Introduction
Osteosarcoma is the most common primary malignant
bone tumor, which predominantly affects young adults and
individuals aged >60 years old. Osteosarcoma typically arises in
the metaphyseal regions of long bones, including the distal femur,
proximal tibia and proximal humerus (1). Despite advancements in treatment, the
5-year survival rate remains at ~60% for localized cases and 20%
for metastatic disease (2,3). Conventional treatment involves a
combination of surgery and chemotherapy. Surgery aims to achieve
complete resection of the tumor, and chemotherapy is administered
both preoperatively (as a neoadjuvant) to shrink the tumor and
postoperatively (as an adjuvant) to eliminate residual disease
(4). However, chemotherapy for
osteosarcoma faces notable challenges, including drug resistance
and the capacity of cancer cells to evade apoptosis (5). Cancer cells can evade apoptosis
through various mechanisms, including the upregulation of
anti-apoptotic proteins (such as Bcl-2), mutations in pro-apoptotic
genes (including p53) and activation of survival pathways (6). These factors contribute to the
limited long-term efficacy of chemotherapy and highlight the
necessity for novel therapeutic approaches (7,8).
Ferroptosis is a programmed form of cell death that
is driven by iron-dependent lipid peroxidation. Unlike apoptosis or
necrosis, ferroptosis is characterized by the accumulation of
lethal lipid reactive oxygen species (ROS) and the depletion of
glutathione (a crucial antioxidant) (9). Key markers of ferroptosis include the
suppression of glutathione peroxidase 4 (GPX4), elevated levels of
lipid peroxides and the presence of iron-dependent ROS (10,11).
In cancer treatment, ferroptosis offers a promising therapeutic
strategy, particularly for overcoming drug resistance. Cancer cells
often develop resistance to conventional therapies by evading
apoptosis; however, inducing ferroptosis can bypass these
resistance mechanisms (9).
Ferroptosis inducers, such as erastin and RSL3, have demonstrated
effectiveness in triggering ferroptosis in various cancer cell
lines, including those resistant to apoptosis (12). Additionally, ferroptosis can
improve the effectiveness of treatments such as chemotherapy,
radiotherapy and immunotherapy by sensitizing cancer cells to these
modalities (13). Therefore,
identifying natural compounds that can induce ferroptosis in cancer
cells is crucial; these compounds may function as chemotherapy
drugs or adjuncts, potentially improving treatment outcomes and
overcoming drug resistance.
Shikonin and acetylshikonin are naphthoquinone
derivatives extracted from the roots of the plant Lithospermum
erythrorhizon (14). Shikonin
has been extensively studied for its anticancer properties; it has
been shown to induce apoptosis, necroptosis and ferroptosis in
various cancer cell lines by generating ROS, inhibiting the EGFR
and PI3K/Akt signaling pathways, and suppressing angiogenesis
(15). Via these mechanisms,
shikonin also enhances the efficacy of conventional therapies such
as chemotherapy and radiotherapy by sensitizing cancer cells to
these treatments (15).
Acetylshikonin shares similar anticancer mechanisms, but has shown
distinct advantages in certain contexts. The acetylation of
shikonin enhances its stability and bioavailability, facilitating
more effective delivery and sustained action within the body
(15). This modification enhances
its ability to induce apoptosis and inhibit cancer cell
proliferation. Additionally, acetylshikonin has been identified as
a novel tubulin polymerization inhibitor, demonstrating marked
antitumor activity in hepatocellular carcinoma (16). Furthermore, ROS production from
acetylshikonin is associated with upregulation of apoptosis
pathways in osteosarcoma cells (17). These properties indicate that
acetylshikonin may be a promising candidate for cancer treatment,
with potential applications in overcoming drug resistance and
improving the efficacy of existing therapies. However, the
mechanism underlying the ability of acetylshikonin to induce cell
death in osteosarcoma remains poorly understood. The present study
aimed to investigate whether acetylshikonin induces ferroptosis in
osteosarcoma cells and to elucidate the underlying mechanisms,
thereby assessing its potential as a novel therapeutic agent.
Materials and methods
Chemicals
Primary antibodies against GPX4 (cat. no. GTX03194),
Bcl-2 (cat. no. GTX100064), Bcl-xl (cat. no. GTX637939), Bak (cat.
no. GTX100063), Bax (cat. no. GTX109683) and β-actin (cat. no.
GTX109639) were acquired from GeneTex, Inc. The secondary
HRP-conjugated anti-rabbit monoclonal (cat. no. sc-2357) antibody
was purchased from Santa Cruz Biotechnology, Inc. The other
chemicals and reagents used in the present study were obtained from
MilliporeSigma, unless otherwise specified.
Cell culture
U2OS is an epithelial-like osteosarcoma cell line
originally derived from the tibial tumor of a 15-year-old Caucasian
female patient. This cell line exhibits a hypertriploid karyotype
with extensive chromosomal rearrangements, including stable marker
chromosomes involving chromosomes 1, 7, 9 and 17. HOS is a
fibroblast- and epithelial-like cell line established from the
osteosarcoma lesion of a 13-year-old Caucasian female patient. This
cell line displays a flat morphology, low saturation density and is
highly sensitive to transformation. MG63 is a fibroblast-like
osteosarcoma cell line derived from the bone of a 14-year-old
Caucasian male patient. This cell line is hypotriploid with a modal
chromosome number of 66, and it consistently exhibits 18–19 marker
chromosomes. All cell lines were authenticated using short tandem
repeat profiling. The human osteosarcoma cell lines (U2OS, HOS and
MG63) and normal human osteoblasts (hFOB 1.19) were sourced from
the American Type Culture Collection. Osteosarcoma cells were
maintained in Dulbecco's Modified Eagle Medium (DMEM) supplemented
with 10% fetal bovine serum, 100 U/ml penicillin and 100 µg/ml
streptomycin. hFOB 1.19 cells were cultured in a 1:1 mixture of
Ham's F-12 Medium and DMEM supplemented with 2.5 mM L-glutamine
(without phenol red), as per the supplier recommendations. All
cells were incubated at 37°C in a humidified atmosphere with 5%
CO2.
Cell viability assays
The hFOB 1.19, U2OS, HOS and MG63 cells were seeded
in 48-well plates at a density of 1×104 cells/well and
were allowed to adhere overnight. The U2OS, HOS and MG63 cells were
then exposed to various concentrations (0, 0.05, 0.1, 0.5, 1, 3, 10
and 20 µM) of acetylshikonin (cat. no. HY-N2181; MedChemExpress)
for 24 or 48 h at 37°C. The hFOB 1.19 cell line was exposed to
different concentrations (0, 0.1, 0.5, 1 and 3 µM) of
acetylshikonin for 24 h at 37°C. Untreated cells were used as
controls. Cell viability was determined using the Cell Counting
Kit-8 (CCK-8; cat. no. 96992; MilliporeSigma), according to the
manufacturer's instructions. Absorbance was measured at 450 nm
using a microplate reader (BioTek; Agilent Technologies, Inc.).
In addition, U2OS, HOS, and MG63 cells were seeded
at 5×104 cells/well on glass coverslips in 24-well
plates and treated with acetylshikonin. (0, 0.5, 1, 2.5, 5 and 10
µM) for 24 h at 37°C. After treatment, the cells were stained with
Hoechst 33342 (5 µg/ml), Calcein-AM (2 µM) and propidium iodide
(PI; 1 µg/ml) at 37°C for 30 min. Hoechst 33342 stained all nuclei
(blue fluorescence), Calcein-AM stained viable cells (green
fluorescence), and PI identified dead cells with compromised
membranes (red fluorescence). Untreated cells were used as
controls. Furthermore, cells treated with acetylshikonin (0 and 3
µM) for 24 h at 37°C were observed using a Nikon Eclipse Ti
fluorescence microscope (Nikon Corporation) to evaluate changes in
morphology and cell density.
To validate the involvement of ferroptosis, U2OS,
HOS and MG63 cells were pretreated with ferrostatin-1 (1 µM; a
ferroptosis inhibitor; cat. no. SML0583; Sigma-Aldrich; Merck KGaA)
or liproxstatin-1 (1 µM; a ferroptosis inhibitor; cat. no. SML1414;
Sigma-Aldrich; Merck KGaA) for 1 h, followed by treatment with
acetylshikonin (3 µM) for 24 h at 37°C. Cell viability was assessed
using the CCK-8 assay, as aforementioned. For comparative analysis,
cells were also treated with erastin (3 µM; a ferroptosis inducer;
cat. no. E7781; Sigma-Aldrich; Merck KGaA) or RSL3 (3 µM; a
ferroptosis inducer; cat. no. SML2234; Sigma-Aldrich) for 24 h at
37°C, and cell viability was measured using the same method. To
differentiate ferroptosis from apoptosis and necroptosis,
osteosarcoma cells (MG63, HOS and U2OS) were pretreated with the
caspase-3 inhibitor z-DEVD-FMK (10 µM; cat. no. 264155;
Sigma-Aldrich), the RIPK1 inhibitor necrostatin-1 (10 µM; cat. no.
480065; Sigma-Aldrich) or the necrosis inhibitor IM-54 (10 µM; cat.
no. SML0412; Sigma-Aldrich) for 1 h prior to treatment with
acetylshikonin (3 µM) for 24 h at 37°C. Untreated cells were used
as controls. Cell viability was subsequently assessed using the
CCK-8 assay, as aforementioned. All experiments were conducted in
quadruplicate (n=4) to ensure reproducibility.
DNA fragmentation analysis
TUNEL enzymatically labels the 3′-ends of fragmented
DNA, a hallmark of apoptosis, using a fluorophore-conjugated
nucleotide (18,19). DNA damage was analyzed using the
TUNEL assay (BD Biosciences). Briefly, cells were treated with
acetylshikonin (0, 0.1, 0.25, 0.5, 1 and 3 µM) for 24 h at 37°C,
followed by fixation with 4% paraformaldehyde in PBS (pH 7.4) for
60 min at room temperature and permeabilization with 0.1% Triton
X-100 in 0.1% sodium citrate on ice for 2 min. TUNEL labeling was
then performed with the reaction mixture containing
fluorescein-dUTP at 37°C for 60 min in a humidified dark chamber.
