Introduction
Hyperlipidemia is a common metabolic disorder, which
has been recognized as an independent risk factor for
cardiovascular disease (CVD) (1).
Individuals with hyperlipidemia have approximately twice the risk
of developing CVD compared with those with normal lipid profiles
(2). Increasing evidence has
suggested that hyperlipidemia causes cardiac damage through
oxidative stress, inflammation, pyroptosis and other mechanisms,
leading to lipid load-induced dysfunction characterized by
myocardial cell death, remodeling, impaired contractility and
autophagy-dependent cell death (3–5).
Despite the benefits of statins in lowering blood cholesterol
levels and preventing CVD, a large number of patients remain at
residual risk of CVD events due to intolerance or side effects
associated with lipid-lowering medications. Statins and other
lipid-lowering agents (e.g., ezetimibe, PCSK9 inhibitors, fibrates,
and niacin) can cause adverse effects such as myopathy, liver
enzyme elevation, gastrointestinal discomfort, and, in rare cases,
new-onset diabetes, which limit their widespread use and patient
adherence (6,7). Therefore, further research is needed
to identify drugs that can alleviate hyperlipidemia-induced cardiac
damage.
As a key intermediate in cellular energy metabolism,
nicotinamide adenine dinucleotide (NAD+) participates in
numerous biochemical reactions in vivo and has garnered
widespread scientific interest (8). Studies have shown that
NAD+ serves a pivotal role in maintaining redox
homeostasis, mitochondrial function and cellular metabolism
(9–11). In metabolic syndrome,
NAD+ deficiency impairs insulin sensitivity and promotes
lipid accumulation (12,13). In heart failure and
ischemia-reperfusion injury, NAD+ enhances mitochondrial
biogenesis and limits oxidative stress through the activation of
sirtuin (SIRT)1 and SIRT3 (9,14).
In arrhythmia, NAD+ influences calcium handling and
electrophysiological stability (15), and in hypertension, it modulates
vascular tone by improving endothelial function and reducing
inflammation (16). Notably,
NAD+ imbalance is associated with a number of diseases,
and is considered a pathogenic factor in numerous human genetic and
acquired conditions, including metabolic disorders such as obesity
and type 2 diabetes, neurodegenerative diseases such as Alzheimer's
and Parkinson's disease, cardiovascular diseases such as heart
failure and atherosclerosis, as well as certain cancers (17). Supplementation with NAD+
has been shown to alleviate the development of heart failure and to
potentially influence lifespan through mitochondrial redox
regulation (18). NAD+
also serves a key role in energy metabolism by acting as an
essential electron carrier in glycolysis and the tricarboxylic acid
cycle, and in cellular repair processes by serving as a substrate
for SIRTs and PARPs, which regulate mitochondrial function, DNA
repair and stress resistance. Exercise promotes aerobic metabolism
and enhances NAD+ biosynthesis through increased
oxidative phosphorylation (14,19).
In addition, increased NAD+ activates signaling pathways
related to cardiac protection, such as SIRT1, which promotes the
expression of antioxidant enzymes, and decreases oxidative stress
and inflammatory responses (20).
Furthermore, mitofusin2 (MFN2), an important protein in
mitochondrial outer membrane fusion, interacts with SIRT1 to reduce
cardiomyocyte autophagy by inhibiting oxidative stress (21). Collectively, these mechanisms
facilitate cardiomyocyte repair and regeneration, thereby providing
a robust foundation for recovery from cardiac injury.
Exercise of varying intensities induces different
levels of NAD+ enhancement (22). The present study conducted a
comparative analysis of moderate-intensity continuous training
(MICT) and high-intensity interval training (HIIT) in promoting
NAD+ levels, aiming to elucidate the underlying
molecular mechanisms involved. Furthermore, exogenous
NAD+ was administered to explore its mechanism of
action. The results of the current study may contribute to an
improved understanding of the role and mechanism of aerobic
exercise in hyperlipidemia-induced cardiac damage in apolipoprotein
E-deficient (ApoE−/−) mice by regulating NAD+
levels.
Materials and methods
Exercise model in mice with
hyperlipidemic cardiac damage
A total of 32 male ApoE−/− mice (age, 8
weeks; body weight, 21–25 g) were obtained from Liaoning Changsheng
Biotechnology Co., Ltd. The mice were randomly assigned to the
following four groups (n=8/group): The control group received a
standard chow diet; the high-fat diet (HFD) group was fed HFD
alone; the HFD + MICT group received the HFD combined with MICT;
and the HFD + HIIT group received the HFD combined with HIIT. The
HFD consisted of 60% fat energy from fat feed [cat. no. XSYT-HFD60;
Xiaoshu Youtai (Beijing) Biotechnology Co., Ltd.], and the HFD
feeding lasted for 12 weeks, followed by 12 weeks of exercise
training. All animals were housed in a controlled environment
maintained at 24–26°C with 40–60% relative humidity and a 12-h
light/dark cycle, with ad libitum access to food and water
throughout the study. At the end of the experiment, or when humane
endpoints (e.g., severe weight loss >20%, inability to eat or
drink, or signs of severe distress), were reached, the mice were
euthanized by intraperitoneal injection of pentobarbital sodium at
a dose of 150 mg/kg. Death was verified by confirming the complete
cessation of heartbeat and respiratory movement, as well as the
absence of corneal reflex, in accordance with institutional ethical
guidelines. Blood was collected by orbital sinus puncture under
anesthesia before sacrifice, with a total volume of ~0.3 ml/mouse,
and stored at −80°C for subsequent analyses. After sacrifice, heart
tissues were collected. Some samples were fixed in 10%
neutral-buffered formalin at room temperature for 24 h for
histological assessment and paraffin embedding, whereas additional
samples were snap-frozen in liquid nitrogen for subsequent protein
expression analysis via western blotting. All experimental animal
procedures were performed in accordance with the Guide for the
Management and Use of Laboratory Animals (23) and were approved by the Animal
Experiment Committee of the Central Hospital of Dalian University
of Technology (Dalian, China; approval no. YN2023-057-55).
Exercise training regimen
ApoE−/− mice in the MICT and HIIT group
underwent a treadmill running test to determine maximum running
speed using the XR-PT-10B treadmill (Shanghai XinRuan Information
Technology Co., Ltd.). The test began at 10 m/min with a 0° incline
for 20 min, with speed increasing by 4 m/min each min until the
mice exhibited signs of exhaustion. In the present study,
exhaustion was defined as either i) the mouse remaining stationary
on the shock grid for 3 consecutive seconds, or ii) the mouse
accumulating a total of 100 electrical stimulations during the
running session, regardless of whether it resumed running
afterward. This criterion was adopted to avoid excessive exposure
to electrical stimulation and to ensure animal welfare by setting a
strict upper limit for the number of shocks (24–26).
The maximum velocity achieved during exercise was defined as the
peak running speed. Before formal training, both groups underwent a
standardized 5-min warm-up at 40% of their respective maximum
running velocities. The HFD + HIIT group underwent nine sets of
high-intensity treadmill exercises, each lasting 1.5 min, with a
1-min rest between sprints, totaling 21.5 min/session at ~85% of
maximum running speed. The HFD + MICT group engaged in continuous
endurance training at 60% of maximum running speed until matching
the HFD + HIIT group distance. After training, both groups had a
5-min flat recovery period at 40% maximum running speed, repeated
five times a week for 12 weeks.
NAD+ therapy model in mice
with hyperlipidemic cardiac damage
Male ApoE−/− mice (age, 8
weeks; body weight, 20–25 g) were obtained from Liaoning Changsheng
Biotechnology Co., Ltd. A total of 32 mice were used in this study,
and these mice were obtained separately from those described in the
first subsection. The mice were housed under standard laboratory
conditions (24–26°C, 40–60% humidity, 12-h light/dark cycle). The
animals were randomly assigned to the following four experimental
groups (n=6/group): i) Normal diet (ND); ii) ND + NAD+
(4 mg/kg/day, intraperitoneally); iii) HFD; and iv) HFD +
NAD+ (4 mg/kg/day, intraperitoneally). The intervention
period lasted for 16 weeks, during which NAD+ was
administered to the designated groups. HFD was formulated using a
commercially available mouse diet enriched with 45%fat Kcal% energy
feed (cat. no. MD12032; Jiangsu Medisen Biomedical Co., Ltd.), HFD
feeding was continued for 12 weeks followed by 16-week
NAD+ intraperitoneal injection (27). NAD+ (cat. no. 53-84-9)
was purchased from Merck KGaA. Anesthesia was induced using 4%
isoflurane in oxygen and maintained at 1.5–2% isoflurane throughout
the orbital sinus puncture procedure; all anesthetic protocols were
performed in accordance with institutional animal care guidelines.
Blood samples were collected from the orbital venous sinus (0.5–0.8
ml/mouse) into serum tubes and immediately centrifuged, with the
resulting serum stored at −80°C for subsequent analysis. At the end
of the experiment or when humane endpoints were reached, the mice
were euthanized by intraperitoneal injection of pentobarbital
sodium at a dose of 150 mg/kg. Death was verified by cessation of
heartbeat and respiratory movement, as well as the absence of
corneal reflex, in accordance with institutional ethical
guidelines. Some of the heart tissues were fixed in 10% formalin
for 24 h at room temperature and subsequently embedded in paraffin
for histological evaluation. The remaining heart tissue was frozen
in liquid nitrogen for western blot analysis. Mice were monitored
daily for general health and behavior. Humane endpoints were
established to minimize animal suffering, including >20% body
weight loss, persistent hunched posture, reduced activity,
inability to access food or water, labored breathing or
unresponsiveness to stimuli. Animals reaching any of these criteria
were humanely euthanized by intraperitoneal injection of
pentobarbital sodium. All experimental animal procedures were
performed in accordance with the Guide for the Management and Use
of Laboratory Animals (23) and
were approved by the Animal Experiment Committee of the Central
Hospital of Dalian University of Technology (ethics approval no.
YN2023-057-55).
Assessment of biochemical
parameters
Blood samples were collected from the orbital venous
plexus and centrifuged at 854 × g for 10 min at 4°C using an
Eppendorf 5424R centrifuge (Eppendorf SE) to obtain serum. The
resulting supernatant (serum) was used for the quantification of a
series of biochemical markers. The serum levels of total
cholesterol (T-C; cat. no. A111-1-1), triglycerides (TG; cat. no.
A110-1-1), and low-density lipoprotein cholesterol (LDL-C; cat. no.
A113-1-1) were measured using colorimetric assay kits (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China) according to
the manufacturer's instructions, and their optical densities (ODs)
were read at 500 nm. Oxidative stress parameters were also
evaluated using serum samples with the following commercial kits
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China),
according to the manufacturer's instructions: superoxide dismutase
(SOD; cat. no. A001-3-2; OD at 450 nm) and malondialdehyde (MDA;
cat. no. A003-1-2; OD at 532 nm). In addition, Levels of coenzyme I
NAD(H) were assessed using a specific enzymatic cycling method
(cat. no. A114-1-1; Nanjing Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer's instructions, and
the OD was measured at 570 nm.
In addition, parameters were additionally measured
in vitro with the following commercial kits (Nanjing
Jiancheng Bioengineering Institute, Nanjing, China), according to
the manufacturer's instructions: Reduced glutathione (GSH; cat. no.
A006-2-1; OD at 405 nm), superoxide dismutase (SOD; cat. no.
A001-3-2; OD at 450 nm), total cholesterol (T-C; cat. no.
A111-1-1), triglycerides (TG; cat. no. A110-1-1), and low-density
lipoprotein cholesterol (LDL-C; cat. no. A113-1-1) were measured
using colorimetric assay kits (Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's instructions, and
optical densities were read at 500 nm.
All measurements were performed using a calibrated
microplate reader and absorbance values were recorded at the
specified wavelengths according to the manufacturer's instructions.
The use of wavelength-specific detection for each analyte ensured
optimal sensitivity and accuracy of the biochemical assays.
Histopathological assessment
Heart tissues were fixed in 10% neutral buffered
formalin at 4°C overnight, followed by sequential dehydration in a
graded ethanol series (50, 75, 85, 95 and 100%; 1 h each). Samples
were then cleared twice in xylene (12 min each) and embedded in
paraffin. Paraffin-embedded cardiac tissues were sectioned at a
thickness of 5 µm, and the sections were deparaffinized with xylene
(twice for 10 min), rehydrated through a descending ethanol
gradient (100, 95, 85 and 75%; 10 min each), and rinsed in PBS
three times, with each rinse lasting 5 min.
Subsequently, the sections underwent hematoxylin and
eosin (H&E) staining (cat. no. G1120; Beijing Solarbio
Technology Co. LTD, Beijing, China), Masson's trichrome staining
(cat. no. G1340; Beijing Solarbio Technology Co. LTD.), fluorescein
isothiocyanate (FITC)-conjugated wheat germ agglutinin (WGA)
staining (cat. no. I3300; Beijing Solarbio Technology Co. LTD,
Beijing, China) and TUNEL assay [One-step TUNEL In Situ
Apoptosis Kit (Green, FITC); cat. no. E-CK-A320; Wuhan Elabscience
Biotechnology Co., Ltd.] to evaluate morphological alterations,
fibrosis, cell membrane boundaries and apoptosis, respectively. All
experiments were performed in accordance with the manufacturers'
instructions. After staining, the sections were dehydrated through
an ascending ethanol series (75, 85, 95 and 100%; 10 min each),
cleared with xylene (twice for 12 min) and mounted with neutral
resin. Sections stained with H&E and Masson's trichrome were
observed under a BX40 upright light microscope (Olympus
Corporation). WGA and TUNEL images were acquired using a STELLARIS
5 laser scanning confocal microscope (Leica Microsystems GmbH).
