Introduction
The thioredoxin (Trx) system constitutes a pivotal
intracellular antioxidant defense network that plays an essential
role in maintaining cellular redox homeostasis and modulating
signaling cascades, impacting numerous cellular processes such as
proliferation, differentiation and apoptosis (1–3).
Recent advances in redox biology have highlighted the Trx system's
involvement not only in redox regulation (4–6) but
also in the modulation of complex cell death pathways (4,7–9).
Despite considerable progress, comprehensive reviews elucidating
the mechanistic underpinnings of Trx's role in programmed cell
death remain scarce, particularly in relation to autophagy and
ferroptosis. Autophagy, an intracellular degradation process, is a
well-recognized double-edged sword, wherein it can both mitigate
oxidative stress damage by removing oxidized biomolecules and
promote cell death under certain conditions (10). Ferroptosis, a recently identified
form of iron-dependent programmed cell death, has garnered
increasing attention due to its pathophysiological relevance in a
variety of diseases, including neurodegeneration, cancer and
ischemic organ injury (11,12).
Autophagy and ferroptosis are distinct yet intimately linked cell
death modalities, with redox status playing a crucial role in their
regulation (13,14). The precise mechanisms through which
the Trx system influences these processes, however, are not fully
elucidated, highlighting a significant area for further
investigation. This review aims to provide an overview of recent
findings on the regulatory roles of the Trx system in cell death
pathways (Fig. 1), with a
particular focus on its interactions with autophagy and
ferroptosis, across various disease contexts. By integrating the
latest research, we aim to provide a conceptual framework for
future studies and therapeutic strategies targeting Trx-mediated
regulation of cell death.
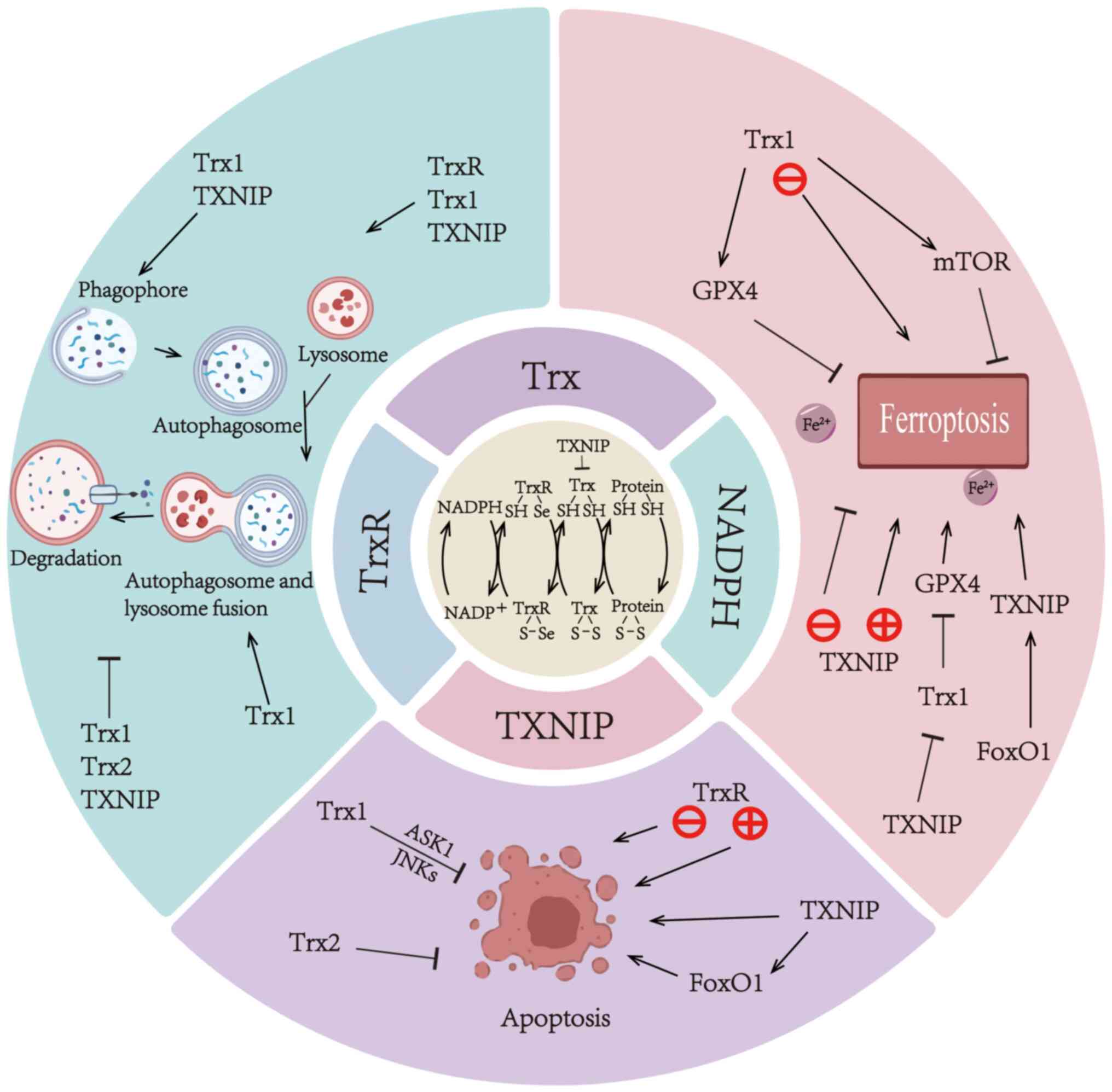
Trx system
The Trx system consists of Trx-interacting protein
(TXNIP), Trx, Trx reductase (TrxR) and NADPH, which serve an
important role in growth promotion, apoptosis, inflammation
regulation, antioxidant defense and maintenance of redox state
homeostasis (15). In a study by
Laurent et al (16) in
1964, Trx was isolated from Escherichia coli. Three
different Trx isoforms were found in mammalian cells, categorized
as Trx1, Trx2 and Trx3 (17).
Trx1, a 105-amino-acid redox protein with a molecular mass of 12
kDa, is an important component of the Trx system and is mainly
localized in the cytoplasm and nucleus (10,18).
Trx2 is present in mitochondria, whereas Trx3 is found only in germ
cells (17).
Trx exhibits broad phylogenetic conservation
spanning prokaryotic to eukaryotic systems. Trx has different
isoforms in different organisms with distinct subcellular
localization patterns, including cytoplasmic, mitochondrial,
endoplasmic reticular, membranous and extracellular distributions.
Notably, these various isoforms have different functions (5). Structural analyses have revealed
conserved architecture across family members, comprising three
α-helical elements encircling a central four-stranded β-sheet core,
with higher eukaryotes demonstrating supplementary secondary
structure elements (19). In
Homo sapiens, the molecular configuration manifests as a
quintuple β-sheet assembly interspersed with four α-helical domains
(20). Evolutionary preservation
is notably observed in the catalytic domain, characterized by a
Cys-Gly-Pro-Cys sequence maintaining redox function across taxa
(18). Trx undergoes a redox
reaction through cysteine residues in its active site. It has been
shown that active site cysteines form disulfide bonds in the
oxidized state, while in the reduced state they exist as thiols
(19). Trx reduces disulfide bonds
in target proteins through this redox exchange, thereby regulating
their activity and function (21).
This shift is important for its catalytic function. The process of
electron transfer through the reversible transformation of active
site sulfhydryl-disulfide (−SH/-S-S-) is the core mechanism of the
Trx system (21).
Biological structure of Trx1 and
Trx2
In addition to the two catalytic cysteine residues
(Cys32 and Cys35) in the catalytic site, human Trx1 contains three
key non-catalytic cysteine residues: Cys62, Cys69 and Cys73. Cys62
and Cys69 are amenable to S-nitrosylation, whereas Cys73 undergoes
multiple modifications, including S-nitrosylation and
glutathionylation (22,23). Given that all these cysteine
residues are subject to post-translational modifications, Trx1
exhibits diverse functional roles.
Trx1 can be proteolytically cleaved by monocytes
in vivo into a truncated form, Trx80, which exhibits
distinct functional properties compared with Trx1 (24,25).
Structurally, Trx1 comprises only a Trx-fold domain containing an
active CXXC motif, whereas Trx2 includes an N-terminal zinc-finger
domain with two additional CXXC motifs alongside the Trx-fold
domain. This structural difference renders Trx2 more complex than
Trx1 (26).
Biological structure of TrxR
As the central enzymatic constituent of the Trx
system, TrxR is classified within the pyridine nucleotide-disulfide
oxidoreductase family (27). TrxR
is a dimer with an N-terminal structural domain that contains the
FAD cofactor, which is responsible for accepting electrons from
NADPH, and a C-terminal structural domain that contains the
conserved selenocysteine (Sec) active site, which is the end-site
for electron transfer (28).
During catalysis, electrons from NADPH are transferred to FAD and
subsequently relayed through a Cys59-Cys64 disulfide bridge to the
Sec residue within the C-terminal domain. This electron flux
culminates in the reduction of oxidized Trx (Trx-S2) to
its dithiol state [Trx-(SH)2], a biochemical process
important for sustaining cellular redox equilibrium (28). Due to the high reactivity of Sec
residues and the accessible position of the C-terminal active site,
TrxR has broad substrate specificity (29).
Human TrxR is a selenoprotein (27). Selenium deficiency leads to
impaired TrxR activity, which disturbs cellular redox homeostasis
and is closely associated with the pathogenesis of multiple
disorders, such as cancer and inflammatory diseases (30,31).
Three isoforms of TrxR exist in humans-TrxR1, TrxR2 and TrxR3-which
are encoded by the TXNRD1, TXNRD2 and TXNRD3 genes, respectively
(32,33). TrxR1 is localized in the cytoplasm
and nucleus, TrxR2 in the mitochondria and TrxR3 in testicular
tissue (34).
Biological structure of NADPH
NADPH, a key coenzyme, comprises three structural
components: i) A reduced nicotinamide ring carrying two hydrogen
atoms, which serves as an electron carrier; ii) an adenine
nucleotide linked to nicotinamide mononucleotide via a
pyrophosphate bond; and iii) a 2′-phosphate group that confers
specificity in enzyme binding, for example to TrxR (35). In the Trx system, NADPH acts as the
primary electron donor, transferring electrons to Trx-S2
via TrxR, thereby driving its reduction to the active form
[Trx-(SH)2] (36).
In addition to its role in the Trx system, NADPH
provides reducing power to enzymes such as glutathione reductase
and glutathione peroxidase (GPX), enabling the regeneration of
antioxidant molecules (37,38).
It also serves as a key coenzyme in fatty acid and cholesterol
synthesis (39). The
NADPH/NADP+ ratio regulates cell proliferation,
apoptosis and inflammatory responses through the modulation of Trx,
nuclear factor E2-related factor 2 and other pathways (40).
Biological structure of TXNIP
TXNIP is an α-arrestin family member encoded by the
TXNIP locus (41). Originally
designated vitamin D3 upregulated protein 1 following
identification in HL-60 promyelocytic cells under
1,25-dihydroxyvitamin D3 treatment (42), TXNIP was subsequently characterized
as Trx-binding protein-2 through yeast two-hybrid screening
(43). Structural characterization
revealed two conserved PPxY motifs that serve as docking platforms
for WW domains of E3 ubiquitin ligases, enabling targeted
ubiquitination-mediated proteolysis (44). TXNIP is able to bind to Trx through
a disulfide bond exchange mechanism, forming a disulfide bond of
TXNIP Cys247-Trx Cys32. This binding results in a structural
rearrangement of TXNIP, unlike other α-arrestin family proteins
(44). This interaction may
explain the negative regulatory effect of TXNIP on Trx
activity.
Translocation of TXNIP
The functional dynamics of TXNIP are intrinsically
linked to its subcellular compartmentalization. Under basal
conditions, TXNIP predominantly localizes to nuclear compartments
and exhibits restricted cytoplasmic translocation (45). Oxidative challenge induces atomic
export of TXNIP to membranous or mitochondrial domains (45). Mechanistically, TXNIP functions as
a Trx1 antagonist, suppressing its redox-regulatory capacity
through direct binding (46).
Cytosolic TXNIP-Trx1 complexes dissociate the Trx1-apoptosis
signal-regulated kinase 1 (ASK1) interaction, triggering the p38
mitogen-activated protein kinase (MAPK) cascade activation, thereby
potentiating oxidative stress responses, cellular damage, apoptotic
execution and senescence pathways (46–48).
