Introduction
Cardiovascular disease (CVD) is recognized as one of
the leading causes of global morbidity and mortality (1,2),
with its associated disease burden posing a substantial public
health challenge. As reported by the American Heart Association,
CVD is responsible for 32.9% of global non-communicable disease
mortality, accounting for ~18 million deaths per year (3). CVD comprises a spectrum of
pathological conditions, including coronary artery disease,
myocardial hypertrophy (MH), hypertension, heart failure (HF) and
peripheral artery disease (4,5). The
prevalence of CVD has been steadily increasing, driven by
population aging, the rising incidence of metabolic disorders and
lifestyle modifications, leading to a marked escalation in
socioeconomic burden (6).
MH serves an important role in the pathological
progression of CVD and serves as a key etiological factor in
numerous severe cardiovascular complications (7). As a compensatory pathological
adaptation, MH is primarily characterized by cardiomyocyte
responses to mechanical stress or neurohumoral stimuli, which
sustain cardiac output and function (8). This pathological remodeling is
typically triggered by chronic pressure overload, such as
hypertension, or volume overload, such as valvular regurgitation
(9). Notably, pathological cardiac
hypertrophy has been identified as a notable contributor to HF
(10), resulting in elevated
global mortality rates. Consequently, an in-depth investigation of
the molecular mechanisms underlying pathological cardiac
hypertrophy is important for the prevention or reversal of HF.
Accumulating evidence indicates that the
phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)
signaling cascade is closely associated with MH (11–13).
The PI3K/AKT pathway not only regulates fundamental biological
processes, including cell migration, protein translation and cell
survival, but also modulates energy metabolism, vascular
homeostasis and thrombosis (14).
In cardiomyocytes, dysregulated activation of this pathway can
markedly influence the progression of MH (15), rendering it a key therapeutic
target in CVD research. Matairesinol has been shown to attenuate
stress-induced MH by upregulating peroxiredoxin-1 expression and
suppressing the PI3K/AKT/forkhead box protein O1 (FoxO1) axis
(16). Similarly, astragaloside
IV, a bioactive compound derived from Traditional Chinese Medicine,
has been reported to inhibit cardiac hypertrophy by blocking
TANK-binding kinase 1/PI3K/AKT signaling (17). Furthermore, a study reported by
Qian et al (18)
demonstrated that hanhuangqin significantly alleviated
isoproterenol (ISO)-induced MH both in vivo and in
vitro by inhibiting the PI3K/AKT hypertrophic pathway. Thus,
downregulation of PI3K/AKT signaling may represent a promising
therapeutic strategy.
ATP-binding cassette (ABC) subfamily C member 9
(ABCC9), an ABC transporter protein (19), is expressed in cardiac tissues,
suggesting its potential involvement in cardiac drug responses or
physiological processes. In cardiomyocytes, ABCC9 encodes
sulfonylurea receptor (SUR) 2A, an ‘atypical’ ABC transporter
protein (20). Despite sharing
structural features with typical ABC proteins, SUR2A does not
function as a transporter; instead, it serves as a regulatory
subunit that binds to the inwardly rectifying potassium channel
Kir6.2 (KCNJ11) (21). Together,
SUR2A and KCNJ11 form ATP-sensitive potassium channels (KATP) in
the ventricular myocyte membrane (22). The physiological functions of KATP
channels are complex. It was found that although SUR2 deficiency
causes vasospasm and increases the risk of sudden death, it also
enhances the resistance of cardiomyocytes to ischemic injury
(23). Additionally, Fahrenbach
et al (24) demonstrated
that ABCC9/SUR2A is important for the transition of the neonatal
heart from fetal glycolytic metabolism to mature oxidative
metabolism. Furthermore, SUR2A has been implicated in regulating
myocardial resistance to metabolic and oxidative stress, as well as
cardiac senescence (25).
Cantu syndrome (CS), a complex disorder caused by
gain-of-function mutations in ABCC9 and ATP-sensitive inward
rectifier potassium channel 8 (26,27),
is characterized by cardiovascular abnormalities, including
vasodilation (28), cardiac
hypertrophy and other structural or functional defects (29). Currently, no targeted therapy
exists for CS, and the reversibility of its cardiovascular
manifestations remains ambiguous. Although ABCC9 has been
implicated as a potential regulator of MH (30), its precise mechanistic role remains
undetermined. Specifically, whether ABCC9 modulates ISO-induced MH
and the underlying molecular pathways requires further
investigation.
The present study aimed to examine the expression
level of ABCC9 in ISO-induced hypertrophic AC16 cardiomyocytes and
evaluate the biological effects of ABCC9 knockdown. Furthermore,
the present study sought to determine whether ABCC9 silencing
attenuated MH and improved cardiac function by suppressing the
PI3K/AKT signaling axis, thereby proposing a novel therapeutic
approach for ISO-induced MH.
Materials and methods
Chemicals and reagents
ISO hydrochloride (cat. no. HY-B0468) was purchased
from MedChemExpress. The Color Reverse Transcription Kit (with gDNA
Remover) (cat. no. A0010CGQ) and the 2× Color SYBR Green qPCR
Master Mix (ROX2; cat. no. A0012-R2) used for qRT-PCR were
purchased from EZBioscience. An anti-ABCC9 antibody (cat. no.
40229) was purchased from Signalway Antibody LLC. Antibodies
against B-cell lymphoma 2 protein (Bcl-2; cat. no. 15071T),
Bcl-2-associated X protein (Bax; cat. no. 2772T), caspase-3 (cat.
no. 9662S), cleaved caspase-3 (cat. no. 9661S), phosphorylated
(p)-PI3K (cat. no. 17366T), PI3K (cat. no. 4292S), p-AKT (cat. no.
9271T), AKT (cat. no. 4691T) and β-actin (cat. no. 3700S) were
purchased from Cell Signaling Technology, Inc. Atrial natriuretic
peptide (ANP; cat. no. ab189921) and brain natriuretic peptide
(BNP; cat. no. ab236101) were purchased from Abcam. GAPDH (cat. no.
sc-32233) and secondary antibodies were purchased from Santa Cruz
Biotechnology, Inc., including mouse anti-rabbit IgG-HRP (cat. no.
sc-2357) and mouse IgGκ light chain binding protein conjugated to
HRP (cat. no. sc-516102). Necessary experimental materials,
including PVDF membrane, RIPA buffer, Actin-Tracker Red-594 (a
microfilament red fluorescent probe),
2′-7′-dichlorodihydrofluorescein diacetate (DCFH-DA), DAPI staining
solution, mitochondrial membrane potential (MMP) assay kit with
JC-1 and BCA protein detection kit were purchased from Beyotime
Biotechnology. Annexin V-FITC/PI apoptosis detection kit was
purchased from Shanghai Yeasen Biotechnology Co., Ltd. iF488-Wheat
Germ Agglutinin (WGA; a green fluorescent dye) was purchased from
Wuhan Servicebio Technology Co., Ltd. 740Y-P (PI3K/AKT activator;
cat. no. HY-P0175) was purchased from MedChemExpress.
Cell culture
The immortalized human cardiomyocyte AC16 cell line
was purchased from Ningbo Mingzhou Biotechnology Co., Ltd. (cat.
no. MZ-4038), and the cells were cultured in DMEM (Shanghai Basal
Media Technologies Co., Ltd.) high-dextran culture medium
supplemented with 10% fetal bovine serum (Vazyme Biotech Co., Ltd.)
and 1% penicillin/streptomycin solution (Shanghai Epizyme
Biomedical Technology Co., Ltd.) at 37°C, with a carbon dioxide
concentration of 5%, in an incubator. Upon reaching logarithmic
growth phase, cells were seeded into 6-well plates at uniform
density and allowed to adhere for 24 h prior to experimental
treatments.
Small interfering RNA (siRNA)-mediated
gene silencing
The ABCC9-targeting siRNA (si-ABCC9) and the
negative control (si-NC) were designed and synthesized by Suzhou
Synbio Technologies Co., Ltd. Silencing of the ABCC9 gene in AC16
cells was accomplished by the use of Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.), a process that
resulted in a notable downregulation of gene expression. When cells
were grown to ~50% confluence, 5 µl Lipofectamine® 3000
and 50 nM siRNA were diluted separately using 125 µl Opti-MEM
(Gibco; Thermo Fisher Scientific, Inc.). After 15 min of incubation
at room temperature, the mixtures were combined. Concurrently,
culture medium was replaced with fresh medium to maintain optimal
cell health during transfection. The prepared
siRNA-Lipofectamine® 3000 complexes were then added to
cells in 6-well plates. Following a 24 h incubation at 37°C with 5%
CO2, transfection efficiency was assessed using western
blotting, and all subsequent experiments were performed 24 h after
transfection. The oligo sequences used for RNA interference (shown
in the 5′-3′ direction) were as follows: ABCC9 siRNA-1, sense
GGUCAGAUUUGCAGUCAAATT, antisense UUUGACUGCAAAUCUGACCTT; ABCC9
siRNA-2, sense CAACGAUGGUGUACUACAATT, antisense
UUGUAGUACACCAUCGUUGTT; ABCC9 siRNA-3, sense GCGUGAUUCUGCUCUAUAATT,
antisense UUAUAGAGCAGAAUCACGCTT; and si-NC, sense
GCGACGAUCUGCCUAAGAUTT, antisense AUCUUAGGCAGAUCGUCGCTT.
Cell viability assay
AC16 cell viability was determined using the Cell
Counting Kit-8 (CCK-8; Beyotime Biotechnology) assay according to
the manufacturer's instructions. Briefly, cells (5,000 cells/well)
were cultured in 96-well culture plates overnight at 37°C and
treated with different concentrations of ISO (0, 5, 10, 20 and 40
µmol/l) for 24 h. Subsequently, cells were rinsed once with
phosphate-buffered saline (PBS), 10 µl CCK-8 solution was added to
each well and cells were incubated at 37°C for 2 h. Finally, the
absorbance of each well was measured at 450 nm using a microplate
reader (PT-3502PC; Bio-Equip.com). A line graph of optical density
(OD) vs. ISO concentration was plotted, the experiment was repeated
three times and cell viability was calculated as follows: Cell
viability=(treated OD value-blank OD value)/(control OD value-blank
OD value) ×100.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted using TRIzol®
reagent (Invitrogen; Thermo Fisher Scientific, Inc.) and RNA
concentration was quantified using a NanoDrop®
spectrophotometer (Thermo Fisher Scientific, Inc.). cDNA synthesis
was carried out using the Color Reverse Transcription Kit (with
gDNA Remover) (cat. no. A0010CGQ; EZBioscience) with the following
thermal profile: 42°C for 15 min followed by 85°C for 5 sec.
Subsequently, qPCR was performed on a Roche
LightCycler®96 Real-time Quantitative Fluorescence PCR
instrument using the 2× Color SYBR Green qPCR Master Mix (ROX2;
cat. no. A0012-R2; EZBioscience) under the following conditions:
Initial denaturation at 95°C for 5 min, followed by denaturation at
95°C for 30 sec, annealing at 58°C for 30 sec and extension at 72°C
for 30 sec; a total of 40 cycles were performed. The primer
sequences were as follows: ANP, forward
5′-CAACGCAGACCTGATGGATTT-3′, reverse 5′-AGCCCCCGCTTCTTCATTC-3; BNP,
forward 5′-TGGAAACGTCCGGGTTACAG-3′, reverse
5′-CTGATCCGGTCCATCTTCCT-3′; β-myosin heavy chain (β-MHC), forward
5′-TCACCAACAACCCCTACGATT-3′, reverse 5′-CTCCTCAGCGTCATCAATGGA-3′;
and β-actin, forward 5′-CACCATTGGCAATGAGCGGTTC-3′, reverse
5′-AGGTCTTTGCGGATGTCCACGT-3′. The 2−ΔΔCq method
(31) was used to assess mRNA
expression levels.
Western blot analysis
After treatment as described previously (ISO 10 µM;
24 h; si-ABCC9 50 nmol/l; 24 h; 740Y-P 10 µM; 24 h; 37°C), cells
from each experimental group (Control; ISO; ISO + si-NC; ISO +
si-ABCC9; ISO + si-ABCC9 + 740Y-P; ISO + 740Y-P) were washed three
times with cold PBS and lysed for 30 min in RIPA buffer (cat. no.
P0013C; Beyotime Biotechnology) supplemented with a protease and
phosphatase inhibitor cocktail (50X; cat. no. P1045; Beyotime
Biotechnology). The mixture was centrifuged at 12,000 × g for 30
min at 4°C, and the supernatant protein concentration was measured
using the BCA Protein Quantification Kit (cat. no. P0009; Beyotime
Biotechnology). Equal amounts of proteins (30 µg per lane) were
separated by 10% SDS-PAGE and transferred onto PVDF membranes. The
membranes were blocked with 5% non-fat dry milk for 1 h at room
temperature and subsequently incubated overnight at 4°C with the
following antibodies: ABCC9 (1:1,000), ANP (1:1,000), BNP
(1:1,000), Bax (1:1,000), Bcl-2 (1:1,000), Caspase-3 (1:1,000),
p-PI3K (1:1,000), PI3K (1:1,000), p-AKT (1:1,000), AKT (1:1,000),
GAPDH (1:5,000) and β-actin (1:5,000). Subsequently, after washing
with TBST (0.1% Tween-20), membranes were incubated with
anti-rabbit (1:50,000) and anti-mouse (1:50,000) secondary
antibodies for 1 h at room temperature. After washing again,
protein bands were visualized using BeyoECL Moon chemiluminescent
substrate (cat. no. P0018FS; Beyotime Biotechnology) and imaged
with a Tanon-4600 chemiluminescence system (Tanon Science and
Technology Co., Ltd.). Band intensities were semi-quantified using
ImageJ software (version 1.53, National Institutes of Health). All
western blot analyses were performed using the same batch of
protein lysates. Within each figure panel, all target proteins and
their corresponding loading controls were obtained from the same
experimental run under identical conditions.
