Introduction
Sepsis represents a serious medical condition marked
by the sudden failure of multiple organ systems due to an
inappropriate response to infection by the host (1). Sepsis has become one of the primary
contributors to severe illness and mortality worldwide (2). Based on a 2020 research report
involving the Intensive Care Units (ICUs) of 44 hospitals
nationwide, the Chinese Expert Consensus on Early Prevention and
Blockade of Sepsis in Emergency Medicine indicated that the
incidence rate in ICUs was 20.6%, with a 90-day mortality rate of
35.5% (3). Notably, nearly 60% of
these patients experience co-infection of the lungs (4), indicating that the lungs may be the
most susceptible and severely affected organ in sepsis. Acute lung
injury (ALI) impacts 25–50% of patients suffering from sepsis and,
if not treated promptly and effectively, can progress to acute
respiratory distress syndrome (ARDS), a more severe condition
(5). Effective sepsis-induced
acute lung injury (SALI) therapies remain limited, prompting urgent
translational research. The pathological characteristics of ALI
involve extensive inflammation resulting in harm to alveolar
epithelial cells (AECs), breakdown of the pulmonary vascular
endothelial barrier and subsequent accumulation of protein-rich
inflammatory edema fluid within the alveolar spaces, ultimately
causing diffuse alveolar damage (6). Apoptosis and necrosis directly
contribute to epithelial and endothelial cell damage in sepsis, as
demonstrated Jiang et al (7). Consequently, modulating apoptosis and
alleviating lung inflammation are promising therapeutic strategies
for managing ALI.
The inflammatory response serves as the body's
defense mechanism against various stimuli, including infections,
pathogenic microorganisms, trauma and metaplasia. In the context of
sepsis-induced ALI, this response is primarily triggered by the
activation of lung macrophages and the infiltration of neutrophils.
The overactive engagement of these immune cells leads to the
increased release of pro-inflammatory substances, which further
exacerbates inflammation and ultimately causes tissue injury.
Apoptosis, often known as programmed cell death, is a genetically
controlled mechanism of systematic and self-directed cell death
that removes damaged, older or excess cells. During infection,
apoptosis serves a key role in limiting pathogen replication,
preventing the spread of infection and maintaining tissue
homeostasis (8). However,
dysregulated apoptosis and excessive apoptotic activity may also
contribute to the pathogenesis of ALI.
The present review examines the combined influence
of apoptosis and inflammation in SALI to offer a thorough
theoretical foundation for a deeper understanding of the mechanisms
underlying this condition.
Activation and regulation of the
inflammatory response of ALI in sepsis
Recruitment and activation of
inflammatory cells
Mechanisms of neutrophil infiltration in lung
tissue
Neutrophils serve as essential elements of the
innate immune system, contributing to the defense against pathogens
that invade the body. During sepsis, neutrophils become activated
and are released into the circulation in large numbers, where they
accumulate in endothelial cells (ECs) at the site of infection
(9,10). Intercellular cell adhesion
molecules (ICAMs) are upregulated under inflammatory conditions
through pro-inflammatory signaling, facilitating the migration of
neutrophils to lung tissue and their firm adhesion to ECs. A
previous study has demonstrated that ICAM-1 levels in lung
capillary cells are markedly elevated within 24 h of the onset of
infection (11). P-selectin on ECs
further mediates neutrophil aggregation and rolling adhesion,
resulting in substantial neutrophil accumulation in the pulmonary
microcirculation and excessive adhesion to the endothelium. This
aberrant alteration contributes to the activation of
pro-inflammatory chemokines [including IL-1, TNF-α and
lipopolysaccharide (LPS)], which further recruit neutrophils to
infected tissues.
Within neutrophils, phagosomes fuse with lysosomes
to form phagolysosomes, which are key in the defense against
invading pathogens. However, during phagocytosis, the contents of
neutrophil granules may be released into the extracellular
environment, particularly hydrolytic enzymes released by lysosomes.
This leakage can lead to local tissue damage and amplify acute
inflammatory signals, ultimately exacerbating inflammatory lung
injury (12). Additionally,
neutrophils can traverse the alveolar epithelium into the lumen and
adhere to the epithelial surface using β2-integrins. In the
inflammatory state, the migration of numerous neutrophils results
in increased alveolar epithelial permeability. Neutrophils that
have been activated secrete various cytokines, such as proteases,
reactive oxygen species (ROS), pro-inflammatory molecules and
agents that facilitate coagulation. Additionally, these harmful
mediators undermine the structural integrity of the tight junctions
(TJs) located between the epithelial cells.
In the context of sepsis, neutrophils migrate from
the intravascular space to inflamed tissues through the
transendothelial migration (TEM) cascade. Previous research
indicates that, during inflammation, the levels of ECs and
extracellular vesicles (EVs) in human blood notably increase
(13). EC-derived EVs facilitate
the reverse TEM (rTEM) of neutrophils from tissues back into the
vasculature. Neutrophils returning to the circulatory system
through rTEM can carry inflammatory signals and pathogen components
acquired from local tissues, thereby promoting the spread of
inflammation to distant organs and exacerbating distal lung injury.
Zi et al (10) demonstrated
that the proportion of rTEM neutrophils in the peripheral blood of
patients with sepsis was elevated, particularly in those who
developed ARDS.
A previous study found that activated neutrophils
can recruit and degrade pathogens by producing neutrophil
extracellular traps (NETs) (14).
NETs are primarily composed of nuclear DNA, histones, antimicrobial
peptides and enzymes such as myeloperoxidase (MPO) and elastase.
The process by which neutrophils form NETs is known as NETosis, a
novel form of programmed neutrophil death. In previous years,
increasing researchs have demonstrated that NETosis is closely
associated with sepsis-induced lung injury and exhibits a complex
dual role (15,16). On the one hand, in the early stages
of sepsis, NETs can help clear pathogens and limit their spread by
capturing and killing bacteria. However, excessive NETosis can lead
to lung tissue damage. Pathogen-associated molecular patterns such
as LPS, induce neutrophils to release NETs by activating toll-like
receptor 4 (TLR4). Meanwhile, MPO and hydrogen peroxide
(H2O2) produced by NETs can further activate
the NF-κB signaling pathway through the TLR signaling pathway of
epithelial cells, thereby enhancing the pro-inflammatory response
of ECs, upregulating the expression of adhesion molecules such as
ICAM-1 and vascular cell adhesion molecule 1 and promoting the
secretion of pro-inflammatory factors, leading to EC damage
(17). Furthermore, the histones
in NETs can directly cause lung epithelial cell and EC death due to
their cytotoxicity, thereby compromising the integrity of cell
membranes. Consequently, histone levels can serve as a clinical
marker for the severity of sepsis-induced lung injury. On the other
hand, the DNA in NETs is associated with coagulation, providing a
scaffold for platelet binding and stimulating platelet aggregation,
activating the extrinsic coagulation pathway, promoting
microthrombus formation and leading to pulmonary microcirculation
disorders and ventilation/perfusion mismatch, thereby exacerbating
lung injury (18).
Functional shifts in macrophages and
inflammatory amplification
Macrophages represent a highly heterogeneous class
of immune cells that serve a pivotal role in the body's immune
system. Macrophages can polarize into different phenotypes
depending on stimuli from various tissue microenvironments. The
primary phenotypes are inflammatory or classically activated
macrophages (M1) and alternatively activated macrophages (M2) that
promote healing. In response to inflammatory stimuli, the actions
of LPS or interferon, whether alone or synergistically, promote
macrophage polarization towards the M1 phenotype, thereby
increasing the release of pro-inflammatory factors [such as TNF-α,
nitric oxide (NO), IL-1β and IL-6], which are essential for
pathogen elimination and defense (19). During the repair phase of late
inflammation, M1 macrophages polarize toward M2 macrophages,
secreting anti-inflammatory factors (including IL-4, IL-10, IL-13
and TGF-β) to inhibit the inflammatory response and facilitate
tissue repair and remodeling (Fig.
1). Dang et al (20)
noted that macrophage polarization within the lungs has a notable
influence on the onset of, and recovery from, septic lung injuries.
An imbalance in the shift between M1 and M2 macrophages, which
leads to ongoing production of pro-inflammatory mediators, can
result in tissue harm. It has previously been reported that, in
cases of SALI, M1 macrophages exacerbate lung damage due to
unchecked inflammatory responses (21). Additionally, Li et al
(22) demonstrated that in
Traditional Chinese Medicine (TCM), the compound Lianhua Qingdian
effectively alleviates SALI by enhancing the infiltration of M2
macrophages. This mechanism may involve promoting the
transformation of macrophages from the M1 to the M2 phenotype by
activating the peroxisome proliferator-activated receptor γ
signaling pathway, inhibiting the NF-κB signaling pathway and
alleviating inflammatory responses. However, it is important to
note that these results were obtained from a mouse model of
LPS-induced ALI and there is still an overall lack of supporting
clinical data in humans.
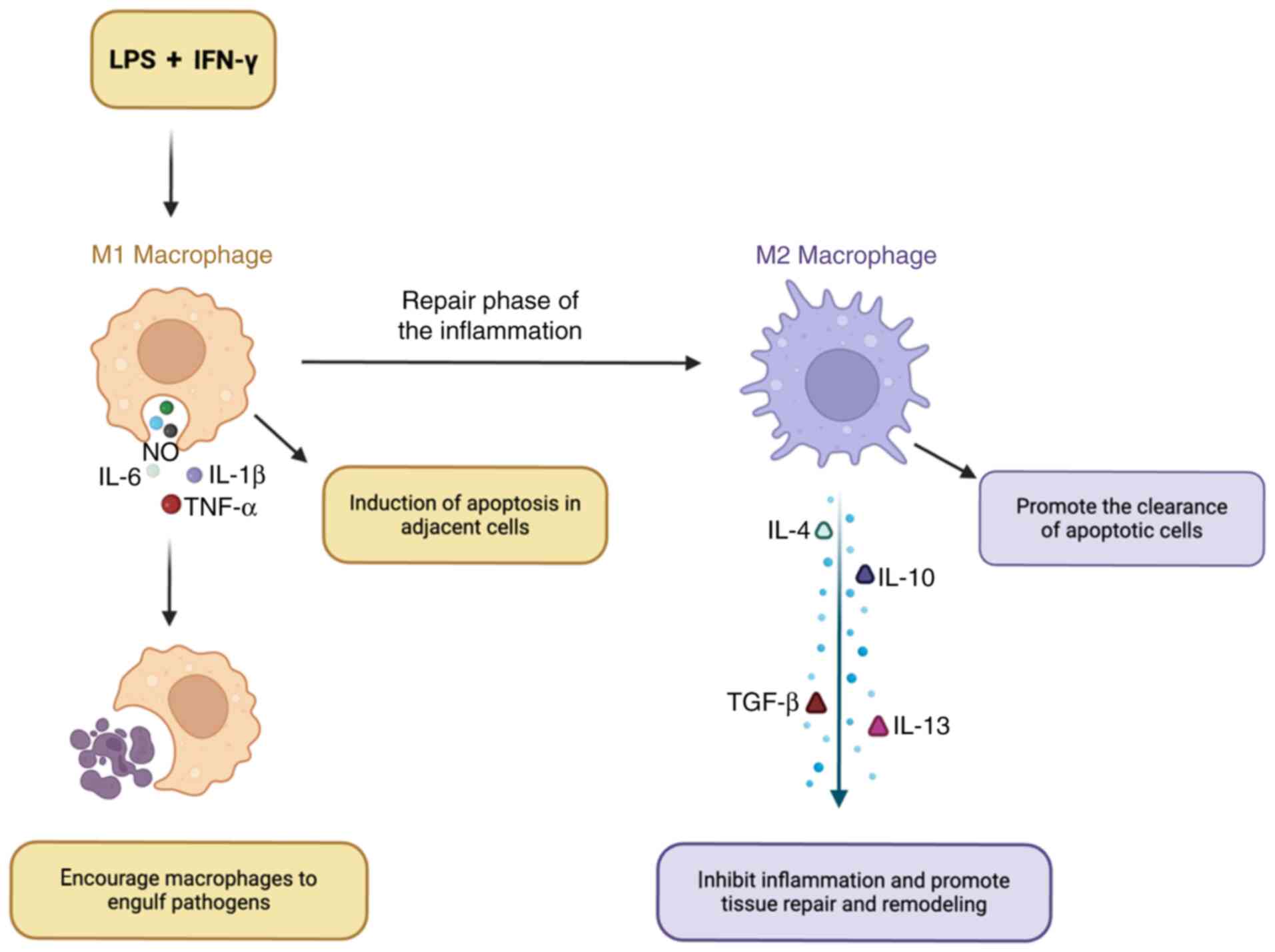 | Figure 1.Functional shifts in macrophages. LPS
or IFN promotes the polarization of macrophages toward the M1
phenotype, resulting in an increased release of pro-inflammatory
factors such as TNF-α, NO, IL-1β and IL-6. In the later stages of
inflammatory repair, M1 macrophages transition into M2 macrophages,
which secrete anti-inflammatory factors such as IL-4, IL-10, IL-13
and TGF-β, thereby suppressing the inflammatory response and
facilitating tissue repair and remodeling. LPS, lipopolysaccharide;
NO, nitric oxide. |
M2 macrophages possess anti-inflammatory and tissue
repair properties, thus serving a key role in the recovery process
of patients with ARDS. Yang et al (23) found that, in LPS-induced ARDS mouse
models, the M2/M1 alveolar macrophage ratio notably decreased as
the severity of lung injury increased. Conversely, the
intratracheal administration of exogenous M2 alveolar
macrophage-derived EVs markedly improved LPS-induced ARDS yet
reduced the levels of inflammatory factors in bronchoalveolar
lavage fluid. Similarly, in patients with ARDS who underwent
continuous mechanical ventilation or succumbed to disease within 28
days, there was found to be a persistent M1 phenotype and
insufficient M2 transformation (24). It may be inferred that the
anti-inflammatory and pro-repair effects mediated by M2 macrophages
are key for alveolar epithelial cell reconstruction and pulmonary
function recovery in patients with ARDS during the recovery
phase.
Release of inflammatory mediators and
signaling pathways
Formation and role of cytokine networks
Cytokines are a class of small proteins with diverse
biological activities. During inflammation, these cytokines can
activate immune cells to synthesize and secrete pro-inflammatory
factors, with the overproduction of TNF-α, IL-1β and IL-6 being
characteristic of systemic inflammatory response syndrome.
