Introduction
Bone homeostasis is maintained through the dynamic
balance between osteoblast-mediated bone formation and
osteoclast-mediated bone resorption (1). The disruption of this balance results
in bone loss, contributing to a variety of skeletal disorders,
including osteoporosis, rheumatoid arthritis, periodontitis and
bone metastases (2–4). Among these disorders, osteoporosis is
the most prevalent, particularly in aging populations, and is
marked by decreased bone mass and a higher risk of fractures
(5). Current therapeutic
strategies for bone loss primarily target osteoclast function,
including bisphosphonates, denosumab and selective estrogen
receptor modulators (SERMs) (6).
However, these treatments are often associated with side effects,
including osteonecrosis of the jaw, atypical femoral fractures and
impaired bone remodeling (7),
highlighting the need for the identification of novel therapeutic
targets with improved safety profiles.
Bone loss typically results from dysregulated
osteoclastogenesis and increased osteoclast-mediated resorption.
Osteoclasts are multinucleated cells derived from macrophage
progenitors, which are important for physiological bone remodeling
and pathological bone resorption (8). The differentiation and activity of
osteoclasts are tightly regulated by a range of signaling
molecules, among which receptor activator of NF-κB ligand (RANKL)
serves a central role (9). The
binding of RANKL to receptor activator of NF-κB (RANK) on
osteoclast precursors initiates multiple intracellular signaling
cascades, including the MAPK and NF-κB pathways, which converge to
activate transcription factors such as nuclear factor of activated
T-cells 1 (NFATc1) and c-Fos (10). These events drive the fusion of
mononuclear precursors into multinucleated osteoclasts capable of
bone resorption. Given the central role of RANKL signaling in
osteoclastogenesis, the inhibition of its downstream pathways has
been considered a promising therapeutic approach for mitigating
pathological bone loss.
Apelin (APLN) is an endogenous peptide and an
adipokine implicated in various physiological processes (11). APLN exerts its effects through
binding to the G protein-coupled receptor APLN receptor, and is
known to regulate angiogenesis, energy homeostasis and
cardiovascular function (12–14).
For example, studies have demonstrated that APLN promotes the
expression of hypoxia-inducible factor-1, VEGF and VEGF receptor 2,
thereby enhancing endothelial cell proliferation and migration, and
ultimately facilitating new blood vessel formation (12,15).
APLN has also been reported to promote inflammatory responses by
upregulating the expression of IL-1β in human synovial fibroblasts,
an effect linked to microRNA (miRNA)-144-3p suppression, and
activation of the PI3K and ERK signaling pathways (16). Emerging evidence has indicated that
adipokines may influence bone metabolism. Leptin is known to
influence bone formation through central and peripheral pathways
(17), whereas adiponectin has
been demonstrated to inhibit osteoclastogenesis and enhance
osteoblast differentiation (18).
Previous evidence has suggested a regulatory role for APLN in
skeletal homeostasis, as APLN deficiency in mice leads to increased
trabecular and cortical bone mass along with elevated osteoblast
activity and bone formation. APLN promotes osteoblast proliferation
but does not enhance mineralized nodule formation, suggesting a
potential disruption of osteoblast maturation. These findings
indicate that APLN may act as an antianabolic factor and contribute
to bone metabolic imbalance (19).
However, the potential influence of APLN on osteoclast lineage
cells, which are primarily responsible for bone resorption, remains
ambiguous. Elucidating whether APLN contributes to osteoclast
formation and function is important for understanding its broader
role in bone remodeling.
To address this, the present study examined the
effects of APLN on RANKL-induced osteoclastogenesis using the
murine RAW264.7 cell model. Specifically, the present study
evaluated the role of APLN in promoting osteoclast differentiation,
regulating cytoskeletal organization and modulating intracellular
signaling pathways associated with osteoclast activation. The
results of the current study may provide new perspectives on the
involvement of APLN in bone metabolism and suggest that the
inhibition of APLN may represent a novel therapeutic strategy for
suppressing excessive bone resorption in osteolytic diseases.
Materials and methods
Materials and reagents
Recombinant APLN was purchased from MyBiosource,
Inc., and recombinant RANKL was obtained from PeproTech, Inc.
(Thermo Fisher Scientific, Inc.). The ERK inhibitor FR180204, and
antibodies against phosphorylated (p)-ERK (cat. no. sc-7383), p-JNK
(cat. no. sc-6254) and p-p38 (cat. no. sc-166182), and total ERK
(cat. no. sc-1647), JNK (cat. no. sc-7345) and p38 (cat. no.
sc-7972) were acquired from Santa Cruz Biotechnology, Inc. The JNK
inhibitor SP600125 was purchased from Enzo Life Sciences, Inc.
whereas the p38 inhibitor SB203580 and NF-κB inhibitor
pyrrolidinedithiocarbamate ammonium (PDTC) were obtained from
MilliporeSigma.
Antibodies against IKK (cat. no. GTX52348), IκB
(cat. no. GTX110521), p65 (cat. no. GTX102090) and β-actin
(1:10,000; cat. no. GT5512) were purchased from GeneTex
International Corporation. Antibodies for p-IKK (cat. no. 2697S)
were from Cell Signaling Technology, Inc., antibodies for p-IκB
(cat. no. sc-8404) were obtained from Santa Cruz Biotechnology,
Inc. and antibodies targeting p-p65 (cat. no. AP0124) were obtained
from ABclonal Biotech Co., Ltd.
Cells and cell cultures
RAW264.7 murine macrophage-like cells were purchased
from the American Type Culture Collection, and were maintained in
Dulbecco's modified Eagle's medium (DMEM; Gibco, Thermo Fisher
Scientific, Inc.) supplemented with 10% characterized fetal bovine
serum (FBS; HyClone™; Cytiva) and the standard antibiotics
penicillin/streptomycin (100 IU/ml). Cells were maintained at 37°C
in a 5% CO2 humidified incubator.
Osteoclast differentiation and
treatment
RAW264.7 cells (2×103/well) were seeded
into 24-well plates and cultured in DMEM supplemented with 10% FBS.
For osteoclast differentiation, cells were treated with RANKL (50
ng/ml), either alone or in combination with APLN (1, 3 or 10
ng/ml), for 5 days at 37°C with 5% CO2. For the
neutralization group, cells were pre-incubated with APLN antibody
(1 µg/ml; cat. no. NBP3-12282; Novus Biologicals; Bio-Techne) for
30 min before the addition of RANKL and APLN. The culture medium
was refreshed every 2 days. The optimal concentration of APLN (10
ng/ml) was determined from a preliminary dose response assay in
RAW264.7 cells, in which this concentration induced maximal
osteoclast differentiation without affecting cell viability. A
previous study also reported that pyroglutamyl-APLN-13, a distinct
peptide fragment of the APLN molecule, at the same concentration
modulated macrophage functions in RAW264.7 cells (20).
For tartrate-resistant acid phosphatase (TRAP)
staining, cells were fixed with 10% paraformaldehyde for 5 min at
room temperature, followed by incubation with the TRAP Staining Kit
(cat. no. PMC-AK04-COS; Cosmo Bio Co., Ltd.) at 37°C for 60 min.
TRAP staining was conducted using a commercial TRAP kit (Cosmo Bio
Co., Ltd.), and TRAP-positive multinucleated cells, defined as
cells containing three or more nuclei, were identified by light
microscopy (KimForest Enterprise Co., Ltd.). Images were analyzed
using ImageJ software (v1.8.0; National Institutes of Health). The
TRAP-positive area in each well was quantified and mean values were
calculated from three independent experiments. For each well,
images from three randomly selected microscopic fields were
captured and averaged. For comparative analysis, TRAP-positive
areas were normalized to the RANKL-only group (100%), which served
as the positive control. All images include a scale bar of 100
µm.
For pathway inhibition, RAW264.7 cells
(1×105/well) were seeded into 6-well plates and
pretreated with specific pathway inhibitors for 30 min at 37°C with
5% CO2 before stimulation with RANKL (50 ng/ml) + APLN
(10 ng/ml) for 5 days. The culture medium was refreshed every 2
days. The final inhibitor concentrations were as follows: ERK
inhibitor FR180204 (10 µM), JNK inhibitor SP600125 (10 µM), p38
inhibitor SB203580 (10 µM) and NF-κB inhibitor PDTC (20 µM).
Immunofluorescence staining
RAW264.7 cells (2×103/well) were seeded
directly into 24-well plates and treated with RANKL (50 ng/ml) with
or without APLN (10 ng/ml) for 5 days at 37°C. Following treatment,
the cells were fixed with 3.7% paraformaldehyde for 15 min and
permeabilized with 0.5% Triton X-100 for 10 min at room
temperature. F-actin staining was performed by incubating the cells
overnight at 4°C with Alexa Fluor 488-conjugated phalloidin (cat.
no. A12379; Thermo Fisher Scientific, Inc.; diluted 1:300 in PBS).