Finally, DNA strand breaks were detected using a flow cytometer
(Accuri C5; BD Biosciences), and flow cytometry data were acquired
and analyzed using BD Accuri C6 Software (version 227.4; BD
Biosciences).
Analysis of apoptotic and necrotic
cells
To assess apoptotic and necrotic cell populations,
Annexin V/PI staining was performed using an apoptosis detection
kit (cat. no. APOAF; Sigma-Aldrich; Merck KGaA). Briefly, cells
were seeded at a density of 5×105 cells/well in 6-well
plates and treated with acetylshikonin at 0, 0.1, 0.25, 0.5, 1 or 3
µM for 24 h at 37°C. Cells were then stained with 1 µg/ml PI and
0.5 µg/ml FITC-conjugated Annexin V for 15 min at 37°C, and
analyzed by flow cytometry (Accuri C5; BD Biosciences). Flow
cytometry data were acquired and analyzed using BD Accuri C6
Software (version 227.4; BD Biosciences). For ferroptosis
inhibition experiments, cells were pretreated with ferrostatin-1
(10 µM) for 1 h at 37°C prior to acetylshikonin treatment (3 µM)
for 24 h at 37°C.
Cell cycle analysis
Cells were seeded in 6-well plates at a density of
5×105 cells/well and treated with acetylshikonin (0,
0.1, 0.5, 1 and 3 µM) for 24 h at 37°C. After treatment, the cells
were fixed with 75% ethanol at −20°C for ≥2 h. After washing, the
cells were stained with PI solution (0.1% Triton X-100, 0.2 mg/ml
RNase A (cat. no. 70856; Merck KGaA), 10 µg/ml PI (cat. no. P4170;
Sigma-Aldrich; Merck KGaA) for 30 min at 37°C and analyzed using
flow cytometry (Accuri C5; BD Biosciences). Untreated cells were
used as controls. Flow cytometry data were acquired and analyzed
using BD Accuri C6 Software (version 227.4; BD Biosciences).
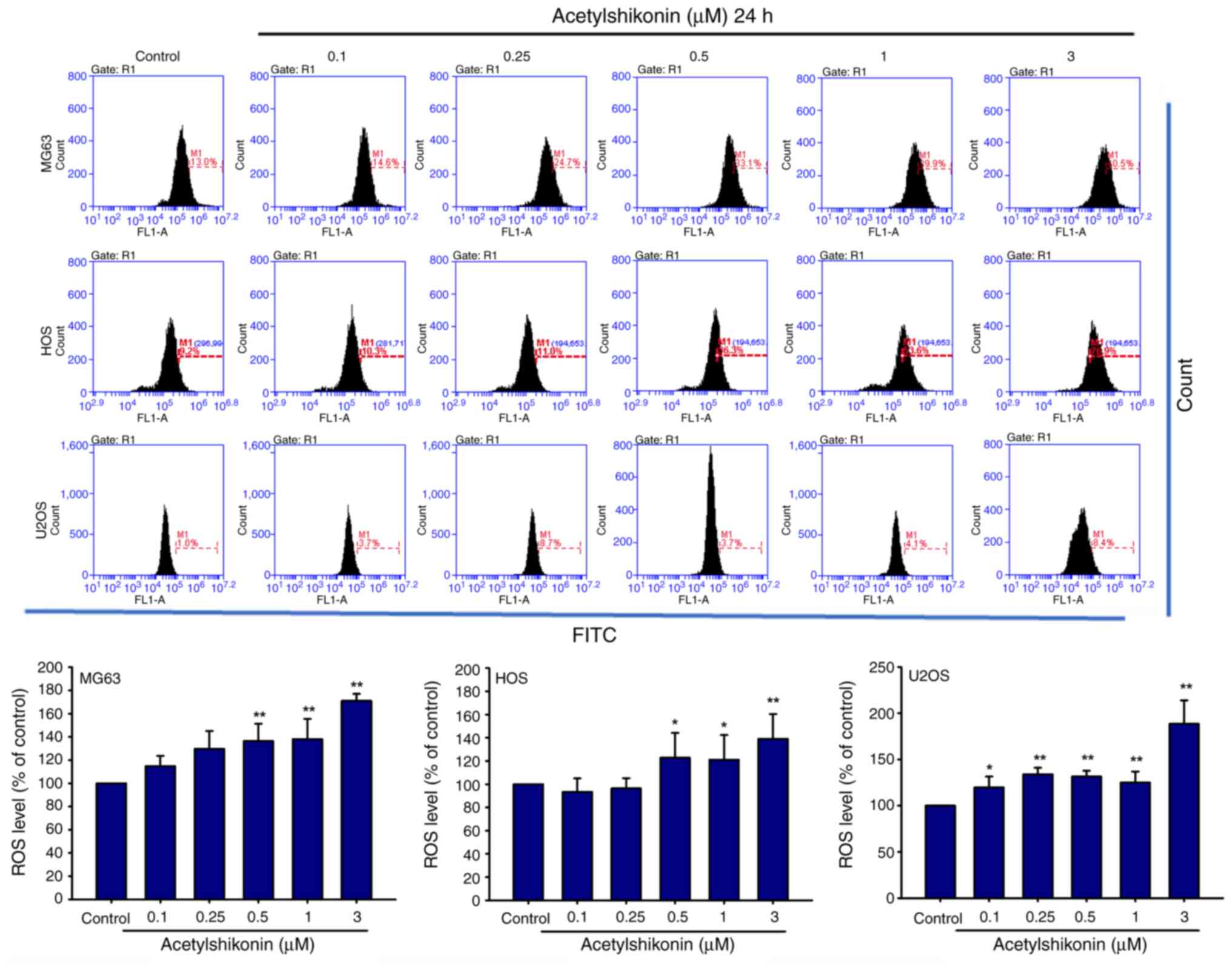
Cellular ROS assay
H2DCFDA (Thermo Fisher Scientific, Inc.)
is a non-fluorescent compound that, after intracellular
deacetylation, emits green fluorescence upon oxidation by ROS
(excitation ~511 nm, emission ~533 nm). Intracellular ROS levels
were determined using H2DCFDA in the present study.
Cells (5×105) were treated with acetylshikonin (0, 0.1,
0.25, 0.5, 1 and 3 µM) for 1 h at 37°C, followed by incubation with
1 µM H2DCFDA at 37°C for 30 min. Untreated cells were
used as controls. ROS levels were analyzed via flow cytometry
(Accuri C5; BD Biosciences). Flow cytometry data were acquired and
analyzed using BD Accuri C6 Software (version 227.4; BD
Biosciences).
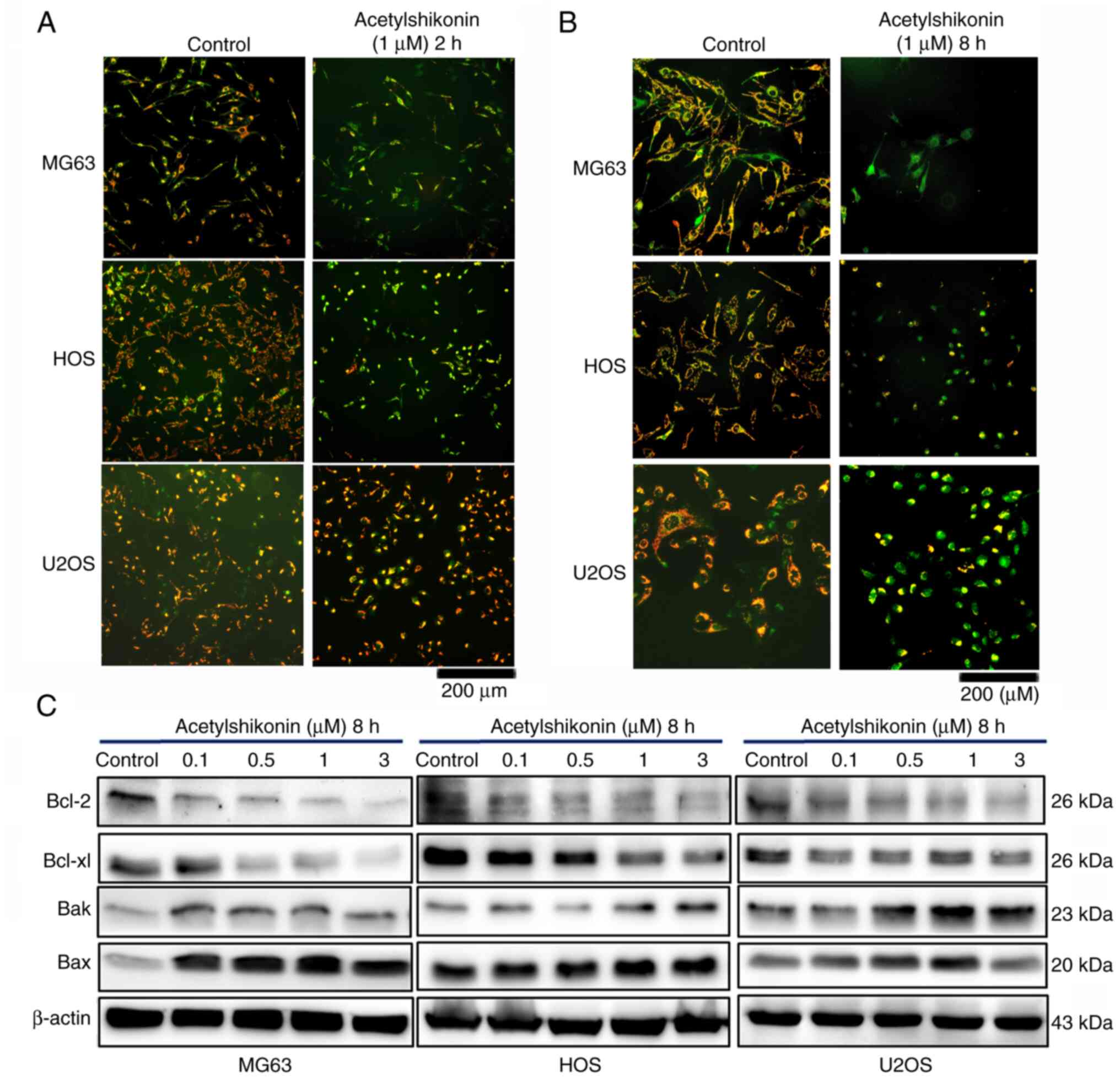
Analysis of mitochondrial membrane
potential
Mitochondrial membrane potential was assessed using
the cationic dye JC-1 (Thermo Fisher Scientific, Inc.); in healthy
mitochondria, it forms red-fluorescent J-aggregates, whereas in
depolarized or apoptotic mitochondria, JC-1 remains in a monomeric
form, emitting green fluorescence. Briefly, cells were incubated
with acetylshikonin (0 and 1 µM) for 2 or 8 h and were subsequently
stained with JC-1 (5 µg/ml) for 30 min at 37°C. Untreated cells
were used as controls. Fluorescence intensity was observed using a
Nikon Eclipse Ti fluorescence microscope (Nikon Corporation).
