Hirschsprung disease (HSCR) is a congenital
neurodevelopmental disorder characterized by the absence of enteric
ganglion cells in the distal bowel, leading to dysmotility and
functional obstruction (1). Its
most severe complication, Hirschsprung-associated enterocolitis
(HAEC), occurs in 17–50% of patients with HSCR. The preoperative
incidence of HAEC peaks at 60% in unscreened cohorts, while
postoperative rates range from 25–37% (2,3). In
total, >95% of cases occur in children <5 years of age in
multicenter studies (4). Asian
cohorts report lower postoperative rates (5,6) than
Western populations with HRSC (7),
with HAEC representing the leading cause of mortality in this
population. Clinically, HAEC manifests as fulminant diarrhea
progressing to sepsis, driven by mucosal barrier failure, dysbiosis
and immune hyperactivation (7,8).
Central to this pathophysiology is tight junction (TJ) dysfunction,
a ‘leaky epithelium’ phenotype that facilitates systemic pathogen
dissemination and fluid loss (9,10),
although its molecular underpinnings remain elusive.
The intestinal epithelial barrier is governed by
TJs, which orchestrate selective permeability to balance nutrient
absorption and pathogen exclusion (11). TJs are assembled from transmembrane
proteins, including claudins, occludin and junctional adhesion
molecules, cytoplasmic scaffolds, such as zonula occludens protein
1 (ZO-1) and cingulin, and the actomyosin cytoskeleton comprising
filamentous-actin (F-actin) and myosin II (12). Claudins form charge-selective
pores, as is the case with claudin-2, or barrier strands, such as
claudin-4, while occludin regulates macromolecular transport via
lipid raft-dependent endocytosis (13,14).
Notably, mechanical coupling between TJ proteins and the actomyosin
cytoskeleton is important for maintaining TJ ultrastructural
stability (15). Dysregulation of
this dynamic assembly system directly precipitates barrier collapse
(16).
Studies have shown that in bowel segments lacking
ganglion cells, intestinal barrier function is impaired and
exhibits decreased expression of ZO-1 and increased expression of
claudin-3 (17). These alterations
in TJ gene expression may result in increased epithelial
permeability, further promoting the development of HAEC. Compared
with other HSCR subgroups and patients with anorectal
malformations, patients with postoperative HAEC exhibit
significantly increased paracellular permeability during radical
surgery (18). Additionally, the
interaction between TJ proteins and the cytoskeleton, composed of
F-actin and myosin, is important for maintaining TJ structure and
function (13,15).
M1-polarized pro-inflammatory macrophages secrete
IL-1β and TNF-α, downregulating occludin and ZO-1 expression and
disrupting actomyosin-TJ coupling (19). This cytokine storm activates the
RhoA/Rho-associated protein kinase (ROCK) axis, which induces
pathological stress fiber assembly. The formation of these stress
fibers dismantles TJ architecture and accelerates luminal toxin
influx, propelling HAEC progression toward toxic megacolon
(20,21).
The pathogenesis of HAEC involves synergistic
defects in tight junction regulation (22) and actomyosin contractility
(23). Targeting TJ dynamic
assembly or modulating cytoskeletal remodeling may offer precision
therapeutic strategies to reverse the ‘leaky barrier’ phenotype,
thereby improving clinical outcomes in HAEC.
The claudin family consists of at least 27 members
that serve as important components of TJs, where they determine
paracellular ion permeability and charge selectivity. Claudins
exhibit tissue-specific expression patterns; for instance,
claudin-5 and −6 are predominant in renal podocytes, claudin-2, −4,
−8, −12 and −13 in bladder urothelium, claudin-2 to −5 in gastric
epithelium and claudin-1 to −19 in murine intestinal epithelium,
while human sigmoid colon primarily expresses claudin-1 to −5, −7
and −8. These proteins collectively contribute to barrier formation
and function (24).
Functionally, claudins are categorized as either
barrier-forming, such as claudin-1, −3 and −4, or pore-forming,
such as claudin-2 (16).
Disruption of this balance is implicated in intestinal pathologies.
For instance, in a benzalkonium chloride (BAC)-induced HSCR model,
claudin-3 is upregulated, compromising barrier integrity (17). Conversely, decreased claudin-4
expression is observed in postoperative patients with HAEC
(22). Although claudin-1 and −2
have been studied in necrotizing enterocolitis (NEC) (25,26),
their roles in HAEC remain to be fully elucidated.
Given their established importance in intestinal
barrier regulation, with claudin-1 enhancing barrier tightness,
claudin-2 modulating fluid and cation flux (27) and the demonstrated dysregulation of