Nuclei were counterstained with DAPI. Fluorescence images were
captured using an ImageXpress Pico imaging system (Molecular
Devices, LLC). The F-actin ring area was quantified using ImageJ
software (v1.8.0) and expressed relative to the RANKL-only group.
All images include a scale bar of 100 µm.
Transcriptome analysis
RNA-sequencing (RNA-seq) data from the GSE21639
dataset were downloaded from the Gene Expression Omnibus database
(https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE21639)
(21). Differentially expressed
genes (DEGs) between control and RANKL-treated RAW264.7 cells on
days 2 and 5 were identified using GEO2R (https://www.ncbi.nlm.nih.gov/geo/info/geo2r.html)
(22), applying a threshold of
absolute log2(fold change)≥2 and an adjusted
P-value≤0.05. Boxplot and uniform manifold approximation and
projection (UMAP) analyses were performed using R software (version
4.2.2) (https://cran.r–project.org/bin/windows/base/old/4.2.2/)
with the GEOquery (https://bioconductor.org/packages/release/bioc/html/GEOquery.html),
limma (https://bioconductor.org/packages/release/bioc/html/limma.html)
and UMAP packages (https://cran.r–project.org/web/packages/umap/index.html)
to evaluate the quality of the GSE21639 dataset, and to confirm
consistent overall gene expression distributions and clustering
patterns. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) pathway enrichment analyses were performed using the
Database for Annotation, Visualization and Integrated Discovery
v.6.8 (https://davidbioinformatics.nih.gov) (23).
Reverse transcription-quantitative PCR
(RT-qPCR)
Cellular RNA was obtained from RAW264.7 cells using
TRIzol® reagent according to the manufacturer's
instructions (cat. no. 12183555; Invitrogen; Thermo Fisher
Scientific, Inc.), and cDNA was generated using the M-MLV Reverse
Transcriptase Kit (Invitrogen; Thermo Fisher Scientific, Inc.) in
accordance with the manufacturer's recommendation. SYBR Green-based
qPCR was performed using SYBR™ Green PCR Master Mix (Applied
Biosystems; Thermo Fisher Scientific, Inc.) on a StepOnePlus
Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific,
Inc.), with GAPDH as the reference gene. The cycling conditions
were as follows: Initial denaturation at 95°C for 20 sec, followed
by 40 cycles at 95°C for 3 sec and 60°C for 30 sec. Subsequently, a
melt curve analysis was performed. Expression levels were
normalized and calculated using the 2−ΔΔCq method
(24). The mouse primer sequences
used were as follows: Cathepsin K (CTSK), forward
5′-AGTAGCCACGCTTCCTATCC-3′, reverse 5′-CCATGGGTAGCAGCAGAAAC-3′;
acid phosphatase 5, tartrate resistant (ACP5), forward
5′-ATGGGCGCTGACTTCATCAT-3′, reverse 5′-GGTCTCCTGGAACCTCTTGT-3′;
matrix metalloproteinase 9 (MMP9), forward
5′-GGACCCGAAGCGGACATTG-3′, reverse 5′-CGTCGTCGAAATGGGCATCT-3′;
NFATc1, forward 5′-GACCCGGAGTTCGACTTCG-3′, reverse
5′-TGACACTAGGGGACACATAACTG-3′; and GAPDH, forward
5′-TGTGTCCGTCGTGGATCTGA-3′, reverse 5′-TTGCTGTTGAAGTCGCAGGAG-3.
Western blot analysis
RAW264.7 cells were lysed in RIPA buffer (cat. no.
P0013; Beyotime Biotechnology) containing protease and phosphatase
inhibitors. Protein levels were measured by BCA assay, and total
proteins (30 µg) were separated by SDS-PAGE on 10% gels and were
subsequently transferred to PVDF membranes. Blocking was performed
with 5% non-fat milk for 1 h at room temperature, followed by
overnight incubation with primary antibodies (1:1,000 for
p-proteins and 1:2,000 for total proteins) at 4°C, and detection
with HRP-conjugated secondary antibodies (goat anti-rabbit IgG,
cat. no. sc-2004 for IKK, IκB, p65, p-IKK, p-IκB, and p-p65;
1:2,000; goat anti-mouse IgG, cat. no. sc-2302 for p-ERK, p-JNK,
p-p38, ERK, JNK, and p38; 1:2,000; both from Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature, followed by
western chemiluminescent HRP substrate (ECL) detection
(MilliporeSigma). Band intensities were measured using ImageJ
software (v1.8.0).
NF-κB reporter assay
NF-κB luciferase reporter assays were conducted
using the pNF-κB-Luc plasmid from the NF-κB cis-Reporting System
(cat. no. 219077; Agilent Technologies, Inc.). The vector contains
five tandem NF-κB response elements upstream of a minimal promoter
driving luciferase expression. RAW264.7 cells were transfected with
NF-κB luciferase constructs using Lipofectamine® 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) and treated with RANKL
(50 ng/ml) or RANKL (50 ng/ml) + APLN (10 ng/ml) 24 h after
transfection for an additional 24 h at 37°C with 5% CO2.
Luciferase activity was assessed using the Dual-Luciferase Reporter
assay (Promega Corporation) and measured with a Fluoroskan Ascent
Microplate Luminometer (Thermo Fisher Scientific, Inc.). The
relative NF-κB luciferase activity was normalized to the control
group and expressed as a percentage of the control value (% of
control).
Statistical analysis
All experiments were performed in triplicate.
Statistical analysis was performed using one-way ANOVA followed by
the Bonferroni post hoc test for pairwise comparisons between
selected groups. All data are presented as the mean ± standard
deviation, and P<0.05 was considered to indicate a statistically
significant difference. Analyses were performed using GraphPad
Prism 9.0 (Dotmatics).
Results
APLN promotes RANKL-induced osteoclast
differentiation and enhances F-actin ring formation
The ability of RANKL to induce osteoclast
differentiation in RAW264.7 cells has been extensively validated
in vitro (25). In the
present study, RAW264.7 cells were stimulated with RANKL (50 ng/ml)
in the presence of increasing concentrations of APLN (1, 3 or 10
ng/ml) for 5 days to assess whether APLN modulated
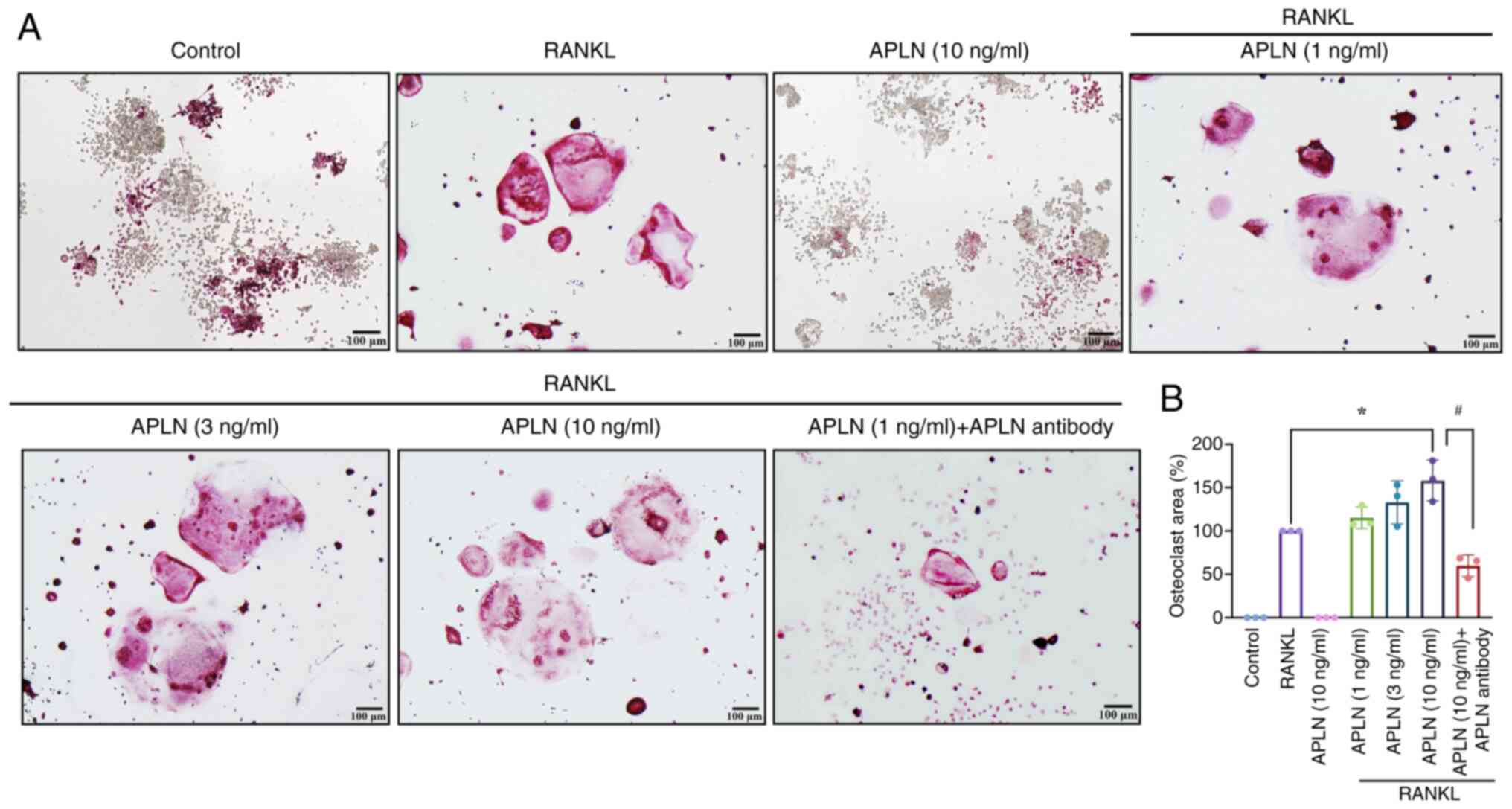
osteoclastogenesis. TRAP staining revealed a marked increase in the
size of multinucleated osteoclasts in cells co-treated with APLN
and RANKL compared with RANKL alone (Fig. 1A). By contrast, APLN treatment
alone did not induce osteoclast formation (Fig. 1A), indicating that APLN enhanced
RANKL-induced osteoclastogenesis as opposed to initiating the
differentiation process independently.