Excessive release of cytokines leads to mutual stimulation among
other different cytokines, forming a complex network. These
cytokines bind to target cells through cell surface receptors,
activating signal transduction pathways and initiating a series of
inflammatory cascade responses, with NF-κB having been identified
as a key target (25). In healthy
cells, NF-κB resides in the cytoplasm, forming a complex with IκB
that then inhibits further NF-κB activity. Upon stimulation by
cytokines, IκB undergoes phosphorylation through the activation of
the inhibitor of κB kinase, followed by its ubiquitination and
degradation. This sequence of events leads to the relocation of
NF-κB into the nucleus. Consequently, this mechanism enhances the
expression of pro-inflammatory cytokines, including IL-6, IL-1β and
TNF-α, thereby intensifying the inflammatory responses.
Synergistic effects of chemokines and
lipid mediators
Chemokines are a class of small molecular weight
cytokines produced by leukocytes in response to external stimuli.
During the inflammatory response, chemokines facilitate the
migration of circulating leukocytes to injured tissues by
recruiting and activating immune cells, thereby forming a
concentration gradient, which is further established by the binding
of chemokines to glycosaminoglycans in the extracellular matrix and
endothelium (26). Neutrophils
tend to be the first immune cells to reach the site of infection,
where they serve a key role in engulfing pathogens and clearing
cellular debris through phagocytosis. Subsequently, monocytes
migrate to the infection, where they transform into macrophages,
continuing the fight against pathogens while releasing various
cytokines, including TNF-α, IL-1β, IL-6, prostaglandins and
leukotrienes. The chemokine family is generally divided into four
categories: Two primary subfamilies (CXC and CC) and two secondary
subfamilies (CX3C and C). The distinct chemokine groups interact
with specific receptors found on various cell types and are
coordinated with the adhesion molecules present, thus contributing
to the inflammatory response (27).
Chemokine receptors are part of the heptameric
transmembrane guanosine triphosphate-binding G protein-coupled
receptor superfamily, which trigger an intracellular signaling
cascade through the associated trimeric G proteins. This mechanism
promotes the adhesion of target cells to the endothelial lining and
directs their migration toward the infection site (28). Among these, monocyte
chemoattractant protein 1 (MCP-1) (29), also known as CC chemokine ligand 2,
is classified within the CC subfamily of chemokines. In the early
stages of inflammation, MCP-1 attaches to the CC motif chemokine
receptor 2 receptor, stimulating the accumulation of monocytes and
their transformation into macrophages (30). MCP-1 serves a key role in directing
the appropriate immune response associated with infection and
inflammation (31). A different
category of CXC chemokines, such as macrophage inflammatory protein
2 [also referred to as C-X-C motif chemokine ligand (CXCL) 2] and
CXCL8 (often called IL-8), are able to promote the infiltration of
innate immune cells and work in conjunction with lipid signaling.
Additionally, prostaglandin E2 (PGE2) and leukotriene B4, produced
by ω-6 polyunsaturated fatty acid, serve as pro-inflammatory lipid
mediators that activate inflammatory vesicles and initiate the
endoplasmic reticulum (ER) stress response (32). These processes serve a key role in
the onset, progression and resolution of inflammation.
Triggering and regulation of apoptosis in
ALI in sepsis
Activation of apoptosis-related
signaling pathways
Death receptor-mediated apoptosis pathway
Death receptor pathways involve specific proteins on
cell membranes binding to ligands that carry apoptotic signals,
subsequently transducing these signals into the cell and ultimately
inducing apoptosis. The activation of the death receptor pathway
mainly occurs through death receptors located on the cell surface,
including the Fas cell surface and TNF receptors. These receptors
initiate the apoptotic process by recruiting junctional proteins,
including Fas-associated via death domain and promoting cysteine
aspartyl proteases, such as caspase-8, upon binding with specific
death ligands such as Fas ligands and TNF-α. Subsequently,
caspase-8 activates downstream effector enzymes, including
caspase-3 and caspase-7, ultimately leading to apoptosis (Fig. 2).
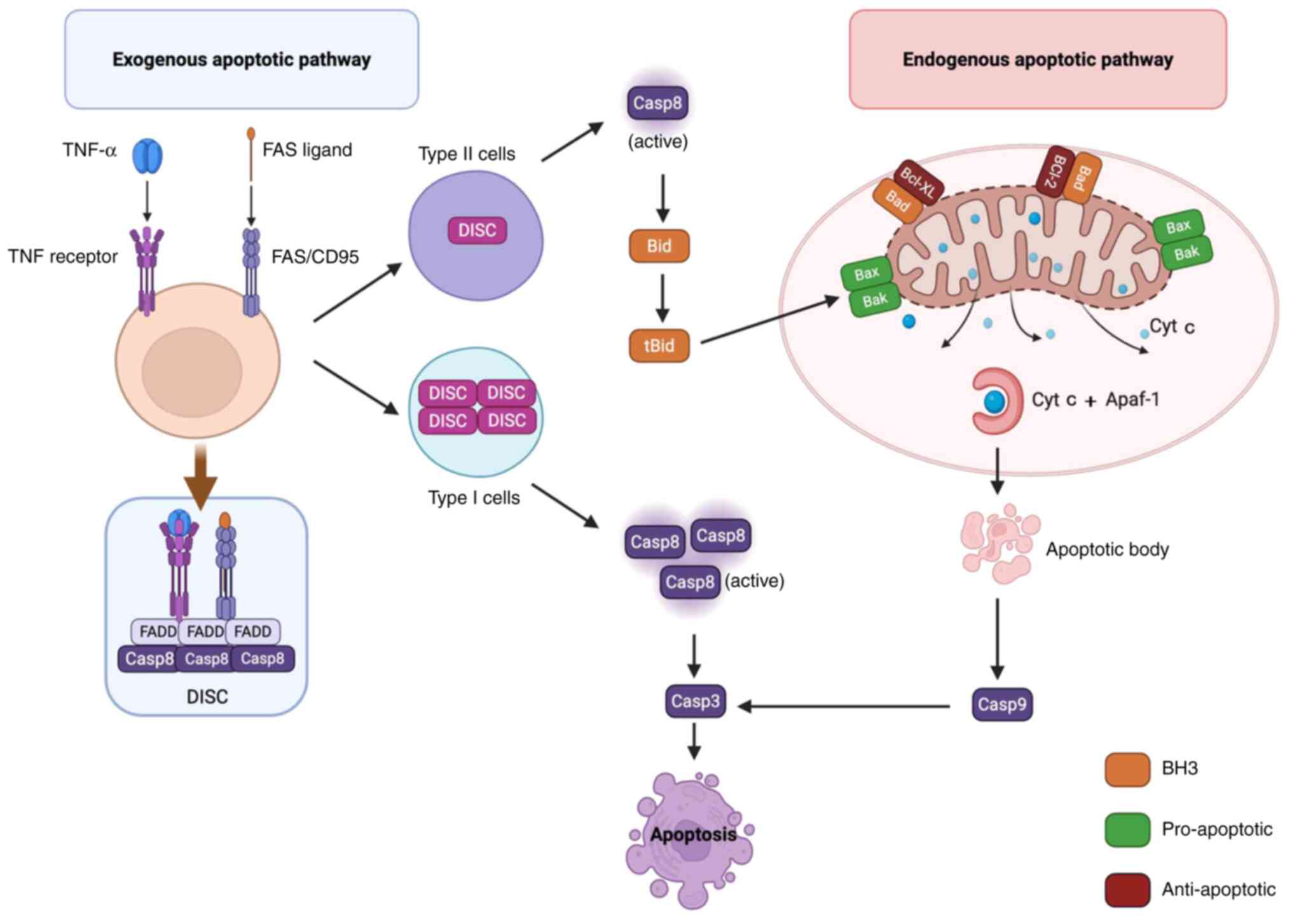 | Figure 2.Activation of apoptosis-related
signalling pathways. The activation of the extrinsic apoptotic
pathway is primarily mediated by the binding of specific ligands to
the Fas receptor and TNF receptor on the cell surface, which is
followed by the recruitment of FADD to form the DISC. Notably,
there are marked differences in the formation of DISC between type
I and II cells. In type I cells, the DISC is formed efficiently and
stably, resulting in the production of a substantial amount of
Casp8. This enzyme directly activates downstream effector enzymes,
including Casp3, thereby triggering apoptosis. Conversely, in type
II cells, DISC formation is inefficient and unstable, yielding only
a limited amount of Casp8, which subsequently cleaves Bid into
tBid. tBid then translocates to the outer mitochondrial membrane,
activating Bax/Bak, which leads to the disruption of mitochondrial
membrane integrity, the release of Cyt c and subsequent binding
with Apaf-1 to form the apoptosome. This process further activates
Casp9, which triggers downstream Casp3 to initiate apoptosis. DISC,
death-inducing signaling complex; Bid, BH3 interacting domain death
agonist; tBid, truncated Bid; Bak, Bcl-2 homologous antagonist
killer; Cyt c, cytochrome c; Apaf-1, apoptotic protease activating
factor 1; FADD, Fas-associated death domain; Casp, caspase; Bad,
Bcl-2-associtaed death protein. |
Caspases, which are cysteine-specific cysteine
proteases, serve a pivotal role in the regulation of apoptosis.
Caspases can be categorized into two groups: i) Initiating caspases
(including caspases-2, −8, −9 and −10); and ii) executioner
caspases (including caspases-3, −6 and −7) (33). When an initiating caspase becomes
activated, it processes and triggers the downstream executioner
caspases, which subsequently cleave cellular proteins at particular
aspartate residues, thus facilitating the process of apoptosis
(34). These executioner caspases
are key in shaping the distinctive characteristics of apoptotic
cells by cleaving a multitude of cellular proteins (including, but
not limited to, caspase-activated inhibitor of DNase, aggrecan and
Pac21), ultimately resulting in DNA fragmentation. This cascade of
events leads to observable apoptotic traits such as nuclear
condensation, shrinkage of the cell and blistering of the membrane.
Generally, caspase enzymes are synthesized as inactive zymogens and
their activation pathways include both exogenous and endogenous
mechanisms, with the death receptor pathway being classified as
exogenous (35).
In the exogenous pathway, caspase-8 not only
activates the execution of caspases, but also facilitates the
cleavage of BH3 interacting domain death agonist (Bid), leading to
its translocation to the mitochondria (25). The CD95 signaling model proposed by
Algeciras-Schimnich et al (36) suggests that the involvement of the
mitochondrial pathway in apoptosis is determined through a
dual-threshold mechanism. As a death receptor, CD95, upon binding
with its ligand, induces the formation of the death-inducing
signaling complex (DISC), the efficiency of which directly
determines the levels of caspase-8 produced. However, there are
notable differences in DISC formation between types I and II cells.
In type I cells, DISC formation is highly efficient and stable,
further generating a substantial quantity of caspase-8 that exceeds
the higher threshold, directly activating caspase-3 to trigger
apoptosis. In type II cells, the formation of DISC is inefficient
and unstable, resulting in only a minimal quantity of caspase-8
that only just exceeds the lower threshold. Apoptosis initiation
can only be triggered by amplifying the cell death signal through
the mitochondrial pathway through the cleavage of Bid (36). Consequently, the process of
apoptosis requires participation from the mitochondrial apoptotic
pathway, wherein Bid acts as a key connection between the death
receptor and mitochondrial apoptotic pathways.
Initiation of the mitochondrial
apoptotic pathway
The mitochondrial apoptotic pathway is endogenous.
When triggered by factors such as DNA damage, metabolic stress, ER
stress or growth factor deprivation, the integrity of the
mitochondrial membrane is disrupted (37,38).
Increased mitochondrial membrane permeability leads to the release
of cytochrome c (Cyt c) from the mitochondria into the cytoplasm.
Cyt c interacts with apoptosis-activating factor 1 to create an
ATP-dependent apoptotic complex. This complex activates caspase-9,
which subsequently triggers the activation of downstream caspases-3
and −7, consequently initiating a cascade of caspase reactions that
leads to apoptosis (Fig. 2).
Furthermore, once released from the mitochondria, Cyt c can bind to
inositol triphosphate receptors located in the ER. This binding
results in an elevation of local calcium levels, which then
enhances the release of Cyt c and triggers the initiation of
apoptosis (39). Conversely,
protein tyrosine phosphatase, which is located between the inner
and outer membranes of the mitochondria, can facilitate the
creation of the mitochondrial permeability transition pore (MPTP),
permitting the movement of molecules of ≤1.5 kDa in size. Abnormal
MPTP opening has been found to impair mitochondrial function and
promote apoptosis (40).
Mitochondria serve as the primary source of cellular
energy and are also the principal site for the production of
intracellular ROS. During apoptosis, mitochondrial damage and ROS
production exacerbate each other, resulting in a vicious cycle
(41). ROS can compromise both the
integrity and functionality of the mitochondrial membrane,
resulting in the release of apoptotic factors, which in turn,
further enhances ROS production, aggravating mitochondrial
impairment (42). Wang et
al (43) induced oxidative
stress in Hepa1-6 cells through the application of fluorine (F),
which led to increased levels of intracellular ROS and
propane-1,2-diol and mitochondrial injury. The oxidative stress
induced by F resulted in marked elevations in intracellular ROS,
malondialdehyde (MDA) and NO concentrations. Furthermore, there was
a notable upregulation in the protein expression of Cyt c,
caspase-9 and caspase-3, along with a considerable increase in
apoptotic cell count and extensive mitochondrial vacuolation, as
evidenced by transmission electron microscopy. This demonstrated
that ROS mediates mitochondrial damage, thereby exacerbating
apoptotic injury.
Role of apoptosis-regulating genes and
proteins
Imbalance in the regulation of Bcl-2 family
proteins
Bcl-2 family proteins are the primary regulators of
the release of mitochondria-associated apoptotic factors and can be
classified into three primary categories based on their biological
functions: i) Anti-apoptotic proteins, such as Bcl-2, Bcl-XL, Bcl-w
and myeloid cell leukemia 1 (Mcl-1); ii) pro-apoptotic proteins,
such as Bax and Bcl-2 homologous antagonist killer (Bak); and iii)
BH3-only proteins, such as Bad, Bid, Bcl-2-interacting mediator of
cell death (Bim), phorbol-12-myristate-13-acetate-induced protein 1
and p53 upregulated modulator of apoptosis (Puma). In healthy
cells, a balance is sustained between anti-apoptotic and
pro-apoptotic proteins. However, in a septic state, the
concentration of free Bad in the cytoplasm increases, allowing Bad
to bind to Bcl-2 and Bcl-XL. This binding leads to the dissociation
of Bax and Bak and the formation of pore-protein complexes through
interactions with other Bax or Bak proteins. These complexes can
insert into the outer mitochondrial membrane, disrupting its
integrity and ultimately resulting in the release of intracellular
apoptosis-inducing factors (Cyt c) and triggering apoptosis
(44).