claudin-3 and −4 in HAEC and HSCR, the present study focused on
claudin-1 to −4 to systematically investigate their collective and
individual roles in HAEC pathogenesis.
The inflammatory microenvironment reprograms the
expression of claudins through cytokines, directly disrupting the
homeostasis of TJ. For example, IL-1β activates multiple
transcription factors, such as NF-κB p50/p65, activating
transcription factor-2 and ETS domain-containing protein Elk-1, and
through the activation of mitogen-activated protein kinase kinase
kinase 1, the catalytic subunit of IKKβ, binds to the minimal
promoter region of myosin light chain kinase (MLCK). This
synergistically activates the MLCK gene and increases the
permeability of TJs (28), thereby
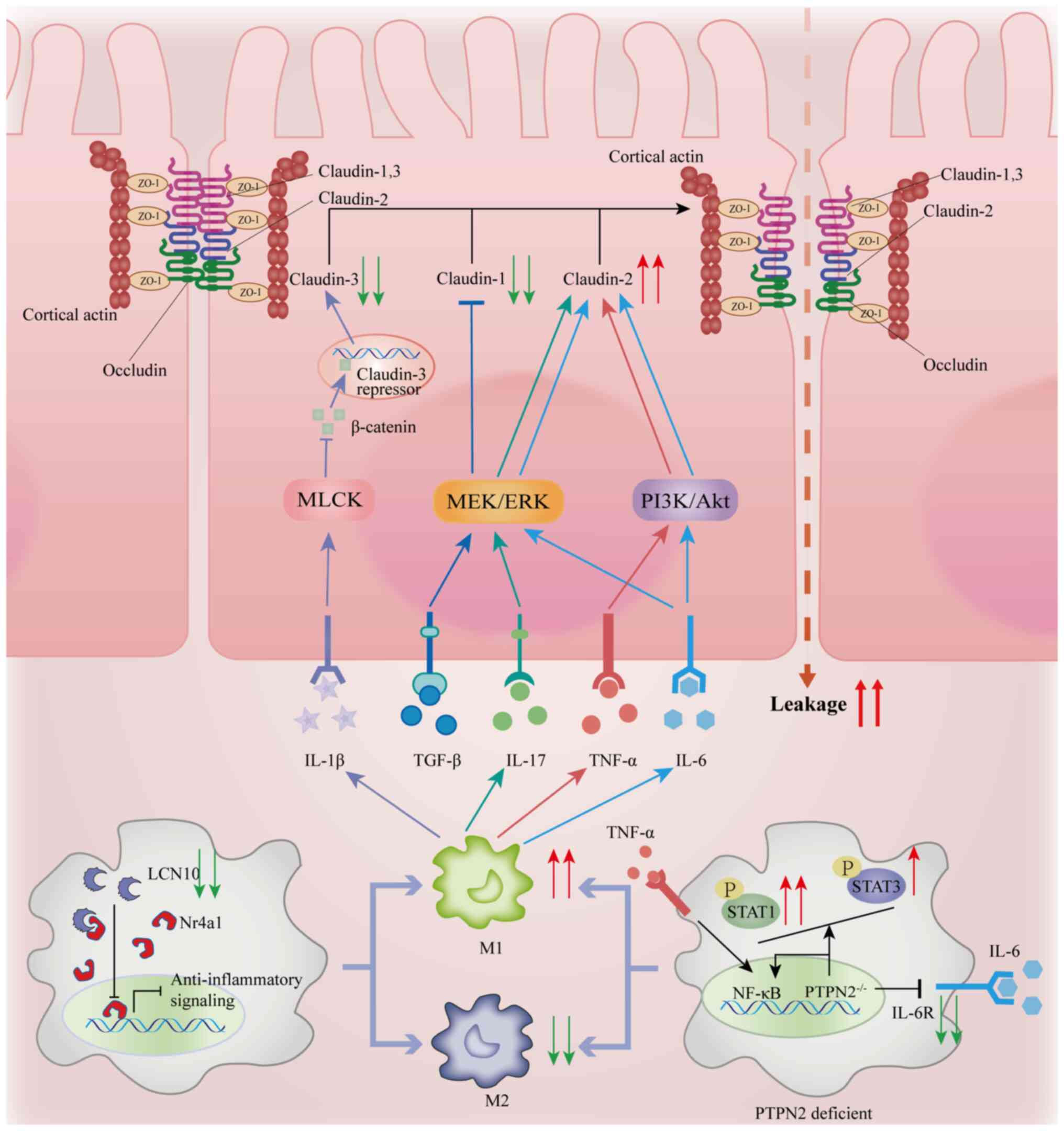
promoting intestinal inflammation (Fig. 1) (29). However, regarding the effect of
IL-1β on claudins, current research results remain inconsistent. A
study by Maria-Ferreira et al (30) found that the increase in TJ
permeability of Caco-2 cells induced by IL-1β is related to a
decrease in claudin-1 expression. By contrast, other studies have
shown that IL-1β activates β-catenin via the Wnt signaling pathway
or through an MLCK-dependent mechanism, thus downregulating the
expression of claudin-3 and leading to an increase in intestinal
permeability (31,32).
The increase in macrophages and the imbalance of the
M1/M2 macrophage ratio can affect intestinal barrier function by
influencing TJ proteins. Classically activated M1 macrophages
usually infiltrate the intestine during infection or inflammation
(37), and an increase in their
proportion has been observed during HAEC episodes (38,39).
M1 macrophages exacerbate intestinal barrier breakdown through a
dual mechanism: i) They secrete TNF-α and IL-6 (Fig. 1), promoting the expression of
claudin-2 and inducing the endocytosis and degradation of claudin-4
(40); and ii) M1 macrophages
activate heparanase to degrade heparan sulfate within the basement
membrane, altering the expression levels of TJ proteins, such as
occludin and ZO-1, and disrupting the TJ-extracellular matrix
interaction (TJ-ECM) anchoring, thus exacerbating intestinal
hyperpermeability (36–38). In macrophages with protein tyrosine
phosphatase non-receptor type 2 deficiency, the overactive
STAT1/NF-κB signaling pathway drives M1 macrophage polarization and
inhibits STAT3-mediated M2 macrophage differentiation by reducing
the expression of the IL-6 receptor (35,39).
The deficiency of lipocalin 10 (LCN10) further enhances this
process by inhibiting the nuclear receptor 4A1 pathway,
exacerbating M1 polarization (41)
and directly disrupting TJ-cytoskeleton coupling, which leads to
barrier leakage (Fig. 1) (42).
Based on the aforementioned mechanisms, multimodal
intervention strategies show clinical promise. In the BAC pig
model, specific small interfering RNA interference of claudin-3 can
reduce intestinal permeability and decrease the release of
inflammatory factors IL-1β and TNF-α (17). The heparanase inhibitor PG545
restores TJ-ECM anchoring and preclinical trials have shown that
the mucosal homeostasis of pediatric patients with HAEC is restored
upon treatment (43). The LCN10
protein increases the TER by regulating macrophage polarization
(41). These findings not only
reveal the multi-level pathogenic mechanisms of HAEC but also
provide a theoretical framework for the development of precise
therapies targeting the TJ network.
Occludin is a four-pass transmembrane protein
consisting of 522 amino acids, and its structural features
determine its dynamic regulatory function in TJs (44). The two extracellular loops mediate
cell-cell adhesion, while the cytoplasmic occludin/ELL (OCEL)
domain comprising 107 amino acids engages in interactions with
ZO-1, actin and kinases such as MLCK, imparting mechanical
stress-response capability to TJs (41,45).
Functional studies have demonstrated that occludin specifically
regulates the paracellular flux of macromolecules (46) without affecting the fundamental
structure of TJs; occludin knockout mice retain intact TJ
morphology (43,44), indicating that the core role of
occludin is as a ‘permeability regulator’ rather than a ‘structural
scaffold.’
The dynamic regulation of occludin in the membrane
is a key mechanism in the plasticity of the intestinal barrier.
Under inflammation or mechanical stress stimuli, occludin can be
directly internalized via vesicle-mediated endocytosis, leading to
the dissociation of the TJ supramolecular structure (47). This process is precisely regulated
by the lipid raft-scaffolding protein caveolin-1, which has an
N-terminus that forms a stable complex with the OCEL domain in the
cytoplasmic tail of occludin and modulates its membrane trafficking
cycle by altering its phosphorylation pattern (46,48).