In addition, quantitative analysis of the mature
osteoclast area supported that APLN co-treatment with RANKL
significantly increased the osteoclast area in a
concentration-dependent manner, with the 10 ng/ml treatment group
having exhibited the most pronounced effect (Fig. 1B). Furthermore, to functionally
validate whether the pro-osteoclastic effect of APLN was
reversible, an APLN-neutralizing antibody was co-administered
together with RANKL and APLN (10 ng/ml). TRAP staining results
demonstrated that the enhanced osteoclast formation observed in the
APLN + RANKL group was markedly attenuated by the addition of APLN
antibody (Fig. 1A). Quantitative
analysis showed a significant reduction in osteoclast area, not
only reversing the APLN-mediated enhancement but further
suppressing osteoclastogenesis, resulting in an osteoclast area
lower than that induced by RANKL alone (Fig. 1B). These findings suggested that
APLN may serve an important modulatory role in osteoclast
differentiation and that its effects could be effectively
neutralized by targeted-antibody blockade.
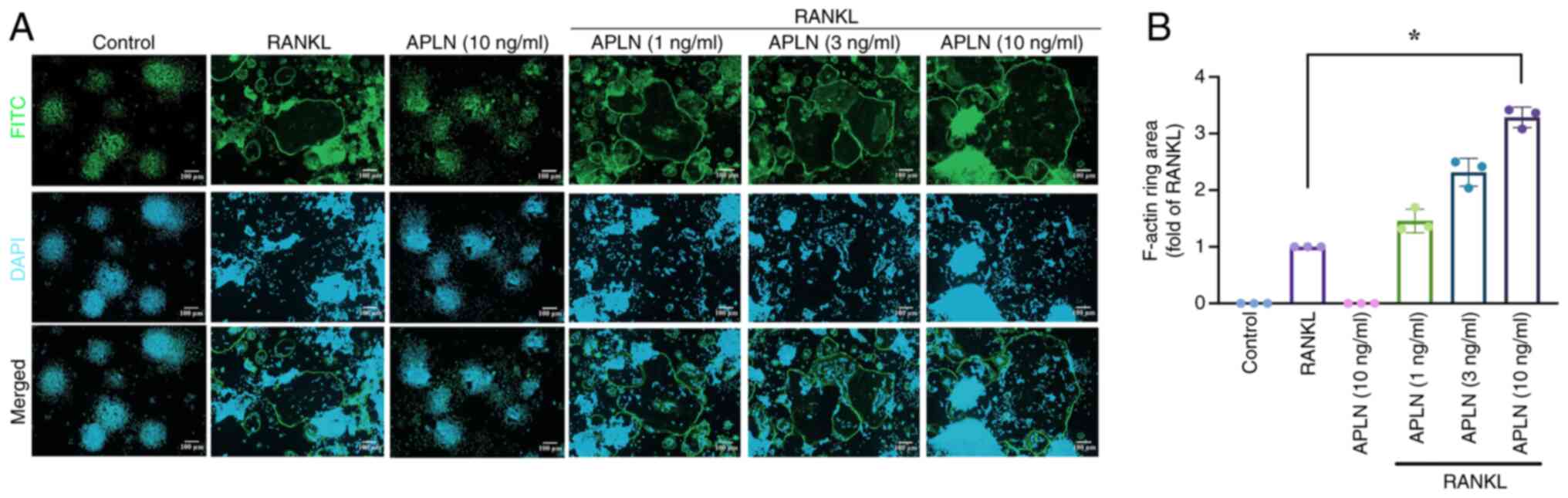
To further investigate whether APLN also affected
the functional activity of mature osteoclasts, the present study
evaluated F-actin ring formation using immunofluorescence staining,
which specifically reflects cytoskeletal organization and the
resorptive capacity of osteoclasts. RAW264.7 cells treated with
RANKL alone exhibited F-actin ring formation. By contrast,
co-treatment with APLN led to a concentration-dependent increase in
the size of F-actin rings (Fig.
2A). Quantitative analysis revealed that the F-actin ring area
was significantly greater in the cells co-treated with RANKL and
APLN, particularly in the cells treated with the 10 ng/ml APLN,
compared with the RANKL-only group (Fig. 2B). These findings allowed for the
comprehensive assessment of the functional activity of osteoclasts
in the presence of APLN and provided further insight into its role
in RANKL-mediated osteoclastogenesis and bone resorption.
APLN facilitates RANKL-induced
osteoclastogenic gene expression
Previous research has demonstrated that osteoclast
marker genes such as ACP5, CTSK, MMP9 and NFATc1 are upregulated
during osteoclast differentiation (26). To support this observation, the
present study analyzed RNA-seq data from the GSE21639 dataset,
which included RAW264.7 cells treated with or without RANKL for 2
and 5 days. The heatmap with hierarchical clustering revealed
increased expression of ACP5, CTSK, MMP9 and NFATc1 in
RANKL-treated groups compared with that in the control groups. Both
control and RANKL groups clustered tightly within their respective
branches, indicating high intra-group consistency and clear
separation between conditions (Fig.
3A). As additional evidence of dataset quality, boxplot and
UMAP analyses showed consistent expression distributions without
outliers and clustering patterns aligned with experimental
conditions (Fig. 3B), indicating
effective normalization, high reproducibility and biological
relevance. Further quantification revealed that the ACP5, CTSK and
MMP9 levels were significantly elevated on both day 2 and 5
following RANKL stimulation compared with those in the control
group. However, NFATc1 exhibited a significant increase in
expression in the RANKL-treated group on day 5 only (Fig. 3C). The present study also validated
the findings of the transcriptome analysis and assessed the effects
of APLN on osteoclast-specific gene expression in RAW264.7 cells.
RT-qPCR analysis demonstrated that co-treatment with RANKL and APLN
increased ACP5, CTSK, MMP9 and NFATc1 mRNA expression relative to
RANKL alone, showing a dose-dependent response, with statistical
significance observed only at 10 ng/ml APLN (Fig. 3D). Notably, APLN treatment alone
had no notable effect. These findings indicated that APLN may
facilitate RANKL-induced osteoclastogenic gene expression, thereby
supporting its functional role as a positive regulator of
osteoclast differentiation.
