novel mechanisms underlying PE
and identify new therapeutic targets. Furthermore, preclinical and
clinical studies are required to assess the safety, efficacy and
translational potential of these emerging therapeutic approaches.
Large-scale clinical trials will be key for determining optimal
dosing regimens, treatment durations and potential adverse effects.
Parallel efforts should also aim to identify and validate
predictive biomarkers to assess patient responses to these
interventions, thereby advancing the application of personalized
medicine in PE management.
In conclusion, leveraging these innovative
therapeutic strategies offers potential to improve clinical
outcomes for both mothers and infants affected by PE. Continued
research into the molecular underpinnings of PE, combined with the
development and clinical validation of targeted therapies, will be
pivotal in addressing the challenges posed by this complex and
multifactorial pregnancy disorder.
Acknowledgements
Not applicable.
Funding
This research was funded by the National Natural Science
Foundation of China (grant no. 82460310) and the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Education (grant no.
RS-2019-NR40073).
Availability of data and materials
Not applicable.
Authors' contributions
CZP collected literature and drafted the manuscript.
XXS helped to draft and modify the manuscript. HX conceived the
topic of the present review and revised the manuscript. KHB offered
guidance and critically reviewed this manuscript. All authors read
and approved the final version of the manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Takahashi M, Makino S, Oguma K, Imai H,
Takamizu A, Koizumi A and Yoshida K: Fetal growth restriction as
the initial finding of preeclampsia is a clinical predictor of
maternal and neonatal prognoses: A single-center retrospective
study. BMC Pregnancy Childbirth. 21:6782021. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Al Sherawi NN, Singhal V and Sarma U:
Chapter 7-Preeclampsia and HELLP syndrome. The kidney of the
critically Ill pregnant woman. Montufar-rueda C, Hidalgo J and
Perez-Fernandez J: Academic Press; pp. 73–83. 2025, View Article : Google Scholar
|
|
3
|
Ali M, Ahmed M, Memon M, Chandio F, Shaikh
Q, Parveen A and Phull AR: Preeclampsia: A comprehensive review.
Clin Chim Acta. 563:1199222024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hu H, Ma J, Peng Y, Feng R, Luo C, Zhang
M, Tao Z, Chen L, Zhang T, Chen W, et al: Thrombospondin-1
regulates trophoblast necroptosis via NEDD4-mediated ubiquitination
of TAK1 in preeclampsia. Adv Sci (Weinh). 11:e23090022024.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Khan B, Allah Yar R, Khakwani AK, Karim S
and Arslan Ali H: Preeclampsia incidence and its maternal and
neonatal outcomes with associated risk factors. Cureus.
14:e311432022.PubMed/NCBI
|
|
6
|
Chakravarty EF and Sammaritano LR:
40-Pregnancy and reproductive health issues in systemic lupus
erythematosus. Dubois' lupus erythematosus and related syndromes.
(tenth edition). Wallace DJ, Hahn BH, Askanase A, et al: Elsevier;
New Delhi, India: pp. 557–579. 2025, View Article : Google Scholar
|
|
7
|
Serrano NC: Immunology and genetic of
preeclampsia. Clin Dev Immunol. 13:197–201. 2006.PubMed/NCBI
|
|
8
|
Stephens J, Grande ED, Roberts T, Kerr M,
Northcott C, Johnson T, Sleep J and Ryder C: Factors associated
with preeclampsia and the hypertensive disorders of pregnancy
amongst Indigenous women of Canada, Australia, New Zealand, and the
United States: A systematic review and meta-analysis. Curr
Hypertens Rep. 27:102025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Giannubilo SR, Marzioni D, Tossetta G,
Montironi R, Meccariello ML and Ciavattini A: The ‘Bad Father’:
Paternal role in biology of pregnancy and in birth outcome. Biology
(Basel). 13:1652024.PubMed/NCBI
|
|
10
|
Zhu Y, Liu X, Xu Y and Lin Y:
Hyperglycemia disturbs trophoblast functions and subsequently leads
to failure of uterine spiral artery remodeling. Front Endocrinol
(Lausanne). 14:10602532023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Annesi L, Tossetta G, Borghi C and Piani
F: The role of xanthine oxidase in pregnancy complications: A
systematic review. Antioxidants (Basel). 13:12342024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tossetta G, Fantone S, Giannubilo SR,
Ciavattini A, Senzacqua M, Frontini A and Marzioni D: HTRA1 in
placental cell models: A possible role in preeclampsia. Curr Issues
Mol Biol. 45:3815–3828. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vilotić A, Nacka-Aleksić M, Pirković A,
Bojić-Trbojević Ž, Dekanski D and Jovanović Krivokuća M: IL-6 and
IL-8: An overview of their roles in healthy and pathological
pregnancies. Int J Mol Sci. 23:145742022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Wang H, Yang M, Liang Y, Jiang L,
Sun S and Fan S: Downregulation of cAMP-dependent protein kinase
inhibitor-b promotes preeclampsia by decreasing phosphorylated Akt.