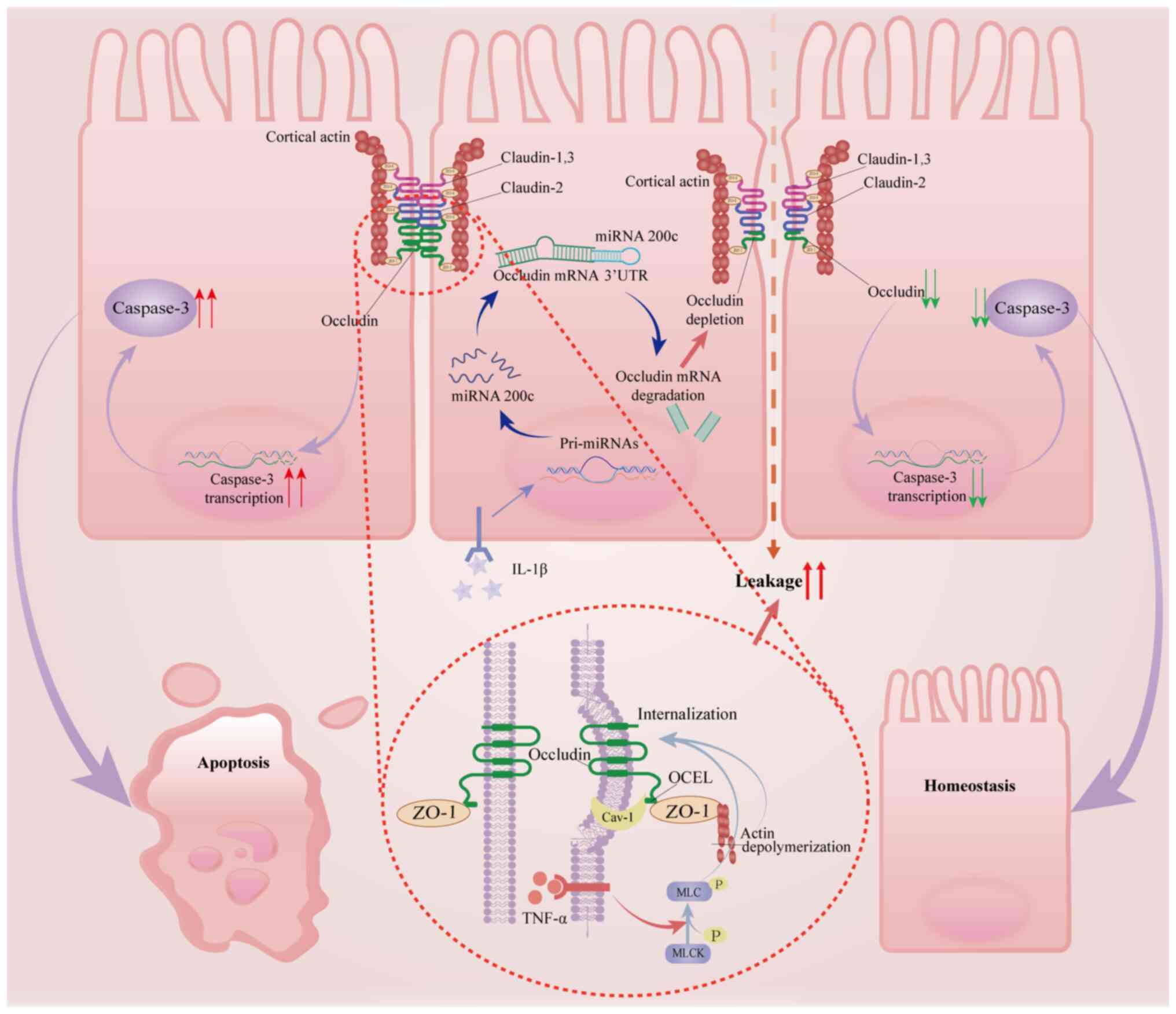
Inflammatory cytokines, such as TNF-α and IL-1β, trigger
hyperphosphorylation of occludin at Ser408 (49). This dysregulation promotes
clathrin-mediated endocytic internalization of occludin while
disrupting ZO-1 anchoring to the actin cytoskeleton,
synergistically increasing paracellular permeability in aganglionic
bowel segments (Fig. 2) (50). A study reported by Van Itallie
et al (51) revealed that
knockout of either occludin or caveolin-1 in Madin-Darby canine
kidney cells attenuated the disruption of the tight junction
barrier induced by inflammatory factors such as TNF-α. In
vivo experiments support that the absence of caveolin-1
significantly reduces the changes in TJ permeability mediated by
TNF-α, suggesting that the pathological function of occludin
depends on caveolin-1-mediated lipid raft-signaling microdomain
remodeling (51).
Notably, TNF-α triggers a caveolin-1-dependent
endocytic cascade by activating the MLCK pathway, a necessary
condition for TNF-α regulation of TJ structure and function
(50,52). Furthermore, actin depolymerization
can exacerbate intestinal barrier breakdown by promoting the
clathrin-mediated endocytosis of TJ components, including occludin,
and further destabilizing the barrier (53). IL-1β activates microRNA-200c-3p
through the MLCK/NF-κB pathway, which binds to the 3′UTR of
occludin mRNA and induces its degradation. This degradation results
in reduced occludin protein levels, thereby increasing intestinal
permeability (Fig. 2) (29). These cooperative mechanisms
together form the molecular basis of HAEC (54,55),
suggesting that targeting the caveolin-1-occludin axis or the MLCK
signaling network could be a potential new strategy for reversing
barrier damage.
Dysregulated expression of occludin is a common
feature of various intestinal diseases. During the active phase of
inflammatory bowel disease (IBD), occludin expression is reduced in
the colonic epithelium, which negatively correlates with barrier
permeability (56). A decrease in
occludin protein levels has also been observed in intestinal
samples from pediatric patients with NEC (57). In response to this pathological
mechanism, ROCK inhibitors can stabilize occludin membrane
localization and increase its expression, improving intestinal
barrier resistance in the BAC rat model (58).
As a representative member of the
membrane-associated guanylate kinase protein family, ZO-1 mediates
multidimensional regulation of TJs through its modular structure:
i) Three PDZ domains guide the topological localization of
claudin/occludin; ii) the SRC homology 3 (SH3) domain recruits
signaling kinases to regulate downstream pathways; and iii) the
actin-binding region (ABR) at the carboxyl terminus forms the
physical-functional coupling interface between TJs and the
cytoskeleton (60,61). Notably, in ZO-1 knockout models,
although TJ ultrastructure can spontaneously assemble (62), its maturation is significantly
delayed and accompanied by increased macromolecular transmembrane
leakage (63), suggesting that
ZO-1 is not a rigid scaffold for TJ assembly. Rather, ZO-1
dynamically regulates the liquid-liquid phase transition of protein
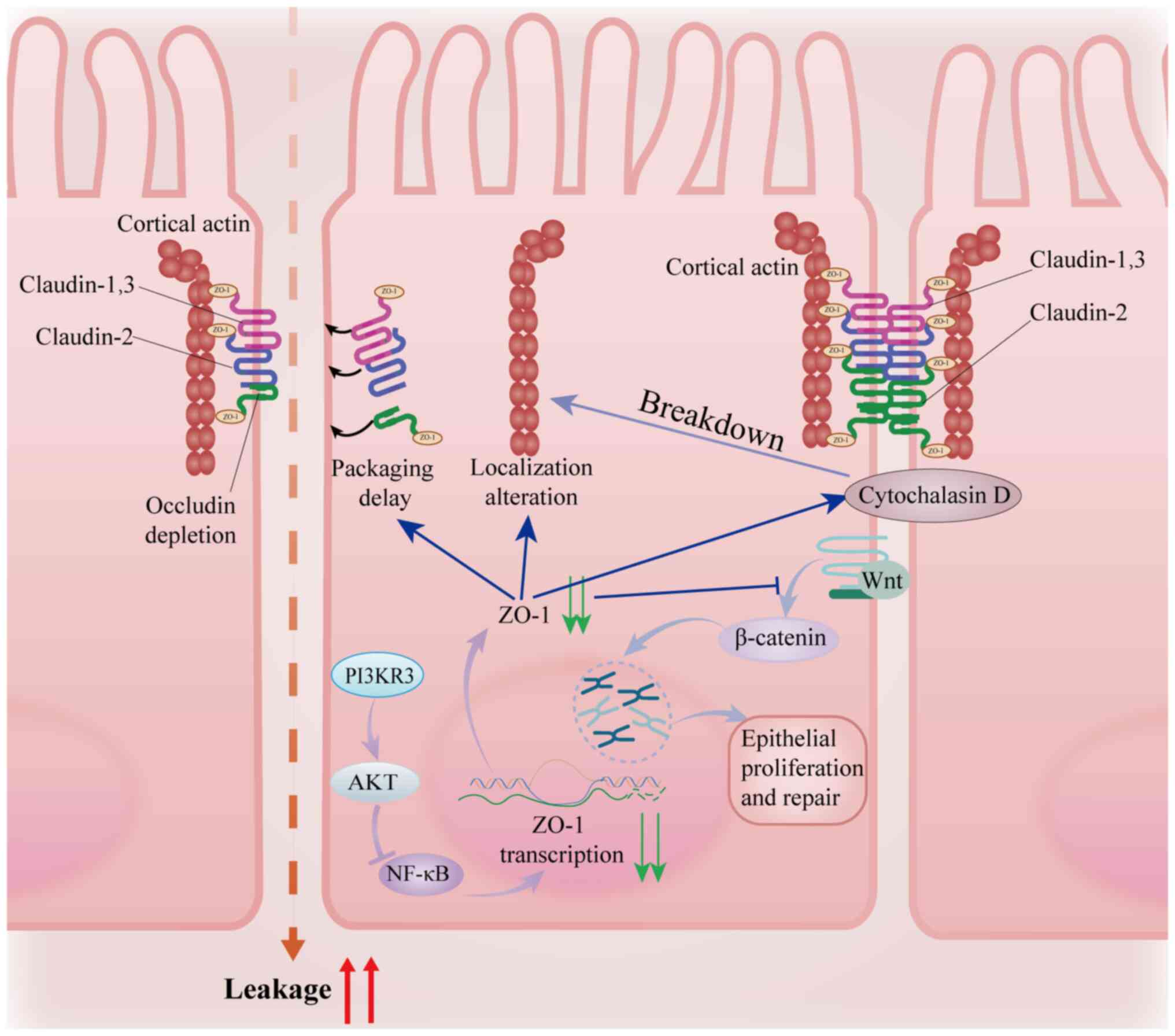
complexes through a phase separation mechanism. ZO-1 phase
separation, mediated through PDZ-SH3 domain-driven biomolecular
condensate formation, orchestrates rapid TJ assembly by
concentrating scaffolding proteins (Fig. 3) (60). Notably, pro-inflammatory cytokines
such as TNF-α disrupt this process by inducing hyperphosphorylation