Rhodamine-phalloidin staining
Cellular surface area was assessed using
rhodamine-phalloidin staining. Briefly, AC16 cardiomyocytes were
washed twice with PBS and fixed with 4% paraformaldehyde for 20 min
at room temperature. After three PBS washes, cells were
permeabilized with 0.1% Triton X-100 in PBS for 15 min at room
temperature. Following additional PBS washes, cells were incubated
with rhodamine-phalloidin working solution (diluted 1:100 in PBS
containing 1% BSA and 0.1% Triton X-100; cat. no. GC305010; Wuhan
Servicebio Technology Co., Ltd.) for 60 min at room temperature.
Nuclei were counterstained with DAPI for 5 min at 37°C. Fluorescent
images were captured using an Olympus fluorescence microscope
(Olympus Corporation). Finally, cell surface areas were quantified
using ImageJ software (version 1.53; National Institutes of
Health).
WGA staining
Cells were cultured on slides under standard
conditions (ISO 10 µM; 24 h; si-ABCC9 50 nmol/l; 24 h; 740Y-P 10
µM; 24 h; 37°C) using the same culture medium as described for the
other experiments. Once the cells reached the appropriate density,
the medium was discarded, the culture medium was discarded and
cells were washed twice with 1X PBS preheated to 37°C.
Subsequently, an appropriate amount of 4% paraformaldehyde was
added to fix the slides at room temperature for 15 min. The cells
were then washed three times with PBS to remove residual fixative.
After the coverslips were dried, the central area was circled with
a brush and a WGA staining solution diluted with PBS (G1730-100UL;
Wuhan Servicebio Technology Co., Ltd.) was added. The solution was
incubated at 37°C in the dark for 30 min to label the cell
membranes. After incubation, the cells were thoroughly washed three
times with PBS to remove any unbound WGA. Subsequently, an
anti-fluorescence quenching mounting medium containing DAPI (cat.
no. P0131; Beyotime Biotechnology) was added, the sample was
covered with a coverslip and images were observed under a laser
confocal microscope (LSM 800; Zeiss AG). Finally, the ImageJ
software (version 1.53; National Institutes of Health) was used to
perform quantitative analysis of the cell surface area.
MMP assessment
MMPs were assessed using an enhanced matrix
metalloproteinase detection kit containing JC-1 (cat. no. C2006;
Beyotime Biotechnology). After removing the culture medium, cells
(30,000 cells per well) seeded in a 6-well plate were supplemented
with 1 ml DMEM (Shanghai Basal Media Technologies Co., Ltd.) and 1
ml JC-1 staining solution. The cells were then incubated at 37°C
for 20 min. After removing the staining solution, the cells were
washed twice with JC-1 staining buffer and observed under a
fluorescence microscope (Olympus Corporation). Fluorescence
intensity analysis was performed using ImageJ software (version
1.53, National Institutes of Health).
Flow cytometry of apoptosis
Cell apoptosis was assessed using an Annexin
V-FITC/PI Apoptosis Detection Kit (cat. no. 40302; Shanghai Yeasen
Biotechnology Co., Ltd.). AC16 cells in logarithmic growth phase
were seeded into 6-well plates at a density of 3.5×105
cells per well and cultured overnight at 37°C. Following
experimental treatments, cells were treated differently according
to the experimental groups (ISO, 10 µM; 24 h; si-ABCC9 50 nmol/l;
24 h; 740Y-P 10 µM; 24 h; 37°C). AC16 cells were digested using
EDTA-free trypsin at 37°C for 1 min, collected, and centrifuged at
300 × g for 5 min at 4°C to remove the supernatant. The cells were
then resuspended twice with PBS. In addition, the cells were
incubated with 100 µl binding buffer (cat. no. 40302; Shanghai
Yeasen Biotechnology Co., Ltd.) and 5 µl Annexin V/FITC for 10 min
at room temperature in the dark. Then, 400 µl binding buffer and 5
µl PI were added and mixed for 5 min in the dark. This was followed
and immediately analyzed using a BD LSRFortessa™ Fusion flow
cytometer (BD Biosciences). Apoptosis rate was analyzed using
FlowJo software (version 10.8.1; BD Biosciences). Cells were
divided into four sections: Q1 (necrotic cells), Q2 (late apoptosis
cells), Q3 (early apoptosis cells) and Q4 (live cells). The
apoptosis rate=early apoptosis rate + late apoptosis rate.
Reactive oxygen species (ROS) level
assessment
Intracellular ROS levels were measured using
DCFH-DA. AC16 cells were plated in 12-well plates at
1.5×105 cells per well and treated as described (ISO 10
µM, 24 h; si-ABCC9 50 nmol/l, 24 h; 740Y-P 10 µM, 24 h; 37°C).
After removing the culture medium, cells were incubated with 500 µl
DCFH-DA (1:1,000 dilution in PBS) for 20 min at 37°C in the dark.
After washing three times with PBS, the cells were observed under
an inverted fluorescence microscope (Olympus Corporation).
Fluorescence intensity was analyzed using ImageJ software (version
1.53, National Institutes of Health).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism (version 9.1.1; Dotmatics). Data are expressed as
the mean ± standard deviation of three independent replications of
the experiment. Comparisons between groups were made using
two-tailed unpaired Student's t-tests and one-way ANOVA, followed
by Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
ABCC9 expression is elevated in the MH
model
To investigate the expression of ABCC9 in MH, an
in vitro model of ISO-induced cardiomyocyte hypertrophy was
established. AC16 cardiomyocytes were treated with different
concentrations of ISO (0, 5, 10, 20 and 40 µmol/l), and the
inhibitory effect of ISO treatment on AC16 cells was observed in a
concentration-dependent manner. After 24 h treatment with 10 µM
ISO, no significant cytotoxicity was observed in AC16 cells, and
cell viability showed no significant reduction compared with the
control group (Fig. 1A). Based on
previous studies (32,33) and the present preliminary
experiment, a concentration of 10 µM ISO was considered non-toxic
to cardiomyocytes, and therefore this concentration of ISO was
selected to establish the MH model in subsequent experiments. To
further investigate the effects of ISO-induced dynamic temporal
changes on ABCC9 protein expression in AC16 cells, cardiomyocytes
were treated with 10 µM ISO for different time periods (0, 12, 24
and 48 h). Western blot analysis revealed that ABCC9 expression
began to significantly increase at 12 h, peaked at 24 h and
remained at a significantly high level at 48 h (Fig. 1B and C). The 24-h treatment with 10
µM ISO appeared to be associated with significantly elevated ABCC9
expression, suggesting a potential role in the progression of
ISO-induced cardiomyocyte hypertrophy. RT-qPCR was used to quantify
the mRNA expression levels of cardiomyopathy markers ANP, BNP and
β-MHC. The results showed that the mRNA expression levels of ANP,
BNP and β-MHC in the ISO group were significantly higher compared
with those in the control group (Fig.
1D). Changes in the surface area of AC16 cardiomyocytes were
observed using confocal microscopy. AC16 cells were stained with
rhodamine-phalloidin and WGA. Compared with the control group, the
surface area of AC16 cells in the ISO group was significantly
larger (Fig. 1E-H). Overall, the
aforementioned findings indicated that the ISO-induced MH model had
been successfully established and ABCC9 was significantly highly
expressed in MH, suggesting that ABCC9 was closely related to the
development of MH disease.
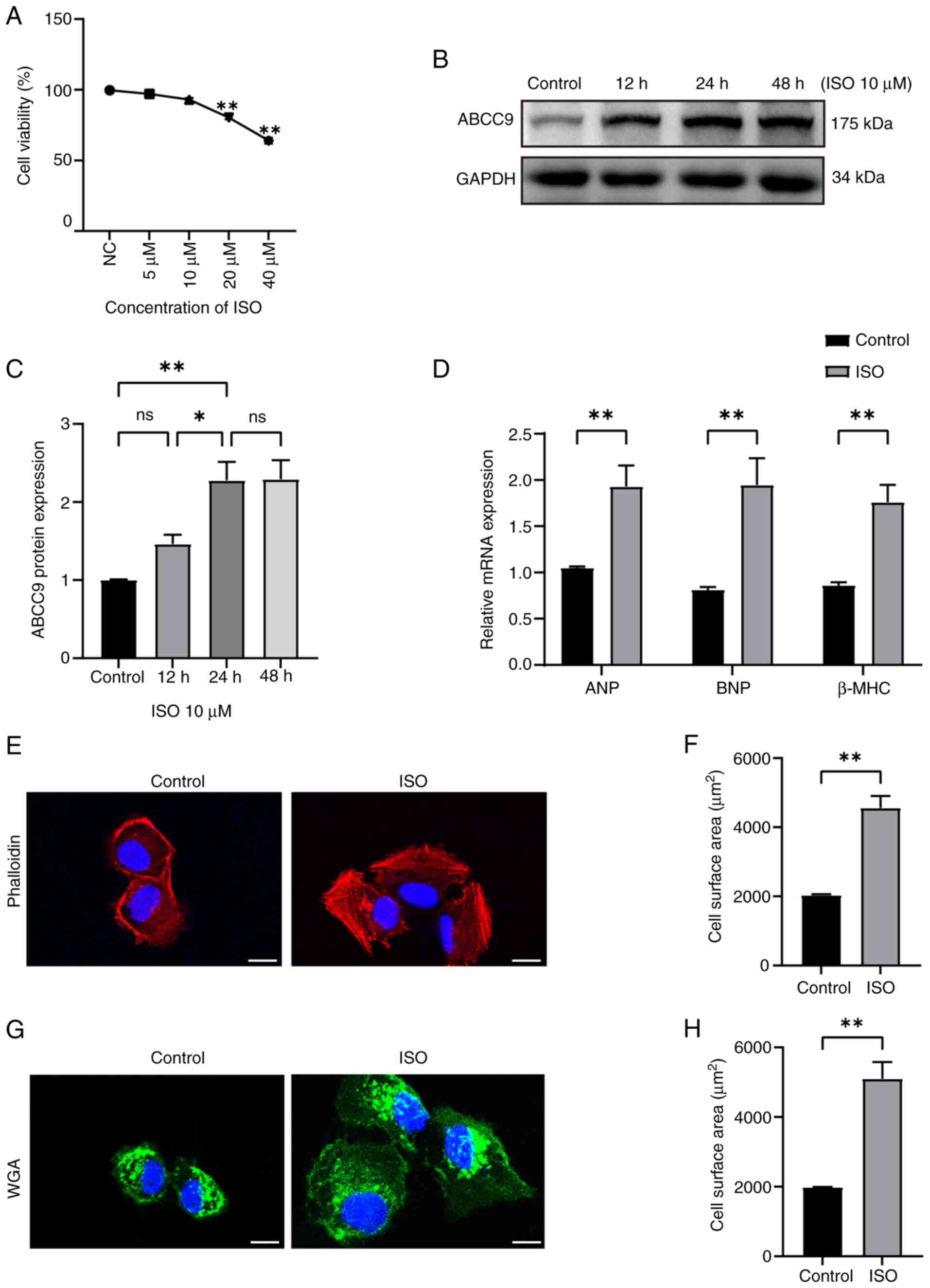 | Figure 1.Elevated expression of ABCC9 in the
ISO-induced myocardial hypertrophy model. (A) Cell Counting Kit-8
was used to determine the viability of ISO-induced AC16 cells.
After treating AC16 cardiomyocytes with 10 µM ISO for different
time periods, western blot analysis was performed to assess changes
in ABCC9 protein expression. (B) Representative western blot image
of ABCC9 and (C) semi-quantitative detection of ABCC9 protein
expression levels using ImageJ software. (D) Evaluation of mRNA
expression levels of ANP, BNP and β-MHC in ISO-treated AC16 cells
using reverse transcription-quantitative PCR. After ISO
stimulation, cardiomyocytes were stained with (E) rhodamine
phalloidin and (F) cell surface area was analyzed. After ISO
stimulation, cardiomyocytes were stained with (G) WGA and (H) cell
surface area was analyzed. Fluorescent images of ISO-treated AC16
cells were obtained using a laser confocal microscope, and cell
surface area was quantified using ImageJ software. Scale bar, 20
µm. AC16 cells were treated with ISO (10 µmol/l) for 24 h. n=3;
*P<0.05 and **P<0.01 vs. control. ABCC9, ATP-binding cassette
subfamily C member 9; NC, negative control; ns, not significant;
ISO, isoproterenol; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; β-MHC, β-myosin heavy chain; WGA, wheat germ
agglutinin. |
Effects of ABCC9 silencing on
myocardial function and structure
Since ABCC9 was upregulated in AC16 hypertrophic
cardiomyocytes, ABCC9 may regulate the progression of cardiomyocyte
hypertrophy. To investigate the potential effects of ABCC9 on
cardiac hypertrophy, ABCC9 loss-of-function was established using
siRNA interference-mediated gene silencing. First, the knockdown
rate of ABCC9-siRNA3 in cells treated with ISO was significant,
compared with the non-significant reductions of ABCC9 expression
mediated by ABCC9-siRNA1 and ABCC9-siRNA2, as observed by western
blotting (Fig. 2A and B).
Therefore, ABCC9-siRNA3 was selected for subsequent studies.
Subsequently, western blotting results further validated the gene
silencing efficiency of ABCC9-siRNA3 at up to 85% under control
conditions (Fig. 2C and D), and
ABCC9 knockdown significantly inhibited the overexpression of ABCC9
protein in AC16 cells induced by ISO (Fig. 2E and F). Notably, ABCC9 knockdown
inhibited ISO-induced cardiomyocyte hypertrophic growth. The
protein expression levels of ANP and BNP were significantly higher
in the ISO and ISO + si-NC groups compared with the NC group,
whereas ABCC9 knockdown significantly reduced the protein
expression levels of ANP and BNP in AC16 cells (Fig. 2G-I). Rhodamine-phalloidin and WGA
staining showed that the surface area of ISO-induced AC16 cells was
significantly increased compared with the NC group, whereas the
surface area of AC16 cardiomyocytes was significantly reduced after
ABCC9 knockdown (Fig. 2J-M).
