Introduction
Chronic kidney disease (CKD) is a serious
progressive disorder and a major public health concern, with a
global prevalence of 8–14%, affecting an estimated 700–840 million
individuals worldwide (1–3). The disease typically manifests with
subtle symptoms in its early stages. However, as it progresses,
patients experience a gradual deterioration of kidney function,
which establishes it as the third most rapidly increasing cause of
mortality globally. In parallel, the economic burden of CKD has
increased markedly (4).
CKD is a multifactorial chronic condition
characterized by structural damage and dysfunction of the kidney
(5). The renal unit is the
fundamental structural and functional unit of the kidney,
comprising two principal components: The glomerulus and the renal
tubule. The key factors accelerating CKD progression include the
loss of renal units, chronic inflammation, myofibroblast activation
and extracellular matrix deposition (6). In addition, mitochondrial dysfunction
and the disruption of cellular redox homeostasis play important
roles in CKD pathogenesis (7).
The treatment of CKD involves lifestyle
modifications and pharmacological interventions. Standard
treatments include renin-angiotensin-aldosterone system inhibitors,
sodium-glucose cotransporter 2 inhibitors and glucagon-like
peptide-1 receptor agonists. However, these medications have
limited clinical efficacy in preventing disease progression
(8,9). Therefore, the development of drugs
targeting mitochondrial dysfunction and redox imbalance may be of
considerable value for the improvement of CKD outcomes.
The primary function of mitochondria is adenosine
triphosphate (ATP) synthesis, which is essential for the
maintenance of various cellular functions and the regulation of
critical processes, including cell growth, senescence and
apoptosis. Impairment of this crucial function substantially
affects renal physiology, contributing to the development of renal
diseases (10). The kidney is
among the most mitochondria-rich organs and is highly vulnerable to
mitochondrial damage. Such damage compromises ATP production,
causes cellular energy deficiency, and ultimately results in
tubular epithelial cell atrophy or dedifferentiation (11).
Damaged mitochondria undergo depolarization, which
results in a reduction of the mitochondrial membrane potential
(MMP) (12). Several mechanisms,
including abnormalities in mitochondrial dynamics, mutations in
nuclear or mitochondrial DNA (mtDNA), nutrient deficiencies and
bursts of reactive oxygen species (ROS), contribute to CKD
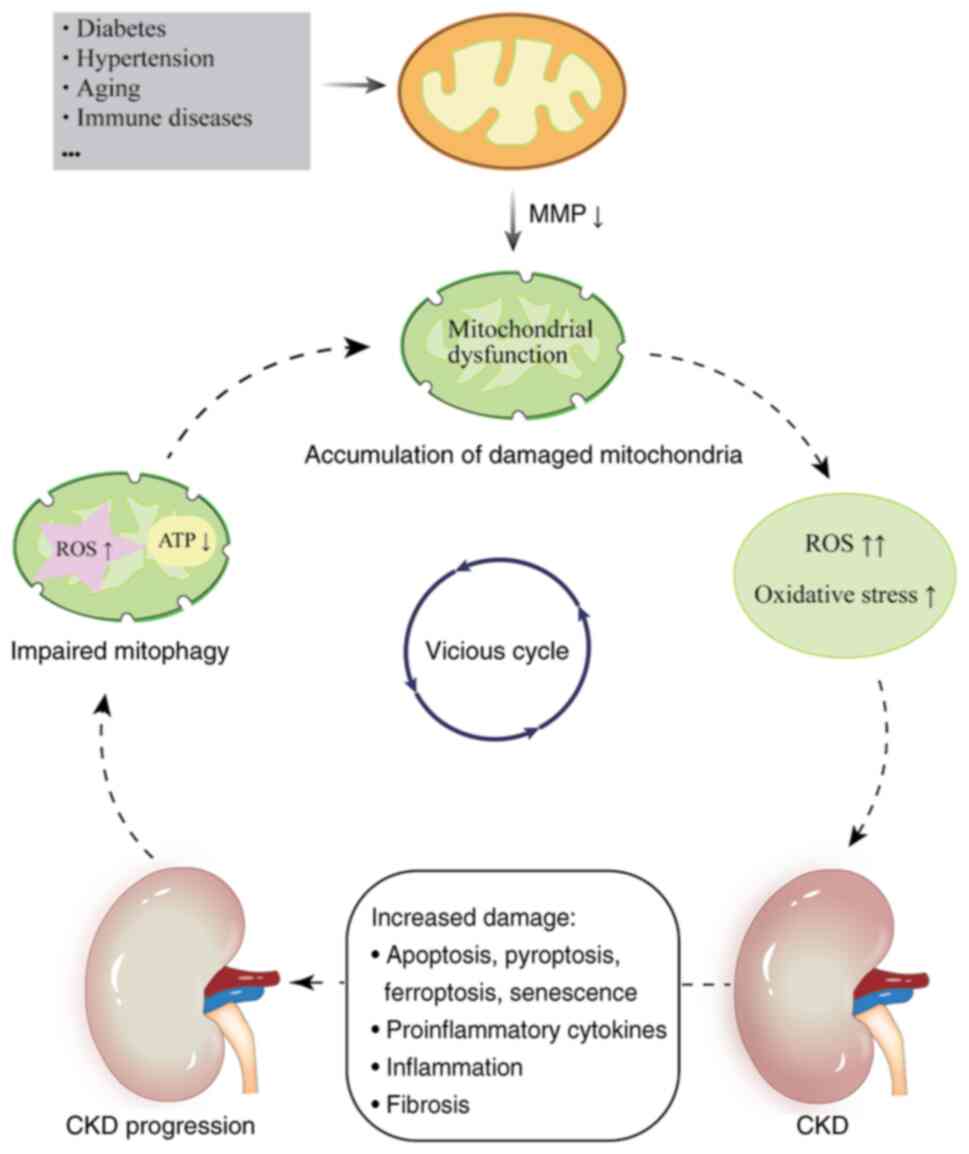
progression, as do certain systemic conditions, such as diabetes,
hypertension, aging and immune diseases (13). Mitochondrial dysfunction disrupts
multiple cellular processes, leading to increased oxidative stress,
chronic inflammation and fibrosis. It also triggers renal
epithelial cell death and promotes microvascular damage, which
collectively impair kidney function (14). Mitochondrial homeostasis and
function are critically dependent on tightly regulated
mitochondrial dynamics (15).
Disruption of these dynamics may reduce the activity of the
mitochondrial respiratory chain complex, diminish ATP generation
and increase mitochondrial ROS (mROS) levels, thereby inducing
oxidative damage (16).
Beyond their role as energy producers, mitochondria
serve as critical regulators of immunity (17). Elevated ROS levels directly induce
oxidative damage in mitochondria via the activation of
proinflammatory signaling molecules, including Toll-like receptors
and the NOD-like receptor protein 3 (NLRP3) inflammasome, thereby
contributing to renal injury (18). Mitochondrial dysfunction regulates
multiple programmed pathways, including apoptosis, pyroptosis and
ferroptosis, thereby promoting tubular cell loss, inflammation and
kidney dysfunction. Apoptosis is the predominant mechanism of cell
death in renal tubular epithelial cells (RTECs) and podocytes
(11). In addition, mitochondrial
dysfunction promotes cellular senescence and influences the
downstream senescence-associated phenotype (19). Senescent RTECs lose their capacity
for self-repair and secrete proinflammatory cytokines, the
continuous accumulation of which substantially contributes to CKD
progression (20) (Fig. 1).
Mitochondria play a key role in essential cellular
processes, including the tricarboxylic acid cycle, electron
transport system, fatty acid β-oxidation and calcium homeostasis.
These activities inherently lead to ROS generation (21). At physiological levels, ROS
function as signaling molecules in various signaling pathways,
while high levels are toxic to mitochondria and cells (22). Elevated ROS activate multiple
inflammatory factors, and excessive inflammation promotes further
ROS production, creating a mutually reinforcing cycle that
accelerates CKD progression (23).
Oxidative stress results from an imbalance in redox homeostasis
between oxidative and antioxidant systems, which adversely affects
all renal components (24). A
primary source of oxidative stress is the overproduction of
strongly oxidizing free radicals, such as ROS and reactive nitrogen
species. Mitochondria serve as predominant intracellular producers
of ROS and as one of their main targets, making them important in
oxidative injury (25). Under
oxidative stress conditions, ROS activate mitophagy, a process that
removes damaged or excessive mitochondria. This helps to reduce
further ROS production, thereby mitigating oxidative stress and
safeguarding cellular components against oxidative injury (26). Although oxidative stress, mitophagy
and ROS have been studied in the context of renal diseases, their
interdependent roles in modulating the progression of CKD are not
fully understood. By synthesizing current evidence, the present
review aims to offer novel insights into the pathophysiological
crosstalk among these mechanisms and to explore promising
therapeutic strategies targeting these processes to ameliorate
CKD.
Mitophagy: An overview
Definition of mitophagy
As the ‘power station’ of the cell, mitochondria
orchestrate multiple intracellular signaling pathways,
participating in amino acid metabolism, pyridine nucleotide
synthesis and phospholipid modifications, which collectively
influence cell survival, senescence and death (27,28).
Mitochondria are dynamic organelles with a high degree of
plasticity, and this adaptability is encompassed by the concept of
mitochondrial dynamics. Mitochondria continuously undergo
coordinated cycles of biogenesis, fusion, fission and autophagy.
Mitophagy is a selective form of autophagy that eliminates damaged
or superfluous mitochondria. These processes determine the
morphology, quality, quantity, distribution and function of
mitochondria within cells (29).
The primary structural components of mitochondria are the outer
mitochondrial membrane (OMM), inner mitochondrial membrane (IMM)
and intermembrane space (IMS). The IMM surrounds the mitochondrial
matrix and folds into invaginated cristae, markedly increasing the
surface area of IMM, thereby efficiently generating ATP (28). The key mediators of mitophagy are
PTEN-induced kinase 1 (PINK1), Parkin and ubiquitin chains, which
promote the engulfment of damaged mitochondria by autophagosomes
for lysosomal degradation (30).
By contrast, mitochondrial biogenesis, a complex biological process
essential for mitochondrial self-renewal, is primarily regulated by
peroxisome proliferator-activated receptor-g coactivator-1 a; this
process protects mtDNA and promotes dynamic cellular homeostasis
(31). Mitochondrial fusion is
mediated by mitofusin 1 or 2 (MFN1/2) and optic atrophy protein 1.
This fusion process involves the merging of the OMM and IMM from
adjacent mitochondria to create an enlarged organelle.
Dynamin-related protein 1 (Drp1) is the primary regulator of
mitochondrial fission, a process in which the OMM and IMM of a
single mitochondrion are constricted and divided to produce two
daughter mitochondria (32).
Notably, MFN2 functions as a mitochondrial fusion protein in its
non-phosphorylated state, whereas PINK1-mediated phosphorylation
converts MFN2 into a key mediator of the translocation of Parkin to
damaged mitochondria (30).
Mitochondrial fusion is a process of repair and renewal for the
retention of mitochondria, whereas mitophagy ensures the removal of
irreparably damaged mitochondria. Therefore, mitophagy, rather than
mitochondrial fission, is considered the biological antithesis of
mitochondrial fusion (33). The
aforementioned processes of mitochondrial dynamics do not function
independently; they are tightly interconnected and dynamically
regulated as adaptations to cellular life activities. This
integrated network of mitochondrial dynamics is essential for
mitochondrial-cellular interactions and homeostasis (Fig. 2).
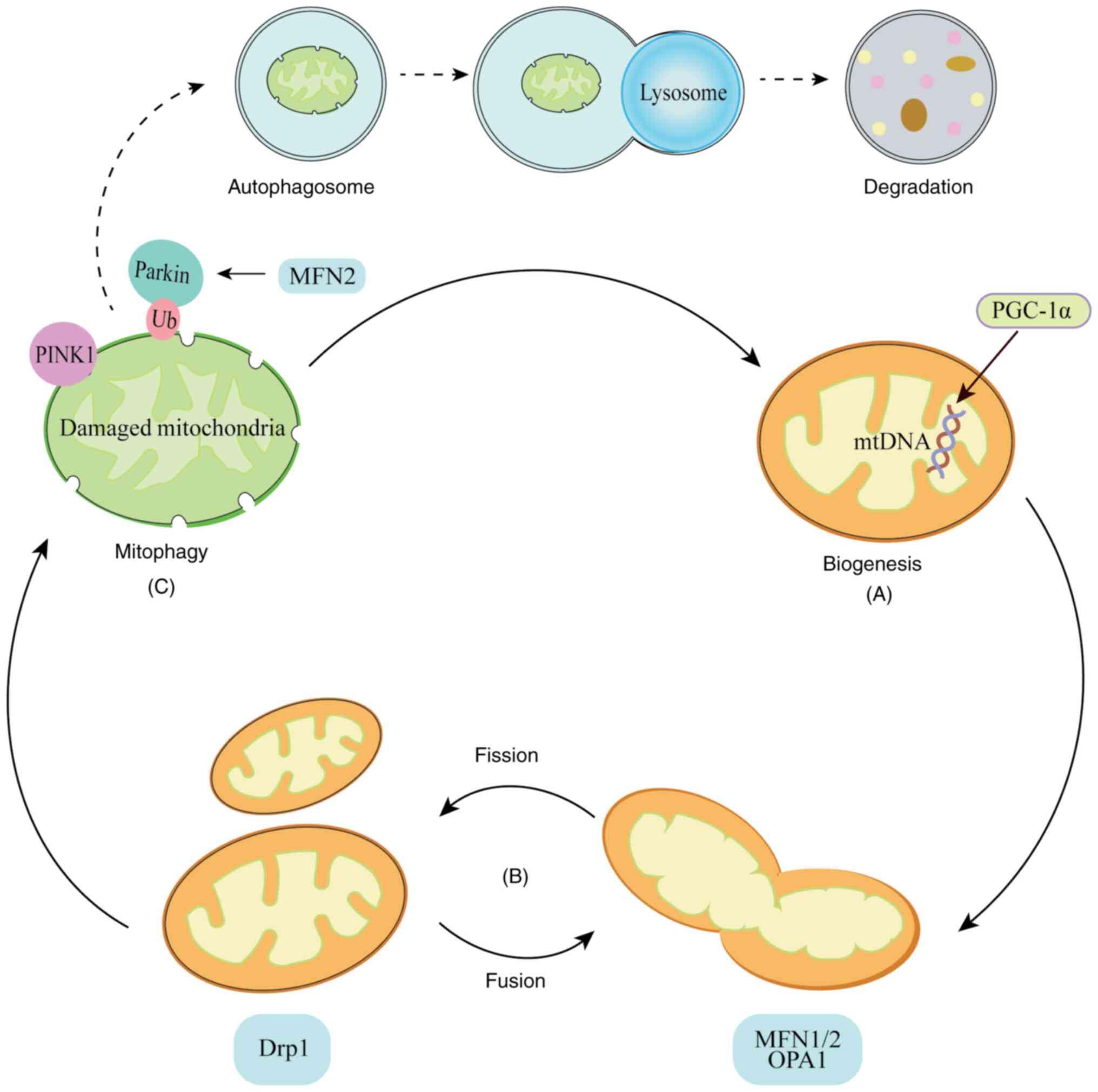 | Figure 2.Schematic diagram of mitochondrial
dynamics. (A) Mitochondrial biogenesis. PGC-1α is a master
regulator of mitochondrial biogenesis, which controls mitochondrial
self-renewal and protects mtDNA. (B) Mitochondrial fission and
fusion. Fission is mainly mediated by Drp1, which severs the outer
and inner mitochondrial membranes, resulting in division of the
organelle into two daughter mitochondria. The primary mediators of
mitochondrial fusion are MFN1, MFN2 and OPA1. (C) Mitophagy.