Periodic acid-Schiff (PAS)
staining
PAS staining was performed using Glycogen PAS
staining kit (with hematoxylin); cat. no. G1281; Beijing Solarbio
Science & Technology Co., Ltd.). Heart tissue fixation and
dehydration were performed in the same manner as described above.
Sections were then treated with periodic acid solution (0.5%) at
room temperature for 5 min to oxidize polysaccharides, followed by
two washes in distilled water (1 min each). Slides were incubated
with Schiff reagent for 15 min in the dark and washed with
distilled water for 1 min. Nuclei were counterstained with
hematoxylin for 1 min. Sections were briefly differentiated in acid
alcohol (5 s) and then blued in running water for 10 min or in 1×
PBS for 5 min. Sections were dehydrated through a graded ethanol
series (75, 85, 95, 100%, 10 min each), cleared in xylene (2×1
min), and mounted with neutral resin. PAS-positive areas,
representing polysaccharides, glycogen, mucosubstances, and
glycoproteins, appear magenta under light microscopy. Slides were
observed and imaged using a BX40 upright light microscope. Staining
intensity was graded on a scale of 0–3 (0=negative, 1=weak,
2=moderate, 3=strong), and the percentage of PAS-positive area was
graded on a scale of 0–4 (0=<5%, 1=5–25%, 2=26–50%, 3=51–75%,
4=>75%). The final PAS score was obtained by multiplying the two
scores, yielding a total score ranging from 0 to 12. Quantitative
evaluation was performed by two independent blinded observers in
five randomly selected fields of view/section.
Western blotting
Mouse heart tissues frozen at −80°C were removed
from the freezer, and an appropriate amount of tissue was placed in
1.5-ml microcentrifuge tube containing a pre-cooled mixture of
protease [and phosphatase inhibitors]. The tissues were cut up on
ice, and then broken and cracked by ultrasound with 4°C ultrasonic
crushing for 3 sec and then stop for 3 seconds, a total of 20 sec;
power, 30% and 4°C overnight to extract total proteins. The next
day, the samples were centrifuged at 13,700 × g for 10 min at 4°C,
the resulting supernatant was carefully transferred to a fresh
1.5-ml microcentrifuge tube and the volume was recorded. Protein
concentration was determined using a bicinchoninic acid assay kit
(Beyotime Institute of Biotechnology) in a 96-well plate, alongside
a series of gradient standard protein solutions (2, 1, 0.5, 0.25,
0.125 and 0.0625 mg/ml). The absorbance at 562 nm was measured
using a microplate reader, and a standard calibration curve was
generated for quantification. Equal amounts of protein were then
aliquoted into clean 1.5-ml tubes, denatured in a boiling water
bath for 5 min, and stored at −80°C for subsequent use. Equal
amounts of protein (10 µg/lane) were separated by SDS-PAGE (10%)
and transferred onto polyvinylidene fluoride membranes (Immobilon;
MilliporeSigma). The membranes were blocked for 1 h at room
temperature in 5% skim milk diluted in Tris-buffered saline-0.1%
Tween-20 (TBST) and were subsequently incubated overnight at 4°C
with the following primary antibodies: Rabbit anti-SOD2 (1:5,000;
cat. no. K106586P; Beijing Solarbio Science & Technology Co.,
Ltd.), rabbit anti-heme oxygenase 1 (HO-1; 1:5,000; cat. no.
10701-1-AP; Proteintech Group, Inc.), rabbit anti-SIRT3 (1:1,000;
cat. no. 10099-1-AP; Proteintech Group, Inc.), mouse anti-SIRT1
(1:5,000; cat. no. 60303-1-Ig; Proteintech Group, Inc.), rabbit
anti-MFN2 (1:5,000; cat. no. 12186-1-AP; Proteintech Group, Inc.),
rabbit anti-PI3K p85 (1:1,000; cat. no. 20584-1-AP; Proteintech
Group, Inc.), rabbit anti-phosphorylated (p)-PI3K p85 (1:1,000;
cat. no. AF3242; Affinity Biosciences), anti-AKT (1:1,000; cat. no.
AF0836; Affinity Biosciences), rabbit anti-LC3 II (1:500; cat. no.
GB115766; Wuhan Servicebio Technology Co., Ltd.), rabbit anti-P62
(1:5,000; cat. no. 18420-1-AP; Proteintech Group, Inc.), rabbit
anti-p-AKT (1:1,000; cat. no. AF0016; Affinity Biosciences), rabbit
anti-mTOR (1:1,000; cat. no. AF6308; Affinity Biosciences), rabbit
anti-p-mTOR (1:1,000; cat. no. AF3308; Affinity Biosciences),
rabbit anti-tubulin β (1:2,000; cat. no. AF7011; all Affinity
Biosciences) and mouse anti-β-actin (1:10,000; cat. no. sc-8432;
Santa Cruz Biotechnology, Inc.). The next day, the membranes were
washed three times in TBST (10 min each) and then incubated for 1 h
at room temperature with the appropriate horseradish
peroxidase-conjugated secondary antibodies: Anti-rabbit IgG
(1:5,000; cat. no. SA00001-2; Proteintech Group, Inc.) or
anti-mouse IgG (1:10,000; cat. no. SA00001-1; Proteintech Group,
Inc.). Protein bands were visualized [High-sensitivity ECL
chemiluminescence detection kit; cat. no. SW134-01; Seven
Innovation (Beijing) Biotechnology Co., LTD,] and semi-quantified
using ImageJ software (version 1.53t; National Institutes of
Health). Tubulin β and β-actin were used as internal loading
controls, and relative protein expression levels were normalized
accordingly.
Immunohistochemistry
Coronal heart tissue sections were fixed in 10%
neutral buffered formalin overnight at 4°C. The following day, the
samples were sequentially dehydrated through a graded ethanol
series (50, 70, 85, 90, 95 and 100%, 1 h each), cleared in xylene
(twice for 12 min) and embedded in paraffin for histological
analysis. Paraffin-embedded tissues were sectioned at a thickness
of 5 µm, and were dewaxed twice in xylene (12 min each), rehydrated
through a descending ethanol series (100, 95, 85 and 75%, 10 min
each), and rinsed in PBS three times for 5 min each. For antigen
retrieval, slides were immersed in citrate buffer (pH 6.0) and
heated in a microwave oven at high power for 3–5 min, followed by
1–2 min on low power. The slides were then allowed to cool at room
temperature for 40 min and rinsed again with PBS (three times, 5
min each). Immunohistochemistry was conducted using a commercial
immunohistochemical kit (cat. no. KIT-9720; Fuzhou Maixin Biotech
Co., Ltd.). Endogenous peroxidase activity was blocked by
incubating the sections with a peroxidase blocking solution for 10
min, followed by three washes with PBS (5 min each). Non-specific
binding was blocked by incubating the slides in 5% sheep serum
(cat. no. FSP 500; Suzhou Excellent Biotechnology Co., LTD; Suzhou,
China) at room temperature for 1 h. After blocking, the sections
were incubated overnight at 4°C with the following primary
antibodies: Rabbit anti-SOD (1:100; cat. no. K106586P; Beijing
Solarbio Science & Technology Co., Ltd.), rabbit anti-HO-1
(1:100; cat. no. 10701-1-AP; Proteintech Group, Inc.), rabbit
anti-SIRT3 (1:100; cat. no. K008232P; Beijing Solarbio Science
& Technology Co., Ltd.), rabbit anti-TGF-β1 (1:300; cat. no.
81746-2-RR; Proteintech Group, Inc.), mouse anti-collagen I
(1:3,000; cat. no. 67288-1-Ig; Proteintech Group, Inc.), rabbit
anti-collagen III (1:1,000; cat. no. 22734-1-AP; Proteintech Group,
Inc.), mouse anti-SIRT1 (1:300; cat. no. 60303-1-Ig; Proteintech
Group, Inc.), and rabbit anti-MFN2 (1:300; cat. no. 12186-1-AP;
Proteintech Group, Inc.).
The following day, the sections were washed three
times in PBS (5 min each), then incubated at room temperature for 1
h with HRP-conjugated goat anti-rabbit IgG secondary antibody
provided in the N-Histofine Simple Stain Kit (cat. no. KIT-9720;
Fuzhou Maixin Biotech Co., Ltd.). Signal development was performed
using the metal-enhanced DAB substrate kit (cat. no. DA1015;
Beijing Solarbio Science & Technology Co., Ltd.) for 10 min at
room temperature, followed by rinsing in distilled water. Sections
were counterstained with hematoxylin for 1–2 min with room
temperature and washed under running tap water for 30 min.
Differentiation was carried out in 1% hydrochloric acid ethanol for
3 sec, followed by an additional rinse under running water for 3
min. The stained slides were then dehydrated in ascending
concentrations of ethanol (75, 85, 95 and 100%, 10 min each),
cleared in xylene twice (12 min each) and mounted with neutral
resin. All stained sections were examined using an Olympus BX40
upright light microscope (Olympus Corporation) for morphological
analysis was performed using ImageJ software (version 1.53,
National Institutes of Health).
KEGG Pathway Analysis
Differentially expressed genes identified from our
experiments were subjected to pathway enrichment analysis using the
Kyoto Encyclopedia of Genes and Genomes (KEGG) database (kegg.jp/
using the default parameters. Pathways with a P-value <0.05 were
considered significantly enriched.
Protein-Protein Interaction (PPI)
Network Analysis
Protein-protein interaction (PPI) networks were
constructed to explore the potential interactions of SIRT1 with
other proteins involved in mitochondrial dynamics and oxidative
stress. Analyses were performed using the STRING database
(string-db.org/). The list of relevant proteins was uploaded, and
interaction scores were calculated using the default medium
confidence threshold (0.4). The resulting network was visualized to
identify central nodes and functional connections. The potential
interactions among PI3K, AKT, and mTOR were investigated using the
STRING database (Search Tool for the Retrieval of Interacting
Genes/Proteins; string-db.org/). The analysis was performed by
inputting the respective gene symbols, with the species limited to
Mus musculus. A high-confidence interaction score (0.7) was
applied as the cutoff. The resulting network was visualized to
identify functional associations relevant to myocardial
protection.
Tissue-specific expression analysis of
MFN2
Tissue-specific expression analysis of MFN2 was
performed using publicly available transcriptomic data from the
Genotype-Tissue Expression (GTEx) database (accession no.
phs000424.v9.p2; gtexportal.org/) and validated with the Human
Protein Atlas (HPA; accession no. ENSG00000116688; http://www.proteinatlas.org/ENSG00000116688-MFN2).
Immunofluorescence
Paraffin-embedded tissue sections were
deparaffinized, subjected to antigen retrieval and blocked
according to the aforementioned standard immunohistochemical
procedures. For immunofluorescence staining, the following primary
antibodies were applied to the tissue sections: Rabbit anti-SIRT1
(1:100; cat. no. BF0189; Affinity Biosciences) was used for cell
staining, mouse anti-SIRT1 (1:300; cat. no. 60303-1-Ig; Proteintech
Group, Inc.) was used for double immunofluorescence staining and
rabbit anti-MFN2 (1:300; cat. no. 12186-1-AP; Proteintech Group,
Inc.). The slides were incubated overnight at 4°C and washed three
times with PBS (5 min each). In the dark, the following appropriate
fluorescent secondary antibodies were added for 1 h at room
temperature: Alexa Fluor® 488-conjugated anti-rabbit IgG
(H+L), F(ab')2 fragment (1:800; cat. no. 4412S; Cell
Signaling Technology, Inc.) and Alexa Fluor 594-conjugated
anti-mouse IgG (H+L), F(ab')2 fragment (1:800; cat. no.
8890S; Cell Signaling Technology, Inc.). The slides were then
washed three times with PBS (5 min each). For nuclear
counterstaining, the sections were treated with DAPI (Wuhan
Servicebio Technology Co., Ltd.); the slides were washed with PBS
three times (5 min each), stained with DAPI for 1 min and rinsed
again three times with PBS in the dark. After staining, the
sections were mounted using an anti-fade fluorescence mounting
medium (Wuhan Servicebio Technology Co., Ltd.), and fluorescence
signals were visualized and captured using a Leica TCS SP8 confocal
laser scanning microscope (Leica Microsystems GmbH).
Cell culture
The HL-1 mouse cardiomyocyte cell line (cat. no.
MYC-TCM783; Dalian Yiran Mingyu Biotechnology Co., Ltd.) was
authenticated using short tandem repeat (STR) profiling upon
receipt. The STR results confirmed that the cell line used
corresponds to HL-1 and does not match the STR profile of C2C12 or
any known contaminant cell lines. HL-1 cells exhibit adherent
growth characteristics and were cultured in DMEM with high glucose
(Gibco; Thermo Fisher Scientific, Inc.) supplemented with 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) and 1%
penicillin-streptomycin (all Gibco; Thermo Fisher Scientific,
Inc.). The cells were maintained in a humidified incubator at 37°C
with 5% CO2 and 70% relative humidity. Subculturing was
performed at a 1:3 ratio when cells reached 80–90% confluence,
using standard enzymatic dissociation with 0.25% trypsin-EDTA
(Gibco; Thermo Fisher Scientific, Inc.).