Collectively, these findings suggest that silencing ABCC9
attenuated cardiomyocyte hypertrophy.
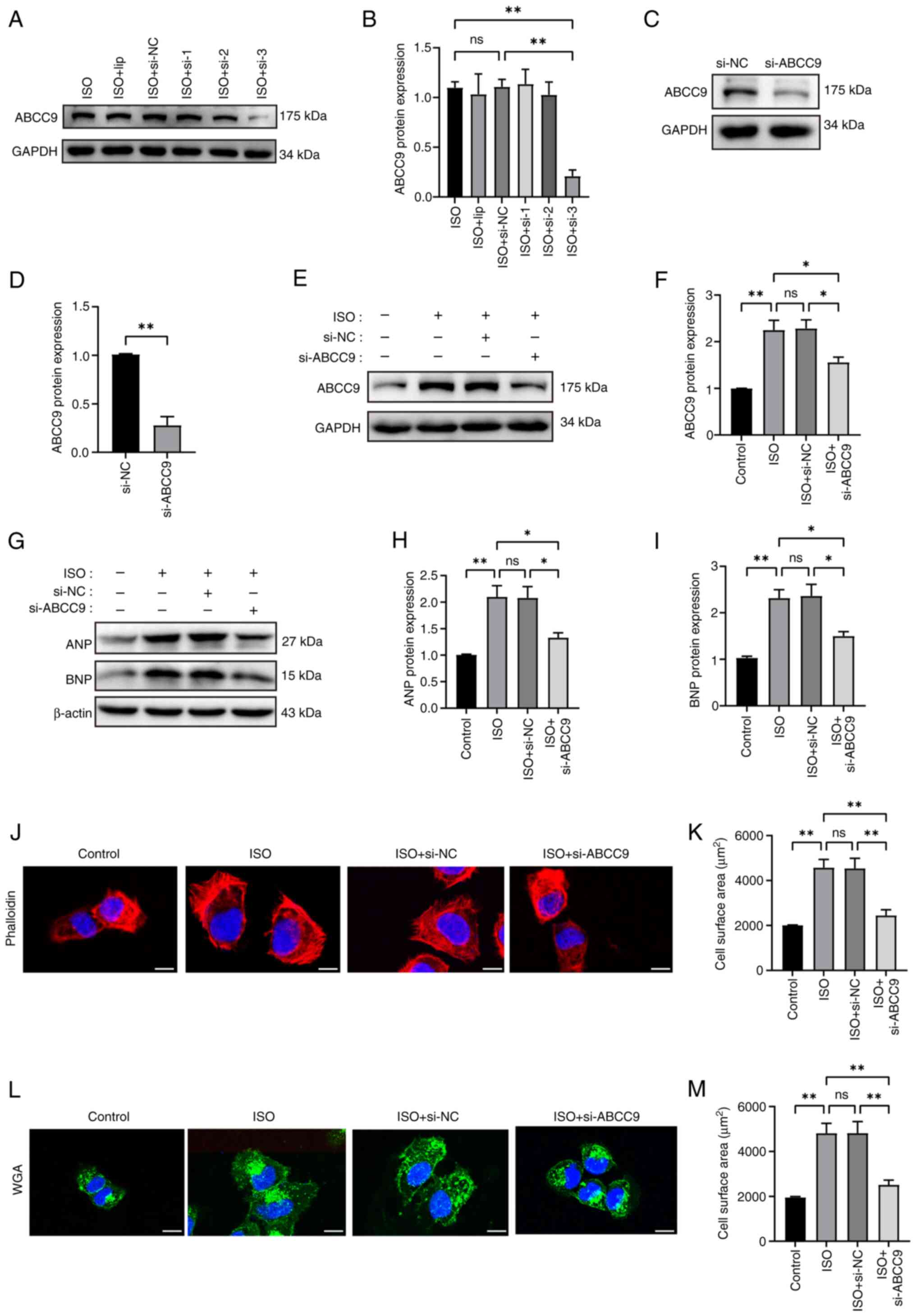 | Figure 2.Silencing of ABCC9 attenuates
ISO-induced myocardial hypertrophy in AC16 cells. (A) Western blot
analysis assessed the transfection efficiency of ABCC9 knockdown.
(B) ImageJ was used for semi-quantitative analysis of ABCC9 protein
expression levels. (C) Western blotting and (D) semi-quantitative
analysis confirmed the knockdown efficiency of ABCC9 si-3
transfection in normal cardiomyocytes. (E) Western blot analysis
was used to assess the protein expression levels of ABCC9 in
ISO-treated AC16 cells after ABCC9 knockdown. (F) ImageJ was used
for semi-quantitative analysis of ABCC9 protein expression levels.
(G) Representative western blot images of ANP and BNP. (H) ANP and
(I) BNP protein expression levels were semi-quantified using
ImageJ. Myocardial cells transfected with ABCC9-siRNA for 24 h were
treated with (J) rhodamine-phalloidin and (K) analyzed
quantitatively. Myocardial cells transfected with ABCC9-siRNA for
24 h were treated with (L) WGA staining and (M) analyzed
quantitatively. The ISO-induced surface area of AC16 myocardial
cells was detected using a laser confocal microscope. The cell
surface area was quantified using ImageJ software. Scale bar, 20
µm. AC16 cells were treated with ISO (10 µmol/l) for 24 h. n=3;
*P<0.05 and **P<0.01. NC, negative control; ns, not
significant; ISO, isoproterenol; ABCC9, ATP-binding cassette
subfamily C member 9; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; si, small interfering RNA; WGA, wheat germ
agglutinin; lip, Lipofectamine® 3000 transfection
reagent. |
ABCC9 knockdown attenuates
cardiomyocyte apoptosis, oxidative stress and mitochondrial
dysfunction
The present study further investigated the role of
silencing ABCC9 in ISO-induced AC16 cells using western blotting
and quantified apoptosis-related proteins in different groups of
AC16 cells. The results showed that, compared with the control
group, the expression levels of Bax, caspase-3 and cleaved
caspase-3 protein were significantly increased in ISO-induced AC16
cells, while the expression level of Bcl-2 was significantly
decreased. Compared with the ISO group, the expression levels of
Bax, caspase-3 and cleaved caspase-3 proteins were significantly
reduced in the si-ABCC9 group, and Bcl-2 was significantly
increased (Fig. 3A-E). To evaluate
caspase-3 activation, the ratio of cleaved caspase-3/caspase-3 was
analyzed. Notably, while both forms were elevated by ISO treatment,
the cleaved caspase-3/caspase-3 ratio showed no significant
difference (Fig. 3F), suggesting
concurrent upregulation of caspase-3 protein alongside its
activation. ABCC9 knockdown significantly reduced the levels of
cleaved caspase-3, indicating suppression of the apoptotic signal.
Flow cytometry analysis further supported the association between
ABCC9 and apoptosis. The apoptosis rate, defined as the sum of
early apoptotic (Annexin V+/PI−) and late
apoptotic (Annexin V+/PI+) cells, was
significantly higher in ISO-treated AC16 cells compared with in the
control group. Specifically, early and late apoptotic cells
accounted for 7.32 and 13.70%, respectively, compared with 1.54 and
3.28% in control cells. Knockout of ABCC9 significantly attenuated
ISO-induced cardiomyocyte apoptosis, reducing the proportions of
early and late apoptotic cells to 2.49 and 7.51%, respectively
(Fig. 3G and H).
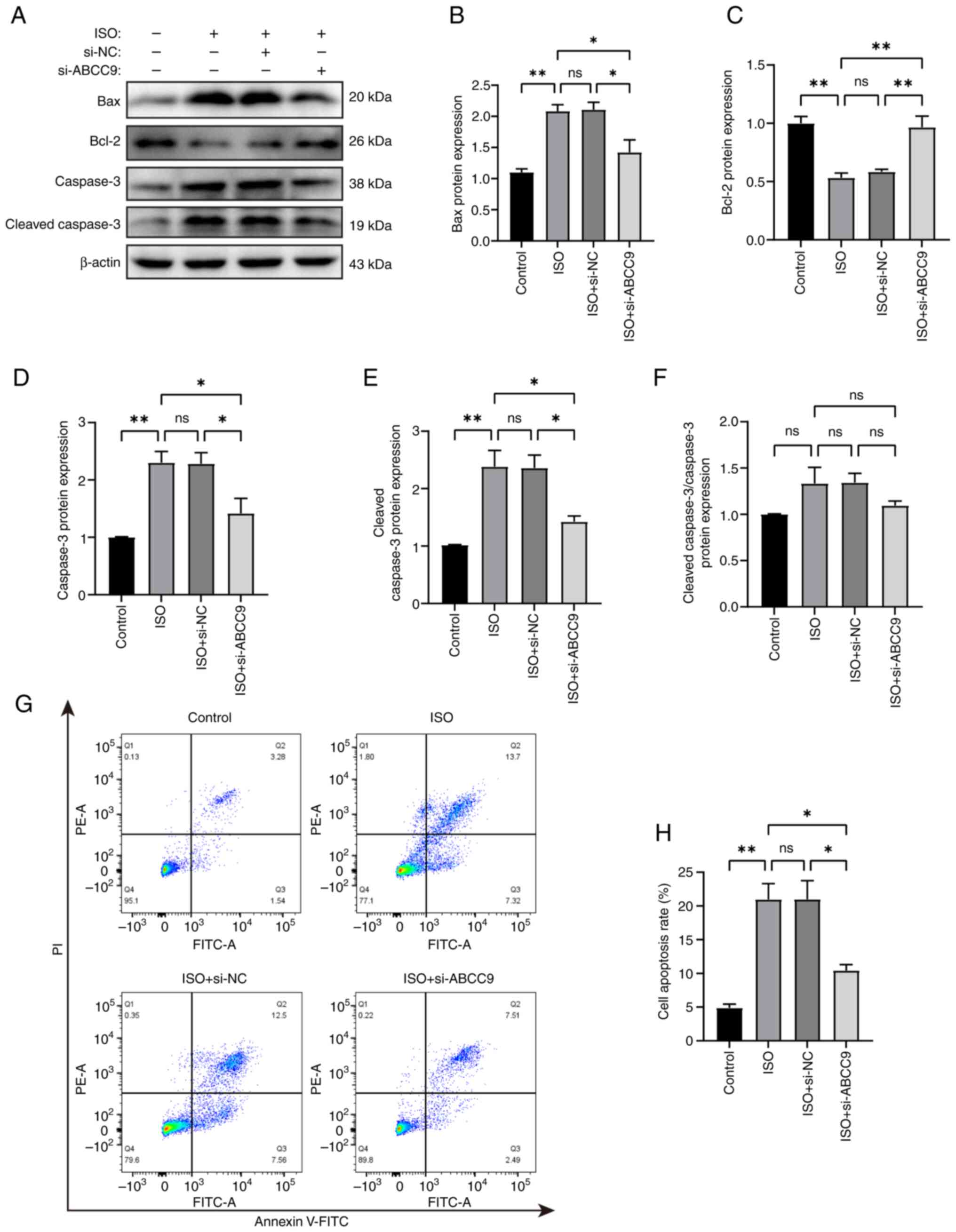 | Figure 3.ABCC9 knockdown inhibits apoptosis in
cardiomyocytes. (A) Western blot analysis was used to assess
changes in the expression of apoptosis-related proteins Bax, Bcl-2,
caspase-3 and cleaved caspase-3 in AC16 cells. ImageJ was used for
semi-quantitative analysis of the expression levels of (B) Bax, (C)
Bcl-2, (D) caspase-3, (E) cleaved caspase-3 proteins and (F)
cleaved caspase-3/caspase-3 ratio. (G) Representative flow
cytometry images and (H) quantitative analysis of Annexin V-FITC/PI
staining in AC16 cells. AC16 cells were treated with ISO (10
µmol/l) for 24 h. n=3; *P<0.05 and **P<0.01. NC, negative
control; ns, not significant; ISO, isoproterenol; ABCC9,
ATP-binding cassette subfamily C member 9; Bax, Bcl-2 associated X
protein; Bcl-2, B-cell lymphoma 2 protein; si, small interfering
RNA. |
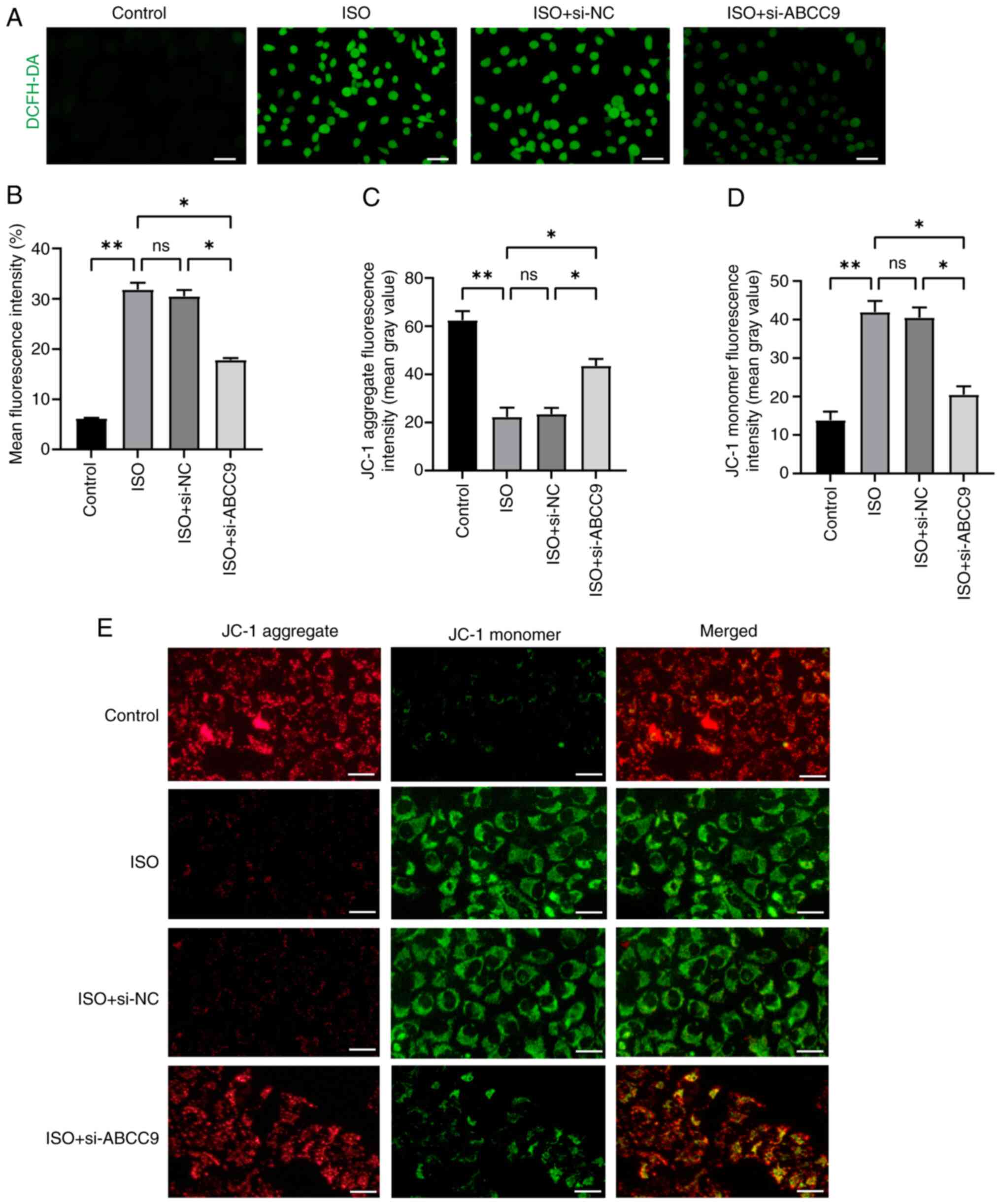
Furthermore, considering the role of oxidative
stress in MH, the levels of ROS were measured in different groups
of AC16 cells using DCFH-DA staining (Fig. 4A and B). The results of the present
study showed that ISO stimulation significantly increased ROS
production in AC16 cells. However, silencing of ABCC9 in AC16 cells
reduced the stimulatory effect of ISO and significantly decreased
ROS levels. To further investigate the effect of ABCC9 knockdown on
mitochondrial function, JC-1 staining was used to assess MMP.