of the intrinsically disordered regions of ZO-1, thereby dissolving
condensates and impairing barrier repair (64). In intestinal epithelia, TNF-α
reduces ZO-1 expression while increasing phosphorylation of its
intrinsically disordered regions, directly impairing barrier
function through delayed TJ assembly (65). This multifaceted
structural-functional characteristic positions ZO-1 as both a
molecular adapter and a signaling hub, with conformational changes
potentially regulating barrier plasticity through allosteric
effects.
ZO-1 regulates TJ plasticity through two synergistic
axes: i) ZO-1 directly interacts with claudin proteins and guides
the localization of their polymerization sites (66), coordinating the assembly of
transmembrane proteins; and ii) ZO-1 couples TJs to the
cytoskeletal network via its ABR, with actin depolymerization
having been shown to significantly increase paracellular
permeability (67). It is worth
noting that although ZO-1 deletion does not alter the total actin
content of the cell, it disrupts its spatial organization (68), highlighting the role of ZO-1 in the
topological regulation of the cytoskeleton rather than providing
only mechanical anchorage. This dual regulatory mechanism may
provide insights into how ZO-1 coordinates acute barrier remodeling
and chronic fibrosis during mucosal repair.
Upon injury, ZO-1 coordinates epithelial repair
through the Wnt/β-catenin signaling pathway (Fig. 3) (69). ZO-1 knockdown reduces β-catenin
nuclear translocation and impairs wound healing (69). Clinical cohort studies have shown a
strong positive correlation between ZO-1 expression in intestinal
epithelium and endoscopic healing scores in patients with IBD
(69), while ZO-1 mRNA levels in
exosomes from fecal samples of neonates with NEC can predict the
risk of intestinal perforation (57). These findings collectively suggest
that ZO-1 not only mediates barrier integrity but also acts as an
important node linking homeostasis maintenance and regenerative
repair through mechanical-chemical signaling. Its functional
depletion may trigger a cycle of barrier defect-induced chronic
inflammation.
Although ZO-1 plays a central role in barrier
regulation, its pathological mechanism in congenital
megacolon-associated fatal complications, such as HAEC, remains yet
to be fully elucidated. Given that HAEC is characterized by the
disruption of TJ structures (17),
targeting the regulation of ZO-1 interactions offers a promising
avenue for therapeutic innovation. Future research may explore the
spatiotemporal analysis of ZO-1 dynamics in HAEC models to identify
important nodes of dysfunction, for example ABR-actin uncoupling
and other pathological processes. Furthermore, peptides derived
from the ABR could be designed to selectively block pathological
cytoskeletal remodeling while preserving barrier integrity. The
systematic integration of these research strategies will drive
translational medical progress from mechanism elucidation to
precise intervention in HAEC.
The plasticity of the intestinal epithelial cell
barrier relies on the spatiotemporal reorganization of the actin
cytoskeleton. The steady-state conversion between globular-actin
monomers and F-actin polymers is finely regulated by the
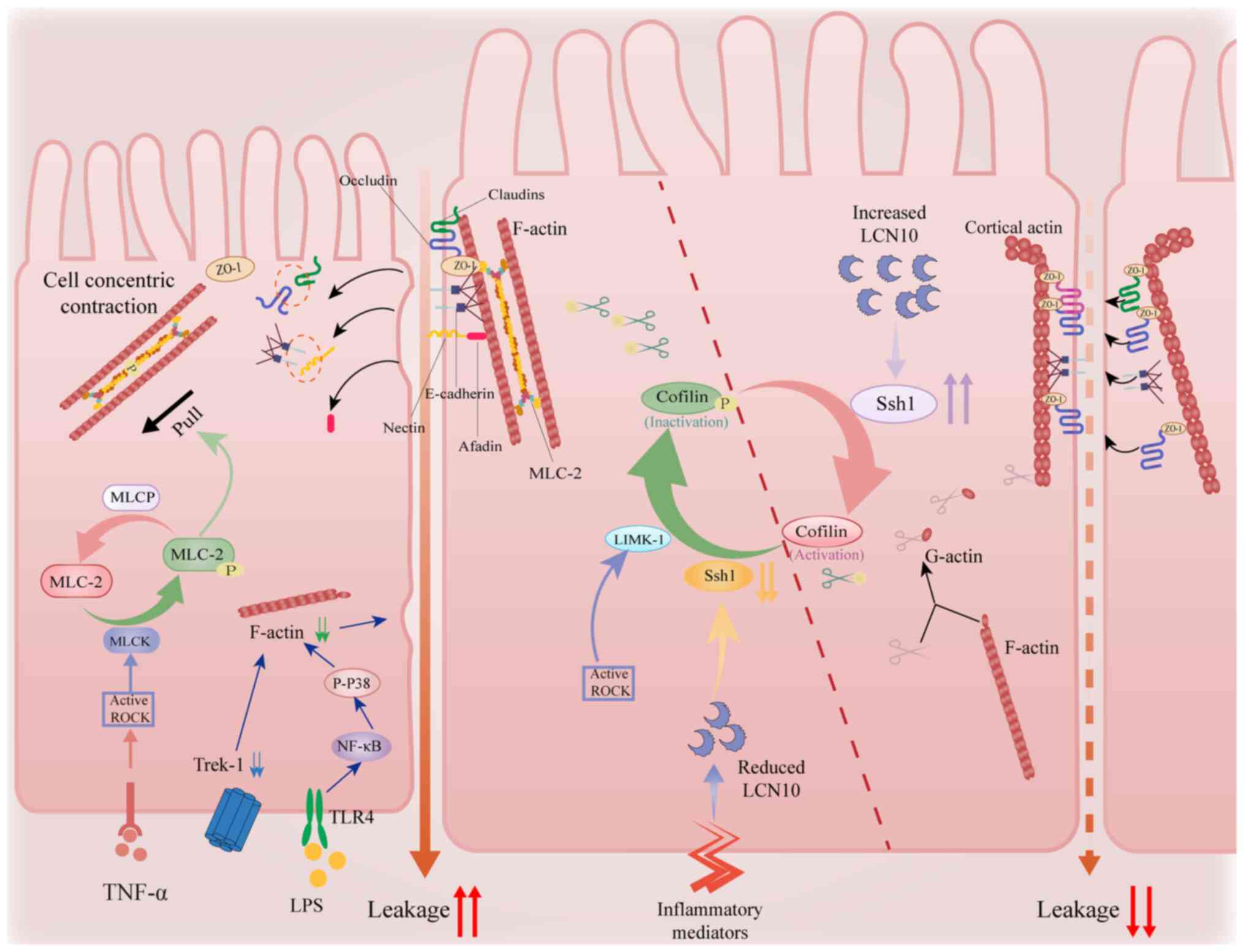
phosphorylation cycle of cofilin. LIM kinase (LIMK) mediates
cofilin phosphorylation, thereby inhibiting its activity, while the
phosphatase slingshot homologue 1 (Ssh1) activates cofilin through
dephosphorylation, promoting the disassembly of F-actin (Fig. 4) (70,71).