Reprod Sci. 28:178–185. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kamrani A, Alipourfard I, Ahmadi-Khiavi H,
Yousefi M, Rostamzadeh D, Izadi M and Ahmadi M: The role of
epigenetic changes in preeclampsia. Biofactors. 45:712–724. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li A, Wang T, Zhou S, Han J and Wu W:
USP17 regulates preeclampsia by modulating the NF-κB signaling
pathway via deubiquitinating HDAC2. Placenta. 145:9–18. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park HB, Hwang S and Baek KH: USP7
regulates the ERK1/2 signaling pathway through deubiquitinating
Raf-1 in lung adenocarcinoma. Cell Death Dis. 13:6982022.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang HR, Wang YH, Xiao ZP, Yang G, Xu YR,
Huang ZT, Wang WZ and He F: E3 ubiquitin ligases: Key regulators of
osteogenesis and potential therapeutic targets for bone disorders.
Front Cell Dev Biol. 12:14470932024. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Song L and Luo ZQ: Post-translational
regulation of ubiquitin signaling. J Cell Biol. 218:1776–1786.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang X, Yan K, Zhan Q, Chen H, Pei CZ and
Zhu L: Exploration of diagnostic deubiquitinating enzymes in
endometriosis and its immune infiltration. Biochem Genet.
62:4359–4379. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li X, Shao LZ, Li ZH, Wang YH, Cai QY,
Wang S, Chen H, Sheng J, Luo X, Chen XM, et al: STK40 inhibits
trophoblast fusion by mediating COP1 ubiquitination to degrade
P57Kip2. J Transl Med. 22:8522024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Song HL, Liu TH, Wang YH, Li FF, Ruan LL,
Adu-Gyamfi EA, Hu SC, Chen XM, Ding YB and Fu LJ: Appropriate
expression of P57kip2 drives trophoblast fusion via cell cycle
arrest. Reproduction. 161:633–644. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Choi HH, Guma S, Fang L, Phan L, Ivan C,
Baggerly K, Sood A and Lee MH: Regulating the stability and
localization of CDK inhibitor p27(Kip1) via CSN6-COP1 axis. Cell
Cycle. 14:2265–2273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ferguson JE III, Wu Y, Smith K, Charles P,
Powers K, Wang H and Patterson C: ASB4 is a hydroxylation substrate
of FIH and promotes vascular differentiation via an
oxygen-dependent mechanism. Mol Cell Biol. 27:6407–6419. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Townley-Tilson WHD, Wu Y, Ferguson JE III
and Patterson C: The ubiquitin ligase ASB4 promotes trophoblast
differentiation through the degradation of ID2. PLoS One.
9:e894512014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li F, Fushima T, Oyanagi G, Townley-Tilson
HW, Sato E, Nakada H, Oe Y, Hagaman JR, Wilder J, Li M, et al:
Nicotinamide benefits both mothers and pups in two contrasting
mouse models of preeclampsia. Proc Natl Acad Sci USA.
113:13450–13455. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kayashima Y, Townley-Tilson WHD, Vora NL,
Boggess K, Homeister JW, Maeda-Smithies N and Li F: Insulin
elevates ID2 expression in trophoblasts and aggravates preeclampsia
in obese ASB4-null mice. Int J Mol Sci. 24:21492023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ling F, Kang B and Sun XH: Id proteins:
Small molecules, mighty regulators. Curr Top Dev Biol. 110:189–216.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lasorella A, Rothschild G, Yokota Y,
Russell RG and iavarone A: Id2 mediates tumor initiation,
proliferation, and angiogenesis in Rb mutant mice. Mol Cell Biol.
