Introduction
Tuberculosis (TB) is an infectious disease caused by
Mycobacterium tuberculosis (MTB) infection. Globally, there
are ~10 million new cases of TB and 1.6 million TB-related deaths
annually, making it the leading source of infection and one of the
top ten causes of death. Notably, China is one of 30 countries with
a high morbidity rate of TB (1).
The Chinese Center for Disease Control and Prevention has shown
that the geographical distribution of TB epidemics in China is
characterized by low incidence in the eastern region and high
prevalence in the western region, and the incidence rate of TB is
higher in the Xinjiang region than in other provinces (2,3). The
average rate of culture-positive TB is 119/100,000 people in China,
whereas it is 433/100,000 in the Xinjiang region (4). The high prevalence rate of TB in
Xinjiang may be due to unique climatic, geographic and demographic
characteristics (5), as well as
the high incidence of HIV infection, the growing floating
population, unhealthy habits and poor living conditions in rural
areas; however, the exact reason is unclear.
MTB is an intracellular pathogen and macrophages are
major innate immune cells, which are the first line of defense
against MTB infection and the primary effector cells for clearing
this pathogen (6,7). To avoid becoming reservoirs of MTB,
host macrophages have evolved a number of defense mechanisms,
including autophagy (8) and
apoptosis (9), to kill or prevent
the growth of MTB. Immune cell apoptosis is an important mechanism
against bacterial and viral infections, and some studies have
suggested that macrophage apoptosis induced by MTB infection can
kill MTB (10,11). However, other studies have
suggested that excessive apoptosis is conducive to MTB survival
(12,13). Thus, a clear understanding of the
interaction of MTB with host macrophages may help develop targeted
strategies for controlling TB.
Cyclin-dependent kinase 1 (CDK1) is a member of the
CDK protein family, and it controls all aspects of cell division,
including cell cycle entry from quiescence, G1/S phase
transition, DNA replication in the S phase, nuclear rupture,
chromosome cohesion and segregation, and cytoplasmic division
(14–16). CDK1 is a proline-directed kinase
that phosphorylates a large number of proteins to drive cell cycle
progression and specific processes associated with different cell
cycle phases (15). Huang et
al (17) showed that CDK1 can
regulate the degree of cell cycle arrest during the G2/M
phase. Furthermore, previous studies have shown a close association
between CDK1 and tumor cell apoptosis in glioblastoma (18), human hepatocellular carcinoma
(19), cervical carcinoma
(20), colorectal carcinoma
(21,22) and ovarian carcinoma (23). However, the role of CDK1 in the
immune response of macrophages after TB infection is unclear.
The transcription factor p53, once activated by
phosphorylation and acetylation, binds directly to specific DNA
sequences in the promoter region of target genes to regulate their
expression (24,25). Notably, p53 target genes are
involved in the regulation of apoptosis, DNA repair, cell cycle
arrest, senescence and programmed cell death (25–27).
It has been shown that promoting phosphorylation of the p53 protein
can enhance macrophage apoptosis and suppress the survival of
MTB-infected macrophages (28).
The present study utilized bioinformatics analysis
to investigate the effects of the CDK1 gene on the apoptosis and
cell cycle progression of macrophages infected with different
sources of MTB in vitro. The current study aimed to reveal
the specific immune response involving CDK1 and phosphorylated
(p)-p53 in macrophages after infection with MTB clinical isolates,
as well as the mechanism by which CDK1 and p-p53 regulate
macrophage cycle arrest and apoptosis after infection with MTB from
Xinjiang (XJMTB). CDK1 and p-p53 may be considered novel treatment
targets for TB in Xinjiang and lay the foundation for formulating
TB treatment strategies.
Materials and methods
Sample collection, and MTB
resuscitation, isolation and culture
The sputum samples of 20 cases were derived from 103
patients with sputum smear-positive TB in the Xinjiang region
during a previous epidemiological survey conducted by the
cooperative team (29). All cases
were diagnosed according to China's national diagnostic criteria
(30). Demographic,
epidemiological and clinical data were obtained from the medical
records of the patients using a unified epidemiological survey
methodology. Table I provides the
details of the 20 patients.
 | Table I.Patient's basic information. |
Table I.
Patient's basic information.
| Number | Sex | Age, years | Ethnicity | Separation and
amplification | TCH | PNB |
|---|
| L1 | Male | 35 | Han | Yes | + | + |
| L2 | Male | 32 | Uighur | Yes | - | - |
| L3 | Male | 25 | Uighur | Yes | + | - |
| L4 | Female | 52 | Han | Yes | + | - |
| L5 | Female | 38 | Han | Yes | + | - |
| L6 | Male | 42 | Uighur | Yes | + | - |
| L7 | Male | 56 | Han | Yes | + | - |
| L8 | Female | 43 | Uighur | Yes | + | - |
| L9 | Male | 44 | Uighur | Yes | + | - |
| L10 | Male | 47 | Han | No | / | / |
| L11 | Female | 37 | Uighur | No | / | / |
| L12 | Male | 55 | Uighur | Yes | + | - |
| L13 | Female | 28 | Uighur | No | / | / |
| L14 | Male | 37 | Han | Yes | + | - |
| L15 | Female | 45 | Han | No | / | / |
| L16 | Male | 29 | Uighur | Yes | + | - |
| L17 | Female | 26 | Han | Yes | + | - |
| L18 | Male | 53 | Han | Yes | + | - |
| L19 | Male | 28 | Uighur | Yes | - | - |
| L20 | Female | 57 | Han | Yes | + | - |
The sputum was mixed with 1–3 times the volume of a
4% NaOH (Beijing Chemical Works) solution, shaken well until
dissolved and maintained at room temperature for 20 min. Once the
sample was completely liquefied, 100 µl sample was aspirated and
dropped evenly on the slant of modified acidic Roche's medium
(Celnovte Biotechnology Co., Ltd.). Each sample was inoculated
three times and placed in a 37°C incubator. After a single white
colony was grown, it was picked off, placed in a 37°C incubator
containing 40 ml Mycobacterium liquid medium (Shanghai
Jingnuo Biotechnology Co., Ltd.) and shaken at 120 rpm for
amplification. In addition, 50–100 µl frozen H37Rv (cat. no. 27294;
American Type Culture Collection) bacterial solution was aspirated,
added to 40 ml Mycobacterium liquid medium and amplified by
shaking at 120 rpm.
Acid-fast staining
An inoculation loop was dipped into the XJMTB
amplified bacterial solution, applied onto a slide and fixed by
heating alone. Subsequently, carbol fuchsin was added dropwise to
the solution, the solution was slowly heated (but not boiled) for
3–5 min and washed with water. Hydrochloric acid-ethanol solution
(3%) was then added dropwise to the solution to decolorize it for
30 sec. Finally, methylene blue was added dropwise to the solution
for 0.5–1 min, after which the slide was washed, dried and observed
under an optical microscope with oil immersion.
Strain typing and identification of
clinical isolates of MTB
Briefly, 100 µl amplified bacilli were dropped
evenly onto para-nitrobenzoic acid (PNB) and thiophene-2-carboxylic
acid hydrazide (TCH) media (Celnovte Biotechnology Co., Ltd.), with
each sample inoculated three times, and cultured at 37°C with 5%
CO2. Observations were made weekly, and the results were
determined after 4 weeks. A single colony growing on the culture
medium indicates a positive result, with negative PNB and positive
TCH considered MTB infection.
For strain typing, the DNA of MTB was extracted
using a bacterial DNA extraction kit (cat. no. R0017S; Beyotime
Institute of Biotechnology), and the MIRU 26 fragment was amplified
using a PCR kit with Taq (cat. no. D7232; Beyotime Institute of
Biotechnology). The PCR thermocycling conditions were as follows:
i) 94°C for 3 min (1 cycle); ii) 30 cycles at 94°C for 30 sec, 55°C
for 30 sec and 72°C for 60 sec; iii) 72°C for 10 min (1 cycle), iv)
maintained at 4°C. The MIRU 26 primer sequences were: Forward
primer 5′-3′: TAGGTCTACCGTCGAAATCTGTGAC, reverse primer 5′-3′:
CATAGGCGACCAGGCGAATAG. Agarose (cat. no. ST118; Beyotime Institute
of Biotechnology) and Gel-Green (cat. no. D0143; Beyotime Institute
of Biotechnology) were used to prepare 1% agarose gel, and the PCR
products were electrophoresed at 130 V and observed using a
Tanon-1600 Gel Image system (Tianneng Science and Technology Co.,
Ltd.).
Cell culture and modeling of MTB
infection
The THP-1 human monocyte cell line was purchased
from The Cell Bank of Type Culture Collection of The Chinese
Academy of Sciences. The cells were inoculated at a concentration
of 1×106 cells/ml in 6-well plates (1 ml cell
suspension/well) in RPMI 1640 medium (Gibco; Thermo Fisher
Scientific, Inc.) supplemented with 10% fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), and serum-free RPMI 1640 medium
with an equal amount of phorbol-12-myristate-13-acetate (PMA;
MedChemExpress) at a concentration of 400 ng/ml was added to induce
cell differentiation into macrophages for 24 h. After removing the
culture medium containing PMA, an equal volume of fresh RPMI 1640
complete culture medium was added. H37Rv and XJMTB were diluted to
an OD600 of 0.6 in Mycobacterium liquid medium, and 100 µl
of the fluid was added to each well to infect cells for 12, 24 and
36 h. Finally, MTB samples isolated from the sputum samples of
patients numbered L3-L6 (Beijing family) were used for L group
modeling in the current study. H37Rv was used for V group modeling
in the current study. THP-1 cells used for transcriptome sequencing
were divided into three groups: XJMTB clinical isolate-infected
group (L group), H37Rv standard strain-infected group (V group) and
blank control group not infected with any bacteria (C group). All
cell culture conditions were as follows: 37°C, 5%
CO2.
Enzyme-linked immunosorbent assay
(ELISA)
Culture supernatants were centrifuged at 300 × g for
10 min at room temperature. Subsequently, the secretion levels of
cytokines in the cell culture supernatants were measured using
sandwich ELISA detection kits for IL-12p70 (cat. no. EK112), TNF-α
(cat. no. EK182), IL-6 (cat. no. EK1153), IL-1β (cat. no. EK101B)
and IL-10 (cat. no. EK110) [Multisciences (Lianke) Biotech, Co.,
Ltd.]. All operations were performed according to the
manufacturer's protocol. The absorbance was measured using a
microplate reader at 450 nm and compared to a standard curve.
Nitric oxide (NO) measurement
assay
To evaluate NO levels during MTB infection, cell
culture supernatants were analyzed using the Griess assay. Culture
supernatants were centrifuged at 300 × g for 10 min at room
temperature. Subsequently, the supernatant (100 µl) was incubated
with Griess reagent (cat. no. G4410; Sigma-Aldrich; Merck KGaA)
(100 µl) at room temperature for 10 min, and the absorbance was
then measured at 541 nm. Sodium nitrite was used to create a
standard concentration curve.
RNA isolation and library
preparation
Total RNA was extracted from the aforementioned
three groups of THP-1 cells using Trizol reagent (cat. no. R0016;
Beyotime Institute of Biotechnology). The extracted RNA was
quantified and assessed for purity and integrity using a NanoDrop
2000 spectrophotometer (NanoDrop; Thermo Fisher Scientific, Inc.)
and an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.).
Subsequently, library construction was performed using TruSeq
Stranded Total RNA Library Prep Kit with Ribo-Zero Gold (cat. no.
RS-122-2301; Illumina, Inc.). The amount of RNA used to construct
the library was 1 µg. Ribosomal RNA (rRNA) removal was performed
using the Ribo-Zero Gold rRNA Removal Kit (Illumina, Inc.).
Subsequently, sequencing libraries were constructed using
rRNA-depleted samples. Finally, products were purified (Agencourt
AMPure XP; Beckman Coulter, Inc.), and library quality was assessed
using the Agilent 2100 Bioanalyzer system.
Transcriptome sequencing and
differentially expressed gene (DEG) analysis
The libraries were sequenced on the Illumina HiSeq X
Ten platform (Illumina, Inc.) to produce 150 bp paired-end reads.
In this step, clean data (clean reads) were obtained by removing
reads containing adapters and ploy-N (sequencing errors or
insufficient sequencing quality) or low-quality reads from the raw
data. Sequencing reads were mapped to the human genome (GRCh38)
using HISAT2 (31). For mRNA, FPKM
was calculated for each gene using Cufflinks (1.2) (32), and read counts were obtained for
each gene using HTSeq-Count (0.12.3) (33). RNA expression profiling data were
analyzed using the R (3.6.3) (https://www.r-project.org/) software package DESeq2
(1.26.0) (https://bioconductor.org/packages/release/bioc/html/DESeq2.html).
The package pheatmap (1.0.12) (https://cran.r-project.org/package=pheatmap) in R
(3.6.3) was used to create heat maps, whereas the package ggplot2
(3.3.5) (https://cran.r-project.org/web/packages/ggplot2/index.html)
was used to create volcano maps. Bioinformatics & Evolutionary
Genomics (http://bioinformatics.psb.ugent.be/webtools/Venn/) was
used for creating Venn diagrams. Principal component analysis (PCA)
was performed using the OECloud tools at https://cloud.oebiotech.com. The |log2 fold change|≥1
and P≤0.05 were used as criteria to screen significant DEGs for
subsequent analysis. STRING (https://cn.string-db.org/cgi/input?sessionId=bTEPyYc5wXuA&input_page_show_search=off)
was used to perform the protein-protein interaction (PPI) network
analysis, and Cytoscape (3.8.1) (https://cytoscape.org/) and its MCODE plugin (34) was used to perform subnetwork
screening and confirmation.
Gene ontology (GO) and kyoto
encyclopedia of genes and genomes (KEGG) enrichment analyses
The Database for Annotation, Visualization, and
Integrated Discovery (https://davidbioinformatics.nih.gov/) was used for
functional analysis of DEGs. The significant GO biological
processes and KEGG pathways with an adjusted P<0.05 were
selected to analyze their biological function. The significant
enrichment results were visualized using the ggplot2 (3.3.5)
(https://cran.r-project.org/web/packages/ggplot2/index.html)
package of R software (3.6.3).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from cultured cells using
TRIzol reagent. mRNA was reverse transcribed into cDNA using the
HiScript II 1st Strand cDNA Synthesis Kit (Vazyme Biotech Co.,
Ltd.), according to manufacturer's protocol. qPCR was performed to
measure mRNA using the ABI 7300 Plus Real-Time PCR System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) using the FastStart
Universal SYBR Green Master Mix (Roche Diagnostics). The qPCR
thermocycling conditions were as follows: i) 50°C for 2 min (1
cycle); ii) 95°C for 10 min (1 cycle); iii) 40 cycles of
denaturation at 95°C for 15 sec, and annealing and extension at
60°C for 60 sec. β-actin was used as the endogenous control for
mRNA expression and relative fold changes were calculated using the
2−ΔΔCq method (35).