Autophagosomes engulf the damaged mitochondria, which are targeted
by PINK, Parkin and Ub, and then delivered to lysosomes for
degradation. MFN2 promotes the translocation of Parkin to damaged
mitochondria when it is phosphorylated by PINK1. PGC-1α, peroxisome
proliferator-activated receptor-γ coactivator-1α; mtDNA,
mitochondrial DNA; Drp1, dynamin-related protein l; MFN1/2,
mitofusin 1/2; OPA1, optic atrophy protein l; PINK1, PTEN-induced
kinase 1; Ub, ubiquitin. |
Autophagic processes can be categorized into three
forms: Macroautophagy, microautophagy and chaperone-mediated
autophagy (34). In the present
review, the term autophagy refers specifically to macroautophagy,
the most extensively studied form. In this process, cargo proteins
are sequestered within a double-membraned autophagosome, either
non-selectively, termed bulk autophagy, or through the precise
elimination of specific cellular components, termed selective
autophagy. Subsequently, the autophagosome is transported to the
lysosomal compartment for degradation and recirculation of its
contents. This process plays a crucial role in the maintenance of
cellular and organismal homeostasis via a complex molecular pathway
(35).
Under several pathological conditions, including
elevated ROS levels, nutrient deficiencies and cellular senescence,
intracellular mitochondria may undergo depolarization, a feature of
mitochondrial dysfunction. To eliminate dysfunctional or excess
mitochondria and maintain intracellular homeostasis, mitophagy is
initiated as a cytoprotective mechanism. As discussed above,
mitophagy is a selective form of autophagy in which impaired
mitochondria are translocated to the lysosomal compartment, where
they undergo degradation. This prevents the accumulation of
dysfunctional mitochondria and downstream molecular events that
lead to disease progression, including oxidative stress (36).
Molecular mechanisms of mitophagy
PINK1/Parkin pathway
In 2008, Narendra et al (37) discovered that Parkin, an E3
ubiquitin ligase, is involved in mitochondrial depolarization and
recruited to accelerate mitochondrial autophagic degradation.
Subsequent studies identified key mitophagy receptors involved in
this process, including sequestosome 1 (also known as p62),
optineurin (OPTN) and nuclear dot protein 52 (NDP52) (36). Parkin, encoded by PRKN, functions
in conjunction with PINK1, a mitochondrial-targeted serine kinase.
Upon mitochondrial depolarization, Parkin is recruited to scavenge
damaged mitochondria and promote mitophagy. This finding is
considered a landmark discovery in mitophagy research (38). The PINK1/Parkin pathway is the most
extensively studied mechanism of mitophagy, and comprises three
main components: PINK1 as the mitochondrial damage sensor, Parkin
as the signal amplifier and ubiquitin chains as the signal
effectors (39). Upon
mitochondrial damage, PINK1 accumulates at the OMM and
phosphorylates ubiquitin. This phosphorylated ubiquitin activates
Parkin, which subsequently adds more ubiquitin chains to OMM
proteins. These chains serve as labels for autophagy receptors in
damaged mitochondria, thereby triggering selective autophagy
(40) (Fig. 3).
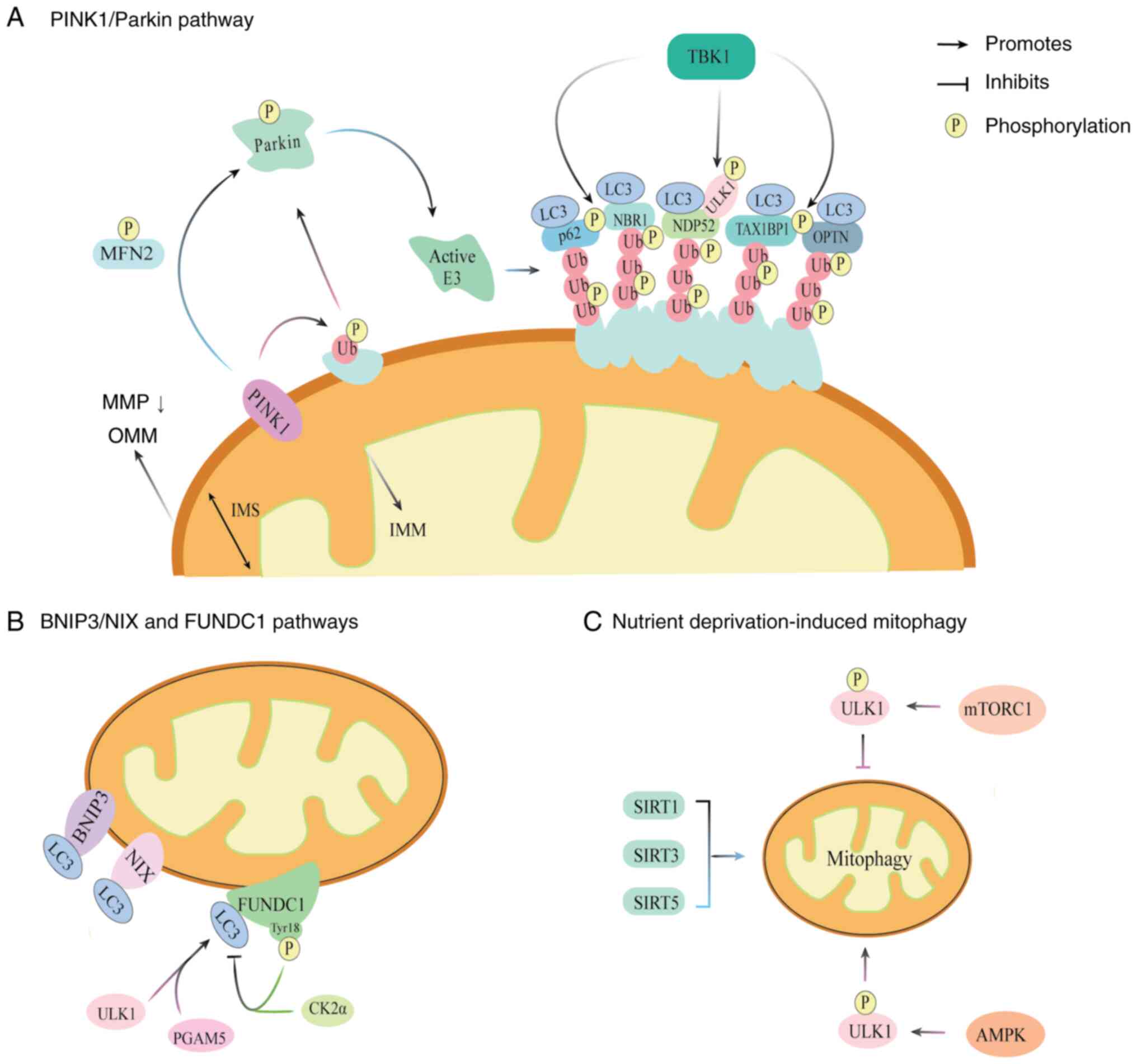 | Figure 3.Molecular mechanisms of mitophagy.
(A) PINK1/Parkin pathway. Among the various mitophagy pathways, the
PINK1/Parkin pathway is the most well-characterized. Loss of MMP
causes PINK1 to accumulate on the OMM of the depolarized
mitochondria. and the recruitment of Parkin. PINK1 phosphorylates
Ub and Parkin, which converts Parkin into an active E3 Ub ligase.
PINK1 also phosphorylates MFN2, further promoting the recruitment
of Parkin to damaged mitochondria. Ub chains are tethered to
autophagy receptors, including p62, NBR1, NDP52, TAX1BP1 and OPTN,
that interact with LC3 on the autophagosome. NDP52 also binds to
the ULK1 complex, a process that is facilitated by the
TBK1-mediated phosphorylation of autophagy receptors. (B) BNIP3/NIX
and FUNDC1 pathways. The receptors BNIP3, NIX and FUNDC1 bind to
LC3, to induce mitophagy. Phosphorylation of the Tyr18 residue of
FUNDC1 by CK2α weakens the binding between LC3 and FUNDC1. By
contract, PGAM5 and ULK1 enhance the interaction of FUNDC1 with LC3
to promote mitophagy. (C) Nutrient deprivation-induced mitophagy.
Mitophagy induced by nutrient deprivation is primarily regulated by
mTOR, AMPK and SIRTs. Active mTORC1 suppresses mitophagy by
phosphorylating ULK1 while AMPK directly activates ULK1 through
phosphorylation. SIRT1, SIRT3 and SIRT5 have been shown to promote
mitophagy in animal models. PINK1, PTEN induced kinase l; MMP,
mitochondrial membrane potential; OMM, outer mitochondrial
membrane; Ub, ubiquitin; MFN2, mitofusin 2; p62, sequestosome 1;
NBR1, neighbor of BRCA1 gene l; NDP52, nuclear dot protein 52;
TAX1BP1, T-cell leukemia virus type I binding protein 1; OPTN,
optineurin; LC3, microtubule-associated protein 1 light chain 3;
ULK1, Unc-51 like autophagy activating kinase l; TBK1, TANK-binding
kinase 1; IMM, inner mitochondrial membrane; IMS, intermembrane
space; BNIP3, BCL2 interacting protein 3; NIX, Nip3-like protein X;
FUNDC1, FUN14 domain-containing protein l; Tyr18, tyrosine 18;
CK2α, casein kinase 2α; PGAM5, phosphoglycerate mutase family
member 5; mTOR, serine/threonine protein kinase mammalian target of
rapamycin; AMPK, AMP-activated protein kinase; SIRT, sirtuin
mTORC1, mTOR complex 1. |
PINK1 acts as a mitochondrial damage sensor. Its
levels on mitochondria are regulated by voltage-dependent
proteolysis, which ensures low expression on healthy, polarized
mitochondria (41). PINK1
functions upstream of Parkin translocation and Parkin-mediated
mitophagy. In dysfunctional mitochondria with low MMP, the
selective accumulation of PINK1 on the OMM recruits Parkin, which
initiates a series of degradation processes to clear the damaged
mitochondria (41). The
translocase of the outer membrane (TOM) complex, a multimeric
protein assembly at the OMM, mediates the mitochondrial targeting
of PINK1 (42). Under
physiological conditions, TOM20 recognizes the mitochondrial
targeting sequence of PINK1 and facilitates its import into the
TOM40-formed translocation pore, with assistance from TOM22 and
TOM70. PINK1 is ultimately delivered to the translocase of inner
mitochondria membrane 23 complex at the IMM (43). Once inside, PINK1 undergoes
sequential cleavage by mitochondrial processing peptidase and
presenilin-associated rhomboid-like, followed by ubiquitin
proteasome system-mediated degradation via the N-end rule pathway
(44,45). Due to a steady balance between
PINK1 import and degradation, healthy mitochondria maintain an
extremely low or undetectable level of PINK1 accumulation (46). However, mitochondrial dysfunction,
such as the loss of MMP induced by mtDNA mutations, mitochondrial
depolarization, protein misfolding and increased ROS production,
impairs the degradation process, resulting in the accumulation of
PINK1 on the depolarized OMM and Parkin recruitment (47). PINK1 also phosphorylates MFN2,
Parkin and ubiquitin, leading to further Parkin activation
(48).
Parkin, a signal amplifier in mitophagy and a member
of the RING-between-RING E3 ligase family, participates in the
ubiquitin-mediated degradation and trafficking of proteins. It
includes a C-terminal ubiquitin ligase domain and an N-terminal
ubiquitin-like (UBL) domain (49).
In healthy mitochondria, Parkin remains in an inactive conformation
as a self-inhibiting dormant enzyme in the cytosolic compartment.
Following mitochondrial damage, PINK1 accumulates on the OMM and
phosphorylates the serine (Ser)65 residue of ubiquitin, even when
present at low levels (50).
Parkin is activated by two key steps. First, its UBL domain is
phosphorylated at Ser65 by PINK1 kinase. Second, it binds to
phosphorylated ubiquitin, These modifications induce substantial
conformational changes, wherein the ubiquitin-E2 binding site of
Parkin is repositioned on the RING1 domain proximal to the cysteine
(Cys)431 acceptor site on the RING2 domain. This structural
remodeling transforms Parkin into an active E3 ligase (51). Activated Parkin ubiquitinates
various OMM proteins, which are subsequently phosphorylated by
PINK1 to form phospho-ubiquitin chains that recruit autophagy
receptors to induce mitophagy (52). At present, ubiquitin and the UBL
domain of Parkin are the only known direct substrates of PINK1
(53).
Ubiquitin chains function as signal effectors in
autophagy. When these chains are attached to OMM proteins, they are
recognized by autophagy receptors, which mediate the encapsulation
of damaged mitochondria into autophagosomes for lysosomal
degradation. The ubiquitin chains also stimulate PINK1 kinase
activity, creating an efficient feedforward loop that ensures
effective mitophagy (46). Lazarou
et al (40) demonstrated
that p62 and neighbor of BRCA1 gene 1 (NBR1) are not essential for
Parkin-mediated mitophagy. Instead, OPTN and NDP52 are the primary,
yet redundant receptors in this pathway. Knockout of NDP52 or OPTN
alone causes no defect in mitophagy. Since ubiquitin chains cannot
directly bind to autophagic membranes or their associated
autophagy-related protein 8 (ATG8) family proteins, molecular
bridges are required to physically connect them (54). The ATG8 autophagy-related
ubiquitin-like protein family includes microtubule-associated
protein 1 light chain 3 (MAP1LC3, most commonly referred to as LC3)
and γ-aminobutyric acid receptor-associated protein (GABARAP)
subfamilies (55). LC3 shows
binding affinity for multiple proteins containing a LC3-interacting
region (LIR) motif, which marks ubiquitinated protein aggregates
and binds to LC3 to facilitate autophagosome recruitment (56). These receptors contain a
ubiquitin-binding domain to recognize ubiquitin-labeled
mitochondria and a LIR motif to interact with ATG8 family proteins,
forming a functional bridge between damaged mitochondria and the
autophagic structures (57).
Among selective autophagy receptors, p62 was the
first to be identified as playing an important role in
Parkin-mediated mitophagy. Its accumulation at
polyubiquitin-positive mitochondrial clusters is dependent on
Parkin (36). For effective
degradation, cargo proteins must be clustered into discrete
compartments to enable their encapsulation by autophagosomes. p62
promotes this in vivo by forming droplets in which ubiquitin
chains are sequestered, thereby mediating the concentration of
autophagic cargo proteins and facilitating association with LC3 for
delivery to lysosomes (58). While
early research (59) indicated
that deletion of p62 completely blocked the final clearance of
damaged mitochondria, Lazarou et al (40) later demonstrated that p62 is
dispensable for Parkin-mediated mitophagy. NBR1 is an autophagy
receptor that is structurally similar to p62 and assists p62 in
this process (60). The Kelch-like
ECH-associated protein 1 (Keap1)-NF-E2-related factor 2 (Nrf2)
system is essential for protection against oxidative and
electrophilic insults (61). p62
directly interacts with Keap1 and competitively inhibits the
Keap1-Nrf2 interaction, which stabilizes Nrf2 and promotes its
accumulation in the nucleus. The nuclear accumulation of Nrf2
activates the transcription of numerous cytoprotective genes,
including antioxidants (61,62).