Bax is a crucial pro-apoptotic protein. Upon
receiving apoptotic signals, Bax, which initially exists as a
monomer in the cytosol, translocates to the mitochondrial surface,
where it forms trans-mitochondrial membrane pores, resulting in
increased membrane permeability and further facilitating the
release of apoptotic factors (45). Simultaneously, when the Bcl-2/Bax
imbalance disrupts the TJ proteins between alveolar epithelial
cells, it directly or indirectly induces the apoptosis of alveolar
epithelial cells, leading to the destruction of the alveolar
epithelial barrier and increased permeability, which ultimately
causes alveolar edema, alveolar collapse and refractory hypoxemia.
Previous research has shown that polydeoxyribonucleotide (PDRN)
extracted from salmon sperm is able to promote tissue healing and
reduce apoptosis and inflammation. An et al (46) treated a rat model of LPS-induced
lung injury with intraperitoneal injections of PDRN and observed a
notable reduction in lung injury scores. In addition, the ratio of
the pro-apoptotic protein Bax relative to the anti-apoptotic
protein Bcl-2 was reduced, suggesting that PDRN treatment
alleviated lung damage through the inhibition of apoptosis.
Regulation of apoptosis by p53
The p53 protein functions as a transcription factor,
triggering the expression of various target genes and serving a key
role in maintaining genomic stability, regulating the cell cycle,
facilitating DNA repair and promoting apoptosis. Under standard
physiological circumstances, p53 is targeted for degradation by
murine double minute 2 homolog (MDM2) and murine double minute X
(MDMX), which ubiquitinate it, thereby keeping its intracellular
levels low through proteasomal degradation. By contrast, when cells
face stressors such as DNA damage, hypoxia or infection, the
process of p53 ubiquitination is suppressed, causing a rapid
increase in its cellular concentrations (47). Various sensor proteins, such as the
ataxia-telangiectasia-mutated protein, ataxia-telangiectasia and
Rad3-related protein, checkpoint kinase 1, checkpoint kinase 2,
DNA-dependent protein kinase and the p14ARF protein, become
activated and p53 stability is enhanced through post-translational
modifications, including acetylation, methylation and
phosphorylation. Stabilized p53 proteins form tetramers in the
nucleus that bind to p53 response elements on target DNA, thereby
regulating gene transcription (48). On one side, p53 is involved in
apoptosis mediated by mitochondria through activating the
transcription of pro-apoptotic proteins such as Bax, Puma and Bid.
Conversely, p53 can trigger apoptosis without relying on
transcription. The p53 protein, in a manner that does not depend on
its transcriptional functions, translocates to the mitochondria,
where it competes with Mcl-1 for binding to Bak through protein
interactions, leading to the release of Bak from Mcl-1 and
initiates Bak oligomerization (49). Furthermore, p53 interacts with
Bcl-XL, prompting the detachment of Bax from the Bax/Bcl-XL
complex, subsequently enhancing oligomerization. The
oligomerization of Bax and Bak modifies the permeability of the
outer mitochondrial membrane, allowing the release of Cyt c into
the cytoplasm, which then activates caspase proteases, ultimately
prompting apoptosis (50).
In addition, the activation of p53 can lead to the
disruption of the EC cytoskeleton, increased vascular permeability
and the induction of apoptosis in alveolar type II epithelial
cells, severely affecting the synthesis and secretion of pulmonary
surfactant (PS). This results in increased alveolar surface
tension, alveolar collapse and ventilator-associated lung injury.
Previous research has found that MDM2 promotes the degradation of
p53 protein through the ubiquitination pathway, maintaining p53 at
low levels. When MDM2 function is lost, uncontrolled activation of
p53 leads to apoptosis, barrier disruption and amplification of
inflammation, ultimately exacerbating lung injury (51).
Mechanisms of synergy between inflammation
and apoptosis
Induction of apoptosis by inflammatory
mediators
Molecular crosstalk of cytokines to apoptotic
signaling pathways
When sepsis manifests, there is an enhanced release
of pro-inflammatory agents in the body (such as TNF-α, IL-1β and
IL-6) and the NF-κB pathway is dysregulated, which exacerbates the
inflammatory response or transcriptionally activates the Bcl-2
family, thus inducing apoptosis through an ‘inflammatory storm’.
Conversely, activation of NF-κB further induces the production of
TNF-α and IL-1β, forming a positive feedback loop. Furthermore,
elevated levels of activated cytokines such as IL-6 attach to the
membrane-associated IL-6 receptor (IL-6R), resulting in the
formation of the IL-6/IL-6R complex. This complex then associates
with glycoprotein 130 (gp130) to establish a signaling complex that
triggers the MAPK cascade through the recruitment of gp130.
Similarly, locally released cytokines (such as IL-1β or TNF-α) have
been shown to activate MAPK pathways, including JNK and ERK
(52,53). Abnormal activation of these
signaling pathways further promotes the activation of inflammatory
cells and disrupts the stress response, thus inducing cell
apoptosis, ultimately leading to tissue injury.
Inflammation-induced oxidative stress
and apoptosis
Oxidative stress influences apoptosis through
various pathways. An important component of this process is the
action of ROS, which function as ‘redox messengers’ and are crucial
for redox regulation and cellular signaling. Generally, ROS are
understood to encompass free radicals derived from oxygen
(O2), including superoxide anion
(O2−), hydroxyl radical (HO−),
peroxyl radical and alkoxyl radical, along with non-free radical
O2 derivatives such as hydrogen peroxide
(H2O2). However, ROS, once produced in
excess, may lead to oxidative modification of cellular
macromolecules, which in turn may cause notable damage to cellular
proteins and DNA. Mokrá (54)
suggested that oxidative stress is not only an important causative
factor in ALI but may also contribute to extensive damage to lung
tissues and worsening inflammatory responses through activation of
apoptotic pathways.
Mitochondria serve as the primary location for the
production of ROS, with 1–2% of the O2 consumed by
mitochondria being utilized for the generation of ROS, particularly
during the electron transfer process to O2 within the
electron transport chain. In the presence of inflammation and
tissue injury, a marked increase in the release of mitochondrial
ROS (mtROS) occurs. This increase may result in mitochondrial
membrane depolarization that relies on the pore-forming protein
gasdermin D (GSDMD). Consequently, there is a reduction in the
mitochondrial membrane potential, prompting GSDMD to associate with
the mitochondrial membrane, thereby forming a pore. This process
ultimately enhances the permeability of the mitochondrial membrane,
facilitates the release of apoptotic factors and triggers the
activation of mitochondrial apoptotic pathways. In addition, it has
been found that ROS can also inhibit the degradation and transport
of mitochondrial proteins, which is the main cause of mitochondrial
dysfunction (55). In addition,
oxidative stress can trigger the activation of transcription
factors such as NF-κB and p53 (56), influencing the expression and
functionality of proteins associated with apoptosis (such as
Bcl-2). Previous research has demonstrated that, in cells treated
with H2O2, there is a marked reduction in
Bcl-2 protein levels (57),
thereby modulating the apoptotic process.
Exacerbation of the inflammatory
response by apoptotic cells
Damage-associated molecular patterns (DAMPs)
released by apoptotic cells
Alveolar macrophages, as the primary immune cells in
the lungs, encounter infections, injuries and stress, leading to
the transformation of specific endogenous molecules into DAMPs.
These DAMPs can activate the immune system and exhibit
pro-inflammatory characteristics. In a steady-state environment,
endogenous molecules, including nucleic acids, proteins, ions,
glycans and metabolites, usually do not initiate an immune
response. However, during stress, these molecules can be
transformed into DAMPs through three mechanisms: i) Positional
substitution; ii) alterations in properties; or iii) changes in
concentration, with positional substitution being the predominant
mechanism (58). It is evident
that, during infection or stress, the mitochondrial membrane
sustains damage, leading to increased membrane permeability and the
release of intracellular substances that can induce apoptosis.
Proteins or peptides released into the extracellular space by
apoptotic cells can be converted into DAMPs such as nuclear
proteins like high mobility group box 1 (HMGB1) (59), histones (60), heat shock proteins (HSPs) (61) and oxidized phospholipids (62). Pattern recognition receptors (PRRs)
identify these molecules, leading to the subsequent release of
chemokines and pro-inflammatory factors that initiate and worsen
the inflammatory response (63,64).
PRRs are a class of innate immune receptors that detect endogenous
molecules released following tissue injury and serve as sensors for
DAMPs, including TLRs, C-type lectin receptors, retinoic
acid-inducible gene I-like receptors, nucleotide-binding
oligomerization domain-like receptors and DNA sensors (65,66).
For example, HMGB1 is recognized by TLR2 and TLR4, while HSPs are
recognized by TLR2. Stimulation of TLR2 or TLR4 triggers the
activation of downstream MAPK and NF-κB pathways through the
intermediary protein myeloid differentiation primary response 88
(Myd88), which subsequently enhances the activation of inflammatory
cells and the secretion of inflammatory mediators (Fig. 3). Furthermore, oxidized lipids
serve as important DAMPs involved in the inflammatory response by
interacting with immune and ECs. For example, high concentrations
of cyclopentenone prostaglandins can activate NLR family pyrin
domain containing 3 inflammatory vesicles, which form a protein
complex with the articulin ASC and pro-caspase-1, thereby mediating
caspase-1-dependent IL-1β production, which in turn exacerbates the
inflammatory response (58). Thus,
inflammation can induce the onset of apoptosis and, conversely,
apoptosis can further exacerbate the inflammatory response, with
the two interacting, resulting in a vicious cycle (Table I).
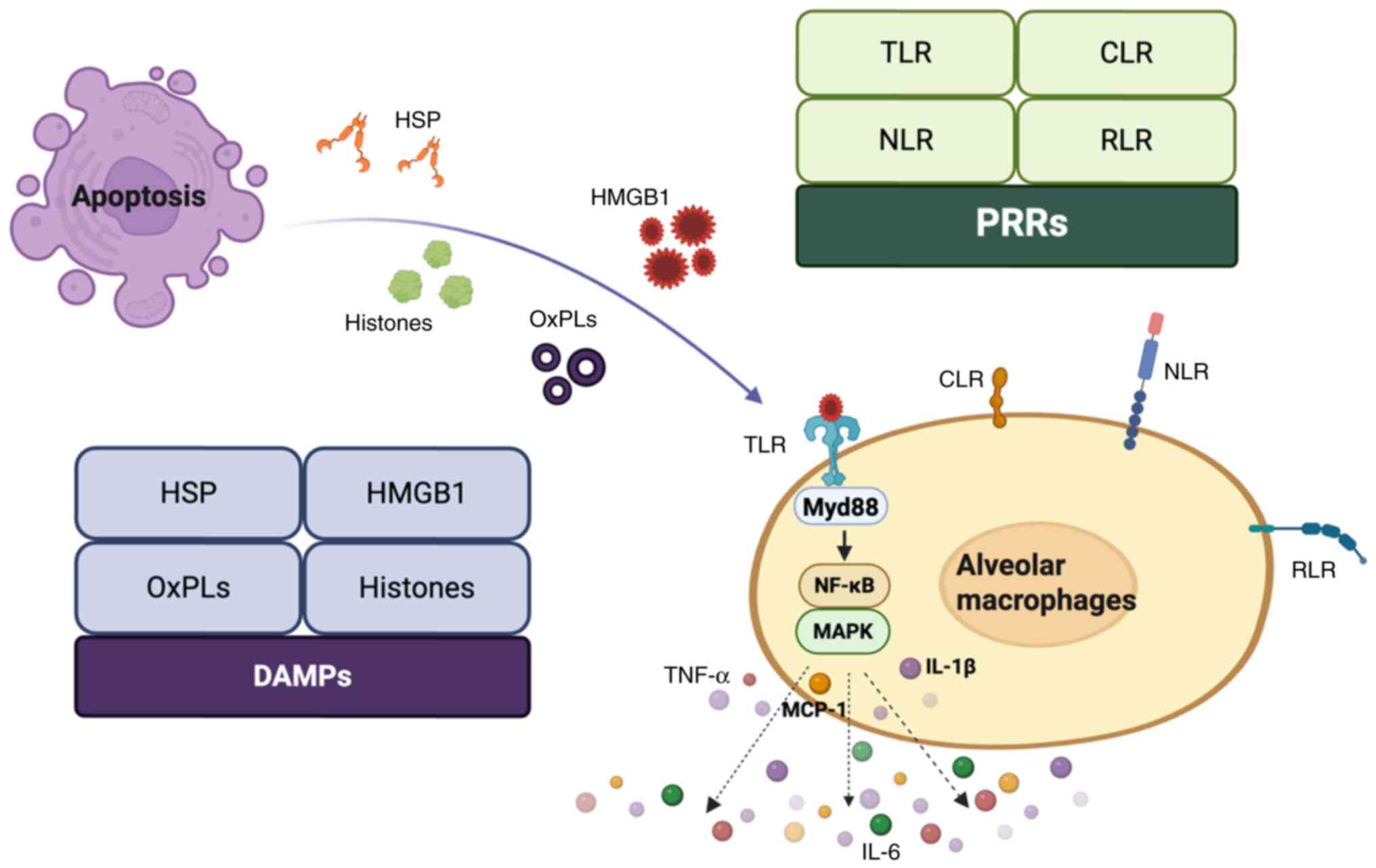 | Figure 3.DAMPs released by apoptotic cells.
Proteins or peptides released from apoptotic cells into the
extracellular space can be transformed into DAMPs, including
nuclear proteins such as HMGB1, histones, HSPs and oxidized
phospholipids. These DAMPs can be recognized by PRRs, which
subsequently activate downstream MAPK and NF-κB pathways through
the adaptor protein Myd88, resulting in the secretion of
inflammatory mediators. DAMPs, damage-associated molecular
patterns; HMGB1, high mobility group box 1; HSPs, heat shock
proteins; PRRs, pattern recognition receptors; MAPK,
mitogen-activated protein kinase; Myd88, myeloid differentiation
primary response 88; OxPLs, oxidized phospholipids; TLR, toll-like
receptors; CLR, C-type lectin receptors; MCP-1, monocyte
chemoattractant protein 1; RLR, RIG-I-like receptors; NLR, NOD-like
receptors. |
 | Table I.Key DAMPs and their clinical
significance in ALI/ARDS. |
Table I.