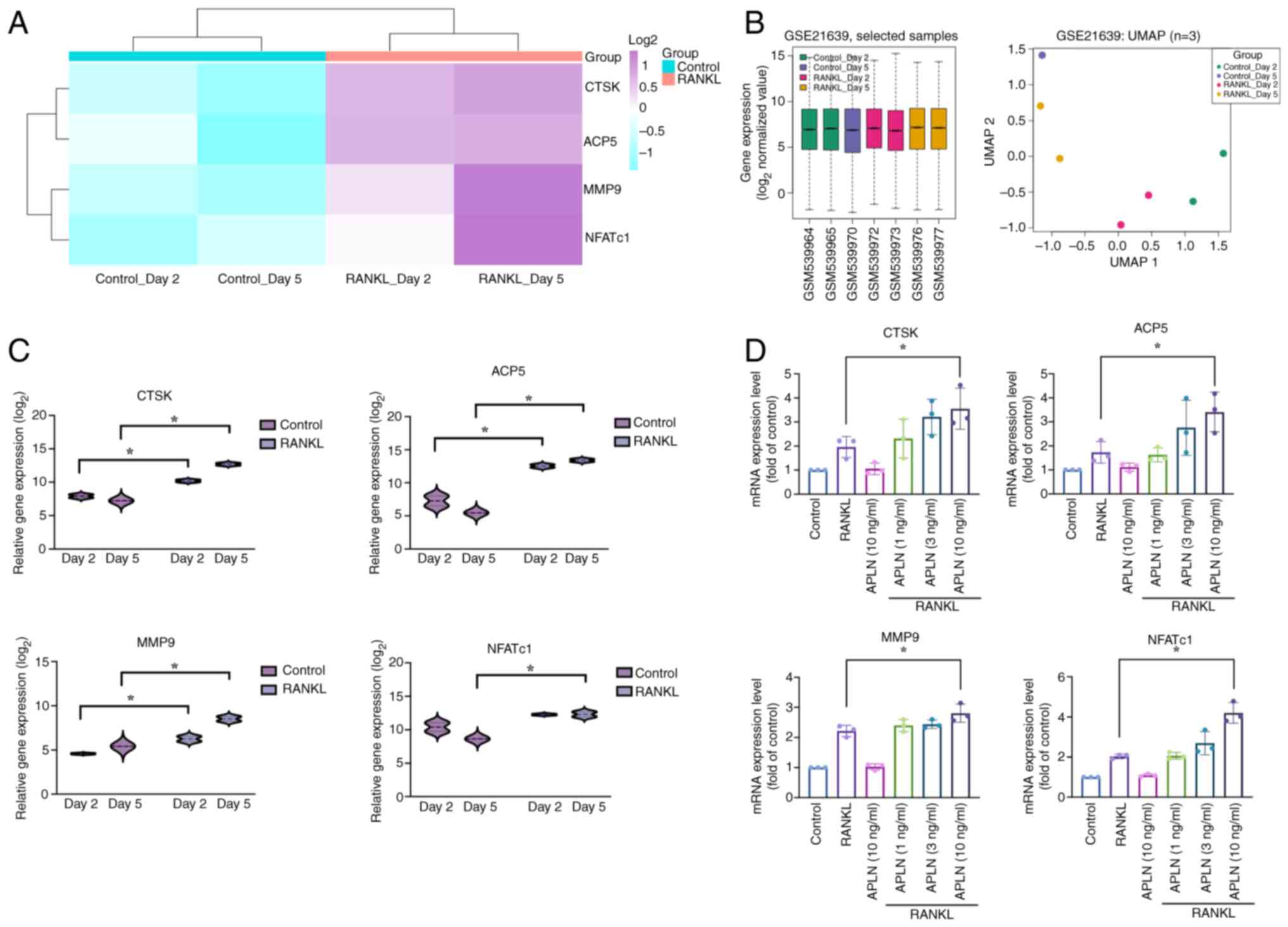 | Figure 3.APLN enhances osteoclast marker gene
expression under RANKL stimulation. (A) Heatmap analysis of
osteoclast-related gene expression (ACP5, CTSK, MMP9 and NFATc1)
from the GSE21639 dataset. RAW264.7 cells were treated with or
without RANKL for 2 or 5 days. (B) Boxplot and UMAP analyses of the
GSE21639 dataset. The boxplot shows consistent overall gene
expression distributions across all samples without outliers, while
the UMAP plot illustrates the clustering of samples according to
experimental conditions (Control or RANKL treatment on days 2 and
5). These analyses were conducted to assess dataset quality and
comparability prior to differential expression analysis. (C) Violin
plot visualization of the transcript levels of each gene. Data were
log2-transformed and grouped by treatment condition. (D)
RAW264.7 cells were treated with RANKL (50 ng/ml) and increasing
concentrations of APLN (1, 3 or 10 ng/ml) for 5 days. The mRNA
expression of osteoclast markers was determined using reverse
transcription-quantitative PCR. *P<0.05 vs. RANKL-treated group.
UMAP, uniform manifold approximation and projection analyses;
RANKL, receptor activator of NF-κB ligand; APLN, apelin; ACP5, acid
phosphatase 5, tartrate resistant; CTSK, cathepsin K; MMP9, matrix
metalloproteinase 9; NFATc1, nuclear factor of activated T-cells
1. |
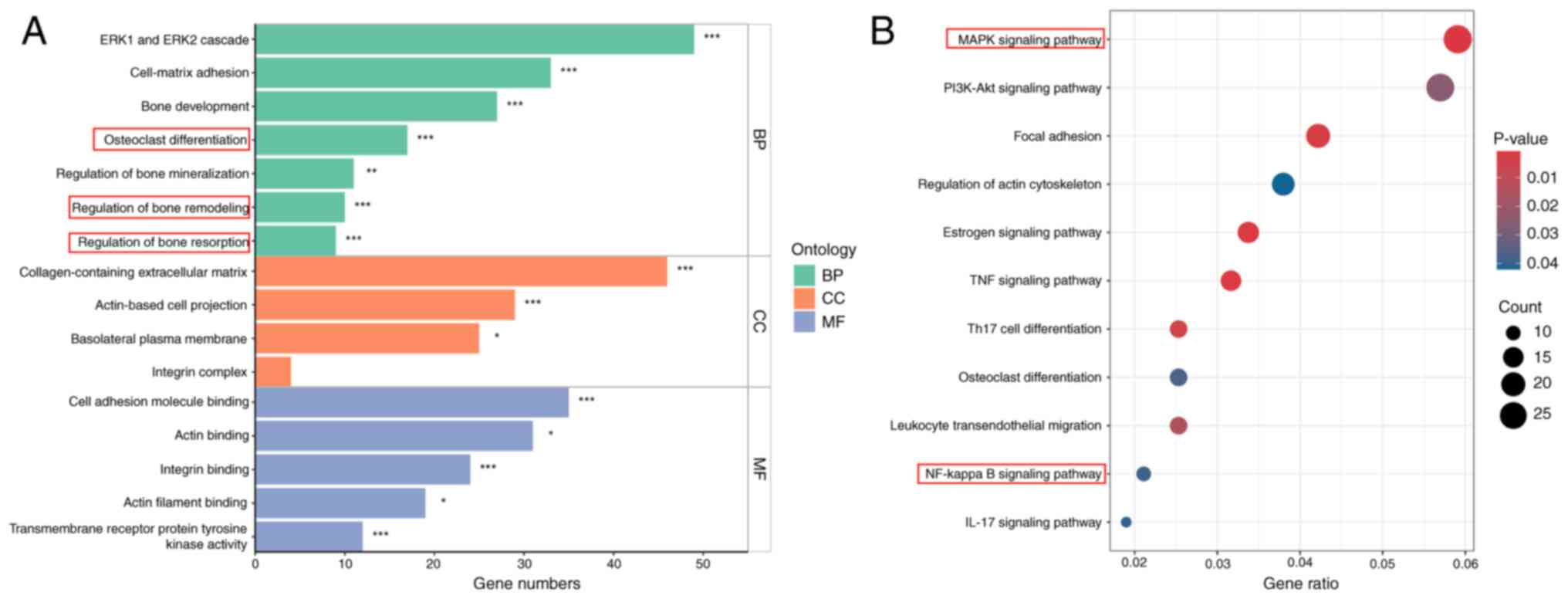
Transcriptome enrichment analysis
reveals osteoclast-related pathways activated by RANKL
To provide further mechanistic insights into the
biological processes and signaling pathways involved in
APLN-enhanced osteoclastogenesis, the present study analyzed DEGs
using RNA-seq data from the GSE21639 dataset. This dataset contains
data on RAW264.7 cells cultured with RANKL for 5 days. DEGs were
defined using thresholds of |log2(fold change)|>2 and
P<0.05. GO enrichment analysis revealed that upregulated DEGs
were significantly associated with biological processes relevant to
osteoclast activity, including ‘osteoclast differentiation’,
‘regulation of bone remodeling’ and ‘regulation of bone resorption’
(Fig. 4A). Furthermore, KEGG
pathway analysis demonstrated significant enrichment in key
signaling cascades, including the ‘MAPK signaling pathway’ and ‘NF-
kappa B signaling pathway’ (Fig.