Imbalance in this system can directly disrupt the membrane
localization of TJ proteins, such as ZO-1 and occludin, leading to
an increase in paracellular permeability (23,72).
ZO-1 forms a mechanical coupling interface with
F-actin through its carboxy-terminal ABR. Loss of ZO-1 function
results in the disruption of actin filament integrity (68). MLCK and ROCK mediate actomyosin
contraction through phosphorylation of myosin light chains, which,
in turn, dissociates the ZO-1 ABR from F-actin, ultimately
triggering the opening of the paracellular pathway (73–75).
Notably, MLCK inhibitors completely reverse the ZO-1
internalization phenotype, while ROCK inhibitors partially restore
barrier function, indicating the signaling pathway specificity of
the ZO-1/F-actin interaction (76,77).
This mechanical transduction property positions ZO-1 as a molecular
sensor linking the mechanical microenvironment with barrier
plasticity.
Pro-inflammatory factors exacerbate barrier damage
via multifaceted regulation of actin dynamics: i) IFN-γ enhances
actomyosin contractility via a ROCK-dependent pathway, leading to
the internalization of ZO-1 and occluding (78); ii) a dynamic interaction exists
between Twik-related K+ channel-1 (Trek-1) and the actin
cytoskeleton. In colonic epithelia, Trek-1 stabilizes cortical
actin via direct binding to F-actin. Trek-1 deficiency reduces
F-actin density, increasing paracellular permeability (79). Hypoganglionic segments in HSCR show
lower Trek-1 expression compared with normoganglionic bowels. This
deficit impairs mechanotransduction in the neurogenic
microenvironment, exacerbating barrier dysfunction during HAEC
flares (Fig. 4) (80). Similarly, activation of the
Toll-like receptor 4/phosphorylated p38/NF-κB signaling pathway
also results in reduced F-actin expression (81), influencing the opening of
intercellular gaps (Fig. 4); and
(iii) lipopolysaccharides (LPS) inhibit EGFR phosphorylation,
thereby hindering its activity in protective actin reorganization
(82). Collectively, these
pathways form a positive feedback loop of inflammatory
signals/cytoskeletal remodeling/barrier breakdown, providing a
mechanistic explanation for the chronic progression of IBD.
The lipid carrier protein LCN10, a secreted protein
expressed in macrophages, endothelial and epithelial cells
(64), serves as an important
regulator of cytoskeletal dynamics. Mechanistically, LCN10
activates Ssh1 phosphatase to dephosphorylate cofilin, thereby
enhancing F-actin depolymerization, a process important for TJ
protein redistribution in barrier dysfunction (42,83).
Notably, this LCN10/Ssh1/cofilin pathway-dependent regulation is
conserved across vascular and intestinal barriers, where LCN10
deficiency exacerbates inflammation-induced permeability (42). Pathologically, pro-inflammatory
stimuli, such as LPS and IFN-γ, suppress LCN10 expression by
>80% in intestinal macrophages (41), which may drive HAEC progression by
disrupting actin-mediated TJ stability.
Furthermore, in cohorts of pediatric patients with
HAEC (n=75), stenotic bowel segments exhibit 73% lower
phosphorylated cofilin levels vs. controls (84,85).
Mechanistically, LCN10 activates Ssh1 via high-affinity binding to
low-density lipoprotein receptor-related protein 2, restoring
cofilin-mediated actin dynamics to reduce barrier leakage (42). These findings nominate the
LCN10/Ssh1/cofilin axis as a theragnostic target for HAEC. Based on
these findings, small molecule agonists targeting LCN10 may
overcome the current lack of cytoskeletal targets in HAEC
therapies. Furthermore, we hypothesize that combinatory strategies
involving ZO-1 ABR stabilizers could produce synergistic barrier
repair effects.
The present study elucidates the molecular
pathogenesis of HAEC, revealing that intestinal barrier dysfunction
stems from TJ protein dysregulation, including claudin-2/4
imbalance, occludin endocytosis via the MLCK/NF-κB pathway and
ZO-1-cytoskeleton uncoupling, coupled with inflammatory
mediator-driven actin remodeling through the cofilin
phosphorylation cycle. The identification of the LCN10/Ssh1/cofilin
axis and TJ-cytoskeleton interactions provides mechanistic insights
into HAEC pathogenesis and potential therapeutic targets. The
scarcity of targeted research on TJ and cytoskeletal proteins in
HAEC underscores the translational significance of the present
study. Future studies should investigate subcellular TJ protein
dynamics, develop small-molecule modulators of the LCN10/Ssh1
pathway, validate their pharmacokinetic and toxicological profiles
and explore combination therapies targeting both barrier repair and
inflammation, ultimately translating these findings into clinical
strategies for HAEC prevention and treatment.
Not applicable.
The present study was supported by the National Natural Science
Foundation of China (grant nos. 81700497 and 81873848) and the
Hubei Natural Science Foundation (grant nos. 2024AFB668 and
2021CFB264).
Not applicable.
ST, YW and LL contributed to the conception and
design of the study, and provided administrative support. LZ, ShaC,
YZ, DY, KL, YL and ShuC participated in data analysis and
visualization, including the creation and interpretation of
graphical figures. SL and CW were major contributors in drafting
and revising the manuscript. Data authentication is not applicable.