Each RT-qPCR assay was repeated three times. The primer sequences
used are shown in Table II.
| Table II.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
| Primer name | Forward primer
5′-3′ | Reverse primer
5′-3′ |
|---|
| CCNA2 |
AGAAACAGCCAGACATCACTAA |
TTCAAACTTTGAGGCTAACAGC |
| CDK1 |
CACAAAACTACAGGTCAAGTGG |
GAGAAATTTCCCGAATTGCAGT |
| β-actin |
GGCCAACCGCGAGAAGATGAC |
GGATAGCACAGCCTGGATAGCAAC |
Western blotting
Cells were lysed using RIPA buffer (MilliporeSigma)
supplemented with EDTA-Free 1× Halt™ protease and phosphatase
inhibitor (Thermo Fisher Scientific, Inc.). The cell lysate was
then centrifuged at 14,000 × g for 15 min at 4°C and the protein
concentration was determined using a bicinchoninic acid assay kit
(Beyotime Institute of Biotechnology). Proteins (30 µg) were
separated by SDS-PAGE on 12% gels and transferred to PVDF
membranes. After blocking at room temperature for 20 min with
protein-free blocking solution (Beyotime Institute of
Biotechnology) and incubating overnight with antibodies against the
target proteins at 4°C, as follows: BCL2 (1:2,000; cat. no.
ab182858; Abcam), BAX (1:2,000; cat. no. ab32503; Abcam), caspase 3
(1:5,000; cat. no. ab32351; Abcam), cleaved-caspase 3 (1:500; cat.
no. ab32042; Abcam), PARP (1:1,000; cat. no. ab191217; Abcam),
cleaved-PARP (1:1,000; cat. no. ab32561; Abcam), β-actin (1:500;
cat. no. ab205; Abcam), CDK1 (1:10,000; cat. no. ab133327; Abcam),
p-CDK1 (Thr161) (1:1,000; cat. no. ab47329; Abcam), p53 (1:1,000;
cat. no. ab32049; Abcam), p-p53 (Ser315) (1:1,000; cat. no.
WL02504; Wanleibio Co., Ltd.). Horseradish peroxidase-conjugated
AffiniPure goat anti-rabbit immunoglobulin G (IgG) (1:1,000; cat.
no. A0277; Beyotime Institute of Biotechnology) and goat anti-mouse
IgG (1:1,000; cat. no. A0216; Beyotime Institute of Biotechnology)
were used as the secondary antibody and incubated at room
temperature for 1 h. Primary antibody diluent (cat. no. P0023A) and
secondary antibody diluent (cat. no. P0023D) were from Beyotime
Institute of Biotechnology. Subsequently, after adding an
appropriate amount of electrochemiluminescence chromogenic
substrate (BeyoECL Star; Beyotime Institute of Biotechnology), a
chemiluminescence imaging system (GeneGnome XRQ; Syngene) was used
to detect the chemiluminescence, and the intensity was measured
using ImageJ software (1.54f; National Institutes of Health).
Inhibitors and activators
Ro3306 (cat. no. SC6673; Beyotime Institute of
Biotechnology) was used as a CDK1 inhibitor at a working
concentration of 20 nM. Pifithrin-α (pft-α; cat. no. S1816;
Beyotime Institute of Biotechnology) was employed as a p53
phosphorylation inhibitor at a working concentration of 10 µM. TC11
(cat. no. HY-129478; MedChemExpress) was used as a CDK1 activator
at a concentration of 5 µM. These were dissolved in DMSO (cat. no.
ST038; Beyotime Institute of Biotechnology). Briefly, after
inducing THP-1 cells to differentiate into macrophages and adhere
to the bottom of 6-well plates, the culture medium containing PMA
was removed, and the same volume of fresh RPMI 1640 complete
culture medium containing the aforementioned concentrations of the
inhibitors and activator was added, followed by the addition of the
bacterial solution for infection for 24 h at 37°C and 5%
CO2.
Cell cycle analysis
The modeling of MTB cell infection was performed as
described previously (using ~1×106 cells/well).
Subsequently, the cells were collected, washed twice with PBS and
fixed in 70% ethanol at 4°C for 24 h. The cells were then stained
with 500 µl propidium iodide/RNase staining buffer (BD Biosciences)
at 37°C for 15 min and cell cycle progression was detected by flow
cytometry (BD FACSVerse; BD Biosciences). FlowJo (10.0.7r2; BD
Biosciences) was used to analyze the results.
Apoptosis analysis
The apoptosis detection kit (cat. no. 556547; BD
Biosciences) was used to assess the apoptosis of THP-1 cells. The
modeling of MTB cell infection was performed as described
previously (using ~1×106 cells/well). Briefly, the cells
were collected, washed twice with pre-cooled PBS, and resuspended
in 1X binding buffer. Subsequently, a 100-µl cell suspension
(~1×105 cells) was incubated with 5 µl Annexin V-FITC
and 5 µl propidium iodide at room temperature in the dark for 15
min, followed by resuspension in 400 µl 1X binding buffer and flow
cytometry (BD FACSVerse; BD Biosciences). FlowJo (10.0.7r2) was
used to analyze the results.
Co-immunoprecipitation (Co-IP)
THP-1-derived macrophages infected with XJMTB were
homogenized in 0.5% Triton X-100 (Beyotime Institute of
Biotechnology) with 1X Halt™ Protease and Phosphatase Inhibitor
SingleUse Cocktail (Thermo Fisher Scientific, Inc.). The cell
lysate was centrifuged at 4°C and 13,680 × g for 20 min to obtain a
clear lysate. For IP, 5 µg rabbit IgG antibody (cat. no. HY-P80879;
MedChemExpress), 5 µg rabbit CDK1 antibody (cat. no. HY-P80611;
MedChemExpress) and 5 µg rabbit p53 antibody (cat. no. HY-P86169;
MedChemExpress) were mixed with 500 µg protein solution, and
incubated overnight at 4°C. The protein A/G magnetic beads (Merck
KGaA) were washed with 500 µl Cell Lysis Buffer for Western or IP
(cat. no. P0013; Beyotime Institute of Biotechnology) three times.
Afterwards, the mixture was mixed with 50 µl protein A/G magnetic
beads and the antibody-protein A/G magnetic beads complex was
gently shaken for 4 h at 4°C, and 500 µl Cell Lysis Buffer for
Western or IP was then added to the precipitate. The precipitate
was then eluted with 100 µl 1X SDS loading buffer at 100°C for 10
min. Finally, SDS-PAGE was performed with eluent and whole cell
lysate (input), and western blotting was conducted as
aforementioned.
Survival analysis of MTB in cells
Briefly, XJMTB-infected macrophage specimens were
prepared according to the aforementioned method. The XJMTB
specimens were washed with PBS to remove the extracellular
bacteria. Subsequently, PBS containing 0.5% Triton X-100 was added
to the cell culture dish to lyse the XJMTB-infected macrophages,
and PBS containing the lysate of XJMTB-infected macrophage was
diluted for 10, 100, 1,000 and 10,000 times, and inoculated onto
7H10 agar plates. The plates were incubated at 37°C for 2–3 weeks,
after which the colonies were counted.
Statistical analysis
Each experiment was repeated three times. Data are
presented as the mean ± SEM. Statistical comparisons were performed
using two-way ANOVA or one-way ANOVA, followed by Tukey's post hoc
test, or using unpaired Student's t-test. All statistical analyses
were conducted using SPSS version 24.0 (IBM Corp.). P<0.05 was
considered to indicate a statistically significant difference.
Results
Study subjects
The isolation results in the modified acidic Roche's
medium are shown in Fig. 1A, and
the results of acid-fast staining are shown in Fig. 1B. The specific information and
sputum features of the 20 patients are shown in Table I, including 12 male patients and
eight female patients, aged 25–57 years old, with a median age of
40 years old. All patients had no history of smoking or other lung
diseases, and tested negative for HIV. As shown in Table I, among the 20 patients, samples
were successfully isolated and amplified from 16 patients. After
PNB and TCH medium identification, 13 of them were isolated as MTB.
A previous study conducted multiple-locus variable-number tandem
repeat analysis strain typing and cluster analysis on the strains
isolated from 103 sputum samples collected in the epidemiological
survey (29), and it was revealed
that most of the MTB strains prevalent in Xinjiang belonged to the
Beijing MTB family (29,36,37).
Rao et al (38) reported
that Beijing family strains have seven repeat fragments at the MIRU
26 site (the size of the seven repeated segments is ~642 bp);
therefore, this locus can be used for rapid identification of
Beijing family strains. Fig. S1A
shows the agarose gel electrophoresis patterns of the 13 MTB
samples isolated after PCR of the MIRU 26 site, and Fig. S1B shows the cluster analysis of
all strains collected by the collaborative team during the
aforementioned epidemiological investigation.
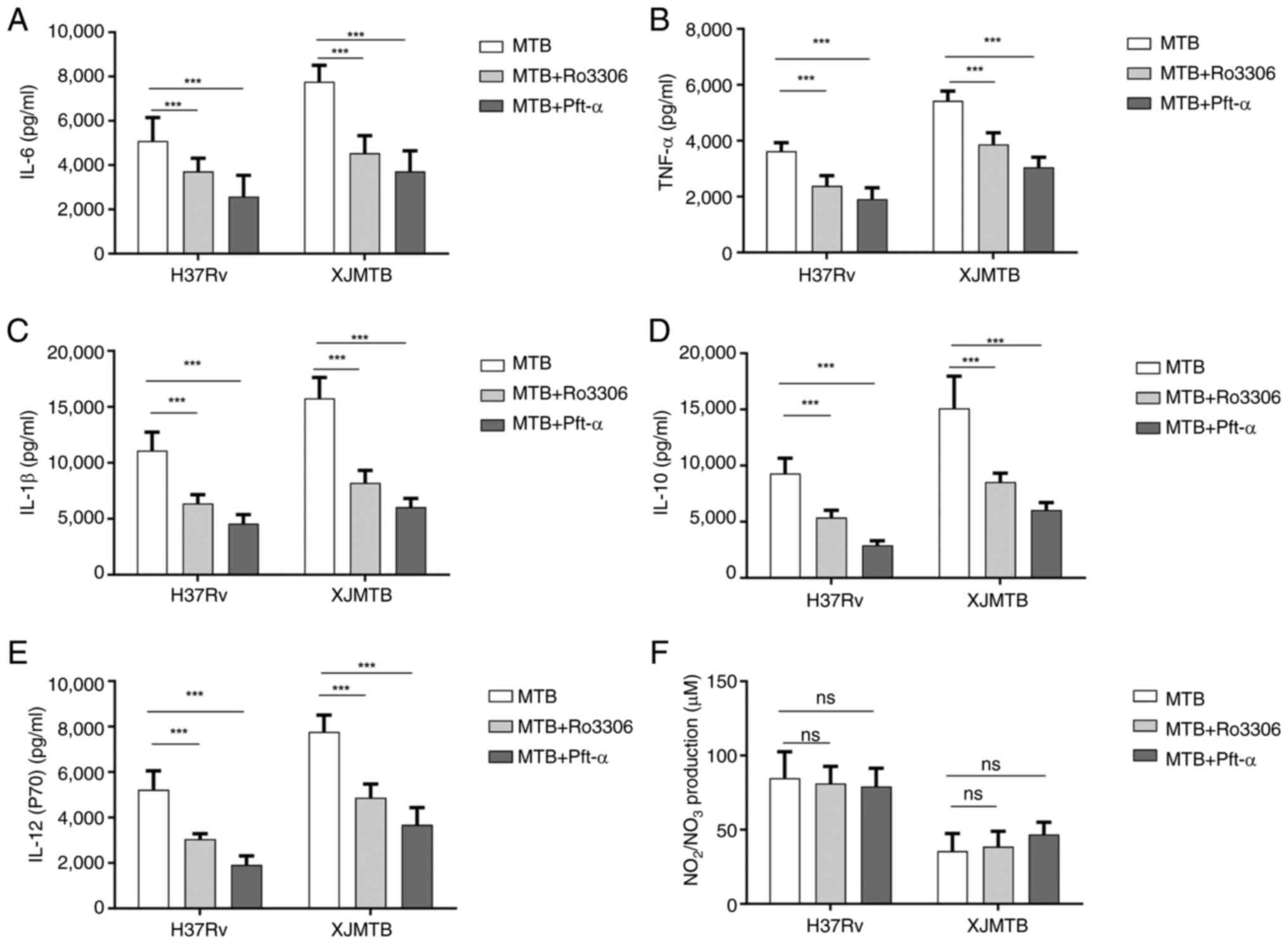
Infection with MTB leads to increased
secretion of inflammatory cytokines and NO
The induction of NO and pro-inflammatory cytokines
produced by macrophages infected with MTB can lead to DNA damage,
oxidative stress and persistent inflammation (28). Therefore, the current study
detected the secretion of the cytokines TNF-α, IL-1β, IL-6, IL-10
and IL-12 in macrophages infected with different MTB strains at 12,
24 and 36 h. As shown in Fig. 2,
compared with those in the C group without any MTB infection, the
secretion levels of these cytokines in the V and L groups were
significantly increased in a dose-dependent manner. After
comprehensively observing the secretion levels of cytokines at
various time points, 24 h was ultimately selected as the infection
time for subsequent experiments, since there was no significant
difference in the secretion of most cytokines between 24 and 36 h
after infection.