25:3563–3574. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsunedomi R, Iizuka N, Tamesa T, Sakamoto
K, Hamaguchi T, Somura H, Yamada M and Oka M: Decreased ID2
promotes metastatic potentials of hepatocellular carcinoma by
altering secretion of vascular endothelial growth factor. Clin
Cancer Res. 14:1025–1031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu Y, Sun Y, Han S, Guo Y, Tian Q, Ma Q
and Zhang S: CHIP promotes the activation of NF-κB signaling
through enhancing the K63-linked ubiquitination of TAK1. Cell Death
Discov. 7:2462021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ulu İ, Çekmez Y, Yıldırım Köpük Ş, Özer N,
Yoğurtçuoğlu EE, Anğın P and Kıran G: Maternal serum
thrombospondin-1 is significantly altered in cases with established
preeclampsia. J Matern Fetal Neonatal Med. 32:2543–2546. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y, Chen Y, Ding C, Zhu X, Song X, Ren
Y, Qang Q, Zhang Y and Sun X: TRIM56 positively regulates
TNFα-induced NF-κB signaling by enhancing the ubiquitination of
TAK1. Int J Biol Macromol. 219:571–578. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Murao A, Aziz M, Wang H, Brenner M and
Wang P: Release mechanisms of major DAMPs. Apoptosis. 26:152–162.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Manning JA and Kumar S: Physiological
functions of Nedd4-2: Lessons from knockout mouse models. Trends
Biochem Sci. 43:635–647. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Busst CJ: Blood pressure regulation via
the epithelial sodium channel: From gene to kidney and beyond. Clin
Exp Pharmacol Physiol. 40:495–503. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leung PYM, Katerelos M, Choy S, Cook N,
Lee M, Paizis K, Abboud A, Manning JA, Mount PF and Power DA:
Expression of NEDD4L and ENaC in urinary extracellular vesicles in
pre-eclampsia. Hypertens Pregnancy. 42:22320292023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Gu X, Sun X, Yu Y and Li L: MiR-218-5p
promotes trophoblast infiltration and inhibits endoplasmic
reticulum/oxidative stress by reducing UBE3A-mediated degradation
of SATB1. J Cell Commun Signal. 17:993–1008. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang J, Lou SS, Wang T, Wu RJ, Li G, Zhao
M, Lu B, Li YY, Zhang J, Cheng X, et al: UBE3A-mediated PTPA
ubiquitination and degradation regulate PP2A activity and dendritic
spine morphology. Proc Natl Acad Sci USA. 116:12500–12505. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Rao H, Bai Y, Li Q, Zhuang B, Yuan Y, Liu
Y, Peng W, Baker PN, Tong C, Luo X and Qi H: SATB1 downregulation
induced by oxidative stress participates in trophoblast invasion by
regulating β-catenin. Biol Reprod. 98:810–820. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Rao H, Bai Y, Zhang F, Li Q, Zhuang B, Luo
X and Qi H: The role of SATB1 in HTR8/SVneo cells and pathological
mechanism of preeclampsia. J Matern Fetal Neonatal Med.
32:2069–2078. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Cele SB, Odun-Ayo F, Onyangunga OA,
Moodley J and Naicker T: Analysis of hepatocyte growth factor
immunostaining in the placenta of HIV-infected normotensive versus
preeclamptic pregnant women. Eur J Obstet Gynecol Reprod Biol.
227:60–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lv B, Wang G, Pan Y, Yuan G and Wei L:
Construction and evaluation of machine learning-based predictive
models for early-onset preeclampsia. Pregnancy Hypertens.
39:1011982025. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Birchmeier C, Birchmeier W, Gherardi E and
Vande Woude GF: Met, metastasis, motility and more. Nat Rev Mol
Cell Biol. 4:915–925. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Uehara Y, Minowa O, Mori C, Shiota K, Kuno
J, Noda T and Kitamura N: Placental defect and embryonic lethality
in mice lacking hepatocyte growth factor/scatter factor. Nature.
373:702–705. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Genbacev O, Joslin R, Damsky CH, Polliotti
BM and Fisher SJ: Hypoxia alters early gestation human
cytotrophoblast differentiation/invasion in vitro and models the
placental defects that occur in preeclampsia. J Clin Invest.
97:540–550. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li G, Wang Y, Cao G, Ma Y, Li YX, Zhao Y,
Shao X and Wang YL: Hypoxic stress disrupts HGF/Met signaling in
human trophoblasts: Implications for the pathogenesis of
preeclampsia. J Biomed Sci. 29:82022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Park EC, Ghose P, Shao Z, Ye Q, Kang L, Xu
XZ, Powell-Coffman JA and Rongo C: Hypoxia regulates glutamate
receptor trafficking through an HIF-independent mechanism. EMBO J.