Western blotting
Cells were seeded at a density of 5×105
cells/well in 6-well plates and treated with acetylshikonin (0,
0.1, 0.5, 1, and 3 µM) for 24 h at 37°C. After treatment, the cells
were lysed with RIPA buffer [50 mM Tris-HCl (pH 7.4), 150 mM NaCl,
1% NP-40, 0.25% sodium deoxycholate, 0.1–2% SDS], supplemented with
a protease inhibitor mix (1 mM PMSF, 10 ng/ml leupeptin, 1 ng/ml
aprotinin). Protein concentration was determined using the BCA
Protein Assay Kit (cat. no. 71285-M; Merck KGaA). Equal amounts of
protein (50 µg/lane) were loaded and separated by SDS-PAGE on 15%
gels, followed by transfer to PVDF membranes (MilliporeSigma).
Membranes were blocked with 5% non-fat milk in TBS-0.05% Tween-20
for 1 h at room temperature and incubated overnight at 4°C with
primary antibodies (1:1,000 dilution). After washing, the membranes
were incubated with HRP-conjugated secondary antibodies (1:10,000
dilution) for 1 h at room temperature. Protein bands were
visualized using an enhanced chemiluminescence detection system
(Analytik Jena US, LLC).
Transmission electron microscopy
analysis
Following treatment with acetylshikonin (0 and 3 µM)
for 24 h at 37°C, HOS cells (5×105) were collected,
fixed in 70% Karnovsky fixative at 4°C, and processed for
ultrastructural examination. Samples were dehydrated through a
graded ethanol series, embedded in Epon 812 resin (polymerized at
60°C for 48 h) and sectioned into ultrathin slices (70–90 nm) using
an ultramicrotome. Sections were stained with 2% uranyl acetate for
20 min at room temperature, followed by Reynolds's lead citrate for
10 min at room temperature. Images were obtained using a JEOL
JEM-1400 transmission electron microscope (JEOL, Ltd.).
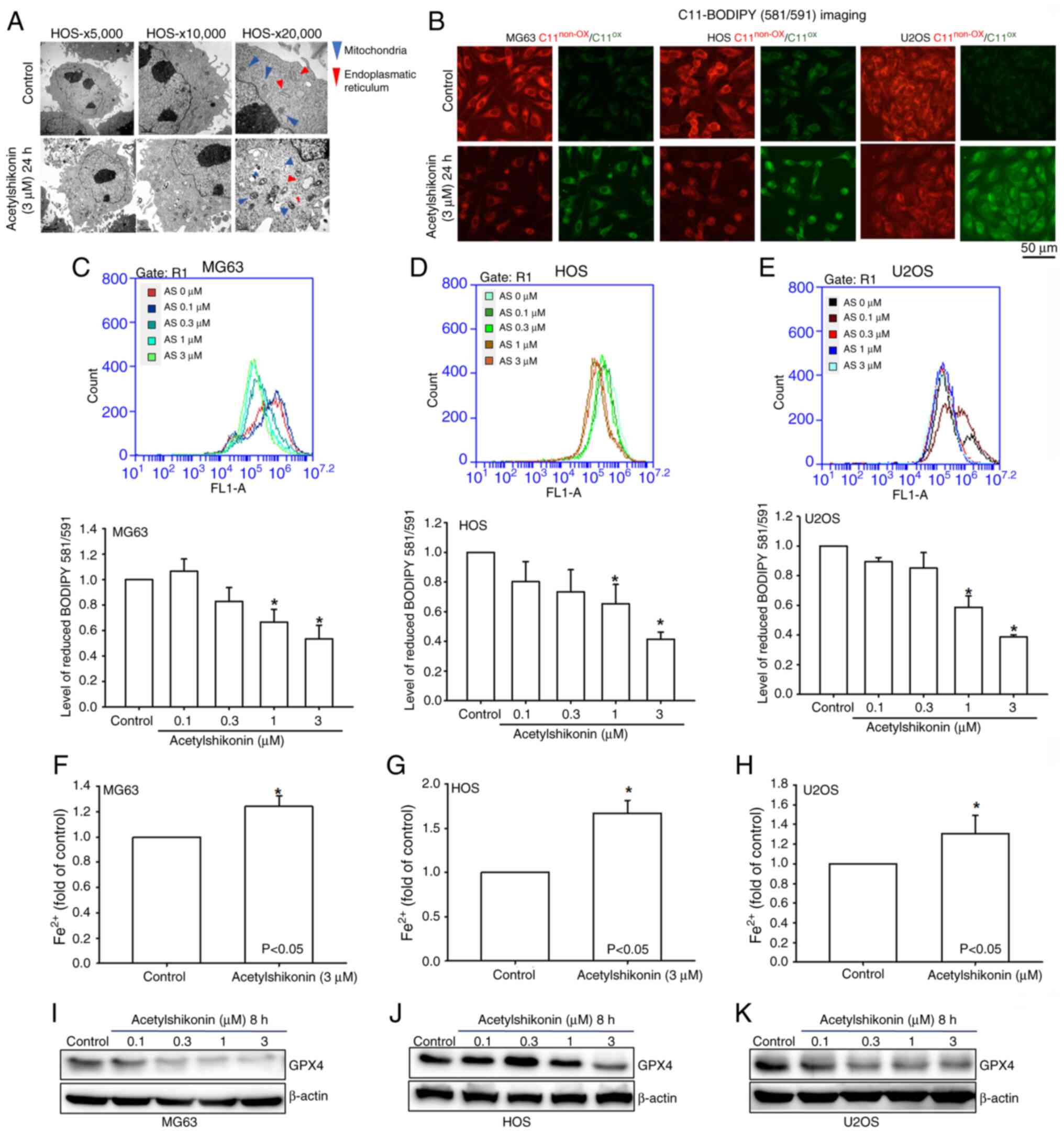
Analysis of lipid peroxidation
C11-BODIPY™ 581/591 (Thermo Fisher Scientific, Inc.)
is a lipid peroxidation sensor dye that shifts its fluorescence
from red (~590 nm) to green (~510 nm) upon oxidation. Imaging and
quantification of C11-BODIPY 581/591 staining were performed using
fluorescence microscopy and flow cytometry in the current study.
Briefly, cells (5×104) were incubated with
acetylshikonin (0, 0.1, 0.3, 1 and 3 µM for 30 min at 37°C and C11
BODIPY dye at 37°C for 30 min. Lipid peroxidation was subsequently
determined based on fluorescence shifts from 591 nm (non-oxidized)
to 510 nm (oxidized) using fluorescence microscopy and flow
cytometry (Accuri C5; BD Biosciences). Untreated cells were used as
controls. Flow cytometry data were acquired and analyzed using BD
Accuri C6 Software (version 227.4; BD Biosciences).
Analysis of intracellular ferrous ion
(Fe2+) concentrations
Osteosarcoma cells (HOS, MG63 and U2OS) were treated
with acetylshikonin (0 and 3 µM) for 24 h at 37°C. After treatment,
the cells were lysed with the buffer solution provided in the
Fe2+ Colorimetric Assay Kit (cat. no. E-BC-K881-M;
Elabscience Bionovation Inc.), which contains Tris-based buffer
components, according to the manufacturer's protocol. For each
assay, 1×106 cells were lysed in 200 µl buffer on ice
for 10 min with gentle mixing every 5 min. Lysates were centrifuged
at 15,000 × g for 10 min at 4°C, and the supernatant was incubated
with the Fe2+ detection reagent for 30 min at room
temperature. Untreated cells were used as controls. Absorbance was
measured at 593 nm using a microplate reader (Thermo Fisher
Scientific, Inc.).