NBR1 is stress-inducible and indispensable for activation of the
p62-Keap1-Nrf2 system (63). It
induces the phase separation of p62, thereby enhancing the
formation of p62 liquid droplets, while Nrf2 increases p62
transcription, establishing a positive feedback loop that
continuously regulates p62 levels (63). The ATG8-binding capacity of NDP52
and OPTN is weaker than that of p62 and NBR1 (40). OPTN consists of several structural
domains, including two coiled-coil (CC) domains, a leucine zipper,
an LIR, a ubiquitin-binding domain and a zinc finger (ZF) domain
(64). The accumulation of
ubiquitin chains on damaged mitochondria triggers the activation of
TANK-binding kinase 1 (TBK1), which physically associates with p62,
OPTN and NDP52, and phosphorylates sites in the ubiquitin-binding
domain of p62 and residues of OPTN neighboring the LIR motifs,
thereby strengthening the ability of p62 and OPTN to interact with
LC3 (65,66). NDP52 is a multi-domain autophagy
receptor, with a N-terminal SKIP carboxyl homology (SKICH) domain,
central CC region and C-terminal ZF ubiquitin-binding domain
(67). The SKICH domain binds to
the Unc-51 like autophagy activating kinase 1 (ULK1) complex
subunit RB1-inducible coiled-coil protein 1, also known as FAK
family kinase-interacting protein of 200 kDa (FIP200), thereby
activating cargo recruitment and autophagosome formation. TBK1
facilitates NDP52-ULK1 complex formation, further supporting the
activation of mitophagy (68).
Notably in addition to the four aforementioned
receptors, there is a fifth autophagy receptor, human T-cell
leukemia virus type I binding protein 1 (TAX1BP1), which is a
paralog of NDP52 (53). Both NDP52
and TAX1BP1 have an atypical C-LIR motif located between the SKICH
and CC domains. The atypical C-LIR motif in NDP52 preferentially
binds with LC3C (69). Following
OMM protein ubiquitination, TAX1BP1 is recruited to damaged
mitochondria, where it interacts with LC3 via its LIR motif to
initiate autophagosome formation and mitochondrial clearance
(70). It has been revealed that
TAX1BP1 and NBR1 colocalize, with TAX1BP1 serving as a critical
organizer that directs the recruitment of upstream autophagy
factors, including FIP200 and TBK1, to form autophagosomes around
NBR1-marked cargo. Via these interactions, TAX1BP1 helps to tether
ubiquitinated mitochondria to autophagic membranes via LC3, thereby
ensuring efficient degradation (71).
BCL2 interacting protein 3
(BNIP3)/Nip3-like protein X (NIX) pathway
A secondary mitophagy mechanism, regulated by the
expression thresholds of BNIP3 and NIX (also known as BNIP3 like),
is known to contribute substantially to mitophagy across multiple
tissues (72). Unlike other BCL2
family members, BNIP3 and NIX contain a divergent BH3 domain that
is not essential for inducing apoptosis (73).
BNIP3 is an OMM protein consisting of a large
N-terminal region containing an LIR motif and a characteristic
C-terminal transmembrane (TM) domain (74). Bruick (75) identified that BNIP3 acts as a
hypoxia-responsive gene. Under hypoxic conditions, BNIP3 promotes
mitophagy by triggering mitochondrial depolarization (76). BNIP3 also interacts with PINK1 to
enhance the accumulation of full-length PINK1 on the OMM, whereas
the inhibition of BNIP3 promotes PINK1 proteolytic processing and
suppresses PINK1/Parkin-mediated mitophagy (77). In addition, BNIP3 actively
suppresses Drp1-mediated mitochondrial fusion, thereby facilitating
the fragmentation and segregation of damaged mitochondria (78).
NIX is structurally similar to BNIP3, and is a
single-pass OMM protein whose TM domain is essential for correct
OMM targeting (79). During
erythroid differentiation, NIX expression is upregulated at the
transcriptional level. The deletion of NIX disrupts erythroid
maturation, resulting in anemia, reticulocytosis and
erythroid-myeloid hyperplasia (80). It also plays essential roles in
retinal ganglion cell differentiation and in the reprogramming of
somatic cells into induced pluripotent stem cells (81,82).
NIX participates in mitophagy induced by hypoxia and elevated
oxidative phosphorylation activity (83). It acts as a substrate of Parkin and
functions downstream of the PINK1/Parkin pathway to promote
mitophagy (84).
In resting cells, low levels of NIX and BNIP3 are
maintained by ubiquitination and proteasomal degradation through an
OMM-localized protein called SCFFBXL4 E3 ubiquitin
ligase complex (85). Their
activity in mitophagy is regulated by phosphorylation: The
phosphorylation of BNIP3 at Ser17 and Ser24 within its LIR motif is
essential for LC3B interaction (86), while the phosphorylation of NIX at
Ser34 and Ser35 enables it to bind with GABARAP family proteins
(87). Generally, the BNIP3/NIX
pathway demonstrates stronger associations with development and
differentiation than other mitophagy pathways (88).
FUN14 domain-containing protein 1
(FUNDC1) pathway
FUNDC1 is an integral OMM protein with a C-terminus
that extends into the mitochondrial IMS and a cytosolic N-terminus
containing a typical LIR (89).
Under hypoxic conditions, FUNDC1 is dephosphorylated, which
enhances its binding to LC3 through its typical LIR and promotes
mitophagy (90), while
phosphorylation inhibits this binding. Therefore, the dynamic
balance between FUNDC1 phosphorylation and dephosphorylation
functions as a critical switch that controls its LC3 binding
capacity and subsequent regulation of mitophagy (36). A key regulatory site of FUNDC1 is
tyrosine (Tyr)18, which is phosphorylated by Src kinase. This
weakens the binding between LC3 and FUNDC1 due to electrostatic
repulsion, thereby inhibiting mitophagy (91).
In addition to Tyr18 phosphorylation, three
potential regulators of FUNDC1 activity are currently known. These
are phosphoglycerate mutase family member 5 (PGAM5) and ULK1, which
promote mitophagy, and casein kinase 2α (CK2α), which inhibits
mitophagy (92–94). CK2α, a constitutive Ser/threonine
(Thr) kinase, is a suppressor of FUNDC1 that promotes its
phosphorylation at Ser13, thus effectively preventing its
association with LC3 and inhibiting mitophagy (92). Chen et al (93) demonstrated that PGAM5, a
mitochondrial phosphatase, dephosphorylates the Ser13 residue of
FUNDC1, resulting in an increased association with LC3 during
hypoxia or after treatment with a mitochondrial oxidative
phosphorylation uncoupler (FCCP). FUNDC1 is a novel substrate of
ULK1. ULK1 directly phosphorylates FUNDC1 at Ser17 under hypoxic
conditions or FCCP exposure, following its recruitment to damaged
mitochondria. This promotes the interaction of ULK1 with LC3,
forming a bridge that links damaged mitochondria to autophagosomes
(94).
Nutrient deprivation-induced
mitophagy
Under conditions of nutrient or energy deprivation,
mitophagy is predominantly regulated by three key pathways: Ser/Thr
protein kinase mammalian target of rapamycin (mTOR), AMP-activated
protein kinase (AMPK) and sirtuins (SIRTs) (95). AMPK and mTOR function as nutrient
sensors in the kidney. In addition, mTOR complex 1 (mTORC1) serves
as the primary negative modulator of mitophagy, while AMPK and
SIRTs are positive regulators (95,96).
As a key regulator of anabolism, mTORC1 promotes
cell growth and proliferation while concurrently inhibiting
mitophagy. Under nutrient-abundant conditions, it phosphorylates
ULK1 to suppress mitophagy, whereas under nutrient-limited
conditions, mTORC1 is inactivated, allowing AMPK to activate ULK1
by phosphorylation, thereby inducing mitophagy (97). AMPK is a heterotrimeric complex
comprising a catalytic α-subunit and regulatory β and γ subunits
(98). During glucose deprivation,
AMPK directly activates ULK1 via the phosphorylation of Ser317 and
Ser777, to enhance mitophagy (99).
SIRTs play major roles in various physiological
functions of the kidney, including DNA repair, mitochondrial
metabolism, mitophagy, oxidative stress control and inflammation.
SIRT dysfunction contributes to CKD pathophysiology and progression
(100). Reduced SIRT1 expression
or activity is associated with diminished mitophagy and exacerbated
kidney disease (101,102). SIRT3, the primary mitochondrial
deacetylase, is transformed from an inactive 44-kDa enzyme
precursor to an active 28-kDa protein that translocates to
mitochondria (103). In a sepsis
model created by cecal ligation and puncture (CLP) in mice,
decreased SIRT1 and SIRT3 activity was associated with
mitochondrial damage (104).
Another study revealed that the activation of SIRT3 preserves
mitophagy and maintains mitochondrial homeostasis in both CLP model
mice and lipopolysaccharide (LPS)-treated RTECs (105). SIRT5 expression in the kidney is
minimal. Polletta et al (106) demonstrated that the main function
of SIRT5 in non-hepatic cells is the regulation of glutamine
metabolism, which controls ammonia production and ammonia-induced
mitophagy.
Mitophagy in CKD
Mitophagy is as a key mechanism for mitochondrial
quality control that helps to maintain cellular homeostasis and
protects cells by enabling them to adapt to external environmental
changes. It plays a pivotal role in CKD by modulating factors
associated with inflammation and fibrosis (107). Renal tubule injury is a major
contributor to CKD progression (108). As the primary cellular
constituents of renal tubules, RTECs are essential for both kidney
repair and CKD progression, and critically determine the overall
condition of the kidneys. Their innate immune functions allow them
to recognize diverse stimuli and produce biologically active
mediators that drive interstitial inflammation and fibrosis
(108).
The unilateral ureteral obstruction (UUO) model is
commonly used to study tubulointerstitial fibrosis and CKD
(109). In this model, PINK1 or
Parkin deficiency disrupts mitophagy, resulting in excessive mROS
production, mitochondrial damage, increased TGF-β1 expression and
Drp1 recruitment in RTECs, all of which promote renal fibrosis.
Hypoxia-induced damage to RTECs and renal fibrosis in the UUO model
are ameliorated by Drp1-Parkin-dependent mitophagy, highlighting
the essential role of this process (110). In another study, it was reported
that PINK1 or Parkin knockout in macrophages leads to the
accumulation of damaged mitochondria and promotes their conversion
to a profibrotic phenotype, which aggravates kidney fibrosis
(111). In the kidneys of
patients with CKD, the expression levels of the mitophagy-related
proteins PINK1, Parkin and MFN2 are downregulated, which inhibits
mitophagy. Furthermore, the circulating levels of C-C motif
chemokine ligand 2 are increased, which recruits macrophages and
exacerbates renal fibrosis. UUO models have also shown that Parkin
and MFN2 expression levels are downregulated during kidney fibrosis
(111). Together, these findings
underscore the anti-fibrotic role of mitophagy in the repair of
kidney injury and its potential to slow CKD progression.
Acute kidney injury (AKI) is a major risk factor for
the development and progression of CKD (112). Following AKI, the maladaptive
repair of injured tubules typically results in a sparse
microvascular bed, persistent inflammation, tubular cell loss and
renal fibrosis, leading to the transition of AKI to CKD (113). Proximal tubular cells contain
abundant mitochondria, the dysfunction of which is a major
contributor to AKI pathogenesis. Regardless of etiology, mitophagy
is a vital protective mechanism in AKI (36). If mitochondrial dysfunction
persists after AKI, it promotes progression to CKD via inflammatory
and fibrotic pathways (114).
Cisplatin is a commonly prescribed alkylating
chemotherapeutic agent that demonstrates broad-spectrum anticancer
activity; however, its therapeutic potential is limited by
nephrotoxic, neurotoxic and ototoxic side effects. In particular,
AKI is a recognized complication of cisplatin-based chemotherapy
(115). Mapuskar et al
(116) revealed that
cisplatin-induced pathological changes in mitochondrial metabolism
occur during the repair phase. These are characterized by
superoxide anion radical (O2•−) accumulation,
which predisposes the kidney to future injury and contribute to CKD
progression. In vitro, Zhao et al (117) demonstrated that in human renal
proximal tubular cells the knockdown of PINK1 or Parkin exacerbates
mitochondrial dysfunction by impairing mitophagy, resulting in the
accumulation of damaged mitochondria and increased cellular injury.
By contrast, the overexpression of PINK1 or Parkin activates
mitophagy, which alleviates cisplatin-induced mitochondrial
dysfunction and cellular injury.
The NLRP3 inflammasome plays a key role in renal
inflammation and pyroptosis (118). In both UUO models and hypoxic
conditions, elevated mROS levels, increased accumulation of damaged
mitochondria, activation of the NLRP3 inflammasome and
significantly increased expression of α-SMA and TGF-β1 are
observed. These changes are further intensified following BNIP3
gene deletion or silencing (119).
The activation of mitophagy can reduce pyroptosis in
renal cells (120). For instance,
Zn2+ has been shown to enhance Parkin expression by
inhibiting SIRT7 activity, thereby promoting mitophagy, suppressing
NLRP3 inflammasome activation and pyroptosis, and consequently
protecting against sepsis-induced AKI (121).
Hypoxia-inducible factor 1α (HIF1α) is upregulated
in UUO models, and functions as an upstream regulator of BNIP3.
Knockout of HIF1α leads to impaired mitophagy, aggravated apoptosis
and increased ROS production (122). These findings suggest that
HIF1α-BNIP3-mediated mitophagy protects renal tubular cells against
hypoxia-induced injury and fibrosis by reducing mROS and inhibiting
NLRP3 inflammasome activation.
In the context of cisplatin-induced nephrotoxicity,
Zhou et al (123) observed
the downregulation of PINK1 and Parkin and upregulation of NIX in
the kidney, indicating that the BNIP3/NIX pathway may be
particularly important in cisplatin-induced mitophagy. The study
also indicated that PINK1 knockout ameliorated cisplatin-induced
kidney injury in rats.
Another key mitophagy pathway involves FUNDC1, which
protects against mitochondrial dysfunction, cell death and renal
fibrosis in CKD. In end-stage renal disease, chronic inflammation
and renal injury suppress FUNDC1-mediated mitophagy, resulting in
defective mitochondrial clearance, aggravated renal fibrosis and
deteriorating renal function (124). Notably, the balance of mitophagy
homeostasis is preserved when FUNDC1 alone is deficient, while the
concurrent deficiency of both FUNDC1 and Parkin disrupts the
homeostasis. Parkin compensates for the lack of FUNDC1 via
compensatory upregulation, thereby sustaining basal mitophagy,
which explains why baseline FUNDC1 knockout alone has little impact
on renal function (125).
Oxidative stress: An overview
Definition of oxidative stress
The term oxidative stress was first coined by Sies
and Cadenas (126) in 1985, and
later defined as an imbalance between oxidants and antioxidants in
favor of oxidants, leading to a disruption in redox signaling and
metabolic regulation (127).
Oxidative stress contributes to disease through two distinct but
interrelated mechanisms. The first involves the generation of
reactive species that directly oxidize macromolecules, such as
membrane lipids, structural proteins, functional enzymes and
nucleic acids, resulting in abnormal cellular function or death.
The second is non-physiological redox signaling, particularly
involving the aberrant production of hydrogen peroxide
(H2O2), which can disrupt redox signaling
(128). Sustained oxidative
stress in the kidney initiates a cascade of cellular damage,
including renal ischemia, glomerular lesions, cell death, fibrosis
and exacerbated inflammation. These processes are also associated
with multiple CKD comorbidities, including hypertension, diabetes
and atherosclerosis (129).