Normal control cells exhibited red fluorescence, while cells with
damaged mitochondrial membranes exhibited green fluorescence. Green
fluorescence was clearly observed in cells from the ISO group,
indicating a decrease in MMP, and the ISO-induced decrease in MMP
was significantly alleviated by ABCC9 knockdown (Fig. 4C-E). These findings suggested that
ABCC9 deficiency directly alleviated ISO-induced apoptosis and
oxidative stress and improved mitochondrial function in
cardiomyocytes in vitro.
ABCC9 knockdown attenuates MH by
inhibiting the PI3K/AKT pathway
The PI3K/AKT pathway has been reported to serve a
key role in the regulation of MH (34). To determine the potential
mechanisms underlying the effects of ABCC9 on ISO-induced MH, the
expression levels of PI3K/AKT signaling pathway proteins were
examined. Protein phosphorylation of PI3K and AKT was significantly
elevated in the ISO group compared with the control group. By
contrast, ABCC9 knockdown significantly downregulated protein
phosphorylation of PI3K and AKT, and neither ISO nor ABCC9
knockdown treatment affected the total level of PI3K or AKT,
suggesting that PI3K/AKT signaling activity was affected by ABCC9
(Fig. 5A-C). In addition, to
further elucidate whether the protective effect of silencing ABCC9
on cardiomyocytes was related to the inhibition of the PI3K/AKT
signaling pathway, ISO-induced cardiomyocytes were treated with the
PI3K/AKT pathway activator 740Y-P. Silencing of ABCC9-mediated
reduction in PI3K/AKT expression was significantly reversed by
740Y-P (Fig. 5D-F). The present
study found that p-PI3K/p-AKT protein expression was significantly
elevated in the activator 740Y-P-treated group alone compared with
the ISO + si-NC group. Thus, the protective effect of knocking down
ABCC9 against ISO-induced MH was mediated by inhibiting the
PI3K/AKT signaling pathway.
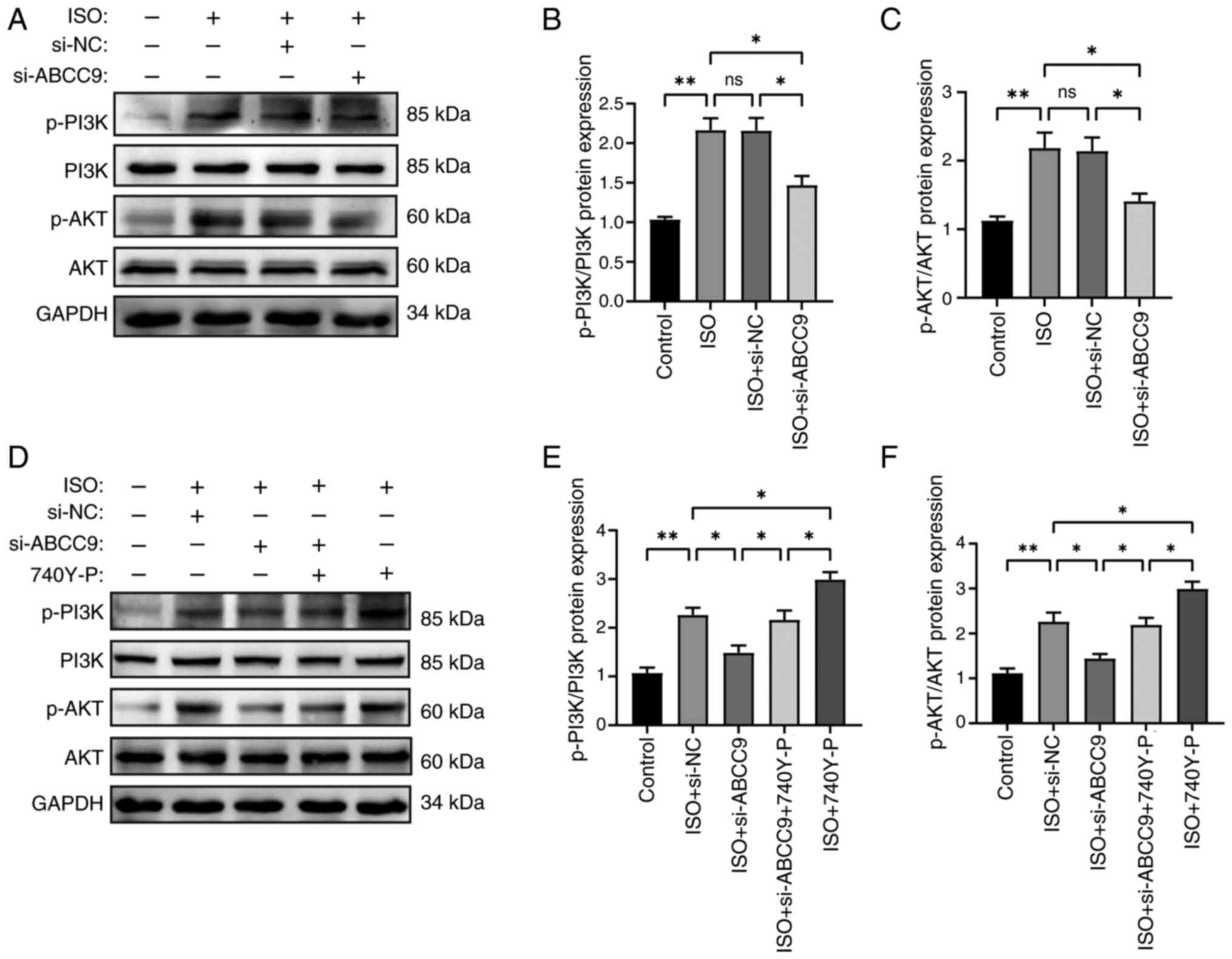 | Figure 5.Effect of 740Y-P and ABCC9 knockdown
on PI3K/AKT signaling pathway proteins. (A) Western blot bands of
PI3K, p-PI3K, AKT and p-AKT in AC16 cells. Semi-quantitative
analysis of (B) p-PI3K and (C) p-AKT protein expression using
ImageJ. (D) Western blotting with 740Y-P showed a reversal of the
protective effect of ABCC9 knockdown on ISO-induced AC16 cells. (E)
Semi-quantitative analysis of p-PI3K and (F) p-AKT protein
expression with 740Y-P treatment. AC16 cells were treated with ISO
(10 µmol/l) for 24 h. n=3; *P<0.05 and **P<0.01. NC, negative
control; ns, not significant; ABCC9, ATP-binding cassette subfamily
C member 9; ISO, isoproterenol; si, small interfering RNA; PI3K,
phosphatidylinositol-3-kinase; AKT, protein kinase B; p-,
phosphorylated. |
Notably, 740Y-P also blocked the effects of ABCC9
knockdown on hypertrophic gene expression and cardiomyocyte surface
area size. The protein and mRNA expression levels of ANP, BNP and
β-MHC were assessed using western blot analysis and RT-qPCR. The
results showed that ABCC9 knockdown significantly reduced
ISO-induced hypertrophic gene mRNA and protein expression compared
with the si-NC group. Notably, co-treatment of the PI3K activator
740Y-P with si-ABCC9 significantly reversed the aforementioned
inhibitory effects (Fig. 6A-F),
and treatment with 740Y-P and ISO alone further aggravated the
expression of hypertrophic markers. Rhodamine-phalloidin and WGA
staining assays showed that the surface area of AC16 cardiomyocytes
was significantly increased in the ISO + si-NC group compared with
the NC group, but ABCC9 knockdown significantly reduced the
ISO-induced increase in AC16 surface area. Similarly, co-treatment
with 740Y-P significantly rescued the reduction in cell surface
area caused by ABCC9 knockdown (Fig.
6G-J), whereas treatment with the activator 740Y-P and ISO
alone significantly increased the cell surface area compared with
the control group and thus exacerbated the MH. The present results
suggested that ABCC9 knockdown attenuated ISO-induced MH by
inhibiting PI3K/AKT signaling, whereas the PI3K activator 740Y-P
partially reversed the protective effect. Overall, the
aforementioned findings suggested that the protective effect of
ABCC9 knockdown on MH was predominantly triggered by PI3K/AKT
inhibition.
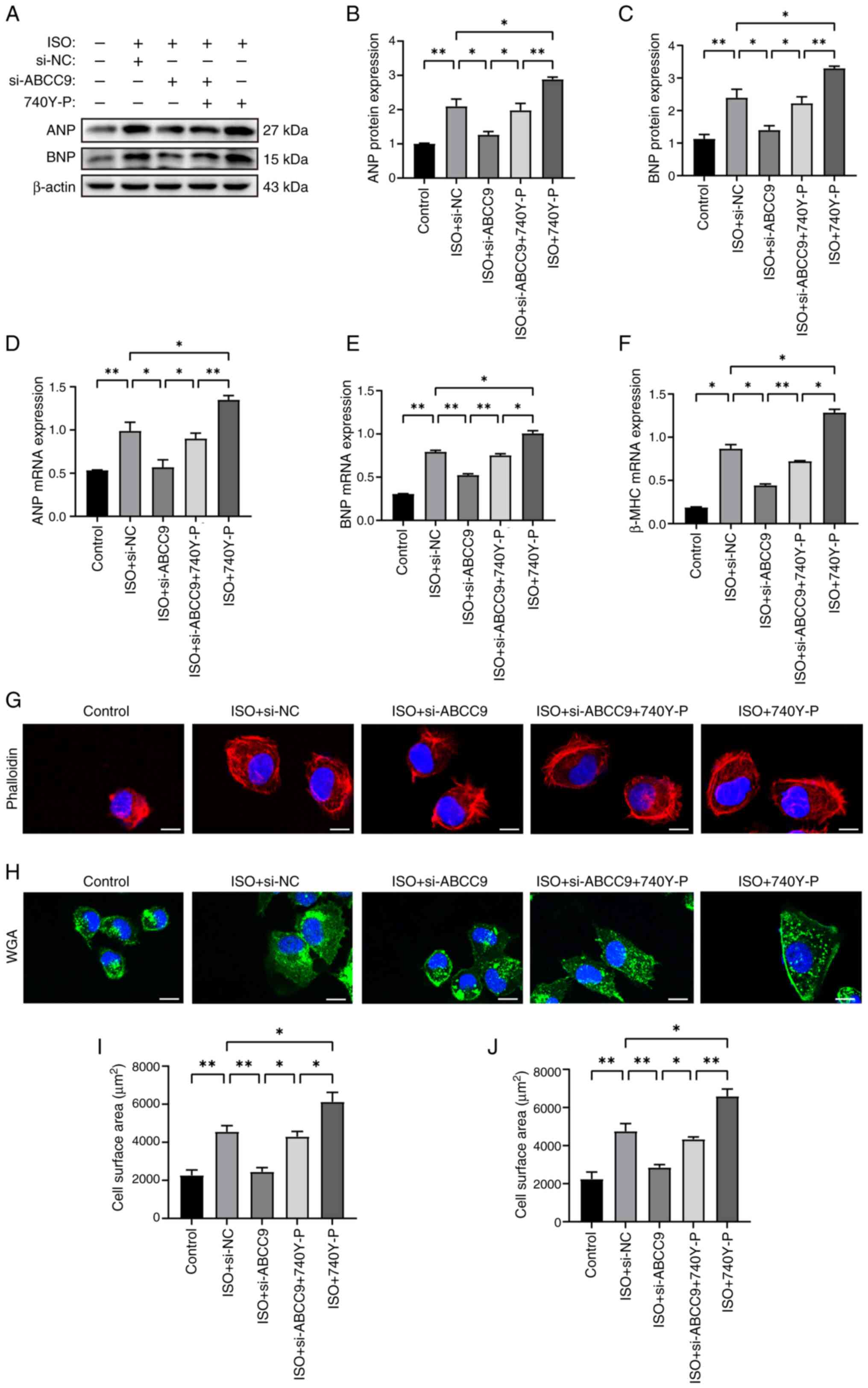 | Figure 6.ABCC9 knockdown attenuates myocardial
hypertrophy by inhibiting PI3K/AKT signaling pathway. (A) Western
blot bands showing the effect of 740Y-P on ANP and BNP protein
expression. (B) Semi-quantitative analysis of ANP and (C) BNP
protein expression. Reverse transcription-quantitative PCR was used
to assess the mRNA levels of the hypertrophic genes (D) ANP, (E)
BNP and (F) β-MHC in 740Y-P treated AC16 cells. After transfection
and 740Y-P pretreatment, AC16 cells were stained with (G) rhodamine
phalloidin and (H) WGA, and fluorescence images of si-ABCC9 and
740Y-P-treated AC16 cells were obtained using a laser confocal
microscope. Quantitative analysis of cell surface area in (I)
rhodamine phalloidin and (J) WGA stained samples using ImageJ
software. Scale bar, 20 µm. AC16 cells were treated with ISO (10
µmol/l) for 24 h. n=3; *P<0.05 and **P<0.01. NC, negative
control; ABCC9, ATP-binding cassette subfamily C member 9; ISO,
isoproterenol; ANP, atrial natriuretic peptide; BNP, brain
natriuretic peptide; β-MHC, β-myosin heavy chain. si, small
interfering RNA; WGA, wheat germ agglutinin. |
ABCC9 knockdown attenuates
cardiomyocyte apoptosis and oxidative stress by inhibiting the
PI3K/AKT signaling pathway and restoring MMP
To further investigate the protective effect of
ABCC9 knockdown on ISO-stimulated AC16 cells, the present study
tested whether activation of PI3K activity counteracted the
amelioration of apoptosis and oxidative stress by ABCC9 knockdown.