Statistical analysis
Data are presented as the mean ± SD from at least
four independent experiments. Statistical comparisons between
groups were conducted using one-way ANOVA followed by Tukey's
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Acetylshikonin reduces the viability
of osteosarcoma cell lines and enhanced membrane permeability
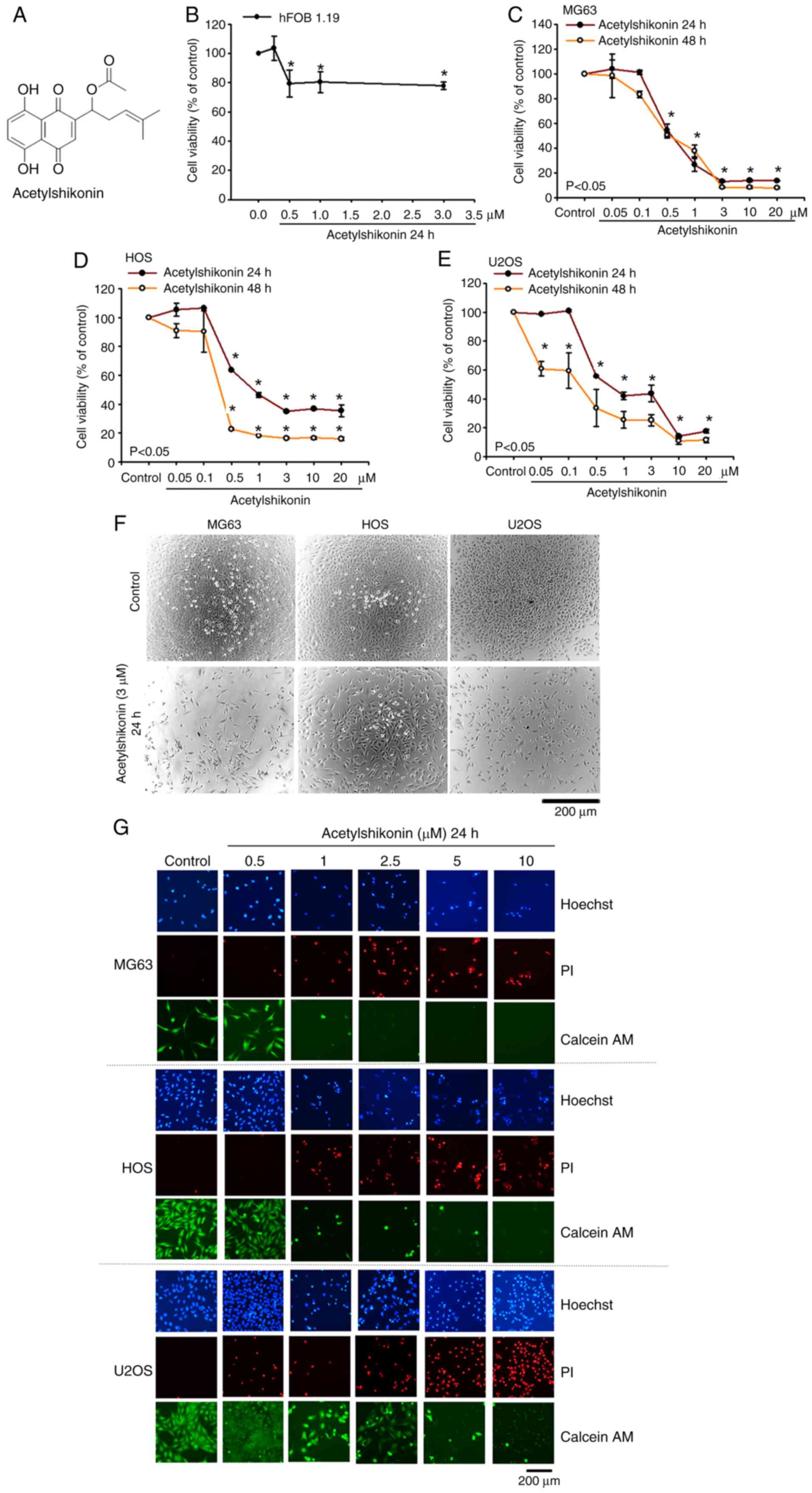
The present study first assessed the effect of
acetylshikonin (Fig. 1A) on the
viability of both normal osteoblasts (hFOB 1.19) and osteosarcoma
cells. After treating hFOB 1.19 cells with acetylshikonin (0.5–3
µM) for 24 h, cell viability remained at ~80% (Fig. 1B), indicating low toxicity in
normal cells. By contrast, the IC50 values for MG63, HOS
and U2OS osteosarcoma cells after 24 h of treatment with
acetylshikonin (0.5–20 µM) were 0.45, 0.83 and 0.69 µM,
respectively (Fig. 1C-E). After 48
h of treatment, the IC50 values decreased to 0.40, 0.28
and 0.17 µM, respectively. In addition, phase-contrast microscopy
revealed a marked reduction in the confluence of osteosarcoma cell
lines following acetylshikonin (3 µM) treatment (Fig. 1F). Hoechst 33342 staining also
showed a marked decrease in the number of MG63, HOS and U2OS cells
after treatment with acetylshikonin (0.5–10 µM). Hoechst 33342
staining revealed dose-dependent increases in apoptotic nuclear
condensation (brighter, punctate nuclei) in U2OS cells treated with
acetylshikonin, which is associated with reduced cell viability
(Fig. 1G). Additionally,
acetylshikonin enhanced the uptake of PI dye into cells in a
dose-dependent manner, indicating compromised cell membrane
integrity. Calcein-AM staining further confirmed that
acetylshikonin notably reduced the viability of osteosarcoma cells.
These results indicated that acetylshikonin has a potent cytotoxic
effect on osteosarcoma cell lines, primarily through mechanisms
associated with cell permeability. Notably, acetylshikonin
demonstrates low toxicity to normal cells, highlighting its
potential as a promising therapeutic agent for osteosarcoma
treatment.
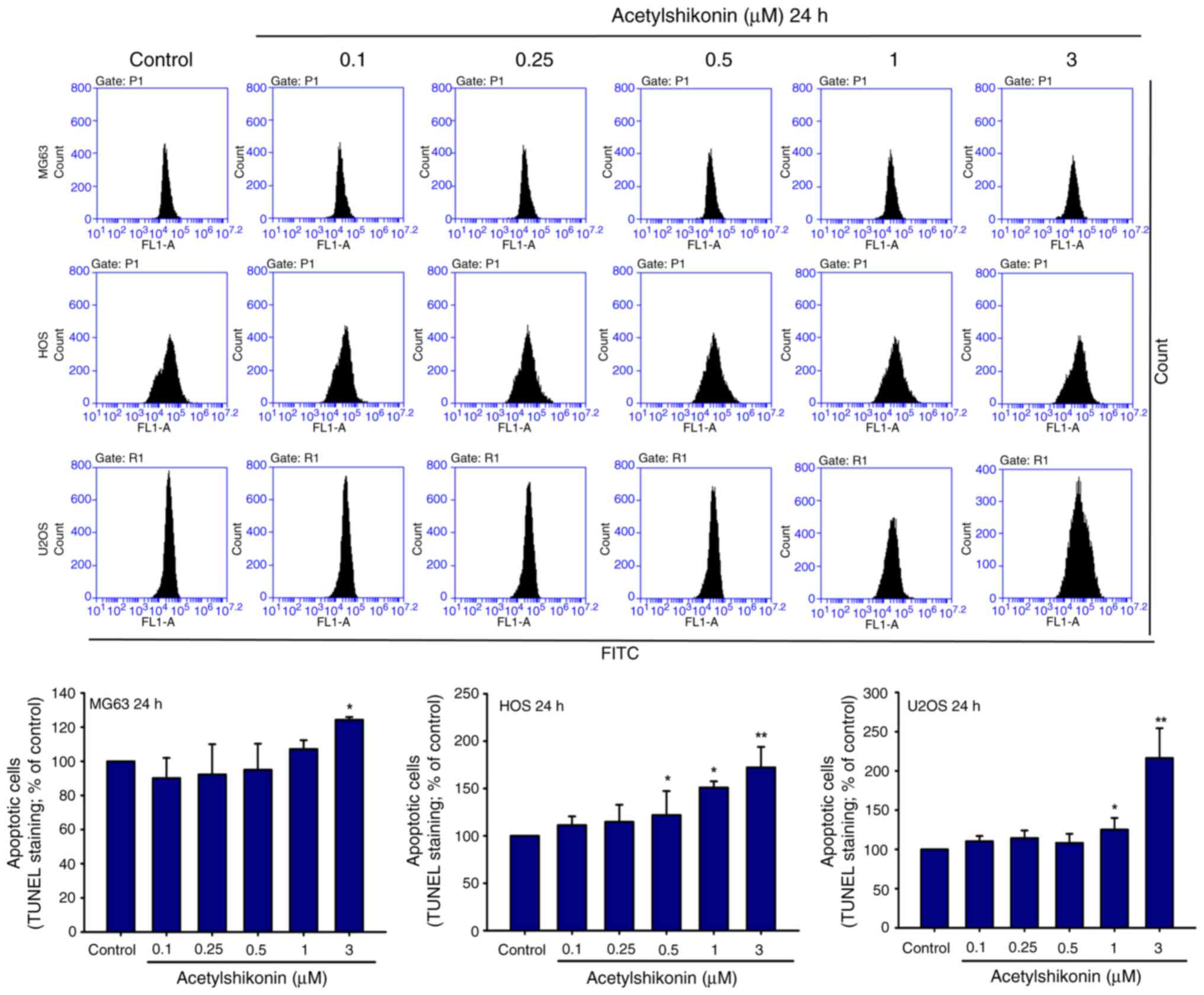
Acetylshikonin induces DNA
fragmentation and cell death in osteosarcoma cells
To elucidate the mechanism by which acetylshikonin
affects osteosarcoma cell viability, MG63, HOS and U2OS cells were
treated with acetylshikonin (0.1–3 µM) for 24 h, and DNA
fragmentation was assessed using the TUNEL assay. A significant
increase in DNA fragmentation was observed in all three cell lines
following treatment with acetylshikonin (Fig. 2). Treatment with 3 µM
acetylshikonin significantly increased the percentage of
TUNEL-positive cells compared with that in the untreated control
group.