Mitochondria-related oxidative
stress
Oxidative stress involves chemical reactions
triggered by various reactive species, with ROS exerting the
greatest biological impact and intracellular ROS predominantly
originating from mitochondria (130). The kidney has high mitochondrial
density, which renders it particularly vulnerable to oxidative
stress-induced damage (11). Under
physiological conditions, ROS generation and scavenging are
strictly balanced. At low levels, ROS are known to affect specific
signaling pathways to regulate cell proliferation, differentiation
and death (129). However, ROS
are generated at high levels when mitochondrial function is
impaired, which creates a signal for the self-elimination of
mitochondria via mitophagy. The excessive production of ROS and
inability of endogenous antioxidants to remove them leads to
oxidative stress (131).
The ROS family includes radical species such as
O2•− and hydroxyl radicals (•OH),
and non-radical oxidants, including H2O2 and
singlet oxygen. Among these, O2•− and
H2O2 are central to redox signaling and can
exert beneficial effects at controlled levels (22,131,132). In the kidney, the major sources
of ROS are NADPH oxidases (NOXs) and the mitochondrial respiratory
chain (133). NOX enzymes
catalyze the transfer of electrons from NADPH to molecular oxygen,
which produce ROS. In addition to NOX4, which serves as a major
source of ROS in the kidney, NOX1 and NOX2 contribute to vascular
dysfunction and fibrosis in CKD by increasing oxidative stress
(134). Furthermore, angiotensin
II (AngII) serves as an early key mediator of kidney disease, which
induces tubular hypertrophy and apoptosis in RTECs through
ROS-mediated molecular mechanisms (135).
The maintenance of redox homeostasis is crucial for
cellular function, and its disruption is a major contributor to
human diseases (136). Oxidative
stress in CKD arises not only from excessive ROS production but
also from diminished antioxidant capacity, particularly due to
impaired Nrf2 activation (137).
The antioxidant system is the primary cellular defense against
oxidative damage. It comprises enzymatic antioxidants, including
superoxide dismutase (SOD) and glutathione peroxidase (GPX), and
nonenzymatic antioxidants such as α-tocopherol (vitamin E),
glutathione and bilirubin (138).
These components are largely regulated by the Keap1-Nrf2 pathway.
Keap1 functions as a sensor of oxidative stress, while Nrf2 is the
principal regulator driving the transcription of antioxidant
enzyme-encoding genes. Under oxidative stress, ROS reacts with
redox-sensitive cysteine residues (Cys273 and Cys288) of Keap1
(139), altering its conformation
and preventing it from binding to Nrf2, thereby affecting the
autophagy receptor p62. The affinity between p62 and Keap1 is
enhanced, forming a stable p62-Keap1 complex, which is subsequently
degraded (140). The
p62-Keap1-Nrf2 system is essential for mitophagy. The degradation
of Keap1 disrupts its polyubiquitination and proteasomal
degradation of Nrf2, thereby enabling Nrf2 stabilization and
accumulation, leading to the sustained transcriptional activation
of antioxidant genes (61).
Mitochondria are major sources of ROS due to
electron leakage, primarily from respiratory chain complexes I and
III. This results in incomplete oxygen reduction and the generation
of O2•−, which is rapidly converted into
H2O2 either spontaneously or via SOD-mediated
catalysis (26). The distribution
of SOD is compartment-specific: SOD1 exists in both the cytosol and
IMS, while SOD2 is localized in the mitochondrial matrix (141). GPX converts
H2O2 to water. However, if not neutralized,
H2O2 can form •OH radicals by
reacting with metal ions (142).
Due to its extreme oxidative capacity, •OH is the most
destructive oxidant, which is capable of inducing oxidative DNA
damage (129) (Fig. 4).
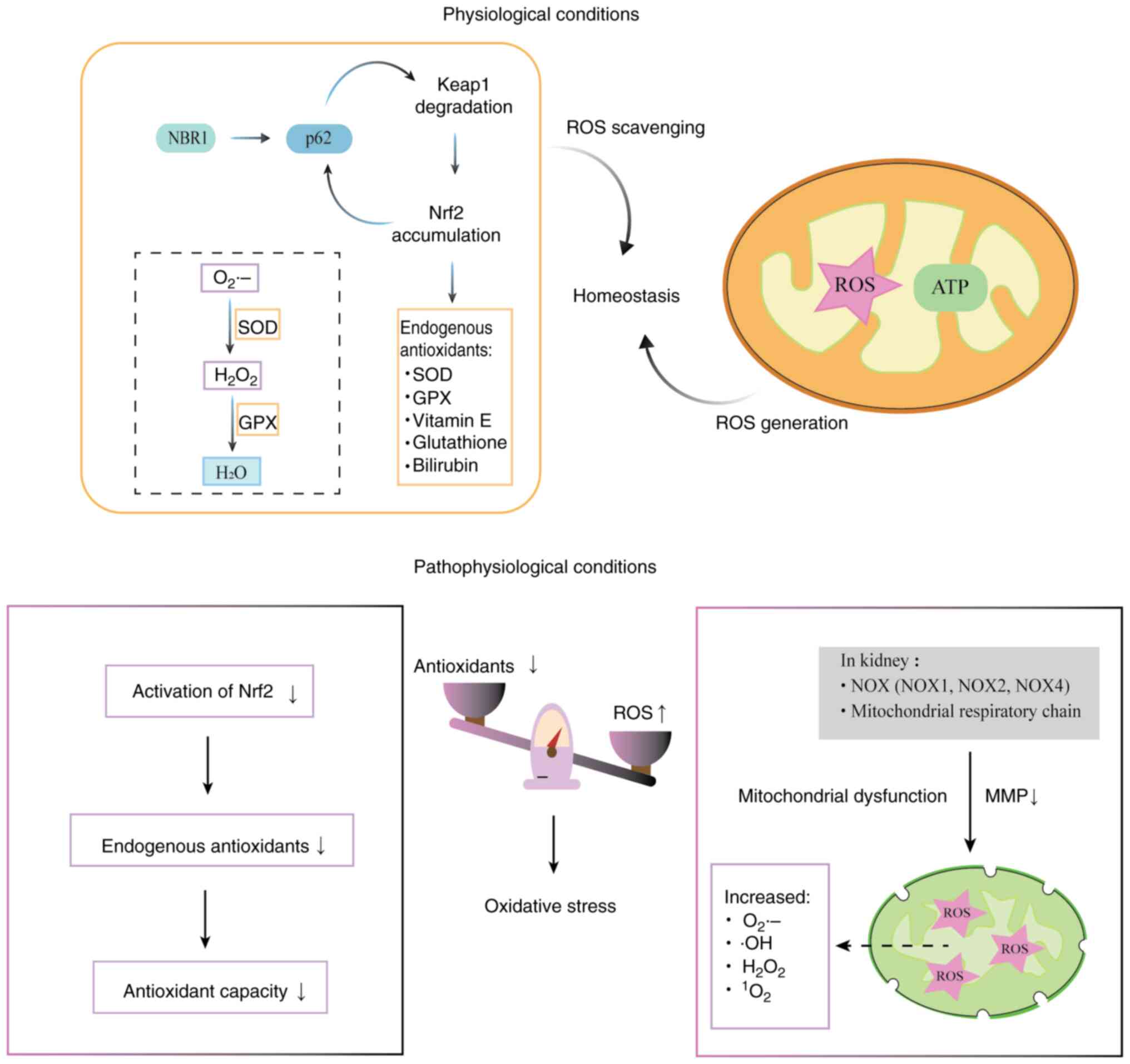 | Figure 4.Diagram of mitochondria-related
oxidative stress. Oxidative stress contributes to disease by two
major mechanisms: ROS overproduction and the disruption of redox
homeostasis. Mitochondria are the primary source of intracellular
ROS. In the kidney, NOXs (NOX1, NOX2 and NOX4) and the
mitochondrial respiratory chain are the major contributors to the
production of ROS, which include O2•−,
•OH, H2O2 and
1O2. Diminished antioxidant capacity, due to
impaired activation of Nrf2, also contributes to oxidative stress.
NBR1 induces the phase separation of p62 and enhances the formation
of p62-containing liquid droplets. A positive feedback mechanism
exists, in which the p62-dependent degradation of Keap1 leads to
the accumulation of Nrf2, which activates the transcription of
antioxidant genes, including p62. The Keap1-Nrf2 pathway regulates
endogenous antioxidants, such as SOD and GPX. SOD rapidly converts
O2•− into H2O2, which
is then converted to water by GPX. However, if unquenched,
H2O2 can generate •OH radicals by reacting
with metal ions. ROS, reactive oxygen species; NOX, NADPH oxidase;
O2•−, superoxide anion radical;
•OH, hydroxyl radical; H2O2,
hydrogen peroxide; 1O2, singlet oxygen; Nrf2,
NF-E2-related factor 2; NBR1, neighbor of BRCA1 gene l; p62,
sequestosome 1; Keap1, Kelch-like ECH-associated protein 1; SOD,
superoxide dismutase; GPX, glutathione peroxidase; ATP, adenosine
triphosphate; MMP, mitochondrial membrane potential. |
Redox regulation of mitophagy
pathways
The regulation of mitophagy by redox imbalance is a
dynamic, biphasic and intensity-dependent. A mild redox imbalance
can activate mitophagy. In this process, the activity of the
mitochondrial damage sensor PINK1 is directly regulated and
activates Parkin, which initiates the ubiquitin chain labeling of
outer membrane proteins, enabling them to be recognized by
autophagy receptor proteins. Elevated ROS levels due to
mitochondrial damage promote the oxidation and oligomerization of
NDP52, an autophagy receptor essential for the efficient clearance
of ubiquitin-labeled mitochondria. This enhances the initiation and
efficiency of mitophagy by facilitating the selective clearance of
damaged mitochondria (143).
Oxidative stress activates PGAM5, which promotes the
dephosphorylation of FUNDC1, enabling it to bind to LC3 and induce
mitophagy (124). However, redox
imbalance under sustained oxidative stress suppresses
mitophagy.
In the chronic and intense oxidative stress
environment of CKD, key mitophagy-associated proteins undergo
excessive oxidative modification, which can lead to their
inactivation. For example, PINK1 and Parkin expression is
suppressed, impairing ubiquitin chain formation and consequently
impeding mitophagy. This crosstalk between mitophagy and oxidative
stress establishes a reinforcing cycle of damage.
Evidence from animal experiments and patient samples
indicates that PINK1/Parkin pathway-mediated mitophagy is impaired
in kidney fibrosis (111).
Furthermore, decreased levels of mitophagy mediators, including
PINK1, Parkin, p62, LC3-II and BNIP3, in muscle-derived
mitochondria from patients with CKD, are associated with poor
oxidative capacity (144).
In contrast with the ubiquitin-dependent pathway,
receptor-mediated mitophagy is more directly regulated by oxidative
stress and is independent of Parkin. Under the hypoxic, nutrient
starvation and oxidative stress environment of CKD, the expression
levels of BNIP3 and NIX are significantly upregulated. However,
high expression of BNIP3 and NIX can lead to the formation of
homodimers, which disrupts the permeability of the OMM. This
exacerbates ROS leakage and directly triggers
mitochondrial-mediated apoptosis (73). In proteinuric mice, NIX-dependent
mitophagy is considerably suppressed and associated with increased
mitochondrial fragmentation, tubular epithelial cell apoptosis and
loss of kidney function (145).
While the activation of mitophagy within a tolerable range is
protective by the removal of damaged mitochondria, excessive
mitochondria injury triggers the overactivation of mitophagy and
subsequent cell death.
Notably, honokiol (HKL), a pharmacologically active
component of Magnolia officinalis, exhibits diverse
therapeutic properties. Wei et al (146) discovered that HKL suppresses
BNIP3, NIX and FUNDC1 expression and reduces AMPK activation levels
in CKD models, resulting in the reduction of excessive mitophagy
and induction of protective effects on the kidneys.
Crosstalk between mitophagy and oxidative
stress in CKD.
Considerable evidence supports the crucial role of
mitophagy in CKD progression, particularly in the removal of
impaired mitochondria, reduction of oxidative stress and
alleviation of inflammation and fibrosis in the kidney, all of
which delay CKD progression (147). However, under CKD conditions,
mitophagy is impaired and mitochondrial dynamics shift toward
excessive fission. Antioxidants increase mitophagy flux in animal
models of CKD, supporting the impairment of mitophagy in CKD
(148). In particular, five-sixth
nephrectomy (5/6Nx) model animals, which are commonly used to study
CKD pathophysiology, exhibit increased levels of p62. Since p62
accumulation is regarded as a marker of defective mitophagy, this
further implicates impaired mitophagy in the context of CKD
(149).
Although mitophagy initially acts as an adaptive,
protective mechanism, it becomes dysfunctional under sustained
oxidative stress. Conversely, impaired mitophagy leads to excessive
ROS production. This harmful feedback cycle is a pivotal
intermediary link between various initial pathologies, such as
diabetes, hypertension, aging and immune diseases, and the
progression of CKD (150).
Diabetic nephropathy (DN) is a major contributor to end-stage
kidney failure; however, current treatment strategies for DN are
limited (151). There is evidence
to suggest that mitophagy in tubular cells is defective in the
diabetic kidney. Han et al (152) found that PINK1, Parkin, LC3-II
and ATG5 expression levels are downregulated in the kidneys of
diabetic mice, suggesting impaired mitophagy. Supporting this,
patients with DN exhibit diminished circulating mtDNA levels,
increased mtDNA damage and deficient mitophagy (153). The renal expression of NOX5 is
upregulated in patients with DN, which induces ROS generation in
podocytes and contributes to DN progression (154). Experimental models suggest that
improving mitochondrial quality control can reduce
tubulointerstitial fibrosis and suppress epithelial-to-mesenchymal
transition (EMT)-like phenotype changes in tubular cells (155).
In hypertensive CKD, oxidative stress induces EMT
and renal fibrosis. A study revealed that AdipoRon, an adiponectin
receptor agonist, alleviated renal fibrosis in a mouse model of
hypertension by promoting mitophagy (156). Similarly, Long et al
(157) found that the kidneys of
aging rats exhibited signs of degenerative pathology, increased
oxidative stress and reduced mitophagy. Treatment with a
combination of Epimedium dried leaves and Ligustrum
lucidum fruits improved mitochondrial dynamics and restored
mitophagy via activation of the AMPK/ULK1 pathway. Furthermore, in
rats with passive Heymann nephritis, Fangji Huangqi (FJHQ)
decoction enhanced mitophagy in podocytes by upregulating the
expression of BNIP3, thereby ameliorating membranous nephropathy
(158). In systemic lupus
erythematosus, the mitophagy-inducing agent UMI-77 promoted
mitophagy, suppressed autoimmunity, and reduced renal inflammation
and ROS production, thereby preventing the progression of lupus
nephritis both in patients and model mice (159). However, excessive oxidative
stress can lead to excessive mitochondrial fragmentation, exceeding
the mitophagic clearance capacity and leading to the accumulation
of damaged mitochondria. One study showed that the activation of
Drp1-dependent mitochondrial fission promoted fibrogenesis, while
decreased mitochondrial fission attenuated fibrogenesis (160).
Regulated oxidative stress triggers signaling
pathways that promote mitochondrial fission and mitophagy to clear
damaged mitochondria and cells. This process prevents the spread of
damage to neighboring mitochondria and cells. Efficient mitophagy
enables the timely clearance of damaged mitochondria, characterized
by reduced MMP and increased electron leakage, which are primary
intracellular sources of ROS. By eliminating these mitochondria,
mitophagy directly reduces ROS production at its source (161). However, because mitophagy is
impaired in CKD, damaged mitochondria accumulate, leading to
excessive ROS generation. Elevated ROS levels further damage
healthy mitochondria and other organelles, activate inflammasomes,
and promote the expression of pro-fibrotic factors, thereby
exacerbating tubulointerstitial inflammation and fibrosis. This
oxidative damage promotes CKD progression and is associated with a
significant decline in renal function (162).