Under oxidative stress conditions, mitochondrial-localized TXNIP
further engages Trx2 via analogous binding, liberating
Trx2-conjugated ASK1 to initiate apoptosis through ASK1 activation
(49).
TXNIP is also transcriptionally regulated in a cell
type-specific manner by nuclear receptors such as peroxisome
proliferator-activated receptor (PPAR), farnesol X receptor,
vitamin D receptor and glucocorticoid receptor (50).
TXNIP translocation has been observed under
high-glucose conditions. Elevated intracellular glucose induces
TXNIP translocation to the cytoplasmic membrane, where it promotes
the endocytosis of glucose transporter 1 (GLUT1), reduces GLUT1
expression and consequently decreases GLUT1 abundance on the cell
membrane, thereby reducing glucose uptake into the cell (51). Nephrotic syndrome is characterized
by notable proteinuria or albuminuria. Albuminuria can induce
endoplasmic reticulum stress (ERS), during which TXNIP translocates
from the nucleus to mitochondria, promoting mitochondrial reactive
oxygen species (ROS) production and activating NOD-like receptor
pyrin domain-containing 3 (NLRP3) inflammasomes (52). Beyond these mechanisms, TXNIP
demonstrates mitochondrial translocation in hyperuricemic
inflammation, where its interaction with NLRP3 induces inflammasome
assembly and subsequent pro-inflammatory cytokine secretion
(53). Furthermore, under
high-glucose conditions, elevated levels of translocated TXNIP in
retinal Müller cells suggest that the TXNIP-Drp1-Parkin axis may be
involved in mediating mitophagy in retinal cells under diabetic
conditions (54).
TXNIP serves multifaceted roles in cancer
metastasis, closely linked to its subcellular localization and
disease context. In renal cancer, nuclear TXNIP in tumor-supporting
endothelial cells is associated with elevated ROS and favorable
prognosis, whereas cytoplasmic TXNIP is associated with abnormal
angiogenesis, necrosis and recurrence (55). Functionally, TXNIP may promote or
suppress metastasis depending on cancer type and cellular
metabolism. For example, TXNIP upregulation enhances migration in
hepatocellular carcinoma (HCC) by increasing ROS levels (56). By contrast, decreased TXNIP
expression is associated with enhanced migration, invasion and
metastatic potential in colorectal cancer, cervical cancer,
melanoma and pancreatic cancer (56).
Furthermore, TXNIP shows context-dependent
subcellular localization and expression changes under conditions
such as high-glucose exposure, proteinuria-induced ERS, and uric
acid-mediated inflammation, which in turn modulate oxidative
stress, inflammation and cell fate (51–53).
These disease-specific localization and expression patterns not
only offer mechanistic insights but also highlight promising
avenues for therapeutic targeting. Modulating TXNIP or its
associated pathways may serve as a novel strategy for treating
tumor metastasis and a broad spectrum of other diseases (56).
Mechanisms of the Trx system involved in
ferroptosis in different diseases
Ferroptosis constitutes a distinctive cell death
modality characterized by iron-dependent lipid peroxidation
(57). A recent study has revealed
the three key processes in the pathogenesis of ferroptosis
following cerebral ischemia: Iron deposition, the accumulation of
lipid peroxides and inhibition of the anti-ferroptosis pathway,
with the GPX4-mediated antioxidant system serving a central role
(58). As a selenoprotein, GPX4
catalyzes the reduction of lipid hydroperoxides, establishing its
enzymatic centrality in ferroptosis regulation (59). This cell death paradigm manifests
distinct ultrastructural features, including mitochondrial volume
reduction with cristae diminution and elevated membrane electron
density, while the nuclear architecture remains unaltered.
Concomitant biochemical alterations involve progressive
accumulation of lipid peroxides and the elevation of ROS (60). The resultant oxidative milieu leads
to protein modification and lipid peroxidation, resulting in
cellular dysfunction, which is associated with numerous diseases
(61).
Linkage between Trx1, TrxR and
ferroptosis
Trx1 functions as an antioxidant factor, maintaining
redox homeostasis and regulating ferroptosis across diverse
pathological conditions. Kelch-like ECH-associated protein 1
(Keap1) serves as an important regulatory component in cellular
redox homeostasis (62,63). The cited pharmacological study has
demonstrated that curdione modulates redox complexes by disrupting
Keap1-Trx1 interactions while enhancing Trx1-GPX4 binding, thereby
attenuating post-infarction ferroptosis through Keap1/Trx1/GPX4
axis regulation (64).
Complementary mechanistic evidence has emerged from
neurodegenerative research: In Parkinson's disease (PD) models
characterized by substantia nigra dopaminergic neuron loss, Trx1
exerts ferroptosis inhibition via the modulation of GPX4 activity
and glutathione (GSH) (65).
Ferroptosis is also closely associated with cancer
development, progression and suppression. The apoptotic resistance
of malignant cells underscores the therapeutic value of
non-apoptotic death modalities, with ferroptosis induction emerging
as a promising oncological intervention strategy (66–68).
Cellular redox homeostasis is jointly maintained by two
thiol-dependent antioxidant systems, the GSH and Trx systems, which
exhibit redundant functions via inter-system electron transfer
capabilities (69). Malignant
cells exhibit marked elevation of redox regulators including Trx1
and GSH, creating therapeutic barriers by neutralizing ROS-mediated
cytotoxicity and impairing Fenton reaction-driven ferroptosis
(34,69). The Fenton process, wherein
Fe2+ catalyzes H2O2 conversion to
hydroxyl radicals, initiates lipid peroxidation cascades that can
be exploited for targeted tumor ferroptosis induction (70,71).
Trx1 demonstrates marked upregulation across
multiple solid malignanciesand is associated with poor cancer
prognosis (72). Radiotherapeutic
interventions potentiate ferroptosis through ionizing
radiation-generated ROS that drive lipid peroxidation cascades
(73). Therapeutic synergy is
achieved when combining radiotherapy with a Trx1 inhibitor, which
enhances tumor-selective ferroptosis induction (74).
Similarly, in chronic myeloid leukaemia (CML),
co-inhibition of Trx1 and glutamate-cysteine ligase may serve as a
therapeutic strategy for both imatinib-sensitive and drug-resistant
patients by triggering ferroptosis (75).
Notably, in other diseases such as PD and myocardial
infarction, Trx1 upregulation inhibits ferroptosis to confer
cellular protection (64,65). By contrast, in tumors such as CML,
Trx1 inhibition promotes ferroptosis, enhancing therapeutic
efficacy and overcoming treatment resistance (74,75).
Therapeutic induction of ferroptosis through radiotherapy,
chemotherapy and immunotherapeutic regimens demonstrates
synergistic augmentation of oncological treatment efficacy
(68,73,76,77).
Overall, Trx1 may negatively regulate ferroptosis; however,
ferroptosis exhibits opposite effects in different diseases. This
dichotomy likely arises from variations in cell type, metabolic
state and pathological microenvironment (78,79).
Furthermore, the diversity and complexity of Trx1
target proteins are highlighted by the identification of 112 Trx1
target proteins in mouse primary cortical neurons, with 77 of these
Trx1-interacting proteins being modulated by rapamycin (80). Among these, the mechanistic target
of rapamycin (mTOR) is a major downstream target of Trx1, together
with AMP-activated protein kinase (AMPK), nuclear factor κB (NF-κB)
and histone deacetylase 4 (75).
mTOR serves as a regulatory kinase governing cellular
proliferation, metabolic programming and survival signaling
(81). Oxidative stress promotes
direct interactions between Trx1 and mTOR, inducing intermolecular
disulfide bond formation within mTOR kinase and thereby leading to
its functional inhibition (81,82).
Overexpression of Trx1 prevents mTOR oxidation and preserves its
catalytic activity, whereas Trx1 knockdown conversely promotes mTOR
oxidation and suppresses mTOR signaling (81,83).
However, the C1483F mutation in mTOR confers resistance to
oxidation-mediated inactivation even under Trx1-deficient
conditions (82). Emerging
evidence has further demonstrated that exosomal Trx1 derived from
hypoxic human stem cells activates mTOR complex 1 (mTORC1)
signaling upon cellular internalization, exhibiting
anti-ferroptotic and cardioprotective effects against
doxorubicin-induced cardiotoxicity (84). These findings collectively
establish the Trx1-mTOR axis as an important regulatory mechanism
in ferroptosis suppression. Complementary studies have revealed
that pharmacological inhibition of TrxR induces ferroptosis in
malignant cells, suggesting its therapeutic potential in cancer
treatment (85,86).
Summarily, Trx1 inhibits ferroptosis primarily
through its antioxidative capacity and regulation of
redox-sensitive signaling cascades. In the context of ferroptosis,
Trx1 supports GPX4 activity and GSH synthesis, and protects mTOR
function under oxidative stress, supporting its role as a negative
regulator of lipid peroxidation and cell death. In various tumor
settings, inhibition of Trx1-mediated suppression of ferroptosis
may enhance the efficacy of anticancer therapies.
Linkage between TXNIP and
ferroptosis
TXNIP has been characterized as a
ferroptosis-associated gene across multiple pathological contexts,
including oncogenesis (87,88),
osteonecrosis of the femoral head (89) and hepatic dysfunction (90).
Functionally, TXNIP acts as a tumor suppressor
through its downregulation in malignant cells, where it
concurrently suppresses proliferation and metastatic potential and
promotes apoptotic pathways (91–93).
In HCC models, the protein inhibitor of activated
STAT3 potentiates ferroptosis through activation of the TGF-β
pathway, which is mechanistically linked to TXNIP transcriptional
upregulation (94). Complementary
multi-omics investigations by Zheng et al (95), employing RNA sequencing and liquid
chromatography-tandem mass spectrometry in lung tumor cell
populations, revealed TXNIP as a ferroptosis-associated prognostic
biomarker. Experimental validation further demonstrated that TXNIP
expression was markedly downregulated in lung cancer stem cells and
correlated with advanced disease progression (95).
While TXNIP downregulation exhibits
ferroptosis-suppressive effects across both neoplastic and
non-neoplastic pathologies, disease-specific regulatory mechanisms
govern its activity.
In high-fructose-induced nephropathy,
carbohydrate-responsive element-binding protein-β-mediated
transcriptional activation of TXNIP leads to lipid peroxidation and
drives ferroptosis in renal tubular epithelial cells, thereby
exacerbating renal injury (96).
By contrast, in diabetic retinopathy, 1,8-eudesmol alleviates
retinal ferroptosis by suppressing TXNIP expression in a
PPAR-γ-dependent manner, thereby reducing oxidative stress and
restoring the integrity of the blood-retinal barrier (97). Mechanistically, TXNIP may inhibit
both Trx1 and Trx2, disrupting redox homeostasis and promoting
ferroptosis. Forkhead box O1 (FoxO1) further exhibits
context-dependent regulation by promoting TXNIP transcription and
GSH metabolic disruption, thereby inducing satellite cell
ferroptosis and contributing to sarcopenia. Notably, TXNIP
silencing rescues these ferroptotic phenotypes, alleviating muscle
atrophy (98). Furthermore,
hypothermic hypoxia-reoxygenation stress elevates TXNIP expression
in ex vivo liver transplantation models. This ferroptosis
cascade is pharmacologically mitigated by dexmedetomidine-argon
co-administration, which blocks TXNIP mitochondrial translocation
to preserve redox homeostasis (99). These findings position TXNIP as a
conserved positive regulator of ferroptosis across multiple
diseases.
TXNIP demonstrates prognostic predictive capacity as
a ferroptosis-related biomarker across malignancies. HCC specimens
exhibit notable TXNIP downregulation, with diminished expression
levels associated with adverse clinical outcomes in HCC progression
(100). In lung adenocarcinoma
stem cell models, this multifunctional regulator serves dual roles
in ferroptosis modulation and prognostic prediction, where reduced
expression patterns are associated with three notable
clinicopathological parameters: Immunosuppressive tumor
microenvironment establishment, advanced tumor, node and metastasis
staging, and impaired histodifferentiation status (95). TXNIP is identified as a
prognostically notable ferroptosis-associated gene implicated in
the pathogenesis of bladder cancer and femoral-head necrosis,
offering potential biomarkers for precision medicine applications
(89,101).