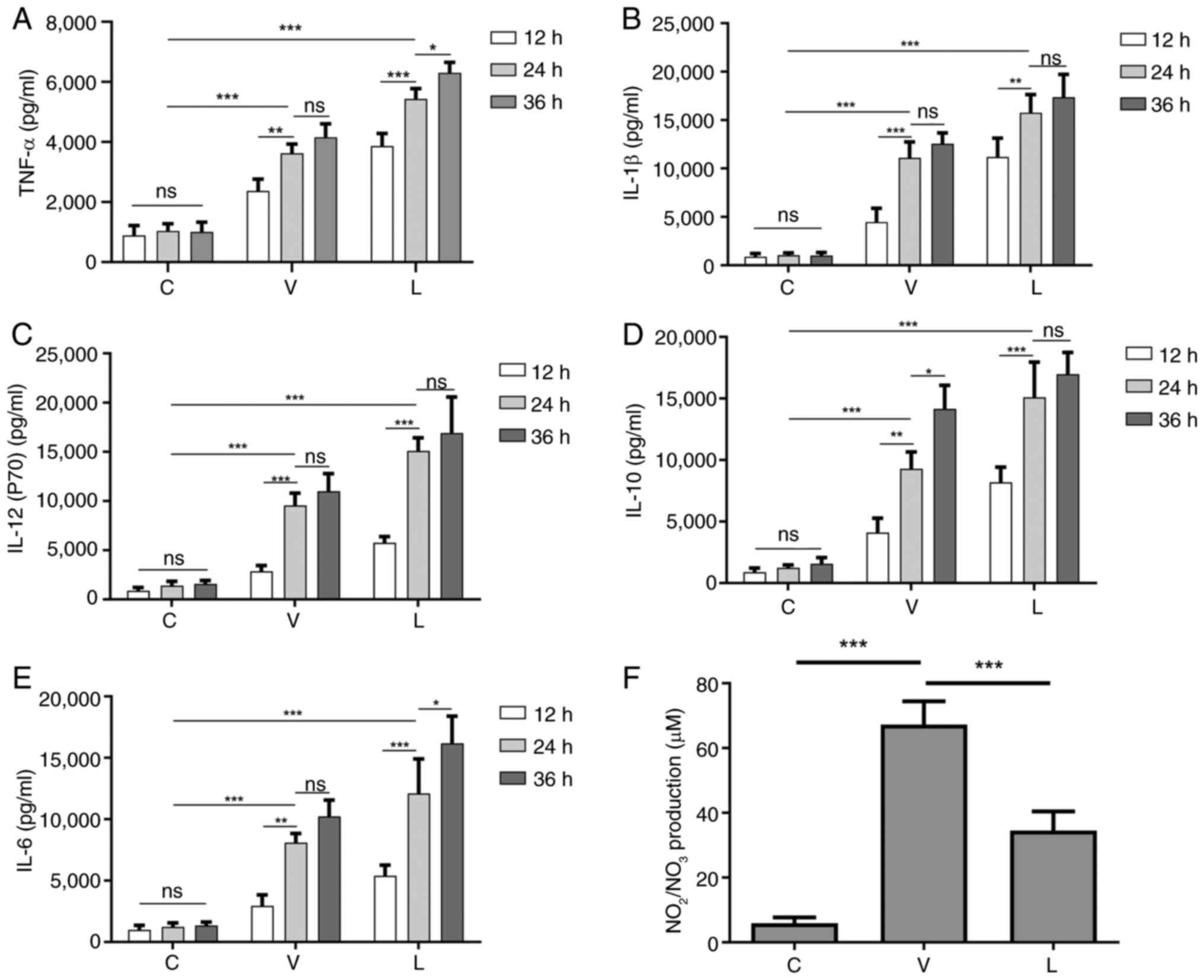 | Figure 2.Secretion levels of cytokines at
different time points after MTB infection of macrophages and the
secretion level of NO 24 h after infection. Levels of (A) TNF-α,
(B) IL-1β, (C) IL-12 (p70), (D) IL-10 and (E) IL-6 secretion at
different time points after MTB infection in macrophages. (F)
Levels of NO secretion in macrophages infected with MTB for 24 h.
Data are presented as the mean ± SEM. *P<0.05, **P<0.01,
***P<0.001. C group, blank control group not infected with any
bacteria; L group, MTB from Xinjang clinical isolate-infected
group; MTB, Mycobacterium tuberculosis; NO, nitric oxide;
ns, not significant; V group, H37Rv standard strain-infected
group. |
NO has a killing effect on MTB (39); therefore, the secretion of NO was
assessed 24 h after infection. As shown in Fig. 2F, NO levels were lower in
macrophages in the L group compared with those in macrophages in
the V group, which may lead to enhanced survival of MTB in the L
group.
CDK1 and cyclin A2 (CCNA2) expression
levels are significantly elevated after MTB infection
To identify DEGs, transcriptome sequencing data
between groups C, L and V were analyzed. The results showed that
the expression levels of 2,848 mRNAs were significantly altered in
the L group compared with the C group, of which 1,737 mRNAs were
upregulated and 1,111 were downregulated (Fig. 3A and D). In group V compared with
the C group, the expression levels of 1,678 mRNAs were
significantly altered, with 1,011 upregulated and 667 downregulated
mRNAs (Fig. 3B and E). The results
of PCA between the three groups are shown in Fig. 3C. Furthermore, a Venn analysis was
performed on the two sets of DEGs (Fig. 3F) and a follow-up analysis for the
1,527 DEGs between groups L and C was performed, since these unique
1,527 DEGs between groups L and C may reveal the specific
biological processes triggered by XJMTB. GO term (Fig. 3G-I) and KEGG pathway (Fig. 3J) enrichment analyses revealed that
multiple biological functions and pathways enriched by the 1,527
DEGs were associated with MTB infection, such as ‘DNA conformation
change’ (40), ‘phagocytic
vesicle’ (41), ‘phagocytic
vesicle membrane’ (42),
‘exonuclease activity’ (43),
‘endocytosis’ (44,45), ‘neutrophil extracellular trap
formation’ (46–48), ‘cellular senescence’ (49,50),
‘natural killer cell mediated cytotoxicity’ (51), ‘p53 signaling pathway’ (28,52)
and ‘nucleotide excision repair’ (53).
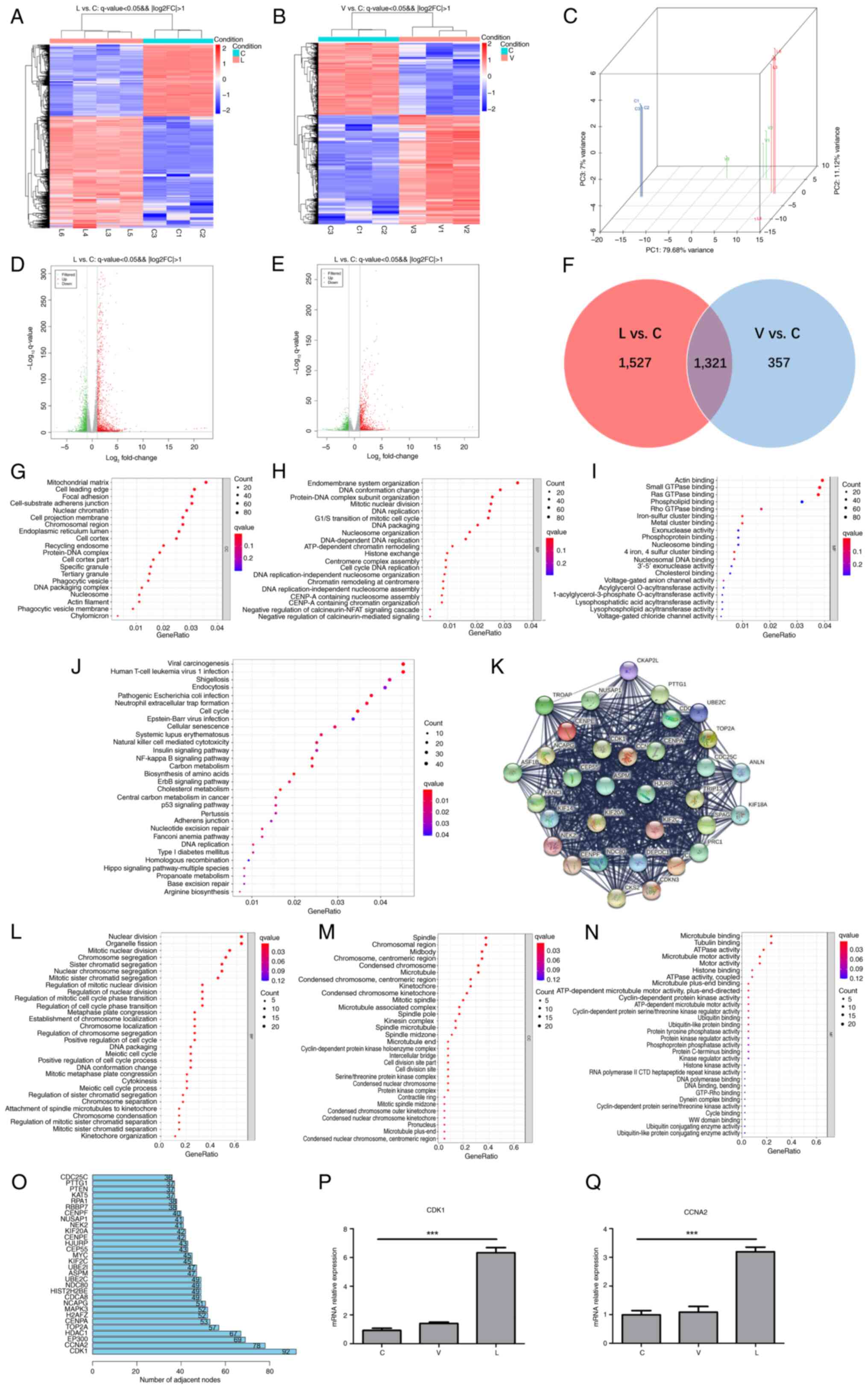 | Figure 3.CDK1 expression is upregulated in
response to infection with MTB. Heatmap of DEGs in (A) L and (B) V
groups vs. C group. Red, high expression; blue, low expression. (C)
Principal component analysis of group L, group V and group C.
Volcano plots of DEGs in the (D) L and (E) V groups (|log2 fold
change|≥1; P<0.05). Green, downregulated expression; gray, no
differential expression; red, upregulated expression. (F) Venn
diagram of L vs. C DEGs and V vs. C DEGs. GO functional enrichment
analysis of 1,527 mRNAs: (G) Cellular components, (H) biological
processes and (I) molecular functions. (J) Kyoto Encyclopedia of
Genes and Genomes pathway enrichment analysis of 1,527 mRNAs. (K)
Protein-protein interaction network top-level module containing 35
genes modularized by the plug-in MCODE. GO functional enrichment
analysis of 35 key mRNAs: (L) Biological processes, (M) cellular
components and (N) molecular functions. (O) Top 30 genes of the
1,527 mRNAs contained in the module that interacted most closely
with other genes in the module. Expression of (P) CDK1 and (Q)
CCNA2 in each group was verified through reverse
transcription-quantitative polymerase chain reaction. Data are
presented as the mean ± SEM. ***P<0.001. C group, blank control
group not infected with any bacteria; CCNA2, cyclin A 2; CDK1,
cyclin-dependent kinase; DEG, differentially expressed gene; GO,
Gene Ontology; L group, MTB from Xinjang clinical isolate-infected
group; MTB, Mycobacterium tuberculosis; V group, H37Rv
standard strain-infected group. |
To further explore the functions of the 1,527 mRNAs
at the protein level, a PPI network (high confidence, 0.7) was
established based on the results of the STRING database analysis,
which consisted of 1,382 nodes and 14,264 edges. Subsequently, the
plug-in MCODE in Cytoscape was used to deeply analyze the network,
and the top module (score=29) was selected for subsequent research
(Fig. 3K). The 33 mRNAs contained
in this top module were regarded as critical mediators, and a
functional analysis was performed. GO functional enrichment
analysis showed that the 33 critical mediators were closely
associated with ‘cell mitosis’ and the ‘cell cycle’ (Fig. 3L-N). R (3.6.3) software was applied
to analyze the core genes in the PPI formed by the 1,527 genes. The
top two genes were found to be CDK1 and CCNA2, and these genes were
also found in the 33 key mRNAs. It has previously been shown that
CDK1 and CCNA2 are closely related to the cell cycle (54). Furthermore, the previous GO and
KEGG enrichment analyses showed that the unique 1,527 DEGs between
groups L and C were enriched in terms of ‘DNA replication’ and
other cell cycle related terms and pathways. Subsequently, RT-qPCR
was used to detect the mRNA expression levels of CDK1 and CCNA2
(Fig. 3P), which showed
significant differences between the L and C groups. Given that the
mRNA expression levels of CDK1 in each group were consistent with
the results of the bioinformatics analysis, and that the number of
interaction nodes between CDK1 and other genes ranked first in
Fig. 3O, CDK1 was identified as
the target gene for subsequent studies.
Inhibition of CDK1 alleviates
G2/M cell cycle block and apoptosis caused by MTB
infection
CDK1 is an enzyme necessary for initiating mitosis
after the end of S-phase, and it can regulate the G2/M
checkpoint (17) and is closely
related to apoptosis (18).
Therefore, flow cytometry was performed to detect differential
changes in the cell cycle of macrophages before and after infection
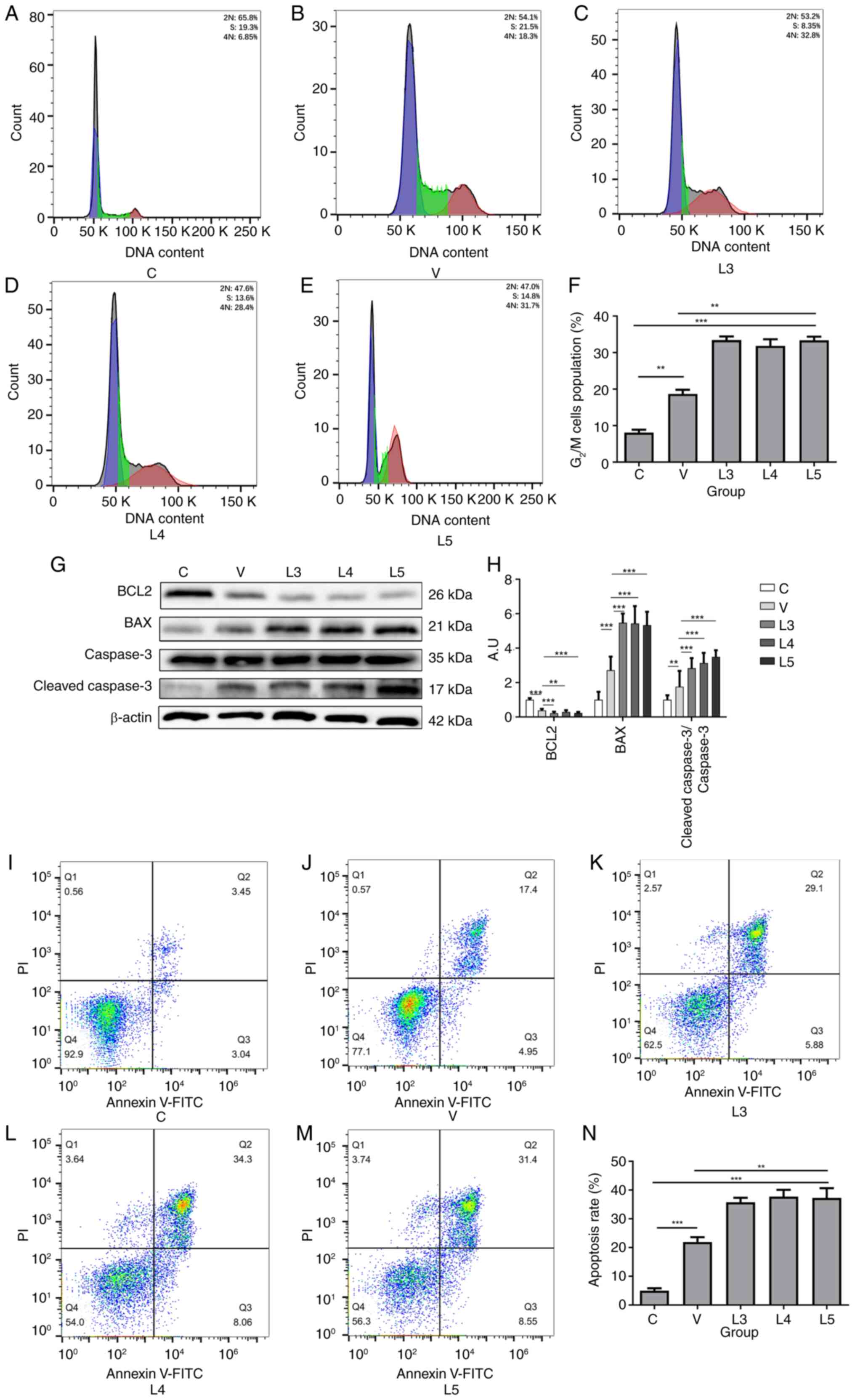
with different sources of MTB. As shown in Fig. 4A-F, compared with in group C,
groups V and L exhibited significantly higher G2/M cell
cycle blockage, and the ratio of cell cycle blockage was
significantly higher in groups L3-5 than that in in group V,
suggesting that macrophages infected with MTB could trigger
G2/M cell cycle block and that different sources of MTB
could induce distinct degrees of cell cycle arrest. Notably,
macrophages infected with XJMTB may trigger a more severe
G2/M cell cycle block compared with those infected with
H37Rv.
 | Figure 4.Infection with XJMTB causes stronger
G2/M cell cycle arrest and apoptosis than infection with
H37Rv. Cell cycle profile of macrophages in the (A) C group, (B) V
group, (C) L3 group, (D) L4 group and (E) L5 group. (F)
G2/M cell cycle ratio in each group. (G) Protein
expression levels of BCL2, BAX, BCL-2, caspase 3 and
cleaved-caspase 3 were determined by western blotting. (H)
Statistical analysis of apoptosis-related proteins detected by
western blotting. Apoptosis of macrophages in the (I) C group, (J)
V group, (K) L3 group, (L) L4 group and (M) L5 group. (N) Apoptosis
rate in each group. Data are presented as the mean ± SEM.