Li et al (110) demonstrated that mitophagy
protects against uncontrolled oxidative stress in a UUO model.
Whether triggered by acute or chronic injury, mitochondrial damage
results in a decline in mtDNA content, ROS overproduction and
reduced ATP generation, subsequently causing oxidative stress and
cell injury (163).
In summary, oxidative stress can inhibit mitophagy,
while impaired mitophagy exacerbates oxidative stress. This harmful
feedback loop drives CKD progression.
Potential therapeutic strategies
Modulators of mitophagy
Oxidative stress is exacerbated by impaired
mitophagy during CKD progression. Therefore, the simultaneous
targeting of both aspects of this feedback loop presents a novel
therapeutic strategy with potential for the treatment of CKD
(Table I).
 | Table I.Therapeutic agents targeting
mitophagy and oxidative stress. |
Table I.
Therapeutic agents targeting
mitophagy and oxidative stress.
| Compound | Experimental
model | Mechanism | Current status | (Refs.) |
|---|
| Quercetin | UUO model mice | Promotes the
expression of SIRT1/PINK1 to attenuate RTEC senescence and kidney
fibrosis | Preclinical | (102) |
| Melatonin | CLP-treated
mice | Enhances the
degradation of p62 via the inhibition of TFAM acetylation and
subsequent SIRT3 activation | Clinical use for
sleep disorders | (105) |
| Honokiol | Rats with
adenine-induced CKD | Suppresses
BNIP3/NIX, FUNDC1 and AMPK expression, reduces excessive mitophagy
and protects the kidneys | Preclinical | (146) |
| Metformin | High-fat and
streptozotocin-induced diabetic mice | Activates AMPK and
promotes mitophagy through the p-AMPK-PINK1-Parkin pathway | Clinical use for
type 2 diabetes | (152) |
| Epimedii
folium and Ligustri lucidi fructus | Aged rats | Delay renal aging
by activating the AMPK/ULK1 pathway | Preclinical
stage | (157) |
| Fangji
huangqi decoction | Passive Heymann
nephritis model rats | Enhances mitophagy
in podocytes by promoting the expression of BNIP3 | Clinical use for
edema and dysuria | (158) |
| UMI-77 | Lupus nephritis
model mice, and clinical samples | Enhances mitophagy,
suppresses autoimmunity and limits renal inflammation | Preclinical | (159) |
| Magnoflorine | High-fat and
high-fructosefed mice | Promotes
Parkin/PINK1-mediated mitophagy and inhibits
NLRP3/caspase-1-mediated pyroptosis | Preclinical | (164) |
| Ruxolitinib | UUO model mice | Activates
Parkin/PINK1 mitophagy, reduces inflammatory responses and
oxidative stress | Clinical use for
myeloproliferative disorders | (165) |
| Chicoric acid | High-fat diet-fed
mice | Activates the Nrf2
pathway and increases PINK1 and Parkin expression, thereby
enhancing mitophagy | Preclinical | (166) |
| Paeoniflorin | RAW264.7 cells
stimulated with lipopolysaccharide | Upregulates PINK1,
Parkin, BNIP3, p62 and LC3, increases mitochondrial membrane
potential and reduces ROS accumulation | Preclinical | (167) |
| Tongluo
yishen decoction | UUO model rats | Modulates
PINK1/Parkin-mediated mitophagy and alleviates renal fibrosis | Clinical use for
CKD | (168) |
| Rapamycin,
everolimus and temsirolimus | UUO model mice | Suppresses mTORC1
and promotes AMPK-induced ULK1 activation to induce mitophagy | Preclinical | (171) |
| Resveratrol | 5/6-nephrectomy and
CLP-treated rats | Activates SIRT1/3
and reduces acetylated SOD2 levels to ameliorate oxidative stress
and mitochondrial function in RTECs | Preclinical | (104, 173) |
| Roxadustat | Rats with renal I/R
injury | Increases HIF1α
protein expression and promotes HIF1α/FUNDC1-mediated
mitophagy | Clinical use for
renal anemia in CKD and dialysis | (174) |
| Uridine | Aged mice | Reduces
intracellular oxidative stress, promotes the synthesis of
high-energy compounds and protects cells from hypoxic damage | Preclinical | (179) |
| Lycopene | Mice with
aristolochic acidinduced nephropathy | Activates the
antioxidant system and mitophagy, upregulates levels of LC3-II and
p62, exerts anti-inflammatory and anti-apoptotic effects | Preclinical | (180) |
| Mitoquinone | Mice with I/R
injury, or infused with AngII | Decreases oxidative
damage and reduces the severity of I/R injury to the kidney | Preclinical | (184,185) |
| SS-31 | Uninephrectomy and
streptozotocin-treated mice; mice with cisplatin-induced AKI | Anti-oxidative and
anti-apoptotic action via the inhibition of ROS production | Preclinical | (187,188) |
| Curcumin-loaded
nanodrug delivery system | Cisplatin-induced
AKI | Inhibits oxidation,
activates autophagy and reduces apoptosis | Preclinical | (190) |
Several pharmacological agents have been identified
that modulate mitophagy and thereby attenuate oxidative
stress-induced renal injury. For example, magnoflorine promotes
PINK1/Parkin-mediated mitophagy, thereby inhibiting
NLRP3/caspase-1-mediated pyroptosis. This effects is reversed by
either the genetic deletion of Parkin or use of a chemical
mitophagy inhibitor, confirming that the ability of magnoflorine to
inhibit NLRP3 inflammasome activation depends on mitophagy in
cultured cells (164).
Ruxolitinib also activates PINK1/Parkin-mediated mitophagy, thereby
reducing oxidative stress, inflammation and renal fibrosis
(165). Chicoric acid, a key
bioactive component of chicory, stimulates Nrf2 signaling and
upregulates PINK1 and Parkin expression levels in high-fat diet
(HFD)-fed mice (166). Similarly,
Cao et al (167) revealed
that paeoniflorin (PF) increases the expression levels of PINK1,
Parkin, BNIP3, p62 and LC3, which repairs damaged mitochondria by
restoring the MMP and clearing accumulated ROS. In addition, PF
ameliorates kidney inflammation by inducing the transition of
macrophages to the M2 phenotype and activating mitophagy in
LPS-induced murine RAW264.7 CKD models
Traditional Chinese medicine (TCM) has also been
shown to regulate mitophagy. In addition to the previously
mentioned FJHQ decoction, which exhibited efficacy in the treatment
of membranous nephropathy, Tongluo Yishen decoction was found to
improve mitochondrial dynamics and alleviate renal fibrosis by
modulating PINK1/Parkin-mediated mitophagy and reducing ROS levels
in UUO model rats (168). Unlike
most mitophagy regulators, whose efficacy has been demonstrated
solely in animal and in vitro models, TCMs are the only
mitophagy modulators supported by clinical data.
The inhibition of mTORC1 or activation of the
AMPK/SIRT1 by genetic or pharmacological approaches has shown
protective effects against the progression of DN in animal models
(169,170). Well-known mTORC1 inhibitors
include rapamycin and its analogs, everolimus and temsirolimus
(171). Han et al
(152) demonstrated that the AMPK
activator metformin promotes mitophagy through the
phospho-AMPK-PINK1-Parkin pathway, while concurrently attenuating
oxidative stress and tubulointerstitial fibrosis in the kidneys of
HFD/streptozotocin-induced diabetic mice. Transcription factor A,
mitochondrial (TFAM) is essential for the initiation of mtDNA
transcription (172). Melatonin
has been identified as a positive regulator of PINK1 and Parkin
expression, which activates upstream mitophagy. Deng et al
(105) demonstrated that
melatonin promotes p62 degradation by suppressing TFAM acetylation
through SIRT3 activation, thereby enhancing mitophagy flux.
Quercetin, an activator of SIRT1, attenuates RTEC senescence and
kidney fibrosis by upregulating the expression of SIRT1 and PINK1
to promote mitophagy in vivo, as shown in an UUO model, and
also enhances mitophagy in AngII-treated RTECs (102). Similarly, resveratrol activates
both SIRT1 and SIRT3 to promote mitophagy (104). In another study, the
administration of resveratrol to rats following 5/6Nx improved
kidney function, as indicated by decreased proteinuria and lower
cystatin C levels (173).
The HIF1α-FUNDC1 mitophagy axis plays a key role in
hypoxia/reoxygenation (H/R)-promoted mitophagy in renal tubular
cells. H/R treatment upregulates the expression of FUNDC1, while
the selective inhibition of HIF1α reduces H/R-induced mitophagy
(174). Notably, treatment with
roxadustat further increases the expression of HIF1α in
vivo. Zhang et al (174) suggested that Roxadustat may serve
as a potential therapeutic agent to block the transition of AKI to
CKD in future clinical applications. Currently, roxadustat is used
clinically to treat renal anemia in patients with CKD and those on
dialysis, and shows considerable promise as a novel
mitophagy-targeting therapeutic agent for CKD.
Antioxidants
Antioxidants prevent oxidative stress by
neutralizing oxidizing agents and blocking harmful oxidation by
converting free radicals into nonreactive compounds (175). Uridine, a pyrimidine nucleoside
composed of uracil and ribose, is important in the maintenance of
cellular function and energy metabolism. It promotes the synthesis
of high-energy compounds and reduces intracellular oxidative
stress, thereby preventing hypoxia-induced cellular damage
(176,177). In animal models of acute ischemia
and ischemia/reperfusion (I/R), uridine administration has been
shown to protect against injury by restoring redox balance and
activating mitochondrial ATP-dependent channels (178). The levels of inflammatory factors
and ROS also decrease significantly following uridine treatment
(179).
Lycopene (LYC), a potent carotenoid antioxidant,
exhibits multiple therapeutic effects, including anti-inflammatory,
anti-apoptotic and mitophagy-modulating properties. Wang et
al (180) demonstrated that
LYC activates the Nrf2 antioxidant system and induces mitophagy to
ameliorate renal fibrosis. The study showed that LYC attenuated the
aristolochic acid I-induced intracellular expression of PINK1,
Parkin and TGF-β; upregulated LC3-II and p62 levels; decreased MMP;
and mitigated renal fibrosis in mice by suppressing mTORC1 and
promoting ULK1 activation.
Mitochondria-targeted
antioxidants
Growing evidence suggests that
mitochondria-targeted antioxidants have substantial therapeutic
potential. Mitoquinone (MitoQ), a mitochondria-targeted
antioxidant, shows therapeutic potential against the oxidative
stress mediated by excessive ROS production (142). MitoQ comprises a ubiquinone
moiety conjugated to a lipophilic triphenyl phosphonium (TPP)
cation, which targets mitochondria. The MMP facilitates efficient
cellular internalization of the TPP cation, leading to its
accumulation in the mitochondrial matrix. Once inside,
mitochondrial complex II reduces ubiquinone to ubiquinol, its
bioactive antioxidant form, thereby counteracting oxidative damage
(181,182). Due to its combination of
extensive mitochondrial uptake, antioxidant recycling and
localization on the matrix-facing surface of the IMM, MitoQ is
substantially more effective than untargeted antioxidants in
preventing oxidative damage (183). Other studies have also confirmed
the ability of MitoQ to prevent MMP reduction and reduce excessive
ROS production (184,185).
SS-31, another mitochondria-targeted antioxidant,
contains dimethyl tyrosine residues that react with oxygen
radicals, forming inactive tyrosine radical species and scavenging
ROS (186). Following its
accumulation on the IMM, SS-31 preserves mitochondrial structure,
promotes ATP production, and inhibits ROS generation and apoptosis
(187). In murine models, SS-31
demonstrates nephroprotective effects against cisplatin-induced
renal damage. It exerts both antioxidant and antiapoptotic
activities by modulating the mROS-NLRP3 signaling axis (188). However, preclinical studies
regarding the effectiveness of SS-31 in the treatment of renal
diseases are limited. Large-scale, multicenter clinical studies
with a diverse range of patients are necessary to assess the
efficacy, safety and tolerability of SS-31 for clinical application
(189).
Nanotechnology-based delivery systems can be used
to encapsulate antioxidants, with surface modifications to enable
their specific uptake by mitochondria. For example, a
curcumin-loaded nanodrug delivery system demonstrated protective
effects on mitochondrial function and kidney tissue by scavenging
excess ROS, reducing apoptosis and activating autophagy (190).
Despite these findings, the therapeutic
applicability of mitochondria-targeted antioxidants in CKD warrants
further investigation. The therapeutic efficacy of these
antioxidants should be evaluated in multiple animal models with
diverse CKD etiologies. Prior to human clinical trials, these
therapies should be tested in models that most closely resemble
human disease to validate their safety and efficacy, providing
robust evidence to support their clinical translation.
Challenges and prospects
CKD is a global health issue with a complex
pathogenesis involving multiple cellular and molecular processes.
Attention has been focused on the roles of mitophagy and oxidative
stress in CKD. A considerable body of evidence emphasizes the
importance of mitophagy and oxidative stress in kidney function in
healthy and disease states. Therefore, targeting mitophagy and
oxidative stress has emerged as a promising approach to improve
renal function and delay CKD progression.
Although targeting mitophagy and oxidative stress
are promising approaches for the treatment CKD, limitations remain,
and multiple challenges must be overcome before these findings can
be translated into clinically effective therapies. Most current
evidence is derived from animal models such as those involving UUO,
I/R injury or aged mice. While these models replicate certain
pathological features of CKD, they cannot fully reproduce the
multifactorial course of human CKD and its long-term progression,
which may involve the complex interplay of hypertension, diabetes
and aging. Differences between animals and humans in drug
metabolism, immune response and renal cell biology may cause
strategies that are effective in animals to be ineffective or even
toxic in humans. In addition, mitophagy and oxidative stress form a
dynamic regulatory cycle. However, clinical methods that can
dynamically and quantitatively monitor mitophagy flux in the kidney
are currently lacking. Therefore, it is not yet possible to
determine the optimal regulatory window for intervention in
mitophagy. The excessive activation of mitophagy may lead to the
unnecessary removal of healthy mitochondria, triggering cellular
energy depletion, while insufficient inhibition of mitophagy fails
to eliminate dysfunctional mitochondria, allowing dysfunction to
persist.
To overcome these challenges, future research
should focus on several aspects. First, to screen drugs and
validate the underlying mechanisms, disease models that more
closely resemble human physiology should be established. Kidney
organoids generated from patient-derived induced pluripotent stem
cells are a promising platform to simulate human-specific CKD
pathological processes in vitro. Second, novel biomarkers
that reflect in vivo mitophagy status and oxidative stress
levels, such as mtDNA or specific mitochondrial proteins detected
in blood or urine exosomes, should be identified and validated. The
development of kidney-targeted nanodelivery systems is also
critical, with the aim of enhancing drug accumulation in the renal
tissue while reducing systemic exposure and off-target effects.
Another area worthy of exploration is the integration of TCM with
modern nanotechnology to design delivery carriers that, according
to TCM theory, have ‘kidney meridian affinity’. Finally, greater
efforts should be made to discover highly specific mitophagy
modulators that precisely regulate specific pathways, providing
more controlled effects than those of broad-spectrum autophagy
inducing agents or antioxidants.
Acknowledgements
Not applicable.