31:1379–1393. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gómez-Gutiérrez AM, Parra-Sosa BE and
Bueno-Sánchez JC: Glycosylation profile of the transferrin receptor
in gestational iron deficiency and early-onset severe preeclampsia.
J Pregnancy. 2019:95145462019. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Uder C, Brückner S, Winkler S, Tautenhahn
HM and Christ B: Mammalian MSC from selected species: Features and
applications. Cytometry A. 93:32–49. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhaoer Y, Mingming G, Wei Z, Dan Y, Yating
Q and Ruizhe J: Extracellular vesicles for the treatment of
preeclampsia. Tissue Cell. 77:1018602022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li Y, Wang C, Xi HM, Li WT, Liu YJ, Feng
S, Chu YJ and Wang YH: Chorionic villus-derived mesenchymal stem
cells induce E3 ligase TRIM72 expression and regulate cell
behaviors through ubiquitination of p53 in trophoblasts. FASEB J.
35:e220052021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Moindjie H, Santos ED, Gouesse RJ,
Swierkowski-Blanchard N, Serazin V, Barnea ER, Vialard F and
Dieudonné MN: Preimplantation factor is an anti-apoptotic effector
in human trophoblasts involving p53 signaling pathway. Cell Death
Dis. 7:e25042016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Sun M, Gao J, Meng T, Liu S, Chen H, Liu
Q, Xing X, Zhao C and Luo Y: Cyclin G2 upregulation impairs
migration, invasion, and network formation through RNF123/Dvl2/JNK
signaling in the trophoblast cell line HTR8/SVneo, a possible role
in preeclampsia. FASEB J. 35:e211692021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Gordon MD and Nusse R: Wnt signaling:
Multiple pathways, multiple receptors, and multiple transcription
factors. J Biol Chem. 281:22429–22433. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
van Amerongen R and Nusse R: Towards an
integrated view of Wnt signaling in development. Development.
136:3205–3214. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Lerner UH and Ohlsson C: The WNT system:
Background and its role in bone. J Intern Med. 277:630–649. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Orian A, Gonen H, Bercovich B, Fajerman I,
Eytan E, Israël A, Mercurio F, Iwai K, Schwartz AL and Ciechanover
A: SCF(beta)(−TrCP) ubiquitin ligase-mediated processing of
NF-kappaB p105 requires phosphorylation of its C-terminus by
IkappaB kinase. EMBO J. 19:2580–2591. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Vinas-Castells R, Beltran M, Valls G,
Gómez I, García JM, Montserrat-Sentís B, Baulida J, Bonilla F, de
Herreros AG and Díaz VM: The hypoxia-controlled FBXL14 ubiquitin
ligase targets SNAIL1 for proteasome degradation. J Biol Chem.
285:3794–3805. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu D, Shi L, Chen X, Cen H and Mao D:
β-TrCP suppresses the migration and invasion of trophoblast cells
in preeclampsia by down-regulating Snail. Exp Cell Res.
395:1122302020. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xu Y, Lee SH, Kim HS, Kim NH, Piao S, Park
SH, Jung YS, Yook JI, Park BJ and Ha NC: Role of CK1 in
GSK3beta-mediated phosphorylation and degradation of snail.