Cell passage
Cells were maintained in complete growth medium
(DMEM supplemented with 10% fetal bovine serum and 1%
penicillin-streptomycin) at 37°C in a humidified atmosphere
containing 5% CO2. When cultures reached 80–90%
confluency, cells were washed twice with phosphate-buffered saline
(PBS), detached using 0.25% trypsin-EDTA solution, and collected by
centrifugation at 300 × g for 5 min. Cells were resuspended in
fresh medium and seeded at an appropriate density for subsequent
experiments. Passages were performed every 2–3 days, and only cells
within passages 3–10 were used for experiments.
Palmitic acid (PA) Treatment in HL-1
Cells
HL-1 cells were treated with PA; MedChemExpress,
Cat. No. HY-N0830) to induce lipotoxicity and mimic metabolic
stress in vitro. PA was prepared as a 5 mM stock solution in 0.1 M
NaOH and complexed with 10% fatty-acid free bovine serum albumin
(BSA) to a final working concentration of 200 µM. Cells were
incubated with PA at 37°C in a 5% CO2 incubator for 24
h. Following treatment, autophagy markers LC3 II and p62 were
assessed by western blotting to evaluate autophagic flux under
lipotoxic conditions.
ROS detection
Reactive oxygen species (ROS) levels in adherent
cells were measured using the fluorescent probe CM-H2DCFDA (cat no.
S0035S, Beyotime Biotechnology Co., Ltd.). CM-H2DCFDA was diluted
(1:1,000 in PBS to a final concentration of 5 µM. Culture medium
was removed, and cells were incubated with sufficient volume of the
diluted CM-H2DCFDA to fully cover the cell layer at 37°C in a
CO2 incubator for 30 min. After incubation, cells were
washed three times with PBS to remove excess probe that had not
entered the cells. For positive control of ROS production, cells
can be stimulated for 20–30 min to achieve a detectable increase in
ROS levels. Fluorescence images were captured immediately using a
laser-scanning confocal microscope (Leica TCS STELLARIS 5), and
signal quantification was performed using ImageJ software (version
1.53).
JC-1 staining for mitochondrial
membrane potential
Mitochondrial membrane potential in adherent cells
was assessed using the JC-1 fluorescent probe kit ((cat no. C2006,
Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. Briefly, cells were washed with PBS
and incubated with JC-1 staining solution at 37°C for 20–30 min.
Following incubation, cells were washed twice with JC-1 buffer to
remove excess dye. Fluorescence images were immediately acquired
using a confocal microscope (Leica TCS STELLARIS 5) with
excitation/emission settings for JC-1 monomers (green, ~514/529 nm)
and aggregates (red, ~585/590 nm). The ratio of red to green
fluorescence intensity was quantified using ImageJ software
(version 1.53) to assess changes in mitochondrial membrane
potential.
EX-527 treatment
HL-1 cardiomyocytes were seeded at a density of
1×105 cells/cm2 in 10 cm culture dishes and
cultured under standard conditions (37°C, 5% CO2) until
reaching the desired confluency for subsequent experiments. To
inhibit SIRT1 activity, cells were treated with 10 µM EX-527 (cat.
no. HY-15452; MedChemExpress) for 24 h (37°C, 5% CO2).
After treatment, the cells were collected for subsequent
assays.
Gene silencing via RNA
interference
Cells were seeded in 6-cm dishes as in the passage
procedure, and the cell confluence was ~60% at the time of
transfection. According to the manufacturer's instructions, the
ApoE small interfering (si)RNA was used to silence ApoE expression
in HL-1 cells using a transfection reagent kit (Lipofectamine 8000;
cat no. C0533, Gibco; Thermo Fisher Scientific, Inc.). Cells were
transfected with siRNA (20 µM, 37°C) for 48 h. Culture medium was
replaced with fresh medium containing NAD+, and the
cells were incubated for 48 h under standard conditions before
subsequent experiments. The efficiency of ApoE knockdown in HL-1
cells was verified by western blotting. Three siRNAs targeting
SIRT1 were designed (sequences provided in Table I). HL-1 cells were transfected with
20 µM of each siRNA using Lipofectamine 8000 (Thermo Fisher
Scientific) according to the manufacturer's protocol. Knockdown
efficiency was assessed by western blot, and ApoE-Mus-415 was used
for subsequent experiments. A non-targeting scramble siRNA was used
as a negative control. The siRNA target sequences are shown in
Table I.
 | Table I.Small interfering RNA target
sequences. |
Table I.
Small interfering RNA target
sequences.
| Name | Forward, 5′→3′ | Reverse, 5′→3′ |
|---|
| ApoE-Mus-217 |
GCCGUGCUGUUGGUCACAUTT |
AUGUGACCAACAGCACGGCTT |
| ApoE-Mus-415 |
GCACUGAUGGAGGACACUATT |
UAGUGUCCUCCAUCAGUGCTT |
| ApoE-Mus-1052 |
GCCAGUGGGCAAACCUGAUTT |
AUCAGGUUUGCCCACUGGCTT |
| Negative
control |
UUCUCCGAACGUGUCACGUTT |
ACGUGACACGUUCGGAGAATT |
MTS colorimetric cell viability
assay
Logarithmic-phase HL-1 cells were collected and
resuspended in complete growth medium. Cell concentration was
determined using a Cell counter instrument after trypan blue
staining to assess cell viability. Based on the counted cell
number, the suspension was adjusted to a concentration of
1×105 cells/ml. Subsequently, 100 µl cell suspension was
seeded into each well of a 96-well flat-bottom plate. The plate was
placed in a humidified incubator at 37°C with 5% CO2 for
24 h. When cells had adhered to the well surface, the medium within
each insert was gently aspirated following a 4-h incubation in
serum-free medium. Subsequently, NAD+ solutions at
varying concentrations (0, 1, 5, 7 and 10 mM) (cat. no. HY-B0445;
MedChemExpress) were added to the wells, and the cells were
incubated for 48 h in a humidified cell incubator at 37°C with 5%
CO2. Following treatment, 20 µl MTS reagent (cat. no.
G3582; Promega Corporation) was added to each well in the dark.
After an additional 2-h incubation, absorbance was measured at 490
nm using a microplate reader. Cell viability under different
NAD+ concentrations was calculated based on OD values at
490 nm. Each experimental condition was performed in triplicate,
and all assays were independently repeated three times. Statistical
analysis was conducted to evaluate differences between groups.
Statistical analysis
All quantitative data are presented as the mean ±
standard error of the mean. Statistical analyses were conducted
using SPSS software (version 23.0; IBM Corp.). Intergroup
differences were assessed by one-way analysis of variance, followed
by Tukey's post hoc test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
HIIT enhances NAD+
metabolism and decreases oxidative stress in ApoE−/−
hyperlipidemic mice
The experimental design is shown in Fig. 1A. Significant differences in body
weight and heart-to-body weight ratio were observed among the
groups (Fig. 1B and C). Mice in
the HFD group exhibited the highest body weight and heart-to-body
weight ratio, whereas both the HFD + MICT and HFD + HIIT groups
demonstrated reductions in these parameters, with the HFD + HIIT
group showing the most pronounced improvement. These findings
suggested that HIIT may alleviate HFD-induced obesity and cardiac
hypertrophy. Serum analysis showed a significant decrease in SOD
activity in the HFD group, which was partially reversed by exercise
interventions, with HIIT demonstrating the greatest effect
(Fig. 1D). Conversely, MDA levels,
which were elevated in the HFD group, were significantly reduced by
exercise, particularly following HIIT (Fig. 1E). Furthermore, in
ApoE−/− mice, Exercise increased the
NAD+/NADH ratio in both cardiac tissue and serum, with
HIIT exerting the most pronounced effect (Fig. 1F and G). Myocardial hypertrophy and
fibrosis induced by HFD) were alleviated by exercise, particularly
in the HFD + HIIT group, as evidenced by histological analyses.
Heart sections were stained with hematoxylin and eosin (H&E)
for general morphology, Masson's trichrome for fibrosis, and
periodic acid-Schiff (PAS) for glycogen deposition (Fig. 1H and I). PAS staining showed
decreased glycogen accumulation in both exercise groups, with the
most notable reduction in the HFD + HIIT group (Fig. 1H and J). These results indicated
that HIIT provided more pronounced protection against HFD-induced
myocardial hypertrophy, fibrosis and glycogen accumulation compared
with MICT.
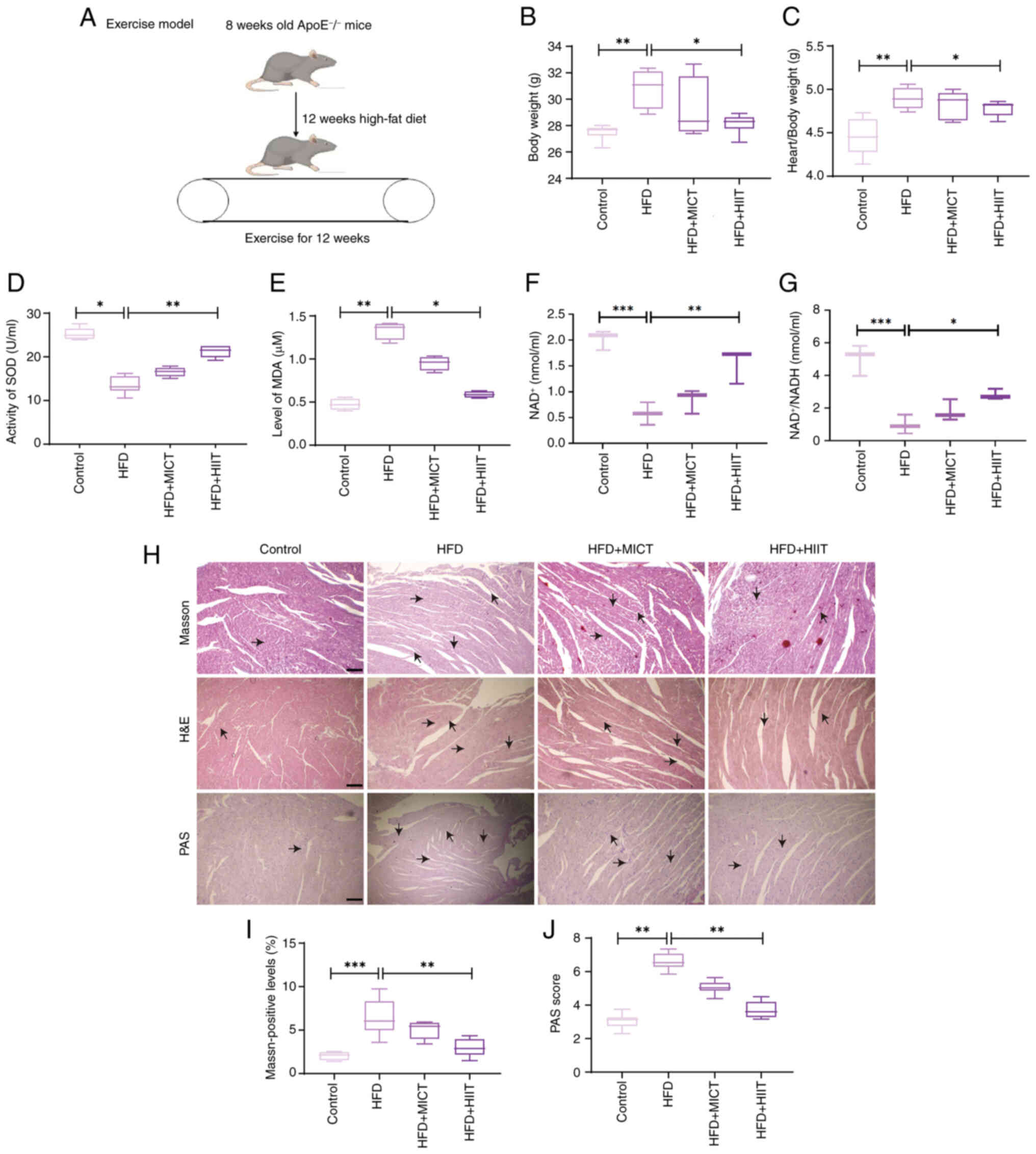 | Figure 1.Metabolic data in each group after 12
weeks of exercise training. (A) Schematic diagram of the
experimental model. (B) Changes in the body weight of mice. (C)
Quantitative analysis of heart/body weight ratio in each group. (D)
SOD levels in the serum of mice in each group (n=6). (E) MDA levels
in the serum of mice in each group (n=4). Serum (F) NAD+
and (G) NAD+/NADH levels in each group of mice (n=3).
(H) H&E, Masson and PAS staining heart tissue sections, with
arrows indicating positively stained cells. Scale bar, 100 µm;
magnification, ×40 (n=3). (I) Masson's trichrome staining of
myocardial tissue showing collagen deposition. (J) PAS) staining of
myocardial tissue showing glycogen deposition. Data are presented
as the mean ± standard error of the mean; statistical analysis was
performed using one-way ANOVA followed by Tukey's post hoc test.