Western blotting results showed that ABCC9 knockdown significantly
alleviated the apoptotic effects of ISO stimulation on AC16 cells,
as evidenced by reduced expression levels of Bax, cleaved caspase-3
and caspase-3 protein, along with increased expression levels of
Bcl-2 protein compared with the ISO + si-NC group. Pretreatment
with the activator 740Y-P significantly reversed this inhibitory
effect. Notably, treatment with 740Y-P and ISO significantly
increased the expression of these pro-apoptotic proteins and
significantly reduced Bcl-2 expression, indicating that PI3K
activation intensified ISO-induced injury (Fig. 7A-E). Consistent with the
observations in Fig. 3, the ratio
of cleaved caspase-3/caspase-3 also showed no significant
difference among groups treated with ISO, si-ABCC9 and 740Y-P
(Fig. 7F). In addition, changes in
apoptotic cell quantity were assessed using flow cytometry. The
results showed that the rate of ISO-induced apoptosis in AC16
cardiomyocytes was significantly increased, and silencing ABCC9
effectively inhibited ISO-induced apoptosis in AC16 cells. Compared
with the si-ABCC9 group, cardiomyocyte apoptosis was significantly
aggravated by pretreatment with the PI3K activator 740Y-P.
Similarly, treatment with activator 740Y-P alone in ISO-treated
cells further increased the number of apoptotic cells (Fig. 7G and H). These results indicate
that the activation of PI3K activity by 740Y-P significantly
reversed the therapeutic effect of knocking down ABCC9.
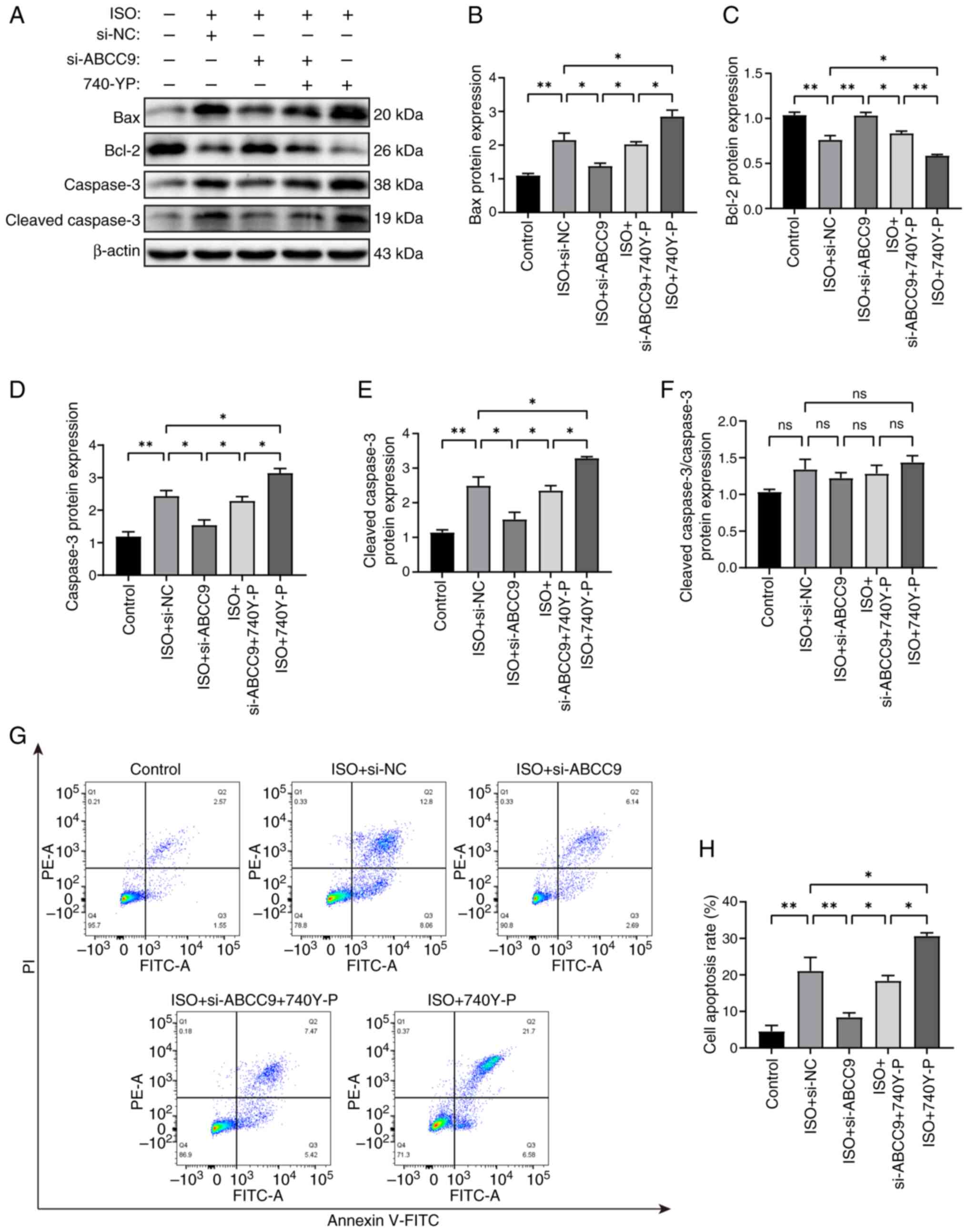 | Figure 7.ABCC9 knockdown attenuates apoptosis
in cardiomyocytes by inhibiting the PI3K/AKT pathway and
alleviating mitochondrial dysfunction. (A) Western blot bands
showing the effect of 740Y-P on the expression of the
apoptosis-related proteins Bax, Bcl-2, caspase-3 and cleaved
caspase-3 in AC16 cells. Semi-quantitative analysis of (B) Bax, (C)
Bcl-2, (D) caspase-3, (E) cleaved caspase-3 proteins expression
levels and (F) cleaved caspase-3/caspase-3 ratio was performed
using ImageJ. (G) Representative flow cytometry images of Annexin
V/PI staining and (H) quantification of apoptosis rate in AC16
cells. AC16 cells were treated with ISO (10 µmol/l) for 24 h. n=3;
*P<0.05 and **P<0.01. NC, negative control; ns, not
significant; ISO, isoproterenol; ABCC9, ATP-binding cassette
subfamily C member 9; Bax, Bcl-2 associated X protein; Bcl-2,
B-cell lymphoma 2 protein; DCFH-DA,
2′,7′-dichlorodihydrofluorescein diacetate; si, small interfering
RNA. |
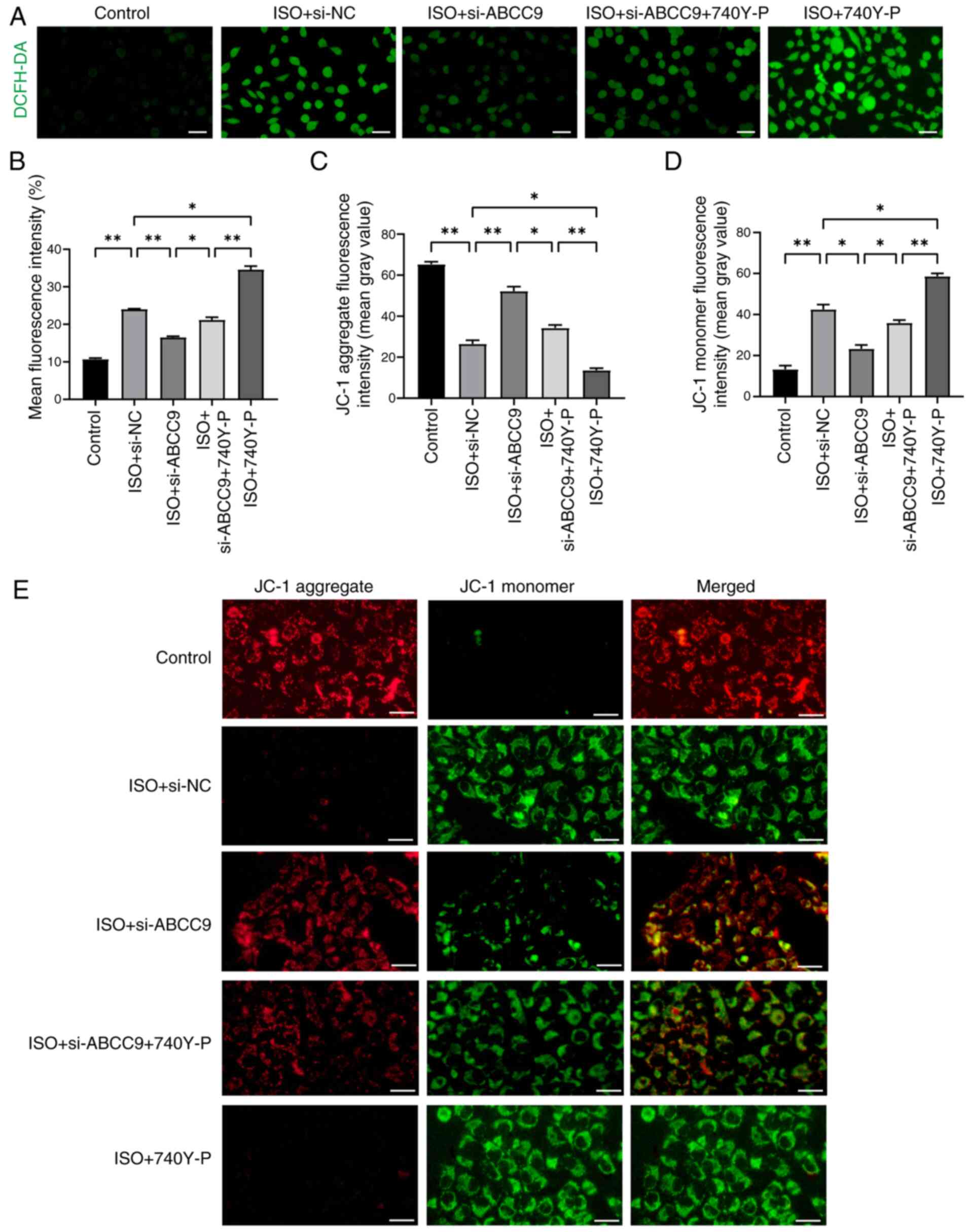
Meanwhile, ROS assay results showed that ABCC9
knockdown significantly reduced ROS levels in ISO-treated AC16
cells, but its effect was attenuated in the presence of 740Y-P
(Fig. 8A and B). To further
investigate the protective mechanism of ABCC9 knockdown against
apoptosis, MMP was analyzed in cardiomyocytes treated with 740Y-P.
Notably, compared with the ISO + si-ABCC9 group, pre-treatment with
the PI3K activator 740Y-P significantly reversed the protective
effect of ABCC9 knockdown on mitochondrial function (Fig. 8C-E). Furthermore, compared with the
si-NC group, the use of activator 740Y-P and ISO also significantly
exacerbated ROS production and mitochondrial dysfunction. The
present results suggested that ABCC9 knockdown protected the
myocardium by inhibiting the PI3K/AKT signaling pathway and
alleviating mitochondrial dysfunction, while the PI3K activator
740Y-P partially reversed this effect. Overall, ABCC9 knockdown
inhibited the PI3K/AKT signaling pathway, restored the MMP and
thereby alleviated cardiomyocyte apoptosis and oxidative
stress.