As a negative regulator of the antioxidant protein
Trx1, TXNIP is an important modulator of cellular redox homeostasis
(102,103). Experimental evidence has
demonstrated that TXNIP inhibits Trx1 activity, thereby promoting
ferroptosis. In a neonatal rat model of hypoxic-ischemic injury,
hippocampal neuron ferroptosis was shown to be triggered via
TXNIP/Trx1/GPX4 pathway activation, which was directly associated
with hypoxic-ischemic brain damage severity (104). Similarly, curcumin inhibits
ferroptosis by inhibiting the TXNIP/Trx1/GPX4 pathway, thereby
attenuating septic lung injury (105).
In summary, TXNIP serves an important regulatory
role in ferroptosis across diverse pathological conditions,
underscoring its potential as a therapeutic target.
Mechanistically, TXNIP antagonizes Trx1 by forming a disulfide bond
with its Cys32 residue, leading to Trx1 inactivation and increased
ROS levels, thereby facilitating lipid peroxidation and ferroptotic
signaling. In addition, TXNIP contributes to ferroptosis by
disrupting GSH metabolism and destabilizing redox homeostasis.
TXNIP demonstrates notable prognostic associations across multiple
malignancies, with expression patterns showing potential utility as
a biomarker for cancer diagnosis and prognostic stratification.
Mechanisms of the Trx system involved in
autophagy in different diseases
The autophagic process comprises four
mechanistically distinct phases: Autophagosome
nucleation/maturation, cargo sequestration, autolysosomal fusion
and lysosomal degradation (106,107). Notably, microtubule-associated
protein light chain 3 (LC3) undergoes conjugation with
phosphatidylethanolamine during autophagosome biogenesis, leading
to the conversion of LC3 to its lipidated form LC3-II, which serves
as a principal biomarker for autophagic flux quantification
(108–110). Mechanistically, p62 orchestrates
selective autophagy execution through ubiquitinated cargo
recognition and autophagosomal translocation, ultimately enabling
substrate proteolysis (111,112). Lysosome-associated membrane
proteins (LAMPs), particularly LAMP1 and LAMP2, are important for
maintaining lysosomal acidity and facilitating autophagic flux
(113,114). Reduced LAMP2 expression impairs
autophagosome clearance through lysosomal dysfunction (115). LAMP2 dysregulation disrupts
lysosomal homeostasis, and also impairs the activity of Cathepsin D
(CTSD), a key protease for autophagosomal cargo degradation
(116,117).
Mitophagy, a selective autophagy subtype,
specifically regulates mitochondrial quality control via targeted
organelle degradation (118). The
PTEN-induced kinase 1 (PINK1)/Parkin axis constitutes an important
mitophagy regulatory mechanism. PINK1 selectively accumulates on
depolarized mitochondrial membranes, orchestrating Parkin
recruitment and activating its E3 ubiquitin-protein ligase activity
to initiate ubiquitin-dependent organelle tagging (119). Subsequent recognition of
ubiquitinated mitochondria by p62 facilitates autophagosomal
engulfment and lysosomal elimination of defective organelles
(120). PINK1 and Parkin are also
key proteins associated with PD (121).
The mechanisms by which the Trx system regulates
autophagy in various diseases are summarized in Table I, highlighting its molecular
interactions and bidirectional regulatory effects on autophagy.
 | Table I.Effect of the Trx system on
autophagy. |
Table I.
Effect of the Trx system on
autophagy.
| A, Trx1 |
|---|
|
|---|
| First author,
year | Autophagy
regulation | Effects on
autophagy | Diseases | (Refs.) |
|---|
| Gu, 2024 | Positive | Trx1 upregulation
improves autophagic flux and promotes clearance of damaged
mitochondria by regulating autophagy-lysosomal processes. | Parkinson's
disease | (125) |
| Ren, 2022 | Positive | Trx1 activates
autophagy and ameliorates impaired autophagic flux. | Diabetic
retinopathy | (126) |
| Hu, 2023 | Positive | Trx1 promotes
autophagy induction mainly at the initiation stage of autophagy and
protects LECs from oxidative damage. | Cataracts | (127) |
| Ren, 2021 | Positive | Trx1 upregulation
activates autophagy and improves autophagic flux by inhibiting
TXNIP. | Diabetes-induced
hearing impairment | (128) |
| Sánchez-Villamil,
2016 | Positive | Trx1 may promote
mitochondrial autophagy, but it remains yet to be fully elucidated
whether autophagic flux is blocked. | Sepsis-induced
myocardial injury | (129) |
| Nagarajan,
2023 | Positive | Trx1 enhances
myocardial ischemia-induced autophagy through ATG7
transnitrosylation, thereby playing an important role in mediating
protection of the heart. | Myocardial
ischemia | (141) |
| Wang, 2020 | Negative | Trx1 antagonizes
the induction of autophagy by acrylamide. | Acrylamide-induced
neurotoxicity | (132) |
| Ren, 2018 | Negative | Trx1 inhibits
autophagy and improves retinal function through the TXNIP/mTOR
pathway. | Diabetic
retinopathy | (133) |
|
| B, Trx2 |
|
| First author,
year | Autophagy
regulation | Effects on
autophagy |
Diseases | (Refs.) |
|
| He, 2021 | Negative | Trx2 knockdown
causes excessive mitochondrial autophagy and accelerates disease
progression. | Type 2
diabetes | (143) |
| Li, 2017 | Negative | Trx2 may inhibit
ROS-mediated autophagy through the Akt/mTOR and AMPK/mTOR signaling
pathways. | Myocardial
ischemia/reperfusion injury in vitro | (144) |
| Li, 2017 | Negative | Trx2 overexpression
reduces cell death via ASK1-dependent mitochondrial apoptosis and
inhibits autophagy through the mTOR pathway. | Myocardial
ischemia/reperfusion injury in vivo | (145) |
|
| C, TrxR |
|
| First author,
year | Autophagy
regulation | Effects on
autophagy |
Diseases | (Refs.) |
|
| Lei, 2018 | Positive | TrxR inhibitors
induce ROS-independent autophagy inhibition and exhibit anticancer
effects. | Hepatocellular
carcinoma | (147) |
| Nagakannan,
2016 | Positive | TrxR inhibition
leads to impaired autophagic flux by interrupting the
autophagy-lysosomal degradation pathway, which in turn inhibits
autophagy. | Neurodegenerative
disease | (148) |
|
| D,
TXNIP |
|
| First author,
year | Autophagy
regulation | Effects on
autophagy |
Diseases | (Refs.) |
|
| Ren, 2018 | Positive | Inhibition of TXNIP
may lead to autophagy inhibition and improved retinal
function. | Diabetic
retinopathy | (133) |
| Gao, 2020 | Positive | TXNIP can stimulate
autophagy by interacting with DNA damage-inducible transcript 4
protein, causing an excessive accumulation of autophagosomes. | Myocardial
ischemia/reperfusion injury | (154) |
| Ao, 2021 | Positive | TXNIP upregulation
positively regulates autophagy and enhances autophagic flux by
inactivating PI3K/Akt/mTOR signaling. | Diabetic
retinopathy | (155) |
| Park, 2021 | Positive | TXNIP induces
autophagy by directly binding to phosphorylated protein kinase
AMP-activated catalytic subunit α and regulating mTOR complex 1 and
TFEB. |
Steatohepatitis | (156) |
| Huang, 2016 | Negative | TXNIP deficiency
stimulates autophagy by inhibiting mTOR activation, attenuates
diabetes-induced autophagic flux blockage and enhances
mitochondrial autophagy. | Diabetic
nephropathy | (149) |
| Huang, 2014 | Negative | Silencing TXNIP
enhances autophagic flux and reduces autophagosome
accumulation. | Diabetic
nephropathy | (150) |
| Du, 2024 | Negative | TXNIP knockdown
attenuates mTOR activation and restores nuclear translocation of
TFEB, thereby stimulating autophagy and ameliorating impaired
autophagic flux. | Diabetic
nephropathy | (153) |
Linkage between Trx1 and
autophagy
PD develops an abnormal accumulation of α-synuclein
(α-syn) (122). Pathological
α-syn aggregates exhibit dual neurotoxic effects: Synaptic
transmission impairment (123)
and autophagic-lysosomal system dysfunction that promotes cytotoxic
substrate accumulation (124).
Autophagy-lysosomal pathway impairment induces
molecular signatures, including elevated LC3-II expression with
concomitant LC3-II/LC3-I ratio upregulation, p62 accumulation and
reduced histone D levels (125).
Trx1 enhances α-syn clearance in
1-methyl-4-phenyl-1,2,3, 6-tetrahydropyridine (MPTP)-induced PD
models by modulating autophagic-lysosomal degradation pathways.
Furthermore, MPTP exposure upregulates PINK1 and Parkin expression
to activate mitophagy. Notably, Trx1 overexpression attenuates this
MPTP-induced overaccumulation of damaged mitochondria,
demonstrating that Trx1 may restore autophagic flux to eliminate
dysfunctional mitochondria (125).
Multiple studies have corroborated the role of Trx1
in enhancing autophagic flux. LC3 accumulation in diabetic
retinopathy (DR), accompanied by p62 upregulation, indicates
lysosomal dysfunction and subsequent autophagosome accumulation.
Trx1 activates autophagy and restores impaired flux by accelerating
lysosomal degradation and suppressing excessive autophagosome
formation, as demonstrated by autophagic flux assays (126). Furthermore, Trx1 overexpression
protects human lens epithelial cells (LECs) from damage by
promoting beneficial autophagy under normal conditions while
preventing its excessive activation during mild oxidative stress
(127). Similarly, Trx
upregulation mitigates diabetes-induced hearing loss by preserving
cochlear hair cells through TXNIP inhibition and autophagy
activation. Elevated LC3-II and reduced p62 expression further
support the ameliorating effect of Trx1 on autophagic flux
(128).
Cardiac-specific Trx1 expression increases the
LC3-II/LC3-I ratio and PPAR-γ coactivator 1-α levels, indicating
enhanced mitochondrial autophagy to eliminate dysfunctional
mitochondria and mitigate sepsis-induced myocardial dysfunction
(129). However, whether
autophagic flux is blocked remains yet to be fully elucidated, as
the elevated LC3-II/LC3-I ratio may reflect either autophagic
activation or impaired flux leading to autophagosome accumulation
(130). Further investigation is
warranted to clarify these mechanisms.
Evidence indicates that pharmacological inhibition
of autophagosome-lysosome fusion with bafilomycin A1, combined with
blockade of autophagosome formation using wortmannin, reveals Trx1
primarily regulates autophagy at the initiation stage and promotes
its induction (127). This
parallels TXNIP-mediated autophagic flux regulation at the
initiation phase of autophagy (131).
However, conflicting evidence exists in the
literature indicating that Trx1 may negatively regulate autophagy,
contradicting the aforementioned findings. Acrylamide (ACR), a
neurotoxic chemical, primarily exerts its effects through ROS
elevation. Trx1 overexpression downregulates ATG4B, LC3-II, histone
D and LAMP2a, and counteracts ACR-induced autophagy, while Trx1
suppression triggers autophagy (132). Consistent with these findings,
Trx1 also suppresses autophagy via the TXNIP/mTOR pathway. DR
represents a severe diabetic complication linked to advanced
glycation end-product (AGE) accumulation (133). Experimental evidence has
demonstrated that Trx upregulation attenuates AGE-mediated
neurodegeneration, potentially through TXNIP/mTOR axis suppression
and subsequent autophagic inhibition (133). Analogously, in Streptococcus
suis serotype 2 models, TrxC modulates AGE-induced
neurotoxicity via phosphatidylinositol-4,5-bisphosphate 3-kinase
(PI3K)/protein kinase B (Akt)/mTOR pathway regulation, thereby
controlling macrophage autophagic responses (134). Streptococcus suis is a
common porcine pathogen (135).
TrxC is a protein that is widely distributed in microorganisms
(134). This evidence indicates
that Trx1 exhibits dual regulatory effects on autophagy,
influencing either initiation or the autophagy-lysosomal pathway,
likely contingent on cellular context and oxidative stress
intensity.
Trx1 may undergo autophagic degradation.
Methylglyoxal (MGO) accumulation, which promotes AGE formation
in vivo, induces neuronal autophagy via AMPK/mTOR pathway
activation. Inhibition of autophagy through bafilomycin treatment
or ATG5 knockdown prevents Trx1 degradation, indicating
AMPK-dependent autophagy mediates Trx1 degradation (136). However, in hyperosmotic
stress-treated neuronal cells, Trx1 reduction occurs independently
of autophagy and AMPK (137),
contrasting with MGO-induced AMPK activation and autophagy
(137). These discrepancies
highlight the need for disease-specific investigation into Trx1
degradation mechanisms.
