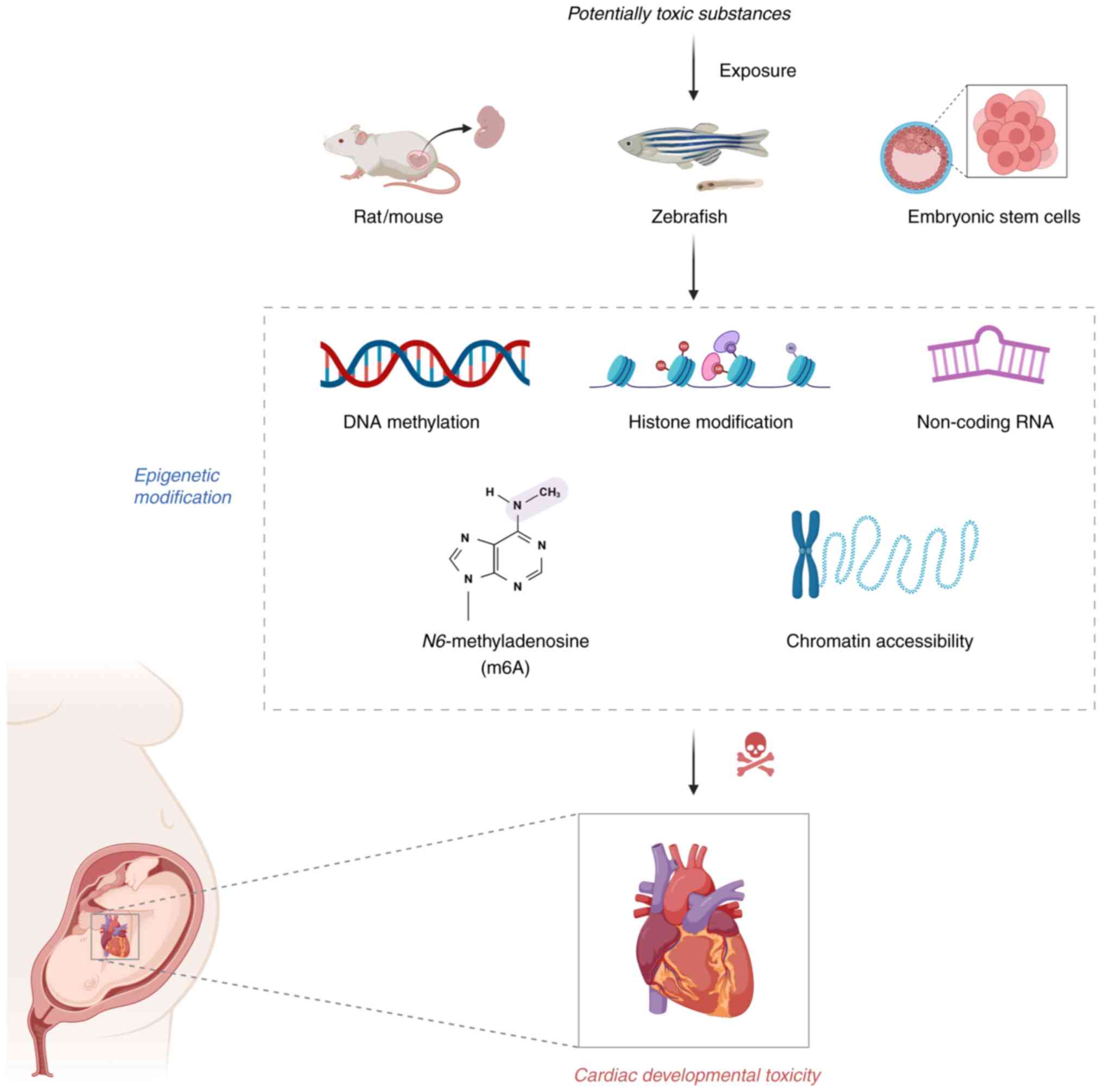
Introduction
Cardiac developmental toxicity refers to harmful
effects induced by external factors, including chemical substances,
pharmaceuticals, environmental pollutants or infectious agents,
which impair normal cardiac morphogenesis, cellular differentiation
or functional maturation during key phases of embryonic or fetal
heart development (1). This
disruption may result in structural cardiac anomalies (such as
ventricular septal defects or patent ductus arteriosus) or
functional impairments (such as cardiac arrhythmias and impaired
cardiac function). Epidemiological studies demonstrate that
congenital heart disease (CHD) affects >1% of live births and
accounts for the majority of prenatal miscarriages (2–4). In
the United States alone, ~500,000 adults currently live with CHD
(5). Notably, although >80,000
chemical substances are commercially available in the United
States, the cardiac teratogenic potential of the majority of these
substances remains uncharacterized, creating notable gaps in
prenatal exposure risk assessment. Beyond genetic predispositions,
maternal exposure to cardiac teratogens during the key cardiac
morphogenesis period, spanning 3 months pre-conception to
gestational weeks 2–7, markedly elevates the risk for developmental
cardiac defects (6). Therefore,
elucidating the molecular mechanisms of xenobiotic-induced cardiac
developmental toxicity is important for developing targeted
clinical surveillance programs and formulating evidence-based
preventive public health measures.