**P<0.01, ***P<0.001. A.U., arbitrary units; C group, blank
control group not infected with any bacteria; L group, XJMTB
clinical isolate-infected group; MTB, Mycobacterium
tuberculosis; V group, H37Rv standard strain-infected group;
XJMTB, MTB from Xinjang. |
The current study next examined the apoptosis of
macrophages after infection with different sources of MTB using
western blotting and flow cytometry. Compared with those in group
C, the protein expression levels of BCL2 were decreased in groups L
and V, whereas the protein expression levels of BAX and
cleaved-caspase 3 (Fig. 4G and H),
and cleaved-PARP (Fig. S2) were
increased. Compared with that in group C (Fig. 4I), groups V (Fig. 4J) and L (Fig. 4K-M) exhibited marked apoptosis,
with the degree of apoptosis being significantly stronger in group
L than that in group V (Fig. 4N).
These findings suggested that MTB could induce macrophage
apoptosis, but the degree of macrophage apoptosis after infection
with different sources of MTB varied, with the proportion of
apoptotic macrophages infected with XJMTB being higher compared
with those infected with H37Rv.
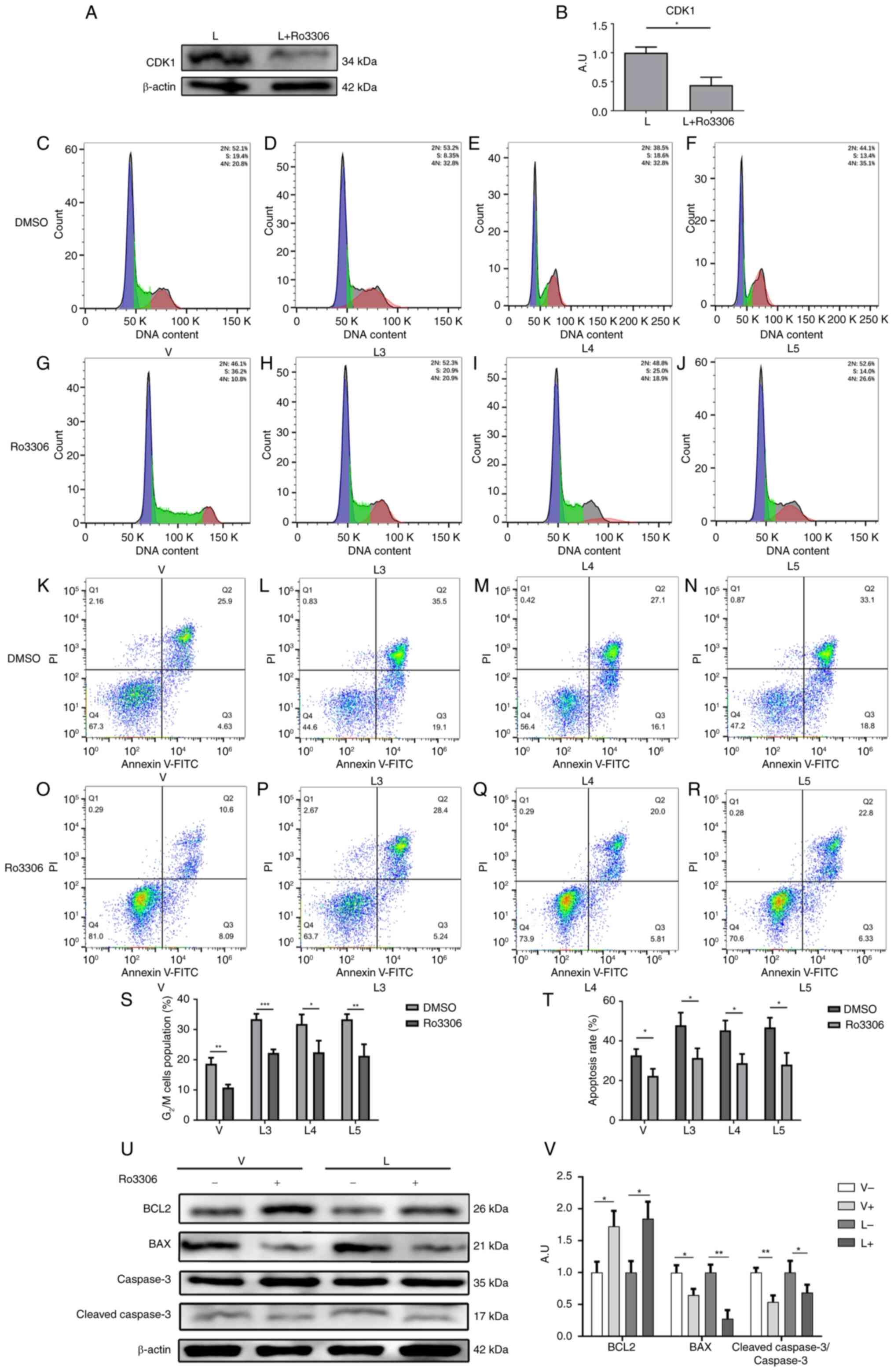
Next, the CDK1 inhibitor Ro3306 was used to inhibit
the expression of CDK1 in macrophages after infection with
different sources of MTB. The expression of CDK1 was first detected
in group L before and after the addition of Ro3306, as shown in
Fig. 5A and B. The findings
indicated that the working concentration provided in the Ro3306
manual could effectively inhibit the expression of CDK1. The
results of the flow cytometric analysis showed that the percentages
of G2/M cell cycle arrest in macrophages in the V group
(Fig. 5C and G), L3 group
(Fig. 5D and H), L4 group
(Fig. 5E and I) and L5 group
(Fig. 5F and J) were significantly
decreased after the addition of Ro3306 (Fig. 5S). These results suggested that
inhibiting CDK1 expression could attenuate the G2/M cell
cycle block of macrophages caused by infection with MTB.
 | Figure 5.Inhibition of CDK1 alleviates
macrophage G2/M cycle arrest and apoptosis caused by MTB
infection. (A) Protein expression levels of CDK1 were determined by
western blotting. (B) Statistical analysis of CDK1 protein
expression detected by western blotting. Cell cycle profiles of
macrophages in the (C) V group, (D) L3 group, (E) L4 group and (F)
L5 group when Ro3306 was not added. Cell cycle profiles of
macrophages in the (G) V group, (H) L3 group, (I) L4 group and (J)
L5 group after the addition of Ro3306. Apoptosis of macrophages in
the (K) V group, (L) L3 group, (M) L4 group and (N) L5 group when
Ro33006 was not added. Apoptosis of macrophages in the (O) V group,
(P) L3 group, (Q) L4 group and (R) L5 group after the addition of
Ro3306. (S) G2/M cell cycle ratio and (T) apoptosis rate
in each group before and after the addition of Ro3306. (U) Protein
expression levels of BCL2, BAX, Bcl-2, caspase 3 and
cleaved-caspase 3 were determined by western blotting before and
after the addition of Ro3306. (V) Statistical analysis of
apoptosis-related protein expression detected by western blotting
before and after the addition of Ro3306. Data are presented as the
mean ± SEM. *P<0.05, **P<0.01, ***P<0.001. A.U., arbitrary
units; CDK1, cyclin-dependent kinase; L group, MTB from Xinjang
clinical isolate-infected group; MTB, Mycobacterium
tuberculosis; V group, H37Rv standard strain-infected
group. |
Similarly, the effects of the CDK1 inhibitor Ro3306
on macrophage apoptosis were examined after infection with
different sources of MTB. The results of flow cytometry showed that
the percentages of apoptotic macrophages in the V group (Fig. 5K and O), L3 group (Fig. 5L and P), L4 group (Fig. 5M and Q) and L5 group (Fig. 5N and R) were significantly
decreased after the addition of Ro3306 (Fig. 5T). Furthermore, the results of
western blotting showed that the protein expression levels of BCL2
were increased by Ro3306, whereas the protein expression levels of
BAX and cleaved-caspase 3 (Fig. 5U and
V), and cleaved-PARP (Fig.
S3) were decreased. These findings suggested that inhibiting
CDK1 expression could attenuate macrophage apoptosis caused by MTB
infection.
The aforementioned results indicated that
macrophages infected with MTB could induce G2/M cell
cycle block and apoptosis, but the degree of cell cycle arrest and
apoptosis induced by different sources of MTB varied. The
proportion of G2/M cell cycle block and apoptosis in
macrophages infected with XJMTB was significantly higher compared
with that in macrophages infected with H37Rv. Furthermore, the
degree of apoptosis and G2/M cell cycle arrest in
macrophages infected with H37Rv or XJMTB was alleviated after
inhibiting CDK1 expression, which was consistent with the predicted
result of the bioinformatics analyses, suggesting that CDK1 may be
a key regulatory gene for G2/M cell cycle arrest and
apoptosis in macrophages with MTB infection.
Inhibition of p53 (Ser315)
phosphorylation attenuates G2/M cell cycle arrest and
apoptosis caused by MTB infection
Since the ‘p53 signaling pathway’ was revealed to be
enriched in the aforementioned KEGG analysis, and it is well known
that CDK1 can interact with p53 to affect cell cycle progression
and apoptosis (55), the current
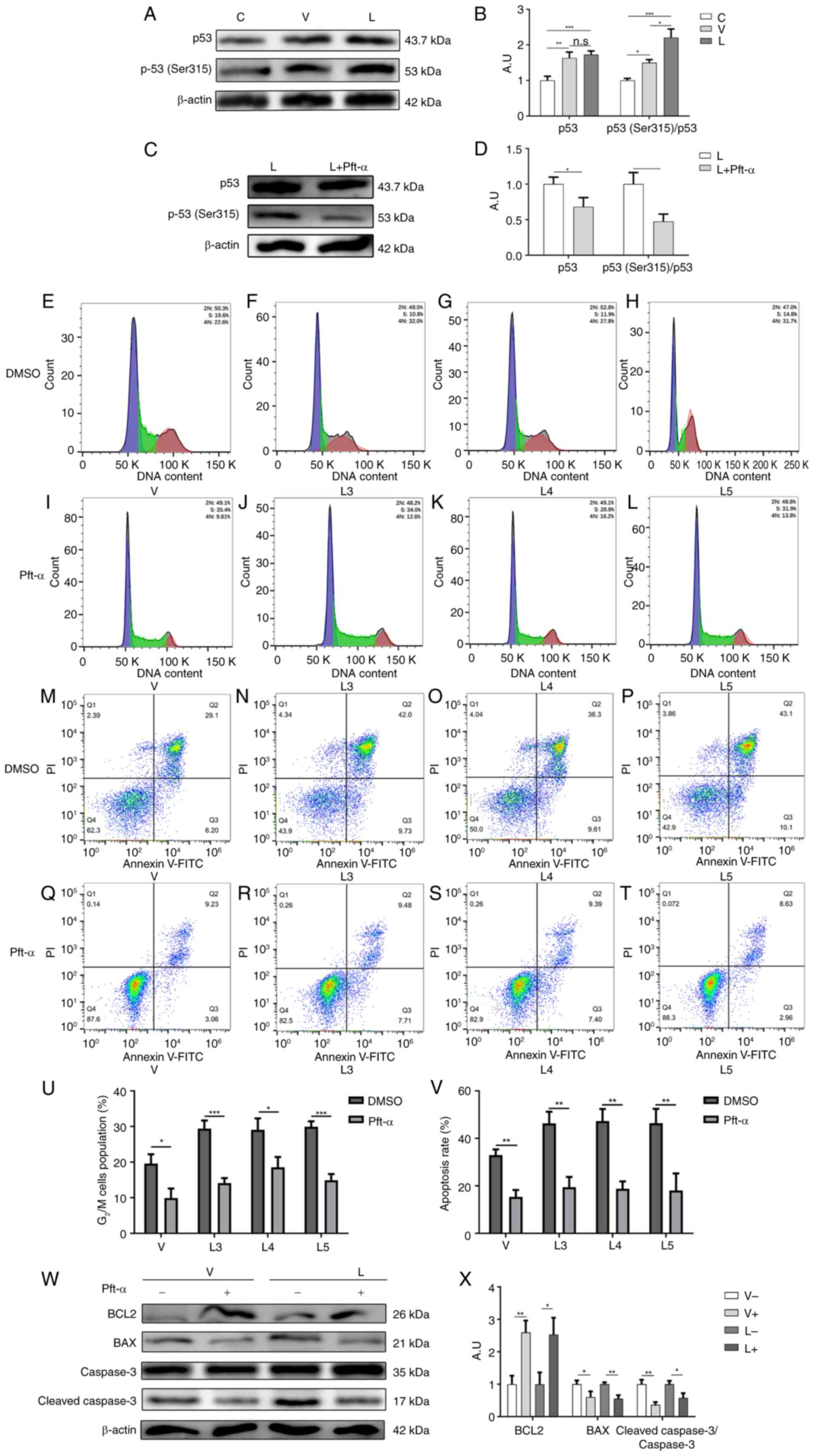
study examined the expression of p53 in macrophages after infection
with different sources of MTB. As shown in Fig. 6A and B, the expression levels of
p53 were significantly elevated after infection with different
sources of MTB, but there was no significant difference between the
two infection groups, suggesting that the G2/M cell
cycle arrest and apoptosis caused by high expression of CDK1 after
MTB infection was not achieved by directly affecting p53
expression.
 | Figure 6.Inhibition of p53 (Ser315)
phosphorylation attenuates macrophage G2/M cycle block
and apoptosis caused by MTB infection. (A) Protein expression
levels of total p53 and p-p53 (Ser315) after MTB infection were
determined by western blotting. (B) Statistical analysis of total
p53 and p-p53 (Ser315) expression detected by western blotting
after MTB infection. (C) Protein expression levels of total p53 and
p-p53 (Ser315) were determined by western blotting before and after
the addition of Pft-α. (D) Statistical analysis of total p53 and
p-p53 (Ser315) expression determined by western blotting before and
after the addition of Pft-α. Cell cycle profiles of macrophages in
the (E) V group, (F) L3 group, (G) L4 group and (H) L5 group when
Pift-α was not added. Cell cycle profiles of macrophages in the (I)
V group, (J) L3 group, (K) L4 group and (L) L5 group after the
addition of Pft-α. Apoptosis of macrophages in the (M) V group, (N)
L3 group, (O) L4 group and (P) L5 group when Pft-α was not added.