Autophagy-related proteins ATG4 and ATG7, important
for autophagosome formation, are regulated by Trx (138,139). Yeast ATG4 is activated by Trx
(140), while Trx1 enhances
myocardial ischemia-induced autophagy through ATG7
transnitrosylation (141).
Linkage between Trx2 and
autophagy
Multiple experiments have indicated that Trx2
suppresses mitochondrial ROS generation, preserving organelle
integrity and function (142).
Trx2 knockdown disrupts systemic metabolic homeostasis, exacerbates
hyperglycemia and promotes excessive mitochondrial autophagy
(143). Specifically, Trx2
deficiency in adipocytes triggers NF-κB-dependent p62 accumulation,
which targets damaged mitochondria for excessive autophagy via
polyubiquitination (143).
Consistent with these findings, in vivo and in vitro
experiments have demonstrated that Trx2 reduces myocardial
ischemia-reperfusion injury by inhibiting ROS-mediated autophagy
through the Akt/mTOR and AMPK/mTOR signaling pathways (144,145). Collectively, Trx2 exerts dual
regulatory effects by suppressing both mitochondrial-specific and
general autophagy processes.
Linkage between TrxR and
autophagy
TrxR regenerates functional Trx through reductive
reactivation, establishing a catalytic cycle that drives iterative
redox transitions (146). TrxR
inhibitors not only display antitumor effects by inhibiting
apoptosis in HCC cells but are also capable of inducing
ROS-independent inhibition of autophagy. Pharmacological autophagy
inhibition enhances HCC susceptibility to TrxR suppression,
positioning TrxR as a promising chemotherapeutic target for HCC
management (147). In serum
deprivation models, TrxR is associated with lysosomal maturation
and regulates the terminal stage of autophagy. Specifically, TrxR
inhibition disrupts autophagosome-lysosome fusion, impairing
autophagic flux and blocking autophagic degradation (148). This mechanism parallels the role
of TrxR as a positive regulator of autophagy in HCC.
Linkage between TXNIP and
autophagy
Inhibition of TXNIP enhances autophagy in diabetic
nephropathy models. Elevated LC3 and p62 levels in the kidneys of
patients with diabetic nephropathy and diabetic rats indicate
autophagy dysregulation. TXNIP deficiency stimulates autophagy by
inhibiting mTOR activation, alleviates diabetes-induced tubular
autophagic flux blockage and promotes tubular mitochondrial
autophagy in diabetic rat kidneys (149). Consistently, in vitro
studies have shown that silencing TXNIP reduces autophagic vesicle
accumulation and the expression of LC3-II and p62 in renal tubular
cells exposed to high glucose, thereby enhancing autophagic flux
and resolving autophagic dysfunction (150). Transcription factor EB (TFEB), a
key regulator of autophagy and lysosomal biogenesis (151,152), is negatively regulated by mTOR,
which suppresses TFEB nuclear translocation (152). TXNIP knockdown attenuates mTOR
activation, restoring TFEB nuclear translocation and stimulating
autophagy to improve impaired flux (153).
However, TXNIP may also exert context-dependent
pro-autophagic effects. For example, in DR, TXNIP inhibition
paradoxically suppresses autophagy (133), highlighting the tissue-specific
and disease context-dependent nature of the autophagic regulation
of TXNIP. TXNIP positively modulates autophagic activity via
complex formation with stress sensor DNA damage-inducible
transcript 4 protein (REDD1). This TXNIP-REDD1 interaction
amplifies autophagosome biogenesis, resulting in cytoplasmic
autophagic vesicle overload that precipitates cardiomyocyte death
through excessive autophagy during myocardial ischemia-reperfusion
injury (154).
TXNIP-mediated autophagy regulation extends to mTOR
pathway inhibition through other mediators under metabolic stress
conditions. In DR models, TXNIP upregulation potentiates Müller
glial autophagic flux via PI3K/Akt/mTOR signaling suppression
(155). Similarly, TXNIP
ameliorates steatohepatitis through activation of autophagy and
suppression of lipotoxic fatty acid oxidation. Mechanistically,
TXNIP induces autophagy by directly binding to phosphorylated
protein kinase AMP-activated catalytic subunit α. and regulating
mTORC1 and TFEB (156).
Crosstalk between apoptosis and
ferroptosis with autophagy
Multiple studies have documented the interplay
between autophagy and apoptosis (157,158). In pancreatic islet cells, TXNIP
overexpression concurrently drives autophagy and apoptosis, whereas
pharmacological autophagy inhibition via 3-methyladenine attenuates
TXNIP-induced apoptosis (159).
Trx upregulation reduces intracellular ROS levels and suppresses
apoptosis by inhibiting autophagy (133).
Autophagy and ferroptosis also exhibit crosstalk in
various disease contexts (160).
Previous evidence has indicated that TXNIP-mediated autophagy
regulates ferroptosis. Specifically, TXNIP suppresses both
canonical autophagy and mitochondrial autophagy in patients with
diabetes (149). In renal
pathology, PINK1/Parkin-driven mitochondrial autophagy protects
tubular epithelial cells from ferroptotic death through the
ROS/heme oxygenase 1/GPX4 axis (161). In diabetic nephropathy,
upregulation of the epigenetic regulator UHRF1 inhibits TXNIP
expression, thereby promoting PINK1-mediated mitochondrial
autophagy and suppressing ferroptosis (162).
Overall, the regulation of autophagy by TXNIP
exhibits a dual nature, capable of either suppressing or promoting
autophagy. This dichotomy may arise from several factors: i)
TXNIP-mediated regulation of autophagy is context-dependent,
varying across different disease settings (163); ii) the function of TXNIP is
influenced by its subcellular localization and interactions with
distinct intracellular factors, which modulate its effects on
autophagy; and iii) this duality may reflect the inherent dual
nature of autophagy itself, a process that maintains cellular
homeostasis by degrading damaged components but becomes detrimental
when excessive.
Furthermore, whether TXNIP possesses a threshold
mechanism that governs the initiation or suppression of autophagy
remains to be elucidated. Given the complexity of autophagy
regulation and the multifaceted role of TXNIP, further
investigation into these mechanisms is warranted to elucidate the
precise regulatory frameworks governing this relationship.
Notably, the Trx system co-regulates autophagy and
other cell death pathways, such as ferroptosis and apoptosis,
primarily through four key signaling axes: i) The PINK1/Parkin
pathway, linking mitophagy and ferroptosis (161,162); ii) the TXNIP-FoxO1 pathway,
linking autophagy to apoptosis (164); iii) the ASK1/c-Jun N-terminal
kinase (JNK) pathway, mediating redox-sensitive apoptosis and
autophagy (165); and iv) the
Trx1/mTOR pathway, linking Trx1 to ferroptosis suppression
(81–84). Such crosstalk reflects the key
regulatory position of the Trx system in balancing stress signaling
and influencing cell survival outcomes.
Mechanisms of the Trx system involved in
apoptosis in different diseases
Apoptosis, a form of programmed cell death, is
executed through tightly regulated gene activation cascades and
molecular signaling networks, exerting important regulatory
functions in both physiological homeostasis and disease
pathogenesis (166).
Trx1 exerts anti-apoptotic effects across various
diseases, often involving ASK1 and JNKs (167,168). Specifically, Trx1 inhibition
abolishes preconditioning-induced cardioprotection by promoting
apoptosis (169). Additionally,
Trx1 suppresses apoptosis triggered by ERS (170), which arises from redox
environment imbalance and Ca2+ homeostasis disruption in
the ER. ERS activation, in turn, stimulates excessive ROS
production (171). However, the
role of Trx1 in apoptosis regulation varies in tumorigenesis. In
HCC, Trx1 upregulation inhibits arsenide-induced apoptosis,
potentially due to mutations in its active site (172). Similarly, Trx2 has been shown to
negatively regulate apoptosis across multiple studies (173,174).
The relationship between TrxR and apoptosis has
been markedly investigated in cancer research, where TrxR
inhibitors have been shown to induce apoptosis and exert antitumor
effects (86,175,176). Beyond pharmacological inhibition,
the upregulation of TrxR itself can promote apoptosis in
non-neoplastic diseases. For example, selenite alleviates
bleomycin-induced idiopathic pulmonary fibrosis by upregulating
TrxR, thereby increasing ROS production and promoting apoptosis in
mouse lung fibroblasts (177).
TXNIP interacts with NLRP3 to activate inflammatory
responses and subsequent pyroptosis (178). Beyond its role in inflammation,
TXNIP markedly contributes to apoptosis induction (179). Specifically, TXNIP upregulation
activates apoptotic pathways (155) and modulates intracellular ROS
generation, which in turn induces oxidative damage and apoptosis
(180). Additionally, TXNIP
upregulates FoxO1 expression via ROS accumulation and enhances
FoxO1 acetylation by inhibiting sirtuin 1 activity, thereby
promoting autophagic apoptosis in diabetic cardiomyopathy (164).
In summary, while the role of the Trx system in
apoptosis regulation is well-documented, its manifestations across
different disease contexts and interactions with other
intracellular mechanisms warrant further in-depth investigation.
TXNIP promotes apoptosis by disrupting the Trx1-ASK1 and Trx2-ASK1
complexes, leading to ASK1 activation, activation of the p38 MAPK
and JNK pathways, and initiation of apoptotic cascades (46,48,49).
TXNIP-induced ROS production further amplifies apoptotic signaling
and activates inflammasomes (52,180). By contrast, Trx1 protects against
apoptosis through ASK1 and JNK inhibition, demonstrating their
opposing regulatory roles in cell fate decisions (167,168). Beyond its roles in autophagy,
ferroptosis and apoptosis, TXNIP has recently emerged as a
modulator of immune responses. Programmed cell death protein 1
(PD-1) and its ligand programmed cell death 1 ligand 1 (PD-L1) are
key immune checkpoint regulators that play a pivotal role in
modulating immune activity. Combination regimens incorporating
PD-1/PD-L1 inhibitors with modalities such as chemotherapy,
radiotherapy, complementary immunotherapies or targeted agents have
demonstrated enhanced therapeutic efficacy (181). Given its role in shaping the
tumor immune microenvironment and promoting inflammasome
activation, TXNIP-targeted modulation may enhance responses to
immune checkpoint inhibitors such as PD-1/PD-L1 blockade. A recent
study suggests that TXNIP depletion could improve T cell-mediated
immunity and may serve as a promising combinatorial strategy in
immunotherapy, warranting further investigation (182).
Challenges and perspectives of targeting the
Trx system in neurodegenerative diseases
Synergistic and antagonistic
interactions in complex disease networks
The Trx system interacts dynamically with multiple
signaling pathways related to oxidative stress, inflammation,
apoptosis and cell survival. These interactions include both
synergistic and antagonistic effects (183). For example, targeting TXNIP can
suppress NLRP3 inflammasome activation, offering a promising
anti-inflammatory strategy (184). Furthermore, Trx/GSH system
inhibitors have demonstrated synergistic antitumor effects in
cancer therapy (185). Therefore,
therapies targeting the Trx system in isolation may be insufficient
or potentially imbalanced. Trx-based diagnosis and treatment should
preferably be combined with other oxidative stress markers or
standard therapies to improve therapeutic efficacy and ensure
system-level coordination.
Genetic and environmental influences
enabling personalized treatment
The expression and activity of Trx/TXNIP are highly
sensitive to environmental stressors, such as glucose, ROS and ERS,
and are further modulated by genetic polymorphisms and
environmental exposures such as nutrition, metals and toxins. These
factors influence individual responses to redox-targeted
interventions. Therefore, personalized therapeutic strategies
integrating genetic and environmental context may reduce off-target
effects and overcome the risk of systemic redox imbalance (183,186).
Spatial and temporal precision in
delivery and regulation
Jia et al (185) demonstrated that adeno-associated
virus-mediated Trx1 overexpression restricted to the hippocampus in
APP/PS1 mice improved cognitive function without disrupting
systemic redox homeostasis. This may support the notion that
region-specific delivery can confine Trx system modulation to
affected tissues, helping avoid systemic imbalance. In cancer
therapy, Trx-targeted treatments have also been combined with
precision delivery systems, such as nanoparticles or exosomes, to
selectively target diseased tissues or cell populations (49,63).
Additionally, the role of the Trx system may vary across different
disease stages; thus, temporal precision and disease-phase
specificity may also be important for therapeutic success.
These considerations are important for developing
precise and safe Trx-based interventions in complex neurological
conditions.