All authors read and approved the final version of the
manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Montalva L, Cheng LS, Kapur R, Langer JC,
Berrebi D, Kyrklund K, Pakarinen M, de Blaauw I, Bonnard A and
Gosain A: Hirschsprung disease. Nat Rev Dis Primers. 9:542023.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gosain A, Frykman PK, Cowles RA, Horton J,
Levitt M, Rothstein DH, Langer JC and Goldstein AM; American
Pediatric Surgical Association Hirschsprung Disease Interest Group,
: Guidelines for the diagnosis and management of
Hirschsprung-associated enterocolitis. Pediatr Surg Int.
33:517–521. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Nakamura H, Tomuschat C, Coyle D, O'Donnel
AM, Lim T and Puri P: Altered goblet cell function in
Hirschsprung's disease. Pediatr Surg Int. 34:121–128. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lewit RA, Veras LV, Cowles RA, Fowler K,
King S, Lapidus-Krol E, Langer JC, Park CJ, Youssef F, Vavilov S
and Gosain A: Reducing Underdiagnosis of Hirschsprung-Associated
Enterocolitis: A Novel Scoring System. J Surg Res. 261:253–260.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Feng W, Zhang B, Fan L, Song A, Hou J, Die
X, Liu W, Wang Y and Guo Z: Clinical characteristics and influence
of postoperative Hirschsprung-associated enterocolitis:
Retrospective study at a tertiary children's hospital. Pediatr Surg
Int. 40:1062024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wu L, Gao Y, Zhou R, Xiao P, Zhang Z, Li
B, Pierro A, Li L, Jiang Q and Li Q: Predictive value of plasma
zonulin for postoperative Hirschsprung-associated enterocolitis.
World J Pediatr Surg. 8:e0010572025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hagens J, Reinshagen K and Tomuschat C:
Prevalence of Hirschsprung-associated enterocolitis in patients
with Hirschsprung disease. Pediatr Surg Int. 38:3–24. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Li S, Zhang Y, Li K, Liu Y, Chi S, Wang Y
and Tang S: Update on the pathogenesis of the
hirschsprung-associated enterocolitis. Int J Mol Sci. 24:46022023.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jiao CL, Chen XY and Feng JX: Novel
Insights into the Pathogenesis of Hirschsprung's-associated
Enterocolitis. Chin Med J (Engl). 129:1491–1497. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cong X and Kong W: Endothelial tight
junctions and their regulatory signaling pathways in vascular
homeostasis and disease. Cell Signal. 66:1094852020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chelakkot C, Ghim J and Ryu SH: Mechanisms
regulating intestinal barrier integrity and its pathological
implications. Exp Mol Med. 50:1–9. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Suzuki T: Regulation of intestinal
epithelial permeability by tight junctions. Cell Mol Life Sci.
70:631–659. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
König J, Wells J, Cani PD, García-Ródenas
CL, MacDonald T, Mercenier A, Whyte J, Troost F and Brummer RJ:
Human intestinal barrier function in health and disease. Clin
Transl Gastroenterol. 7:e1962016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsukita S, Furuse M and Itoh M:
Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol.
2:285–293. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Van Itallie CM and Anderson JM:
Architecture of tight junctions and principles of molecular
composition. Semin Cell Dev Biol. 36:157–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Garcia-Hernandez V, Quiros M and Nusrat A:
Intestinal epithelial claudins: Expression and regulation in
homeostasis and inflammation. Ann N Y Acad Sci. 1397:66–79. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Arnaud AP, Hascoet J, Berneau P, LeGouevec
F, Georges J, Randuineau G, Formal M, Henno S and Boudry G: A
piglet model of iatrogenic rectosigmoid hypoganglionosis reveals
the impact of the enteric nervous system on gut barrier function
and microbiota postnatal development. J Pediatr Surg. 56:337–345.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dariel A, Grynberg L, Auger M, Lefèvre C,
Durand T, Aubert P, Le Berre-Scoul C, Venara A, Suply E, Leclair
MD, et al: Analysis of enteric nervous system and intestinal
epithelial barrier to predict complications in Hirschsprung's
disease. Sci Rep. 10:217252020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen X, Meng X, Zhang H, Feng C, Wang B,
Li N, Abdullahi KM, Wu X, Yang J, Li Z, et al: Intestinal
proinflammatory macrophages induce a phenotypic switch in
interstitial cells of Cajal. J Clin Invest. 130:6443–6456. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pall H: Advances in pediatric
gastroenterology. Pediatr Clin North Am. 68:xix–xx. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Roorda D, Oosterlaan J, van Heurn E and
Derikx JPM: Risk factors for enterocolitis in patients with
Hirschsprung disease: A retrospective observational study. J
Pediatr Surg. 56:1791–1798. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Abe K, Takeda M, Ishiyama A, Shimizu M,
Goto H, Iida H, Fujimoto T, Ueda-Abe E, Yamada S, Fujiwara K, et
al: Impact of epithelial claudin-4 and leukotriene B4 receptor 2 in
normoganglionic hirschsprung disease colon on post pull-through
enterocolitis. J Pediatr Surg. 60:1619002025. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Perrin L and Matic Vignjevic D: The
emerging roles of the cytoskeleton in intestinal epithelium
homeostasis. Semin Cell Dev Biol. 150-151:23–27. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Günzel D and Yu AS: Claudins and the
modulation of tight junction permeability. Physiol Rev. 93:525–569.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bu C, Hu M, Su Y, Yuan F, Zhang Y, Xia J,
Jia Z and Zhang L: Cell-permeable JNK-inhibitory peptide regulates
intestinal barrier function and inflammation to ameliorate
necrotizing enterocolitis. J Cell Mol Med. 28:e185342021.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gunasekaran A, Eckert J, Burge K, Zheng W,
Yu Z, Kessler S, de la Motte C and Chaaban H: Hyaluronan 35 kDa
enhances epithelial barrier function and protects against the
development of murine necrotizing enterocolitis. Pediatr Res.
87:1177–1184. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ganapathy AS, Saha K, Suchanec E, Singh V,
Verma A, Yochum G, Koltun W, Nighot M, Ma T and Nighot P: AP2M1
mediates autophagy-induced CLDN2 (claudin 2) degradation through
endocytosis and interaction with LC3 and reduces intestinal
epithelial tight junction permeability. Autophagy. 18:2086–2103.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Al-Sadi R, Ye D, Said HM and Ma TY:
Cellular and molecular mechanism of interleukin-1β modulation of
Caco-2 intestinal epithelial tight junction barrier. J Cell Mol
Med. 15:970–982. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rawat M, Nighot M, Al-Sadi R, Gupta Y,
Viszwapriya D, Yochum G, Koltun W and Ma TY: IL1B increases
intestinal tight junction permeability by Up-regulation of
MIR200C-3p, which degrades occludin mRNA. Gastroenterology.
159:1375–1389. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Maria-Ferreira D, Nascimento AM, Cipriani
TR, Santana-Filho AP, Watanabe PDS, Sant Ana DMG, Luciano FB,
Bocate KCP, van den Wijngaard RM, Werner MFP and Baggio CH:
Rhamnogalacturonan, a chemically-defined polysaccharide, improves
intestinal barrier function in DSS-induced colitis in mice and
human Caco-2 cells. Sci Rep. 8:122612018. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Haines RJ, Beard RS Jr, Chen L, Eitnier RA
and Wu MH: Interleukin-1β mediates β-Catenin-driven downregulation
of claudin-3 and barrier dysfunction in caco2 cells. Dig Dis Sci.