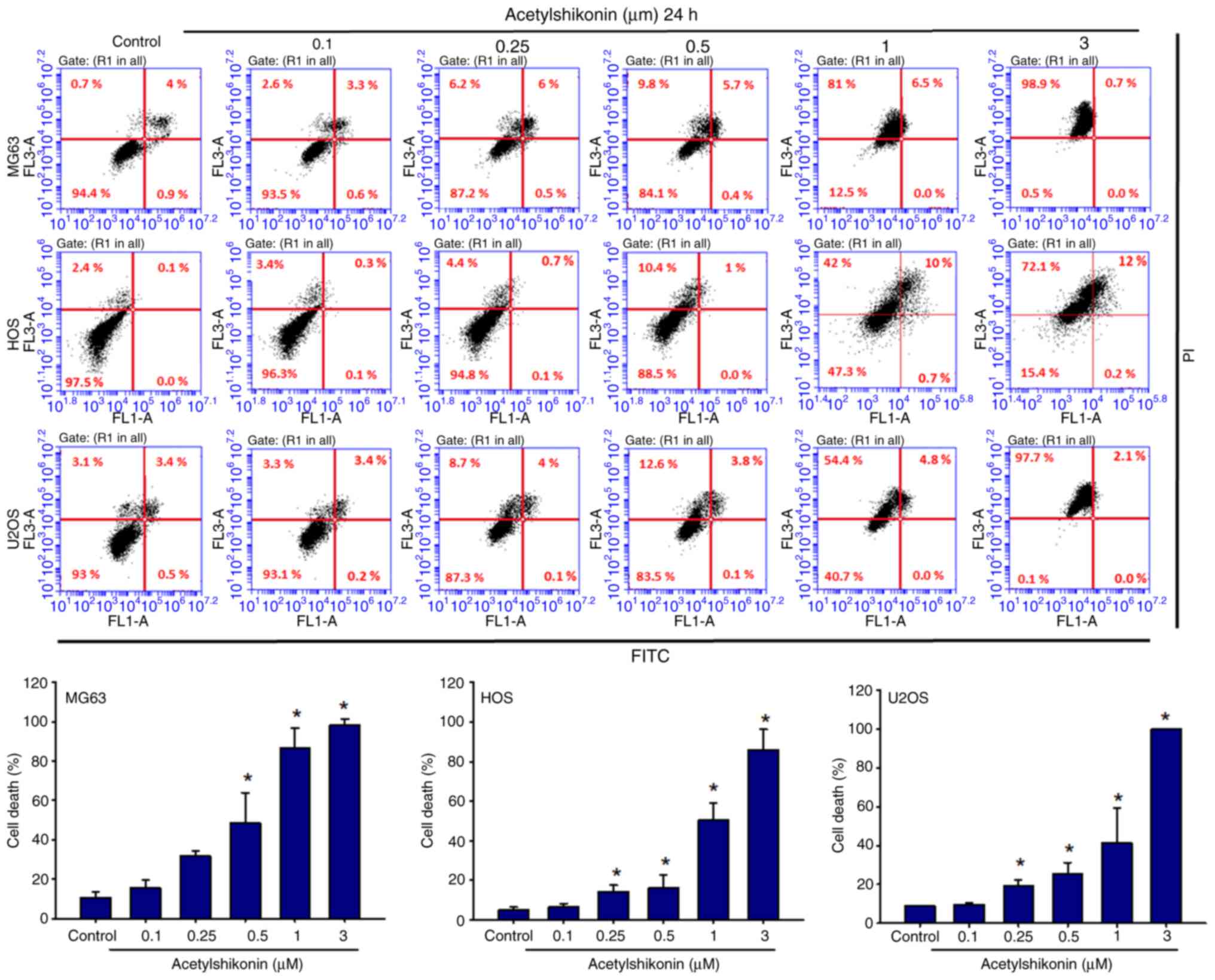
Flow-cytometric apoptosis profiling after 24 h of
acetylshikonin treatment (0.0-0.5 µM) showed increases in
upper-right (UR; Annexin V+/PI+) and
upper-left (UL; PI+ only) quadrant populations. In
particular, following treatment with 0.5 µM acetylshikonin, in MG63
cells, the percentage of UL quadrant cells increased from 0.7 to
9.8% and UR quadrant cells increased from 4.0 to 5.7%); in HOS
cells the percentage of UL quadrant cells increased from 2.4 to
10.4% and UR quadrant cells increased from 0.1 to 1.0%; and in U2OS
cells the percentage of UL quadrant cells increased from 3.1 to
12.6% and UR quadrant cells increased from 3.4 to 3.8%, these
findings indicated that the cells were undergoing apoptosis. By
contrast, at higher doses (1–3 µM), the major population shifted to
PI+/Annexin V− cells in the UL quadrant, with
a concomitant decrease in Annexin V+/PI+
cells in the UR quadrant, indicating a dose-dependent increase in
necrotic cell death rather than late apoptosis/secondary necrosis
(Fig. 3). Quantification in
Fig. 3 was performed by combining
Annexin V+/PI+ and Annexin
V−/PI+ populations. Statistical analysis
showed that acetylshikonin treatment at 1–3 µM significantly
increased the proportion of PI+ cells, mainly due to the
enhanced Annexin V−/PI+ population.
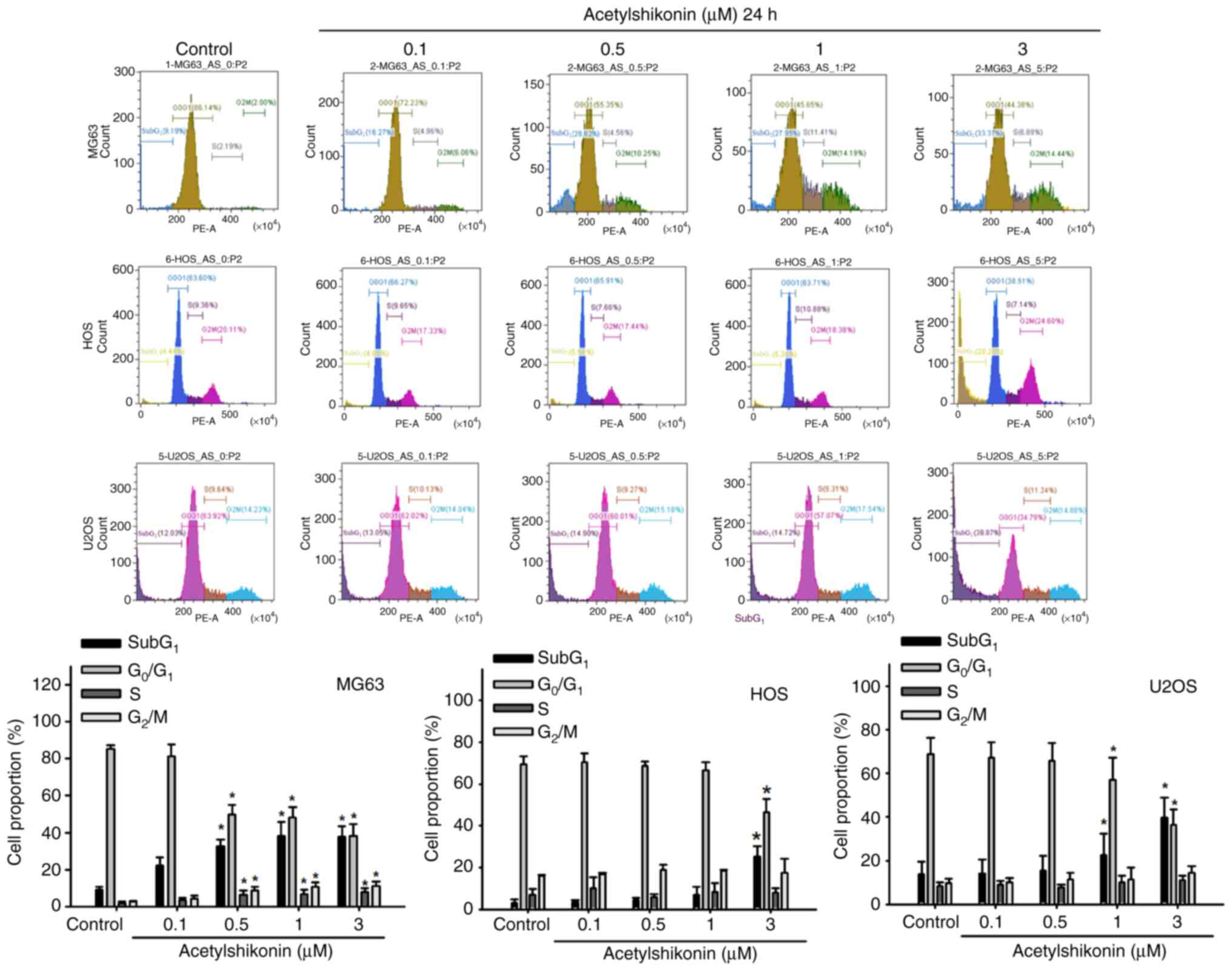
The current study also examined the effect of
acetylshikonin on cell cycle distribution. Acetylshikonin treatment
significantly increased the number of sub-G1 cells,
indicative of DNA fragmentation, and decreased the number of
G0/G1 cells in osteosarcoma cell lines
(Fig. 4). A significant increase
in the sub-G1 population was observed in all cell lines
following 3 µM acetylshikonin treatment. Taken together, the
increase in DNA fragmentation (TUNEL assay), the predominance of
PI+ cells at higher doses (Annexin V/PI analysis), and
the accumulation of sub-G1 cells (cell cycle analysis)
indicated that acetylshikonin may disrupt cellular integrity and
induce cell death in osteosarcoma cells through mechanisms that may
differ from conventional apoptosis.
Acetylshikonin induces ROS production
and loss of mitochondrial membrane potential
Given the established anticancer effects of ROS
generation induced by shikonin and acetylshikonin in various cancer
cells (15,17), the current study investigated the
effect of acetylshikonin on intracellular ROS levels in
osteosarcoma cells. Treatment with acetylshikonin at 0.5–3 µM
resulted in a significant increase in ROS production in
osteosarcoma cell lines (Fig. 5).
Acetylshikonin at 3 µM significantly increased intracellular ROS
levels compared with those in the untreated control group
(P<0.01). This increase in ROS may be strongly associated with
cell death and mitochondrial dysfunction (20,21).
The present study next examined how acetylshikonin influences
mitochondrial membrane potential in osteosarcoma cells to further
elucidate its effects on mitochondrial function. Administration of
acetylshikonin (1 µM) disrupted the mitochondrial membrane
potential, assessed by fluorescence microscopy, as indicated by a
reduction in JC-1 red fluorescence (Fig. 6A and B). A marked reduction in
mitochondrial membrane potential, indicated by the JC-1 red/green
fluorescence ratio, was observed after acetylshikonin
treatment.