Conclusion
Being a central regulator of intracellular redox
homeostasis, the Trx system is markedly involved in autophagy,
ferroptosis and apoptosis regulation via its electron transfer
function and diverse signaling pathways. The present review
summarizes the biological structure and function of the Trx system,
with a focus on its dual roles in autophagy and ferroptosis, and
its complex influence on cell fate across different
pathophysiological contexts.
In autophagy regulation, the Trx system exhibits
dual functionality, either inhibiting or enhancing autophagy. It
modulates autophagy at both the initiation and autophagy-lysosome
binding stages, likely contingent on disease-specific environments
and cell types. TXNIP, in particular, achieves bidirectional
autophagy regulation by engaging with distinct target proteins,
such as the metabolic regulators mTOR and AMPK, the stress sensor
REDD1, the master transcription factor TFEB, and by influencing the
PI3K/Akt/mTOR signaling pathway. Notably, when observing autophagy
inhibition, the status of autophagic flux should be carefully
assessed to determine if it is blocked.
Regarding ferroptosis, Trx predominantly exerts
negative regulation. Trx1 inhibition induces ferroptosis in tumors,
offering potential antitumor strategies (86,173,174). Conversely, TXNIP positively
regulates ferroptosis across various diseases, including diabetic
retinopathy, high-fructose-induced nephropathy, sarcopenia and
ex vivo liver transplantation models, and serves as a
ferroptosis-related gene for cancer diagnosis and prognosis
prediction.
In the regulation of apoptosis, Trx and TrxR
generally exhibit anti-apoptotic properties, whereas TXNIP and TrxR
in non-neoplastic diseases show pro-apoptotic effects. Furthermore,
crosstalk between autophagy and ferroptosis, as well as between
autophagy and apoptosis, has gained research attention. These
interplays reflect cellular adaptive strategies to complex stress
conditions, and provide novel therapeutic targets and concepts for
disease intervention.
Acknowledgements
Not applicable.
Funding
The present review was supported by the Shandong Provincial
Medical and Health Science and Technology Project (grant no.
202303070601) and the Weifang Municipal Health Commission
Scientific Research Project (grant no. WFWSJK-2024-160).
Availability of data and materials
Not applicable.
Authors' contributions
WHW, DLL and SSX were responsible for designing the
study. WHW and YDM both contributed to writing the manuscript. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
Trx
|
thioredoxin
|
|
TXNIP
|
thioredoxin-interacting protein
|
|
TrxR
|
thioredoxin reductase
|
|
GPX
|
glutathione peroxidase
|
|
ASK1
|
apoptosis signal-regulated kinase
1
|
|
MAPK
|
mitogen-activated protein kinase
|
|
GLUT1
|
glucose transporter 1
|
|
ERS
|
endoplasmic reticulum stress
|
|
ROS
|
reactive oxygen species
|
|
NLRP3
|
NOD-like receptor family pyrin
domain-containing 3
|
|
PPAR
|
peroxisome proliferator-activated
receptor
|
|
Keap1
|
Kelch-like ECH-associated protein
1
|
|
PD
|
Parkinson's disease
|
|
CML
|
chronic myeloid leukaemia
|
|
AMPK
|
AMP-activated protein kinase
|
|
NF-κB
|
nuclear factor κB
|
|
mTOR
|
mechanistic target of rapamycin
|
|
mTORC1
|
mTOR complex 1
|
|
HCC
|
hepatocellular carcinoma
|
|
FoxO1
|
forkhead box O1
|
|
LC3
|
microtubule-associated protein light
chain 3
|
|
LAMPs
|
lysosome-associated membrane
proteins
|
|
PINK1
|
PTEN-induced kinase 1
|
|
α-syn
|
α-synuclein
|
|
MPTP
|
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
|
|
DR
|
diabetic retinopathy
|
|
LECs
|
lens epithelial cells
|
|
ACR
|
acrylamide
|
|
AGE
|
advanced glycation end-product
|
|
PI3K
|
phosphatidylinositol-4,5-bisphosphate
3-kinase
|
|
Akt
|
protein kinase B
|
|
MGO
|
methylglyoxal
|
|
TFEB
|
transcription factor EB
|
|
JNK
|
c-Jun N-terminal kinase
|
References
|
1
|
Hasan AA, Kalinina E, Tatarskiy V and
Shtil A: The thioredoxin system of mammalian cells and its
modulators. Biomedicines. 10:17572022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lee S, Kim SM and Lee RT: Thioredoxin and
thioredoxin target proteins: From molecular mechanisms to
functional significance. Antioxid Redox Signal. 18:1165–1207. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seitz R, Tümen D, Kunst C, Heumann P,
Schmid S, Kandulski A, Müller M and Gülow K: Exploring the
thioredoxin system as a therapeutic target in cancer: Mechanisms
and implications. Antioxidants (Basel). 13:10782024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Muri J and Kopf M: The thioredoxin system:
Balancing redox responses in immune cells and tumors. Eur J
Immunol. 53:e22499482023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang B, Lin Y, Huang Y, Shen YQ and Chen
Q: Thioredoxin (Trx): A redox target and modulator of cellular
senescence and aging-related diseases. Redox Biol. 70:1030322024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dagah OMA, Silaa BB, Zhu M, Pan Q, Qi L,
Liu X, Liu Y, Peng W, Ullah Z, Yudas AF, et al: Exploring immune
redox modulation in bacterial infections: Insights into
thioredoxin-mediated interactions and implications for
understanding host-pathogen dynamics. Antioxidants (Basel).
13:5452024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lu J and Holmgren A: Thioredoxin system in
cell death progression. Antioxid Redox Signal. 17:1738–1747. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jiang C, Krzyzanowski G, Chandel DS, Tom
WA, Fernando N, Olou A and Fernando MR: Inhibition of thioredoxin
reductase activity and oxidation of cellular thiols by
antimicrobial agent, 2-bromo-2-nitro-1,3-propanediol, causes
oxidative stress and cell death in cultured noncancer and cancer
cells. Biology (Basel). 14:5092025.PubMed/NCBI
|
|
9
|
Oberacker T, Kraft L, Schanz M, Latus J
and Schricker S: The importance of thioredoxin-1 in health and
disease. Antioxidants (Basel). 12:10782023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yu G and Klionsky DJ: Life and death
decisions-the many faces of autophagy in cell survival and cell
death. Biomolecules. 12:8662022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi
AA and Lei P: Ferroptosis: Mechanisms and links with diseases.
Signal Transduct Target Ther. 6:492021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yang Y, Cheng J, Lin Q and Ni Z:
Autophagy-dependent ferroptosis in kidney disease. Front Med
(Lausanne). 9:10718642023. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu X, Tuerxun H and Zhao Y, Li Y, Wen S,
Li X and Zhao Y: Crosstalk between ferroptosis and autophagy:
Broaden horizons of cancer therapy. J Transl Med. 23:182025.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mahmood DFD, Abderrazak A, El Hadri K,
Simmet T and Rouis M: The thioredoxin system as a therapeutic
target in human health and disease. Antioxid Redox Signal.
19:1266–1303. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Laurent TC, Moore EC and Reichard P:
Enzymatic synthesis of deoxyribonucleotides. IV. isolation and
characterization of thioredoxin, the hydrogen donor from
Escherichia coli B. J Biol Chem. 239:3436–3444. 1964.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Xinastle-Castillo LO and Landa A:
Physiological and modulatory role of thioredoxins in the cellular
function. Open Med (Wars). 17:2021–2035. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Eklund H, Gleason FK and Holmgren A:
Structural and functional relations among thioredoxins of different
species. Proteins. 11:13–28. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hanschmann EM, Godoy JR, Berndt C,
Hudemann C and Lillig CH: Thioredoxins, glutaredoxins, and
peroxiredoxins-molecular mechanisms and health significance: From
cofactors to antioxidants to redox signaling. Antioxid Redox
Signal. 19:1539–1605. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Forman-Kay JD, Clore GM, Wingfield PT and
Gronenborn AM: High-resolution three-dimensional structure of
reduced recombinant human thioredoxin in solution. Biochemistry.
30:2685–2698. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Collet JF and Messens J: Structure,
function, and mechanism of thioredoxin proteins. Antioxid Redox
Signal. 13:1205–1216. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Barglow KT, Knutson CG, Wishnok JS,
Tannenbaum SR and Marletta MA: Site-specific and redox-controlled
S-nitrosation of thioredoxin. Proc Natl Acad Sci USA.
108:E600–E606. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ungerstedt J, Du Y, Zhang H, Nair D and
Holmgren A: In vivo redox state of human thioredoxin and redox
shift by the histone deacetylase inhibitor suberoylanilide
hydroxamic acid (SAHA). Free Radic Biol Med. 53:2002–2007. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cortes-Bratti X, Bassères E,
Herrera-Rodriguez F, Botero-Kleiven S, Coppotelli G, Andersen JB,
Masucci MG, Holmgren A, Chaves-Olarte E, Frisan T and Avila-Cariño
J: Thioredoxin 80-activated-monocytes (TAMs) inhibit the
replication of intracellular pathogens. PLoS One. 6:e169602011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
King BC, Nowakowska J, Karsten CM, Köhl J,
Renström E and Blom AM: Truncated and full-length thioredoxin-1
have opposing activating and inhibitory properties for human
complement with relevance to endothelial surfaces. J Immunol.
188:4103–4112. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim MK, Zhao L, Jeong S, Zhang J, Jung JH,
Seo HS, Choi JI and Lim S: Structural and biochemical
characterization of thioredoxin-2 from deinococcus radiodurans.
Antioxidants (Basel). 10:18432021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gencheva R, Cheng Q and Arnér ESJ:
Thioredoxin reductase selenoproteins from different organisms as
potential drug targets for treatment of human diseases. Free Radic
Biol Med. 190:320–338. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Arnér ES and Holmgren A: Physiological
functions of thioredoxin and thioredoxin reductase. Eur J Biochem.
267:6102–6109. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Papp LV, Lu J, Holmgren A and Khanna KK:
From selenium to selenoproteins: Synthesis, identity, and their
role in human health. Antioxid Redox Signal. 9:775–806. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lu J, Zhong L, Lönn ME, Burk RF, Hill KE
and Holmgren A: Penultimate selenocysteine residue replaced by
cysteine in thioredoxin reductase from selenium-deficient rat
liver. FASEB J. 23:2394–2402. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zeisel L, Felber JG, Scholzen KC, Poczka
L, Cheff D, Maier MS, Cheng Q, Shen M, Hall MD, Arnér ESJ, et al:
Selective cellular probes for mammalian thioredoxin reductase
TrxR1: Rational design of RX1, a modular 1,2-thiaselenane redox
probe. Chem. 8:1493–1517. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Muri J, Heer S, Matsushita M, Pohlmeier L,
Tortola L, Fuhrer T, Conrad M, Zamboni N, Kisielow J and Kopf M:
The thioredoxin-1 system is essential for fueling DNA synthesis
during T-cell metabolic reprogramming and proliferation. Nat
Commun. 9:18512018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cebula M, Moolla N, Capovilla A and Arnér
ESJ: The rare TXNRD1_v3 (‘v3’) splice variant of human thioredoxin
reductase 1 protein is targeted to membrane rafts by N-acylation
and induces filopodia independently of its redox active site
integrity. J Biol Chem. 288:10002–10011. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang J, Li X, Han X, Liu R and Fang J:
Targeting the thioredoxin system for cancer therapy. Trends
Pharmacol Sci. 38:794–808. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pollak N, Dölle C and Ziegler M: The power
to reduce: Pyridine nucleotides-small molecules with a multitude of
functions. Biochem J. 402:205–218. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Holmgren A and Lu J: Thioredoxin and
thioredoxin reductase: current research with special reference to
human disease. Biochem Biophys Res Commun. 396:120–124. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gaber A, Tamoi M, Takeda T, Nakano Y and
Shigeoka S: NADPH-dependent glutathione peroxidase-like proteins
(Gpx-1, Gpx-2) reduce unsaturated fatty acid hydroperoxides in
Synechocystis PCC 6803. FEBS Lett. 499:32–36. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Frasier CR, Moukdar F, Patel HD, Sloan RC,
Stewart LM, Alleman RJ, La Favor JD and Brown DA: Redox-dependent
increases in glutathione reductase and exercise preconditioning:
Role of NADPH oxidase and mitochondria. Cardiovasc Res. 98:47–55.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Koh HJ, Lee SM, Son BG, Lee SH, Ryoo ZY,
Chang KT, Park JW, Park DC, Song BJ, Veech RL, et al: Cytosolic
NADP+-dependent isocitrate dehydrogenase plays a key role in lipid
metabolism. J Biol Chem. 279:39968–39974. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Santos CXC, Raza S and Shah AM: Redox
signaling in the cardiomyocyte: From physiology to failure. Int J
Biochem Cell Biol. 74:145–151. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Masutani H: Thioredoxin-interacting
protein in cancer and diabetes. Antioxid Redox Signal.