Funding
This study received support from the following sources: Sichuan
Science and Technology Program (grant no. 2022YFS0621), Southwest
Medical University Technology Program (grant no. 2023QN019),
National Natural Science Foundation of China (grant no. 82205002),
Science and Technology Research Special Project of Sichuan
Administration of Traditional Chinese Medicine (grant no.
2024MS524) and Special Project of Integrated Chinese and Western
Medicine, Southwest Medical University (grant no. 2023ZYQJ04).
Availability of data and materials
Not applicable.
Authors' contributions
QM was involved in writing, reviewing and editing
the manuscript, and creating visualizations. YT drafted the
original manuscript and worked on visualization. JL contributed to
writing, reviewing and editing, visualization and supervision. YQ
and HF contributed to writing the original draft of the manuscript.
QH and QZ helped to write, review and edit the manuscript, and also
provided supervision and secured funding. Data authentication is
not applicable. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
CKD
|
chronic kidney disease
|
|
ROS
|
reactive oxygen species
|
|
mROS
|
mitochondrial ROS
|
|
ATP
|
adenosine triphosphate
|
|
mtDNA
|
mitochondrial DNA
|
|
NLRP3
|
NOD-like receptor protein 3
|
|
Drp1
|
dynamin-related protein l
|
|
MFN1/2
|
mitofusin 1/2
|
|
MMP
|
mitochondrial membrane potential
|
|
OMM
|
outer mitochondrial membrane
|
|
IMM
|
inner mitochondrial membrane
|
|
IMS
|
intermembrane space
|
|
HIF1α
|
hypoxia-inducible factor 1α
|
|
ULK1
|
Unc-51 like autophagy activating
kinase l
|
|
PINK1
|
PTEN-induced kinase l
|
|
p62
|
sequestosome 1
|
|
NBR1
|
neighbor of BRCA1 gene l
|
|
OPTN
|
optineurin
|
|
NDP52
|
nuclear dot protein 52
|
|
LC3
|
microtubule-associated protein 1
light chain 3
|
|
TBK1
|
TANK-binding kinase 1
|
|
BNIP3
|
BCL2 interacting protein 3
|
|
NIX
|
Nip3-like protein X
|
|
FUNDC1
|
FUN14 domain-containing protein l
|
|
mTOR
|
serine/threonine protein kinase
mammalian target of rapamycin
|
|
AMPK
|
AMP-activated protein kinase
|
|
Keap1
|
Kelch-like ECH-associated protein
1
|
|
Nrf2
|
NF-E2-related factor 2
|
|
SOD
|
superoxide dismutase
|
|
GPX
|
glutathione peroxidase
|
|
AKI
|
acute kidney injury
|
|
DN
|
diabetic nephropathy
|
|
RTECs
|
renal tubular epithelial cells
|
|
UUO
|
unilateral ureteral obstruction
|
|
EMT
|
epithelial-to-mesenchymal
transition
|
References
|
1
|
Kovesdy CP: Epidemiology of chronic kidney
disease: An update 2022. Kidney Int Suppl (2011). 12:7–11. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Flythe JE and Watnick S: Dialysis for
chronic kidney failure: A review. JAMA. 332:1559–1573. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yang W, Wu X, Zhao M, Hu J, Lin C, Mei Z,
Chen J, Zhou XJ, Nie S, Nie J, et al: Kidney function and
cardiovascular disease: Evidence from observational studies and
mendelian randomization analyses. Phenomics. 4:250–253. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Jha V, Al-Ghamdi SMG, Li G, Wu MS,
Stafylas P, Retat L, Card-Gowers J, Barone S, Cabrera C and Garcia
Sanchez JJ: Global economic burden associated with chronic kidney
disease: A pragmatic review of medical costs for the inside CKD
research programme. Adv Ther. 40:4405–4420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dan Hu Q, Wang HL, Liu J, He T, Tan RZ,
Zhang Q, Su HW, Kantawong F, Lan HY and Wang L: Btg2 promotes focal
segmental glomerulosclerosis via Smad3-dependent
podocyte-mesenchymal transition. Adv Sci (Weinh). 10:e23043602023.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Panizo S, Martínez-Arias L, Alonso-Montes
C, Cannata P, Martín-Carro B, Fernández-Martín JL, Naves-Díaz M,
Carrillo-López N and Cannata-Andía JB: Fibrosis in chronic kidney
disease: Pathogenesis and consequences. Int J Mol Sci. 22:4082021.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ho HJ and Shirakawa H: Oxidative stress
and mitochondrial dysfunction in chronic kidney disease. Cells.
12:882022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kushner P, Khunti K, Cebrián A and Deed G:
Early identification and management of chronic kidney disease: A
narrative review of the crucial role of primary care practitioners.
Adv Ther. 41:3757–3770. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu HZ, Li CY, Liu LJ, Tong JB, Lan ZH,
Tian SG, Li Q, Tong XL, Wu JF, Zhu ZG, et al: Efficacy and safety
of qingfei huatan formula in the treatment of acute exacerbation of
chronic obstructive pulmonary disease: A multi-centre, randomised,
double-blind, placebo-controlled trial. J Integr Med. 22:561–569.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aranda-Rivera AK, Cruz-Gregorio A,
Aparicio-Trejo OE and Pedraza-Chaverri J: Mitochondrial redox
signaling and oxidative stress in kidney diseases. Biomolecules.
11:11442021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Doke T and Susztak K: The multifaceted
role of kidney tubule mitochondrial dysfunction in kidney disease
development. Trends Cell Biol. 32:841–853. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lu Y, Li Z, Zhang S, Zhang T, Liu Y and
Zhang L: Cellular mitophagy: Mechanism, roles in diseases and small
molecule pharmacological regulation. Theranostics. 13:736–766.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hoogstraten CA, Hoenderop JG and de Baaij
JHF: Mitochondrial dysfunction in kidney tubulopathies. Annu Rev
Physiol. 86:379–403. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Che R, Yuan Y, Huang S and Zhang A:
Mitochondrial dysfunction in the pathophysiology of renal diseases.
Am J Physiol Renal Physiol. 306:F367–F378. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhan M, Brooks C, Liu F, Sun L and Dong Z:
Mitochondrial dynamics: Regulatory mechanisms and emerging role in
renal pathophysiology. Kidney Int. 83:568–581. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Palorini R, De Rasmo D, Gaviraghi M, Sala
Danna L, Signorile A, Cirulli C, Chiaradonna F, Alberghina L and
Papa S: Oncogenic K-ras expression is associated with derangement
of the cAMP/PKA pathway and forskolin-reversible alterations of
mitochondrial dynamics and respiration. Oncogene. 32:352–362. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mills EL, Kelly B and O'Neill LAJ:
Mitochondria are the powerhouses of immunity. Nat Immunol.
18:488–498. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Shimada K, Crother TR, Karlin J, Dagvadorj
J, Chiba N, Chen S, Ramanujan VK, Wolf AJ, Vergnes L, Ojcius DM, et
al: Oxidized mitochondrial DNA activates the NLRP3 inflammasome
during apoptosis. Immunity. 36:401–414. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miwa S, Kashyap S, Chini E and von
Zglinicki T: Mitochondrial dysfunction in cell senescence and
aging. J Clin Invest. 132:e1584472022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen K, Dai H, Yuan J, Chen J, Lin L,
Zhang W, Wang L, Zhang J, Li K and He Y: Optineurin-mediated
mitophagy protects renal tubular epithelial cells against
accelerated senescence in diabetic nephropathy. Cell Death Dis.
9:1052018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bhargava P and Schnellmann RG:
Mitochondrial energetics in the kidney. Nat Rev Nephrol.
13:629–646. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Holmström KM and Finkel T: Cellular
mechanisms and physiological consequences of redox-dependent
signalling. Nat Rev Mol Cell Biol. 15:411–421. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yuan Q, Tang B and Zhang C: Signaling
pathways of chronic kidney diseases, implications for therapeutics.
Signal Transduct Target Ther. 7:1822022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Podkowińska A and Formanowicz D: Chronic
kidney disease as oxidative stress- and inflammatory-mediated
cardiovascular disease. Antioxidants (Basel). 9:7522020. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu X, Hussain R, Mehmood K, Tang Z, Zhang
H and Li Y: Mitochondrial-endoplasmic reticulum
communication-mediated oxidative stress and autophagy. Biomed Res
Int. 2022:64595852022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Quintana-Cabrera R and Scorrano L:
Determinants and outcomes of mitochondrial dynamics. Mol Cell.
83:857–876. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kühlbrandt W: Structure and function of
mitochondrial membrane protein complexes. BMC Biol. 13:892015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen W, Zhao H and Li Y: Mitochondrial
dynamics in health and disease: Mechanisms and potential targets.
Signal Transduct Target Ther. 8:3332023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Dorn GW and Kitsis RN: The mitochondrial
dynamism-mitophagy-cell death interactome: Multiple roles performed
by members of a mitochondrial molecular ensemble. Circ Res.
116:167–182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dumesic PA, Wilensky SE, Bose S, Van
Vranken JG, Gygi SP and Spiegelman BM: RBM43 controls PGC1α
translation and a PGC1α-STING signaling axis. Cell Metab.
37:742–757 .e8. 2025.
|
|
32
|
Pernas L and Scorrano L: Mito-morphosis:
Mitochondrial fusion, fission, and cristae remodeling as key
mediators of cellular function. Annu Rev Physiol. 78:505–531. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dorn GW: Evolving concepts of
mitochondrial dynamics. Annu Rev Physiol. 81:1–17. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Garza-Lombó C, Pappa A, Panayiotidis MI
and Franco R: Redox homeostasis, oxidative stress and mitophagy.
Mitochondrion. 51:105–117. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Klionsky DJ, Petroni G, Amaravadi RK,
Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K,
Cecconi F, Choi AMK, et al: Autophagy in major human diseases. EMBO
J. 40:e1088632021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduct Target Ther.
8:3042023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Narendra D, Tanaka A, Suen DF and Youle
RJ: Parkin is recruited selectively to impaired mitochondria and
promotes their autophagy. J Cell Biol. 183:795–803. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han R, Liu Y, Li S, Li XJ and Yang W:
PINK1-PRKN mediated mitophagy: Differences between in vitro and in
vivo models. Autophagy. 19:1396–1405. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Harper JW, Ordureau A and Heo JM: Building
and decoding ubiquitin chains for mitophagy. Nat Rev Mol Cell Biol.
19:93–108. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lazarou M, Sliter DA, Kane LA, Sarraf SA,
Wang C, Burman JL, Sideris DP, Fogel AI and Youle RJ: The ubiquitin
kinase PINK1 recruits autophagy receptors to induce mitophagy.
Nature. 524:309–314. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Narendra DP, Jin SM, Tanaka A, Suen DF,
Gautier CA, Shen J, Cookson MR and Youle RJ: PINK1 is selectively
stabilized on impaired mitochondria to activate parkin. PLoS Biol.
8:e10002982010. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wiedemann N and Pfanner N: Mitochondrial
machineries for protein import and assembly. Annu Rev Biochem.
86:685–714. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rasool S, Veyron S, Soya N, Eldeeb MA,
Lukacs GL, Fon EA and Trempe JF: Mechanism of PINK1 activation by
autophosphorylation and insights into assembly on the TOM complex.
Mol Cell. 82:44–59.e6. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Lysyk L, Brassard R, Touret N and Lemieux
MJ: PARL protease: A glimpse at intramembrane proteolysis in the
inner mitochondrial membrane. J Mol Biol. 432:5052–5062. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yamano K and Youle RJ: PINK1 is degraded
through the N-end rule pathway. Autophagy. 9:1758–1769. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pickrell AM and Youle RJ: The roles of
PINK1, parkin, and mitochondrial fidelity in Parkinson's disease.
Neuron. 85:257–273. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang R, Zhu Y, Ren C, Yang S, Tian S, Chen
H, Jin M and Zhou H: Influenza A virus protein PB1-F2 impairs
innate immunity by inducing mitophagy. Autophagy. 17:496–511. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
McLelland GL, Goiran T, Yi W, Dorval G,
Chen CX, Lauinger ND, Krahn AI, Valimehr S, Rakovic A, Rouiller I,
et al: Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent
release of ER from mitochondria to drive mitophagy. Elife.
7:e328662018. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Gao XY, Yang T, Gu Y and Sun XH:
Mitochondrial dysfunction in parkinson's disease: From mechanistic
insights to therapy. Front Aging Neurosci. 14:8855002022.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Yamano K, Matsuda N and Tanaka K: The
ubiquitin signal and autophagy: An orchestrated dance leading to
mitochondrial degradation. EMBO Rep. 17:300–316. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zheng X and Hunter T: Parkin mitochondrial
translocation is achieved through a novel catalytic activity
coupled mechanism. Cell Res. 23:886–897. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Malpartida AB, Williamson M, Narendra DP,
Wade-Martins R and Ryan BJ: Mitochondrial dysfunction and mitophagy
in Parkinson's disease: From mechanism to therapy. Trends Biochem
Sci. 46:329–343. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Matsuda N: Phospho-ubiquitin: Upending the
PINK-parkin-ubiquitin cascade. J Biochem. 159:379–385. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Johansen T and Lamark T: Selective
autophagy: ATG8 family proteins, LIR motifs and cargo receptors. J
Mol Biol. 432:80–103. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Deretic V and Lazarou M: A guide to
membrane atg8ylation and autophagy with reflections on immunity. J
Cell Biol. 221:e2022030832022. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Birgisdottir ÅB, Lamark T and Johansen T:
The LIR motif-crucial for selective autophagy. J Cell Sci.
126:3237–3247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stolz A, Ernst A and Dikic I: Cargo
recognition and trafficking in selective autophagy. Nat Cell Biol.
16:495–501. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Sun D, Wu R, Zheng J, Li P and Yu L:
Polyubiquitin chain-induced p62 phase separation drives autophagic
cargo segregation. Cell Res. 28:405–415. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Geisler S, Holmström KM, Skujat D, Fiesel
FC, Rothfuss OC, Kahle PJ and Springer W: PINK1/parkin-mediated
mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol.
12:119–131. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Deosaran E, Larsen KB, Hua R, Sargent G,
Wang Y, Kim S, Lamark T, Jauregui M, Law K, Lippincott-Schwartz J,
et al: NBR1 acts as an autophagy receptor for peroxisomes. J Cell
Sci. 126:939–952. 2013.PubMed/NCBI
|
|
61
|
Taguchi K, Fujikawa N, Komatsu M, Ishii T,
Unno M, Akaike T, Motohashi H and Yamamoto M: Keap1 degradation by
autophagy for the maintenance of redox homeostasis. Proc Natl Acad
Sci USA. 109:13561–13566. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Komatsu M, Kurokawa H, Waguri S, Taguchi
K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, et
al: The selective autophagy substrate p62 activates the stress
responsive transcription factor Nrf2 through inactivation of Keap1.
Nat Cell Biol. 12:213–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Sánchez-Martín P, Sou YS, Kageyama S,
Koike M, Waguri S and Komatsu M: NBR1-mediated p62-liquid droplets
enhance the Keap1-Nrf2 system. EMBO Rep. 21:e489022020. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Qiu Y, Wang J, Li H, Yang B, Wang J, He Q
and Weng Q: Emerging views of OPTN (optineurin) function in the
autophagic process associated with disease. Autophagy. 18:73–85.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Matsumoto G, Wada K, Okuno M, Kurosawa M
and Nukina N: Serine 403 phosphorylation of p62/SQSTM1 regulates
selective autophagic clearance of ubiquitinated proteins. Mol Cell.