Key DAMPs and their clinical
significance in ALI/ARDS.
| First author,
year | DAMP | Release
mechanism | Recognition
receptors | Clinical relevance
(ALI/ARDS in sepsis) | (Refs.) |
|---|
| Deng et al,
2022 | HMGB1 | Nucleoprotein
leakage from necrotic cells | TLR2/4 | HMGB1 levels are
notably elevated in serum and BALF from patients with sepsis, but
their association with the extent of lung injury and prognosis
remains controversial | (59) |
| Dutta et al,
2025 | Histone | Neutrophil
NETosis | NLRP3 | Extracellular
histones associate with ARDS severity and mortality and are
expected to be early diagnostic markers of ARDS. In particular,
histones H3 and H4 may serve as effective biomarkers and
therapeutic targets in inflammatory diseases | (60) |
| Pei et al,
2022 | HSP70 | Stress cell
cytoplasmic protein release | TLR2 | Enhanced HSP70
expression attenuates sepsis-induced lung injury by inhibiting
inflammation and apoptosis | (61) |
| Karki et al,
2023 | Oxidized
phospholipids | Apoptotic cell
membrane lipid peroxidation | NLRP3 | Oxidized
phospholipids levels are elevated in patients with sepsis and ARDS,
which can be used for early identification of high-risk patients
and guide clinical decision-making | (62) |
Impaired clearance of apoptotic cells
and persistence of inflammation
Under normal physiological conditions, apoptotic
cells are cleared through phagocytosis, primarily by macrophages,
thereby preventing the initiation of an inflammatory response.
Effective clearance of apoptotic cells is key for managing
inflammatory diseases, preventing secondary necrosis and restoring
normal tissue function. Phagocytes are guided by chemokine
‘find-me’ signals in order to migrate towards apoptotic cells
(67). The current four identified
signals that facilitate locating cells are chemokine C-X3-C motif
ligand 1 (CX3CL1; fractalkine), nucleotide triphosphates [including
ATP and uridine triphosphate (UTP)], sphingosine-1 phosphate (S1P)
and lysophosphatidylcholine (LysoPC). Among these, the release of
ATP, UTP and CX3CL1 occurs during the early stages of apoptosis,
while LysoPC and S1P are lipid chemokines produced in the later
stages of apoptosis (67).
Phagocytes near apoptotic cells recognize specific
ligands on their surface, referred to as ‘eat-me’ signals,
including cell-surface calreticulin, ICAM-1 and complement
component 1q, which activate signal transduction pathways and
facilitate the phagocytosis of apoptotic cells (68). Following phagocytosis, apoptotic
cells undergo cytosolic burial, maturing gradually and secreting
anti-inflammatory and pro-tissue healing factors. However, when the
clearing process is hindered, apoptosis can advance to necrotic
apoptosis, which is marked by the rupture of the cell membrane and
a marked release of intracellular DAMPs, thereby intensifying
inflammation and causing tissue injury.
In normal lung tissue, the process of cytosolic
burial helps to avert tissue injury and regulate the inflammatory
response, largely facilitated by alveolar macrophages. Compared
with other macrophage types, alveolar macrophages exhibit a longer
lifespan and an enhanced self-renewal capacity. Under steady-state
conditions, even during acute lung inflammation, the efficient
cellular clearance of alveolar macrophages results in a minimal
presence of apoptotic cells in the airways. When the function of
alveolar macrophages is compromised and when apoptotic cells are
generated in large quantities and cannot be rapidly cleared through
cytosolic burial, a sustained release of DAMPs is prompted,
perpetuating the inflammatory response (69). In certain patients with bacterial
pneumonia, failure to resolve inflammation promptly or the
occurrence of an inflammatory storm, results in impaired lung
function and the subsequent development of ALI or ARDS, which
further prolongs the inflammatory response in the lungs (70). A previous study has indicated that
an increased quantity of uncleared apoptotic cells is found in the
airways of patients with ARDS, respiratory failure or chronic lung
inflammation, all of which are conditions frequently marked by
impairments in the cellular clearance capabilities of alveolar
macrophages (71). Therefore, the
timely and effective clearance of apoptotic cells is key in
modulating the inflammatory response and protecting lung
function.
Effects of inflammation and apoptosis:
Pathophysiological functions of lung tissue
Alveolar epithelial cell damage and
dysfunction
Alveolar epithelial barrier disruption
Alveolar epithelial cell damage is a key determinant
of the severity of ALI and ARDS. Therefore, protecting the function
and barrier integrity of AECs is of great importance (72). AECs, the structural components of
the lung, comprise alveolar type I and II epithelial cells. Type I
epithelial cells, which serve as the main location for gas
exchange, experience higher vulnerability to inflammation,
oxidative stress and various injuries (mechanical injury, chemical
injury, hyperoxic injury, etc.), resulting in an increased
likelihood of cell death. Conversely, type II epithelial cells
exhibit greater resilience, possessing the ability to proliferate
and convert into type I epithelial cells, thus serving a role in
the repair and preservation of alveolar epithelial barrier
integrity (73). Furthermore, AECs
regulate ion transport through the
Na+/K+-ATPase on the cell surface,
establishing an osmotic gradient between the intracellular and
extracellular environments. This regulates the alveolar fluid
clearance process, preventing the accumulation of alveolar edema
fluid (74).
The integrity of the AEC barrier is also supported
by the glycocalyx, a layer of glycosaminoglycans and proteoglycans
covering the alveolar surface and the basement membrane shared with
ECs (75). However, during sepsis,
the organism releases a large quantity of pro-inflammatory factors
and chemokines, leading to an exaggerated and dysregulated
inflammatory response. Mitochondria, essential organelles in AECs,
not only generate ATP but also function in intracellular and
extracellular signaling under stress (76). Evidence indicates that sustained
mitochondrial dysfunction serves an important role in the process
of necrotic apoptosis within AECs (77,78).
Inflammatory stimuli activate the mitochondria-associated apoptotic
pathway, which further releases apoptotic factors and upregulates
pro-inflammatory factor levels. This excessive cytokine production
can directly or indirectly induce epithelial cell apoptosis by
recruiting leukocytes to migrate to the lungs and disrupting TJ
proteins (such as connexin, sealin and E-calmodulin) between AECs.
Consequently, this results in damage to epithelial structures,
detachment of the glycocalyx, impairment of the alveolar epithelial
barrier integrity, increased permeability, decreased production of
surface-active substances and reduced ion and fluid transport
capacity from the alveolar lumen. These changes lead to dysfunction
in gas exchange, ultimately causing alveolar edema, alveolar
collapse and refractory hypoxemia. Lei et al (79) demonstrated that the treatment of
SALI mice with 3-methyladenine (3-MA), a drug that regulates
apoptosis, significantly enhanced the integrity of the alveolar
epithelial barrier. This study found that it effectively inhibited
lung inflammation and epithelial cell apoptosis in mice, improved
pulmonary pathological changes and alleviated lung injury.
Conversely, activated epithelial cells contribute to increased
secretion of chemokines and adhesion molecules, leading to cell
death and inflammatory responses, therefore resulting in a vicious
cycle.
Therefore, the repair of the alveolar epithelial
barrier is a critical indicator for the prognosis of lung injury. A
previous clinical study has shown that therapeutic strategies
targeting epithelial repair, such as mesenchymal stem cell
(MSC)-secreted keratinocyte growth factor, can improve the
oxygenation index in patients with ARDS (80).
Abnormal synthesis and secretion of
alveolar surface-active substances
Alveolar type II epithelial cells are capable of
secreting the alveolar surface-active substance pulmonary
surfactant (PS). The functions of PS include reducing alveolar
surface tension, maintaining the relative stability of the alveolar
structure, facilitating the absorption of alveolar fluid,
maintaining fluid balance in the lungs, preventing pulmonary edema
and atelectasis and enhancing the lung's defense function (81,82).
PS is a complex formulation that consists primarily of 90%
phospholipids and 10% proteins (83). It contains four surfactant proteins
(SPs) that are linked to surface-active agents, namely SP-A, SP-B,
SP-C and SP-D. Among these, SP-A and SP-D are hydrophilic proteins
that are part of the collectin family and are key in innate immune
defense. Conversely, SP-B and SP-C are highly hydrophobic
apolipoproteins that are key in the biophysical functionalities of
surfactants (84). A previous
study found that SP-A knockout mice exhibited a higher severity of
lung injury and mortality in a severe acute respiratory syndrome
coronavirus (COVID) 2-induced mouse ALI model, indicating that SP-A
serves a key role in pathogen defense (85).
Under physiological conditions, 80–90% of PS is
distributed in large aggregates (LAs) with high surface activity.
However, during infection, the upregulation of inflammatory
mediators and activation of apoptosis-inducing signals leads to the
disruption or inhibition of PS. The content of PS in LAs is
markedly reduced, shifting to small aggregates with markedly low
surface activity, which are mainly products of PS degradation.
Furthermore, activated immune cells generate ROS, which may disrupt
surfactant functionality by diminishing the synthesis of SP and
phospholipids (86). Additionally,
high levels of NO free radicals and TNF-α produced during
inflammation can directly impede the production of SP-A, SP-B and
SP-C (87). As a result, the
synthesis and activity of alveolar surface-active substances are
reduced, leading to an increase in alveolar surface tension. On the
other hand, the increased alveolar surface tension fails to
maintain the normal alveolar structure, leading to alveolar
collapse. During mechanical ventilation, atelectatic alveoli may be
damaged by the force generated by the cyclic opening and closing of
the ventilator, ultimately exacerbating respiratory failure.
Dargaville et al (88),
through a 2-year follow-up study of preterm infants with ARDS
treated with minimally invasive surfactant application, found that
infants treated with surfactant had a lower incidence of adverse
respiratory outcomes within a period of 2 years. This finding, to
some extent, suggests that alveolar surfactant can improve lung
injury and maintain normal alveolar function.
Pulmonary vascular EC damage and
vascular dysfunction
Vascular endothelial barrier disruption and
increased permeability
The endothelium acts as a physical barrier that
separates blood, gases and stromal tissues, serving a vital role in
preventing inflammation and coagulation, aiding in gas exchange,
controlling vascular tone and engaging in endocrine signaling
(89). In particular, the
pulmonary endothelium functions as a semi-permeable barrier key for
gas exchange at the alveolar-capillary interface and in managing
the flow of fluids and solutes between the blood and the
interstitial compartments of the lungs. The connections among lung
ECs include adherens junctions (AJs), TJs and gap junctions. Among
these, AJs are mainly composed of VE-cadherin, which bind to
intracellular connexins (such as p120-connexin), waveform proteins
and other proteins in the actin cytoskeleton to maintain the
integrity of the pulmonary endothelial barrier. During an
inflammatory response, various factors such as the activation of
pro-inflammatory factors (including TNF-α and IL-1β) and increased
ROS production can lead to the phosphorylation of VE-cadherin and
its associated proteins. This results in the detachment of
VE-cadherin from the actin cytoskeleton and the breakdown of AJ
proteins. At the same time, the contraction of actin stress fibers
creates a pulling force on VE-cadherin, compelling it to dissociate
from the proteins it is bound to. This results in impaired
inter-endothelial connections, increased permeability and damage to
the endothelial barrier. Therefore, VE-cadherin has become an
important biomarker for endothelial barrier disruption and is key
for maintaining the integrity of the endothelial barrier. Previous
research has suggested that the concentration of serum VE-cadherin
in patients with ARDS is notably elevated compared with healthy
controls. With this, its levels are negatively associated with the
pulmonary vascular permeability and oxygenation indexes (90).
In addition, TJ proteins (such as claudin and
occludin) interact with zonula occludens (ZO-1) in the cytoplasm.
ZO-1 binds to α-catenin and the actin cytoskeleton to stabilize
endothelial barrier function (91). When inflammatory mediators such as
histamine are present, TJ proteins experience phosphorylation
dependent on Src and undergo depletion, resulting in a compromised
endothelial barrier function and increased endothelial permeability
(92). Previous research indicates
that the expression levels of TJ proteins are markedly reduced in
the lung endothelium of patients with ARDS (93). Meanwhile, damaged or dead ECs
further release toxic cellular components, allowing more leukocytes
to migrate from the circulation to the site of inflammation, thus
exacerbating the inflammatory response. Additionally, inflammatory
mediators may further activate the apoptotic pathway, leading to EC
damage and death.
Vasodilatory dysfunction and
microthrombosis
A healthy pulmonary endothelium largely suppresses
inflammation, maintains blood flow and prevents thrombosis. Some
studies have shown that various stimuli, including hypoxia,
cytokines, chemokines, thrombin, LPS, DAMPs and apoptosis, can lead
to the activation and damage of ECs (94,95).
On the one hand, ECs produce endothelium-derived diastolic factors
(EDRFs) such as NO and prostacyclin as well as endothelium-derived
contractile factors such as endothelin, epoxyeicosatrienoic acid
and thromboxane A2 to regulate vascular tone (96). Activated ECs exhibit impaired
synthesis or release of EDRFs, resulting in sustained
vasoconstriction, reduced vessel diameter, slowed blood flow and
impaired pulmonary microcirculation. On the other hand, activated
ECs recruit activated neutrophils and form neutrophil extracellular
traps with activated platelets, shifting to a pro-coagulant
phenotype. Anticoagulant molecules such as thrombomodulin (TM) are
other important markers of endothelial injury. Under normal
conditions, TM is expressed on the surface of ECs, where it exerts
anticoagulant effects by activating the protein C system through
binding to thrombin (97). During
endothelial injury, TM is shed from the cell surface into
circulation, forming soluble TM (sTM), exposing collagen fibers and
other subendothelial matrix proteins. The expression of platelet
adhesion molecules is upregulated and platelets are activated and
rapidly adhere to form a microthrombus, initiating the hemostatic
process. Previous research has shown that patients with ARDS who
exhibit high levels of sTM have a 3.5-fold increased risk of 60-day
mortality, which can serve as a biomarker for early prognostic
assessment, and perhaps provide a reference for clinical risk
stratification and treatment decision-making (98).
Coagulation factors interact with tissue factor
(TF), which is present on AECs and macrophages, initiating an
exogenous coagulation cascade. Within the intact vessel wall, TF
remains hidden. Following damage to the vascular endothelium, TF
becomes accessible to blood and can bind directly to coagulation
factor VII to create the TF-VIIa complex, which is capable of
activating clotting factors IX and X in the bloodstream, resulting
in the formation of active enzymes (coagulation factors IXa and Xa)
that subsequently convert plasminogen to thrombin. Thrombin then
cleaves fibrinogen into fibrin, which polymerizes to create a
fibrin network that forms a thrombus, incorporating aggregated
platelets (99). Naderpour et
al (100) studied patients
with COVID-19 during the pandemic and found that the combined use
of tissue plasminogen activator and heparin in patients with severe
respiratory failure improved oxygenation and reduced overall
mortality. Thus, impaired pulmonary microcirculation further
exacerbates tissue ischemia and hypoxia, promoting EC injury,
pro-inflammatory factor release and activation of apoptotic
pathways, thereby exacerbating lung tissue injury.