Oncogene. 29:3124–3133. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wu D, Shi L, Hong L, Chen X and Cen H:
MiR-135a-5p promotes the migration and invasion of trophoblast
cells in preeclampsia by targeting β-TrCP. Placenta. 99:63–69.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Anson-Cartwright L, Dawson K, Holmyard D,
Fisher SJ, Lazzarini RA and Cross JC: The glial cells missing-1
protein is essential for branching morphogenesis in the
chorioallantoic placenta. Nat Genet. 25:311–314. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Yu C, Shen K, Lin M, Chen P, Lin C, Chang
GD and Chen H: GCMa regulates the syncytin-mediated trophoblastic
fusion. J Biol Chem. 277:50062–50068. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chang M, Mukherjea D, Gobble RM, Groesch
KA, Torry RJ and Torry DS: Glial cell missing 1 regulates placental
growth factor (PGF) gene transcription in human trophoblast. Biol
Reprod. 78:841–851. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Chiang MH, Liang FY, Chen CP, Chang CW,
Cheong ML, Wang LJ, Liang CY, Lin FY, Chou CC and Chen H: Mechanism
of hypoxia-induced GCM1 degradation: Implications for the
pathogenesis of preeclampsia. J Biol Chem. 284:17411–17419. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wang B, Xu W, Cai Y, Chen J, Guo C, Zhou G
and Yuan C: DUXAP8: A promising lncRNA with carcinogenic potential
in cancer. Curr Med Chem. 29:1677–1686. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Wei XH, Liao LY, Yin YX, Xu Q, Xie SS, Liu
M, Gao LB, Chen HQ and Zhou R: Overexpression of long noncoding RNA
DUXAP8 inhibits ER-phagy through activating AKT/mTOR signaling and
contributes to preeclampsia. Cell Mol Life Sci. 81:3362024.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lim Y, Rubio-Peña K, Sobraske PJ, Molina
PA, Brookes PS, Galy V and Nehrke K: Fndc-1 contributes to paternal
mitochondria elimination in C. elegans. Dev Biol. 454:15–20. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Chen G, Chen L, Huang Y, Zhu X and Yu Y:
Increased FUN14 domain containing 1 (FUNDC1) ubiquitination level
inhibits mitophagy and alleviates the injury in hypoxia-induced
trophoblast cells. Bioengineered. 13:3620–3633. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
van Dijk M, Mulders J, Poutsma A, Könst
AA, Lachmeijer AM, Dekker GA, Blankenstein MA and Oudejans CB:
Maternal segregation of the Dutch preeclampsia locus at 10q22 with
a new member of the winged helix gene family. Nat Genet.
37:514–519. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
72
|
Zhao X, Gan L, Pan H, Kan D, Majeski M,
Adam SA and Unterman TG: Multiple elements regulate
nuclear/cytoplasmic shuttling of FOXO1: Characterization of
phosphorylation- and 14-3-3-dependent and -independent mechanisms.
Biochem J. 378:839–849. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
van Dijk M, van Bezu J, van Abel D, Dunk
C, Blankenstein MA, Oudejans CB and Lye SJ: The STOX1 genotype
associated with pre-eclampsia leads to a reduction of trophoblast
invasion by alpha-T-catenin upregulation. Hum Mol Genet.
19:2658–2667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Hualong M, Liu J, Yin T, Cao X, Su Z, Zhao
DG and Ma YY: Discovery of a selective and orally bioavailable RET
degrader with effectiveness in various mutations. J Med Chem.
68:2657–2679. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Raina K and Crews CM: Targeted protein
knockdown using small molecule degraders. Curr Opin Chem Biol.
39:46–53. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Jin Y and Lee Y: Proteolysis targeting
chimeras (PROTACs) in breast cancer therapy. ChemMedChem.
19:2024002672024. View Article : Google Scholar
|
|
77
|
Wang T, Shi XZ and Wu WH: Crosstalk
analysis of dysregulated pathways in preeclampsia. Exp Ther Med.
17:2298–2304. 2019.PubMed/NCBI
|
|
78
|
Liang X, Ren H, Han F, Liang R, Zhao J and
Liu H: The new direction of drug development: Degradation of
undruggable targets through targeting chimera technology. Med Res
Rev. 44:632–685. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Liu J, Wang Y, Zhang S, Sun L and Shi Y:
ADAM9 deubiquitination induced by USP22 suppresses proliferation,
migration, invasion, and epithelial-mesenchymal transition of
trophoblast cells in preeclampsia. Placenta. 146:50–57. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chou CW, Huang YK, Kuo TT, Liu JP and Sher
YP: An overview of ADAM9: structure, activation, and regulation in
human diseases. Int J Mol Sci. 21:77902020. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Wei N and Deng XW: The COP9 signalosome.
Annu Rev Cell Dev Biol. 19:261–286. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wei N, Serino G and Deng XW: The COP9
signalosome: More than a protease. Trends Biochem Sci. 33:592–600.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Cope GA, Suh GS, Aravind L, Schwarz SE,
Zipursky SL, Koonin EV and Deshaies RJ: Role of predicted
metalloprotease motif of Jab1/Csn5 in cleavage of Nedd8 from Cul1.
Science. 298:608–611. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cayli S, Demirturk F, Ocakli S, Aytan H,
Caliskan AC and Cimsir H: Altered expression of COP9 signalosome
proteins in preeclampsia. Gynecol Endocrinol. 28:488–491. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Bianchi E, Denti S, Catena R, Rossetti G,
Polo S, Gasparian S, Putignano S, Rogge L and Pardi R:
Characterization of human constitutive photomorphogenesis protein
1, a RING finger ubiquitin ligase that interacts with Jun
transcription factors and modulates their transcriptional activity.