Apoptosis of macrophages in the (Q), V group, (R) L3 group, (S) L4
group and (T) L5 group after the addition of Pft-α. (U)
G2/M cell cycle ratio and (V) apoptosis rate of each
group before and after the addition of Pft-α. (W) Protein
expression levels of BCL2, BAX, caspase 3 and cleaved-caspase 3
were determined by western blotting before and after the addition
of Pft-α. (X) Statistical analysis of apoptosis-related protein
expression detected by western blotting before and after the
addition of Pft-α. Data are presented as the mean ± SEM.
*P<0.05, **P<0.01, ***P<0.001. A.U., arbitrary units; L
group, MTB from Xinjang clinical isolate-infected group; MTB,
Mycobacterium tuberculosis; n.s., not significant; p-,
phosphorylated; Pft-α, pifithrin-α; V group, H37Rv standard
strain-infected group. |
Fogal et al (56) demonstrated that CDK1 could mediate
the phosphorylation of p53 at the Ser315 site to affect apoptosis,
and indicated that the expression of total p53 may not change
during this process. Therefore, the present study examined the
phosphorylation of p53 at the Ser315 site in macrophages infected
with different sources of MTB. The results showed that the
phosphorylation of p53 at the Ser315 site was higher in macrophages
infected with XJMTB than in those infected with H37Rv (Fig. 6A and B), suggesting that the G2/M
cell cycle arrest and apoptosis caused by high expression of CDK1
after infection with MTB might be mediated by the phosphorylation
of p53 at the Ser315 site.
Subsequently, the p53 inhibitor pft-α was used to
inhibit the phosphorylation of p53. The expression of p53 and the
phosphorylation of p53 (Ser315) before and after the addition of
pft-α is shown in Fig. 6C and D.
The findings indicated that the working concentration recomeded in
the pft-α instruction manual could effectively inhibit the
phosphorylation of p53 (Ser315). The results of flow cytometric
analysis revealed that the percentages of G2/M cell
cycle block in macrophages in the V group (Fig. 6E and I), L3 group (Fig. 6F and J), L4 group (Fig. 6G and K) and L5 group (Fig. 6H and L) were decreased after the
addition of pft-α (Fig. 6U). These
findings indicated that inhibiting the phosphorylation of p53
(Ser315) could attenuate the G2/M cell cycle arrest
caused by MTB infection.
The current study then examined the effect of pft-α
on the apoptosis of macrophages after infection with different
sources of MTB. The results of flow cytometric analysis showed that
the percentages of apoptotic macrophages in the V group (Fig. 6M and Q), L3 group (Fig. 6N and R), L4 group (Fig. 6O and S) and L5 group (Fig. 6P and T) were decreased after the
addition of pft-α (Fig. 6V). The
western blot analysis showed that the protein expression levels of
BCL2 were increased after the addition of pft-α, whereas the
protein expression levels of BAX and cleaved-caspase 3 (Fig. 6W and X), and cleaved-PARP (Fig. S4) were decreased. The two
experiments suggested that inhibiting the phosphorylation of p53
(Ser315) could attenuate macrophage apoptosis due to infection with
MTB.
The aforementioned results indicated that infection
with MTB could induce elevated p53 expression; however, there was
no significant difference in p53 expression among macrophages
infected with different sources of MTB, whereas there was a
difference in the phosphorylation of p53 (Ser315). The degree of
macrophage apoptosis and G2/M cell cycle arrest caused
by infection with different sources of MTB was alleviated by
inhibiting the phosphorylation of p53 (Ser315), which was
consistent with the predicted result of the bioinformatics
analysis, suggesting that p-p53 may be a key regulatory protein for
G2/M cell cycle block and apoptosis in macrophages
infected with MTB. Therefore, it was hypothesized that a regulatory
relationship may exist between CDK1 and p-p53.
Effect of inhibiting CDK1 expression
or p53 (Ser315) phosphorylation on cytokine and NO secretions in
MTB-infected macrophages
The present study revealed that the secretion levels
of TNF-α, IL-6, IL-12, IL-10 and IL-1β were increased after MTB
infection of macrophages. Therefore, the secretions of TNF-α, IL-6,
IL-12, IL-10, IL-1β and NO were examined before and after
inhibiting CDK1 expression or p53 (Ser315) phosphorylation. As
shown in Fig. 7, in macrophage
models infected with MTB from different sources, inhibition of CDK1
expression (MTB + Ro3306) or p53 (Ser315) phosphorylation (MTB +
Pft-α) resulted in a significant decrease in the secretion of
TNF-α, IL-6, IL-12, IL-10 and IL-1β, but no change in NO secretion.
These results indicated that inhibiting CDK1 expression or p53
(Ser315) phosphorylation may suppress the release of inflammatory
cytokines in THP-1-derived macrophages infected with MTB.
CDK1 mediates G2/M cell
cycle arrest and apoptosis induced by MTB infection and MTB
survival in macrophages by regulating p53 (Ser315)
phosphorylation
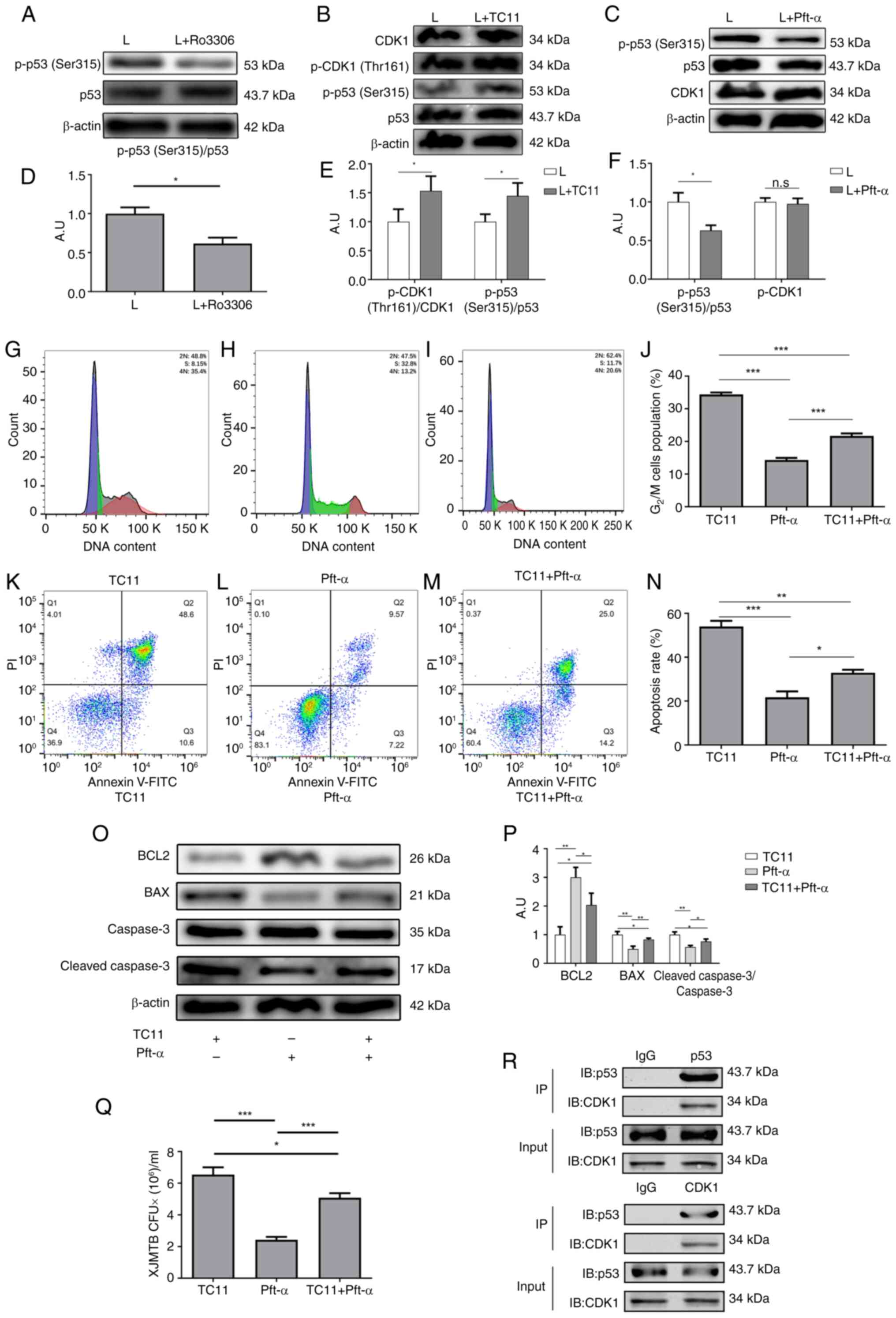
To verify whether CDK1 could affect G2/M
cell cycle arrest and apoptosis due to MTB infection by regulating
the phosphorylation of p53 (Ser315), western blotting was performed
to examine the phosphorylation of p53 (Ser315) following the
addition of Ro3306 to XJMTB-infected macrophages to inhibit the
expression of CDK1. As shown in Fig.
8A and D, after the addition of Ro3306, the phosphorylation of
p53 (Ser315) was subsequently decreased.
 | Figure 8.CDK1 mediates G2/M cell
cycle arrest and apoptosis caused by infection with XJMTB through
regulation of p53 (Ser315) phosphorylation. (A) Phosphorylation of
p53 (Ser315) after inhibition of CDK1. (B) Phosphorylation of CDK1
(Thr161) and of p53 (Ser315) following treatment with TC11. (C)
Expression of CDK1 after inhibition of p53 (Ser315)
phosphorylation. (D) Statistical analysis of total p53 and p-p53
(Ser315) expression determined by western blotting before and after
the addition of Ro3306. (E) Statistical analysis of total p53,
p-p53 (Ser315), total CDK1 and p-CDK1 (Thr161) expression
determined by western blotting before and after the addition of
TC11. (F) Statistical analysis of total p53, p-p53 (Ser315) and
total CDK1 expressiondetermined by western blotting before and
after the addition of Pft-α. Cell cycle progression of
XJMTB-infected macrophages in the (G) TC11, (H) Pft-α and (I) TC11
+ Pft-α groups. (J) G2/M cell cycle ratio in the various
groups. Apoptosis of XJMTB-infected macrophages in the (K) TC11,
(L) Pft-α and (M) TC11 + Pft-α groups. (N) Apoptosis rate in the
various groups. (O and P) Protein expression levels of BCL2, BAX,
caspase 3 and cleaved-caspase 3 were determined by western blotting
in the TC11, Pft-α and TC11 + Pft-α groups. (Q) CFU analysis was
performed to determine the survival rate of XJMTB in macrophages in
the TC11, Pft-α and TC11 + Pft-α groups. (R) Co-IP results of CDK1
and p53. Data are presented as the mean ± SEM. *P<0.05,
**P<0.01, ***P<0.001. A.U., arbitrary units; CDK1,
cyclin-dependent kinase; CFU, colony-forming unit; IB,
immunoblotting; IgG, immunoglobulin G; IP, immunoprecipitation; L
group, XJMTB clinical isolate-infected group; MTB, Mycobacterium
tuberculosis; n.s., not significant; p-, phosphorylated; Pft-α,
pifithrin-α; XJMTB, MTB from Xinjang. |
Subsequently, TC11 (an activator of CDK1) was used
to activate CDK1. TC11 functions as a phenylacetylamide derivative
and is structurally related to immunomodulatory active drugs, such
as thalidomide, lenalidomide and pomalidomide (57). TC11 can induce MCL1 degradation and
activate caspase 9 and CDK1, leading to cell apoptosis (58,59).
Fig. 8B and E showed that TC11
could enhance the phosphorylation of CDK1 (Thr161), and the
phosphorylation of p53 (Ser315) was also increased. Moreover,
inhibiting the phosphorylation of p53 (Ser315) did not alter CDK1
expression (Fig. 8C and F).
Therefore, it was hypothesized that CDK1 could regulate p53
(Ser315) phosphorylation in macrophages infected with XJMTB.
Next, cell cycle progression and apoptosis were
examined in three situations: i) Activation of CDK1 only; ii)
inhibition of p53 (Ser315) phosphorylation only; iii) and
activation of CDK1 + inhibition of p53 (Ser315) phosphorylation.
The aforementioned results revealed that elevated CDK1 expression
and enhanced p53 phosphorylation levels led to G2/M cell
cycle arrest. TC11 can activate CDK1, whereas pft-α can inhibit p53
(Ser315) phosphorylation. The results of flow cytometry showed that
the proportion of G2/M cell cycle arrest in macrophages
with CDK1 activation and p-p53 (Ser315) inhibition (Fig. 8I) was higher than that in
macrophages with p-p53 (Ser315) inhibition alone (Fig. 8H), but lower than that in those
with CDK1 activation (Fig. 8J).
These findings indicated that the inhibitory ability of pft-α on
p53 (Ser315) phosphorylation was weakened by TC11, thus suggesting
that the CDK1 activation caused by TC11 could reverse the reduction
in the proportion of G2/M cell cycle block caused by the
inhibition of p53 (Ser315) phosphorylation in macrophages infected
with XJMTB.
As expected, flow cytometry results showed that the
proportion of apoptosis in macrophages in which CDK1 was activated
and p53 phosphorylation was inhibited (Fig. 8M) was higher than that in
macrophages in which only p53 phosphorylation was inhibited
(Fig. 8L), but lower than that in
macrophages in which only CDK1 was activated (Fig. 8K). Fig. 8N shows the apoptosis ratio in each
group. Furthermore, the results of western blotting were consistent
with those obtained by flow cytometry (Figs. 8O, P and S5). These findings suggested that
TC11-induced CDK1 activation could reverse the reduction in the
proportion of apoptosis caused by the inhibition of p53 (Ser315)
phosphorylation in macrophages infected with XJMTB.