44:279–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Heo JM, Ordureau A, Paulo JA, Rinehart J
and Harper JW: The PINK1-PARKIN mitochondrial ubiquitylation
pathway drives a program of OPTN/NDP52 recruitment and TBK1
activation to promote mitophagy. Mol Cell. 60:7–20. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yamano K and Kojima W: Molecular functions
of autophagy adaptors upon ubiquitin-driven mitophagy. Biochim
Biophys Acta Gen Subj. 1865:1299722021. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Vargas JNS, Wang C, Bunker E, Hao L, Maric
D, Schiavo G, Randow F and Youle RJ: Spatiotemporal control of ULK1
activation by NDP52 and TBK1 during selective autophagy. Mol Cell.
74:347–362.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
von Muhlinen N, Akutsu M, Ravenhill BJ,
Foeglein Á, Bloor S, Rutherford TJ, Freund SM, Komander D and
Randow F: LC3C, bound selectively by a noncanonical LIR motif in
NDP52, is required for antibacterial autophagy. Mol Cell.
48:329–342. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Turco E, Savova A, Gere F, Ferrari L,
Romanov J, Schuschnig M and Martens S: Reconstitution defines the
roles of p62, NBR1 and TAX1BP1 in ubiquitin condensate formation
and autophagy initiation. Nat Commun. 12:52122021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Ohnstad AE, Delgado JM, North BJ, Nasa I,
Kettenbach AN, Schultz SW and Shoemaker CJ: Receptor-mediated
clustering of FIP200 bypasses the role of LC3 lipidation in
autophagy. EMBO J. 39:e1049482020. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Uoselis L, Nguyen TN and Lazarou M:
Mitochondrial degradation: Mitophagy and beyond. Mol Cell.
83:3404–3420. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Field JT and Gordon JW: BNIP3 and nix:
Atypical regulators of cell fate. Biochim Biophys Acta Mol Cell
Res. 1869:1193252022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Chen G, Ray R, Dubik D, Shi L, Cizeau J,
Bleackley RC, Saxena S, Gietz RD and Greenberg AH: The E1B
19K/Bcl-2-binding protein Nip3 is a dimeric mitochondrial protein
that activates apoptosis. J Exp Med. 186:1975–1983. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Bruick RK: Expression of the gene encoding
the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad
Sci USA. 97:9082–9087. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Twig G, Elorza A, Molina AJ, Mohamed H,
Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al:
Fission and selective fusion govern mitochondrial segregation and
elimination by autophagy. EMBO J. 27:433–446. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Zhang T, Xue L, Li L, Tang C, Wan Z, Wang
R, Tan J, Tan Y, Han H, Tian R, et al: BNIP3 protein suppresses
PINK1 kinase proteolytic cleavage to promote mitophagy. J Biol
Chem. 291:21616–21629. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Lee Y, Lee HY, Hanna RA and Gustafsson ÅB:
Mitochondrial autophagy by Bnip3 involves Drp1-mediated
mitochondrial fission and recruitment of parkin in cardiac
myocytes. Am J Physiol Heart Circ Physiol. 301:H1924–H1931. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Marinković M, Šprung M and Novak I:
Dimerization of mitophagy receptor BNIP3L/NIX is essential for
recruitment of autophagic machinery. Autophagy. 17:1232–1243. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sandoval H, Thiagarajan P, Dasgupta SK,
Schumacher A, Prchal JT, Chen M and Wang J: Essential role for nix
in autophagic maturation of erythroid cells. Nature. 454:232–235.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xiang G, Yang L, Long Q, Chen K, Tang H,
Wu Y, Liu Z, Zhou Y, Qi J, Zheng L, et al: BNIP3L-dependent
mitophagy accounts for mitochondrial clearance during 3
factors-induced somatic cell reprogramming. Autophagy.
13:1543–1555. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Esteban-Martínez L and Boya P:
BNIP3L/NIX-dependent mitophagy regulates cell differentiation via
metabolic reprogramming. Autophagy. 14:915–917. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Melser S, Chatelain EH, Lavie J, Mahfouf
W, Jose C, Obre E, Goorden S, Priault M, Elgersma Y, Rezvani HR, et
al: Rheb regulates mitophagy induced by mitochondrial energetic
status. Cell Metab. 17:719–730. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Gao F, Chen D, Si J, Hu Q, Qin Z, Fang M
and Wang G: The mitochondrial protein BNIP3L is the substrate of
PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum Mol Genet.
24:2528–2538. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Cao Y, Zheng J, Wan H, Sun Y, Fu S, Liu S,
He B, Cai G, Cao Y, Huang H, et al: A mitochondrial SCF-FBXL4
ubiquitin E3 ligase complex degrades BNIP3 and NIX to restrain
mitophagy and prevent mitochondrial disease. EMBO J.
42:e1130332023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Zhu Y, Massen S, Terenzio M, Lang V,
Chen-Lindner S, Eils R, Novak I, Dikic I, Hamacher-Brady A and
Brady NR: Modulation of serines 17 and 24 in the LC3-interacting
region of Bnip3 determines pro-survival mitophagy versus apoptosis.
J Biol Chem. 288:1099–1113. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Rogov VV, Suzuki H, Marinković M, Lang V,
Kato R, Kawasaki M, Buljubašić M, Šprung M, Rogova N, Wakatsuki S,
et al: Phosphorylation of the mitochondrial autophagy receptor nix
enhances its interaction with LC3 proteins. Sci Rep. 7:11312017.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Clague MJ and Urbé S: Diverse routes to
mitophagy governed by ubiquitylation and mitochondrial import.
Trends Cell Biol. 35:527–538. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Zhang W: The mitophagy receptor FUN14
domain-containing 1 (FUNDC1): A promising biomarker and potential
therapeutic target of human diseases. Genes Dis. 8:640–654. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Liu L, Feng D, Chen G, Chen M, Zheng Q,
Song P, Ma Q, Zhu C, Wang R, Qi W, et al: Mitochondrial
outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in
mammalian cells. Nat Cell Biol. 14:177–185. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kuang Y, Ma K, Zhou C, Ding P, Zhu Y, Chen
Q and Xia B: Structural basis for the phosphorylation of FUNDC1 LIR
as a molecular switch of mitophagy. Autophagy. 12:2363–2373. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zhou H, Zhu P, Wang J, Zhu H, Ren J and
Chen Y: Pathogenesis of cardiac ischemia reperfusion injury is
associated with CK2α-disturbed mitochondrial homeostasis via
suppression of FUNDC1-related mitophagy. Cell Death Differ.
25:1080–1093. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Chen G, Han Z, Feng D, Chen Y, Chen L, Wu
H, Huang L, Zhou C, Cai X, Fu C, et al: A regulatory signaling loop
comprising the PGAM5 phosphatase and CK2 controls receptor-mediated
mitophagy. Mol Cell. 54:362–377. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Wu W, Tian W, Hu Z, Chen G, Huang L, Li W,
Zhang X, Xue P, Zhou C, Liu L, et al: ULK1 translocates to
mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO
Rep. 15:566–575. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tang C, Livingston MJ, Liu Z and Dong Z:
Autophagy in kidney homeostasis and disease. Nat Rev Nephrol.
16:489–508. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Hosokawa N, Hara T, Kaizuka T, Kishi C,
Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N, et
al: Nutrient-dependent mTORC1 association with the
ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell.
20:1981–1991. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Herzig S and Shaw RJ: AMPK: Guardian of
metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol.
19:121–135. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Perico L, Remuzzi G and Benigni A:
Sirtuins in kidney health and disease. Nat Rev Nephrol. 20:313–329.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu S, Liu Y, Li J, Wang M, Chen X, Gan F,
Wen L, Huang K and Liu D: Arsenic exposure-induced acute kidney
injury by regulating SIRT1/PINK1/mitophagy axis in mice and in HK-2
cells. J Agric Food Chem. 71:15809–15820. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Liu T, Yang Q, Zhang X, Qin R, Shan W,
Zhang H and Chen X: Quercetin alleviates kidney fibrosis by
reducing renal tubular epithelial cell senescence through the
SIRT1/PINK1/mitophagy axis. Life Sci. 257:1181162020. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jin L, Galonek H, Israelian K, Choy W,
Morrison M, Xia Y, Wang X, Xu Y, Yang Y, Smith JJ, et al:
Biochemical characterization, localization, and tissue distribution
of the longer form of mouse SIRT3. Protein Sci. 18:514–525. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Xu S, Gao Y, Zhang Q, Wei S, Chen Z, Dai
X, Zeng Z and Zhao KS: SIRT1/3 activation by resveratrol attenuates
acute kidney injury in a septic rat model. Oxid Med Cell Longev.
2016:72960922016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Deng Z, He M, Hu H, Zhang W, Zhang Y, Ge
Y, Ma T, Wu J, Li L, Sun M, et al: Melatonin attenuates
sepsis-induced acute kidney injury by promoting mitophagy through
SIRT3-mediated TFAM deacetylation. Autophagy. 20:151–165. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Polletta L, Vernucci E, Carnevale I,
Arcangeli T, Rotili D, Palmerio S, Steegborn C, Nowak T,
Schutkowski M, Pellegrini L, et al: SIRT5 regulation of
ammonia-induced autophagy and mitophagy. Autophagy. 11:253–270.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cui X, Zhou Z, Tu H, Wu J, Zhou J, Yi Q,
Liu O and Dai X: Mitophagy in fibrotic diseases: Molecular
mechanisms and therapeutic applications. Front Physiol.
15:14302302024. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Liu BC, Tang TT, Lv LL and Lan HY: Renal
tubule injury: A driving force toward chronic kidney disease.
Kidney Int. 93:568–579. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Ohashi R, Shimizu A, Masuda Y, Kitamura H,
Ishizaki M, Sugisaki Y and Yamanaka N: Peritubular capillary
regression during the progression of experimental obstructive
nephropathy. J Am Soc Nephrol. 13:1795–1805. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li S, Lin Q, Shao X, Zhu X, Wu J, Wu B,
Zhang M, Zhou W, Zhou Y, Jin H, et al: Drp1-regulated
PARK2-dependent mitophagy protects against renal fibrosis in
unilateral ureteral obstruction. Free Radic Biol Med. 152:632–649.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Bhatia D, Chung KP, Nakahira K, Patino E,
Rice MC, Torres LK, Muthukumar T, Choi AM, Akchurin OM and Choi ME:
Mitophagy-dependent macrophage reprogramming protects against
kidney fibrosis. JCI Insight. 4:e1328261328262019. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Chawla LS, Eggers PW, Star RA and Kimmel
PL: Acute kidney injury and chronic kidney disease as
interconnected syndromes. N Engl J Med. 371:58–66. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Venkatachalam MA, Weinberg JM, Kriz W and
Bidani AK: Failed tubule recovery, AKI-CKD transition, and kidney
disease progression. J Am Soc Nephrol. 26:1765–1776. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Pavlović N, Križanac M, Kumrić M,
Vukojević K and Božić J: Mitochondrial dysfunction: The silent
catalyst of kidney disease progression. Cells. 14:7942025.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Bhat ZY, Cadnapaphornchai P, Ginsburg K,
Sivagnanam M, Chopra S, Treadway CK, Lin HS, Yoo G, Sukari A and
Doshi MD: Understanding the risk factors and long-term consequences
of cisplatin-associated acute kidney injury: An observational
cohort study. PLoS One. 10:e01422252015. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Mapuskar KA, Wen H, Holanda DG, Rastogi P,
Steinbach E, Han R, Coleman MC, Attanasio M, Riley DP, Spitz DR, et
al: Persistent increase in mitochondrial superoxide mediates
cisplatin-induced chronic kidney disease. Redox Biol. 20:98–106.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zhao C, Chen Z, Xu X, An X, Duan S, Huang
Z, Zhang C, Wu L, Zhang B, Zhang A, et al: Pink1/Parkin-mediated
mitophagy play a protective role in cisplatin induced renal tubular
epithelial cells injury. Exp Cell Res. 350:390–397. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Ludwig-Portugall I, Bartok E, Dhana E,
Evers BD, Primiano MJ, Hall JP, Franklin BS, Knolle PA, Hornung V,
Hartmann G, et al: An NLRP3-specific inflammasome inhibitor
attenuates crystal-induced kidney fibrosis in mice. Kidney Int.
90:525–539. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Li J, Lin Q, Shao X, Li S, Zhu X, Wu J,
Mou S, Gu L, Wang Q, Zhang M, et al: HIF1α-BNIP3-mediated mitophagy
protects against renal fibrosis by decreasing ROS and inhibiting
activation of the NLRP3 inflammasome. Cell Death Dis. 14:2002023.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Huang Y and Yang L: Regulation of
pyroptosis and ferroptosis by mitophagy in chronic kidney disease.
Zhong Nan Da Xue Xue Bao Yi Xue Ban. 49:1769–1776. 2024.(In
English, Chinese). PubMed/NCBI
|
|
121
|
Guo J, Yuan Z and Wang R: Zn2+
improves sepsis-induced acute kidney injury by upregulating
SIRT7-mediated parkin acetylation. Am J Physiol Renal Physiol.
327:F184–F197. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Fu ZJ, Wang ZY, Xu L, Chen XH, Li XX, Liao
WT, Ma HK, Jiang MD, Xu TT, Xu J, et al: HIF-1α-BNIP3-mediated
mitophagy in tubular cells protects against renal
ischemia/reperfusion injury. Redox Biol. 36:1016712020. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Zhou L, Zhang L, Zhang Y, Yu X, Sun X, Zhu
T, Li X, Liang W, Han Y and Qin C: PINK1 deficiency ameliorates
cisplatin-induced acute kidney injury in rats. Front Physiol.
10:12252019. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Li K, Xia X and Tong Y: Multiple roles of
mitochondrial autophagy receptor FUNDC1 in mitochondrial events and
kidney disease. Front Cell Dev Biol. 12:14533652024. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Bi Y, Liu S, Qin X, Abudureyimu M, Wang L,
Zou R, Ajoolabady A, Zhang W, Peng H, Ren J and Zhang Y: FUNDC1
interacts with GPx4 to govern hepatic ferroptosis and fibrotic
injury through a mitophagy-dependent manner. J Adv Res. 55:45–60.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Sies H and Cadenas E: Oxidative stress:
Damage to intact cells and organs. Philos Trans R Soc Lond B Biol
Sci. 311:617–631. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sies H, Berndt C and Jones DP: Oxidative
stress. Annu Rev Biochem. 86:715–748. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Forman HJ and Zhang H: Targeting oxidative
stress in disease: Promise and limitations of antioxidant therapy.
Nat Rev Drug Discov. 20:689–709. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Modlinger PS, Wilcox CS and Aslam S:
Nitric oxide, oxidative stress, and progression of chronic renal
failure. Semin Nephrol. 24:354–365. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Murphy MP: How mitochondria produce
reactive oxygen species. Biochem J. 417:1–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS
release. Physiol Rev. 94:909–950. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Sies H and Jones DP: Reactive oxygen
species (ROS) as pleiotropic physiological signalling agents. Nat
Rev Mol Cell Biol. 21:363–383. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Sureshbabu A, Ryter SW and Choi ME:
Oxidative stress and autophagy: Crucial modulators of kidney
injury. Redox Biol. 4:208–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Sedeek M, Nasrallah R, Touyz RM and Hébert
RL: NADPH oxidases, reactive oxygen species, and the kidney: Friend
and foe. J Am Soc Nephrol. 24:1512–1518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Leung JCK, Chan LYY, Tang SCW, Lam MF,
Chow CW, Lim AI and Lai KN: Oxidative damages in tubular epithelial
cells in IgA nephropathy: Role of crosstalk between angiotensin II
and aldosterone. J Transl Med. 9:1692011. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Ursini F, Maiorino M and Forman HJ: Redox
homeostasis: The golden mean of healthy living. Redox Biol.