*P<0.05, **P<0.01, ***P<0.001. ApoE−/−,
apolipoprotein E-deficient; H&E, hematoxylin and eosin; HFD,
high-fat diet; HIIT, high-intensity interval training; MDA,
malondialdehyde; MICT, moderate-intensity continuous training;
NAD+, nicotinamide adenine dinucleotide; PAS, Periodic
acid-Schiff; SOD, superoxide dismutase. |
NAD+ supplementation
ameliorates HFD-induced obesity, dyslipidemia and oxidative
stress
The experimental design is shown in Fig. 2A. Significant differences in body
weight and lipid profiles were observed among the groups (Fig. 2B-E). The HFD group showed a
pronounced increase in body weight compared with that in the ND
group, whereas NAD+ supplementation (HFD +
NAD+) significantly lowered body weight, nearing that of
the ND group (Fig. 2B). In terms
of lipid levels, the HFD group had significantly higher TC, TG, and
LDL-C levels compared with those in the ND group. NAD+
supplementation in the HFD + NAD+ group led to a
significant decrease in serum lipid levels, including TC), (TG),
and (LDL-C), compared with the HFD group (Fig. 2C-E). Immunohistochemistry analysis
(Fig. 2F-I) revealed that the ND
group exhibited significantly higher expression of SOD, HO-1, and
SIRT3 compared with the HFD group. The HFD group showed decreased
expression of these markers, whereas the HFD + NAD+
group exhibited increased staining for SOD, HO-1 and SIRT3, similar
to the ND group. Semi-quantification of immunohistochemistry
results (Fig. 2F-H) confirmed the
significantly increased expression of these proteins in the HFD +
NAD+ group compared with that in the HFD group. Western
blot analysis (Fig. 2J) further
supported these findings, showing a marked increase in SOD, HO-1
and SIRT3 expression in the HFD + NAD+ group compared
with those in the HFD group. Semi-quantitative analysis of western
blotting data confirmed the enhanced expression of these proteins
in the NAD+ supplemented group. Notably, NAD+
supplementation did not produce significant effects in mice fed a
ND.
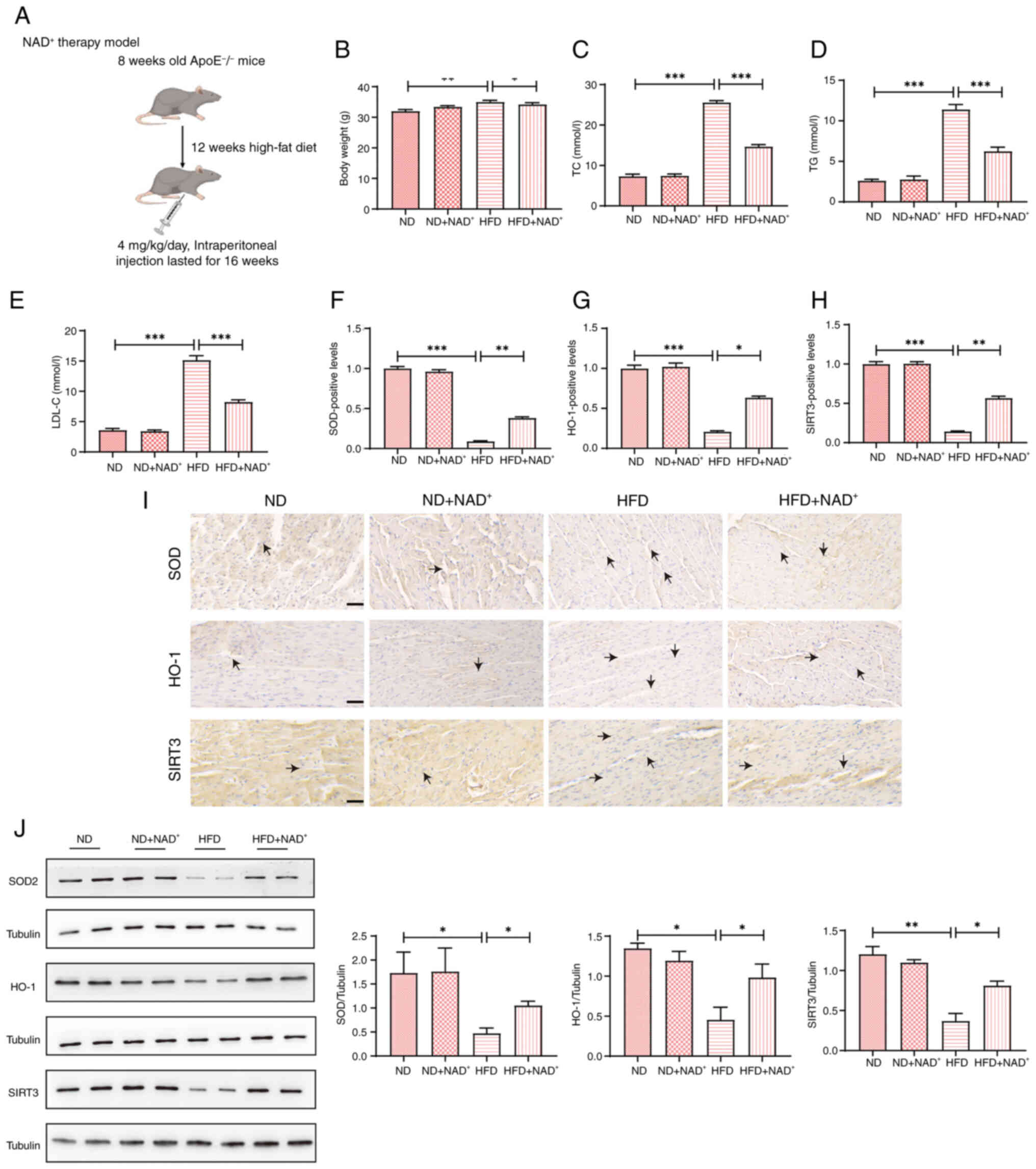 | Figure 2.Effects of NAD+
supplementation on body weight, lipid profiles and antioxidant
proteins in HFD-induced mice. (A) Schematic diagram of the
experimental model. (B) Body weight changes in the ND, ND +
NAD+, HFD and HFD + NAD+ groups. Lipid
profiles of the mice in all groups, including (C) TC, (D) TG and
(E) LDL-C (n=6). Semi-quantification of immunohistochemical results
showing significant upregulation of (F) SOD, (G) HO-1 and (H) SIRT3
in the HFD + NAD+ compared with in the HFD group (n=4).
(I) Immunohistochemical staining of the antioxidant proteins SOD,
HO-1 and SIRT3 in heart tissue; arrows indicate positively stained
cells. Magnification, ×40; scale bar, 100 µm (n=3). (J) Western
blot analysis of SOD, HO-1 and SIRT3 expression in heart tissue,
and semi-quantification of western blotting data, confirming
increased expression of SOD, HO-1 and SIRT3 in the HFD +
NAD+ group. Data are presented as the mean ± standard
error of the mean; statistical analysis was performed using one-way
ANOVA followed by Tukey's post hoc test. *P<0.05, **P<0.01,
***P<0.001. ApoE−/−, apolipoprotein E-deficient; HFD,
high-fat diet; HO-1, heme oxygenase 1; LDL-C, low-density
lipoprotein cholesterol; NAD+, nicotinamide adenine
dinucleotide; ND, normal diet; SIRT3, sirtuin 3; SOD, superoxide
dismutase; TC, total cholesterol; TG, triglycerides. |
NAD+ supplementation
attenuates myocardial fibrosis and extracellular matrix (ECM)
remodeling in HFD-induced mice
Masson's trichrome staining (Fig. 3A) revealed substantial collagen
deposition in the HFD group, indicative of fibrosis, while
NAD+ supplementation (HFD + NAD+) reduced
this deposition to levels similar to those in the ND group.
Immunohistochemical staining of the fibrosis-related markers TGF-β,
collagen I and III (Fig. 3B-D)
exhibited elevated expression in the HFD group, which was markedly
reduced in response to NAD+ supplementation, approaching
the levels observed in the ND group. H&E staining (Fig. 3E) demonstrated structural
disorganization and injury in the HFD group, whereas
NAD+ supplementation improved myocardial architecture,
especially in the HFD + NAD+ group. WGA staining
(Fig. 3F) confirmed enhanced
myocardial cell integrity in both the ND + NAD+ and HFD
+ NAD+ groups. Semi-quantitative analysis further
confirmed that NAD+ supplementation significantly
reduced the Masson-positive area, and TGF-β, collagen I and
collagen III expression (Fig.
3G-J) levels, indicating a protective effect against myocardial
fibrosis and ECM remodeling in the HFD + NAD+ group.
Notably, NAD+ supplementation did not induce significant
changes in the ND group, suggesting that its protective effects are
specific to pathological conditions such as HFD-induced cardiac
injury.
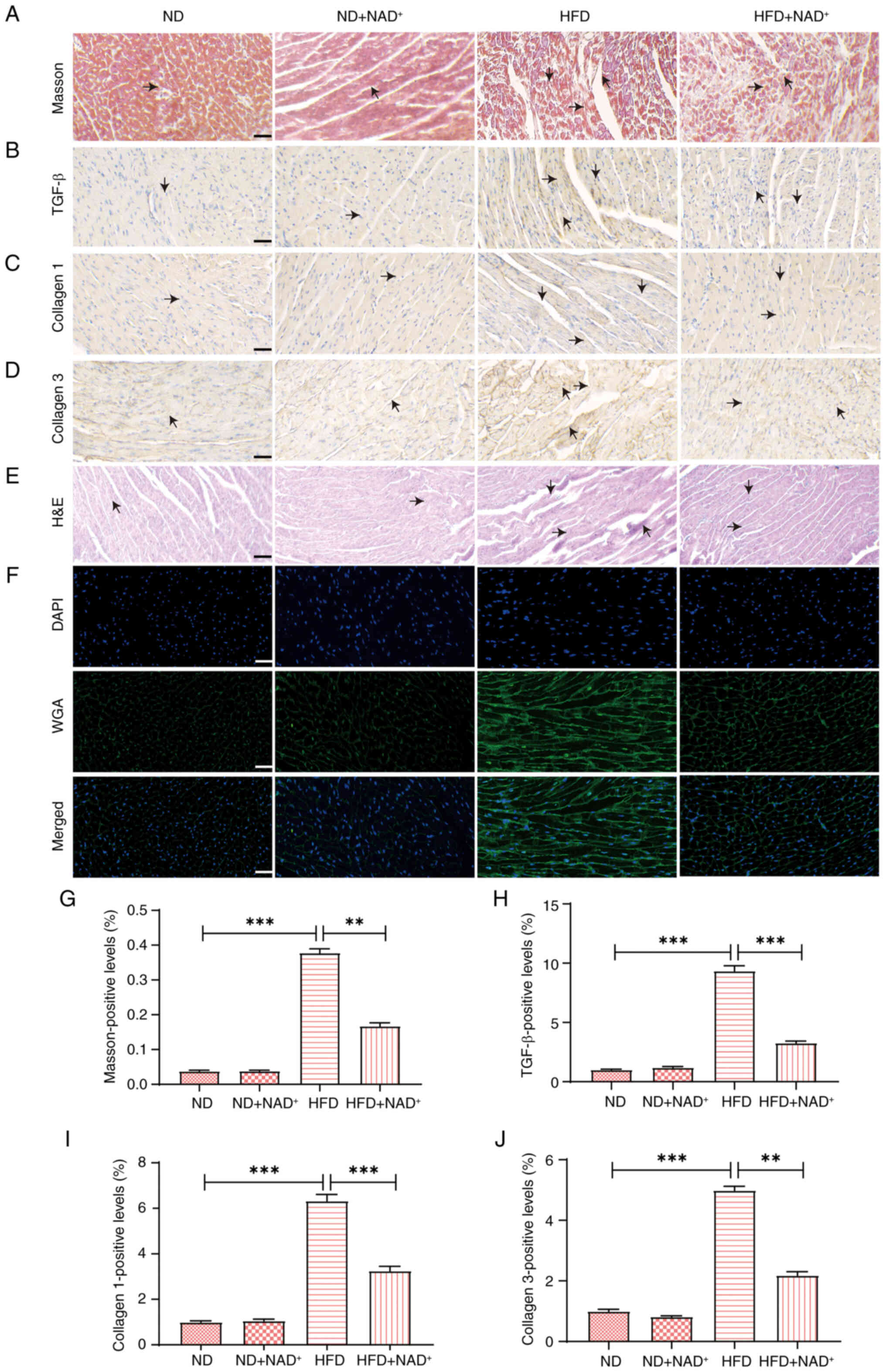 | Figure 3.NAD+ supplementation
attenuates myocardial fibrosis and ECM remodeling in HFD-induced
mice. (A) Masson's trichrome staining of myocardial tissue.
Blue-stained regions indicate collagen deposition (fibrotic areas),
and red indicates myocardial fibers. Black arrows point to areas of
increased collagen accumulation. Scale bar, 100 µm.
Immunohistochemical staining of fibrosis-related markers: (B)
TGF-β, (C) collagen I, and (D) collagen III. Brown staining
indicates positive expression, and black arrows highlight regions
with enhanced marker expression. Scale bar, 100 µm. (E) H&E
staining of myocardial tissue showing general histological
structure. Black arrows indicate areas of myocardial hypertrophy or
structural abnormalities. Scale bar, 100 µm. (F) WGA staining
showing improved myocardial cell integrity in the HFD +
NAD+ group. Scale bar, 100 µm. Semi-quantification of
the (G) Masson-positive area, and (H) TGF-β, (I) collagen I and (J)
collagen III levels. NAD+ supplementation significantly
reduced fibrosis and ECM remodeling in the HFD + NAD+
group compared with in the HFD group. Data are presented as the
mean ± standard error of the mean, n=3; statistical analysis was
performed using one-way ANOVA followed by Tukey's post hoc test.