36:1001–1022. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen KS and DeLuca HF: Isolation and
characterization of a novel cDNA from HL-60 cells treated with
1,25-dihydroxyvitamin D-3. Biochim Biophys Acta. 1219:26–32. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nishiyama A, Matsui M, Iwata S, Hirota K,
Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y and Yodoi J:
Identification of thioredoxin-binding protein-2/vitamin D(3)
up-regulated protein 1 as a negative regulator of thioredoxin
function and expression. J Biol Chem. 274:21645–21650. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hwang J, Suh HW, Jeon YH, Hwang E, Nguyen
LT, Yeom J, Lee SG, Lee C, Kim KJ, Kang BS, et al: The structural
basis for the negative regulation of thioredoxin by
thioredoxin-interacting protein. Nat Commun. 5:29582014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Saxena G, Chen J and Shalev A:
Intracellular shuttling and mitochondrial function of
thioredoxin-interacting protein. J Biol Chem. 285:3997–4005. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen CL, Lin CF, Chang WT, Huang WC, Teng
CF and Lin YS: Ceramide induces p38 MAPK and JNK activation through
a mechanism involving a thioredoxin-interacting protein-mediated
pathway. Blood. 111:4365–4374. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li Y, Deng W, Wu J, He Q, Yang G, Luo X,
Jia Y, Duan Y, Zhou L and Liu D: TXNIP exacerbates the senescence
and aging-related dysfunction of β cells by inducing cell cycle
arrest through p38-p16/p21-CDK-Rb pathway. Antioxid Redox Signal.
38:480–495. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Song S, Qiu D, Wang Y, Wei J, Wu H, Wu M,
Wang S, Zhou X, Shi Y and Duan H: TXNIP deficiency mitigates
podocyte apoptosis via restraining the activation of mTOR or p38
MAPK signaling in diabetic nephropathy. Exp Cell Res.
388:1118622020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Pan M, Zhang F, Qu K, Liu C and Zhang J:
TXNIP: A double-edged sword in disease and therapeutic outlook.
Oxid Med Cell Longev. 2022:78051152022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yoshihara E: TXNIP/TBP-2: A master
regulator for glucose homeostasis. Antioxidants (Basel). 9:7652020.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wu N, Zheng B, Shaywitz A, Dagon Y, Tower
C, Bellinger G, Shen CH, Wen J, Asara J, McGraw TE, et al:
AMPK-dependent degradation of TXNIP upon energy stress leads to
enhanced glucose uptake via GLUT1. Mol Cell. 49:1167–1175. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Park SJ, Kim Y, Li C, Suh J, Sivapackiam
J, Goncalves TM, Jarad G, Zhao G, Urano F, Sharma V and Chen YM:
Blocking CHOP-dependent TXNIP shuttling to mitochondria attenuates
albuminuria and mitigates kidney injury in nephrotic syndrome. Proc
Natl Acad Sci USA. 119:e21165051192022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kim SK, Choe JY and Park KY:
TXNIP-mediated nuclear factor-κB signaling pathway and
intracellular shifting of TXNIP in uric acid-induced NLRP3
inflammasome. Biochem Biophys Res Commun. 511:725–731. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Devi TS, Somayajulu M, Kowluru RA and
Singh LP: TXNIP regulates mitophagy in retinal Müller cells under
high-glucose conditions: Implications for diabetic retinopathy.
Cell Death Dis. 8:e27772017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Meszaros M, Yusenko M, Domonkos L, Peterfi
L, Kovacs G and Banyai D: Expression of TXNIP is associated with
angiogenesis and postoperative relapse of conventional renal cell
carcinoma. Sci Rep. 11:172002021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Deng J, Pan T, Liu Z, McCarthy C, Vicencio
JM, Cao L, Alfano G, Suwaidan AA, Yin M, Beatson R and Ng T: The
role of TXNIP in cancer: A fine balance between redox, metabolic,
and immunological tumor control. Br J Cancer. 129:1877–1892. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Fearnhead HO, Vandenabeele P and Vanden
Berghe T: How do we fit ferroptosis in the family of regulated cell
death? Cell Death Differ. 24:1991–1998. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Tuo QZ and Lei P: Ferroptosis in ischemic
stroke: Animal models and mechanisms. Zool Res. 45:1235–1248. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Xie Y, Kang R, Klionsky DJ and Tang D:
GPX4 in cell death, autophagy, and disease. Autophagy.
19:2621–2638. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Cheng Q, Chen M, Liu M, Chen X, Zhu L, Xu
J, Xue J, Wu H and Du Y: Semaphorin 5A suppresses ferroptosis
through activation of PI3K-AKT-mTOR signaling in rheumatoid
arthritis. Cell Death Dis. 13:6082022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yodoi J, Matsuo Y, Tian H, Masutani H and
Inamoto T: Anti-inflammatory thioredoxin family proteins for
medicare, healthcare and aging care. Nutrients. 9:10812017.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sastre J, Pérez S, Sabater L and
Rius-Pérez S: Redox signaling in the pancreas in health and
disease. Physiol Rev. 105:593–650. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Shen K, Wang X, Wang Y, Jia Y, Zhang Y,
Wang K, Luo L, Cai W, Li J, Li S, et al: miR-125b-5p in adipose
derived stem cells exosome alleviates pulmonary microvascular
endothelial cells ferroptosis via Keap1/Nrf2/GPX4 in sepsis lung
injury. Redox Biol. 62:1026552023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang H, Xie B, Shi S, Zhang R, Liang Q,
Liu Z and Cheng Y: Curdione inhibits ferroptosis in
isoprenaline-induced myocardial infarction via regulating
Keap1/Trx1/GPX4 signaling pathway. Phytother Res. 37:5328–5340.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bai L, Yan F, Deng R, Gu R, Zhang X and
Bai J: Thioredoxin-1 rescues MPP+/MPTP-induced
ferroptosis by increasing glutathione peroxidase 4. Mol Neurobiol.
58:3187–3197. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Bebber CM, Müller F, Prieto Clemente L,
Weber J and von Karstedt S: Ferroptosis in cancer cell biology.
Cancers (Basel). 12:1642020. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Ye Z, Liu W, Zhuo Q, Hu Q, Liu M, Sun Q,
Zhang Z, Fan G, Xu W, Ji S, et al: Ferroptosis: Final destination
for cancer? Cell Prolif. 53:e127612020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Su Y, Zhao B, Zhou L, Zhang Z, Shen Y, Lv
H, AlQudsy LHH and Shang P: Ferroptosis, a novel pharmacological
mechanism of anti-cancer drugs. Cancer Lett. 483:127–136. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Lu J and Holmgren A: The thioredoxin
antioxidant system. Free Radic Biol Med. 66:75–87. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Tang Z, Liu Y, He M and Bu W: Chemodynamic
therapy: Tumour microenvironment-mediated fenton and fenton-like
reactions. Angew Chem Int Ed Engl. 58:946–956. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Mohammadi F, Soltani A, Ghahremanloo A,
Javid H and Hashemy SI: The thioredoxin system and cancer therapy:
A review. Cancer Chemother Pharmacol. 84:925–935. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lei G, Mao C, Yan Y, Zhuang L and Gan B:
Ferroptosis, radiotherapy, and combination therapeutic strategies.
Protein Cell. 12:836–857. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Lin Y, Chen X, Yu C, Xu G, Nie X, Cheng Y,
Luan Y and Song Q: Radiotherapy-mediated redox
homeostasis-controllable nanomedicine for enhanced ferroptosis
sensitivity in tumor therapy. Acta Biomater. 159:300–311. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Sun X, Zhang C, Fan B, Liu Q, Shi X, Wang
S, Chen T, Cai X, Hu C, Sun H, et al: Cotargeting of thioredoxin 1
and glutamate-cysteine ligase in both imatinib-sensitive and
imatinib-resistant CML cells. Biochem Pharmacol. 233:1167632025.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhao L, Zhou X, Xie F and Zhang L, Yan H,
Huang J, Zhang C, Zhou F, Chen J and Zhang L: Ferroptosis in cancer
and cancer immunotherapy. Cancer Commun (Lond). 42:88–116. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hassannia B, Vandenabeele P and Vanden
Berghe T: Targeting ferroptosis to iron out cancer. Cancer Cell.
35:830–849. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang S, Guo Q, Zhou L and Xia X:
Ferroptosis: A double-edged sword. Cell Death Discov. 10:2652024.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhu W, Liu X, Yang L, He Q, Huang D and
Tan X: Ferroptosis and tumor immunity: In perspective of the major
cell components in the tumor microenvironment. Eur J Pharmacol.
961:1761242023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Islam MI, Sultana S, Padmanabhan N, Rashid
MU, Siddiqui TJ, Coombs KM, Vitiello PF, Karimi-Abdolrezaee S and
Eftekharpour E: Thioredoxin-1 protein interactions in neuronal
survival and neurodegeneration. Biochim Biophys Acta Mol Basis Dis.
1871:1675482025. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Oka SI, Hirata T, Suzuki W, Naito D, Chen
Y, Chin A, Yaginuma H, Saito T, Nagarajan N, Zhai P, et al:
Thioredoxin-1 maintains mechanistic target of rapamycin (mTOR)
function during oxidative stress in cardiomyocytes. J Biol Chem.
292:18988–19000. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Oka SI, Chin A, Park JY, Ikeda S,
Mizushima W, Ralda G, Zhai P, Tong M, Byun J, Tang F, et al:
Thioredoxin-1 maintains mitochondrial function via mechanistic
target of rapamycin signalling in the heart. Cardiovasc Res.
116:1742–1755. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Oka SI, Mizushima W, Huan C and Sadoshima
J: Abstract 877: Thioredoxin-1 maintains cardiac function and
metabolic gene expression via mTOR signaling. Circ Res. 125 (Suppl
1):2019. View Article : Google Scholar
|
|
84
|
Yu Y, Wu T, Lu Y, Zhao W, Zhang J, Chen Q,
Ge G, Hua Y, Chen K, Ullah I and Zhang F: Exosomal thioredoxin-1
from hypoxic human umbilical cord mesenchymal stem cells inhibits
ferroptosis in doxorubicin-induced cardiotoxicity via mTORC1
signaling. Free Radic Biol Med. 193:108–121. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Yang Z, Huang S, Liu Y, Chang X, Liang Y,
Li X, Xu Z, Wang S, Lu Y, Liu Y and Liu W: Biotin-targeted Au(I)
radiosensitizer for cancer synergistic therapy by intervening with
redox homeostasis and inducing ferroptosis. J Med Chem.
65:8401–8415. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Xi J, Tian LL, Xi J, Girimpuhwe D, Huang
C, Ma R, Yao X, Shi D, Bai Z, Wu QX and Fang J: Alterperylenol as a
novel thioredoxin reductase inhibitor induces liver cancer cell
apoptosis and ferroptosis. J Agric Food Chem. 70:15763–15775. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Guo Y, Han Y, Zhang J, Zhou Y, Wei M and
Yu L: Identification and experimental validation of prognostic
miRNA signature and ferroptosis-related key genes in cervical
squamous cell carcinoma. Cancer Med. 13:e704152024. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Jin W, Liu J, Yang J, Feng Z, Feng Z,
Huang N, Yang T and Yu L: Identification of a key ceRNA network
associated with ferroptosis in gastric cancer. Sci Rep.
12:200882022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Chen N, Meng Y, Zhan H and Li G:
Identification and validation of potential ferroptosis-related
genes in glucocorticoid-induced osteonecrosis of the femoral head.