Advances in therapeutic strategies targeting
the synergistic effects of inflammation and apoptosis
Drug intervention targets and
strategies
Inhibitors of inflammatory and apoptotic
signaling pathways
The activation of essential signaling pathways, such
as NF-κB and MAPK, is closely linked to ALI associated with sepsis,
which subsequently triggers the apoptotic pathway and exacerbates
lung damage. NF-κB, a major regulator of inflammation, serves a
pivotal role in the onset and progression of numerous inflammatory
diseases. Inhibitors of NF-κB reduce the production of inflammatory
factors while also suppressing the expression of apoptosis-related
genes, thus presenting a potential therapeutic avenue for lung
injury driven by synergistic inflammation and apoptosis (Table II). For example, non-steroidal
anti-inflammatory medications (such as aspirin) specifically
prevent the phosphorylation and breakdown of IκBα, which in turn
reduces the activity of NF-κB. In the lung tissue of mice treated
with aspirin, NF-κB activation was notably inhibited, leading to
improved pulmonary edema (101).
Glucocorticoid receptor agonists, such as dexamethasone, can
inhibit NF-κB through direct interaction with its RELA
enhancer-binding protein like arrestin subunit, effectively
blocking its functional activity. Furthermore, fibroblast growth
factor (FGF) 18, which belongs to the FGF family, has demonstrated
the ability to reduce cellular inflammation (102). Previous research has shown that
FGF18 suppresses the phosphorylation of NF-κB p65 and reduces its
translocation to the nucleus in both in vivo and in
vitro settings. This activity consequently inhibits the
activation of the NF-κB pathway, reduces lung injury and supports
lung repair (103). In TCM, it is
considered that numerous herbs are related to this mechanism, among
which quercetin is a well-researched example (104).
| Table II.Inhibitors of inflammatory and
apoptotic signaling pathways. |
Table II.
Inhibitors of inflammatory and
apoptotic signaling pathways.
| Drug class | Representative
drugs | Mechanism of
action | Effect | (Refs.) |
|---|
| NF-κB
inhibitor | Aspirin | Blocks IκBα
phosphorylation and degradation, inhibits NF-κB activation | Reduces pulmonary
edema, inhibits inflammatory factor release | (101) |
|
| FGF18 | Inhibits NF-κB p65
phosphorylation and nuclear translocation | Reduces
inflammatory response, promotes lung repair | (103) |
| MAPK inhibitor | SB203580 (p38
inhibitor) | Inhibits p38 MAPK
phosphorylation | Reduces TNF-α,
IL-1β level, inhibits caspase-3 activation | (105) |
|
| SP600125 (JNK
inhibitor) | Blocks JNK
signaling | Inhibits Bax/Bak
activation, reduces mitochondrial apoptosis | (106) |
| Apoptosis pathway
inhibitor | Soluble Fas | Fas/FasL
inhibitor | Blocks exogenous
apoptotic pathway, attenuates alveolar epithelial cell injury | (107) |
|
| Z-VAD-FMK | Broad-spectrum
caspase inhibitor | Blocks apoptosis
execution stage, attenuates lung tissue damage | (108) |
| Herbal active
ingredient | Quercetin
pathway | Inhibits
NF-κB-related | Attenuates
SALI | (104) |
The MAPK pathway (including p38, JNK and ERK) also
serves a key role in inflammatory responses and apoptosis. Previous
research indicates that p38 inhibitors such as SB203580 can
mitigate LPS-induced lung injury through various mechanisms,
including inhibition of TNF-α and IL-1β release and reduction of
caspase-3 activity (105).
SP600125 is an orally active, reversible ATP-competitive JNK
inhibitor that can influence the expression of key proteins in the
mitochondrial apoptotic pathway, upregulate the expression of the
anti-apoptotic protein Bcl-2 and downregulate expression of the
pro-apoptotic protein Bax, thereby reducing apoptosis (106).
In addition, specific inhibitors of apoptotic
pathways may also become therapeutic targets for SALI. Fas
receptor/Fas ligand inhibitors (such as soluble Fas), as inhibitors
of the death receptor pathway, can block the extrinsic apoptotic
pathway and reduce alveolar epithelial cell damage (107). As a factor in apoptosis, the
broad-spectrum caspase inhibitor Z-Val-Ala-Asp-fluoromethylketone
can block the execution phase of apoptosis and alleviate lung
tissue damage (108).
Antioxidants and free radical
scavengers
Free radicals serve a key role in intracellular
signal transduction and various physiological processes at optimal
concentrations. However, excess of these free radicals can lead to
oxidative damage to proteins. The body's antioxidant defense system
comprises both endogenous antioxidants and exogenous free radical
scavenging mechanisms. Endogenous antioxidants primarily eliminate
excess free radicals and are categorized into enzymatic and
non-enzymatic ROS scavengers. Enzymatic antioxidants, including
superoxide dismutase (SOD) (109), catalase (CAT) (110) and glutathione peroxidase (GPX)
(111), facilitate the conversion
of O2 radicals into H2O2 and
O2, which are further catalyzed into H2O and
O2. Non-enzymatic ROS scavengers include coenzyme Q10
(also known as ubiquinone) (112,113) and acetylated phospholipids
(plasmalogens) (114), among
others. Coenzyme Q10 is integral to the mitochondrial electron
transport chain, functioning as a component of the mitochondrial
respiratory chain while scavenging lipid peroxidation free radicals
(113,115). Exogenous free radical scavengers,
primarily sourced from dietary intake, encompass hydrophilic
agents, such as vitamin C (116)
and glutathione, as well as lipophilic agents, including vitamin E
(117), flavonoids (118,119) and carotenoids (120), which effectively scavenge
O2− and HO−.
Mitochondria, as the primary site of ROS production,
are often subjected to high levels of ROS, leading to oxidative
damage of mitochondrial DNA. This damage subsequently activates the
mitochondrial-associated apoptotic pathway, thus exacerbating lung
injury. Antioxidants serve a key role in mitigating
inflammation-induced oxidative stress by scavenging free radicals
and maintaining the body's redox balance. This action effectively
inhibits the mitochondrial apoptotic pathway, thereby protecting
lung tissue from the combined detrimental effects of inflammation
and apoptosis. Numerous antioxidant drugs have been employed in
clinical treatments, including recombinant SOD, vitamin C, vitamin
E, α-lipoic acid (121),
bioflavonoids, selenium (122),
glutathione, coenzyme Q10 (123)
and various TCM herbal medicines such as Curcuma longa
(124), Astragalus
(125) and Rhodiola rosea
(126). Curcumin (124), through its β-diketone group and
Astragalus (125), through
active components such as polysaccharides, saponins and flavonoids,
directly scavenge ROS while simultaneously activating the
endogenous antioxidant enzyme system, which includes SOD, CAT and
GPX. This dual action results in a multi-level, multi-target
antioxidant protective effect. Similarly, study has demonstrated
that Rhodiola injection not only exhibits direct ROS
scavenging capabilities, but also protects mitochondrial function,
regulates the AMP-activated protein kinase/mTOR autophagy signaling
pathway and maintains the balance between cell apoptosis and
survival (126). This creates a
comprehensive protective network ranging from oxidative stress
prevention to cellular damage repair.
However, there is currently very few related
clinical trial available (127).
These therapeutic agents face important challenges in clinical
application. Previous research has indicated that vitamin E
supplementation does not reduce all-cause mortality in patients and
high doses of vitamin E may even increase overall mortality
(128). A similar issue is
observed with vitamin C, whereby excessively high levels in the
body can induce oxidative stress-related DNA damage, similar to the
effects of lower levels. Potential limitations of antioxidant
therapy may arise from the need to preserve an equilibrium between
oxidation and reduction. Elevated levels of ROS can activate
endogenous antioxidant defenses to protect damaged tissues.
Notably, scavenging ROS may increase the risk of infection and
disrupt ROS-dependent signaling pathways. Consequently, the timing,
concentration and dosage of antioxidants, along with their
bioavailability and effective targeted delivery to specific organs,
are important factors that are often challenging to regulate in
clinical practice.
Cell therapy and biological
agents
MSC therapy
There is increasing evidence to suggest that therapy
involving MSCs shows considerable potential for treating SALI and
ARDS and it is currently being evaluated in clinical trials
(129,130). MSCs have multiple functions and
originate from various sources including adipose tissue, bone
marrow, umbilical cord blood and placental tissue. These cells are
able to differentiate into a wide variety of cell types belonging
to the mesenchymal lineage (131,132). As a novel therapeutic agent for
ALI, MSCs may exhibit immunomodulatory, angiogenic and regenerative
effects (133–135). Mechanistically, MSCs can
differentiate into lung cells, directly replacing damaged cells and
tissues, thereby facilitating repair at the injury site.
Additionally, MSCs can migrate to damaged lung areas and reduce the
permeability of lung ECs and epithelial cells by secreting various
paracrine factors that promote neoangiogenesis and tissue repair, a
phenomenon validated in multiple preclinical ALI models (136–138).
Furthermore, MSCs possess complex immunomodulatory
functions. In a pro-inflammatory setting, MSCs have the ability to
release anti-inflammatory cytokines, including IL-10, TGF-β1 and
PGE2 (118), release exosomes
with microRNAs with anti-inflammatory effects (136), inhibit the activation of NF-κB,
reduce the release of pro-inflammatory factors (139) and upregulate the expression of
anti-apoptotic molecules, thereby exerting direct anti-inflammatory
and anti-apoptotic effects. Concurrently, MSCs also reduce
mitochondrial division, mitigate oxidative stress damage in
macrophages and induce macrophage polarization towards
anti-inflammatory phenotypes, indirectly contributing to
anti-inflammatory and antioxidant effects (140,141) (Fig.
4).
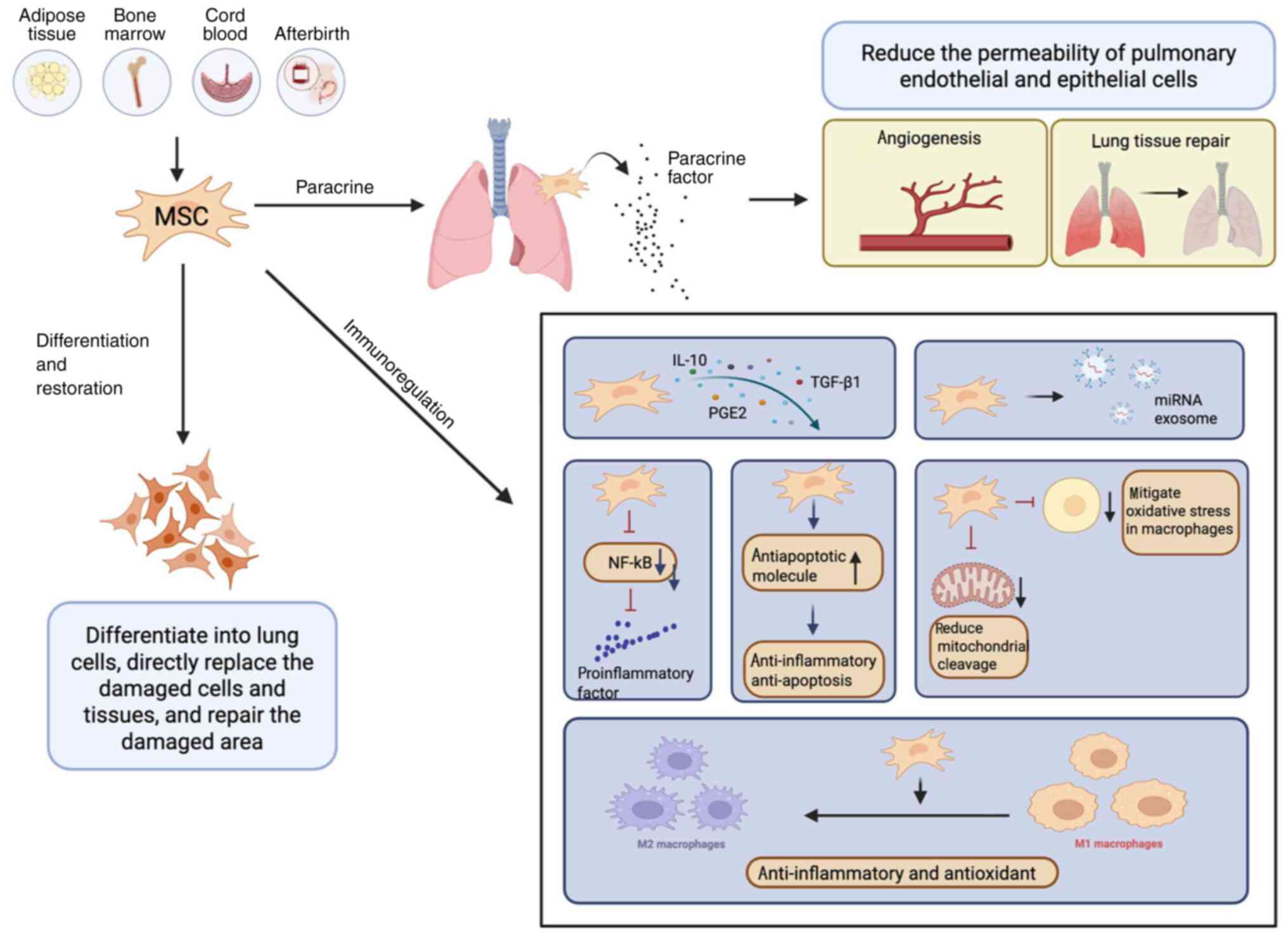 | Figure 4.MSC therapy. MSCs are derived from
various sources, including adipose tissue, bone marrow, umbilical
cord blood and placental tissue. On the one hand, MSCs can
differentiate into lung cells, directly replacing damaged cells and
tissues, thereby promoting the repair of injured sites.
Additionally, MSCs can migrate to the damaged lung area, where they
reduce the permeability of pulmonary endothelial and epithelial
cells by secreting various paracrine factors that promote
angiogenesis and tissue repair. On the other hand, MSCs release
anti-inflammatory cytokines and miRNA exosomes, which inhibit NF-κB
activation, decrease the release of pro-inflammatory factors and
upregulate the expression of anti-apoptotic molecules.