61:2252–2261. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahmad R, Kumar B, Chen Z, Chen X, Müller
D, Lele SM, Washington MK, Batra SK, Dhawan P and Singh AB: Loss of
claudin-3 expression induces IL6/gp130/Stat3 signaling to promote
colon cancer malignancy by hyperactivating Wnt/β-catenin signaling.
Oncogene. 36:6592–6604. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mankertz J, Amasheh M, Krug SM, Fromm A,
Amasheh S, Hillenbrand B, Tavalali S, Fromm M and Schulzke JD:
TNFalpha up-regulates claudin-2 expression in epithelial HT-29/B6
cells via phosphatidylinositol-3-kinase signaling. Cell Tissue Res.
336:67–77. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Barmeyer C, Fromm M and Schulzke JD:
Active and passive involvement of claudins in the pathophysiology
of intestinal inflammatory diseases. Pflugers Arch. 469:15–26.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee SH: Intestinal permeability regulation
by tight junction: Implication on inflammatory bowel diseases.
Intest Res. 13:11–18. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Raju P, Shashikanth N, Tsai PY,
Pongkorpsakol P, Chanez-Paredes S, Steinhagen PR, Kuo WT, Singh G,
Tsukita S and Turner JR: Inactivation of paracellular
cation-selective claudin-2 channels attenuates immune-mediated
experimental colitis in mice. J Clin Invest. 130:5197–5208. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bain CC, Scott CL, Uronen-Hansson H,
Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW
and Mowat AM: Resident and pro-inflammatory macrophages in the
colon represent alternative context-dependent fates of the same
Ly6Chi monocyte precursors. Mucosal Immunol. 6:498–510. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Meng X, Xiao J, Wang J, Sun M, Chen X, Wu
L, Feng C, Zhuansun D, Yang J, Wu X, et al: Mesenchymal stem cells
attenuates hirschsprung diseases-associated enterocolitis by
reducing M1 macrophages infiltration via COX-2 dependent mechanism.
J Pediatr Surg. 59:1498–1514. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zheng Z, Lin L, Lin H, Zhou J, Wang Z,
Wang Y, Chen J, Lai C, Li R, Shen Z, et al: Acetylcholine from tuft
cells promotes M2 macrophages polarization in
Hirschsprung-associated enterocolitis. Front Immunol.
16:15599662025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Spalinger MR, Sayoc-Becerra A, Santos AN,
Shawki A, Canale V, Krishnan M, Niechcial A, Obialo N, Scharl M, Li
J, et al: PTPN2 regulates interactions between macrophages and
intestinal epithelial cells to promote intestinal barrier function.
Gastroenterology. 159:1763–1777.e14. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li Q, Li Y, Huang W, Wang X, Liu Z, Chen
J, Fan Y, Peng T, Sadayappan S, Wang Y and Fan GC: Loss of
lipocalin 10 exacerbates diabetes-induced cardiomyopathy via
disruption of Nr4a1-mediated anti-inflammatory response in
macrophages. Front Immunol. 13:9303972022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhao H, Wang P, Wang X, Du W, Yang HH, Liu
Y, Cui SN, Huang W, Peng T, Chen J, et al: Lipocalin 10 is
essential for protection against inflammation-triggered vascular
leakage by activating LDL receptor-related protein 2-slingshot
homologue 1 signalling pathway. Cardiovasc Res. 119:1981–1996.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Capozzi A, Riitano G, Recalchi S,
Manganelli V, Costi R, Saccoliti F, Pulcinelli F, Garofalo T,
Misasi R, Longo A, et al: Effect of heparanase inhibitor on tissue
factor overexpression in platelets and endothelial cells induced by
anti-β2-GPI antibodies. J Thromb Haemost. 19:2302–2313. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Furuse M, Hirase T, Itoh M, Nagafuchi A,
Yonemura S and Tsukita S and Tsukita S: Occludin: A novel integral
membrane protein localizing at tight junctions. J Cell Boil. 123((6
Pt 2)): 1777–1788. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tugal D, Liao X and Jain MK:
Transcriptional control of macrophage polarization. Arterioscler,
Thromb, Vasc Biol. 33:1135–1144. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Buckley A and Turner JR: Cell biology of
tight junction barrier regulation and mucosal disease. Cold Spring
Harb Perspect Biol. 10:a0293142018. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Shimizu Y, Shirasago Y, Kondoh M, Suzuki
T, Wakita T, Hanada K, Yagi K and Fukasawa M: Monoclonal antibodies
against occludin completely prevented hepatitis C virus infection
in a mouse model. J Virol. 92:e02258–17. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nusrat A, Chen JA, Foley CS, Liang TW, Tom
J, Cromwell M, Quan C and Mrsny RJ: The coiled-coil domain of
occludin can act to organize structural and functional elements of
the epithelial tight junction. J Biol Chem. 275:29816–29822. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Srivastava AK, Venkata BS, Sweat YY, Rizzo
HR, Jean-François L, Zuo L, Kurgan KW, Moore P, Shashikanth N, Smok
I, et al: Serine 408 phosphorylation is a molecular switch that
regulates structure and function of the occludin α-helical bundle.
Proc Natl Acad Sci USA. 119:e22046181192022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Buschmann MM, Shen L, Rajapakse H, Raleigh
DR, Wang Y, Wang Y, Lingaraju A, Zha J, Abbott E, McAuley EM, et
al: Occludin OCEL-domain interactions are required for maintenance
and regulation of the tight junction barrier to macromolecular
flux. Mol Biol Cell. 24:3056–3068. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Van Itallie CM, Fanning AS, Holmes J and
Anderson JM: Occludin is required for cytokine-induced regulation
of tight junction barriers. J Cell Sci. 123:2844–2852. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Marchiando AM, Shen L, Graham WV, Weber
CR, Schwarz BT, Austin JR II, Raleigh DR, Guan Y, Watson AJ,
Montrose MH and Turner JR: Caveolin-1-dependent occludin
endocytosis is required for TNF-induced tight junction regulation
in vivo. J Cell Biol. 189:111–126. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shen L and Turner JR: Actin
depolymerization disrupts tight junctions via caveolae-mediated
endocytosis. Mol Biol Cell. 16:3919–3936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Budianto IR, Kusmardi K, Maulana AMuh,
Arumugam S, Afrin R and Soetikno V: Paneth-like cells disruption
and intestinal dysbiosis in the development of enterocolitis in an
iatrogenic rectosigmoid hypoganglionosis rat model. Front Surg.
11:14079482024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nakamura H, O'Donnell AM, Tomuschat C,
Coyle D and Puri P: Altered expression of caveolin-1 in the colon
of patients with Hirschsprung's disease. Pediatr Surg Int.
35:929–934. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zeissig S, Bürgel N, Günzel D, Richter J,
Mankertz J, Wahnschaffe U, Kroesen AJ, Zeitz M, Fromm M and
Schulzke JD: Changes in expression and distribution of claudin 2, 5
and 8 lead to discontinuous tight junctions and barrier dysfunction
in active Crohn's disease. Gut. 56:61–72. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhang X, Zhang Y, He Y, Zhu X, Ai Q and
Shi Y: β-glucan protects against necrotizing enterocolitis in mice
by inhibiting intestinal inflammation, improving the gut barrier,
and modulating gut microbiota. J Transl Med. 21:142023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Grothaus JS, Ares G, Yuan C, Wood DR and
Hunter CJ: Rho kinase inhibition maintains intestinal and vascular
barrier function by upregulation of occludin in experimental
necrotizing enterocolitis. Am J Physiol Gastrointest Liver Physiol.