Medicina (Kaunas). 59:2972023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zheng Y, Song J, Qian Q and Wang H: Silver
nanoparticles induce liver inflammation through ferroptosis in
zebrafish. Chemosphere. 362:1426732024. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Han SH, Jeon JH, Ju HR, Jung U, Kim KY,
Yoo HS, Lee YH, Song KS, Hwang HM, Na YS, et al: VDUP1 upregulated
by TGF-beta1 and 1,25-dihydorxyvitamin D3 inhibits tumor cell
growth by blocking cell-cycle progression. Oncogene. 22:4035–4046.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Nasoohi S, Ismael S and Ishrat T:
Thioredoxin-interacting protein (TXNIP) in cerebrovascular and
neurodegenerative diseases: Regulation and implication. Mol
Neurobiol. 55:7900–7920. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Morrison JA, Pike LA, Sams SB, Sharma V,
Zhou Q, Severson JJ, Tan AC, Wood WM and Haugen BR: Thioredoxin
interacting protein (TXNIP) is a novel tumor suppressor in thyroid
cancer. Mol Cancer. 13:622014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Bao W, Wang J, Fan K, Gao Y and Chen J:
PIAS3 promotes ferroptosis by regulating TXNIP via TGF-β signaling
pathway in hepatocellular carcinoma. Pharmacol Res. 196:1069152023.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zheng Y, Yang W, Wu W, Jin F, Lu D, Gao J
and Wang S: Diagnostic and predictive significance of the
ferroptosis-related gene TXNIP in lung adenocarcinoma stem cells
based on multi-omics. Transl Oncol. 45:1019262024. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Guo H, Fang T, Cheng Y, Li T, Qu JR, Xu
CF, Deng XQ, Sun B and Chen LM: ChREBP-β/TXNIP aggravates
frucose-induced renal injury through triggering ferroptosis of
renal tubular epithelial cells. Free Radic Biol Med. 199:154–165.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Liu Z, Gan S, Fu L, Xu Y, Wang S, Zhang G,
Pan D, Tao L and Shen X: 1,8-Cineole ameliorates diabetic
retinopathy by inhibiting retinal pigment epithelium ferroptosis
via PPAR-γ/TXNIP pathways. Biomed Pharmacother. 164:1149782023.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Maimaiti Y, Abulitifu M, Ajimu Z, Su T,
Zhang Z, Yu Z and Xu H: FOXO regulation of TXNIP induces
ferroptosis in satellite cells by inhibiting glutathione
metabolism, promoting sarcopenia. Cell Mol Life Sci. 82:812025.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chen Q, Sun J, Liu X, Qin Z, Li J, Ma J,
Xue Z, Li Y, Yang Z, Sun Q, et al: Dexmedetomidine and argon in
combination against ferroptosis through tackling TXNIP-mediated
oxidative stress in DCD porcine livers. Cell Death Discov.
10:3192024. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li J, Yue Z, Xiong W, Sun P, You K and
Wang J: TXNIP overexpression suppresses proliferation and induces
apoptosis in SMMC7221 cells through ROS generation and MAPK pathway
activation. Oncol Rep. 37:3369–3376. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Yi K, Liu J, Rong Y, Wang C, Tang X, Zhang
X, Xiong Y and Wang F: Biological functions and prognostic value of
ferroptosis-related genes in bladder cancer. Front Mol Biosci.
8:6311522021. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
He L, He T, Farrar S, Ji L, Liu T and Ma
X: Antioxidants maintain cellular redox homeostasis by elimination
of reactive oxygen species. Cell Physiol Biochem. 44:532–553. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mohamed IN, Li L, Ismael S, Ishrat T and
El-Remessy AB: Thioredoxin interacting protein, a key molecular
switch between oxidative stress and sterile inflammation in
cellular response. World J Diabetes. 12:1979–1999. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Zhang XY, Liu CM, Ma YH, Meng N, Jiang JY,
Yu XH and Wang XL: The TXNIP/Trx-1/GPX4 pathway promotes
ferroptosis in hippocampal neurons after hypoxia-ischemia in
neonatal rats. Zhongguo Dang Dai Er Ke Za Zhi. 24:1053–1060.
2022.(In Chinese). PubMed/NCBI
|
|
105
|
Chen K, Meng Z, Min J, Wang J, Li Z, Gao Q
and Hu J: Curcumin alleviates septic lung injury in mice by
inhibiting TXNIP/TRX-1/GPX4-mediated ferroptosis. Nan Fang Yi Ke Da
Xue Xue Bao. 44:1805–1813. 2024.(In Chinese). PubMed/NCBI
|
|
106
|
Bento CF, Renna M, Ghislat G, Puri C,
Ashkenazi A, Vicinanza M, Menzies FM and Rubinsztein DC: Mammalian
autophagy: How does it work? Annu Rev Biochem. 85:685–713. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Yoshimori T: Autophagy: A regulated bulk
degradation process inside cells. Biochem Biophys Res Commun.
313:453–458. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Chueh KS, Lu JH, Juan TJ, Chuang SM and
Juan YS: The molecular mechanism and therapeutic application of
autophagy for urological disease. Int J Mol Sci. 24:148872023.
View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Jiang P and Mizushima N: LC3- and
p62-based biochemical methods for the analysis of autophagy
progression in mammalian cells. Methods. 75:13–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Esteban-Martínez L and Boya P: Autophagic
flux determination in vivo and ex vivo. Methods. 75:79–86. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Kumar AV, Mills J and Lapierre LR:
Selective autophagy receptor p62/SQSTM1, a pivotal player in stress
and aging. Front Cell Dev Biol. 10:7933282022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Chaudhry N, Sica M, Surabhi S, Hernandez
DS, Mesquita A, Selimovic A, Riaz A, Lescat L, Bai H, MacIntosh GC
and Jenny A: Lamp1 mediates lipid transport, but is dispensable for
autophagy in Drosophila. Autophagy. 18:2443–2458. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zhang J, Zeng W, Han Y, Lee WR, Liou J and
Jiang Y: Lysosomal LAMP proteins regulate lysosomal pH by direct
inhibition of the TMEM175 channel. Mol Cell. 83:2524–2539.e7. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Yadin D, Petrover Z, Shainberg A, Alcalai
R, Waldman M, Seidman J, Seidman CE, Abraham NG, Hochhauser E and
Arad M: Autophagy guided interventions to modify the cardiac
phenotype of Danon disease. Biochem Pharmacol. 204:1152292022.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Abokyi S, Shan SW, Lam CH, Catral KP, Pan
F, Chan HHL, To CH and Tse DYY: Targeting lysosomes to reverse
hydroquinone-induced autophagy defects and oxidative damage in
human retinal pigment epithelial cells. Int J Mol Sci. 22:90422021.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Bunk J, Prieto Huarcaya S, Drobny A,
Dobert JP, Walther L, Rose-John S, Arnold P and Zunke F: Cathepsin
D variants associated with neurodegenerative diseases show
dysregulated functionality and modified α-synuclein degradation
properties. Front Cell Dev Biol. 9:5818052021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Mary A, Eysert F, Checler F and Chami M:
Mitophagy in Alzheimer's disease: Molecular defects and therapeutic
approaches. Mol Psychiatry. 28:202–216. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wang H, Luo W, Chen H, Cai Z and Xu G:
Mitochondrial dynamics and mitochondrial autophagy: Molecular
structure, orchestrating mechanism and related disorders.
Mitochondrion. 75:1018472024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Shang C, Liu Z, Zhu Y, Lu J, Ge C, Zhang
C, Li N, Jin N, Li Y, Tian M and Li X: SARS-CoV-2 causes
mitochondrial dysfunction and mitophagy impairment. Front
Microbiol. 12:7807682022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Bitar S, Baumann T, Weber C, Abusaada M,
Rojas-Charry L, Ziegler P, Schettgen T, Randerath IE, Venkataramani
V, Michalke B, et al: Iron-sulfur cluster loss in mitochondrial
CISD1 mediates PINK1 loss-of-function phenotypes. Elife.
13:e970272024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Tong B, Ba Y, Li Z, Yang C, Su K, Qi H,
Zhang D, Liu X, Wu Y, Chen Y, et al: Targeting dysregulated lipid
metabolism for the treatment of Alzheimer's disease and Parkinson's
disease: Current advancements and future prospects. Neurobiol Dis.
196:1065052024. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Lamonaca G and Volta M: Alpha-synuclein
and LRRK2 in synaptic autophagy: Linking early dysfunction to
late-stage pathology in Parkinson's disease. Cells. 9:11152020.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Calabresi P, Mechelli A, Natale G,
Volpicelli-Daley L, Di Lazzaro G and Ghiglieri V: Alpha-synuclein
in Parkinson's disease and other synucleinopathies: From overt
neurodegeneration back to early synaptic dysfunction. Cell Death
Dis. 14:1762023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Gu R, Bai L, Yan F, Zhang S, Zhang X, Deng
R, Zeng X, Sun B, Hu X, Li Y and Bai J: Thioredoxin-1 decreases
alpha-synuclein induced by MPTP through promoting
autophagy-lysosome pathway. Cell Death Discov. 10:932024.
View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ren X, Lv J, Wang N, Liu J, Gao C, Wu X,
Yu Y, Teng Q, Dong W, Kong H and Kong L: Thioredoxin upregulation
delays diabetes-induced photoreceptor cell degeneration via
AMPK-mediated autophagy and exosome secretion. Diabetes Res Clin
Pract. 185:1097882022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Hu J, Liu J, Chen S, Zhang C, Shen L, Yao
K and Yu Y: Thioredoxin-1 regulates the autophagy induced by
oxidative stress through LC3-II in human lens epithelial cells.
Clin Exp Pharmacol Physiol. 50:476–485. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Ren X, Lv J, Fu Y, Zhang N, Zhang C, Dong
Z, Chudhary M, Zhong S, Kong L and Kong H: Upregulation of
thioredoxin contributes to inhibiting diabetic hearing impairment.
Diabetes Res Clin Pract. 179:1090252021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Sánchez-Villamil JP, D'Annunzio V,
Finocchietto P, Holod S, Rebagliati I, Pérez H, Peralta JG, Gelpi
RJ, Poderoso JJ and Carreras MC: Cardiac-specific overexpression of
thioredoxin 1 attenuates mitochondrial and myocardial dysfunction
in septic mice. Int J Biochem Cell Biol. 81:323–334. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Shojaei S, Barzegar Behrooz A, Cordani M,
Aghaei M, Azarpira N, Klionsky DJ and Ghavami S: A non-fluorescent
immunohistochemistry method for measuring autophagy flux using
MAP1LC3/LC3 and SQSTM1 as core markers. FEBS Open Bio. 15:898–905.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zhou J, Yao K, Zhang Y, Chen G, Lai K, Yin
H and Yu Y: Thioredoxin binding protein-2 regulates autophagy of
human lens epithelial cells under oxidative stress via inhibition
of Akt phosphorylation. Oxid Med Cell Longev. 2016:48564312016.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Wang W, Huang L, Thomas ER, Hu Y, Zeng F
and Li X: Notoginsenoside R1 protects against the
acrylamide-induced neurotoxicity via upregulating Trx-1-mediated
ITGAV expression: Involvement of autophagy. Front Pharmacol.
11:5590462020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Ren X, Wang NN, Qi H, Qiu YY, Zhang CH,
Brown E, Kong H and Kong L: Up-regulation thioredoxin inhibits
advanced glycation end products-induced neurodegeneration. Cell
Physiol Biochem. 50:1673–1686. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Ji C, Pan Y, Liu B, Liu J, Zhao C, Nie Z,
Liao S, Kuang G, Wu X, Liu Q, et al: Thioredoxin C of streptococcus
suis serotype 2 contributes to virulence by inducing antioxidative
stress and inhibiting autophagy via the MSR1/PI3K-Akt-mTOR pathway
in macrophages. Vet Microbiol. 298:1102632024. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Vötsch D, Willenborg M, Weldearegay YB and
Valentin-Weigand P: Streptococcus suis-the ‘two faces’ of a
pathobiont in the porcine respiratory tract. Front Microbiol.
9:4802018. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Dafre AL, Schmitz AE and Maher P:
Methylglyoxal-induced AMPK activation leads to autophagic
degradation of thioredoxin 1 and glyoxalase 2 in HT22 nerve cells.
Free Radic Biol Med. 108:270–279. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Dafre AL, Schmitz AE and Maher P:
Hyperosmotic stress initiates AMPK-independent autophagy and AMPK-
and autophagy-independent depletion of thioredoxin 1 and glyoxalase
2 in HT22 nerve cells. Oxid Med Cell Longev. 2019:27158102019.