To clarify whether this regulatory relationship
affected the survival of XJMTB in macrophages, XJMTB survival was
assessed in macrophages under the aforementioned three conditions.
As shown in Fig. 8Q, the survival
rate of XJMTB in macrophages with CDK1 activation and p-p53
(Ser315) inhibition was higher than that in macrophages with p-p53
(Ser315) inhibition, but lower than that in those with CDK1
activation. These data strongly suggested that CDK1 activity
influenced p53 phosphorylation levels.
To clarify the relationship between CDK1 and p53 in
macrophages infected with XJMTB, Co-IP experiments were performed
to observe whether they could interact with each other. The results
showed that there was an interaction between CDK1 and p53 (Fig. 8R). CDK1 is a kinase that can
transfer phosphate groups, thus it may be hypothesized that CDK1
can directly regulate p53 phosphorylation. Based on the
aforementioned results in an in vitro model of XJMTB
infected-macrophages, the high expression of CDK1 could enhance the
phosphorylation of p53 (Ser315), affect the secretion of TNF-α,
IL-6, IL-10, IL-1β and IL-12, promote G2/M cell cycle
arrest and apoptosis of macrophages, and enhance the survival of
XJMTB in macrophages.
Discussion
TB is a complex disease involving multiple
mechanisms (60), and macrophages
are the first immune mechanism against MTB infection and the main
effector cells to eliminate MTB (61). In the early stages of TB, MTB
predominantly adopts an intracellular parasitic mode. The release
of inflammatory factors (62), the
production of reactive oxygen species (ROS) (63) and inducible NO synthase (iNOS)
(62), and macrophage autophagy
(64) and apoptosis (65) after MTB infection affect the
ability of macrophages to clear MTB. However, their effects on the
ability of macrophages to clear MTB is not very clear, especially
autophagy and apoptosis. The morbidity of TB in Xinjiang is
markedly higher than the national average (4), bringing great economic pressure to
the majority of patients, and seriously affecting their physical
and mental health. The search for specific immune responses
triggered by XJMTB may help develop targeted control strategies for
TB. Therefore, the present study used transcriptome sequencing to
identify specific immune responses triggered by XJMTB.
Cell cycle progression is tightly regulated by
cyclins and their catalytic partners, CDKs (66–68).
Cyclin and CDKs can regulate the progression of the G1,
S, G2 and M phases of the cell cycle through various
mechanisms, which may include periodic synthesis and degradation,
post-translational modification and subcellular localization
(69). Cyclins are a large class
of proteins characterized by the presence of a CDK-binding domain
called the ‘cyclin box’, of which the cell cycle regulator CCNA2 is
a member. The results of the present bioinformatics analysis showed
that the expression levels of CDK1 and CCNA2 were significantly
elevated in macrophages infected with XJMTB compared with in those
infected with H37Rv, suggesting that XJMTB could lead to more
severe cell cycle arrest, affect macrophage proliferation and
influence the effectiveness of the host immune defense. There have
been numerous reports on the role of CDK1 in various diseases
(70–74); however, to the best of our
knowledge, no reports have described its role in TB. According to
the present results, the number of interaction nodes between CDK1
and other genes ranked higher than CCNA2, and since kinases serve
an important role in the post-translational modification of
proteins CDK1 was selected as the primary research target.
Notably, the regulatory relationship between CDK1
and the cell cycle and apoptosis has been widely reported in a
number of studies (18,75,76).
The present findings suggested that elevated CDK1 expression in
macrophages infected with MTB could lead to the exacerbation of
G2/M cell cycle block and a higher proportion of
apoptotic cells. When the expression of CDK1 was inhibited by
Ro3306, the proportion of G2/M cell cycle block and
macrophage apoptosis was reduced, suggesting that CDK1 may be
essential in MTB infection-induced cell cycle arrest and apoptosis.
It has previously been shown that p53 can promote cell cycle
arrest, induce DNA repair, and participate in various cell
death-related pathways and metabolic changes (77–79).
Notably, macrophages infected with MTB have been shown to exhibit
increased p53 (Ser315) phosphorylation and enhanced apoptosis
(80). Similarly, the current
study revealed that an increase in p53 (Ser315) phosphorylation led
to G2/M cell cycle arrest and apoptosis in MTB-infected
macrophages. Yuan et al (81) showed that circular RNA CEA could
promote CDK1-mediated phosphorylation of p53 at the Ser315 site,
which was in line with the results of the present study, which
revealed that elevated CDK1 expression could regulate p53 (Ser315)
phosphorylation.
Infection with MTB leads to macrophage apoptosis
(82–85), which may be mediated by surface or
secreted proteins. MTB Rv3261 protein can induce ROS production to
inhibit MTB growth in macrophages, activate caspase 3/9-dependent
pathways, and induce macrophage apoptosis (86). Medha et al (87) showed that the C-terminal and
N-terminal sequences of MTB Rv0335c were similar to those of the
BCL2 protein, and in vitro experiments confirmed that it
could lead to apoptosis. The present study observed elevated
cleaved-caspase 3 expression and decreased BCL2 expression
following infection with MTB, and this phenomenon was more
pronounced in samples infected with XJMTB than in those infected
with H37Rv.
There is a view that although apoptosis is usually
a non-inflammatory form of cell death, excessive apoptosis may lead
to the propagation of MTB, which is beneficial for the pathogen
rather than the host (12,13). Host infection can be exacerbated by
the elimination of important host defense cells, penetration of the
epithelial barrier and transmission of pathogens to immature host
phagocytes that engulf apoptotic bodies (88). It has been shown that Fas-mediated
apoptosis does not affect the survival of macrophages and MTB
(89). Kelly et al
(90) showed that infected
macrophages can induce apoptosis of uninfected macrophages in a
cell-to-cell contact-dependent manner, leading to exacerbation of
MTB infection. In addition, Lee et al (91) reported that bacteria released from
apoptotic macrophages infected with MTB were still alive and could
subsequently grow extracellularly. Furthermore, H37Rv has been
reported to induce more cell death compared with H37Ra, and this
mode of death was initially characterized by apoptosis (91–93).
Gan et al (92) infected
mice in vivo with the MTB H37Rv strain or attenuated H37Ra
strain, and showed that H37Rv induced a higher macrophage death
rate and notably depleted resident alveolar macrophages, whereas
H37Ra resulted in a marked reduction in cell death and only
partially depleted the resident macrophages. Afriyie-Asante et
al (94) revealed that MTB
inhibited ROS production by decreasing focal adhesion kinase
expression, leading to apoptosis and persistent lung injury. One
hypothesis is that apoptosis in the late stages of infection during
granuloma formation may be a pathogenic process that increases the
persistence and future spread of MTB (95). In addition, macrophage apoptosis
caused by MTB may evolve into macrophage necrosis, which releases
cellular contents and triggers inflammation (96,97),
and sustained inflammation exacerbates tissue damage and leads to
disease progression (98,99). Compared with other forms of cell
death, apoptotic cells may be less likely to activate the specific
immune responses required to control MTB infection (100). As aforementioned, H37Rv can
induce more severe apoptosis than H37Ra, and H37Rv is more
pathogenic than H37Ra. This is consistent with the present findings
that XJMTB, which is more pathogenic, induces stronger macrophage
apoptosis compared with H37Rv. Nevertheless, there is also a view
that activating host cell apoptosis is an important defense
strategy for pathogens that utilize host cell resources for
survival and replication, meaning that the more severe the
apoptosis, the better the bactericidal effect (101). Moreover, the virulence of MTB has
been reported to be negatively associated with the degree of
classical apoptosis, and the attenuated strain H37Ra has been
suggested to induce more apoptosis compared with the highly
virulent strain H37Rv (102).
The aforementioned studies suggest that the
macrophage apoptosis induced by MTB infection can be beneficial for
both the host and MTB, which may be influenced by the viability of
the bacteria and host cells, as well as the duration of the
infection (103). Therefore, it
is hypothesized that apoptosis can be considered as having a dual
role. Apoptosis is a means of controlling the growth of
intracellular pathogens and is capable of killing MTB only under
the appropriate conditions (89,104–108). The fate of cells may depend on
the balance between pro- and anti-apoptotic signals emitted by the
cell or pathogen, as well as other variables, including host
species, origin and state of the host cell differentiation, in
vitro culture conditions, MTB strains and multiplicity of
infection. MTB can induce macrophage apoptosis directly when
objective conditions change (109), regardless of the killing effect
of MTB (110). In addition, in
the presence of large numbers of mycobacteria and when large
numbers of apoptotic cells exceed the local phagocytic clearance
capacity of macrophages, macrophages are considered to be critical
for the clearance of apoptotic vesicles, and thus ineffective
clearance of apoptotic cells may lead to tissue damage (111). Moreover, unabsorbed apoptotic
cells may undergo secondary necrosis, thereby favoring MTB
dissemination (112). These
results suggest that when infected with MTB, the occurrence of
massive apoptosis in macrophages does not necessarily represent
notable clearance of MTB, but rather indicates an exacerbation of
the infection, which explains the phenomenon that clinical isolates
of XJMTB were more likely to induce macrophage apoptosis compared
with clinical isolates of H37Rv.
NO is a free radical mediator that serves an
important role in the immune system. Long et al (39) concluded that exogenous NO has a
time- and dose-dependent killing effect on MTB, and that short-term
exposure of extracellular MTB to low concentrations of NO (<100
ppm) can kill MTB. During macrophage infection, NO may act directly
or combine with superoxide to form peroxynitrite, which kills MTB
in phagosomes (113). NOS
converts L-arginine to NO, which is protective against parasitic
infections and a variety of infectious organisms, such as MTB
(114,115). Mice deficient in iNOS have been
shown to be highly susceptible to MTB infection, permitting MTB
infection and leading to early death (116–118). Furthermore, the expression levels
of iNOS are higher in the peripheral monocytes and alveolar
macrophages of patients with TB compared with those in healthy
individuals (119). These studies
indicated that NO may have a strong bactericidal effect on
MTB-infected macrophages. In the present study, XJMTB induced a
lower secretion of NO than H37Rv, which may explain its enhanced
survival in macrophages.
It has been shown that in the J774.1 macrophage
model of mice infected with Mycobacterium bovis BCG, IFN-γ
and lipopolysaccharide treatment enhance the expression of the
arginine permease MCAT2B; however, this does not explain the
observed increase in L-arginine uptake, suggesting that the
activity of the L-arginine transporter may also be altered in
response to macrophage activation, which in turn affects NO
production (120). In addition,
expression of the solute carrier family 11 member 1 gene affects
the ability of macrophages to produce NO, resulting in differential
susceptibility or resistance to MTB (121). NO production by alveolar
macrophages in patients with TB may have an autoregulatory role,
which can promote proinflammatory cytokine production through
activation of the transcription factor NF-κB (122–124). Upregulation of the inhibitor of
NF-κB has confirmed that the IκBα kinase-NF-κB signaling pathway
enhances IFN-γ- and MTB lipoarabinomannan-induced iNOS promoter
activity and NO expression (124). These studies suggested that NO
production is regulated by multiple factors and does not come from
a single source, which may be one of the reasons why the regulation
of the CDK1-p53 axis had no effect on NO secretion in the current
study.
One limitation of the present study is the
relatively small sample size, with only four strains of XJMTB used
for infection and sequencing, which may limit the generalizability
of the findings. Additionally, the study was conducted using a
single cell line, THP-1, which might not reflect the broader
population. The use of human monocyte-derived macrophages may
enhance the physiological relevance of the study. However, primary
monocyte-derived macrophages are difficult to obtain and isolate.
We previously considered validating the relevant conclusions in the
RAW264.7 cell line, but considering that RAW264.7 cells are
mouse-derived cells, this cell line was not used due to species
differences between humans and mice. In addition, the conclusions
should be verified in animal experiments; however, animal
experiments related to TB require extremely stringent experimental
conditions and environments.
CDK1 was the target of the present study due to its
high bioinformatics score and verifiability, as well as its novelty
in the field of MTB. The identification of the ‘p53 signaling
pathway’ entry in the bioinformatics analysis process attracted
clarification as to whether a regulatory relationship exists
between CDK1 and p53 in the process of MTB infection of
macrophages, thus the upstream molecules that regulate the high
expression of CDK1 were overlooked. Future research should include
larger and more diverse samples to validate these results,
including validating this mechanism in MTBs from other regions.
The present study showed that MTB infection of
macrophages resulted in high CDK1 expression and increased levels
of p53 (Ser315) phosphorylation, and that the level of p53 (Ser315)
phosphorylation increased with CDK1 activation, leading to
increased macrophage G2/M cell cycle arrest and
apoptosis. Compared with H37Rv, XJMTB infection typically led to a
greater elevation of CDK1 and p-p53. Furthermore, Co-IP analysis
showed that there was indeed a protein interaction between CDK1 and
p53. The activation of CDK1 could reverse the reduction of
G2/M cell cycle arrest and apoptosis caused by p-p53
inhibition. These results suggested that the increase in p53
(Ser315) phosphorylation in MTB-infected macrophages may be caused
by elevated CDK1 expression. In an in vitro model of
XJMTB-infected macrophages, high activation of CDK1 can enhance the
phosphorylation of p53 (Ser315), affect the secretion of TNF-α,
IL-6, IL-10, IL-1β and IL-12, promote G2/M cell cycle
arrest and apoptosis of macrophages, and enhance the survival of
XJMTB in macrophages. These findings lay the foundation for
formulating treatment strategies targeting the prognosis of TB in
Xinjiang, China. Future studies should explore the potential of
targeting CDK1 and p53 phosphorylation in clinical settings.
Regulating the macrophage cycle or apoptosis by adjusting the
activity of CDK1 or p53 may be considered a potential treatment for
TB.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Ms. Jing Wang
(Department of Respiratory Medicine, The Second Affiliated Hospital
of Hainan Medical College) for their contribution to specimen
collection.
Funding
The present study received funding from the National Natural
Science Foundation of China (grant no. 81672109).
Availability of data and materials
The sequencing data generated in the present study
may be found in the Gene Expression Omnibus database under
accession number GSE275640 or at the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE275640.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
BS, QM, ZZ and FL conceptualized the study. XJ, SL
and FC collected the data in the study and conducted statistical
analysis. FL acquired funding. BS, QM, LP, ZZ and FL designed the
study. FL conducted project administration. ZZ and FL provided
materials and resources for the study. BS, QM and LP conducted
bioinformatics analysis using software. FL supervised the study.
BS, LP and XJ validated the research results obtained. FC and ZZ
visualized the content and data to be published. BS and ZZ
conducted the initial draft writing. QiM and FL reviewed and edited
the manuscript. BS and FL confirm the authenticity of all the raw
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
All protocols performed in the present study were
approved by the Ethics Committee of Jilin University (approval no.