Simultaneously, MSCs also reduce mitochondrial fission, alleviate
oxidative stress damage in macrophages and induce the polarization
of macrophages toward an anti-inflammatory phenotype. MSCs,
mesenchymal stem cells; miRNA, micro-RNA; PGE2, prostaglandin
E2. |
The effects of MSCs on reducing inflammation,
modulating the immune response and promoting tissue repair have
been validated, showing considerable effectiveness in preclinical
studies involving animals. However, the marked individual
variability and prevalence of non-responsive patients observed in
clinical trials pose substantial challenges for the broader
application of MSC therapy in clinical practice (142). An initial clinical study suggests
that MSC therapy is generally safe and does not remain in the body
for a prolonged period of time (143). However, a certain experimental
study has reported the detection of MSCs ≤120 days
post-administration (144). This
persistence raises concerns regarding potential tumorigenesis, an
area where conclusive findings and solutions remain elusive.
Consequently, further accumulation of safety data regarding
long-term treatment and the large-scale application of MSC therapy
is important. Additionally, the optimal therapeutic regimen for
MSCs in treating SALI, including therapeutic dosage, timing of
treatment and route of administration, remains to be established.
Despite the encouraging effectiveness shown by MSCs in preliminary
animal research, it is important to recognize that mouse models
frequently do not accurately mimic human diseases. The quantity of
MSCs utilized in these mouse experiments vary greatly from those
administered to clinical patients, along with there being
considerable individual differences among humans, which could
additionally affect treatment results (144). Therefore, the clinical
therapeutic effects of MSCs are yet to be fully elucidated,
necessitating additional follow-up data to accurately assess their
efficacy.
Anti-apoptotic gene therapy and
biologics
Oxidative stress is a key mechanism in SALI.
Traditional antioxidants, such as vitamin C or vitamin E, have
limited clinical efficacy due to their lack of targeting
specificity and dose-dependent effects. To overcome this
bottleneck, progress has been made in recent years with novel
targeted antioxidants and biologics. For instance, developments
have been made in mitochondrial-targeted antioxidants such as
MitoQ, which has been shown to protect cells from apoptosis and
inhibit H2O2-induced growth factor receptor
signaling. This is primarily achieved through its unique
triphenylphosphonium carrier system, which precisely delivers the
compound to the interior of mitochondria, directly scavenging mtROS
and effectively blocking the
mtROS-Ca2+/calmodulin-dependent kinase II-mediated
apoptotic signaling pathway. In an investigation using MitoQ to
intervene in LPS-induced ALI in mice, it was found that, compared
with the model group, MitoQ treatment significantly reduced the
levels of oxidative markers in mouse serum (a 50% decrease in MDA
content; P<0.05), while also improving lung injury in mice
(145).
Edaravone, approved in Japan as a free radical
scavenger for ischemia-reperfusion injury, primarily works by
inhibiting lipid peroxidation and preventing the abnormal opening
of the mitochondrial MPTP, thereby reducing apoptosis in alveolar
epithelial cells. In a randomized clinical trial, edaravone was
used to treat critically ill patients with COVID-19 admitted to the
ICU. Results showed that the edaravone treatment group experienced
a reduction in mechanical ventilation dependency and a shortened
ICU treatment duration (146).
Numerous studies have shown that specific
biological substances can alleviate pulmonary injury by mitigating
inflammatory oxidative stress, offering new avenues for the
clinical management of SALI. For example, the nebulized formulation
of recombinant human SOD can specifically neutralize
O2− within lung tissue, directly targeting
the sites of inflammation. Furthermore, FGF21, which was initially
identified and cloned in 2000, performs various biological roles,
including promoting tissue repair and regulating metabolism
(147). Research conducted by Gao
et al (148) suggested
that the administration of exogenous FGF21 leads to a reduction in
lung inflammation and apoptosis by influencing the TLR4/MyD88/NF-κB
signaling pathway, thereby mitigating SALI. As a result, the
relevance of FGF21 in severe conditions such as ALI and ARDS has
gained interest (149).
Conclusion
The present study systematically summarized the
synergistic mechanisms of inflammation and apoptosis in SALI,
emphasizing the pivotal role of signaling pathways such as
NF-κB/MAPK and the cascade of inflammatory factors (TNF-α, IL-1β
and MCP-1) they mediate, which drive oxidative stress and apoptosis
activation, ultimately leading to the disruption of alveolar
epithelial and pulmonary vascular endothelial barriers. Despite
progress in therapeutic strategies targeting synergistic effects
(such as MSC therapy and targeted antioxidants), key research gaps
and translational challenges remain. For example, the detailed
molecular regulatory networks between inflammation and apoptosis,
as well as the impact of individual differences on their
synergistic effects, have not yet been fully elucidated.
Furthermore, there is a lack of targeted specificity regulation
tools, such as precise identification of biomarkers for different
pathological stages of SALI and insufficient targeting of
lung-specific drug delivery systems to acute inflammatory lesion
areas.
Future research should focus on elucidating the
molecular complexity of the synergistic effects between
inflammation and apoptosis to develop more targeted and effective
therapeutic strategies. Additionally, multicenter, large-sample
clinical studies should be conducted to explore the clinical
translation potential of apoptosis/inflammation pathway inhibitors.
Integrating multidisciplinary resources from immunology,
bioengineering, data science and other fields will advance the
synergistic development of regulatory science and technological
innovation. There is an expectation that such initiatives will
generate innovative concepts and approaches to enhance the
prognosis of patients with SALI.
Acknowledgements
Not applicable.
Funding
The present review was supported by the National Key Research
and Development Program of China (grant no. 2021YFC2501800) and the
6th Shanghai Three-Year Action Plan to Strengthen the Construction
of Public Health System (grant no. GWVI-2.1–3).
Availability of data and materials
Not applicable.
Authors' contributions
LZ contributed to conceptualization, visualization,
writing the original draft, reviewing and editing. HL contributed
to investigation, writing, reviewing and editing the manuscript. DL
contributed to formal analysis, the software used, writing,
reviewing and editing the manuscript. QD contributed to funding
acquisition, project administration, resources, supervision,
writing, reviewing and editing the manuscript. All authors have
read and approved the final manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gao X, Cai S, Li X and Wu G:
Sepsis-induced immunosuppression: Mechanisms, biomarkers and
immunotherapy. Front Immunol. 16:15771052025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xie J, Wang H, Kang Y, Zhou L, Liu Z, Qin
B, Ma X, Cao X, Chen D, Lu W, et al: The epidemiology of sepsis in
Chinese ICUs: A National cross-sectional survey. Crit Care Med.
48:e209–e218. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Dulhunty JM, Brett SJ, De Waele JJ,
Rajbhandari D, Billot L, Cotta MO, Davis JS, Finfer S, Hammond NE,
Knowles S, et al: Continuous vs Intermittent β-lactam antibiotic
infusions in critically Ill patients with sepsis: The BLING III
Randomized clinical trial. JAMA. 332:629–637. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li W, Li D, Chen Y, Abudou H, Wang H, Cai
J, Wang Y, Liu Z, Liu Y and Fan H: Classic signaling pathways in
alveolar injury and repair involved in sepsis-induced ALI/ARDS: New
research progress and prospect. Dis Markers.
2022:63623442022.PubMed/NCBI
|
|
6
|
Vincent JL, Opal SM, Marshall JC and
Tracey KJ: Sepsis definitions: Time for change. Lancet.
381:774–775. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang J, Huang K, Xu S, Garcia JGN, Wang C
and Cai H: Targeting NOX4 alleviates sepsis-induced acute lung
injury via attenuation of redox-sensitive activation of
CaMKII/ERK1/2/MLCK and endothelial cell barrier dysfunction. Redox
Biol. 36:1016382020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Boada-Romero E, Martinez J, Heckmann BL
and Green DR: The clearance of dead cells by efferocytosis. Nat Rev
Mol Cell Biol. 21:398–414. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qu J, Jin J, Zhang M and Ng LG: Neutrophil
diversity and plasticity: Implications for organ transplantation.
Cell Mol Immunol. 20:993–1001. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zi SF, Wu XJ, Tang Y, Liang YP, Liu X,
Wang L, Li SL, Wu CD, Xu JY, Liu T, et al: Endothelial cell-derived
extracellular vesicles promote aberrant neutrophil trafficking and
subsequent remote lung injury. Adv Sci (Weinh). 11:e24006472024.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang R, Zheng T, Xiang H, Liu M and Hu K:
Lung single-cell RNA profiling reveals response of pulmonary
capillary to sepsis-induced acute lung injury. Front Immunol.
15:13089152024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Serhan CN, Chiang N and Van Dyke TE:
Resolving inflammation: Dual anti-inflammatory and pro-resolution
lipid mediators. Nat Rev Immunol. 8:349–361. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
do Nascimento MF, Ferreira LRP, Vieira
Junior JM, Deheinzelin D, Aparecida Santos Nussbaum AC, Toshihiro
Sakamoto LH, Vasconcelos RO, Salomao R, Waisberg J, Azevedo LCP, et
al: Circulating extracellular vesicles as potential biomarkers and
mediators of acute respiratory distress syndrome in sepsis. Sci
Rep. 15:55122025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gupta S and Sahni V: The intriguing
commonality of NETosis between COVID-19 & Periodontal disease.
Med Hypotheses. 144:1099682020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Scozzi D, Liao F, Krupnick AS, Kreisel D
and Gelman AE: The role of neutrophil extracellular traps in acute
lung injury. Front Immunol. 13:9531952022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kumar S, Payal N, Srivastava VK, Kaushik
S, Saxena J and Jyoti A: Neutrophil extracellular traps and organ
dysfunction in sepsis. Clin Chim Acta. 523:152–162. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP
and Miao C: Neutrophil, neutrophil extracellular traps and
endothelial cell dysfunction in sepsis. Clin Transl Med.
13:e11702023. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Fuchs TA, Brill A, Duerschmied D,
Schatzberg D, Monestier M, Myers DD Jr, Wrobleski SK, Wakefield TW,
Hartwig JH and Wagner DD: Extracellular DNA traps promote
thrombosis. Proc Natl Acad Sci USA. 107:15880–15885. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peng Y, Zhou M, Yang H, Qu R, Qiu Y, Hao
J, Bi H and Guo D: Regulatory mechanism of M1/M2 macrophage
polarization in the development of autoimmune diseases. Mediators
Inflamm. 2023:88216102023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dang W, Tao Y, Xu X, Zhao H, Zou L and Li
Y: The role of lung macrophages in acute respiratory distress
syndrome. Inflamm Res. 71:1417–1432. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Z and Wang Z: The role of macrophages
polarization in sepsis-induced acute lung injury. Front Immunol.
14:12094382023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li S, Feng T, Zhang Y, Shi Q, Wang W, Ren
J, Shen G, Gu H, Luo C and Li Y: Lianhua Qingwen protects
LPS-induced acute lung injury by promoting M2 macrophage
infiltration. J Ethnopharmacol. 320:1174672024. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang J, Huang X, Yu Q, Wang S, Wen X, Bai
S, Cao L, Zhang K, Zhang S and Wang X: Extracellular vesicles
derived from M2-like macrophages alleviate acute lung injury in a
miR-709-mediated manner. J Extracell Vesicles. 13:e124372024.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Morrell ED, Bhatraju PK, Mikacenic CR,
Radella F II, Manicone AM, Stapleton RD, Wurfel MM and Gharib SA:
Alveolar macrophage transcriptional programs are associated with
outcomes in acute respiratory distress Syndrome. Am J Respir Crit
Care Med. 200:732–741. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zenga J, Loi GWZ, Saipuljumri EN, Romero
Durán MA, Silva-García O, Perez-Aguilar JM, Baizabal-Aguirre VM and
Lo CH: Peptide-based allosteric inhibitor targets TNFR1
conformationally active region and disables receptor-ligand
signaling complex. Proc Natl Acad Sci USA. 121:e23081321212024.
View Article : Google Scholar
|
|
26
|
Crijns H, Vanheule V and Proost P:
Targeting chemokine-glycosaminoglycan interactions to inhibit
inflammation. Front Immunol. 11:4832020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Karin N and Wildbaum G: The role of
chemokines in shaping the balance between CD4+ T cell subsets and
its therapeutic implications in autoimmune and cancer diseases.
Front Immunol. 6:6092015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang S, Yu J, Dong X, Zeng J, Tan L, Zhang
H, Sun R, Tuo Y, Yang J, Wan C and Bai H: CCR2 signaling regulates
anti-chlamydia T cell immune responses in the airway. PLoS Pathog.
21:e10129122025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cheng PP, He XL, Jia ZH, Hu SH, Feng X,
Jiang YH, Li Q, Zhao LQ, Cui XL, Ye SY, et al: Midkine, a novel
MCP-1 activator mediated PM2.5-aggravated experimental pulmonary
fibrosis. Environ Int. 197:1093542025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Neehus AL, Carey B, Landekic M, Panikulam
P, Deutsch G, Ogishi M, Arango-Franco CA, Philippot Q, Modaresi M,
Mohammadzadeh I, et al: Human inherited CCR2 deficiency underlies
progressive polycystic lung disease. Cell. 187:390–408. e232024.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Molino S, Pisarevsky A, Badu S, Wu Q,
Mingorance FL, Vega P, Stefanolo JP, Repetti J, Ludueña G, Pepa P,
et al: Randomized placebo-controlled trial of oral tannin
supplementation on COVID-19 symptoms, gut dysbiosis and cytokine
response. J Funct Foods. 99:1053562022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hammock BD, Wang W, Gilligan MM and
Panigrahy D: Eicosanoids: The overlooked storm in coronavirus
disease 2019 (COVID-19)? Am J Pathol. 190:1782–1788. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wu J, Ye J, Kong W, Zhang S and Zheng Y:
Programmed cell death pathways in hearing loss: A review of
apoptosis, autophagy and programmed necrosis. Cell Prolif.
53:e129152020. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dho SH, Cho M, Woo W, Jeong S and Kim LK:
Caspases as master regulators of programmed cell death: apoptosis,
pyroptosis and beyond. Exp Mol Med. 57:1121–1132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Carneiro BA and El-Deiry WS: Targeting
apoptosis in cancer therapy. Nat Rev Clin Oncol. 17:395–417. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Algeciras-Schimnich A, Shen L, Barnhart
BC, Murmann AE, Burkhardt JK and Peter ME: Molecular ordering of
the initial signaling events of CD95. Mol Cell Biol. 22:207–220.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fu Y, Sacco O, DeBitetto E, Kanshin E,
Ueberheide B and Sfeir A: Mitochondrial DNA breaks activate an
integrated stress response to reestablish homeostasis. Mol Cell.