4B). These findings highlighted the involvement of
osteoclast-related biological processes and signaling pathways in
RANKL-induced differentiation.
APLN promotes RANKL-mediated
osteoclast differentiation through MAPK and NF-κB signaling
pathways
Given the enrichment of MAPK and NF-κB pathways
observed in transcriptome analysis, the present study then
investigated whether these pathways contributed to the
pro-osteoclastogenic effects of APLN. Western blot analysis
revealed that co-treatment with APLN and RANKL markedly enhanced
the phosphorylation of ERK, JNK and p38 compared with RANKL
treatment alone (Fig. 5A).
Quantitative analysis of the western blots revealed that treatment
with RANKL significantly increased the phosphorylation of ERK, JNK
and p38 compared with the control group (Fig. 5B). This phosphorylation was
significantly enhanced by co-treatment with RANKL and APLN. To
determine whether APLN promoted osteoclast differentiation through
MAPK pathway activation, the effects of selective MAPK inhibitors
on APLN-treated cells were assessed. RAW264.7 cells were treated
with APLN and RANKL in the presence of the ERK inhibitor FR180294,
JNK inhibitor SP600125 or p38 inhibitor SB203580, and the
expression levels of osteoclast marker genes were subsequently
assessed by RT-qPCR analysis. As expected, treatment with RANKL and
APLN significantly upregulated the mRNA expression levels of ACP5,
CTSK, MMP9 and NFATc1 compared with those in the control group.
However, this enhanced expression was significantly suppressed upon
treatment with MAPK inhibitors (Fig.
5C), indicating that the activation of MAPK signaling was
important for APLN activity in promoting RANKL-mediated osteoclast
differentiation.
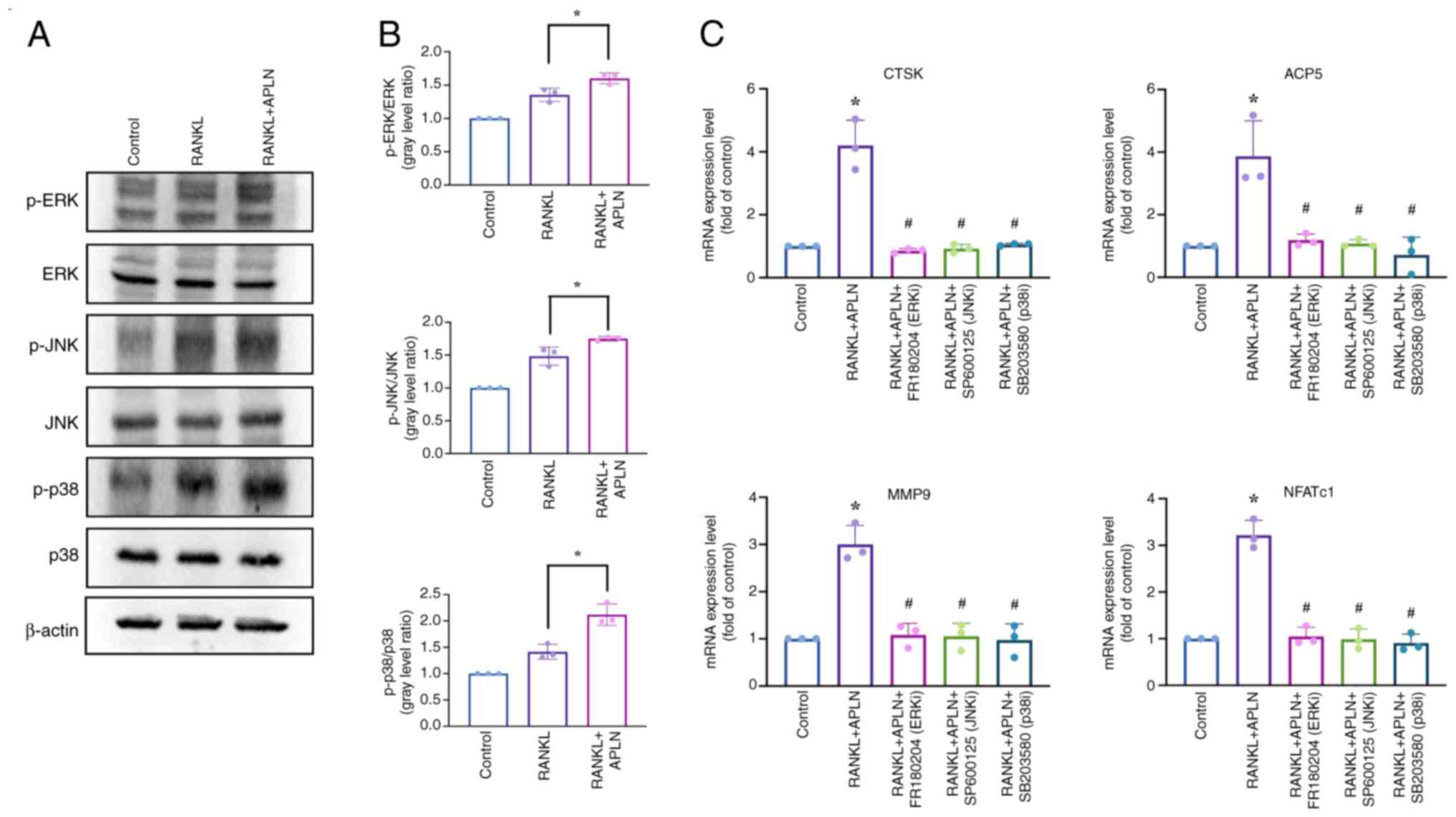 | Figure 5.APLN promotes osteoclast
differentiation through activation of the MAPK pathway. (A)
RAW264.7 cells were exposed to a control medium, RANKL (50 ng/ml)
or RANKL + APLN (10 ng/ml) for 10 min. Cell lysates were subjected
to western blot analysis to assess the phosphorylation of MAPK
family members, including ERK, JNK and p38. (B) Signal intensities
of p-ERK, p-JNK and p-p38 were semi-quantified using ImageJ
software and normalized to their respective total protein levels.
*P<0.05 vs. RANKL-treated group. (C) mRNA expression levels of
osteoclast marker genes (ACP5, CTSK, MMP9 and NFATc1) were assessed
using reverse transcription-quantitative PCR in cells treated with
RANKL + APLN (10 ng/ml), either alone or in combination with MAPK
inhibitors (FR180204 for ERK, SP600125 for JNK and SB203580 for
p38). *P<0.05 vs. Control group; #P<0.05 vs. RANKL
+ APLN group. APLN, apelin; RANKL, receptor activator of NF-κB
ligand; ACP5, acid phosphatase 5, tartrate resistant; CTSK,
cathepsin K; MMP9, matrix metalloproteinase 9; NFATc1, nuclear
factor of activated T-cells 1; p-, phosphorylated-; ERK/JNK/p38i,
ERK/JNK/p38 inhibitor. |
The present study then investigated the role of
NF-κB signaling in APLN-enhanced RANKL-induced osteoclastogenesis.
Western blot analysis and signal semi-quantification revealed that
RANKL treatment significantly increased the phosphorylation of IKK,
IκB and NF-κB p65 compared with that in the control group.
Co-treatment with APLN and RANKL significantly enhanced the
phosphorylation levels of these proteins relative to RANKL alone
(Fig. 6A and B). Consistently,
luciferase reporter assays revealed that APLN significantly
enhanced RANKL-induced NF-κB transcriptional activity (Fig. 6C). In addition, RT-qPCR analysis
demonstrated that co-treatment of RAW264.7 cells with APLN and
RANKL significantly upregulated the expression of osteoclast marker
genes compared with the control group; however, this effect was
significantly reduced by the NF-κB inhibitor PDTC (Fig. 6D). Taken together, these results
suggested that APLN may promote RANKL-mediated osteoclast
differentiation by activating the MAPK and NF-κB signaling
pathways.