2016-057; Changchun, China). Written informed consent for
participation was obtained from the patients.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Abdullah M, Humayun A, Imran M, Bashir MA
and Malik AA: A bibliometric analysis of global research
performance on tuberculosis (2011–2020): Time for a global approach
to support high-burden countries. J Family Community Med.
29:117–124. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Li XX, Zhang H, Jiang SW, Liu XQ, Fang Q,
Li J, Li X and Wang LX: Geographical distribution regarding the
prevalence rates of pulmonary tuberculosis in China in 2010.
Zhonghua Liu Xing Bing Xue Za Zhi. 34:980–984. 2013.(In Chinese).
PubMed/NCBI
|
|
3
|
Wubuli A, Xue F, Jiang D, Yao X, Upur H
and Wushouer Q: Socio-demographic predictors and distribution of
pulmonary tuberculosis (TB) in Xinjiang, China: A spatial analysis.
PLoS One. 10:e01440102015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang L, Zhang H, Ruan Y, Chin DP, Xia Y,
Cheng S, Chen M, Zhao Y, Jiang S, Du X, et al: Tuberculosis
prevalence in China, 1990–2010; a longitudinal analysis of national
survey data. Lancet. 383:2057–2064. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
He X, Cao M, Mahapatra T, Du X, Mahapatra
S, Li Q, Feng L, Tang S, Zhao Z, Liu J and Tang W: Burden of
tuberculosis in Xinjiang between 2011 and 2015: A surveillance
data-based study. PLoS One. 12:e01875922017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Anilkumar U and Prehn JHM: Anti-apoptotic
BCL-2 family proteins in acute neural injury. Front Cell Neurosci.
8:2812014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Behar SM, Divangahi M and Remold HG:
Evasion of innate immunity by Mycobacterium tuberculosis: Is
death an exit strategy? Nat Rev Microbiol. 8:668–674. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deretic V and Wang F: Autophagy is part of
the answer to tuberculosis. Nat Microbiol. 8:762–763. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arnett E and Schlesinger LS: Live and let
die: TB control by enhancing apoptosis. Immunity. 54:1625–1627.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Häcker G: Apoptosis in infection. Microbes
Infect. 20:552–559. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ashida H, Mimuro H, Ogawa M, Kobayashi T,
Sanada T, Kim M and Sasakawa C: Cell death and infection: A
double-edged sword for host and pathogen survival. J Cell Biol.
195:931–942. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hirsch CS, Johnson JL, Okwera A, Kanost
RA, Wu M, Peters P, Muhumuza M, Mayanja-Kizza H, Mugerwa RD,
Mugyenyi P, et al: Mechanisms of apoptosis of T-cells in human
tuberculosis. J Clin Immunol. 25:353–364. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pagán AJ and Ramakrishnan L: Immunity and
immunopathology in the tuberculous granuloma. Cold Spring Harb
Perspect Med. 5:a0184992014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malumbres M and Barbacid M: Cell cycle,
CDKs and cancer: A changing paradigm. Nat Rev Cancer. 9:153–166.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Enserink JM and Kolodner RD: An overview
of Cdk1-controlled targets and processes. Cell Div. 5:112010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Morgan DO: Cyclin-dependent kinases:
Engines, clocks, and microprocessors. Annu Rev Cell Dev Biol.
13:261–291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Huang R, Gao S, Han Y, Ning H, Zhou Y,
Guan H, Liu X, Yan S and Zhou PK: BECN1 promotes radiation-induced
G2/M arrest through regulation CDK1 activity: A potential role for
autophagy in G2/M checkpoint. Cell Death Discov. 6:702020.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Huang S, Xiao J, Wu J, Liu J, Feng X, Yang
C, Xiang D and Luo S: Tizoxanide promotes apoptosis in glioblastoma
by inhibiting CDK1 activity. Front Pharmacol. 13:8955732022.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang Q, Chen L, Gao M, Wang S, Meng L and
Guo L: Molecular docking and in vitro experiments verified that
kaempferol induced apoptosis and inhibited human HepG2 cell
proliferation by targeting BAX, CDK1, and JUN. Mol Cell Biochem.
478:767–780. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu G, Yan X, Hu Z, Zheng L, Ding K, Zhang
Y, Qing Y, Liu T, Cheng L and Shi Z: Glucocappasalin induces
G2/M-phase arrest, apoptosis, and autophagy pathways by targeting
CDK1 and PLK1 in cervical carcinoma cells. Front Pharmacol.
12:6711382021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang YJ, Luo S and Xu ZL: Effects of
miR-490-5p targeting CDK1 on proliferation and apoptosis of colon
cancer cells via ERK signaling pathway. Eur Rev Med Pharmacol Sci.
26:2049–2056. 2022.PubMed/NCBI
|
|
22
|
Tong Y, Huang Y, Zhang Y, Zeng X, Yan M,
Xia Z and Lai D: DPP3/CDK1 contributes to the progression of
colorectal cancer through regulating cell proliferation, cell
apoptosis, and cell migration. Cell Death Dis. 12:5292021.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang Y, Fan Y, Zhao Z, Zhang X, Tucker K,
Staley A, Suo H, Sun W, Shen X, Deng B, et al: Inhibition of CDK1
by RO-3306 exhibits anti-tumorigenic effects in ovarian cancer
cells and a transgenic mouse model of ovarian cancer. Int J Mol
Sci. 24:123752023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ozaki T and Nakagawara A: Role of p53 in
cell death and human cancers. Cancers (Basel). 3:994–1013. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Chen J: The cell-cycle arrest and
apoptotic functions of p53 in tumor initiation and progression.
Cold Spring Harb Perspect Med. 6:a0261042016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zheng SJ, Lamhamedi-Cherradi SE, Wang P,
Xu L and Chen YH: Tumor suppressor p53 inhibits autoimmune
inflammation and macrophage function. Diabetes. 54:1423–1428. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fischer M: Census and evaluation of p53
target genes. Oncogene. 36:3943–3956. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lim YJ, Lee J, Choi JA, Cho SN, Son SH,
Kwon SJ, Son JW and Song CH: M1 macrophage dependent-p53 regulates
the intracellular survival of mycobacteria. Apoptosis. 25:42–55.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gong X, Li Y, Yao L, Aynur M, Liu N, Wang
L and Wang J: Preliminary study on genotype of Mycobacterium
tuberculosis in some areas of Xinjiang by the multiple locus
VNTR analysis. Chin J Lung Dis (Electronic Edition). 10:304–308.
2017.(In Chinese).
|
|
30
|
Chinese Society of Tuberculosis and
Chinese Medical Association, . Guidelines for the Diagnosis and
Treatment of Pulmonary Tuberculosis. Chin J Tuberc Respir Dis.
24:70–74. 2001.(In Chinese).
|
|
31
|
Kim D, Paggi JM, Park C, Bennett C and
Salzberg SL: Graph-based genome alignment and genotyping with
HISAT2 and HISAT-genotype. Nat Biotechnol. 37:907–915. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ghosh S and Chan CKK: Analysis of RNA-seq
data using tophat and cufflinks. Methods Mol Biol. 1374:339–361.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Anders S, Pyl PT and Huber W: HTSeq-a
Python framework to work with high-throughput sequencing data.
Bioinformatics. 31:166–169. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bader GD and Hogue CWV: An automated
method for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4:22003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang J, Mi L, Wang Y, Liu P, Liang H,
Huang Y, Lv B and Yuan L: Genotypes and drug susceptibility of
Mycobacterium tuberculosis Isolates in Shihezi, Xinjiang
Province, China. BMC Res Notes. 5:3092012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yuan L, Mi L, Li Y, Zhang H, Zheng F and
Li Z: Genotypic characteristics of Mycobacterium
tuberculosis circulating in Xinjiang, China. Infect Dis (Lond).
48:108–115. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Rao KR, Ahmed N, Srinivas S, Sechi LA and
Hasnain SE: Rapid identification of Mycobacterium
tuberculosis Beijing genotypes on the basis of the
mycobacterial interspersed repetitive unit locus 26 signature. J
Clin Microbiol. 44:274–277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Long R, Jones R, Talbot J, Mayers I,
Barrie J, Hoskinson M and Light B: Inhaled nitric oxide treatment
of patients with pulmonary tuberculosis evidenced by positive
sputum smears. Antimicrob Agents Chemother. 49:1209–1212. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Butala B, Busada M and Cormican D:
Malignant hyperthermia: Review of diagnosis and treatment during
cardiac surgery with cardiopulmonary bypass. J Cardiothorac Vasc
Anesth. 32:2771–2779. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fatima N, Upadhyay T, Ahmad F, Arshad M,
Kamal MA, Sharma D and Sharma R: Particulate β-glucan activates
early and delayed phagosomal maturation and autophagy within
macrophage in a NOX-2 dependent manner. Life Sci. 266:1188512021.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Majeed M, Perskvist N, Ernst JD, Orselius
K and Stendahl O: Roles of calcium and annexins in phagocytosis and
elimination of an attenuated strain of Mycobacterium
tuberculosis in human neutrophils. Microb Pathog. 24:309–320.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nasir N and Kisker C: Mechanistic insights
into the enzymatic activity and inhibition of the replicative
polymerase exonuclease domain from Mycobacterium
tuberculosis. DNA Repair (Amst). 74:17–25. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Parveen N, Varman R, Nair S, Das G, Ghosh
S and Mukhopadhyay S: Endocytosis of Mycobacterium
tuberculosis heat shock protein 60 is required to induce
interleukin-10 production in macrophages. J Biol Chem.
288:24956–24971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sachdeva K, Goel M and Sundaramurthy V:
Heterogeneity in the endocytic capacity of individual macrophage in
a population determines its subsequent phagocytosis, infectivity
and subcellular trafficking. Traffic. 21:522–533. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Moreira-Teixeira L, Stimpson PJ,
Stavropoulos E, Hadebe S, Chakravarty P, Ioannou M, Aramburu IV,
Herbert E, Priestnall SL, Suarez-Bonnet A, et al: Type I IFN
exacerbates disease in tuberculosis-susceptible mice by inducing
neutrophil-mediated lung inflammation and NETosis. Nat Commun.
11:55662020. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kroon EE, Correa-Macedo W, Evans R, Seeger
A, Engelbrecht L, Kriel JA, Loos B, Okugbeni N, Orlova M, Cassart
P, et al: Neutrophil extracellular trap formation and gene programs
distinguish TST/IGRA sensitization outcomes among Mycobacterium
tuberculosis exposed persons living with HIV. PLoS Genet.
19:e10108882023. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Dang G, Cui Y, Wang L, Li T, Cui Z, Song
N, Chen L, Pang H and Liu S: Extracellular sphingomyelinase Rv0888
of Mycobacterium tuberculosis contributes to pathological
lung injury of mycobacterium smegmatis in mice via inducing
formation of neutrophil extracellular traps. Front Immunol.
9:6772018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bobak CA, Abhimanyu Natarajan H, Gandhi T,
Grimm SL, Nishiguchi T, Koster K, Longlax SC, Dlamini Q, Kahari J,
et al: Increased DNA methylation, cellular senescence and premature
epigenetic aging in guinea pigs and humans with tuberculosis. Aging
(Albany NY). 14:2174–2193. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Parandhaman DK and Narayanan S: Cell death
paradigms in the pathogenesis of Mycobacterium tuberculosis
infection. Front Cell Infect Microbiol. 4:312014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Li S, Wang D, Wei P, Liu R, Guo J, Yang B,
Zhang H, Lu J, Gao M and Pang Y: Elevated natural killer
cell-mediated cytotoxicity is associated with cavity formation in
pulmonary tuberculosis patients. J Immunol Res. 2021:79259032021.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Liang S, Song Z, Wu Y, Gao Y, Gao M, Liu
F, Wang F and Zhang Y: MicroRNA-27b modulates inflammatory response
and apoptosis during Mycobacterium tuberculosis infection. J
Immunol. 200:3506–3518. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Houghton J, Townsend C, Williams AR,
Rodgers A, Rand L, Walker KB, Böttger EC, Springer B and Davis EO:
Important role for Mycobacterium tuberculosis UvrD1 in
pathogenesis and persistence apart from its function in nucleotide
excision repair. J Bacteriol. 194:2916–2923. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Jin M, Li J, Hu R, Xu B, Huang G, Huang W,
Chen B, He J and Cao Y: Cyclin A2/cyclin-dependent kinase
1-dependent phosphorylation of Top2a is required for S phase entry
during retinal development in zebrafish. J Genet Genomics.
48:63–74. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Thorenoor N, Faltejskova-Vychytilova P,
Hombach S, Mlcochova J, Kretz M, Svoboda M and Slaby O: Long
non-coding RNA ZFAS1 interacts with CDK1 and is involved in
p53-dependent cell cycle control and apoptosis in colorectal
cancer. Oncotarget. 7:622–637. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fogal V, Hsieh JK, Royer C, Zhong S and Lu
X: Cell cycle-dependent nuclear retention of p53 by E2F1 requires
phosphorylation of p53 at Ser315. EMBO J. 24:2768–2782. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Aida S, Hozumi M, Ichikawa D, Iida K,
Yonemura Y, Tabata N, Yamada T, Matsushita M, Sugai T, Yanagawa H
and Hattori Y: A novel phenylphthalimide derivative, pegylated
TC11, improves pharmacokinetic properties and induces apoptosis of
high-risk myeloma cells via G2/M cell-cycle arrest. Biochem Biophys
Res Commun. 493:514–520. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ichikawa D, Nakamura M, Murota W, Osawa S,
Matsushita M, Yanagawa H and Hattori Y: A phenylphthalimide
derivative, TC11, induces apoptosis by degrading MCL1 in multiple
myeloma cells. Biochem Biophys Res Commun. 521:252–258. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Shiheido H, Terada F, Tabata N, Hayakawa
I, Matsumura N, Takashima H, Ogawa Y, Du W, Yamada T, Shoji M, et
al: A phthalimide derivative that inhibits centrosomal clustering
is effective on multiple myeloma. PLoS One. 7:e388782012.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Reid M, Agbassi YJP, Arinaminpathy N,
Bercasio A, Bhargava A, Bhargava M, Bloom A, Cattamanchi A,
Chaisson R, Chin D, et al: Scientific advances and the end of
tuberculosis: A report from the lancet commission on tuberculosis.
Lancet. 402:1473–1498. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chandra P, Grigsby SJ and Philips JA:
Immune evasion and provocation by Mycobacterium
tuberculosis. Nat Rev Microbiol. 20:750–766. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jin C, Wu X, Dong C, Li F, Fan L, Xiong S
and Dong Y: EspR promotes mycobacteria survival in macrophages by
inhibiting MyD88 mediated inflammation and apoptosis. Tuberculosis
(Edinb). 116:22–31. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li F, Feng L, Jin C, Wu X, Fan L, Xiong S
and Dong Y: LpqT improves mycobacteria survival in macrophages by
inhibiting TLR2 mediated inflammatory cytokine expression and cell
apoptosis. Tuberculosis (Edinb). 111:57–66. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Feng L, Hu J, Zhang W, Dong Y, Xiong S and
Dong C: RELL1 inhibits autophagy pathway and regulates
Mycobacterium tuberculosis survival in macrophages.