J Biol Chem. 278:19682–19690. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Qi L, Heredia JE, Altarejos JY, Screaton
R, Goebel N, Niessen S, Macleod IX, Liew CW, Kulkarni RN, Bain J,
et al: TRB3 links the E3 ubiquitin ligase COP1 to lipid metabolism.
Science. 312:1763–1766. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Dentin R, Liu Y, Koo SH, Hedrick S, Vargas
T, Heredia J, Yates J III and Montminy M: Insulin modulates
gluconeogenesis by inhibition of the coactivator TORC2. Nature.
449:366–369. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Kato JY and Yoneda-Kato N: Mammalian COP9
signalosome. Genes Cells. 14:1209–1225. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhao Y and Zong F: Inhibiting USP14
ameliorates inflammatory responses in trophoblast cells by
suppressing MAPK/NF-κB signaling. Immun Inflamm Dis. 9:1016–1024.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Tang C, Jin M, Ma B, Cao B, Lin C, Xu S,
Li J and Xu Q: RGS2 promotes estradiol biosynthesis by trophoblasts
during human pregnancy. Exp Mol Med. 55:240–252. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Perschbacher KJ, Deng G, Sandgren JA,
Walsh JW, Witcher PC, Sapouckey SA, Owens CE, Zhang SY, Scroggins
SM, Pearson NA, et al: Reduced mRNA expression of RGS2 (regulator
of G protein signaling-2) in the placenta is associated with human
preeclampsia and sufficient to cause features of the disorder in
mice. Hypertension. 75:569–579. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wu S, Cui Y, Zhao H, Xiao X, Gong L, Xu H,
Zhou Q, Ma D and Li X: Trophoblast exosomal UCA1 induces
endothelial injury through the PFN1-RhoA/ROCK pathway in
preeclampsia: A human-specific adaptive pathogenic mechanism. Oxid
Med Cell Longev. 2022:21989232022. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhang S, Shi Y and Dong P: USP8 targeted
by Mir-874-3p promotes trophoblastic cell invasion by stabilizing
the expression of ENaC on trophoblast membrane. Hum Immunol.
84:618–630. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Segre CV and Chiocca S: Regulating the
regulators: The post-translational code of class I HDAC1 and HDAC2.
J Biomed Biotechnol. 2011:6908482011.PubMed/NCBI
|
|
95
|
Spradlin JN, Zhang E and Nomura DK:
Reimagining druggability using chemoproteomic platforms. Acc Chem
Res. 54:1801–1813. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Henning NJ, Boike L, Spradlin JN, Ward CC,
Liu G, Zhang E, Belcher BP, Brittain SM, Hesse MJ, Dovala D, et al:
Deubiquitinase-targeting chimeras for targeted protein
stabilization. Nat Chem Biol. 18:412–421. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Jan SA, Debnath A, Singh RK, Tyagi PK,
Singh S and Singh AK: Targeting undruggable proteins: The siRNA
revolution beyond small molecules-advances, challenges, and future
prospects in therapeutic innovation. Curr Gene Ther. Feb
4–2025.(Epub ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Ma Z, Zhou M, Chen H, Shen Q and Zhou J:
Deubiquitinase-targeting chimeras (DUBTACs) as a potential
paradigm-shifting drug discovery approach. J Med Chem.
68:6897–6915. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Podder V, Ranjan T, Gowda M, Camacho AM
and Ahluwalia MS: Emerging therapies for brain metastases in NSCLC,
breast cancer, and melanoma: A critical review. Curr Neurol
Neurosci Rep. 25:62024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Lou Y, Chen Y, Chen L, Yang T and He L:
Novel molecular strategies for preeclampsia management: A
pathophysiological and therapeutic perspective. Hypertens
Pregnancy. 44:25402852025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tu Y, Dai G, Chen Y, Tan L, Liu H and Chen
M: Emerging target discovery strategies drive the decoding of
therapeutic power of natural products and further drug development:
A case study of celastrol. Exploration. e202402472025. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Wang H, Tan Y, Liu Q, Yang S and Cui L:
Ubiquitin-proteasome system: A potential participant and
therapeutic target in antiphospholipid syndrome. Front Immunol.
16:15237992025. View Article : Google Scholar : PubMed/NCBI
|