83:3740–3753. e92023. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bilen M, Benhammouda S, Slack RS and
Germain M: The integrated stress response as a key pathway
downstream of mitochondrial dysfunction. Curr Opinion Physiol.
27:1005552022. View Article : Google Scholar
|
|
39
|
Breckenridge DG, Stojanovic M, Marcellus
RC and Shore GC: Caspase cleavage product of BAP31 induces
mitochondrial fission through endoplasmic reticulum calcium
signals, enhancing cytochrome c release to the cytosol. J Cell
Biol. 160:1115–1127. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Du Y, Wang G, Liu B, Guo M, Yan X, Dou M,
Yu F, Ba Y and Zhou G: Naringin alleviates fluoride-induced
neurological impairment: A focus on the regulation of energy
metabolism mediated by mitochondrial permeability transition pore.
Sci Total Environ. 955:1770732024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Xue Y, Wang J, Huang Y, Gao X, Kong L,
Zhang T and Tang M: Comparative cytotoxicity and apoptotic pathways
induced by nanosilver in human liver HepG2 and L02 cells. Hum Exp
Toxicol. 37:1293–1309. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chang X, Niu S, Shang M, Li J, Guo M,
Zhang W, Sun Z, Li Y, Zhang R, Shen X, et al: ROS-Drp1-mediated
mitochondria fission contributes to hippocampal HT22 cell apoptosis
induced by silver nanoparticles. Redox Biol. 63:1027392023.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang HW, Zhang Y, Tan PP, Jia LS, Chen Y
and Zhou BH: Mitochondrial respiratory chain dysfunction mediated
by ROS is a primary point of fluoride-induced damage in Hepa1-6
cells. Environ Pollut. 255((Pt 3)): 1133592019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kushnareva Y, Andreyev AY, Kuwana T and
Newmeyer DD: Bax activation initiates the assembly of a multimeric
catalyst that facilitates bax pore formation in mitochondrial outer
membranes. PLoS Biol. 10:e10013942012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Vandenabeele P, Bultynck G and Savvides
SN: Pore-forming proteins as drivers of membrane permeabilization
in cell death pathways. Nat Rev Mol Cell Biol. 24:312–333. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
An J, Park SH, Ko IG, Jin JJ, Hwang L, Ji
ES, Kim SH, Kim CJ, Park SY, Hwang JJ and Choi CW:
Polydeoxyribonucleotide ameliorates lipopolysaccharide-induced lung
injury by inhibiting apoptotic cell death in rats. Int J Mol Sci.
18:18472017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang H, Guo M, Wei H and Chen Y: Targeting
p53 pathways: Mechanisms, structures, and advances in therapy.
Signal Transduct Target Ther. 8:922023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chen L, Liu S and Tao Y: Regulating tumor
suppressor genes: Post-translational modifications. Signal
Transduct Target Ther. 5:902020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wei H, Qu L, Dai S, Li Y, Wang H, Feng Y,
Chen X, Jiang L, Guo M, Li J, et al: Structural insight into the
molecular mechanism of p53-mediated mitochondrial apoptosis. Nat
Commun. 12:22802021. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Aubrey BJ, Kelly GL, Janic A, Herold MJ
and Strasser A: How does p53 induce apoptosis and how does this
relate to p53-mediated tumour suppression? Cell Death Differ.
25:104–113. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wang S, Zhong S, Huang Y, Zhu S, Chen S,
Wang R, Wangmo S, Peng B, Lv H, Yang J, et al: MDM2 is essential to
maintain the homeostasis of epithelial cells by targeting p53. J
Innate Immun. 16:397–412. 2024.PubMed/NCBI
|
|
52
|
Lane K, Andres-Terre M, Kudo T, Monack DM
and Covert MW: Escalating threat levels of bacterial infection can
be discriminated by distinct MAPK and NF-kappaB signaling dynamics
in single host cells. Cell Syst. 8:183–196. e42019. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
DeCuzzi NL, Oberbauer D, Chmiel KJ,
Pargett M, Ferguson JM, Murphy D, Hardy M, Ram A, Zeki AA and
Albeck JG: Spatiotemporal clusters of extracellular
signal-regulated kinase activity coordinate cytokine-induced
inflammatory responses in human airway epithelial cells. Am J
Respir Cell Mol Biol. 72:520–532. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Mokra D: Acute lung injury - from
pathophysiology to treatment. Physiol Res. 69 (Suppl 3):S353–S366.
2020.PubMed/NCBI
|
|
55
|
McMinimy R, Manford AG, Gee CL,
Chandrasekhar S, Mousa GA, Chuang J, Phu L, Shih KY, Rose CM,
Kuriyan J, et al: Reactive oxygen species control protein
degradation at the mitochondrial import gate. Mol Cell.
84:4612–4628. e132024. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang J, Wei Y, Yue Y, Jiao H, Wu Y, Fu W,
Lin KM, Lu C, Mou S and Zhong Q: RIPK4 promotes oxidative stress
and ferroptotic death through the downregulation of ACSM1. Proc
Natl Acad Sci USA. 121:e24106281212024. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Liang J, Cao R, Wang X, Zhang Y, Wang P,
Gao H, Li C, Yang F, Zeng R, Wei P, et al: Mitochondrial PKM2
regulates oxidative stress-induced apoptosis by stabilizing Bcl2.
Cell Res. 27:329–351. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ma M, Jiang W and Zhou R: DAMPs and
DAMP-sensing receptors in inflammation and diseases. Immunity.
57:752–771. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Deng C, Zhao L, Yang Z, Shang JJ, Wang CY,
Shen MZ, Jiang S, Li T, Di WC, Chen Y, et al: Targeting HMGB1 for
the treatment of sepsis and sepsis-induced organ injury. Acta
Pharmacol Sin. 43:520–528. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Dutta S, Dutta S, Somanath PR, Narayanan
SP, Wang X and Zhang D: Circulating nucleosomes and histones in the
development of lung injury and sepsis. Curr Issues Mol Biol.
47:1332025. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Pei Q, Ni W, Yuan Y, Yuan J, Zhang X and
Yao M: HSP70 Ameliorates Septic Lung Injury via Inhibition of
Apoptosis by Interacting with KANK2. Biomolecules. 12:4102022.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Karki P, Zhang CO, Promnares K, Li Y, Ke
Y, Birukova AA and Birukov KG: Truncated oxidized phospholipids
exacerbate endothelial dysfunction and lung injury caused by
bacterial pathogens. Cell Signal. 109:1108042023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Munemasa Y: Histone H2B induces retinal
ganglion cell death through toll-like receptor 4 in the vitreous of
acute primary angle closure patients. Lab Invest. 100:1080–1089.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Beltran-Garcia J, Osca-Verdegal R,
Perez-Cremades D, Novella S, Hermenegildo C, Pallardó FV and
García-Giménez JL: Extracellular histones activate endothelial
NLRP3 inflammasome and are associated with a severe sepsis
phenotype. J Inflamm Res. 15:4217–4238. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Orecchioni M, Kobiyama K, Winkels H,
Ghosheh Y, McArdle S, Mikulski Z, Kiosses WB, Fan Z, Wen L, Jung Y,
et al: Olfactory receptor 2 in vascular macrophages drives
atherosclerosis by NLRP3-dependent IL-1 production. Science.
375:214–221. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Dong Y and Yong VW: Oxidized phospholipids
as novel mediators of neurodegeneration. Trends Neurosci.
45:419–429. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Elliott MR, Chekeni FB, Trampont PC,
Lazarowski ER, Kadl A, Walk SF, Park D, Woodson RI, Ostankovich M,
Sharma P, et al: Nucleotides released by apoptotic cells act as a
find-me signal to promote phagocytic clearance. Nature.
461:282–286. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yang M, Liu J, Piao C, Shao J and Du J:
ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2
polarization through blockade of efferocytosis. Cell Death Dis.
6:e17802015. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Wang Y, Zhang W, Xu Y, Wu D, Gao Z, Zhou
J, Qian H, He B and Wang G: Extracellular HMGB1 impairs
macrophage-mediated efferocytosis by suppressing the
Rab43-controlled cell surface transport of CD91. Front Immunol.
13:7676302022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Bondue B, Vosters O, de Nadai P, Glineur
S, De Henau O, Luangsay S, Van Gool F, Communi D, De Vuyst P,
Desmecht D and Parmentier M: ChemR23 dampens lung inflammation and
enhances anti-viral immunity in a mouse model of acute viral
pneumonia. PLoS Pathog. 7:e10023582011. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Duvall MG, Bruggemann TR and Levy BD:
Bronchoprotective mechanisms for specialized pro-resolving
mediators in the resolution of lung inflammation. Mol Aspects Med.
58:44–56. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Bos LDJ and Ware LB: Acute respiratory
distress syndrome: Causes, pathophysiology, and phenotypes. Lancet.
400:1145–1156. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Burgess CL, Huang J, Bawa PS, Alysandratos
KD, Minakin K, Ayers LJ, Morley MP, Babu A, Villacorta-Martin C,
Yampolskaya M, et al: Generation of human alveolar epithelial type
I cells from pluripotent stem cells. Cell Stem Cell. 31:657–675.
e82024. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Meyer NJ, Gattinoni L and Calfee CS: Acute
respiratory distress syndrome. Lancet. 398:622–637. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Rizzo AN, Haeger SM, Oshima K, Yang Y,
Wallbank AM, Jin Y, Lettau M, McCaig LA, Wickersham NE, McNeil JB,
et al: Alveolar epithelial glycocalyx degradation mediates
surfactant dysfunction and contributes to acute respiratory
distress syndrome. JCI Insight. 7:e1545732022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Han S, Lee M, Shin Y, Giovanni R,
Chakrabarty RP, Herrerias MM, Dada LA, Flozak AS, Reyfman PA,
Khuder B, et al: Mitochondrial integrated stress response controls
lung epithelial cell fate. Nature. 620:890–897. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Xi Q, Liu L, Zhao Q and Zhu S: KLF13
attenuates lipopolysaccharide-induced alveolar epithelial cell
damage by regulating mitochondrial quality control via binding
PGC-1α. J Interferon Cytokine Res. 45:227–237. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang C, Wu Z, Li Z, Wang Z, Ke H and Huang
X: Beneficial effect of the mitochondrial ATP-sensitive potassium
channel-specific opener nicorandil on the collapsed lung via
inhibition of apoptosis in clinical thoracic surgery. Mol Med Rep.
27:612023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Lei X, Liu X, Yu J, Li K, Xia L, Su S, Lin
P, Zhang D and Li Y: 3-methyladenine ameliorates acute lung injury
by inhibiting oxidative damage and apoptosis. Heliyon.
10:e339962024. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Lanzoni G, Linetsky E, Correa D, Messinger
Cayetano S, Alvarez RA, Kouroupis D, Alvarez Gil A, Poggioli R,
Ruiz P, Marttos AC, et al: Umbilical cord mesenchymal stem cells
for COVID-19 acute respiratory distress syndrome: A double-blind,
phase 1/2a, randomized controlled trial. Stem Cells Transl Med.
10:660–673. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Collada A, Cruz A and Perez-Gil J:
Studying the interfacial activity and structure of pulmonary
surfactant complexes. Chem Phys Lipids. 266:1054592025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Inoue S, Nagao J, Kawamoto K, Kan-O K,
Fukuyama S, Sasaki S, Kudo S, Okamoto I and Sera T: Overstretching
alveolar epithelial type II cells decreases surfactant secretion
via actin polymerization and intracellular trafficking alteration.
Heliyon. 10:e334992024. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Hanusrichterova J, Mokry J, Al-Saiedy MR,
Koetzler R, Amrein MW, Green FHY and Calkovska A: Factors
influencing airway smooth muscle tone: A comprehensive review with
a special emphasis on pulmonary surfactant. Am J Physiol Cell
Physiol. 327:C798–C816. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Bastani MN and Jalilian S: Unraveling the
enigma: The emerging significance of pulmonary surfactant proteins
in predicting, diagnosing, and managing COVID-19. Immun Inflamm
Dis. 12:e13022024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Jacob IB, Lawal AO, Mahmoud SS, Kopsack
EM, Reynolds ES, Meng Q, Fan H, Massa PT, Thangamani S, Jia H and
Wang G: Differential immunoregulation by human surfactant protein A
variants determines severity of SARS-CoV-2-induced lung disease.
Front Immunol. 16:14622782025. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhu Y, Choi D, Somanath PR and Zhang D:
Lipid-laden macrophages in pulmonary diseases. Cells. 13:8892024.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang J, Deng Y, Li G and Sun X: Interplay
of surfactant protein A and tumor necrosis factor α in lung and
intestinal tissues of rats with severe pneumonia. Mol Biotechnol.
Apr 24–2025.(Epub ahead of print). View Article : Google Scholar
|
|
88
|
Dargaville PA, Kamlin COF, Orsini F, Wang
X, De Paoli AG, Kanmaz Kutman HG, Cetinkaya M, Kornhauser-Cerar L,
Derrick M, Özkan H, et al: Two-year outcomes after minimally
invasive surfactant therapy in preterm infants: Follow-Up of the
OPTIMIST-A Randomized clinical trial. JAMA. 330:1054–1063. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Schupp JC, Adams TS, Cosme C Jr, Raredon
MSB, Yuan Y, Omote N, Poli S, Chioccioli M, Rose KA, Manning EP, et
al: Integrated single-cell atlas of endothelial cells of the human
lung. Circulation. 144:286–302. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Whitney JE, Zhang B, Koterba N, Chen F,
Bush J, Graham K, Lacey SF, Melenhorst JJ, Teachey DT, Mensinger
JL, et al: Systemic endothelial activation is associated with early
acute respiratory distress syndrome in children with extrapulmonary
sepsis. Crit Care Med. 48:344–352. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Runkle EA and Mu D: Tight junction
proteins: From barrier to tumorigenesis. Cancer Lett. 337:41–48.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Guo X, Eitnier RA, Beard RS Jr, Meegan JE,
Yang X, Aponte AM, Wang F, Nelson PR and Wu MH: Focal adhesion
kinase and Src mediate microvascular hyperpermeability caused by
fibrinogen- үC-terminal fragments. PLoS One. 15:e02317392020.
View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Garcia-Flores AE, Gross CM, Zemskov EA, Lu
Q, Tieu K, Wang T and Black SM: Loss of SOX18/CLAUDIN5 disrupts the
pulmonary endothelial barrier in ventilator-induced lung injury.