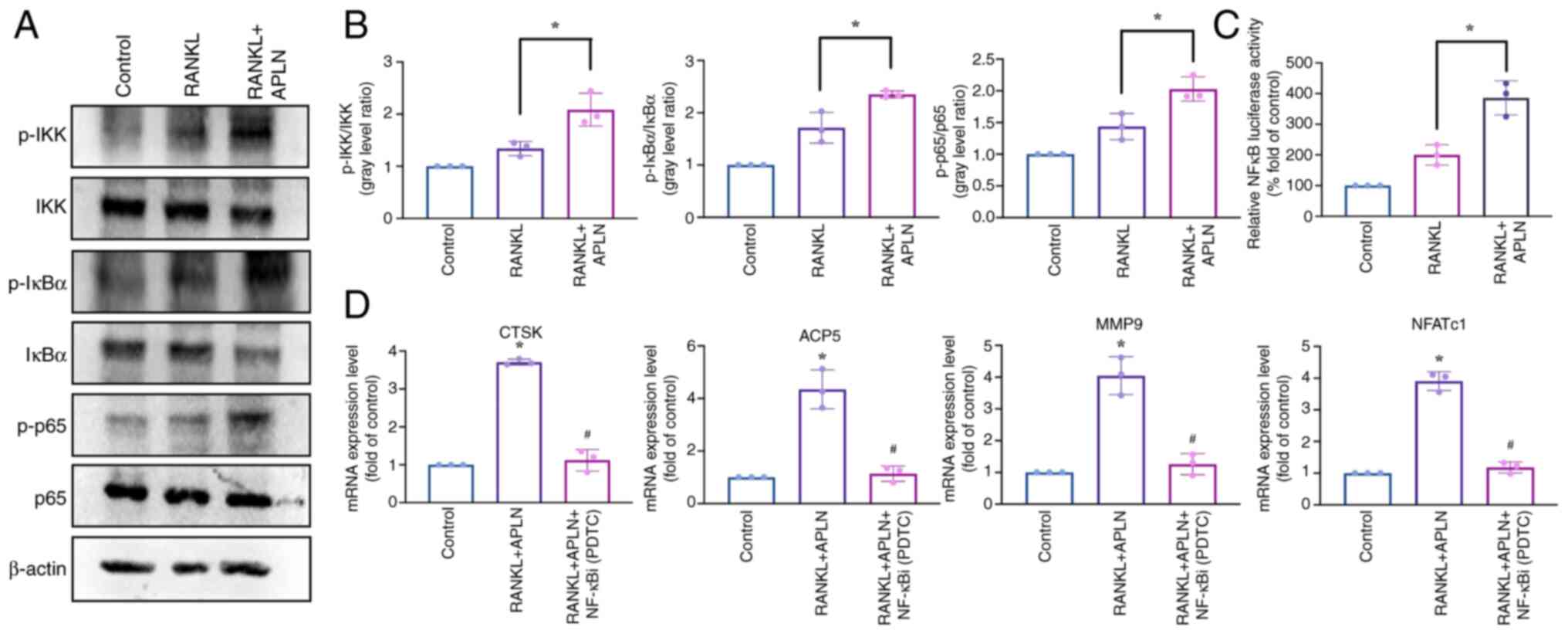 | Figure 6.APLN enhances RANKL-induced
activation of the NF-κB signaling pathway. (A) Western blot
analysis of RAW264.7 cells treated with control medium, RANKL (50
ng/ml) or RANKL + APLN (10 ng/ml) for 10 min. Protein levels of
p-IKK, p-IκB and p-p65 were detected to evaluate pathway
activation. (B) Semi-quantification of p-IKK, p-IκB and p-p65
levels relative to their corresponding total proteins was performed
using ImageJ software. (C) RAW264.7 cells were transfected with an
NF-κB luciferase reporter construct and treated with RANKL (50
ng/ml) or RANKL + APLN (10 ng/ml) for 24 h. Luciferase activity was
measured using cell lysates and reporter buffer mixed at a 1:1
ratio. NF-κB transcriptional activity is shown as the fold change
relative to the RANKL-only group. *P<0.05 vs. RANKL-treated
group. (D) Reverse transcription-quantitative PCR analysis of
osteoclast marker genes (ACP5, CTSK, MMP9 and NFATc1) in cells
treated with RANKL + APLN (10 ng/ml) with or without the NF-κBi
PDTC (20 µM). *P<0.05 vs. Control group; #P<0.05
vs. RANKL + APLN group. APLN, apelin; RANKL, receptor activator of
NF-κB ligand; ACP5, acid phosphatase 5, tartrate resistant; CTSK,
cathepsin K; MMP9, matrix metalloproteinase 9; NFATc1, nuclear
factor of activated T-cells 1; p-, phosphorylated-; NF-κBi, NF-κB
inhibitor; PDTC, pyrrolidinedithiocarbamate ammonium. |
Discussion
Adipokines are bioactive peptides derived from
adipose tissue that regulate key physiological processes, including
metabolism, inflammation and vascular function. Increasing evidence
has indicated that adipokines exert effects on the musculoskeletal
system and also participate in the regulation of bone remodeling
(27,28). Leptin has been shown to induce the
production of oncostatin M in human osteoblasts, thereby promoting
inflammatory signaling that disrupts osteoblast function and bone
homeostasis (29). The
proinflammatory and catabolic effects of visfatin in gingival
fibroblasts may indirectly promote osteoclast activation, thereby
exacerbating alveolar bone resorption in periodontitis (30). However, the roles of numerous
adipokines in bone homeostasis remain yet to be fully elucidated.
APLN has been identified as an adipokine implicated in
cardiovascular control, glucose metabolism and inflammatory
responses (31–33). Despite its broad biological role,
information on the influence of APLN on osteoclast differentiation
or bone resorption is limited.
The present study demonstrated that APLN enhanced
RANKL-induced osteoclast differentiation and function in a
concentration-dependent manner. Co-treatment with APLN
significantly increased TRAP-positive multinucleated cell
formation, F-actin ring size and the expression of osteoclast
marker genes, including ACP5, CTSK, MMP9 and NFATc1. These findings
provided novel insights into the bone-regulatory role of APLN and
suggested that APLN inhibition may serve as a potential strategy to
modulate osteoclast function.
Bone remodeling is a dynamic process that requires
regulation to maintain a balance between osteoblast-mediated bone
formation and osteoclast-mediated bone resorption (1). The disruption of this balance,
particularly when bone resorption outpaces formation, contributes
to the pathogenesis of various skeletal disorders, including
osteoporosis, rheumatoid arthritis and periodontitis (34–36).
Osteoclast differentiation and activation are primarily driven by
RANKL, which initiates a cascade of intracellular signaling events
upon binding to its receptor RANK on osteoclast precursors
(37). Activated pathways include
the NF-κB and MAPK pathways, both of which are important for the
induction of key osteoclastogenic transcription factors such as
NFATc1 (38). The present study
found that APLN co-treatment with RANKL significantly enhanced the
phosphorylation of ERK, JNK and p38 in the MAPK pathway, as well as
IKK, IκB and NF-κB p65 in the canonical NF-κB pathway. The
inhibition of these signaling pathways significantly attenuated the
APLN-induced upregulation of osteoclast-specific genes, indicating
that APLN promoted osteoclastogenesis through the activation of
classical signaling cascades downstream of RANKL. These findings
suggested that APLN may facilitate pathological bone resorption by
amplifying RANKL-induced signals.
Building on these mechanistic insights, targeting
APLN may represent a promising therapeutic strategy for osteolytic
conditions such as osteoporosis, rheumatoid arthritis and
periodontitis. However, given that APLN is a multifunctional
adipokine involved in cardiovascular regulation, angiogenesis and
metabolic homeostasis, systemic inhibition may lead to undesirable
off-target effects (39–42). To overcome these limitations,
localized treatment strategies may offer a more favorable
therapeutic strategy. For example, APLN-silencing molecules, such
as small interfering RNAs or miRNA mimics, could be encapsulated
within engineered exosomes for targeted delivery to the bone
microenvironment. This approach may enable site-specific
suppression of APLN activity, thereby attenuating osteoclast
overactivation while minimizing systemic effects. Further
investigations are warranted to evaluate the feasibility and
therapeutic benefit of exosome-mediated APLN inhibition in
bone-resorptive diseases.
Current pharmacological approaches for the treatment
of bone-resorptive diseases mainly focus on inhibiting osteoclast
activity (43). Bisphosphonates,
such as alendronate and zoledronic acid, are used to suppress
osteoclast-mediated bone resorption by inducing osteoclast
apoptosis (44). While effective
in reducing fracture risk, their prolonged use has been shown to be
associated with complications such as atypical femoral fractures
and osteonecrosis of the jaw (45). Denosumab, a monoclonal antibody
against RANKL, provides the potent and reversible suppression of
osteoclastogenesis, but its discontinuation may lead to rapid bone
loss and an increased risk of multiple vertebral fractures
(46). Furthermore, both
treatments broadly suppress bone turnover, potentially impairing
the physiological remodeling process.
Other anti-resorptive agents, including calcitonin,
hormone replacement therapy (HRT) and SERMs such as raloxifene,
have also been used in clinical practice. However, these options
are limited by suboptimal efficacy and potential systemic risks.
For example, calcitonin has been associated with an increased risk
of developing cancer (47); and
HRT has been a subject of concern regarding cardiovascular disease
and hormone-sensitive cancers (48). In addition, SERMs may increase the
risk of thromboembolism (49).