**P<0.01, ***P<0.001. ECM, extracellular matrix; H&E,
hematoxylin and eosin; HFD, high-fat diet; NAD+,
nicotinamide adenine dinucleotide; ND, normal diet; WGA, what germ
agglutinin. |
NAD+ ameliorates cardiac
damage in hyperlipidemic mice via the SIRT1/MFN2 pathway
KEGG pathway analysis (Fig. S1) indicated that SIRT1 serves a
central role in regulating key pathways involved in cellular
metabolism and stress response. Protein interaction analysis
(Fig. 4A) highlighted potential
interactions between SIRT1 and MFN2 involved in mitochondrial
dynamics and oxidative stress. Tissue specificity analysis of MFN2
(Fig. S2) revealed its
predominant expression in myocardial tissue, suggesting its
critical role in cardiac function. Western blot analysis (Fig. 4B) showed that SIRT1 and MFN2
protein expression levels were significantly reduced in the HFD
group compared with those in the ND group. However, NAD+
supplementation (HFD + NAD+) restored the expression of
both SIRT1 and MFN2 to levels comparable with the HFD group,
suggesting that NAD+ exerts protective effects by
modulating these proteins. Immunohistochemical staining (Fig. 4C) further confirmed that SIRT1 and
MFN2 expression levels were reduced in the HFD group, with marked
restoration in the HFD + NAD+ group. The arrows in the
staining images indicate areas of stained cells, illustrating the
cellular localization of these proteins in myocardial tissue.
Additionally, immunofluorescence double-labeling colocalization
staining (Fig. 4D) showed clear
colocalization of SIRT1 and MFN2 in the heart tissue, confirming
that NAD+ supplementation enhanced the expression of
both proteins in myocardial cells. Notably, NAD+
administration did not significantly affect SIRT1 or MFN2
expression in the ND group, reinforcing that its modulatory effects
are specific to pathological conditions such as HFD-induced cardiac
stress.
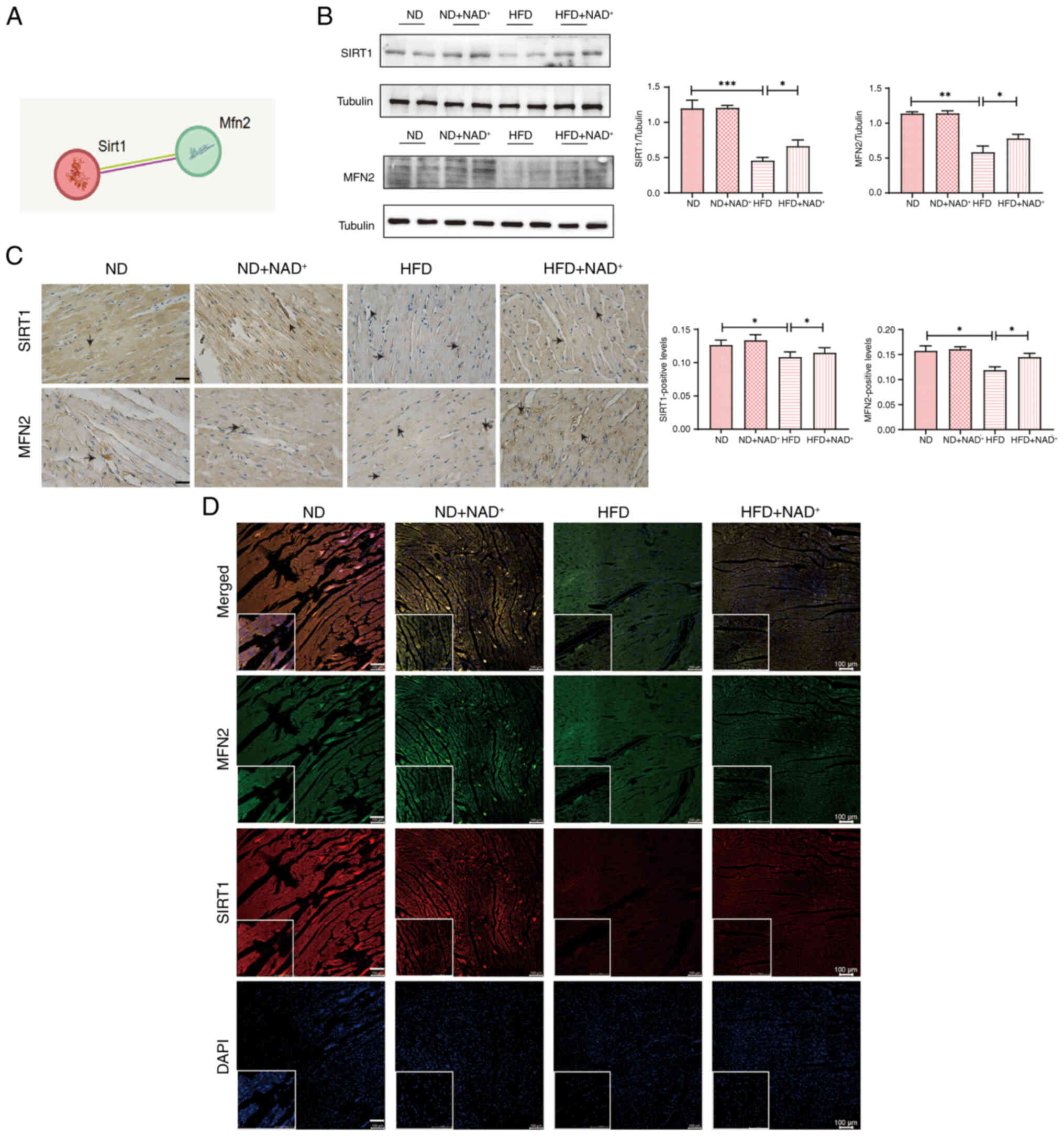 | Figure 4.NAD+ ameliorates cardiac
damage in hyperlipidemic mice by modulating the SIRT1/MFN2 pathway.
(A) Protein interaction analysis. (B) Expression levels of SIRT1
and MFN2 proteins in the heart tissues of mice. (C) Representative
immunohistochemistry images showing the expression of SIRT1 and
MFN2 in myocardial tissue. The arrows indicate areas of stained
cells. Scale bar, 100 µm; magnification, ×40. (D) SIRT1 and MFN 2
were detected by immunofluorescence double staining to assess
colocalization. Scale bars, 100 and 50 µm; magnification, ×40. Data
are presented as the mean ± standard error of the mean (n=3);
statistical analysis was performed using one-way ANOVA followed by
Tukey's post hoc test. *P<0.05, **P<0.01, ***P<0.001. HFD,
high-fat diet; MFN2, mitofusin 2; NAD+, nicotinamide
adenine dinucleotide; ND, normal diet; SIRT1, sirtuin 1. |
HIIT regulates the myocardial
PI3K/AKT/mTOR pathway through NAD+-induced regulation of
the SIRT1/MFN2 pathway
TUNEL staining (Fig.
5A) revealed increased myocardial apoptosis in the HFD group,
which was markedly reduced by NAD+ supplementation in
the HFD + NAD+ group, indicating enhanced cell survival.
Protein interaction analysis (Fig.
5B) identified key interactions between PI3K, AKT and mTOR,
suggesting their involvement in myocardial protection. Western blot
analysis (Fig. 5C) confirmed these
findings, showing decreased p-PI3K/PI3K and p-AKT/AKT levels but
elevated p-mTOR/mTOR levels in the HFD group. Notably,
NAD+ supplementation restored PI3K and AKT
phosphorylation while suppressing the abnormal increase in p-mTOR
expression. In addition, in the HFD group, both LC3 II and P62
levels were increased, suggesting impaired autophagic flux
(Fig. S3A). By contrast, in the
HFD + NAD+ group, the levels of LC3 II and p62 were
decreased, indicating restoration of autophagic degradation and
enhanced autophagic flux following NAD+ supplementation,
as decreased accumulation of both LC3 II and p62 reflects improved
turnover of autophagosomes rather than impaired autophagosome
formation. These results suggested that NAD+ exerts
cardioprotective effects through activation of the PI3K/AKT/mTOR
pathway and increased autophagy by promoting autophagic flux.
Notably, NAD+ treatment did not significantly affect
apoptosis markers or autophagy-related proteins in the ND group,
suggesting that its beneficial effects are specific to the
pathological state induced by HFD.
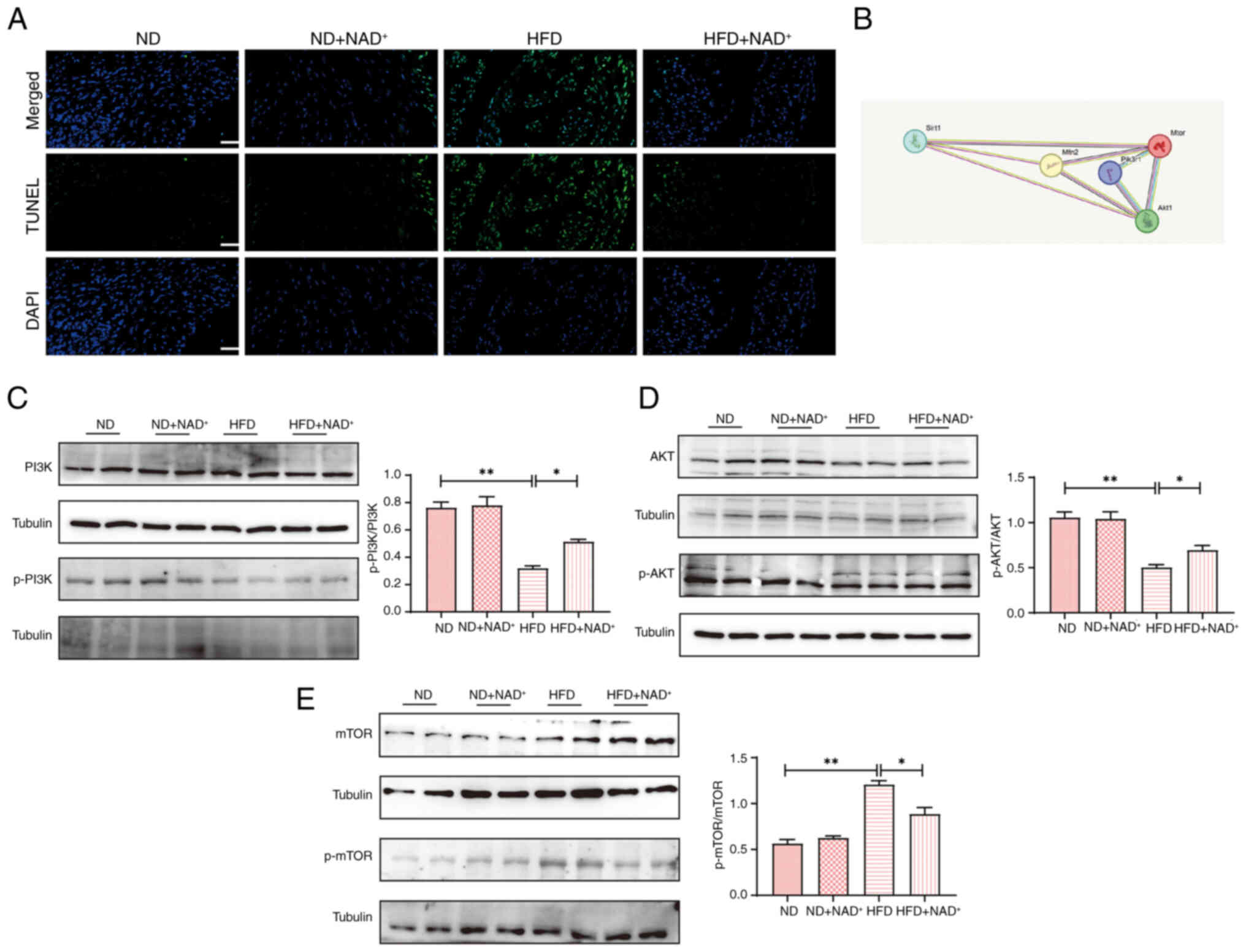 | Figure 5.NAD+ supplementation
activates the PI3K/AKT/mTOR pathway in the myocardium. (A) TUNEL
staining was performed to assess myocardial cell apoptosis in the
heart tissues of mice. Magnification, ×40; scale bar, 100 µm. (B)
Protein interaction analysis. Expression levels of (C) p-PI3K/PI3K,
(D) p-AKT/AKT and (E) p-mTOR/mTOR in the heart tissues of mice.
Data are presented as the mean ± standard error of the mean (n=4);
statistical analysis was performed using one-way ANOVA followed by
Tukey's post hoc test. *P<0.05, **P<0.01. HFD, high-fat diet;
Mfn2, mitofusin 2; NAD+, nicotinamide adenine
dinucleotide; ND, normal diet; p-, phosphorylated; Sirt1, sirtuin
1. |
NAD+ protects
ApoE−/− HL-1 cells from lipid accumulation and oxidative
stress
Western blot analysis confirmed successful knockdown
of ApoE expression in HL-1 cardiomyocytes following transfection
with ApoE-specific siRNA (Fig.