Tuberculosis (Edinb). 120:1019002020. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Dai X, Zhou L, He X, Hua J, Chen L and Lu
Y: Identification of apoptosis-related gene signatures as potential
biomarkers for differentiating active from latent tuberculosis via
bioinformatics analysis. Front Cell Infect Microbiol.
14:12854932024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lim S and Kaldis P: Cdks, cyclins and
CKIs: Roles beyond cell cycle regulation. Development.
140:3079–3093. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Satyanarayana A and Kaldis P: Mammalian
cell-cycle regulation: Several Cdks, numerous cyclins and diverse
compensatory mechanisms. Oncogene. 28:2925–2939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Barnum KJ and O'Connell MJ: Cell cycle
regulation by checkpoints. Methods Mol Biol. 1170:29–40. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Risal S, Adhikari D and Liu K: Animal
models for studying the in vivo functions of cell cycle CDKs.
Methods Mol Biol. 1336:155–166. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zhu Y, Li K, Zhang J, Wang L, Sheng L and
Yan L: Inhibition of CDK1 reverses the resistance of 5-Fu in
colorectal cancer. Cancer Manag Res. 12:11271–11283. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang Z, Shen G and Gao J: CDK1 promotes
the stemness of lung cancer cells through interacting with Sox2.
Clin Transl Oncol. 23:1743–1751. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Dai Y, Hu S, Bai S, Li J, Yang N, Zhai P,
Zhao B, Chen Y and Wu X: CDK1 promotes the proliferation of
melanocytes in Rex rabbits. Genes Genomics. 44:1191–1199. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Chen Q, Xu L, Lu C, Xue Y, Gong X, Shi Y,
Wang C and Yu L: Prognostic significance of CDK1 expression in
diffuse large B-Cell lymphoma. BMC Cancer. 25:202025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Piao J, Zhu L, Sun J, Li N, Dong B, Yang Y
and Chen L: High expression of CDK1 and BUB1 predicts poor
prognosis of pancreatic ductal adenocarcinoma. Gene. 701:15–22.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Malumbres M: Cyclin-dependent kinases.
Genome Biol. 15:1222014. View
Article : Google Scholar : PubMed/NCBI
|
|
76
|
Kang X, Chen H, Zhou Z, Tu S, Cui B, Li Y,
Dong S, Zhang Q and Xu Y: Targeting cyclin-dependent kinase 1
induces apoptosis and cell cycle arrest of activated hepatic
stellate cells. Adv Biol (Weinh). 8:e23004032024. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hager KM and Gu W: Understanding the
non-canonical pathways involved in p53-mediated tumor suppression.
Carcinogenesis. 35:740–746. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Kang R, Kroemer G and Tang D: The tumor
suppressor protein p53 and the ferroptosis network. Free Radic Biol
Med. 133:162–168. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kruiswijk F, Labuschagne CF and Vousden
KH: p53 in survival, death and metabolic health: A lifeguard with a
licence to kill. Nat Rev Mol Cell Biol. 16:393–405. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Bao J, He Y, Yang C, Lu N, Li A, Gao S,
Hosyanto FF, Tang J, Si J, Tang X, et al: Inhibition of
mycobacteria proliferation in macrophages by low cisplatin
concentration through phosphorylated p53-related apoptosis pathway.
PLoS One. 18:e02811702023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yuan Y, Zhang X, Du K, Zhu X, Chang S,
Chen Y, Xu Y, Sun J, Luo X, Deng S, et al: Circ_CEA promotes the
interaction between the p53 and cyclin-dependent kinases 1 as a
scaffold to inhibit the apoptosis of gastric cancer. Cell Death
Dis. 13:8272022. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Klingler K, Tchou-Wong KM, Brändli O,
Aston C, Kim R, Chi C and Rom WN: Effects of mycobacteria on
regulation of apoptosis in mononuclear phagocytes. Infect Immun.
65:5272–5278. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Danelishvili L, McGarvey J, Li YJ and
Bermudez LE: Mycobacterium tuberculosis infection causes
different levels of apoptosis and necrosis in human macrophages and
alveolar epithelial cells. Cell Microbiol. 5:649–660. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ciaramella A, Cavone A, Santucci MB, Garg
SK, Sanarico N, Bocchino M, Galati D, Martino A, Auricchio G,
D'Orazio M, et al: Induction of apoptosis and release of
interleukin-1 beta by cell wall-associated 19-kDa lipoprotein
during the course of mycobacterial infection. J Infect Dis.
190:1167–1176. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
85
|
Arcila ML, Sánchez MD, Ortiz B, Barrera
LF, García LF and Rojas M: Activation of apoptosis, but not
necrosis, during Mycobacterium tuberculosis infection
correlated with decreased bacterial growth: Role of TNF-alpha,
IL-10, caspases and phospholipase A2. Cell Immunol. 249:80–93.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lee KI, Choi S, Choi HG, Kebede SG, Dang
TB, Back YW, Park HS and Kim HJ: Recombinant Rv3261 protein of
Mycobacterium tuberculosis induces apoptosis through a
mitochondrion-dependent pathway in macrophages and inhibits
intracellular bacterial growth. Cell Immunol. 354:1041452020.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
MedhaPriyanka Bhatt P, Sharma S and Sharma
M: Role of C-terminal domain of Mycobacterium tuberculosis
PE6 (Rv0335c) protein in host mitochondrial stress and macrophage
apoptosis. Apoptosis. 28:136–165. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Zychlinsky A: Programmed cell death in
infectious diseases. Trends Microbiol. 1:114–117. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Stenger S, Mazzaccaro RJ, Uyemura K, Cho
S, Barnes PF, Rosat JP, Sette A, Brenner MB, Porcelli SA, Bloom BR
and Modlin RL: Differential effects of cytolytic T cell subsets on
intracellular infection. Science. 276:1684–1687. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Kelly DM, ten Bokum AM, O'Leary SM,
O'Sullivan MP and Keane J: Bystander macrophage apoptosis after
Mycobacterium tuberculosis H37Ra infection. Infect Immun.
76:351–360. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Lee J, Remold HG, Ieong MH and Kornfeld H:
Macrophage apoptosis in response to high intracellular burden of
Mycobacterium tuberculosis is mediated by a novel
caspase-independent pathway. J Immunol. 176:4267–4274. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Gan H, Lee J, Ren F, Chen M, Kornfeld H
and Remold HG: Mycobacterium tuberculosis blocks
crosslinking of annexin-1 and apoptotic envelope formation on
infected macrophages to maintain virulence. Nat Immunol.
9:1189–1197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen M, Gan H and Remold HG: A mechanism
of virulence: Virulent Mycobacterium tuberculosis strain
H37Rv, but not attenuated H37Ra, causes significant mitochondrial
inner membrane disruption in macrophages leading to necrosis. J
Immunol. 176:3707–3716. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Afriyie-Asante A, Dabla A, Dagenais A,
Berton S, Smyth R and Sun J: Mycobacterium tuberculosis
exploits focal adhesion kinase to induce necrotic cell death and
inhibit reactive oxygen species production. Front Immunol.
12:7423702021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Mohareer K, Medikonda J, Vadankula GR and
Banerjee S: Mycobacterial control of host mitochondria:
Bioenergetic and metabolic changes shaping cell fate and infection
outcome. Front Cell Infect Microbiol. 10:4572020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Behar SM, Martin CJ, Booty MG, Nishimura
T, Zhao X, Gan HX, Divangahi M and Remold HG: Apoptosis is an
innate defense function of macrophages against Mycobacterium
tuberculosis. Mucosal Immunol. 4:279–287. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Dallenga T, Repnik U, Corleis B, Eich J,
Reimer R, Griffiths GW and Schaible UE: M. tuberculosis-induced
necrosis of infected neutrophils promotes bacterial growth
following phagocytosis by macrophages. Cell Host Microbe.
22:519–530.e3. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Toossi Z: The inflammatory response in
Mycobacterium tuberculosis infection. Arch Immunol Ther Exp
(Warsz). 48:513–519. 2000.PubMed/NCBI
|
|
99
|
Amaral EP, Costa DL, Namasivayam S, Riteau
N, Kamenyeva O, Mittereder L, Mayer-Barber KD, Andrade BB and Sher
A: A major role for ferroptosis in Mycobacterium
tuberculosis-induced cell death and tissue necrosis. J Exp Med.
216:556–570. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Su H, Zhu S, Zhu L, Huang W, Wang H, Zhang
Z and Xu Y: Recombinant lipoprotein Rv1016c derived from
Mycobacterium tuberculosis is a TLR-2 ligand that induces
macrophages apoptosis and inhibits MHC II antigen processing. Front
Cell Infect Microbiol. 6:1472016. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Schaible UE, Winau F, Sieling PA, Fischer
K, Collins HL, Hagens K, Modlin RL, Brinkmann V and Kaufmann SH:
Apoptosis facilitates antigen presentation to T lymphocytes through
MHC-I and CD1 in tuberculosis. Nat Med. 9:1039–1046. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Velmurugan K, Chen B, Miller JL, Azogue S,
Gurses S, Hsu T, Glickman M, Jacobs WR Jr, Porcelli SA and Briken
V: Mycobacterium tuberculosis nuoG is a virulence gene that
inhibits apoptosis of infected host cells. PLoS Pathog. 3:e1102007.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Nisa A, Kipper FC, Panigrahy D, Tiwari S,
Kupz A and Subbian S: Different modalities of host cell death and
their impact on Mycobacterium tuberculosis infection. Am J
Physiol Cell Physiol. 323:C1444–C1474. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Kornfeld H, Mancino G and Colizzi V: The
role of macrophage cell death in tuberculosis. Cell Death Differ.
6:71–78. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lammas DA, Stober C, Harvey CJ, Kendrick
N, Panchalingam S and Kumararatne DS: ATP-induced killing of
mycobacteria by human macrophages is mediated by purinergic
P2Z(P2X7) receptors. Immunity. 7:433–444. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Kusner DJ and Adams J: ATP-induced killing
of virulent Mycobacterium tuberculosis within human
macrophages requires phospholipase D. J Immunol. 164:379–388. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Laochumroonvorapong P, Paul S, Elkon KB
and Kaplan G: H2O2 induces monocyte apoptosis and reduces viability
of Mycobacterium avium-M. intracellulare within cultured human
monocytes. Infect Immun. 64:452–459. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Fratazzi C, Arbeit RD, Carini C and Remold
HG: Programmed cell death of Mycobacterium avium serovar 4-infected
human macrophages prevents the mycobacteria from spreading and
induces mycobacterial growth inhibition by freshly added,
uninfected macrophages. J Immunol. 158:4320–4327. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Aliprantis AO, Yang RB, Mark MR, Suggett
S, Devaux B, Radolf JD, Klimpel GR, Godowski P and Zychlinsky A:
Cell activation and apoptosis by bacterial lipoproteins through
toll-like receptor-2. Science. 285:736–739. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Brightbill HD, Libraty DH, Krutzik SR,
Yang RB, Belisle JT, Bleharski JR, Maitland M, Norgard MV, Plevy
SE, Smale ST, et al: Host defense mechanisms triggered by microbial
lipoproteins through toll-like receptors. Science. 285:732–736.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Ren Y and Savill J: Apoptosis: The
importance of being eaten. Cell Death Differ. 5:563–568. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Martin CJ, Peters KN and Behar SM:
Macrophages clean up: Efferocytosis and microbial control. Curr
Opin Microbiol. 17:17–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Idh J, Mekonnen M, Abate E, Wedajo W,
Werngren J, Ängeby K, Lerm M, Elias D, Sundqvist T, Aseffa A, et
al: Resistance to first-line anti-TB drugs is associated with
reduced nitric oxide susceptibility in Mycobacterium
tuberculosis. PLoS One. 7:e398912012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Flesch IE and Kaufmann SH: Mechanisms
involved in mycobacterial growth inhibition by gamma
interferon-activated bone marrow macrophages: Role of reactive
nitrogen intermediates. Infect Immun. 59:3213–3218. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Korver AJ: Amputees in a hospital of the
international committee of the red cross. Injury. 24:607–609. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
MacMicking JD, North RJ, LaCourse R,
Mudgett JS, Shah SK and Nathan CF: Identification of nitric oxide
synthase as a protective locus against tuberculosis. Proc Natl Acad
Sci USA. 94:5243–5248. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Denis M: Interferon-gamma-treated murine
macrophages inhibit growth of tubercle bacilli via the generation
of reactive nitrogen intermediates. Cell Immunol. 132:150–157.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Chan J, Xing Y, Magliozzo RS and Bloom BR:
Killing of virulent Mycobacterium tuberculosis by reactive
nitrogen intermediates produced by activated murine macrophages. J
Exp Med. 175:1111–1122. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Jamaati H, Mortaz E, Pajouhi Z, Folkerts
G, Movassaghi M, Moloudizargari M, Adcock IM and Garssen J: Nitric
oxide in the pathogenesis and treatment of tuberculosis. Front
Microbiol. 8:20082017. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Peteroy-Kelly M, Venketaraman V and
Connell ND: Effects of Mycobacterium bovis BCG infection on
regulation of L-arginine uptake and synthesis of reactive nitrogen
intermediates in J774.1 murine macrophages. Infect Immun.
69:5823–5831. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Arias M, Rojas M, Zabaleta J, Rodríguez
JI, París SC, Barrera LF and García LF: Inhibition of virulent
Mycobacterium tuberculosis by Bcg(r) and Bcg(s) macrophages
correlates with nitric oxide production. J Infect Dis.
176:1552–1558. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
122
|
Dlugovitzky D, Bay ML, Rateni L, Vietti L,
Farroni MA and Bottasso OA: Influence of disease severity on
nitrite and cytokine production by peripheral blood mononuclear
cells (PBMC) from patients with pulmonary tuberculosis (TB). Clin
Exp Immunol. 122:343–349. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Ernst WA, Thoma-Uszynski S, Teitelbaum R,
Ko C, Hanson DA, Clayberger C, Krensky AM, Leippe M, Bloom BR, Ganz
T and Modlin RL: Granulysin, a T cell product, kills bacteria by
altering membrane permeability. J Immunol. 165:7102–7108. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Chan ED, Morris KR, Belisle JT, Hill P,
Remigio LK, Brennan PJ and Riches DW: Induction of inducible nitric
oxide synthase-NO* by lipoarabinomannan of Mycobacterium
tuberculosis is mediated by MEK1-ERK, MKK7-JNK, and NF-kappaB
signaling pathways. Infect Immun. 69:2001–2010. 2001. View Article : Google Scholar : PubMed/NCBI
|