Discussion
The present study demonstrated that ABCC9 knockdown
attenuated MH by reducing cardiomyocyte apoptosis and ROS
production. This protective effect was mediated through inhibition
of PI3K/AKT pathway phosphorylation and the subsequent improvement
of mitochondrial function, thereby protecting cardiomyocytes from
ISO-induced pathological changes. The framework of ABCC9-mediated
regulation of the PI3K/AKT signaling pathway in ISO-induced MH is
shown in Fig. 9.
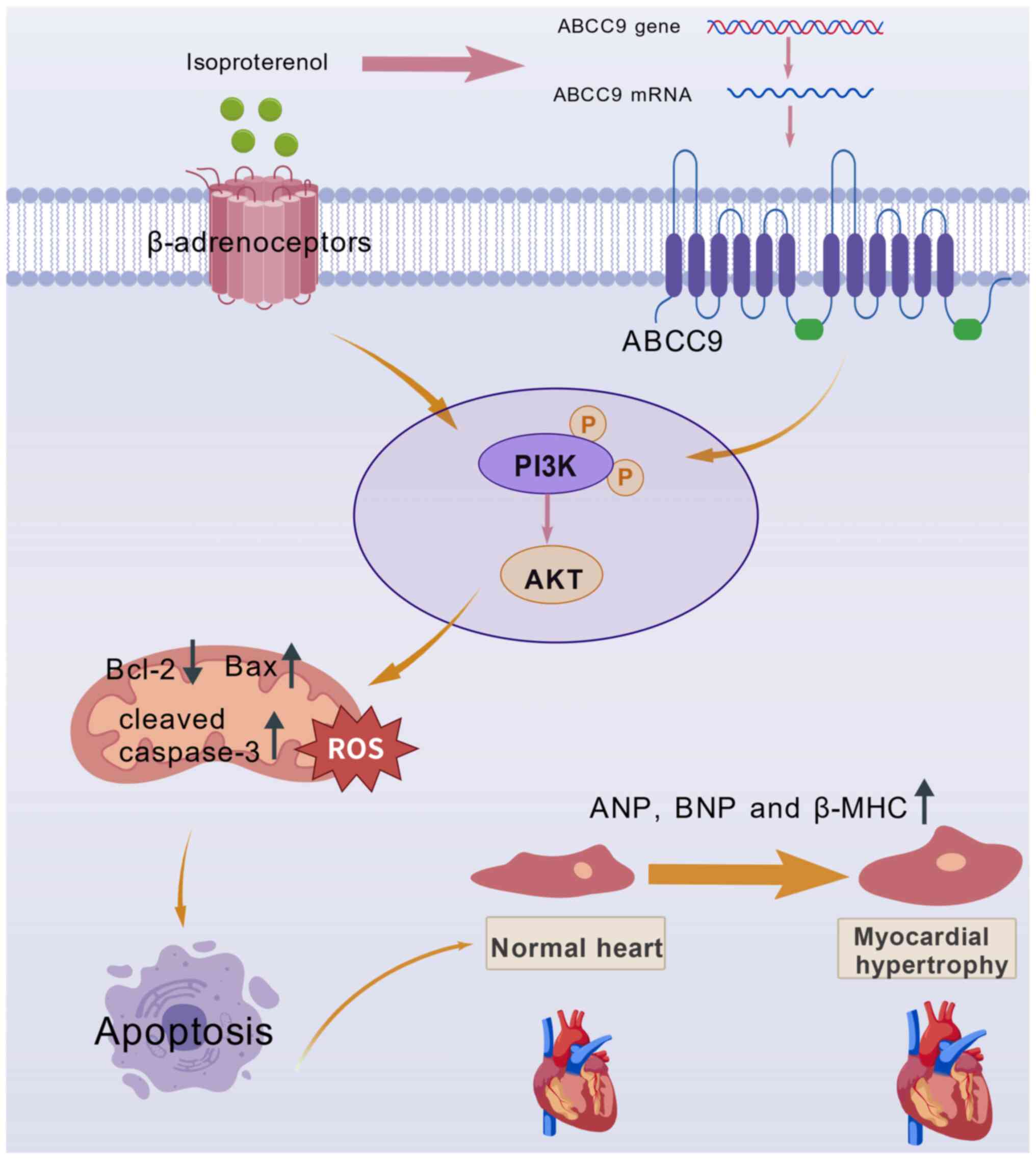 | Figure 9.Schematic diagram describing the
PI3K/AKT signaling pathway mechanism by which ABCC9 acts on MH. ISO
binds to the cellular β-adrenergic surface receptor of
cardiomyocytes, while ABCC9 is highly expressed in ISO-mediated
cardiomyocytes, which triggers apoptosis and oxidative stress
through activation of the PI3K/AKT pathway and results in
mitochondrial dysfunction, ultimately leading to MH. ISO,
isoproterenol; ABCC9, ATP-binding cassette subfamily C member 9;
ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide;
β-MHC, β-myosin heavy chain; Bax, Bcl-2 associated X protein;
Bcl-2, B-cell lymphoma 2 protein; ROS, reactive oxygen species; P,
phosphate group; MH, myocardial hypertrophy. |
MH, a well-established risk factor for CVD, is
associated with markedly increased morbidity and mortality rates.
However, the precise pathophysiological mechanisms of MH are yet to
be fully elucidated. Although numerous pharmacological
interventions for MH exist, including angiotensin-converting enzyme
inhibitors, β-adrenergic receptor (β-AR) blockers, calcium channel
blockers and histone deacetylase inhibitors (35–37),
these therapies present several clinical limitations and an
effective treatment strategy for MH remains elusive.
β-ARs, members of the G protein-coupled receptor
family, serve as the primary cardiac receptors (38). The β-AR signaling pathway serves an
important role in cardiac remodeling progression, with excessive
activation leading to pathological cardiac hypertrophy and fibrosis
(39), ultimately resulting in HF.
While β-AR agonists are non-selective activators of these receptors
(40) that primarily function
through sympathetic nervous system (SNS) activation, chronic SNS
stimulation contributes to progressive cardiac dysfunction and
structural deterioration, further exacerbating MH (41). It has been demonstrated that ISO is
widely used to induce MH (42–44).
In the present study, 10 µM ISO was selected for AC16 cell
treatment to establish the MH model based on the results of the
CCK-8 assay. ISO-induced MH is characterized by markedly elevated
ROS levels in cardiomyocytes (45), increased apoptotic activity
(46) and upregulated
pro-inflammatory cytokine expression (47). Without intervention, severe
prolonged MH progresses to cardiac dysfunction, myocardial
infarction and HF (48),
underscoring the notable need for developing effective therapeutic
strategies for MH patients.
As a member of the ABC family of transporter
proteins, ABCC9 mediates specific cardiovascular phenotypes
characterized by left ventricular dilatation, increased ventricular
mass and enhanced cardiac contractility (49). The ABCC9 gene has been implicated
in various cardiac pathologies (27,28),
and the present investigation revealed a strong association between
ABCC9 expression levels and drug-induced MH, with expression
positively associated with both MH severity and duration. Under
pathological conditions such as myocardial infarction and HF,
cardiomyocytes exhibit hypertrophic responses with a marked
increase in the mRNA and protein expression of atrial ANP, β-MHC
and ventricular BNP (50),
biomarkers that maintain stable expression levels in normal
cardiomyocytes (51). The results
of the present study demonstrated that ISO treatment induced
hypertrophic responses characterized by elevated ANP and BNP
expression. Previous studies have established that MH progression
is initially marked by increased cardiomyocyte surface area
(52), a morphological change that
directly reflects hypertrophic development. Notably, the present
study provided, to the best of our knowledge, the first
demonstration that ABCC9 siRNA transfection in cardiomyocytes
significantly attenuated the expression of the hypertrophic markers
ANP, BNP and β-MHC. Furthermore, ABCC9 knockdown effectively
suppressed hypertrophic cardiomyocyte enlargement, demonstrating
protective effects against ISO-induced MH.
The pathological mechanisms underlying MH primarily
involve cardiomyocyte apoptosis, ROS generation and mitochondrial
dysfunction (53). Due to its
exceptionally high metabolic demands, cardiac tissue generates
substantial ROS levels and remains particularly vulnerable to
oxidative damage (54). Under
pathological conditions, excessive ROS accumulation overwhelms
cellular antioxidant defenses, resulting in oxidative stress and
mitochondrial impairment (55),
thereby accelerating hypertrophic progression. In the present
study, oxidative stress was observed in ISO-treated AC16 cells,
whereas ABCC9 knockdown effectively attenuated the ISO-induced
increase in ROS levels and the proportion of ROS-positive
cells.
Persistent oxidative stress compromises
mitochondrial integrity, inhibits ATP synthesis and triggers
apoptotic pathways, collectively worsening cardiac dysfunction
(56). Cardiomyocyte apoptosis
represents a well-established contributor to HF pathogenesis, with
apoptotic acceleration in hypertrophic cardiomyopathy ultimately
precipitating systolic impairment (57). This programmed cell death process
is regulated through multiple mechanisms, including mitochondrial
dysfunction, caspase activation and modulation of the
apoptosis-related factors Bcl-2, Bax, caspase-3 and cleaved
caspase-3 (58). The findings of
the present study demonstrated that ABCC9 knockdown significantly
reduced ISO-induced apoptosis, as shown by decreased Bax, caspase-3
and cleaved caspase-3 expression and increased Bcl-2 levels, and
supported through Annexin V/PI staining with flow cytometric
analysis. Furthermore, MMP measurements indicated that ABCC9
knockdown alleviated ISO-induced mitochondrial damage in
cardiomyocytes. Notably, the ISO + si-NC group exhibited no
cytotoxic effects at standard doses compared with ISO-treated
cells. The simultaneous elevation of caspase-3 and cleaved
caspase-3 expression levels may have reflected a dynamic
equilibrium in ISO-induced MH. Under stress conditions,
cardiomyocytes may have upregulated caspase-3 transcription via
pathways such as the NF-κB or p53 pathways, while a portion of
caspase-3 was cleaved during apoptosis. This mechanism would have
resulted in sustained caspase-3 synthesis alongside partial
activation, reflecting the coexistence of hypertrophy and apoptosis
during the pathological process. These results positioned ABCC9 as
a promising therapeutic target with a favorable safety profile for
mitigating ISO-induced cardiomyocyte injury and suppressing
hypertrophic progression.
The signaling mechanisms underlying MH are highly
complex. Substantial evidence implicates the PI3K/AKT pathway as a
central regulator of cardiac remodeling through its influence on MH
progression, fibrotic changes, oxidative stress and inflammatory
responses (59). PI3K, in
particular, has been shown to contribute notably to oxidative
stress while also driving the development of cardiac hypertrophy
and HF (60). As an important
downstream effector of PI3K, AKT further participates in
hypertrophic processes by modulating diverse cellular metabolism,
proliferation, survival, growth and angiogenesis (61). Furthermore, activation of the
PI3K/AKT axis by various extracellular ligands such as
lipopolysaccharide or CpG oligodeoxynucleotides enhances dendritic
cell activity, which in turn amplifies immune and inflammatory
reactions (62). Based on these
findings, the present study hypothesized that ABCC9 knockdown might
exert cardioprotective effects through modulation of p-PI3K/p-AKT
levels. By examining the levels of p-PI3K and p-AKT proteins in
AC16 cardiomyocytes, the present study found that the PI3K/AKT
signaling pathway was activated in ISO-induced MH and that ABCC9
knockdown attenuated this effect. These experimental results
supported the hypothesis that ABCC9 regulated MH through PI3K/AKT
signaling pathway modulation.
It is notable that ABCC9 knockdown alleviated MH by
inhibiting the PI3K/AKT signaling pathway, an effect likely
mediated through the regulation of key AKT downstream effector
molecules. Glycogen synthase kinase-3β (GSK-3β), a negative
regulator of cardiac hypertrophy, is a downstream target of AKT;
its activity is suppressed by AKT-mediated phosphorylation
(63). Active GSK-3β has been
demonstrated to directly inhibit pathological MH by preventing the
nuclear translocation of pro-hypertrophic transcription factors,
such as nuclear factor of activated T-cells (64). Furthermore, AKT phosphorylates the
FoxO1 transcription factor (65),
an important regulator of autophagy, metabolism and cell survival.
Consequently, FoxO1 serves an important role in modulating MH and
fibrosis in mice via the PI3K/AKT pathway (66). These processes collectively
constitute an important defense mechanism against MH.
The mammalian target of rapamycin complex 1
(mTORC1), a notable downstream target of AKT, serves as a central
regulator of protein synthesis and cell growth (67). Pharmacological inhibition of the
mTORC1 signaling pathway has been shown to ameliorate pathological
MH (68). The observed inhibition
of AKT in the present study suggested a concomitant reduction in
mTORC1 signaling activity, which would directly suppress the
hypertrophic translational machinery. Additionally, AKT can
activate NF-κB through the IKK pathway (69). As a classic pro-inflammatory and
pro-survival transcription factor, NF-κB contributes to myocardial
dysfunction in advanced HF (70).
Thus, the AKT inhibition resulting from ABCC9 knockdown may have
attenuated NF-κB signaling, thereby mitigating inflammatory
responses and functional impairment in the hearts of hypertrophic
mice. In summary, the inhibition of the PI3K/AKT pathway following
ABCC9 knockdown is proposed to mitigate MH by coordinately
regulating key nodal points downstream of AKT, including GSK-3β,
FoxO1, mTOR and NF-κB. Consequently, future studies directly
assessing the phosphorylation status and activity of these effector
proteins are warranted to precisely elucidate the molecular
mechanisms downstream of ABCC9.
It has been demonstrated that cell survival depends
predominantly on the Bcl-2 family of apoptosis regulators (71) and that Bcl-2 is involved in cardiac
hypertrophy as a key downstream effector of the PI3K/AKT signaling
pathway (72). Subsequently, the
present study further investigated whether ABCC9 knockdown could
mitigate cardiomyocyte apoptosis and oxidative damage by improving
mitochondrial injury through modulation of the PI3K/AKT pathway.