S4). NAD+ treatment at concentrations of 1, 5 and 7
µM significantly improved cell viability compared with the control
group, with 5 µM showing the most pronounced effect (Fig. 6A). By contrast, 10 µM
NAD+ did not significantly affect viability compared
with the control. In the ApoE−/− group, lipid
accumulation was evident, as indicated by elevated TC (Fig. 6B), TG (Fig. 6C) and LDL-C (Fig. 6D) levels, which were significantly
reduced by NAD+ supplementation (ApoE−/− +
NAD+ group). Additionally, NAD+ treatment
increased GSH levels (Fig. 6E) and
enhanced SOD activity (Fig. 6F),
indicating improved antioxidant capacity. ROS) staining (Fig. 6G) showed increased ROS accumulation
in the ApoE−/− group, which was mitigated by
NAD+ treatment. Microscopic images (Fig. 6H) confirmed that NAD+
supplementation improved cell morphology and viability, as
evidenced by more uniform cell shape, increased adherence and
reduced cell shrinkage and detachment compared with the untreated
group. Immunofluorescence staining (Fig. 6J) showed increased expression of
SIRT1 in the ApoE−/− + NAD+ group, suggesting
NAD+ activation of the SIRT1 pathway compared with
ApoE−/− group. JC-1 staining (Fig. 6I) revealed improved mitochondrial
membrane potential in the ApoE−/− + NAD+
group compared with ApoE−/− group, indicating enhanced
mitochondrial function with NAD+ treatment. These
findings demonstrate that NAD+ supplementation may
improve cellular function, lipid metabolism and oxidative stress in
HL-1 cells. Notably, NAD+ had minimal effects on lipid
levels, antioxidant capacity and mitochondrial function in
untransfected control cells, suggesting that its beneficial effects
are specific to ApoE−/−-induced cellular stress.
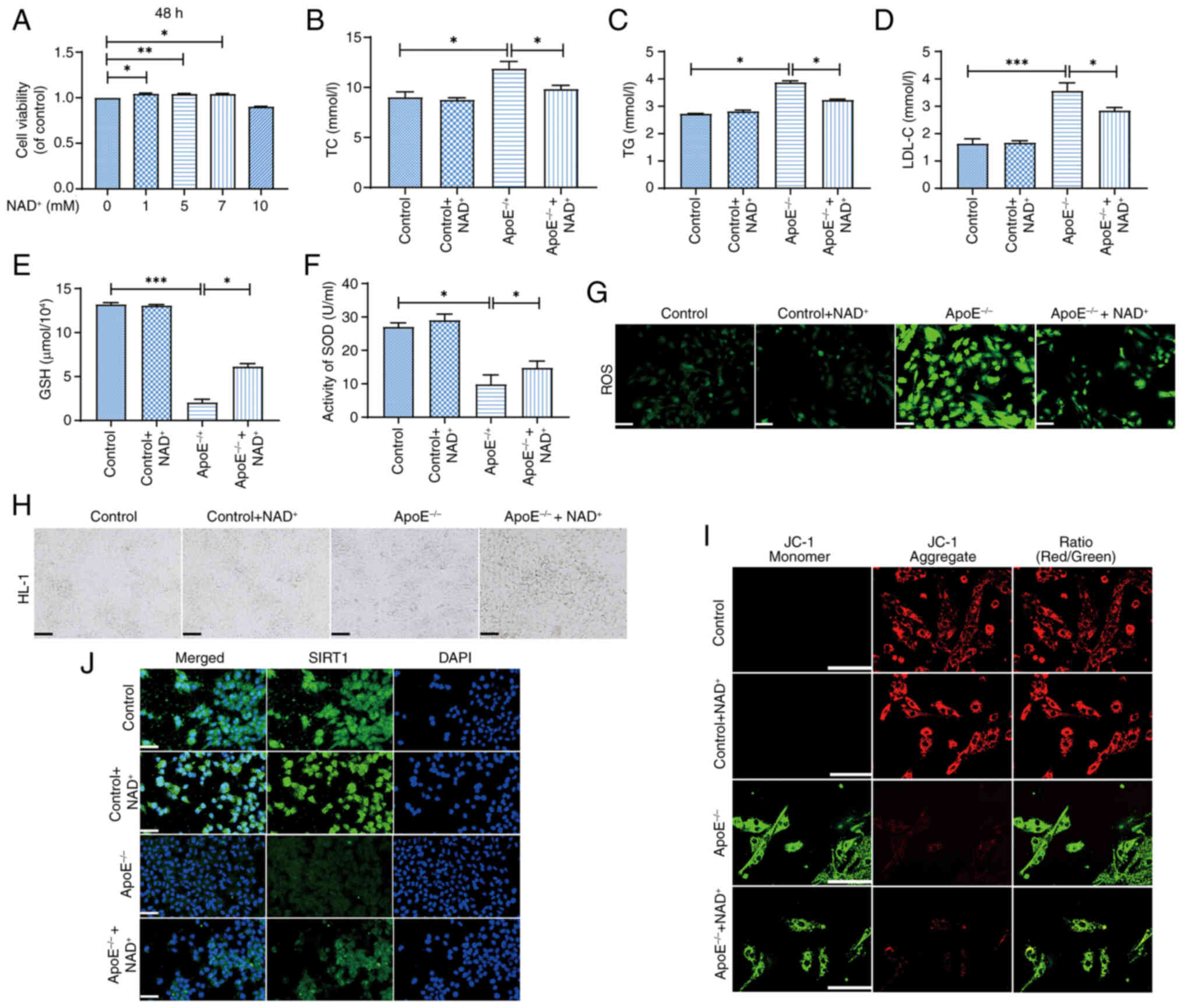 | Figure 6.NAD+ protects
ApoE−/− HL-1 cells from lipid accumulation and oxidative
stress. (A) Cell viability was significantly enhanced in the 5 mM
NAD+ group compared with that in the control group. (B)
NAD+ supplementation reduced TC levels in
ApoE−/−-treated cells. (C) TG levels were also
significantly decreased by NAD+ treatment. (D) LDL-C
levels were also reduced in the ApoE−/− +
NAD+ group, indicating improved lipid metabolism. (E)
NAD+ supplementation increased GSH levels, reflecting
enhanced antioxidant defense. (F) SOD activity was also
significantly elevated in the ApoE−/− + NAD+
group, suggesting improved oxidative stress response. (G) ROS
staining revealed reduced reactive oxygen species in the
ApoE−/− + NAD+ group, indicating a decrease
in oxidative stress. Scale bar, 200 µm. (H) Microscopic images
confirmed improved cell morphology and viability in the
ApoE−/− + NAD+ group. Scale bar, 100 µm. (I)
JC-1 staining showed improved mitochondrial membrane potential in
the ApoE−/− + NAD+ group. Scale bar, 100 µm.
(J) Immunofluorescence staining demonstrated increased expression
of SIRT1 in the ApoE−/− + NAD+ group,
suggesting activation of the SIRT1 pathway. Magnification, ×40;
scale bar, 50 µm. Data are presented as the mean ± standard error
of the mean (n=3); statistical analysis was performed using one-way
ANOVA followed by Tukey's post hoc test. *P<0.05, **P<0.01,
***P<0.001. ApoE−/−, apolipoprotein E-deficient; GSH,
glutathione; LDL-C, low-density lipoprotein cholesterol;
NAD+, nicotinamide adenine dinucleotide; ROS, reactive
oxygen species; SIRT1, sirtuin 1; SOD, superoxide dismutase; TC,
total cholesterol; TG, triglycerides. |
NAD+ enhances the
SIRT1/MFN2 pathway to protect ApoE−/− HL-1 cells through
SIRT1 upregulation
Fig. 7A shows
representative immunofluorescence images of MFN2 and SIRT1 double
staining, with colocalization observed in the cytoplasm of cells,
particularly in the ApoE−/−+NAD+ group,
indicating that NAD+ treatment promoted the
co-expression of these proteins. Furthermore, western blot analysis
showed increased levels of both MFN2 and SIRT1 in the
ApoE−/− + NAD+ group compared with those in
the ApoE−/− group, further supporting the
immunofluorescence results (Fig.
7B). Additionally, activation of the PI3K/AKT pathway was
observed in the ApoE−/− + NAD+
group, indicated by restored phosphorylation of PI3K and AKT and
suppression of the HFD-induced increase in p-mTOR, compared with
the ApoE−/− group. To further validate the
involvement of autophagy in vitro, HL-1 cells were treated
with PA), and the levels of LC3 II and P62 were assessed.
Consistent with the in vivo findings, PA treatment led to a
significant accumulation of both LC3 II and P62, suggesting
impaired autophagic flux (Fig.
S3B). However, NAD+ markedly reduced the levels of
LC3 II and P62 in PA-treated cells, indicating that NAD+
supplementation may restore autophagic degradation and enhance
autophagic flux under lipotoxic stress conditions. To determine
whether NAD+ supplementation activated SIRT1 rather than
merely increased its expression, HL-1 cells were treated with PA
and co-incubated with NAD+ and/or the SIRT1 inhibitor
EX-527. NAD+ treatment significantly increased SIRT1
expression and modulated p-mTOR levels altered by PA, indicative of
enhanced autophagic flux; this effect was abolished by EX-527
Fig. S5). These results suggested
that NAD+ may exert its protective effects through
functional activation of SIRT1.
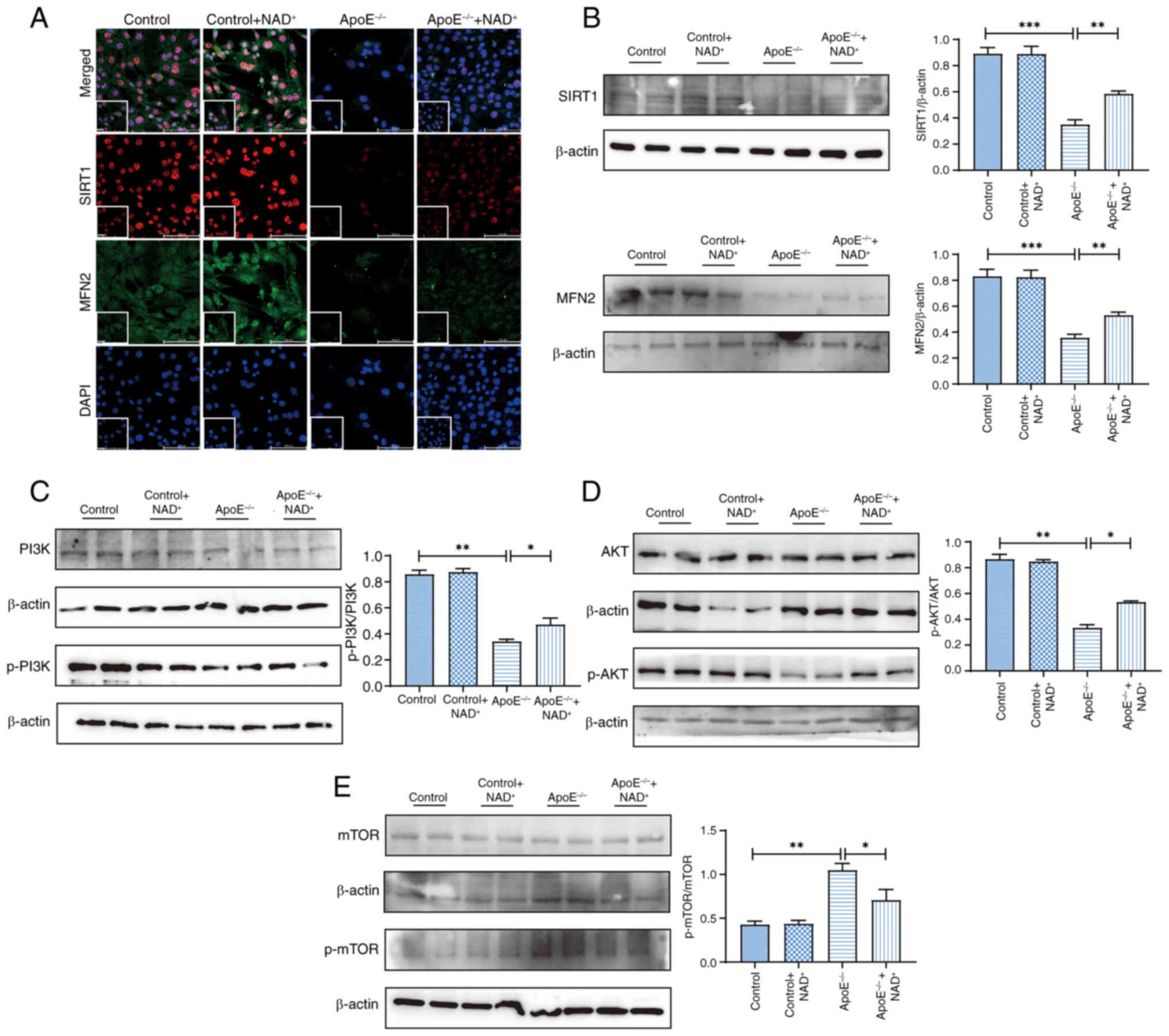 | Figure 7.NAD+ enhances MFN2/SIRT1
expression and activates the PI3K/AKT pathway in HL-1 cells. (A)
Representative immunofluorescence double staining showing
colocalization of MFN2 and SIRT1 in HL-1 cells. Magnification, ×40;
scale bars, 100 and 50 µm. Protein expression levels of (B) SIRT1
and MFN2, (C) PI3K and p-PI3K, (D) AKT and p-AKT, and (E) mTOR and
p-mTOR in HL-1 cells. Data are presented as the mean ± SEM (n=4);
statistical analysis was performed using one-way ANOVA followed by
Tukey's post hoc test. *P<0.05, **P<0.01, ***P<0.001.