Given these limitations, there is growing interest in identifying
novel osteoclastogenic regulatory pathways complementing current
therapies. A co-therapeutic strategy combining adipokine-targeted
interventions with current anti-resorptive agents may improve
outcomes in osteolytic disease treatment.
In conclusion, the present study demonstrated that
APLN may promote RANKL-induced osteoclast differentiation and
function, and upregulate the levels of key osteoclast marker genes.
APLN was found to enhance the MAPK and NF-κB signaling pathways in
the presence of RANKL, and the inhibition of these pathways
attenuated the ability of APLN to promote osteoclast
differentiation (Fig. 7). These
findings provide a novel perspective into the role of APLN in bone
remodeling and identify APLN as an important enhancer of
osteoclastogenesis. Given these observations, therapeutic
strategies aimed at inhibiting APLN signaling may represent a novel
approach for mitigating pathological bone resorption in conditions
such as osteoporosis, inflammatory arthritis and periodontitis.
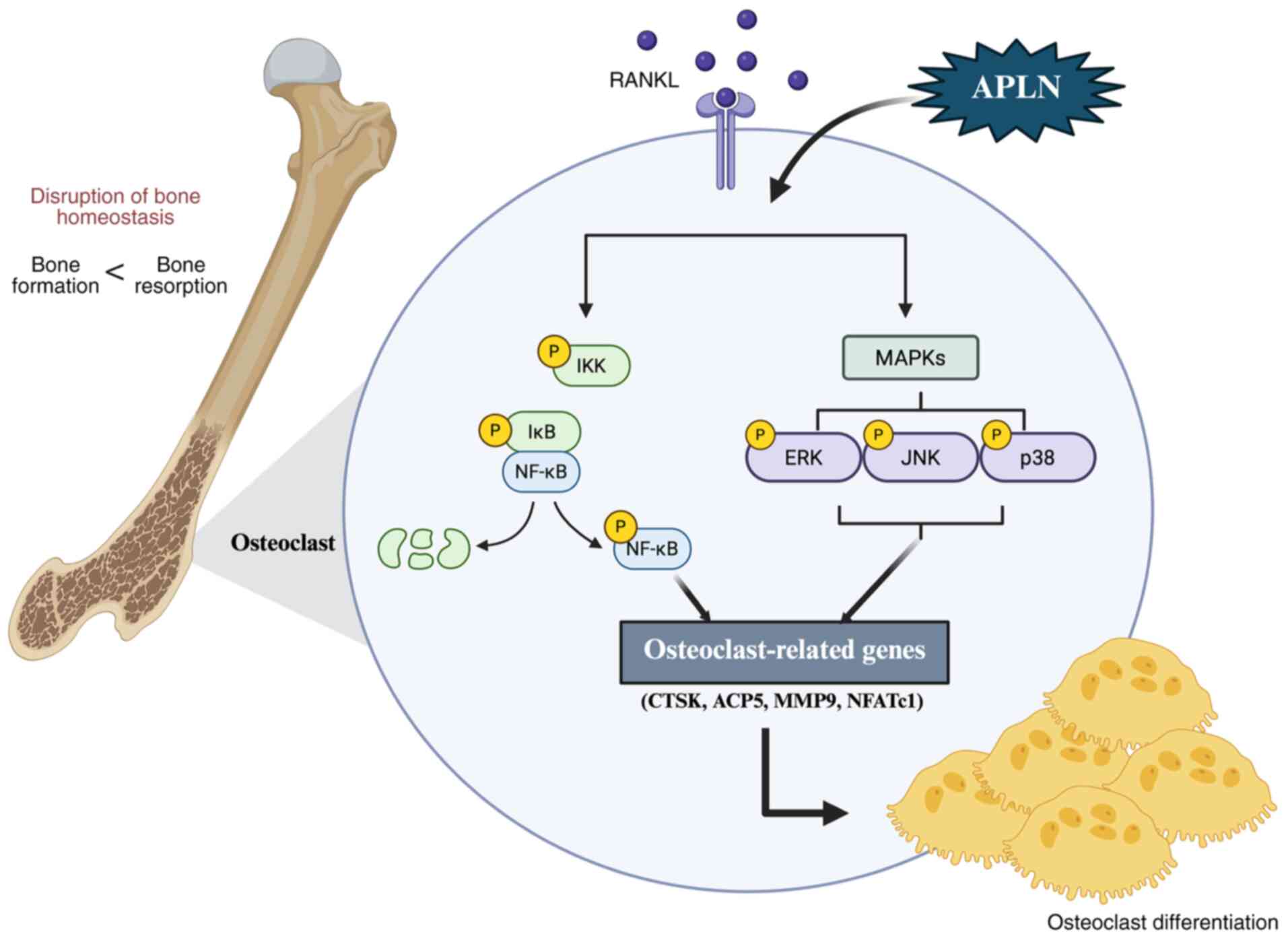 | Figure 7.APLN promotes RANKL-mediated
osteoclastogenesis, and activates MAPK and NF-κB signaling
pathways. APLN augmented RANKL-induced phosphorylation of ERK, JNK,
p38 and NF-κB p65, leading to the increased expression of
osteoclast marker genes and enhanced osteoclast differentiation and
function. APLN, apelin; RANKL, receptor activator of NF-κB ligand;
ACP5, acid phosphatase 5, tartrate resistant; CTSK, cathepsin K;
MMP9, matrix metalloproteinase 9; NFATc1, nuclear factor of
activated T-cells 1. |
Acknowledgements
Not applicable.
Funding
The authors acknowledge financial support from the Ministry of
Science and Technology of Taiwan (grant nos. MOST
110-2320-B-039-022-MY3, NSTC 112-2320-B-039-035-MY3, MOST
111-2314-B-039-048-MY3, NSTC 113-2320-B-039-049-MY3, NSTC
111-2320-B-371-002, NSTC 114-2314-B-039-051-MY3 and NSTC
114-2320-B- 039-016-), the China Medical University Hospital (grant
nos. DMR-114-003 and DMR-114-043) and the China Medical University
(grant no. CMU 113-ASIA-05).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YHW, YYW, CHTs, and CHTa contributed to the
conception and design of the study. YCF, CYK and HTC were
responsible for data acquisition and experimental validation. CHTs
and YYL contributed to data analysis and interpretation. YHW and
CHTa drafted the manuscript and confirm the authenticity of all the
raw data. All authors critically revised the manuscript for
important intellectual content and agreed to be accountable for all
aspects of the work. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Bolamperti S, Villa I and Rubinacci A:
Bone remodeling: An operational process ensuring survival and bone
mechanical competence. Bone Res. 10:482022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhu L, Zhou C, Chen S, Huang D, Jiang Y,
Lan Y, Zou S and Li Y: Osteoporosis and alveolar bone health in
periodontitis niche: A predisposing factors-centered review. Cells.
11:33802022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ashai S and Harvey NC: Rheumatoid
arthritis and bone health. Clin Med (Lond). 20:565–567. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Park JJ and Wong C: Pharmacological
prevention and management of skeletal-related events and bone loss
in individuals with cancer. Semin Oncol Nurs. 38:1512762022.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Salari N, Ghasemi H, Mohammadi L, Behzadi
MH, Rabieenia E, Shohaimi S and Mohammadi M: The global prevalence
of osteoporosis in the world: A comprehensive systematic review and
meta-analysis. J Orthop Surg Res. 16:6092021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Elahmer NR, Wong SK, Mohamed N, Alias E,
Chin KY and Muhammad N: Mechanistic insights and therapeutic
strategies in osteoporosis: A comprehensive review. Biomedicines.
12:16352024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ayers C, Kansagara D, Lazur B, Fu R, Kwon
A and Harrod C: Effectiveness and safety of treatments to prevent
fractures in people with low bone mass or primary osteoporosis: A
living systematic review and network meta-analysis for the American
College of Physicians. Ann Intern Med. 176:182–195. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Weivoda MM and Bradley EW: Macrophages and
bone remodeling. J Bone Miner Res. 38:359–369. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Campbell MJ, Bustamante-Gomez C, Fu Q,
Beenken KE, Reyes-Pardo H, Smeltzer MS and O'Brien CA:
RANKL-mediated osteoclast formation is required for bone loss in a
murine model of Staphylococcus aureus osteomyelitis. Bone.
187:1171812024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Park JH, Lee NK and Lee SY: Current
understanding of RANK signaling in osteoclast differentiation and
maturation. Mol Cells. 40:706–713. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yan J, Wang A, Cao J and Chen L:
Apelin/APJ system: An emerging therapeutic target for respiratory
diseases. Cell Mol Life Sci. 77:2919–2930. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wang YH, Kuo SJ, Liu SC, Wang SW, Tsai CH,
Fong YC and Tang CH: Apelin affects the progression of
osteoarthritis by regulating VEGF-dependent angiogenesis and
miR-150-5p expression in human synovial fibroblasts. Cells.