View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Nakatogawa H: Two ubiquitin-like
conjugation systems that mediate membrane formation during
autophagy. Essays Biochem. 55:39–50. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Arakawa S, Honda S, Yamaguchi H and
Shimizu S: Molecular mechanisms and physiological roles of
Atg5/Atg7-independent alternative autophagy. Proc Jpn Acad Ser B
Phys Biol Sci. 93:378–385. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Pérez-Pérez ME, Zaffagnini M, Marchand CH,
Crespo JL and Lemaire SD: The yeast autophagy protease Atg4 is
regulated by thioredoxin. Autophagy. 10:1953–1964. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Nagarajan N, Oka SI, Nah J, Wu C, Zhai P,
Mukai R, Xu X, Kashyap S, Huang CY, Sung EA, et al: Thioredoxin 1
promotes autophagy through transnitrosylation of Atg7 during
myocardial ischemia. J Clin Invest. 133:e1623262023. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Huang Q, Zhou HJ, Zhang H, Huang Y,
Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G,
Giordano FJ, et al: Thioredoxin-2 inhibits mitochondrial reactive
oxygen species generation and apoptosis stress kinase-1 activity to
maintain cardiac function. Circulation. 131:1082–1097. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
He F, Huang Y, Song Z, Zhou HJ, Zhang H,
Perry RJ, Shulman GI and Min W: Mitophagy-mediated adipose
inflammation contributes to type 2 diabetes with hepatic insulin
resistance. J Exp Med. 218:e202014162021. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Li YY, Xiang Y, Zhang S, Wang Y, Yang J,
Liu W and Xue FT: Thioredoxin-2 protects against oxygen-glucose
deprivation/reperfusion injury by inhibiting autophagy and
apoptosis in H9c2 cardiomyocytes. Am J Transl Res. 9:1471–1482.
2017.PubMed/NCBI
|
|
145
|
Li Y, Xiang Y, Zhang S, Wang Y, Yang J,
Liu W and Xue F: Intramyocardial injection of thioredoxin
2-expressing lentivirus alleviates myocardial ischemia-reperfusion
injury in rats. Am J Transl Res. 9:4428–4439. 2017.PubMed/NCBI
|
|
146
|
Bjørklund G, Zou L, Wang J, Chasapis CT
and Peana M: Thioredoxin reductase as a pharmacological target.
Pharmacol Res. 174:1058542021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Lei H, Wang G, Zhang J and Han Q:
Inhibiting TrxR suppresses liver cancer by inducing apoptosis and
eliciting potent antitumor immunity. Oncol Rep. 40:3447–3457.
2018.PubMed/NCBI
|
|
148
|
Nagakannan P, Iqbal MA, Yeung A, Thliveris
JA, Rastegar M, Ghavami S and Eftekharpour E: Perturbation of redox
balance after thioredoxin reductase deficiency interrupts
autophagy-lysosomal degradation pathway and enhances cell death in
nutritionally stressed SH-SY5Y cells. Free Radic Biol Med.
101:53–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Huang C, Zhang Y, Kelly DJ, Tan CYR, Gill
A, Cheng D, Braet F, Park JS, Sue CM, Pollock CA and Chen XM:
Thioredoxin interacting protein (TXNIP) regulates tubular autophagy
and mitophagy in diabetic nephropathy through the mTOR signaling
pathway. Sci Rep. 6:291962016. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Huang C, Lin MZ, Cheng D, Braet F, Pollock
CA and Chen XM: Thioredoxin-interacting protein mediates
dysfunction of tubular autophagy in diabetic kidneys through
inhibiting autophagic flux. Lab Invest. 94:309–320. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Tang H, Hou H, Song L, Tian Z, Liu W, Xia
T and Wang A: The role of mTORC1/TFEB axis mediated lysosomal
biogenesis and autophagy impairment in fluoride neurotoxicity and
the intervention effects of resveratrol. J Hazard Mater.
467:1336342024. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Martina JA, Chen Y, Gucek M and
Puertollano R: MTORC1 functions as a transcriptional regulator of
autophagy by preventing nuclear transport of TFEB. Autophagy.
8:903–914. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Du Y, Wu M, Song S, Bian Y and Shi Y:
TXNIP deficiency attenuates renal fibrosis by modulating
mTORC1/TFEB-mediated autophagy in diabetic kidney disease. Ren
Fail. 46:23389332024. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Gao C, Wang R, Li B, Guo Y, Yin T, Xia Y,
Zhang F, Lian K, Liu Y, Wang H, et al: TXNIP/Redd1 signalling and
excessive autophagy: A novel mechanism of myocardial
ischaemia/reperfusion injury in mice. Cardiovasc Res. 116:645–657.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Ao H, Li H, Zhao X, Liu B and Lu L: TXNIP
positively regulates the autophagy and apoptosis in the rat müller
cell of diabetic retinopathy. Life Sci. 267:1189882021. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Park HS, Song JW, Park JH, Lim BK, Moon
OS, Son HY, Lee JH, Gao B, Won YS and Kwon HJ: TXNIP/VDUP1
attenuates steatohepatitis via autophagy and fatty acid oxidation.
Autophagy. 17:2549–2564. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Gordy C and He YW: The crosstalk between
autophagy and apoptosis: Where does this lead? Protein Cell.
3:17–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Song S, Tan J, Miao Y, Li M and Zhang Q:
Crosstalk of autophagy and apoptosis: Involvement of the dual role
of autophagy under ER stress. J Cell Physiol. 232:2977–2984. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Wang J, Wang J, Wang JJ, Zhang WF and Jiao
XY: Role of autophagy in TXNIP overexpression-induced apoptosis of
INS-1 islet cells. Sheng Li Xue Bao. 69:445–451. 2017.(In Chinese).
PubMed/NCBI
|
|
160
|
Wang W, Lu D, Shi Y and Wang Y: Exploring
the neuroprotective effects of lithium in ischemic stroke: A
literature review. Int J Med Sci. 21:284–298. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Lin Q, Li S, Jin H, Cai H, Zhu X, Yang Y,
Wu J, Qi C, Shao X, Li J, et al: Mitophagy alleviates
cisplatin-induced renal tubular epithelial cell ferroptosis through
ROS/HO-1/GPX4 axis. Int J Biol Sci. 19:1192–1210. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Ji H, Zhao Y, Ma X, Wu L, Guo F, Huang F,
Song Y, Wang J and Qin G: Upregulation of UHRF1 promotes
PINK1-mediated mitophagy to alleviate ferroptosis in diabetic
nephropathy. Inflammation. 47:718–732. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Li C, Fang Y and Chen YM: Beyond redox
regulation: Novel roles of TXNIP in the pathogenesis and
therapeutic targeting of kidney disease. Am J Pathol. 195:615–625.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Zhang Y, Li B, Fu Y, Cai H and Zheng Y:
Txnip promotes autophagic apoptosis in diabetic cardiomyopathy by
upregulating FoxO1 and its acetylation. Cell Signal.
124:1114692024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Zhao Q, Liu Y, Zhong J, Bi Y, Liu Y, Ren
Z, Li X, Jia J, Yu M and Yu X: Pristimerin induces apoptosis and
autophagy via activation of ROS/ASK1/JNK pathway in human breast
cancer in vitro and in vivo. Cell Death Discov. 5:1252019.
View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Cavalcante GC, Schaan AP, Cabral GF,
Santana-da-Silva MN, Pinto P, Vidal AF and Ribeiro-Dos-Santos Â: A
cell's fate: An overview of the molecular biology and genetics of
apoptosis. Int J Mol Sci. 20:41332019. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Liu Y and Min W: Thioredoxin promotes ASK1
ubiquitination and degradation to inhibit ASK1-mediated apoptosis
in a redox activity-independent manner. Circ Res. 90:1259–1266.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Bao N, Wang J, Yue Q, Cao F, Gu X, Wen K,
Kong W and Gu M: Chrysophanol-mediated trx-1 activation attenuates
renal fibrosis through inhibition of the JNK/Cx43 signaling
pathway. Ren Fail. 46:23987102024. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
D'Annunzio V, Perez V, Boveris A, Gelpi RJ
and Poderoso JJ: Role of thioredoxin-1 in ischemic preconditioning,
postconditioning and aged ischemic hearts. Pharmacol Res.
109:24–31. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Yang L, Guo N, Fan W, Ni C, Huang M, Bai
L, Zhang L, Zhang X, Wen Y, Li Y, et al: Thioredoxin-1 blocks
methamphetamine-induced injury in brain through inhibiting
endoplasmic reticulum and mitochondria-mediated apoptosis in mice.
Neurotoxicology. 78:163–169. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Liu H, Dai L, Wang M, Feng F and Xiao Y:
Tunicamycin induces hepatic stellate cell apoptosis through
calpain-2/Ca2 +-dependent endoplasmic reticulum stress
pathway. Front Cell Dev Biol. 9:6848572021. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Tian C, Gao P, Zheng Y, Yue W, Wang X, Jin
H and Chen Q: Redox status of thioredoxin-1 (TRX1) determines the
sensitivity of human liver carcinoma cells (HepG2) to arsenic
trioxide-induced cell death. Cell Res. 18:458–471. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Pérez VI, Lew CM, Cortez LA, Webb CR,
Rodriguez M, Liu Y, Qi W, Li Y, Chaudhuri A, Van Remmen H, et al:
Thioredoxin 2 haploinsufficiency in mice results in impaired
mitochondrial function and increased oxidative stress. Free Radic
Biol Med. 44:882–892. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Kundumani-Sridharan V, Subramani J, Owens
C and Das KC: Nrg1β released in remote ischemic preconditioning
improves myocardial perfusion and decreases ischemia/reperfusion
injury via ErbB2-mediated rescue of endothelial nitric oxide
synthase and abrogation of Trx2 autophagy. Arterioscler Thromb Vasc
Biol. 41:2293–2314. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Xu Q, Zhang J, Zhao Z, Chu Y and Fang J:
Revealing PACMA 31 as a new chemical type TrxR inhibitor to promote
cancer cell apoptosis. Biochim Biophys Acta Mol Cell Res.
1869:1193232022. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Qian J, Xu Z, Meng C, Liu J, Hsu PL, Li Y,
Zhu W, Yang Y, Morris-Natschke SL and Lee KH: Design and synthesis
of benzylidenecyclohexenones as TrxR inhibitors displaying high
anticancer activity and inducing ROS, apoptosis, and autophagy. Eur
J Med Chem. 204:1126102020. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Lin JH, Liu CC, Liu CY, Hsu TW, Yeh YC,
How CK, Hsu HS and Hung SC: Selenite selectively kills lung
fibroblasts to treat bleomycin-induced pulmonary fibrosis. Redox
Biol. 72:1031482024. View Article : Google Scholar : PubMed/NCBI
|
|
178
|
Jia Y, Cui R, Wang C, Feng Y, Li Z, Tong
Y, Qu K, Liu C and Zhang J: Metformin protects against intestinal
ischemia-reperfusion injury and cell pyroptosis via
TXNIP-NLRP3-GSDMD pathway. Redox Biol. 32:1015342020. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Yang C, Mo J, Liu Q, Li W, Chen Y, Feng J,
Jia J, Liu L, Bai Y and Zhou J: TXNIP/NLRP3 aggravates global
cerebral ischemia-reperfusion injury-induced cognitive decline in
mice. Heliyon. 10:e274232024. View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Huang L, Zhang S, Bian M, Xiang X, Xiao L,
Wang J, Lu S, Chen W, Zhang C, Mo G, et al: Injectable,
anti-collapse, adhesive, plastic and bioactive bone graft
substitute promotes bone regeneration by moderating oxidative
stress in osteoporotic bone defect. Acta Biomater. 180:82–103.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Hashimoto K, Nishimura S and Goto K:
PD1/PDL1 immune checkpoint in bone and soft tissue tumors (Review).
Mol Clin Oncol. 29:312025. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Dubuisson A, Mangelinck A, Knockaert S,
Zichi A, Becht E, Philippon W, Dromaint-Catesson S, Fasquel M,
Melchiore F, Provost N, et al: Glucose deprivation and
identification of TXNIP as an immunometabolic modulator of T cell
activation in cancer. Front Immunol. 16:15485092025. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
Bjørklund G, Zou L, Peana M, Chasapis CT,
Hangan T, Lu J and Maes M: The role of the thioredoxin system in
brain diseases. Antioxidants (Basel). 11:21612022. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Choi EH and Park SJ: TXNIP: A key protein
in the cellular stress response pathway and a potential therapeutic
target. Exp Mol Med. 55:1348–1356. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Jia JJ, Geng WS, Wang ZQ, Chen L and Zeng
XS: The role of thioredoxin system in cancer: strategy for cancer
therapy. Cancer Chemother Pharmacol. 84:453–470. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Tsubaki H, Tooyama I and Walker DG:
Thioredoxin-interacting protein (TXNIP) with focus on brain and
neurodegenerative diseases. Int J Mol Sci. 21:93572020. View Article : Google Scholar : PubMed/NCBI
|