ApoE−/−, apolipoprotein E-deficient; MFN2, mitofusin 2;
NAD+, nicotinamide adenine dinucleotide; p-,
phosphorylated; SIRT1, sirtuin 1. |
The present findings support the notion that
NAD+ could promote autophagy in cardiomyocytes via the
modulation of autophagic flux, potentially contributing to its
cardioprotective effects. In addition, it was suggested that
NAD+ supplementation not only modulated the MFN2/SIRT1
pathway but also activated the PI3K/AKT signaling pathway,
contributing to improved cellular function and mitochondrial
integrity. Notably, NAD+ did not produce significant
effects on autophagy- or metabolism-related markers in untreated
HL-1 cells, indicating that its regulatory role is specific to
lipotoxic or stress-induced conditions (Fig. S6).
Discussion
The present study systematically investigated the
effects of NAD+ supplementation and exercise
intervention on a model of HFD-induced cardiac injury, highlighting
their key roles in alleviating oxidative stress, correcting
metabolic dysfunction and modulating autophagy. ApoE−/−
mice were selected as a model of hyperlipidemia and
atherosclerosis. Owing to the lack of ApoE, these mice develop
spontaneous hypercholesterolemia and exhibit early cardiovascular
alterations under a HFD, closely resembling human cardiometabolic
disorders (28,29). Notably, in the present study
different HFD formulations and suppliers were used for each model;
however, both diets were selected based on their validation in the
literature for reliably inducing hyperlipidemia, obesity and
cardiovascular dysfunction in rodents (30). Moreover, both suppliers are
certified Chinese manufacturers whose products meet national
standards for laboratory animal feed production; although minor
compositional differences (such as fatty acid profiles and
micronutrients) between diet sources could theoretically affect
certain metabolic parameters, consistent phenotypic outcomes were
observed in terms of body weight gain, serum lipid levels and
cardiac pathology. These findings support the reproducibility and
reliability of both models.
The present HFD model offered a sensitive and
reproducible platform for evaluating interventions targeting lipid
metabolism, oxidative stress and myocardial injury. Notably,
ApoE−/− mice represent a hyperlipidemia-prone model with
increased susceptibility to cardiac injury (31). This enabled the present study to
delineate the cardioprotective effects of NAD+ under
metabolic stress, particularly via activation of the SIRT1/MFN2 and
PI3K/AKT/mTOR pathway. Future studies using wild-type mice should
be performed to determine whether NAD+ confers similar
benefits under physiological or less severe metabolic conditions,
thereby assessing its broader translational potential (32,33).
Unlike earlier studies relying on NAD+ precursors, the
approach of the present study directly demonstrated the therapeutic
value of NAD+ in lipid-induced myocardial injury
(34,35).
Exercise is a well-recognized non-pharmacological
intervention that improves metabolic homeostasis and cardiovascular
health, and it has been shown to enhance endogenous NAD+
biosynthesis via AMPK and NAMPT pathways (36,37).
In the present study, although the focus was on exogenous
NAD+ supplementation, the results revealed that HIIT
also elevated cardiac NAD+ levels, and alleviated
oxidative stress, fibrosis and myocardial injury in
ApoE−/− mice. These results underscore the potential of
exercise to mitigate hyperlipidemia-induced cardiac damage,
partially through NAD+-related mechanisms. The findings
demonstrated that NAD+ markedly improved mitochondrial
integrity and reduced excessive ROS accumulation potentially by
activating the SIRT1/MFN2 pathway (Fig. 8), thereby protecting cardiomyocytes
from metabolic stress-induced damage. Furthermore, the combination
of NAD+ and HIIT may amplify these protective effects by
enhancing tissue NAD+/NADH ratios and increasing the
phosphorylation levels of PI3K and AKT, thereby modulating the
PI3K/AKT/mTOR signaling pathway and providing a multilayered
cardioprotective mechanism at both molecular and cellular levels.,
providing a multilayered cardioprotective mechanism at both
molecular and cellular levels. Although prior studies have explored
the independent roles of NAD+ and exercise in cardiac
metabolic regulation (38–40), the present study provides
preliminary evidence suggesting that NAD+
supplementation combined with HIIT may exert complementary
protective effects against HFD-induced cardiac damage, offering
potential theoretical insights and therapeutic implications for
metabolic cardiomyopathy.
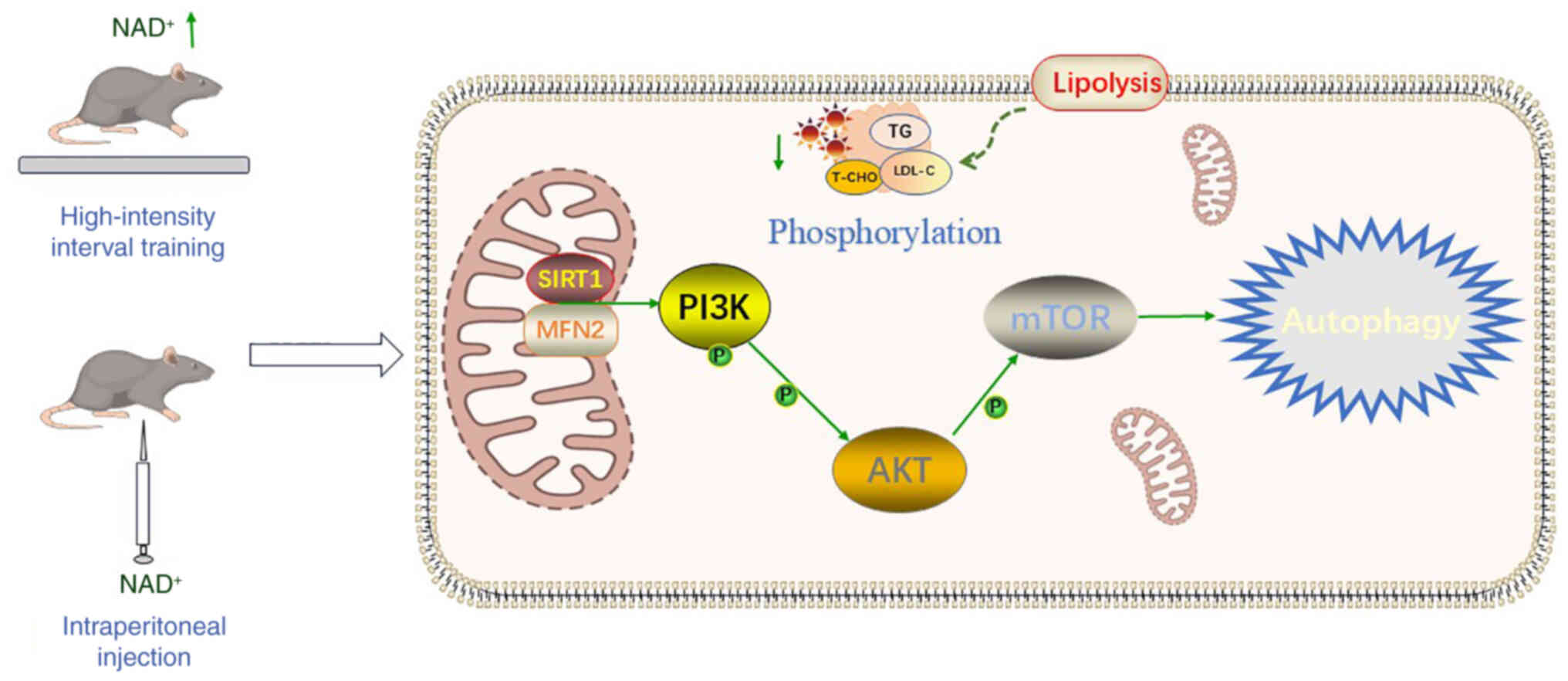 | Figure 8.Mechanistic overview of
NAD+ in improving HFD-induced cardiac dysfunction.
High-intensity interval training elevates cardiac NAD+
levels, and subsequent intraperitoneal NAD+
supplementation further activates SIRT1/MFN2 signaling. This
enhances PI3K/AKT/mTOR phosphorylation, promoting autophagy and
mitochondrial quality control. Interventions also improve lipid
metabolism, reducing TG, T-CHO, and LDL-C. Together, these
mechanisms mitigate mitochondrial dysfunction, and metabolic
disturbances in HFD-induced cardiac injury. LDL-C, low-density
lipoprotein cholesterol; MFN2, mitofusin 2; NAD+,
nicotinamide adenine dinucleotide; T-CHO, total cholesterol; TG,
triglycerides; SIRT1, sirtuin 1. |
SIRT1, a NAD+-dependent deacetylase,
serves a central role in regulating mitochondrial function,
oxidative stress and metabolic homeostasis (41). NAD+ supplementation
markedly upregulated SIRT1 expression, thereby attenuating
oxidative damage through enhanced mitochondrial quality control and
reduced ROS generation. Additionally, SIRT1 deacetylates MFN2, a
critical protein in mitochondrial fusion, stabilizing its function
under metabolic stress conditions (42). In addition to the involvement of
the SIRT1/MFN2 pathway, the present study demonstrated that
exercise increased NAD+ levels, which subsequently
activated the PI3K/AKT/mTOR signaling pathway via SIRT1/MFN2. This
pathway is known to play a crucial role in regulating lipid
accumulation, fibrosis, and metabolic homeostasis (43). Excessive activation of this pathway
in HFD models has been associated with exacerbated cardiac
remodeling. By suppressing this signaling cascade, NAD+
and exercise intervention effectively reduced myocardial fibrosis
and lipid deposition. These findings align with previous metabolic
studies and highlight the beneficial effects of NAD+
supplementation and exercise in improving metabolic cardiomyopathy
(44,45).
The present study highlighted the distinct effects
of HIIT and MICT on cardiac metabolic regulation. HIIT demonstrated
superior benefits in increasing NAD+ levels and reducing
oxidative stress compared with MICT. The acute metabolic stress
induced by HIIT may trigger the activation of NAD+
biosynthetic enzymes such as NAMPT, thereby amplifying NAD
availability and improving mitochondrial function (46,47).
These findings underscore the potential of HIIT as a potent
exercise modality for enhancing cardiac resilience in metabolic
conditions. From a clinical perspective, the specificity of
exercise interventions highlights the importance of tailoring
training regimens to optimize therapeutic outcomes. Future studies
should further explore the dose-response relationship of exercise
intensity and NAD+ supplementation in improving cardiac
function.
The involvement of SIRT1/MFN2 and PI3K/AKT/mTOR
pathways offers mechanistic insights in heart and liver (42,48–50).
The present findings build upon and expand the existing knowledge
on the independent effects of NAD+ and exercise in
cardiac metabolic regulation. Previous studies have predominantly
investigated their individual effects. This dual approach amplifies
the benefits, providing a more comprehensive understanding of how
metabolic and mechanical interventions can jointly enhance cardiac
health. To further validate the signaling relationship, in
vitro experiments were performed using the specific SIRT1
inhibitor EX-527 (51). Treatment
with EX-527 significantly attenuated NAD+-induced
activation of the p-mTOR mTOR pathway, indicating that SIRT1 is an
upstream regulator. These results confirm that the PI3K/AKT/mTOR
pathway functions downstream of the SIRT1/MFN2 pathway. The
additional experiments using EX-527 confirmed that SIRT1 modulates
the PI3K/AKT/mTOR pathway in response to NAD+,
reinforcing the proposed SIRT1/MFN2-PI3K/AKT/mTOR signaling cascade
involved in cardioprotection. Moreover, exogenous NAD+
supplementation reduced the levels of LC3 II and P62 in the
hyperlipidemia model, indicating that autophagic degradation may be
restored and autophagic flow enhanced after NAD+
supplementation. Therefore, it could be hypothesized that
NAD+ treatment promotes the maturation of autophagosomes
and enhances the fusion of autophagosomes with lysosomes, thereby
improving autophagic flow and promoting autophagic degradation in
hyperlipidemia-induced heart injury. These findings establish a
foundation for future translational studies exploring the broader
applications of NAD+ and exercise intervention.
In conclusion, the present study demonstrated that
NAD+ supplementation and exercise, particularly HIIT,
independently protect against HFD-induced cardiac damage by
targeting oxidative stress, mitochondrial dysfunction, and
metabolic disturbances. The protective effects of NAD+
in HFD-induced cardiac injury were mediated primarily via
activation of the SIRT1/MFN2 pathway and the PI3K/AKT/mTOR pathway.
These findings provide new insights into the prevention and
treatment of metabolic cardiomyopathy, supporting further research
and potential clinical applications.
Supplementary Material
Supporting Data
Acknowledgements
Not applicable.