9:5942020. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hu G, Wang Z, Zhang R, Sun W and Chen X:
The role of apelin/apelin receptor in energy metabolism and water
homeostasis: A comprehensive narrative review. Front Physiol.
12:6328862021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chapman FA, Melville V, Godden E, Morrison
B, Bruce L, Maguire JJ, Davenport AP, Newby DE and Dhaun N:
Cardiovascular and renal effects of apelin in chronic kidney
disease: A randomised, double-blind, placebo-controlled, crossover
study. Nat Commun. 15:83872024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wysocka MB, Pietraszek-Gremplewicz K and
Nowak D: The role of apelin in cardiovascular diseases, obesity and
cancer. Front Physiol. 9:5572018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chang TK, Wang YH, Kuo SJ, Wang SW, Tsai
CH, Fong YC, Wu NL, Liu SC and Tang CH: Apelin enhances IL-1beta
expression in human synovial fibroblasts by inhibiting miR-144-3p
through the PI3K and ERK pathways. Aging (Albany NY). 12:9224–9239.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ducy P, Amling M, Takeda S, Priemel M,
Schilling AF, Beil FT, Shen J, Vinson C, Rueger JM and Karsenty G:
Leptin inhibits bone formation through a hypothalamic relay: A
central control of bone mass. Cell. 100:197–207. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tu Q, Zhang J, Dong LQ, Saunders E, Luo E,
Tang J and Chen J: Adiponectin inhibits osteoclastogenesis and bone
resorption via APPL1-mediated suppression of Akt1. J Biol Chem.
286:12542–12553. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wattanachanya L, Lu WD, Kundu RK, Wang L,
Abbott MJ, O'Carroll D, Quertermous T and Nissenson RA: Increased
bone mass in mice lacking the adipokine apelin. Endocrinology.
154:2069–2080. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Izgut-Uysal VN, Gemici B, Birsen I, Acar N
and Ustunel I: The effect of apelin on the functions of peritoneal
macrophages. Physiol Res. 66:489–496. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sharma P, Patntirapong S, Hann S and
Hauschka PV: RANKL-RANK signaling regulates expression of
xenotropic and polytropic virus receptor (XPR1) in osteoclasts.
Biochem Biophys Res Commun. 399:129–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15:5502014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lampiasi N, Russo R, Kireev I, Strelkova
O, Zhironkina O and Zito F: Osteoclasts differentiation from murine
RAW 264.7 cells stimulated by RANKL: Timing and behavior. Biology
(Basel). 10:1172021.PubMed/NCBI
|
|
26
|
Sun W, Li Y, Li J, Tan Y, Yuan X, Meng H,
Ye J, Zhong G, Jin X, Liu Z, et al: Mechanical stimulation controls
osteoclast function through the regulation of Ca(2+)-activated
Cl(−) channel Anoctamin 1. Commun Biol. 6:4072023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Greco EA, Lenzi A and Migliaccio S: The
obesity of bone. Ther Adv Endocrinol Metab. 6:273–286. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Armutcu F, McCloskey E and Ince M: Obesity
significantly modifies signaling pathways associated with bone
remodeling and metabolism. J Cell Signal. 5:183–194. 2024.
View Article : Google Scholar
|
|
29
|
Yang WH, Tsai CH, Fong YC, Huang YL, Wang
SJ, Chang YS and Tang CH: Leptin induces oncostatin M production in
osteoblasts by downregulating miR-93 through the Akt signaling
pathway. Int J Mol Sci. 15:15778–15790. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park KH, Kim DK, Huh YH, Lee G, Lee SH,
Hong Y, Kim SH, Kook MS, Koh JT, Chun JS, et al: NAMPT enzyme
activity regulates catabolic gene expression in gingival
fibroblasts during periodontitis. Exp Mol Med. 49:e3682017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
de Oliveira AA, Vergara A, Wang X, Vederas
JC and Oudit GY: Apelin pathway in cardiovascular, kidney, and
metabolic diseases: Therapeutic role of apelin analogs and apelin
receptor agonists. Peptides. 147:1706972022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang Q, Wang B, Zhang W, Zhang T, Liu Q,
Jiao X, Ye J, Hao Y, Gao Q, Ma G, et al: APLN promotes the
proliferation, migration, and glycolysis of cervical cancer through
the PI3K/AKT/mTOR pathway. Arch Biochem Biophys. 755:1099832024.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen L, Tao Y and Jiang Y: Apelin
activates the expression of inflammatory cytokines in microglial
BV2 cells via PI-3K/Akt and MEK/Erk pathways. Sci China Life Sci.
58:531–540. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tilotta F, Gosset M, Herrou J, Briot K and
Roux C: Association between osteoporosis and periodontitis. Joint
Bone Spine. 92:1058832025. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Llorente I, Garcia-Castaneda N, Valero C,
Gonzalez-Alvaro I and Castaneda S: Osteoporosis in rheumatoid
arthritis: Dangerous liaisons. Front Med (Lausanne). 7:6016182020.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sarhan RS, El-Hammady AM, Marei YM, Elwia
SK, Ismail DM and Ahmed EAS: Plasma levels of miR-21b and miR-146a
can discriminate rheumatoid arthritis diagnosis and severity.
Biomedicine (Taipei). 15:30–41. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takegahara N, Kim H and Choi Y: Unraveling
the intricacies of osteoclast differentiation and maturation:
Insight into novel therapeutic strategies for bone-destructive
diseases. Exp Mol Med. 56:264–272. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee NK: RANK signaling pathways and key
molecules inducing osteoclast differentiation. Biomed Sci Lett.
23:295–302. 2017. View Article : Google Scholar
|
|
39
|
Castan-Laurell I, Dray C and Valet P: The
therapeutic potentials of apelin in obesity-associated diseases.
Mol Cell Endocrinol. 529:1112782021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yasir M, Senthilkumar GP, Jayashree K,
Ramesh Babu K, Vadivelan M and Palanivel C: Association of serum
omentin-1, apelin and chemerin concentrations with the presence and
severity of diabetic retinopathy in type 2 diabetes mellitus
patients. Arch Physiol Biochem. 128:313–320. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Gao S and Chen H: Therapeutic potential of
apelin and Elabela in cardiovascular disease. Biomed Pharmacother.
166:1152682023. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Couvineau P and Llorens-Cortes C:
Metabolically stable apelin analogs: Development and functional
role in water balance and cardiovascular function. Clin Sci (Lond).
139:131–149. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liang B, Burley G, Lin S and Shi YC:
Osteoporosis pathogenesis and treatment: Existing and emerging
avenues. Cell Mol Biol Lett. 27:722022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mbese Z and Aderibigbe BA:
Bisphosphonate-based conjugates and derivatives as potential
therapeutic agents in osteoporosis, bone cancer and metastatic bone
cancer. Int J Mol Sci. 22:68692021. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hussain MA, Joseph A, Cherian VM,
Srivastava A, Cherian KE, Kapoor N and Paul TV:
Bisphosphonate-induced atypical femoral fracture in tandem:
long-term follow-up is warranted. Endocrinol Diabetes Metab Case
Rep. 2022:2022:22–0249. 2022.PubMed/NCBI
|
|
46
|
Tsourdi E, Zillikens MC, Meier C, Body JJ,
Rodriguez EG, Anastasilakis AD, Abrahamsen B, McCloskey E, Hofbauer
LC, Guañabens N, et al: Fracture risk and management of
discontinuation of denosumab therapy: A systematic review and
position statement by ECTS. J Clin Endocrinol Metab.
26:dgaa7562020.
|
|
47
|
Okamoto H, Shibazaki N, Yoshimura T, Uzawa
T and Sugimoto T: Association between elcatonin use and cancer risk
in Japan: A follow-up study after a randomized, double-blind,
placebo-controlled study of once-weekly elcatonin in primary
postmenopausal osteoporosis. Osteoporos Sarcopenia. 6:15–19. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Vinogradova Y, Coupland C and
Hippisley-Cox J: Use of hormone replacement therapy and risk of
breast cancer: Nested case-control studies using the QResearch and
CPRD databases. BMJ. 371:m38732020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Nordstrom BL, Cai B, De Gregorio F, Ban L,
Fraeman KH, Yoshida Y and Gibbs T: Risk of venous thromboembolism
among women receiving ospemifene: A comparative observational
study. Ther Adv Drug Saf. Nov 19–2022.(Epub ahead of print).
View Article : Google Scholar : PubMed/NCBI
|