Furthermore, western blot analysis revealed that
acetylshikonin treatment increased the levels of pro-apoptotic
proteins Bax and Bak, which regulate mitochondrial membrane
permeability, while decreasing the expression levels of
anti-apoptotic proteins Bcl-2 and Bcl-xl (Fig. 6C). These results suggested that
acetylshikonin induces osteosarcoma cell death through ROS
generation and the loss of mitochondrial membrane potential, which
may be associated with apoptosis. However, additional experimental
data are required to fully elucidate the underlying mechanisms.
Acetylshikonin induces ferroptosis in
osteosarcoma cells
Mitochondria are the primary generators of
intracellular ROS and serve a crucial role in regulating iron
homeostasis (22). Using
transmission electron microscopy, the effect of acetylshikonin on
mitochondrial morphology was observed in osteosarcoma cells.
Treatment with acetylshikonin reduced mitochondrial volume without
inducing cell swelling or shrinkage, which are morphological
characteristics of ferroptosis (Fig.
7A) (22). The current study
further investigated whether acetylshikonin induces lipid
peroxidation in osteosarcoma cell lines. Fluorescence microscopy
and flow cytometry revealed that acetylshikonin (1–3 µM)
significantly induced lipid peroxidation, as evidenced by the shift
in BODIPY dye fluorescence from red to green (Fig. 7B-E). Treatment with 3 µM
acetylshikonin significantly increased lipid peroxidation, as
indicated by enhanced green fluorescence of C11-BODIPY compared
with that in the control group. Additionally, treatment with
acetylshikonin (3 µM) significantly increased intracellular
Fe2+ concentrations in osteosarcoma cells compared with
those in untreated cells (Fig.
7F-H). The present study also observed a reduction in GPX4
protein expression in acetylshikonin-treated osteosarcoma cells
compared with that in the untreated control group, as shown by
western blotting (Fig. 7I-K).
These findings suggested that acetylshikonin induces osteosarcoma
cell death through mechanisms involving mitochondrial size
reduction, lipid peroxidation and a decrease in GPX4 levels, which
are indicative of ferroptosis.
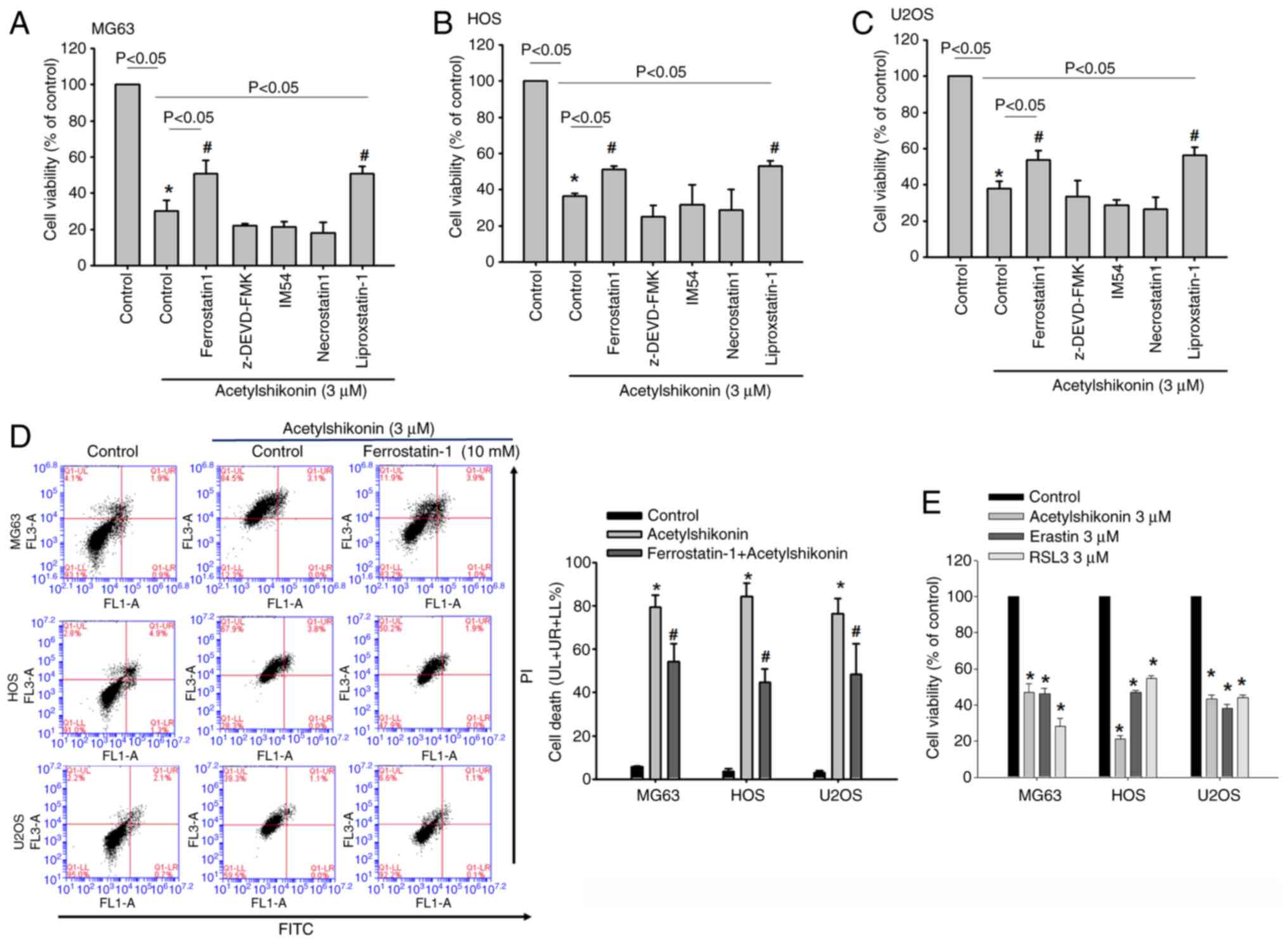
To further confirm that acetylshikonin-induced cell
death is ferroptosis-dependent, the current study compared the
protective effects of two ferroptosis inhibitors, ferrostatin-1 and
liproxstatin-1. Pretreatment with either ferrostatin-1 (1 µM) or
liproxstatin-1 (1 µM) for 1 h significantly reversed the reduction
in cell viability caused by acetylshikonin (3 µM) in osteosarcoma
cells (Fig. 8A-C). To rigorously
exclude apoptosis and necroptosis as alternative cell-death
pathways, cells were treated with the caspase-3 inhibitor
z-DEVD-FMK, the RIPK1 inhibitor necrostatin-1 and the necrosis
inhibitor IM-54. None of these inhibitors rescued
acetylshikonin-induced cytotoxicity (Fig. 8A-C). These results provide
additional pharmacological evidence that the cell death induced by
acetylshikonin is mediated through ferroptosis. Similar results
were observed in flow cytometry with Annexin V/PI staining.
Treatment with acetylshikonin markedly increased the proportion of
PI+ cells, calculated as the combined Annexin
V+/PI+ and Annexin
V−/PI+ populations, whereas pretreatment with
ferrostatin-1 significantly reduced this PI+ population
in osteosarcoma cell lines (Fig.
8D). Although the reduction was mainly attributable to a
decrease in Annexin V−/PI+ cells, this subset
was not analyzed statistically on its own. Additionally, the
cytotoxic potency of acetylshikonin was compared with two classical
ferroptosis inducers, erastin and RSL3. Treatment with erastin (3
µM) or RSL3 (3 µM) for 24 h resulted in a significant decrease in
cell viability, similar in magnitude to that caused by
acetylshikonin (Fig. 8E). These
findings indicated that acetylshikonin exhibits
ferroptosis-inducing activity comparable to established inducers,
further reinforcing its potential as a therapeutic ferroptosis
activator in osteosarcoma cells. These results experimentally
demonstrated that acetylshikonin induces ferroptosis in
osteosarcoma cells. This form of cell death is distinct from
classic necrosis and apoptosis, highlighting the potential of
acetylshikonin as a novel therapeutic agent targeting ferroptosis
in osteosarcoma cells.
Discussion
The present study investigated the potential of
acetylshikonin to induce cell death in osteosarcoma cells. The
results demonstrated that acetylshikonin significantly reduced the
viability of osteosarcoma cell lines while exhibiting low toxicity
to normal cells. Mechanistically, acetylshikonin induced ROS
production, disrupted the mitochondrial membrane potential and
promoted lipid peroxidation, ultimately leading to ferroptosis.
Additionally, treatment with acetylshikonin resulted in decreased
levels of GPX4 and increased intracellular Fe2+
concentrations, further supporting its role in the induction of
ferroptosis. These findings suggested that acetylshikonin may
address the limitations of conventional chemotherapy by targeting
ferroptosis, a unique form of cell death. This highlights the
potential of acetylshikonin as a promising therapeutic agent for
osteosarcoma, offering a novel treatment option that could enhance
patient outcomes and address the limitations of current
chemotherapy.
Chemotherapeutic agents have long been a cornerstone
of cancer treatment, which primarily function by inducing apoptosis
in cancer cells. However, their efficacy is frequently hindered by
substantial challenges, including drug resistance and severe side
effects (23,24). The development of drug resistance
and the ability of cancer cells to evade apoptosis represent key
adaptations that ultimately result in treatment failure and disease
progression (23,24). Ferroptosis, a form of programmed
cell death that is distinct from apoptosis, presents a promising
alternative target for cancer therapy. Characterized by elevated
lipid peroxides dependent on iron, the production of ROS and the
suppression of GPX4, ferroptosis circumvents the conventional
mechanisms associated with apoptosis (9,12,25,26).
Inducing ferroptosis in cancer cells can help overcome drug
resistance and enhance the effectiveness of existing treatments,
including chemotherapy, radiotherapy and immunotherapy (9,12).
The current findings are consistent with these concepts, and
notably, acetylshikonin-induced ferroptosis was further confirmed
by the reversal of cell death following treatment with
ferrostatin-1, underscoring the critical involvement of ferroptosis
processes in acetylshikonin-mediated cytotoxicity.