Front Physiol. 13:10665152022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Rezk-Hanna M, Rossman MJ, Ludwig K, Sakti
P, Cheng CW, Brecht ML, Benowitz NL and Seals DR: Electronic hookah
(waterpipe) vaping reduces vascular endothelial function: The role
of nicotine. Am J Physiol Heart Circ Physiol. 326:H490–H496. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Solomon SD, Lowenstein CJ, Bhatt AS,
Peikert A, Vardeny O, Kosiborod MN, Berger JS, Reynolds HR,
Mavromichalis S, Barytol A, et al: Effect of the P-selectin
inhibitor crizanlizumab on survival free of organ support in
patients hospitalized for COVID-19: A Randomized controlled trial.
Circulation. 148:381–390. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Zhu J, Yang L, Jia Y, Balistrieri A,
Fraidenburg DR, Wang J, Tang H and Yuan JX: Pathogenic mechanisms
of pulmonary arterial hypertension: Homeostasis imbalance of
endothelium-derived relaxing and contracting factors. JACC Asia.
2:787–802. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Boron M, Hauzer-Martin T, Keil J and Sun
XL: Circulating thrombomodulin: Release mechanisms, measurements,
and levels in diseases and medical procedures. TH Open.
6:e194–e212. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Sanchez-Santillan RN, Sierra-Vargas MP,
Gonzalez-Islas D, Aztatzi-Aguilar OG, Pérez-Padilla R, Orea-Tejeda
A, Debray-García Y, Ortega-Romero M, Keirns-Davis C, Loaeza-Roman A
and Rios-Pereda A: Endothelial biomarkers (Von willebrand factor,
BDCA3, urokinase) as predictors of mortality in COVID-19 patients:
Cohort study. BMC Pulm Med. 24:3252024. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Neubauer K and Zieger B: Endothelial cells
and coagulation. Cell Tissue Res. 387:391–398. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Naderpour Z, Aliannejad R, Mehrtash V,
Mollazadeh R, Hosseini SE, Amini S, Pak N, Motlaq TM, Khodaei B,
Jafarzadeh B, et al: Tissue plasminogen activator for
COVID-19-induced severe acute respiratory distress Syndrome: A
controlled clinical trial. Infect Disord Drug Targets. 2025.(Epub
ahead of print). View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Tung YT, Wei CH, Yen CC, Lee PY, Ware LB,
Huang HE, Chen W and Chen CM: Aspirin attenuates hyperoxia-induced
acute respiratory distress Syndrome (ARDS) by suppressing pulmonary
inflammation via the NF-kappaB signaling pathway. Front Pharmacol.
12:7931072022. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Li XG, Song X, Wang JY, Sun CH, Li ZQ,
Meng LL and Chi SH: Fibroblast growth factor 18 alleviates
hyperoxia-induced lung injury in mice by adjusting oxidative stress
and inflammation. Eur Rev Med Pharmacol Sci. 25:1485–1494.
2021.PubMed/NCBI
|
|
103
|
Hu Z, Dai J, Xu T, Chen H, Shen G, Zhou J,
Ma H, Wang Y and Jin L: FGF18 alleviates sepsis-induced acute lung
injury by inhibiting the NF-ĸB pathway. Respir Res. 25:1082024.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Wang T, Lv L, Feng H and Gu W: Unlocking
the potential: Quercetin and its natural derivatives as promising
therapeutics for sepsis. Biomedicines. 12:4442024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Malekinejad Z, Baghbanzadeh A, Nakhlband
A, Baradaran B, Jafari S, Bagheri Y, Raei F, Montazersaheb S and
Farahzadi R: Recent clinical findings on the role of kinase
inhibitors in COVID-19 management. Life Sci. 306:1208092022.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Sun DZ, Song CQ, Xu YM and Dong XS: Role
of the MAPK pathway in human lung epithelial-like A549 cells
apoptosis induced by paraquat. Genet Mol Biol. 43:e201901372020.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Grassme H, Kirschnek S, Riethmueller J,
Riehle A, von Kürthy G, Lang F, Weller M and Gulbins E: CD95/CD95
ligand interactions on epithelial cells in host defense to
Pseudomonas aeruginosa. Science. 290:527–530. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sun R, Jiang K, Zeng C, Zhu R, Chu H, Liu
H and Du J: Synergism of TNF-α and IFN-β triggers human airway
epithelial cells death by apoptosis and pyroptosis. Mol Immunol.
153:160–169. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Luo Z, Wang Q, Fan X, Koh XQ, Loh XJ, Wu
C, Li Z and Wu YL: ROS-driven nanoventilator for MRSA-induced acute
lung injury treatment via in situ oxygen supply, anti-inflammation
and immunomodulation. Adv Sci (Weinh). 12:e24060602025. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Hansberg W: Monofunctional Heme-Catalases.
Antioxidants (Basel). 11:21732022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Flohe L, Toppo S and Orian L: The
glutathione peroxidase family: Discoveries and mechanism. Free
Radic Biol Med. 187:113–122. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chen Y, Yang H, Hu X, Yang T, Zhao Y, Liu
H and Fan H: Coenzyme Q10 ameliorates lipopolysaccharide-induced
acute lung injury by attenuating oxidative stress and NLRP3
inflammation through regulating mitochondrial dynamics. Int
Immunopharmacol. 141:1129412024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Wang Y, Lilienfeldt N and Hekimi S:
Understanding coenzyme Q. Physiol Rev. 104:1533–1610. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Honsho M and Fujiki Y: Asymmetric
distribution of plasmalogens and their roles-A mini review.
Membranes (Basel). 13:7642023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Steenberge LH, Rogers S, Sung AY, Fan J
and Pagliarini DJ: Coenzyme Q(4) is a functional substitute for
coenzyme Q(10) and can be targeted to the mitochondria. J Biol
Chem. 300:1072692024. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Traber MG, Leonard SW, Vasu VT, Morrissey
BM, Lei HJ, Atkinson J and Cross CE: α-Tocopherol pharmacokinetics
in adults with cystic fibrosis: Benefits of supplemental vitamin C
administration. Nutrients. 14:37172022. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Traber MG and Head B: Vitamin E: How much
is enough, too much and why! Free Radic Biol Med. 177:212–225.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Kazemi A, Iraji A, Esmaealzadeh N, Salehi
M and Hashempur MH: Peppermint and menthol: A review on their
biochemistry, pharmacological activities, clinical applications,
and safety considerations. Crit Rev Food Sci Nutr. 65:1553–1578.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Huang M, Liu X, Ren Y, Huang Q, Shi Y,
Yuan P and Chen M: Quercetin: A flavonoid with potential for
treating acute lung injury. Drug Des Devel Ther. 18:5709–5728.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Martini D, Negrini L, Marino M, Riso P,
Del Bo C and Porrini M: What is the current direction of the
research on carotenoids and human health? An overview of registered
clinical trials. Nutrients. 14:11912022. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Shanaida M, Lysiuk R, Mykhailenko O, Hudz
N, Abdulsalam A, Gontova T, Oleshchuk O, Ivankiv Y, Shanaida V,
Lytkin D and Bjørklund G: Alpha-lipoic Acid: An antioxidant with
anti-aging properties for disease therapy. Curr Med Chem. 32:23–54.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Maia LB, Maiti BK, Moura I and Moura JJG:
Selenium-more than just a fortuitous sulfur substitute in redox
biology. Molecules. 29:1202023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Shin JY, Choi JW, Kim DG, Zhou ZQ, Shin
YK, Seo JH, Song HJ, Choi BM, Bae GS and Park SJ: Protective
effects of Coenzyme Q10 against acute pancreatitis. Int
Immunopharmacol. 88:1069002020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Hou J, Fang F, Kang S, Wang Z and Yang Y:
Curcumin from Jianghuang (Rhizoma Curcumae Longae) protects against
exposure to ultraviolet B by antioxidation and attenuating
mitochondrion-dependent apoptosis. J Tradit Chin Med. 40:782–791.
2020.PubMed/NCBI
|
|
125
|
Yao J, Peng T, Shao C and Liu Y, Lin H and
Liu Y: The antioxidant action of astragali radix: Its active
components and molecular basis. Molecules. 29:16912024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Zhao J, Zhang J, Liu Q, Wang Y, Jin Y,
Yang Y, Ni C and Zhang L: Hongjingtian injection protects against
myocardial ischemia reperfusion-induced apoptosis by blocking ROS
induced autophagic-flux. Biomed Pharmacother. 135:1112052021.
View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Shafabakhsh R, Mobini M, Raygan F,
Aghadavod E, Ostadmohammadi V, Amirani E, Mansournia MA and Asemi
Z: Curcumin administration and the effects on psychological status
and markers of inflammation and oxidative damage in patients with
type 2 diabetes and coronary heart disease. Clin Nutr ESPEN.
40:77–82. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
O'Connor EA, Evans CV, Ivlev I, Rushkin
MC, Thomas RG, Martin A and Lin JS: Vitamin and mineral supplements
for the primary prevention of cardiovascular disease and cancer:
Updated evidence report and systematic review for the US preventive
services task force. JAMA. 327:2334–2347. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Monsel A, Zhu YG, Gennai S, Hao Q, Liu J
and Lee JW: Cell-based therapy for acute organ injury: Preclinical
evidence and ongoing clinical trials using mesenchymal stem cells.
Anesthesiology. 121:1099–1121. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Matthay MA, Calfee CS, Zhuo H, Thompson
BT, Wilson JG, Levitt JE, Rogers AJ, Gotts JE, Wiener-Kronish JP,
Bajwa EK, et al: Treatment with allogeneic mesenchymal stromal
cells for moderate to severe acute respiratory distress syndrome
(START study): A randomised phase 2a safety trial. Lancet Respir
Med. 7:154–162. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Xu Q, Hou W, Zhao B, Fan P, Wang S, Wang L
and Gao J: Mesenchymal stem cells lineage and their role in disease
development. Mol Med. 30:2072024. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Muzes G and Sipos F: Mesenchymal stem
cell-derived secretome: A potential therapeutic option for
autoimmune and immune-mediated inflammatory diseases. Cells.
11:23002022. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Zhou T, Yuan Z, Weng J, Pei D, Du X, He C
and Lai P: Challenges and advances in clinical applications of
mesenchymal stromal cells. J Hematol Oncol. 14:242021. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Xu Z, Shi L, Wang Y, Zhang J, Huang L,
Zhang C, Liu S, Zhao P, Liu H, Zhu L, et al: Pathological findings
of COVID-19 associated with acute respiratory distress syndrome.
Lancet Respir Med. 8:420–422. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Mehta P, McAuley DF, Brown M, Sanchez E,
Tattersall RS and Manson JJ; HLH Across Speciality Collaboration
UK, : COVID-19: Consider cytokine storm syndromes and
immunosuppression. Lancet. 395:1033–1034. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Homma K, Bazhanov N, Hashimoto K, Shimizu
M, Heathman T, Hao Q, Nawgiri R, Muthukumarana V, Lee JW, Prough DS
and Enkhbaatar P: Mesenchymal stem cell-derived exosomes for
treatment of sepsis. Front Immunol. 14:11369642023. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Mastrolia I, Foppiani EM, Murgia A,
Candini O, Samarelli AV, Grisendi G, Veronesi E, Horwitz EM and
Dominici M: Challenges in clinical development of mesenchymal
stromal/stem cells: Concise review. Stem Cells Transl Med.
8:1135–1148. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Park WS, Ahn SY, Sung SI, Ahn JY and Chang
YS: Strategies to enhance paracrine potency of transplanted
mesenchymal stem cells in intractable neonatal disorders. Pediatr
Res. 83:214–222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Kakabadze Z, Kipshidze N, Paresishvili T,
Kipshidze N, Vadachkoria Z and Chakhunashvili D: Human placental
mesenchymal stem cells for the treatment of ARDS in Rat. Stem Cells
Int. 2022:84185092022. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Pharoun J, Berro J, Sobh J, Abou-Younes
MM, Nasr L, Majed A, Khalil A, Joseph Stephan and Faour WH:
Mesenchymal stem cells biological and biotechnological advances:
Implications for clinical applications. Eur J Pharmacol.
977:1767192024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Wang J, Huang R, Xu Q, Zheng G, Qiu G, Ge
M, Shu Q and Xu J: Mesenchymal stem cell-derived extracellular
vesicles alleviate acute lung injury via transfer of miR-27a-3p.
Crit Care Med. 48:e599–e610. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Murata M and Teshima T: Treatment of
steroid-refractory acute graft-versus-host disease using commercial
mesenchymal stem cell products. Front Immunol. 12:7243802021.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Levy O, Kuai R, Siren EMJ, Bhere D, Milton
Y, Nissar N, De Biasio M, Heinelt M, Reeve B, Abdi R, et al:
Shattering barriers toward clinically meaningful MSC therapies. Sci
Adv. 6:eaba68842020. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Shan Y, Zhang M, Tao E, Wang J, Wei N, Lu
Y, Liu Q, Hao K, Zhou F and Wang G: Pharmacokinetic characteristics
of mesenchymal stem cells in translational challenges. Signal
Transduct Target Ther. 9:2422024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Cen M, Ouyang W, Zhang W, Yang L, Lin X,
Dai M, Hu H, Tang H, Liu H, Xia J and Xu F: MitoQ protects against
hyperpermeability of endothelium barrier in acute lung injury via a
Nrf2-dependent mechanism. Redox Biol. 41:1019362021. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Moslemi M, Hejazian SM, Shaddelan M,
Javanali F, Mirghaffari A, Sadeghi A, Valizadeh H, Sharifi A,
Haramshahi M, Ardalan M and Zununi Vahed S: Evaluating the effect
of Edaravone on clinical outcome of patients with severe COVID-19
admitted to ICU: A randomized clinical trial. Inflammopharmacology.
30:1277–1282. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Ding P, Yang R, Li C, Fu HL, Ren GL, Wang
P, Zheng DY, Chen W, Yang LY, Mao YF, et al: Fibroblast growth
factor 21 attenuates ventilator-induced lung injury by inhibiting
the NLRP3/caspase-1/GSDMD pyroptotic pathway. Crit Care.
27:1962023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Gao J, Liu Q, Li J, Hu C, Zhao W, Ma W,
Yao M and Xing L: Fibroblast Growth Factor 21 dependent
TLR4/MYD88/NF-ĸB signaling activation is involved in
lipopolysaccharide-induced acute lung injury. Int Immunopharmacol.
80:1062192020. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Yan F, Yuan L, Yang F, Wu G and Jiang X:
Emerging roles of fibroblast growth factor 21 in critical disease.
Front Cardiovasc Med. 9:10539972022. View Article : Google Scholar : PubMed/NCBI
|