Funding
The present study was funded by the Central Hospital of Dalian
University of Technology ‘Climbing Plan’ (grant no. 2023ZZ008).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZP designed the study. SG and WY performed the
experiments. ZP, SG and WY analyzed the data. ZP and SG confirm the
authenticity of all the raw data. SG and WY drafted and wrote the
manuscript. JY and YL analyzed the data and revised the manuscript.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All animal experiments were performed in accordance
with the Guide for the Care and Use of Laboratory Animals and were
approved by the Ethics Committee of Dalian Municipal Central
Hospital (grant no. YN2023-057-55).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kotseva K, Jennings C, Bassett P, Adamska
A, Hobbs R and Wood D; ASPIRE-3-PREVENT Investigators, : Challenge
of cardiovascular prevention in primary care: Achievement of
lifestyle, blood pressure, lipids and diabetes targets for primary
prevention in England-results from ASPIRE-3-PREVENT cross-sectional
survey. Open Heart. 11:e0027042024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han S, Kim NR, Kang JW, Eun JS and Kang
YM: Radial BMD and serum CTX–I can predict the progression of
carotid plaque in rheumatoid arthritis: A 3-year prospective cohort
study. Arthritis Res Ther. 23:2582021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zheng L, Than A, Zan P, Li D, Zhang Z,
Leow MKS and Chen P: Mild-photothermal and nanocatalytic therapy
for obesity and associated diseases. Theranostics. 14:5608–5620.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Narasimhulu CA and Singla DK: BMP-7
Attenuates sarcopenia and adverse muscle remodeling in diabetic
mice via alleviation of lipids, inflammation, HMGB1, and
pyroptosis. Antioxidants (Basel). 12:3312023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Galluzzi L, Vitale I, Aaronson SA, Abrams
JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews
DW, et al: Molecular mechanisms of cell death: Recommendations of
the Nomenclature committee on cell death 2018. Cell Death Differ.
25:486–541. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shoji S and Mentz RJ: Beyond quadruple
therapy: The potential roles for ivabradine, vericiguat, and
omecamtiv mecarbil in the therapeutic armamentarium. Heart Fail
Rev. 29:949–955. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bentivegna E, Galastri S, Onan D and
Martelletti P: Unmet needs in the acute treatment of migraine. Adv
Ther. 41:1–13. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Perryman R, Chau TW, De-Felice J, O'Neill
K and Syed N: Distinct capabilities in NAD metabolism mediate
resistance to NAMPT inhibition in glioblastoma. Cancers (Basel).
16:20542024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abdellatif M, Sedej S and Kroemer G:
NAD+ metabolism in cardiac health, aging, and disease.
Circulation. 144:1795–1817. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chu X and Raju RP: Regulation of NAD(+)
metabolism in aging and disease. Metabolism. 126:1549232022.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lin Q, Zuo W, Liu Y, Wu K and Liu Q:
NAD(+) and cardiovascular diseases. Clin Chim Acta. 515:104–110.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yoshino M, Yoshino J, Kayser BD, Patti GJ,
Franczyk MP, Mills KF, Sindelar M, Pietka T, Patterson BW, Imai SI
and Klein S: Nicotinamide mononucleotide increases muscle insulin
sensitivity in prediabetic women. Science. 372:1224–1229. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cheng L, Deepak RNVK, Wang G, Meng Z, Tao
L, Xie M, Chi W, Zhang Y, Yang M, Liao Y, et al: Hepatic
mitochondrial NAD + transporter SLC25A47 activates AMPKα mediating
lipid metabolism and tumorigenesis. Hepatology. 78:1828–1842. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lopaschuk GD, Karwi QG, Tian R, Wende AR
and Abel ED: Cardiac energy metabolism in heart failure. Circ Res.
128:1487–1513. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Doan KV, Luongo TS, Ts'olo TT, Lee WD,
Frederick DW, Mukherjee S, Adzika GK, Perry CE, Gaspar RB, Walker
N, et al: Cardiac NAD+ depletion in mice promotes
hypertrophic cardiomyopathy and arrhythmias prior to impaired
bioenergetics. Nat Cardiovasc Res. 3:1236–1248. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Qiu Y, Xu S, Chen X, Wu X, Zhou Z, Zhang
J, Tu Q, Dong B, Liu Z, He J, et al: NAD(+) exhaustion by CD38
upregulation contributes to blood pressure elevation and vascular
damage in hypertension. Signal Transduct Target Ther. 8:3532023.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Zhang C, Hu Y, Peng J, Feng Q and Hu
X: Nicotinamide enhances Treg differentiation by promoting Foxp3
acetylation in immune thrombocytopenia. Br J Haematol.
205:2432–2441. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pei Z, Wang F, Wang K and Wang L:
Nicotinamide adenine dinucleotide in the development and treatment
of cardiac remodeling and aging. Mini Rev Med Chem. 22:2310–2317.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Fritzen AM, Lundsgaard AM and Kiens B:
Tuning fatty acid oxidation in skeletal muscle with dietary fat and
exercise. Nat Rev Endocrinol. 16:683–696. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Qi XM, Qiao YB, Zhang YL, Wang AC, Ren JH,
Wei HZ and Li QS: PGC-1α/NRF1-dependent cardiac mitochondrial
biogenesis: A druggable pathway of calycosin against triptolide
cardiotoxicity. Food Chem Toxicol. 171:1135132023. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang H, Wang Y, Wu K, Liu R, Wang H, Yao
Y, Kvietys P and Rui T: miR-141 impairs mitochondrial function in
cardiomyocytes subjected to hypoxia/reoxygenation by targeting
Sirt1 and MFN2. Exp Ther Med. 24:7632022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ji LL and Yeo D: Maintenance of NAD+
homeostasis in skeletal muscle during aging and exercise. Cells.
11:7102022. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
National Research Council Committee for
the Update of the Guide for the C. A. Use of Laboratory. The
National Academies Collection, . Reports funded by National
Institutes of Health, in Guide for the Care and Use of Laboratory
Animals. National Academies Press; Washington, DC: 2011
|
|
24
|
Nishida Y, Nawaz A, Kado T, Takikawa A,
Igarashi Y, Onogi Y, Wada T, Sasaoka T, Yamamoto S, Sasahara M, et
al: Astaxanthin stimulates mitochondrial biogenesis in insulin
resistant muscle via activation of AMPK pathway. J Cachexia
Sarcopenia Muscle. 11:241–258. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brault V, Duchon A, Romestaing C, Sahun I,
Pothion S, Karout M, Borel C, Dembele D, Bizot JC, Messaddeq N, et
al: Opposite phenotypes of muscle strength and locomotor function
in mouse models of partial trisomy and monosomy 21 for the proximal
Hspa13-App region. PLoS Genet. 11:e10050622015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang L, Lavier J, Hua W, Wang Y, Gong L,
Wei H, Wang J, Pellegrin M, Millet GP and Zhang Y: High-Intensity
interval training and moderate-intensity continuous training
attenuate oxidative damage and promote myokine response in the
skeletal muscle of ApoE KO mice on high-fat diet. Antioxidants
(Basel). 10:9922021. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pei Z, Li Y, Yao W, Sun F and Pan X:
NAD+ Protects against hyperlipidemia-induced kidney
injury in apolipoprotein E-deficient mice. Cur Pharm Biotechnol.
25:488–498. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aravani D, Kassi E, Chatzigeorgiou A and
Vakrou S: Cardiometabolic syndrome: An update on available mouse
models. Thromb Haemost. 121:703–715. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Poledne R and Jurčíková-Novotná L:
Experimental models of hyperlipoproteinemia and atherosclerosis.
Physiol Res. 66 (Suppl 1):S69–S75. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park Y, Jang I, Park HY, Kim J and Lim K:
Hypoxic exposure can improve blood glycemic control in high-fat
diet-induced obese mice. Phys Act Nutr. 24:19–23. 2020. View Article : Google Scholar
|
|
31
|
Zhao Y, Qu H, Wang Y, Xiao W, Zhang Y and
Shi D: Small rodent models of atherosclerosis. Biomed Pharmacother.
129:1104262020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhao Y, Zhang J, Zheng Y, Zhang Y, Zhang
XJ, Wang H, Du Y, Guan J, Wang X and Fu J: NAD+ improves
cognitive function and reduces neuroinflammation by ameliorating
mitochondrial damage and decreasing ROS production in chronic
cerebral hypoperfusion models through Sirt1/PGC-1α pathway. J
Neuroinflammation. 18:2072021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo C, Huang Q, Wang Y, Yao Y, Li J, Chen
J, Wu M, Zhang Z, Mingyao E, Qi H, et al: Therapeutic application
of natural products: NAD+ metabolism as potential
target. Phytomedicine. 114:1547682023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Trueblood NA, Ramasamy R, Wang LF and
Schaefer S: Niacin protects the isolated heart from
ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol.
279:H764–H771. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Perry CE, Halawani SM, Mukherjee S, Ngaba
LV, Lieu M, Lee WD, Davis JG, Adzika GK, Bebenek AN, Bazianos DD,
et al: NAD+ precursors prolong survival and improve cardiac
phenotypes in a mouse model of Friedreich's Ataxia. JCI Insight.
9:e1771522024.PubMed/NCBI
|
|
36
|
Chong MC, Silva A, James PF, Wu SSX and
Howitt J: Exercise increases the release of NAMPT in extracellular
vesicles and alters NAD+ activity in recipient cells.
Aging Cell. 21:e136472022. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Morales-Alamo D and Calbet JAL: AMPK
signaling in skeletal muscle during exercise: Role of reactive
oxygen and nitrogen species. Free Radic Biol Med. 98:68–77. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Glancy B, Kane DA, Kavazis AN, Goodwin ML,
Willis WT and Gladden LB: Mitochondrial lactate metabolism: History
and implications for exercise and disease. J Physiol. 599:863–888.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Agorrody G, Peclat TR, Peluso G, Gonano
LA, Santos L, van Schooten W, Chini CCS, Escande C, Chini EN and
Contreras P: Benefits in cardiac function by CD38 suppression:
Improvement in NAD+ levels, exercise capacity, heart
rate variability and protection against catecholamine-induced
ventricular arrhythmias. J Mol Cell Cardiol. 166:11–22. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cabrera ME, Zhou L, Stanley WC and Saidel
GM: Regulation of cardiac energetics: role of redox state and
cellular compartmentation during ischemia. Ann N Y Acad Sci.
1047:259–270. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen Y, Zhang H, Ji S, Jia P, Chen Y, Li Y
and Wang T: Resveratrol and its derivative pterostilbene attenuate
oxidative stress-induced intestinal injury by improving
mitochondrial redox homeostasis and function via SIRT1 signaling.
Free Radic Biol Med. 177:1–14. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hu L, Guo Y, Song L, Wen H, Sun N, Wang Y,
Qi B, Liang Q, Geng J, Liu X, et al: Nicotinamide riboside promotes
Mfn2-mediated mitochondrial fusion in diabetic hearts through the
SIRT1-PGC1α-PPARα pathway. Free Radic Biol Med. 183:75–88. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wu YC, Yan Q, Yue SQ, Pan LX, Yang DS, Tao
LS, Wei ZY, Rong F, Qian C, Han MQ, et al: NUP85 alleviates lipid
metabolism and inflammation by regulating PI3K/AKT signaling
pathway in nonalcoholic fatty liver disease. Int J Biol Sci.
20:2219–2235. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Yu H, Gan D, Luo Z, Yang Q, An D, Zhang H,
Hu Y, Ma Z, Zeng Q, Xu Z and Ren D: α-Ketoglutarate improves
cardiac insufficiency through NAD(+)-SIRT1 signaling-mediated
mitophagy and ferroptosis in pressure overload-induced mice. Mol
Med. 30:152024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ma Y, Kuang Y, Bo W, Liang Q, Zhu W, Cai M
and Tian Z: Exercise training alleviates cardiac fibrosis through
increasing fibroblast growth factor 21 and regulating
TGF-β1-Smad2/3-MMP2/9 signaling in mice with myocardial infarction.
Int J Mol Sci. 22:123412021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Walzik D, Joisten N, Schenk A, Trebing S,
Schaaf K, Metcalfe AJ, Spiliopoulou P, Hiefner J, McCann A, Watzl
C, et al: Acute exercise boosts NAD(+) metabolism of human
peripheral blood mononuclear cells. Brain Behav Immun.
123:1011–1023. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yagi M, Toshima T, Amamoto R, Do Y, Hirai
H, Setoyama D, Kang D and Uchiumi T: Mitochondrial translation
deficiency impairs NAD(+) -mediated lysosomal acidification. EMBO
J. 40:e1052682021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li J, Wang T, Liu P, Yang F, Wang X, Zheng
W and Sun W: Hesperetin ameliorates hepatic oxidative stress and
inflammation via the PI3K/AKT-Nrf2-ARE pathway in oleic
acid-induced HepG2 cells and a rat model of high-fat diet-induced
NAFLD. Food Funct. 12:3898–3918. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Savova MS, Mihaylova LV, Tews D, Wabitsch
M and Georgiev M: Targeting PI3K/AKT signaling pathway in obesity.
Biomed Pharmacother. 159:1142442023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Magaye RR, Savira F, Hua Y, Xiong X, Huang
L, Reid C, Flynn BL, Kaye D, Liew D and Wang BH: Attenuating
PI3K/Akt- mTOR pathway reduces dihydrosphingosine 1 phosphate
mediated collagen synthesis and hypertrophy in primary cardiac
cells. Int J Biochem Cell Biol. 134:1059522021. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Wawruszak A, Luszczki J, Bartuzi D,
Kalafut J, Okon E, Czerwonka A and Stepulak A: Selisistat, a SIRT1
inhibitor, enhances paclitaxel activity in luminal and
triple-negative breast cancer: In silico, in vitro, and in vivo
studies. J Enzyme Inhib Med Chem. 40:24585542025. View Article : Google Scholar : PubMed/NCBI
|