To further distinguish ferroptosis from other forms
of regulated cell death, inhibitor rescue assays were performed
using the caspase-3 inhibitor z-DEVD-FMK (apoptosis), the RIPK1
inhibitor necrostatin-1 (necroptosis) and the necrosis inhibitor
IM-54 across three osteosarcoma cell lines. None of these
inhibitors significantly reversed the cytotoxic effects of
acetylshikonin. By contrast, ferrostatin-1 conferred substantial
protection. These findings strengthen the conclusion that
acetylshikonin-induced cell death is mediated through a
ferroptosis-specific mechanism rather than apoptosis or
necroptosis. These findings suggest that acetylshikonin may address
the limitations of conventional chemotherapy by targeting
ferroptosis, thereby providing a novel approach for treating
osteosarcoma.
Shikonin, a naphthoquinone derivative derived from
Lithospermum erythrorhizon, has demonstrated notable
anticancer properties against various types of cancer, including
breast, lung and colorectal cancer (15). Its mechanisms of action include the
induction of apoptosis, necroptosis and immunogenic cell death, as
well as the inhibition of angiogenesis and key signaling pathways,
such as EGFR and PI3K/Akt (27,28).
Shikonin also enhances the efficacy of conventional therapies by
sensitizing cancer cells to chemotherapy and radiotherapy (28,29).
However, its clinical application is limited by poor stability, low
bioavailability and potential toxicity to normal cells (29). Acetylshikonin, a shikonin
derivative, overcomes these limitations and demonstrates potential
as a more effective candidate for cancer treatment. The acetylation
of shikonin enhances its stability and bioavailability,
facilitating more effective delivery and sustained action within
the body (15). Acetylshikonin has
been reported to trigger ROS-dependent apoptosis in hepatocellular
carcinoma and to activate necroptosis via the RIPK1/RIPK3 signaling
axis in lung cancer cells (16,30).
However, most of these studies have predominantly focused on
classical cell death pathways, and the role of acetylshikonin in
ferroptosis has remained largely unexplored. The present findings
provide novel evidence that acetylshikonin selectively induces
ferroptosis in osteosarcoma cells by promoting lipid peroxidation,
increasing intracellular Fe2+ levels and suppressing
GPX4 expression. These results highlight a previously unrecognized
mechanism for acetylshikonin, which could have important
therapeutic implications in overcoming therapeutic resistance in
osteosarcoma.
Although the present study primarily focused on the
in vitro anticancer effects of acetylshikonin, several
previous investigations have provided valuable insights into its
pharmacokinetic characteristics. In radiolabeled mouse studies,
[3H]-acetylshikonin became detectable in plasma within
15 min of oral administration, with peak concentrations occurring
at ~1 h. Notably, the compound showed widespread tissue
distribution, particularly in the gastrointestinal tract and liver;
however, it exhibited poor systemic absorption, with >80% of the
administered dose recovered in feces within 48 h, suggesting
limited bioavailability (31).
Furthermore, acetylshikonin has demonstrated high plasma protein
binding, and in mice, its parent form was reported to be
undetectable in circulation, complicating direct pharmacokinetic
analysis (30). In a non-human
primate model, Sun et al (32) employed a derivatization-based
liquid chromatography-electrospray ionization-tandem mass
spectrometry assay, and revealed that oral administration of
acetylshikonin resulted in a terminal half-life of 12.3±1.6 h and a
mean residence time of 10.2±0.7 h, indicating prolonged systemic
exposure. In terms of chemical stability, acetylshikonin has been
reported to be relatively stable under acidic aqueous-organic
conditions. For example, in pH 3.0 glycine buffer containing 50%
ethanol, its half-life was measured as 67.0±9.6 h at 40°C and
31.3±3.3 h at 70°C (33). These
findings suggest that while acetylshikonin demonstrates favorable
chemical stability and residence time, its limited oral
bioavailability remains a key limitation. Future in vivo
studies should therefore focus on formulation strategies and
structural modifications to enhance its absorption and therapeutic
potential in osteosarcoma models.
The present study utilized U2OS, HOS and MG63
osteosarcoma cell lines, which represent diverse genetic profiles
of osteosarcoma. In the HOS cell line, a TP53 mutation has been
confirmed, classifying it as a p53-mutated cell line. Therefore,
HOS represents a model of p53-mutated osteosarcoma with a
functional RB pathway (34). The
U2OS cell line is classified as p53 wild-type or expressing,
maintaining functional p53 expression. It harbors a heterozygous
BRCA2 mutation, with likely preservation of one intact allele, and
shows reduced or absent RB1 protein expression, suggesting a
defective RB pathway. Thus, U2OS exhibits partial BRCA2 deficiency
combined with a compromised RB pathway (34,35).
MG63 cells are characterized by a complete absence of TP53
(classified as p53-null), and markedly reduced or absent RB1
protein expression. This cell line demonstrates genomic
instability, homologous recombination deficiency, loss of
heterozygosity and a ‘BRCAness’ phenotype, typical of
BRCA1/2-mutant tumors, marked by disruptive alterations in DNA
repair pathways (34,35). While these cell lines model varying
degrees of tumor aggressiveness and genetic alterations, they may
not fully capture the molecular diversity of clinical osteosarcoma
subtypes, particularly those with metastatic or drug-resistant
phenotypes. To address this limitation, future studies should
include additional cell lines, such as SJSA-1 (TP53-mutated, highly
invasive) and 143B (a metastatic HOS derivative with enhanced
migratory capacity), to better evaluate the therapeutic potential
of acetylshikonin across diverse osteosarcoma subtypes.
From a clinical perspective, the present findings
suggest that acetylshikonin could serve as a promising
complementary agent in osteosarcoma treatment. In traditional
Chinese medicine, natural compounds are often used alone or in
combination with Western therapies to enhance antitumor efficacy
and reduce treatment-related toxicity. The integration of natural
products into conventional treatment regimens is gaining increasing
attention in oncology (36–38).
Given the substantial side effects and the frequent emergence of
drug resistance associated with current standard chemotherapies for
osteosarcoma, including doxorubicin, cisplatin and high-dose
methotrexate, the introduction of a ferroptosis-inducing agents
such as acetylshikonin may provide synergistic benefits (39,40).
Specifically, combining acetylshikonin with standard
chemotherapeutic agents could enhance therapeutic effectiveness by
simultaneously activating multiple, non-redundant cell death
pathways. Moreover, the minimal toxicity of acetylshikonin toward
normal osteoblasts, as observed in the present study, supports its
feasibility as an adjunctive treatment strategy.
To provide a broader context, the present findings
may be compared with those of previous studies on the anticancer
effects of acetylshikonin. Prior studies have demonstrated that
acetylshikonin induces apoptosis in hepatocellular carcinoma and
triggers necroptosis through the RIPK1/RIPK3 pathway in lung cancer
cells (15,30). However, its role in ferroptosis has
remained largely uncharacterized. The current study is among the
first to show that acetylshikonin may promote ferroptosis in
osteosarcoma cells by enhancing lipid peroxidation, increasing
intracellular Fe2+ concentrations and downregulating
GPX4 expression, representing a novel mechanistic contribution to
the understanding of this compound (10,11,41).
To assess clinical feasibility, previous reports have highlighted
the limitations of high-dose MAP regimens (methotrexate,
doxorubicin and cisplatin) in metastatic or recurrent osteosarcoma,
and have identified iron metabolism as a targetable vulnerability
associated with ROS production and tumor resistance (39,42,43).
Furthermore, curcumin enhances methotrexate efficacy by suppressing
the Hedgehog signaling pathway, thereby overcoming resistance and
reducing proliferation (39).
Based on these insights, acetylshikonin may represent a potential
complementary agent to activate non-redundant cell death pathways,
warranting future investigation into its synergistic potential with
conventional chemotherapeutics.
In summary, the present study highlights
acetylshikonin, a naphthoquinone derivative, as a promising
therapeutic candidate for osteosarcoma. Acetylshikonin reduced
osteosarcoma cell viability and selectively promoted ferroptosis by
increasing ROS production, disrupting mitochondrial function,
enhancing lipid peroxidation, downregulating GPX4 and elevating
intracellular iron levels. These findings suggest that
acetylshikonin may help overcome therapeutic resistance and improve
outcomes in osteosarcoma. Future studies should evaluate its
efficacy in vivo, explore its potential in combination with
standard chemotherapy agents, and assess its pharmacokinetic and
safety profiles to facilitate clinical translation.
Acknowledgements
Not applicable.
Funding
This work was supported by a grant from the National Science and
Technology Council of Taiwan (NSTC113-2628-B-038-008-MY3).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JYC was responsible for data collection,
conceptualization, methodology development, investigation and
preparation of the original draft. GSW contributed to
conceptualization, project administration, investigation and
software analysis. TMC performed formal analysis, contributed to
methodology and project administration, and conducted software
analysis. JFL conceptualized the study, developed the methodology,
performed investigation, collected data, acquired funding, and
critically reviewed and edited the manuscript. TMC and JFL confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ottaviani G and Jaffe N: The epidemiology
of osteosarcoma. Cancer Treat Res. 152:3–13. 2009. View Article : Google Scholar
|
|
2
|
Meltzer PS and Helman LJ: New Horizons in
the treatment of osteosarcoma. N Engl J Med. 385:2066–2076. 2021.
View Article : Google Scholar
|
|
3
|
Belayneh R, Fourman MS, Bhogal S and Weiss
KR: Update on osteosarcoma. Curr Oncol Rep. 23:712021. View Article : Google Scholar
|
|
4
|
Lilienthal I and Herold N: Targeting
molecular mechanisms underlying treatment efficacy and resistance
in osteosarcoma: A review of current and future strategies. Int J
Mol Sci. 21:68852020. View Article : Google Scholar
|
|
5
|
Haider T, Pandey V, Banjare N, Gupta PN
and Soni V: Drug resistance in cancer: Mechanisms and tackling
strategies. Pharmacol Rep. 72:1125–1151. 2020. View Article : Google Scholar
|
|
6
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35 (Suppl 1):S78–S103. 2015. View Article : Google Scholar
|
|
7
|
Wang J, Li M, Guo P and He D: Correction:
Survival benefits and challenges of adjuvant chemotherapy for
high-grade osteosarcoma: A population-based study. J Orthop Surg
Res. 18:8342023. View Article : Google Scholar
|
|
8
|
Hiraga H and Ozaki T: Adjuvant and
neoadjuvant chemotherapy for osteosarcoma: JCOG bone and soft
tissue tumor study group. Jpn J Clin Oncol. 51:1493–1497. 2021.