Consistently, the present study showed that ABCC9 knockdown
downregulated PI3K/AKT pathway protein expression while attenuating
ISO-induced mitochondrial dysfunction, oxidative stress and
apoptosis, supporting its potential to alleviate ISO-induced MH by
enhancing the anti-apoptotic and antioxidant systems.
Although AKT has been shown to exert
cardioprotective effects by promoting physiological hypertrophy and
survival signaling, the outcomes of AKT signaling depend on the
duration, frequency and intensity of AKT pathway activation. For
example, short-term AKT activation promotes adaptive hypertrophy,
whereas long-term AKT activation or high-levels of expression have
been associated with pathological hypertrophy and HF (73–75).
In the present study, this hypothesis was driven by the observation
that reactivation of PI3K/AKT signaling with 740Y-P partially
reversed the protective effects of ABCC9 knockdown-restoring
ISO-induced ROS accumulation, mitochondrial dysfunction, and
apoptosis. These results suggest that while short-term AKT
activation may promote cell survival, prolonged excessive
activation may lead to myocardial cell apoptosis, potentially
mediated through mitochondrial dysfunction and increased oxidative
stress. Supporting this view, studies have shown that high levels
of ROS can promote the reversal of MMP by activating MAPK,
resulting in cytochrome c release and the activation of
caspase-3, which in turn trigger apoptosis (76,77).
Under sustained pathological stress, chronic and excessive AKT
signaling may become maladaptive, leading to metabolic dysfunction,
increased oxidative stress and ultimately mitochondrial dysfunction
(78,79), thereby triggering cardiomyocyte
apoptosis.
To further verify whether activation of the PI3K/AKT
signaling pathway eliminated the protective effect of ABCC9
knockdown, the PI3K/AKT pathway was activated using the specific
agonist 740Y-P. 740Y-P is a PI3K/AKT pathway activator that has
been reported to activate the enzyme in vitro by binding to
the Src homology 2 structural domain of the p85 regulatory subunit
of PI3K (80), thereby initiating
the PI3K/AKT signaling pathway. In the present study, oxidative
stress and MMP were partially restored and apoptosis was reduced in
cells treated by transfection with ABCC9 siRNA. Notably, 740Y-P not
only counteracted ABCC9 knockdown-mediated suppression of p-PI3K
and its downstream target p-AKT but also reversed the protective
effects of ABCC9 knockdown on apoptosis, oxidative stress and MMP.
These results provided notable evidence that ABCC9 knockdown
alleviated ISO-induced MH partly by modulating the PI3K/AKT
pathway, which subsequently improved mitochondrial function,
thereby reducing apoptosis and oxidative stress.
Although the data of the present study showed that
knockdown of the ABCC9 gene attenuated MH by inhibiting the
PI3K/AKT signaling pathway, thereby regulating apoptosis, MMP and
oxidative stress, the role of the classic function of ABCC9, as a
regulatory subunit of KATP channels, has not yet been directly
assessed in this process. The SUR2 protein encoded by ABCC9 forms
the KATP channel in conjunction with the Kir6.2 subunit, which
serves a central role in linking cellular metabolic state to
membrane potential, thereby regulating processes such as calcium
influx, vascular tone (81),
insulin secretion and cellular protection (82). The phenotype observed in the
present study could theoretically have also arisen from cellular
electrophysiological changes caused by altered KATP channel
activity (83), so the
contribution of ion channel activity cannot be entirely ruled out.
For example, membrane hyperpolarization caused by KATP channel
opening may have affected the activity of voltage-gated calcium
channels, thereby indirectly influencing MMP, apoptosis and the
PI3K/AKT signaling pathway. To unequivocally distinguish between
the channel-dependent and independent functions of ABCC9, future
studies should employ patch-clamp techniques (84) to directly measure KATP currents in
cardiomyocytes following ABCC9 knockdown. Furthermore,
gain-of-function and loss-of-function experiments using specific
KATP channel openers, such as pinacidil, or inhibitors, such as
glibenclamide, (85) are
warranted. Such investigations would provide more robust support
for the conclusions of the present study and offer a more
comprehensive elucidation of the mechanistic role of ABCC9 in
MH.
However, the present study had some limitations.
First, although ABCC9 knockdown had a significant effect on the
treatment of MH, it would be more convincing if the overexpression
of ABCC9 was used to also obtain the corresponding results.
Nevertheless, since protein expression is often subject to
endogenous saturation, simple overexpression may not necessarily
induce a hypertrophic phenotype, and loss-of-function approaches
could potentially reveal the physiological role of ABCC9 more
clearly. Furthermore, only the core proteins in the PI3K signaling
pathway were evaluated in the present study, and the detailed
molecular mechanisms of downstream regulation of PI3K/AKT signaling
during MH have not been fully elucidated. Finally, the present
study demonstrated that ABCC9 knockdown protected against
pathological cardiac hypertrophy predominantly in ISO-induced AC16
cells; therefore, further evaluation of efficacy in animal models
is needed. In future studies, the role of ABCC9 overexpression in
MH pathogenesis should be investigated, the downstream molecular
targets of the PI3K/AKT signaling pathway should be explored in
greater detail and the therapeutic efficacy of ABCC9 knockdown or
inhibition should be validated in animal models to fully establish
its clinical relevance.
In summary, the present study demonstrated that
ABCC9 knockdown alleviated ISO-induced MH partly by regulating the
PI3K/AKT pathway, which subsequently improved mitochondrial
function and thereby reduced myocardial apoptosis, attenuated
oxidative stress and preserved cardiac function. The present study
provides new evidence for the role of ABCC9 in MH pathogenesis, and
ABCC9 can be regarded as a potential promising candidate target for
clinical intervention.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Shanghai Pudong New Area
Health Commission (grant no. 2025-PWYC-14).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QP, RC, LM and YL contributed to the study
conception and design. QP performed experiments, analyzed the data
and wrote the manuscript. RC analyzed data and revised the
manuscript. YL and LM supervised the work and analyzed data. YL and
LM confirm the authenticity of all the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Jagannathan R, Patel SA, Ali MK and
Narayan KMV: Global updates on cardiovascular disease mortality
trends and attribution of traditional risk factors. Curr Diab Rep.
19:442019. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kahleova H, Levin S and Barnard ND:
Vegetarian dietary patterns and cardiovascular disease. Prog
Cardiovasc Dis. 61:54–61. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Tsao CW, Aday AW, Almarzooq ZI, Alonso A,
Beaton AZ, Bittencourt MS, Boehme AK, Buxton AE, Carson AP,
Commodore-Mensah Y, et al: Heart disease and stroke Statistics-2022
update: A report from the American heart association. Circulation.
145:e153–e639. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tan Y-Q, Chen HW and Li J: Astragaloside
IV: An effective drug for the treatment of cardiovascular diseases.
Drug Des Devel Ther. 14:3731–3746. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Roth GA, Mensah GA, Johnson CO, Addolorato
G, Ammirati E, Baddour LM, Barengo NC, Beaton AZ, Benjamin EJ,
Benziger CP, et al: Global burden of cardiovascular diseases and
risk factors, 1990–2019. J Am Coll Cardiol. 76:2982–3021. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu XY, Shi MQ, Jiang ZM, Xiao Li, Tian JW
and Su FF: Global, regional, and national burden of cardiovascular
diseases attributable to metabolic risks across all age groups from
1990 to 2021: An analysis of the 2021 global burden of disease
study data. BMC Public Health. 25:17042025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yan Z, Zhao W, Zhao N, Liu Y, Yang B, Wang
L, Liu J, Wang D, Wang J, Jiao X, et al: PRMT1 alleviates
isoprenaline-induced myocardial hypertrophy by methylating SRSF1.
Acta Biochim Biophys Sin (Shanghai). 57:1338–1349. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chang CC, Cheng HC, Chou WC, Huang YT,
Hsieh PL, Chu PM and Lee SD: Sesamin suppresses
angiotensin-II-enhanced oxidative stress and hypertrophic markers
in H9c2 cells. Environ Toxicol. 38:2165–2172. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Feng Z, Pan L, Qiao C, Yang Y, Yang X and
Xie Y: Cardamonin intervenes in myocardial hypertrophy progression
by regulating Usp18. Phytomedicine. 134:1559702024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pang Y, Wu L, Xia J, Xu X, Gao C, Hou L
and Jiang L: Trim38 attenuates pressure overload-induced cardiac
hypertrophy by suppressing the TAK1/JNK/P38 signaling pathway. Int
J Mol Med. 55:982025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li D, Guo Y, Cen X, Qiu HL, Chen S, Zeng
XF, Zeng Q, Xu M and Tang QZ: Lupeol protects against cardiac
hypertrophy via TLR4-PI3K-Akt-NF-κB pathways. Acta Pharmacol Sin.
43:1989–2002. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Oudit GY, Crackower MA, Eriksson U, Sarao
R, Kozieradzki I, Sasaki T, Irie-Sasaki J, Gidrewicz D, Rybin VO,
Wada T, et al: Phosphoinositide 3-kinase gamma-deficient mice are
protected from isoproterenol-induced heart failure. Circulation.
108:2147–2152. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou WW, Dai C, Liu WZ, Zhang C, Zhang Y,
Yang GS, Guo QH, Li S, Yang HX and Li AY: Gentianella acuta
improves TAC-induced cardiac remodelling by regulating the Notch
and PI3K/Akt/FOXO1/3 pathways. Biomed Pharmacother. 154:1135642022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ghafouri-Fard S, Khanbabapour Sasi A,
Hussen BM, Shoorei H, Siddiq A, Taheri M and Ayatollahi SA:
Interplay between PI3K/AKT pathway and heart disorders. Mol Biol
Rep. 49:9767–9781. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Yan C, Wang Y, Chen C, Yu H, Liu D,
Huang K and Han Y: GCN5-mediated regulation of pathological cardiac
hypertrophy via activation of the TAK1-JNK/p38 signaling pathway.
Cell Death Dis. 13:4212022. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhang T, Li L, Mo X, Xie S, Liu S, Zhao N,
Zhang H, Chen S, Zeng X, Wang S, et al: Matairesinol blunts adverse
cardiac remodeling and heart failure induced by pressure overload
by regulating Prdx1 and PI3K/AKT/FOXO1 signaling. Phytomedicine.
135:1560542024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu Z, Liu H and Wang J: Astragaloside IV
protects against the pathological cardiac hypertrophy in mice.
Biomed Pharmacother. 97:1468–1478. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qian W, Yu D, Zhang J, Hu Q, Tang C, Liu
P, Ye P, Wang X, Lv Q, Chen M and Sheng L: Wogonin attenuates
isoprenaline-induced myocardial hypertrophy in mice by suppressing
the PI3K/Akt pathway. Front Pharmacol. 9:8962018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Solbach TF, König J, Fromm MF and Zolk O:
ATP-binding cassette transporters in the heart. Trends Cardiovasc
Med. 16:7–15. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Park S, Lim BBC, Perez-Terzic C, Mer G and
Terzic A: Interaction of asymmetric ABCC9-encoded nucleotide
binding domains determines KATP channel SUR2A catalytic activity. J
Proteome Res. 7:1721–1728. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aubert G, Barefield DY, Demonbreun AR,
Ramratnam M, Fallon KS, Warner JL, Rossi AE, Hadhazy M, Makielski
JC and McNally EM: Deletion of sulfonylurea receptor 2 in the adult
myocardium enhances cardiac glucose uptake and is cardioprotective.
JACC Basic Transl Sci. 4:251–268. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Flagg TP, Kurata HT, Masia R, Caputa G,
Magnuson MA, Lefer DJ, Coetzee WA and Nichols CG: Differential
structure of atrial and ventricular KATP: Atrial KATP channels
require SUR1. Circ Res. 103:1458–1465. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Stoller D, Kakkar R, Smelley M, Chalupsky
K, Earley JU, Shi NQ, Makielski JC and McNally EM: Mice lacking
sulfonylurea receptor 2 (SUR2) ATP-sensitive potassium channels are
resistant to acute cardiovascular stress. J Mol Cell Cardiol.
43:445–454. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fahrenbach JP, Stoller D, Kim G, Aggarwal
N, Yerokun B, Earley JU, Hadhazy M, Shi NQ, Makielski JC and
McNally EM: Abcc9 is required for the transition to oxidative
metabolism in the newborn heart. FASEB J. 28:2804–2815. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mohammed Abdul KS, Jovanović S, Du Q,
Sukhodub A and Jovanović A: A link between ATP and SUR2A: A novel
mechanism explaining cardioprotection at high altitude. Int J
Cardiol. 189:73–76. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McClenaghan C and Nichols CG: Kir6.1 and
SUR2B in Cantú syndrome. Am J Physiol Cell Physiol. 323:C920–C935.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
McClenaghan C, Huang Y, Yan Z, Harter TM,
Halabi CM, Chalk R, Kovacs A, van Haaften G, Remedi MS and Nichols
CG: Glibenclamide reverses cardiovascular abnormalities of Cantu
syndrome driven by KATP channel overactivity. J Clin Invest.
130:1116–1121. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
McClenaghan C, Huang Y, Matkovich SJ,
Kovacs A, Weinheimer CJ, Perez R, Broekelmann TJ, Harter TM, Lee
JM, Remedi MS and Nichols CG: The mechanism of High-output cardiac
hypertrophy arising from potassium channel Gain-of-Function in
Cantú syndrome. Function (Oxf). 1:zqaa0042020. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang H, Hanson A, de Almeida TS, Emfinger
C, McClenaghan C, Harter T, Yan Z, Cooper PE, Brown GS, Arakel EC,
et al: Complex consequences of Cantu syndrome SUR2 variant R1154Q
in genetically modified mice. JCI Insight. 6:e1459342021.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fernlund E, Kissopoulou A, Green H,
Karlsson JE, Ellegård R, Årstrand HK, Jonasson J and Gunnarsson C:
Hereditary hypertrophic cardiomyopathy in children and young
Adults-the value of reevaluating and expanding gene panel analyses.