8:205–215. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ruiz S, Pergola PE, Zager RA and Vaziri
ND: Targeting the transcription factor Nrf2 to ameliorate oxidative
stress and inflammation in chronic kidney disease. Kidney Int.
83:1029–1041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Yamamoto M, Kensler TW and Motohashi H:
The KEAP1-NRF2 system: A thiol-based sensor-effector apparatus for
maintaining redox homeostasis. Physiol Rev. 98:1169–1203. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Wakabayashi N, Dinkova-Kostova AT,
Holtzclaw WD, Kang MI, Kobayashi A, Yamamoto M, Kensler TW and
Talalay P: Protection against electrophile and oxidant stress by
induction of the phase 2 response: Fate of cysteines of the Keap1
sensor modified by inducers. Proc Natl Acad Sci USA. 101:2040–2045.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Sykiotis GP and Bohmann D:
Stress-activated Cap'n'collar transcription factors in aging and
human disease. Sci Signal. 3:re32010. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Tretter V, Hochreiter B, Zach ML, Krenn K
and Klein KU: Understanding cellular redox homeostasis: A challenge
for precision medicine. Int J Mol Sci. 23:1062021. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Oyewole AO and Birch-Machin MA:
Mitochondria-targeted antioxidants. FASEB J. 29:4766–4771. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Kataura T, Otten EG, Rabanal-Ruiz Y,
Adriaenssens E, Urselli F, Scialo F, Fan L, Smith GR, Dawson WM,
Chen X, et al: NDP52 acts as a redox sensor in
PINK1/parkin-mediated mitophagy. EMBO J. 42:e1113722023. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Hsu SN, Stephen LA, Phadwal K, Dillon S,
Carter R, Morton NM, Luijten I, Emelianova K, Amin AK, Macrae VE,
et al: Mitochondrial dysfunction and mitophagy blockade contribute
to renal osteodystrophy in chronic kidney disease-mineral bone
disorder. Kidney Int. 107:1017–1036. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Xu D, Chen P, Wang B, Wang Y, Miao N, Yin
F, Cheng Q, Zhou Z, Xie H, Zhou L, et al: NIX-mediated mitophagy
protects against proteinuria-induced tubular cell apoptosis and
renal injury. Am J Physiol Renal Physiol. 316:F382–F395. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wei X, Wang Y, Lao Y, Weng J, Deng R, Li
S, Lu J, Yang S and Liu X: Effects of honokiol protects against
chronic kidney disease via BNIP3/NIX and FUNDC1-mediated mitophagy
and AMPK pathways. Mol Biol Rep. 50:6557–6568. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Yang K, Li T, Geng Y, Zou X, Peng F and
Gao W: The role of mitophagy in the development of chronic kidney
disease. PeerJ. 12:e172602024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Reyes-Fermín LM, Avila-Rojas SH,
Aparicio-Trejo OE, Tapia E, Rivero I and Pedraza-Chaverri J: The
protective effect of alpha-mangostin against cisplatin-induced cell
death in LLC-PK1 cells is associated to mitochondrial function
preservation. Antioxidants (Basel). 8:1332019. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Prieto-Carrasco R, García-Arroyo FE,
Aparicio-Trejo OE, Rojas-Morales P, León-Contreras JC,
Hernández-Pando R, Sánchez-Lozada LG, Tapia E and Pedraza-Chaverri
J: Progressive reduction in mitochondrial mass is triggered by
alterations in mitochondrial biogenesis and dynamics in chronic
kidney disease induced by 5/6 nephrectomy. Biology (Basel).
10:3492021.PubMed/NCBI
|
|
150
|
Wen CP, Cheng TYD, Tsai MK, Chang YC, Chan
HT, Tsai SP, Chiang PH, Hsu CC, Sung PK, Hsu YH and Wen SF:
All-cause mortality attributable to chronic kidney disease: A
prospective cohort study based on 462 293 adults in taiwan. Lancet.
371:2173–2182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Dwivedi S and Sikarwar MS: Diabetic
nephropathy: Pathogenesis, mechanisms, and therapeutic strategies.
Horm Metab Res. 57:7–17. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Han YC, Tang SQ, Liu YT, Li AM, Zhan M,
Yang M, Song N, Zhang W, Wu XQ, Peng CH, et al: AMPK agonist
alleviate renal tubulointerstitial fibrosis via activating
mitophagy in high fat and streptozotocin induced diabetic mice.
Cell Death Dis. 12:9252021. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Czajka A, Ajaz S, Gnudi L, Parsade CK,
Jones P, Reid F and Malik AN: Altered mitochondrial function,
mitochondrial DNA and reduced metabolic flexibility in patients
with diabetic nephropathy. EBioMedicine. 2:499–512. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Holterman CE, Thibodeau JF, Towaij C,
Gutsol A, Montezano AC, Parks RJ, Cooper ME, Touyz RM and Kennedy
CR: Nephropathy and elevated BP in mice with podocyte-specific
NADPH oxidase 5 expression. J Am Soc Nephrol. 25:784–797. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Sang XY, Xiao JJ, Liu Q, Zhu R, Dai JJ,
Zhang C, Yu H, Yang SJ and Zhang BF: Regulators of calcineurin 1
deficiency attenuates tubulointerstitial fibrosis through improving
mitochondrial fitness. FASEB J. doi: 10.1096/fj.202000781RRR.
|
|
156
|
Li Y, Song B, Ruan C, Xue W and Zhao J:
AdipoRon attenuates hypertension-induced epithelial-mesenchymal
transition and renal fibrosis via promoting epithelial autophagy. J
Cardiovasc Transl Res. 14:538–545. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Long Y, Li Y, Ma Z, Xie Y, Zhao H, Zhang M
and Liu R: Epimedii folium and ligustri lucidi fructus
synergistically delay renal aging through
AMPK/ULK1/Bcl2L13-mediated mitophagy. J Ethnopharmacol.
346:1196682025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Wang Y, Ma Y, Ke Y, Jiang X, Liu J, Xiao
Y, Zheng H, Wang C, Chen X and Shi M: Fangji huangqi
decoction ameliorates membranous nephropathy through the
upregulation of BNIP3-mediated mitophagy. J Ethnopharmacol.
324:1177342024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Wang H, Shen M, Ma Y, Lan L, Jiang X, Cen
X, Guo G, Zhou Q, Yuan M, Chen J, et al: Novel mitophagy inducer
alleviates lupus nephritis by reducing myeloid cell activation and
autoantigen presentation. Kidney Int. 105:759–774. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Wang Y, Lu M, Xiong L, Fan J, Zhou Y, Li
H, Peng X, Zhong Z, Wang Y, Huang F, et al: Drp1-mediated
mitochondrial fission promotes renal fibroblast activation and
fibrogenesis. Cell Death Dis. 11:292020. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Dröge W: Free radicals in the
physiological control of cell function. Physiol Rev. 82:47–95.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Dounousi E, Papavasiliou E, Makedou A,
Ioannou K, Katopodis KP, Tselepis A, Siamopoulos KC and Tsakiris D:
Oxidative stress is progressively enhanced with advancing stages of
CKD. Am J Kidney Dis. 48:752–760. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Zhang L, Miao M, Xu X, Bai M, Wu M and
Zhang A: From physiology to pathology: The role of mitochondria in
acute kidney injuries and chronic kidney diseases. Kidney Dis
(Basel). 9:342–357. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Cheng Y, Lu Z, Mao T, Song Y, Qu Y, Chen
X, Chen K, Liu K and Zhang C: Magnoflorine ameliorates chronic
kidney disease in high-fat and high-fructose-fed mice by promoting
parkin/PINK1-dependent mitophagy to inhibit
NLRP3/caspase-1-mediated pyroptosis. J Agric Food Chem.
72:12775–12787. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Li H, Meng P, Meng Y, Yang Y, Zheng W,
Huang H and Zhao Z: Mechanism of ruxolitinib enhancing mitophagy
against renal fibrosis via PINK1/parkin pathway. Biochim Biophys
Acta Mol Basis Dis. 1871:1679782025. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ding XQ, Jian TY, Gai YN, Niu GT, Liu Y,
Meng XH, Li J, Lyu H, Ren BR and Chen J: Chicoric acid attenuated
renal tubular injury in HFD-induced chronic kidney disease mice
through the promotion of mitophagy via the Nrf2/PINK/parkin
pathway. J Agric Food Chem. 70:2923–2935. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Cao Y, Xiong J, Guan X, Yin S, Chen J,
Yuan S, Liu H, Lin S, Zhou Y, Qiu J, et al: Paeoniflorin suppresses
kidney inflammation by regulating macrophage polarization via
KLF4-mediated mitophagy. Phytomedicine. 116:1549012023. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Jia Q, Han L, Zhang X, Yang W, Gao Y, Shen
Y, Li B, Wang S, Qin M, Lowe S, et al: Tongluo yishen decoction
ameliorates renal fibrosis via regulating mitochondrial dysfunction
induced by oxidative stress in unilateral ureteral obstruction
rats. Front Pharmacol. 12:7627562021. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Ren H, Shao Y, Wu C, Ma X, Lv C and Wang
Q: Metformin alleviates oxidative stress and enhances autophagy in
diabetic kidney disease via AMPK/SIRT1-FoxO1 pathway. Mol Cell
Endocrinol. 500:1106282020. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Hong Q, Zhang L, Das B, Li Z, Liu B, Cai
G, Chen X, Chuang PY, He JC and Lee K: Increased podocyte sirtuin-1
function attenuates diabetic kidney injury. Kidney Int.
93:1330–1343. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Galluzzi L, Bravo-San Pedro JM, Levine B,
Green DR and Kroemer G: Pharmacological modulation of autophagy:
Therapeutic potential and persisting obstacles. Nat Rev Drug
Discov. 16:487–511. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Bonekamp NA, Jiang M, Motori E, Garcia
Villegas R, Koolmeister C, Atanassov I, Mesaros A, Park CB and
Larsson NG: High levels of TFAM repress mammalian mitochondrial DNA
transcription in vivo. Life Sci Alliance. 4:e2021010342021.
View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Wang R, Yuan W, Li L, Lu F, Zhang L, Gong
H and Huang X: Resveratrol ameliorates muscle atrophy in chronic
kidney disease via the axis of SIRT1/FoxO1. Phytother Res.
36:3265–3275. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
174
|
Zhang W, Guo C, Li Y, Wang H, Wang H, Wang
Y, Wu T, Wang H, Cheng G, Man J, et al: Mitophagy mediated by
HIF-1α/FUNDC1 signaling in tubular cells protects against renal
ischemia/reperfusion injury. Ren Fail. 46:23324922024. View Article : Google Scholar : PubMed/NCBI
|
|
175
|
Sharma GN, Gupta G and Sharma P: A
comprehensive review of free radicals, antioxidants, and their
relationship with human ailments. Crit Rev Eukaryot Gene Expr.
28:139–154. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
176
|
Harden TK, Sesma JI, Fricks IP and
Lazarowski ER: Signalling and pharmacological properties of the P2Y
receptor. Acta Physiol (Oxf). 199:149–160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
177
|
Bai X, Huang D, Xie P, Sun R, Zhou H and
Liu Y: Effect of uridine on mitochondrial function. Sheng Wu Gong
Cheng Xue Bao. 39:3695–3709. 2023.(In Chinese). PubMed/NCBI
|
|
178
|
Krylova IB, Selina EN, Bulion VV,
Rodionova OM, Evdokimova NR, Belosludtseva NV, Shigaeva MI and
Mironova GD: Uridine treatment prevents myocardial injury in rat
models of acute ischemia and ischemia/reperfusion by activating the
mitochondrial ATP-dependent potassium channel. Sci Rep.
11:169992021. View Article : Google Scholar : PubMed/NCBI
|
|
179
|
Jiang N and Zhao Z: Intestinal aging is
alleviated by uridine via regulating inflammation and oxidative
stress in vivo and in vitro. Cell Cycle. 21:1519–1531. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
180
|
Wang Y, Ping Z, Gao H, Liu Z, Xv Q, Jiang
X and Yu W: LYC inhibits the AKT signaling pathway to activate
autophagy and ameliorate TGFB-induced renal fibrosis. Autophagy.
20:1114–1133. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
181
|
Smith RAJ, Hartley RC and Murphy MP:
Mitochondria-targeted small molecule therapeutics and probes.
Antioxid Redox Signal. 15:3021–3038. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
182
|
Kelso GF, Porteous CM, Coulter CV, Hughes
G, Porteous WK, Ledgerwood EC, Smith RA and Murphy MP: Selective
targeting of a redox-active ubiquinone to mitochondria within
cells: Antioxidant and antiapoptotic properties. J Biol Chem.
276:4588–4596. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
183
|
James AM, Cochemé HM, Smith RAJ and Murphy
MP: Interactions of mitochondria-targeted and untargeted
ubiquinones with the mitochondrial respiratory chain and reactive
oxygen species. Implications for the use of exogenous ubiquinones
as therapies and experimental tools. J Biol Chem. 280:21295–21312.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
184
|
Hamed M, Logan A, Gruszczyk AV, Beach TE,
James AM, Dare AJ, Barlow A, Martin J, Georgakopoulos N, Gane AM,
et al: Mitochondria-targeted antioxidant MitoQ ameliorates
ischaemia-reperfusion injury in kidney transplantation models. Br J
Surg. 108:1072–1081. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
185
|
Zhu Z, Liang W, Chen Z, Hu J, Feng J, Cao
Y, Ma Y and Ding G: Mitoquinone protects podocytes from angiotensin
II-induced mitochondrial dysfunction and injury via the Keap1-Nrf2
signaling pathway. Oxid Med Cell Longev. 2021:13944862021.
View Article : Google Scholar : PubMed/NCBI
|
|
186
|
Reddy PH, Manczak M and Kandimalla R:
Mitochondria-targeted small molecule SS31: A potential candidate
for the treatment of Alzheimer's disease. Hum Mol Genet.
26:15972017. View Article : Google Scholar : PubMed/NCBI
|
|
187
|
Hou Y, Li S, Wu M, Wei J, Ren Y, Du C, Wu
H, Han C, Duan H and Shi Y: Mitochondria-targeted peptide SS-31
attenuates renal injury via an antioxidant effect in diabetic
nephropathy. Am J Physiol Renal Physiol. 310:F547–F559. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
188
|
Yang SK, Han YC, He JR, Yang M, Zhang W,
Zhan M, Li AM, Li L, Na-Song, Liu YT, et al: Mitochondria targeted
peptide SS-31 prevent on cisplatin-induced acute kidney injury via
regulating mitochondrial ROS-NLRP3 pathway. Biomed Pharmacother.
130:1105212020. View Article : Google Scholar : PubMed/NCBI
|
|
189
|
Zhu Y, Luo M, Bai X, Li J, Nie P, Li B and
Luo P: SS-31, a mitochondria-targeting peptide, ameliorates kidney
disease. Oxid Med Cell Longev. 2022:12955092022. View Article : Google Scholar : PubMed/NCBI
|
|
190
|
Lan T, Guo H, Lu X, Geng K, Wu L, Luo Y,
Zhu J, Shen X, Guo Q and Wu S: Dual-responsive curcumin-loaded
nanoparticles for the treatment of cisplatin-induced acute kidney
injury. Biomacromolecules. 23:5253–5266. 2022. View Article : Google Scholar : PubMed/NCBI
|