315:G514–G528. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Kuo WT, Shen L, Zuo L, Shashikanth N, Ong
MLDM, Wu L, Zha J, Edelblum KL, Wang Y, Wang Y, et al:
Inflammation-induced Occludin Downregulation Limits Epithelial
Apoptosis by Suppressing Caspase-3 Expression. Gastroenterology.
157:1323–1337. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Spadaro D, Le S, Laroche T, Mean I, Jond
L, Yan J and Citi S: Tension-dependent stretching activates ZO-1 to
control the junctional localization of its interactors. Curr Biol.
27:3783–3795.e8. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Rouaud F, Sluysmans S, Flinois A, Shah J,
Vasileva E and Citi S: Scaffolding proteins of vertebrate apical
junctions: Structure, functions and biophysics. Biochim Biophys
Acta Biomembr. 1862:1833992020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Umeda K, Matsui T, Nakayama M, Furuse K,
Sasaki H, Furuse M and Tsukita S: Establishment and
characterization of cultured epithelial cells lacking expression of
ZO-1. J Biol Chem. 279:44785–44794. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Otani T, Nguyen TP, Tokuda S, Sugihara K,
Sugawara T, Furuse K, Miura T, Ebnet K and Furuse M: Claudins and
JAM-A coordinately regulate tight junction formation and epithelial
polarity. J Cell Biol. 218:3372–3396. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sun S and Zhou J: Phase separation as a
therapeutic target in tight junction-associated human diseases.
Acta Pharmacol Sin. 41:1310–1313. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhang J, Lei H, Hu X and Dong W:
Hesperetin ameliorates DSS-induced colitis by maintaining the
epithelial barrier via blocking RIPK3/MLKL necroptosis signaling.
Eur J Pharmacol. 873:1729922020. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Umeda K, Ikenouchi J, Katahira-Tayama S,
Furuse K, Sasaki H, Nakayama M, Matsui T and Tsukita S, Furuse M
and Tsukita S: ZO-1 and ZO-2 independently determine where claudins
are polymerized in tight-junction strand formation. Cell.
126:741–754. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bentzel CJ, Hainau B, Ho S, Hui SW,
Edelman A, Anagnostopoulos T and Benedetti EL: Cytoplasmic
regulation of tight-junction permeability: Effect of plant
cytokinins. Am J Physiol. 239:C75–C89. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Van Itallie CM, Fanning AS, Bridges A and
Anderson JM: ZO-1 stabilizes the tight junction solute barrier
through coupling to the perijunctional cytoskeleton. Mol Biol Cell.
20:3930–3940. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kuo WT, Zuo L, Odenwald MA, Madha S, Singh
G, Gurniak CB, Abraham C and Turner JR: The tight junction protein
ZO-1 is dispensable for barrier function but critical for effective
mucosal repair. Gastroenterology. 161:1924–1939. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Grintsevich EE and Reisler E: Drebrin
inhibits cofilin-induced severing of F-actin. Cytoskeleton
(Hoboken). 71:472–483. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Suzuki K, Lareyre JJ, Sánchez D, Gutierrez
G, Araki Y, Matusik RJ and Orgebin-Crist MC: Molecular evolution of
epididymal lipocalin genes localized on mouse chromosome 2. Gene.
339:49–59. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Van Itallie CM, Tietgens AJ, Krystofiak E,
Kachar B and Anderson JM: A complex of ZO-1 and the BAR-domain
protein TOCA-1 regulates actin assembly at the tight junction. Mol
Biol Cell. 26:2769–2787. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Yu D, Marchiando AM, Weber CR, Raleigh DR,
Wang Y, Shen L and Turner JR: MLCK-dependent exchange and actin
binding region-dependent anchoring of ZO-1 regulate tight junction
barrier function. Proc Natl Acad Sci USA. 107:8237–8241. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Turner JR, Rill BK, Carlson SL, Carnes D,
Kerner R, Mrsny RJ and Madara JL: Physiological regulation of
epithelial tight junctions is associated with myosin light-chain
phosphorylation. Am J Physiol. 273:C1378–C1385. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Walsh SV, Hopkins AM, Chen J, Narumiya S,
Parkos CA and Nusrat A: Rho kinase regulates tight junction
function and is necessary for tight junction assembly in polarized
intestinal epithelia. Gastroenterology. 121:566–579. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Odenwald MA, Choi W, Kuo WT, Singh G,
Sailer A, Wang Y, Shen L, Fanning AS and Turner JR: The scaffolding
protein ZO-1 coordinates actomyosin and epithelial apical
specializations in vitro and in vivo. J Biol Chem. 293:17317–17335.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Kuo W, Odenwald MA, Turner JR and Zuo L:
Tight junction proteins occludin and ZO-1 as regulators of
epithelial proliferation and survival. Ann N Y Acad Sci.
1514:21–33. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Ma TY, Boivin MA, Ye D, Pedram A and Said
HM: Mechanism of TNF-{alpha} modulation of Caco-2 intestinal
epithelial tight junction barrier: Role of myosin light-chain
kinase protein expression. Am J Physiol Gastrointest Liver Physiol.
288:G422–G430. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Roan E, Waters CM, Teng B, Ghosh M and
Schwingshackl A: The 2-pore domain potassium channel TREK-1
regulates stretch-induced detachment of alveolar epithelial cells.
PLoS One. 9:e894292014. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Tomuschat C, O'Donnell AM, Coyle D, Dreher
N, Kelly D and Puri P: Altered expression of a two-pore domain
(K2P) mechano-gated potassium channel TREK-1 in Hirschsprung's
disease. Pediatr Res. 80:729–733. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zheng Z, Gao M, Tang C, Huang L, Gong Y,
Liu Y and Wang J: E.coli JM83 damages the mucosal barrier in Ednrb
knockout mice to promote the development of Hirschsprung-associated
enterocolitis via activation of TLR4/p-p38/NF-κB signaling. Mol Med
Rep. 25:1682022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Samak G, Aggarwal S and Rao RK: ERK is
involved in EGF-mediated protection of tight junctions, but not
adherens junctions, in acetaldehyde-treated Caco-2 cell monolayers.
Am J Physiol Gastrointest Liver Physiol. 301:G50–G59. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Krndija D, El Marjou F, Guirao B, Richon
S, Leroy O, Bellaiche Y, Hannezo E and Matic Vignjevic D: Active
cell migration is critical for steady-state epithelial turnover in
the gut. Science. 365:705–710. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Lappalainen P, Kotila T, Jégou A and
Romet-Lemonne G: Biochemical and mechanical regulation of actin
dynamics. Nat Rev Mol Cell Biol. 23:836–852. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhou WK, Qu Y, Liu YM, Gao MJ, Tang CY,
Huang L, Du Q and Yin J: The abnormal phosphorylation of the Rac1,
Lim-kinase 1, and Cofilin proteins in the pathogenesis of
Hirschsprung's disease. Bioengineered. 13:8548–8557. 2022.
View Article : Google Scholar : PubMed/NCBI
|