View Article : Google Scholar
|
|
9
|
Zhang C, Liu X, Jin S, Chen Y and Guo R:
Ferroptosis in cancer therapy: A novel approach to reversing drug
resistance. Mol Cancer. 21:472022. View Article : Google Scholar
|
|
10
|
Zhou N and Bao J: FerrDb: A manually
curated resource for regulators and markers of ferroptosis and
ferroptosis-disease associations. Database (Oxford).
2020:baaa0212020. View Article : Google Scholar
|
|
11
|
Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y,
Sun Y, Zeng F, Chen X and Deng G: Ferroptosis in cancer: From
molecular mechanisms to therapeutic strategies. Signal Transduct
Target Ther. 9:552024. View Article : Google Scholar
|
|
12
|
Nie Q, Hu Y, Yu X, Li X and Fang X:
Induction and application of ferroptosis in cancer therapy. Cancer
Cell Int. 22:122022. View Article : Google Scholar
|
|
13
|
Wu Y, Yu C, Luo M, Cen C, Qiu J, Zhang S
and Hu K: Ferroptosis in cancer treatment: Another way to rome.
Front Oncol. 10:5711272020. View Article : Google Scholar
|
|
14
|
Hayashi M: Pharmacological studies of
Shikon and Tooki. (2) Pharmacological effects of the pigment
components, Shikonin and acetylshikonin. Nihon Yakurigaku Zasshi.
73:193–203. 1977.(In Japanese). View Article : Google Scholar
|
|
15
|
Wang Q, Wang J, Wang J, Ju X and Zhang H:
Molecular mechanism of shikonin inhibiting tumor growth and
potential application in cancer treatment. Toxicol Res (Camb).
10:1077–1084. 2021. View Article : Google Scholar
|
|
16
|
Hu S, Li Y, Zhou J, Xu K, Pang Y,
Weiskirchen R, Ocker M and Ouyang F: Identification of
acetylshikonin as a novel tubulin polymerization inhibitor with
antitumor activity in human hepatocellular carcinoma cells. J
Gastrointest Oncol. 14:2574–2586. 2023. View Article : Google Scholar
|
|
17
|
Cha HS, Lee HK, Park SH and Nam MJ:
Acetylshikonin induces apoptosis of human osteosarcoma U2OS cells
by triggering ROS-dependent multiple signal pathways. Toxicol In
Vitro. 86:1055212023. View Article : Google Scholar
|
|
18
|
Majtnerova P and Rousar T: An overview of
apoptosis assays detecting DNA fragmentation. Mol Biol Rep.
45:1469–1478. 2018. View Article : Google Scholar
|
|
19
|
Loo DT: TUNEL assay. An overview of
techniques. Methods Mol Biol. 203:21–30. 2002.
|
|
20
|
Wang B, Wang Y, Zhang J, Hu C, Jiang J, Li
Y and Peng Z: ROS-induced lipid peroxidation modulates cell death
outcome: Mechanisms behind apoptosis, autophagy, and ferroptosis.
Arch Toxicol. 97:1439–1451. 2023. View Article : Google Scholar
|
|
21
|
Rizwan H, Pal S, Sabnam S and Pal A: High
glucose augments ROS generation regulates mitochondrial dysfunction
and apoptosis via stress signalling cascades in keratinocytes. Life
Sci. 241:1171482020. View Article : Google Scholar
|
|
22
|
Battaglia AM, Chirillo R, Aversa I, Sacco
A, Costanzo F and Biamonte F: Ferroptosis and cancer: Mitochondria
meet the ‘Iron Maiden’ cell death. Cells. 9:15052020. View Article : Google Scholar
|
|
23
|
Fu B, Lou Y, Wu P, Lu X and Xu C: Emerging
role of necroptosis, pyroptosis, and ferroptosis in breast cancer:
New dawn for overcoming therapy resistance. Neoplasia.
55:1010172024. View Article : Google Scholar
|
|
24
|
Sajeev A, Sailo B, Unnikrishnan J,
Talukdar A, Alqahtani MS, Abbas M, Alqahtani A, Sethi G and
Kunnumakkara AB: Unlocking the potential of Berberine: Advancing
cancer therapy through chemosensitization and combination
treatments. Cancer Lett. 597:2170192024. View Article : Google Scholar
|
|
25
|
Chen X, Comish PB, Tang D and Kang R:
Characteristics and biomarkers of ferroptosis. Front Cell Dev Biol.
9:6371622021. View Article : Google Scholar
|
|
26
|
Chen F, Kang R, Tang D and Liu J:
Ferroptosis: Principles and significance in health and disease. J
Hematol Oncol. 17:412024. View Article : Google Scholar
|
|
27
|
Yadav S, Sharma A, Nayik GA, Cooper R,
Bhardwaj G, Sohal HS, Mutreja V, Kaur R, Areche FO, AlOudat M, et
al: Review of shikonin and derivatives: Isolation, chemistry,
biosynthesis, pharmacology and toxicology. Front Pharmacol.
13:9057552022. View Article : Google Scholar
|
|
28
|
Iranzadeh S, Dalil D, Kohansal S and
Isakhani M: Shikonin in breast cancer treatment: A comprehensive
review of molecular pathways and innovative strategies. J Pharm
Pharmacol. 76:967–982. 2024. View Article : Google Scholar
|
|
29
|
Qi K, Li J, Hu Y, Qiao Y and Mu Y:
Research progress in mechanism of anticancer action of shikonin
targeting reactive oxygen species. Front Pharmacol. 15:14167812024.
View Article : Google Scholar
|
|
30
|
Lin SS, Chang TM, Wei AI, Lee CW, Lin ZC,
Chiang YC, Chi MC and Liu JF: Acetylshikonin induces necroptosis
via the RIPK1/RIPK3-dependent pathway in lung cancer. Aging (Albany
NY). 15:14900–14914. 2023. View Article : Google Scholar
|
|
31
|
Zhang Z, Bai J, Zeng Y, Cai M, Yao Y, Wu
H, You L, Dong X and Ni J: Pharmacology, toxicity and
pharmacokinetics of acetylshikonin: A review. Pharm Biol.
58:950–958. 2020. View Article : Google Scholar
|
|
32
|
Sun DX, Tian HF, Meng ZY, Du A, Yuan D, Gu
RL, Wu ZN and Dou GF: Quantitative determination of acetylshikonin
in macaque monkey blood by LC-ESI-MS/MS after precolumn
derivatization with 2-mercaptoethanol and its application in
pharmacokinetic study. Acta Pharmacol Sin. 29:1499–1506. 2008.
View Article : Google Scholar
|
|
33
|
Cho MH, Paik YS and Hahn TR: Physical
stability of shikonin derivatives from the roots of Lithospermum
erythrorhizon cultivated in Korea. J Agric Food Chem. 47:4117–4120.
1999. View Article : Google Scholar
|
|
34
|
Zhang Q, Hao S, Wei G, Liu X and Miao Y:
The p53-mediated cell cycle regulation is a potential mechanism for
emodin-suppressing osteosarcoma cells. Heliyon. 10:e268502024.
View Article : Google Scholar
|
|
35
|
Engert F, Kovac M, Baumhoer D, Nathrath M
and Fulda S: Osteosarcoma cells with genetic signatures of BRCAness
are susceptible to the PARP inhibitor talazoparib alone or in
combination with chemotherapeutics. Oncotarget. 8:48794–48806.
2017. View Article : Google Scholar
|
|
36
|
Wei J, Liu Z, He J, Liu Q, Lu Y, He S,
Yuan B, Zhang J and Ding Y: Traditional Chinese medicine reverses
cancer multidrug resistance and its mechanism. Clin Transl Oncol.
24:471–482. 2022. View Article : Google Scholar
|
|
37
|
Miao K, Liu W, Xu J, Qian Z and Zhang Q:
Harnessing the power of traditional Chinese medicine monomers and
compound prescriptions to boost cancer immunotherapy. Front
Immunol. 14:12772432023. View Article : Google Scholar
|
|
38
|
Yuan J, Liu Y, Zhang T, Zheng C, Ding X,
Zhu C, Shi J and Jing Y: Traditional Chinese medicine for breast
cancer treatment: A bibliometric and visualization analysis. Pharm
Biol. 62:499–512. 2024. View Article : Google Scholar
|
|
39
|
Argenziano M, Tortora C, Pota E, Di Paola
A, Di Martino M, Di Leva C, Di Pinto D and Rossi F: Osteosarcoma in
children: Not only chemotherapy. Pharmaceuticals (Basel).
14:9232021. View Article : Google Scholar
|
|
40
|
Giliberti G, Marrapodi MM, Di Feo G, Pota
E, Di Martino M, Di Pinto D, Rossi F and Di Paola A: Curcumin and
methotrexate: A promising combination for osteosarcoma treatment
via hedgehog pathway inhibition. Int J Mol Sci. 25:113002024.
View Article : Google Scholar
|
|
41
|
Hsu PC, Tsai CC, Lin YH and Kuo CY:
Therapeutic targeting of apoptosis, autophagic cell death,
necroptosis, pyroptosis, and ferroptosis pathways in oral squamous
cell carcinoma: Molecular mechanisms and potential strategies.
Biomedicines. 13:17452025. View Article : Google Scholar
|
|
42
|
Ma X, Zhao J and Feng H: Targeting iron
metabolism in osteosarcoma. Discov Oncol. 14:312023. View Article : Google Scholar
|
|
43
|
Zhao J, Zhao Y, Ma X, Zhang B and Feng H:
Targeting ferroptosis in osteosarcoma. J Bone Oncol. 30:1003802021.
View Article : Google Scholar
|