Genes (Basel). 11:14722020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He K, Wang X, Li T, Li Y and Ma L:
Chlorogenic acid attenuates isoproterenol Hydrochloride-induced
cardiac hypertrophy in AC16 cells by inhibiting the Wnt/β-Catenin
signaling pathway. Molecules. 29:7602024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Wang X, He K, Ma L, Wu L, Yang Y and Li Y:
Puerarin attenuates isoproterenol-induced myocardial hypertrophy
via inhibition of the Wnt/β-catenin signaling pathway. Mol Med Rep.
26:3062022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dorn GW and Force T: Protein kinase
cascades in the regulation of cardiac hypertrophy. J Clin Invest.
115:527–537. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bisserier M, Berthouze-Duquesnes M,
Breckler M, Tortosa F, Fazal L, de Régibus A, Laurent AC, Varin A,
Lucas A, Branchereau M, et al: Carabin protects against cardiac
hypertrophy by blocking calcineurin, Ras, and
Ca2+/calmodulin-dependent protein kinase II signaling. Circulation.
131:390–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
de Lucia C, Eguchi A and Koch WJ: New
Insights in cardiac β-adrenergic signaling during heart failure and
aging. Front Pharmacol. 9:9042018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jeong MY, Lin YH, Wennersten SA,
Demos-Davies KM, Cavasin MA, Mahaffey JH, Monzani V, Saripalli C,
Mascagni P, Reece TB, et al: Histone deacetylase activity governs
diastolic dysfunction through a nongenomic mechanism. Sci Transl
Med. 10:eaao01442018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Singh K, Xiao L, Remondino A, Sawyer DB
and Colucci WS: Adrenergic regulation of cardiac myocyte apoptosis.
J Cell Physiol. 189:257–265. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lymperopoulos A, Rengo G and Koch WJ:
Adrenergic nervous system in heart failure: Pathophysiology and
therapy. Circ Res. 113:739–753. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Su H, Liu M, Wang S, Tian B, Hu H, Ma LK
and Pan J: Co-administration of isoprenaline and phenylephrine
induced a new HFrEF mouse model through activation of both SNS and
RAAS. Front Cardiovasc Med. 12:15315092025. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ferrari R, Ceconi C, Curello S and Visioli
O: The neuroendocrine and sympathetic nervous system in congestive
heart failure. Eur Heart J. 19 (Suppl F):F45–F51. 1998.PubMed/NCBI
|
|
42
|
Abi-Gerges A, Castro L, Leroy J, Domergue
V, Fischmeister R and Vandecasteele G: Selective changes in
cytosolic β-adrenergic cAMP signals and L-type Calcium Channel
regulation by Phosphodiesterases during cardiac hypertrophy. J Mol
Cell Cardiol. 150:109–121. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Xu H, Wang Z, Chen M, Zhao W, Tao T, Ma L,
Ni Y and Li W: YTHDF2 alleviates cardiac hypertrophy via regulating
Myh7 mRNA decoy. Cell Biosci. 11:1322021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Murray DR, Prabhu SD and Chandrasekar B:
Chronic beta-adrenergic stimulation induces myocardial
proinflammatory cytokine expression. Circulation. 101:2338–2341.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Liu BY, Li L, Liu GL, Ding W, Chang WG, Xu
T, Ji XY, Zheng XX, Zhang J and Wang JX: Baicalein attenuates
cardiac hypertrophy in mice via suppressing oxidative stress and
activating autophagy in cardiomyocytes. Acta Pharmacol Sin.
42:701–714. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wei H, Guo X, Yan J, Tian X, Yang W, Cui
K, Wang L and Bingyan G: Neuregulin-4 alleviates isoproterenol
(ISO)-induced cardial remodeling by inhibiting inflammation and
apoptosis via AMPK/NF-κB pathway. Int Immunopharmacol.
143:1133012024. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang J, Wang Z and Chen DL: Shikonin
ameliorates isoproterenol (ISO)-induced myocardial damage through
suppressing fibrosis, inflammation, apoptosis and ER stress. Biomed
Pharmacother. 93:1343–1357. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Yang D, Liu HQ, Liu FY, Guo Z, An P, Wang
MY, Yang Z, Fan D and Tang QZ: Mitochondria in pathological cardiac
hypertrophy research and therapy. Front Cardiovasc Med.
8:8229692022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Singh GK, McClenaghan C, Aggarwal M, Gu H,
Remedi MS, Grange DK and Nichols CG: A unique High-output cardiac
hypertrophy phenotype arising from low systemic vascular resistance
in cantu syndrome. J Am Heart Assoc. 11:e0273632022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Samad M, Malempati S and Restini CBA:
Natriuretic peptides as biomarkers: Narrative review and
considerations in cardiovascular and respiratory dysfunctions. Yale
J Biol Med. 96:137–149. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Edwards JG: Cardiac MHC gene expression:
More complexity and a step forward. Am J Physiol Heart Circ
Physiol. 294:H14–H15. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Xu J, Sun Z, Li J, Li J, Li Y, Huang H,
Yuan F, Liu M and Fang Z: Qian Yang Yu Yin Granule prevents
hypertensive cardiac remodeling by inhibiting NLRP3 inflammasome
activation via Nrf2. J Ethnopharmacol. 337:1188202025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Nakamura M and Sadoshima J: Mechanisms of
physiological and pathological cardiac hypertrophy. Nat Rev
Cardiol. 15:387–407. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Münzel T, Camici GG, Maack C, Bonetti NR,
Fuster V and Kovacic JC: Impact of oxidative stress on the heart
and vasculature: Part 2 of a 3-Part series. J Am Coll Cardiol.
70:212–229. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fandy TE, Jiemjit A, Thakar M, Rhoden P,
Suarez L and Gore SD: Decitabine induces delayed reactive oxygen
species (ROS) accumulation in leukemia cells and induces the
expression of ROS generating enzymes. Clin Cancer Res.
20:1249–1258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dai DF, Johnson SC, Villarin JJ, Chin MT,
Nieves-Cintrón M, Chen T, Marcinek DJ, Dorn GW II, Kang YJ, Prolla
TA, et al: Mitochondrial oxidative stress mediates angiotensin
II-induced cardiac hypertrophy and Galphaq overexpression-induced
heart failure. Circ Res. 108:837–846. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Qi B, Xu R, Jin Y, Wang Y, Cheng T, Liu C,
Ji Y, Guo L, Li J, Gao Y, et al: A critical role for IL-21/IL-21
receptor signaling in isoproterenol-induced cardiac remodeling. Sci
Rep. 15:189852025. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Mustafa M, Ahmad R, Tantry IQ, Ahmad W,
Siddiqui S, Alam M, Abbas K, Moinuddin Hassan MI, Habib S and Islam
S: Apoptosis: A comprehensive overview of signaling pathways,
morphological changes, and physiological significance and
therapeutic implications. Cells. 13:18382024. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Cao H, Zhao L, Yuan Y, Liao C, Zeng W, Li
A, Huang Q, Zhao Y, Fan Y, Jiang L, et al: Lipoamide attenuates
hypertensive myocardial hypertrophy through PI3K/Akt-mediated Nrf2
signaling pathway. J Cardiovasc Transl Res. 17:910–922. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mei L, Chen Y, Chen P, Chen H, He S, Jin
C, Wang Y, Hu Z, Li W, Jin L, et al: Fibroblast growth factor 7
alleviates myocardial infarction by improving oxidative stress via
PI3Kα/AKT-mediated regulation of Nrf2 and HXK2. Redox Biol.
56:1024682022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Maruyama N, Ogata T, Kasahara T, Hamaoka
T, Higuchi Y, Tsuji Y, Tomita S, Sakamoto A, Nakanishi N and Matoba
S: Loss of Cavin-2 destabilizes phosphatase and tensin homologue
and enhances Akt signalling pathway in cardiomyocytes. Cardiovasc
Res. 120:1562–1576. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang Q, Luo Y, Zheng Q, Zhao H, Wei X and
Li X: Itaconate attenuates autoimmune hepatitis via PI3K/AKT/mTOR
pathway-mediated inhibition of dendritic cell maturation and
autophagy. Heliyon. 9:e175512023. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Vainio L, Taponen S, Kinnunen SM,
Halmetoja E, Szabo Z, Alakoski T, Ulvila J, Junttila J, Lakkisto P,
Magga J and Kerkelä R: GSK3β serine 389 phosphorylation modulates
cardiomyocyte hypertrophy and ischemic injury. Int J Mol Sci.
22:135862021. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Heineke J and Molkentin JD: Regulation of
cardiac hypertrophy by intracellular signalling pathways. Nat Rev
Mol Cell Biol. 7:589–600. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Gu J, Qiu M, Lu Y, Ji Y, Qian Z and Sun W:
Piperlongumine attenuates angiotensin-II-induced cardiac
hypertrophy and fibrosis by inhibiting Akt-FoxO1 signalling.
Phytomedicine. 82:1534612021. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Ren J, Yang L, Zhu L, Xu X, Ceylan AF, Guo
W, Yang J and Zhang Y: Akt2 ablation prolongs life span and
improves myocardial contractile function with adaptive cardiac
remodeling: Role of Sirt1-mediated autophagy regulation. Aging
Cell. 16:976–987. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bencun M, Spreyer L, Boileau E, Eschenbach
J, Frey N, Dieterich C and Völkers M: A novel uORF regulates
folliculin to promote cell growth and lysosomal biogenesis during
cardiac stress. Sci Rep. 15:33192025. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Völkers M, Toko H, Doroudgar S, Din S,
Quijada P, Joyo AY, Ornelas L, Joyo E, Thuerauf DJ, Konstandin MH,
et al: Pathological hypertrophy amelioration by PRAS40-mediated
inhibition of mTORC1. Proc Natl Acad Sci USA. 110:12661–12666.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Liu Q, Chen Y, Auger-Messier M and
Molkentin JD: Interaction between NFκB and NFAT coordinates cardiac
hypertrophy and pathological remodeling. Circ Res. 110:1077–1086.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Yu H, Lin L, Zhang Z, Zhang H and Hu H:
Targeting NF-κB pathway for the therapy of diseases: Mechanism and
clinical study. Signal Transduct Target Ther. 5:2092020. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Czabotar PE, Lessene G, Strasser A and
Adams JM: Control of apoptosis by the BCL-2 protein family:
Implications for physiology and therapy. Nat Rev Mol Cell Biol.
15:49–63. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Meng X, Cui J and He G: Bcl-2 is involved
in cardiac hypertrophy through PI3K-Akt pathway. Biomed Res Int.
2021:66155022021. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Condorelli G, Drusco A, Stassi G,
Bellacosa A, Roncarati R, Iaccarino G, Russo MA, Gu Y, Dalton N,
Chung C, et al: Akt induces enhanced myocardial contractility and
cell size in vivo in transgenic mice. Proc Natl Acad Sci USA.
99:12333–12338. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Schiekofer S, Shiojima I, Sato K, Galasso
G, Oshima Y and Walsh K: Microarray analysis of Akt1 activation in
transgenic mouse hearts reveals transcript expression profiles
associated with compensatory hypertrophy and failure. Physiol
Genomics. 27:156–170. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Matsui T, Li L, Wu JC, Cook SA, Nagoshi T,
Picard MH, Liao R and Rosenzweig A: Phenotypic spectrum caused by
transgenic overexpression of activated Akt in the heart. J Biol
Chem. 277:22896–22901. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang T, Xiu YH, Xue H, Li YN, Cao JL, Hou
WS, Liu J, Cui YH, Xu T, Wang Y and Jin CH: A mechanism of
Isoorientin-induced apoptosis and migration inhibition in gastric
cancer AGS cells. Pharmaceuticals (Basel). 15:15412022. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Marchi S, Giorgi C, Suski JM, Agnoletto C,
Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti
F, et al: Mitochondria-Ros crosstalk in the control of cell death
and aging. J Signal Transduct. 2012:3296352012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Manning BD and Toker A: AKT/PKB Signaling:
Navigating the network. Cell. 169:381–405. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Nagoshi T, Matsui T, Aoyama T, Leri A,
Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, et
al: PI3K rescues the detrimental effects of chronic Akt activation
in the heart during ischemia/reperfusion injury. J Clin Invest.
115:2128–2138. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Derossi D, Williams EJ, Green PJ, Dunican
DJ and Doherty P: Stimulation of mitogenesis by a cell-permeable PI
3-kinase binding peptide. Biochem Biophys Res Commun. 251:148–152.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Bryan J, Muñoz A, Zhang X, Düfer M, Drews
G, Krippeit-Drews P and Aguilar-Bryan L: ABCC8 and ABCC9: ABC
transporters that regulate K+ channels. Pflugers Arch. 453:703–718.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Seino S: ATP-sensitive potassium channels:
A model of heteromultimeric potassium channel/receptor assemblies.
Annu Rev Physiol. 61:337–362. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Huang Y, McClenaghan C, Harter TM, Hinman
K, Halabi CM, Matkovich SJ, Zhang H, Brown GS, Mecham RP, England
SK, et al: Cardiovascular consequences of KATP overactivity in
Cantu syndrome. JCI Insight. 3:e1211532018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Li K, Janve VS and Denton JS: Automated
patch clamp analysis of heterologously expressed Kir6.2/SUR1 and
Kir6.1/SUR2B KATP currents. Am J Physiol Cell Physiol. 329:C82–C92.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Winter A, Nepper P, Hermann M, Bayer F,
Riess S, Salem R, Hlavicka J, Prinzing A, Hecker F, Holubec T, et
al: Glibenclamide serves as a potent vasopressor to treat
vasoplegia after cardiopulmonary bypass and reperfusion in a
porcine model. Int J Mol Sci. 26:40402025. View Article : Google Scholar : PubMed/NCBI
|