Epigenetic regulation controls transcriptional
activity through stable but reversible chromatin modifications that
occur independently of DNA sequence alterations (7). Key epigenetic mechanisms, including
DNA methylation, histone modifications (acetylation and
methylation), regulatory non-coding RNAs (ncRNAs), RNA methylation
and chromatin remodeling, act synergistically to precisely modulate
gene expression while preserving the underlying genetic sequence
(8,9). These modifications demonstrate
plasticity in response to diverse extrinsic factors, encompassing
dietary composition, physical activity, pathogenic challenges,
environmental toxicants, senescence processes and psychosocial
stressors (10,11). Maternal exposure to pharmaceuticals
or environmental contaminants during pregnancy may disrupt cardiac
development through epigenetic mechanisms, leading to dysregulation
of cardiac morphogenesis-related genes and impaired cardiac
differentiation. Such perturbations ultimately contribute to
congenital heart defects. The present review summarizes emerging
evidence regarding epigenetic regulation of cardiac developmental
toxicity (Fig. 1), offering
potential avenues for early detection and prevention of
chemical-induced cardiac malformations.
DNA methylation
DNA methylation represents a key epigenetic
mechanism involving the addition of methyl groups to the fifth
carbon position of cytosines, generating 5-methylcytosine (5mC)
(12,13). This reaction is catalyzed by DNA
methyltransferases (DNMTs), with DNMT3A and DNMT3B primarily
mediating de novo methylation at unmethylated CpG sites
(14,15). DNMT1, as the principal maintenance
methyltransferase, serves a key role in preserving epigenetic
patterns during DNA replication. This enzyme specifically
recognizes hemimethylated CpG dinucleotides, ensuring accurate
methylation inheritance from parent to daughter strands and thereby
maintaining epigenetic memory throughout cellular proliferation
(16). The enzymatic transfer of
methyl moieties to the fifth carbon of cytosines induces
conformational constraints in genomic DNA, creating steric
occlusion that competitively inhibits transcription factor docking.
This biochemical blockade constitutes a fundamental paradigm of
epigenetic gene silencing (17,18).
DNA demethylation proceeds through two
mechanistically distinct pathways. Namely, passive
replication-dependent dilution, followed by active enzymatic
removal. Passive demethylation results from the gradual loss of
methylation marks during successive rounds of DNA replication,
whereas active demethylation involves direct enzymatic erasure of
methyl groups independent of DNA synthesis (19). The ten-eleven translocation (TET)
family of dioxygenases (TET1, TET2 and TET3) drive active DNA
demethylation through iterative oxidation reactions, progressively
modifying 5mC to form 5-hydroxymethylcytosine, subsequently
advancing to 5-formylcytosine and concluding with the generation of
5-carboxylcytosine (20).
Following oxidative modification, thymine DNA glycosylase
selectively binds and cleaves these epigenetic intermediates,
enabling their replacement with canonical cytosine through base
excision repair-mediated nucleotide substitution (21). Alternatively, non-oxidative
demethylation pathways exist whereby deaminases such as
activation-induced deaminase/apolipoprotein B mRNA editing
catalytic polypeptide family enzymes can directly convert 5mC to
thymine (22). This initiates
distinct repair processes that ultimately result in
demethylation.
With regard to DNA methylation dysregulation-induced
cardiac developmental toxicity, recent methodological advances in
whole-genome bisulfite sequencing (WGBS), coupled with newly
developed single-cell methylome analysis technologies, now
facilitate genome-wide DNA methylation profiling with unprecedented
resolution and comprehensiveness (23,24).
Mounting evidence indicates that DNA methylation serves as a key
epigenetic regulator governing embryonic development, cellular
differentiation and responses to environmental toxicants (25–27).
Notably, aberrant alterations in DNA methylation are associated
with a number of birth defects and developmental disorders, such as
cardiac malformations and abnormal heart rates, highlighting its
importance in developmental toxicity. Table I summarizes the mechanisms by which
potential toxic substances induce cardiac developmental toxicity
through abnormal DNA methylation.
 | Table I.DNA methylation dysregulation-induced
cardiac developmental toxicity. |
Table I.
DNA methylation dysregulation-induced
cardiac developmental toxicity.
| First author,
year | Toxicant | Embryo model | Methylation
status | Potential
mechanism | Cardiac toxicity
manifestations | (Refs.) |
|---|
| Jiang et al,
2019 | PM2.5 | Zebrafish | Widespread
methylation dysregulation (both hyper- and hypomethylation) | Dysregulated
cardiac developmental gene expression | Increased incidence
of cardiac malformations and decreased heart rate | (25) |
| Mu et al,
2020 | Phthalates | Zebrafish | Hypomethylation of
NPPA and cTnT, hypermethylation of TBX5b | Dysregulated
cardiac developmental gene expression | Abnormal heart rate
and pericardial edema | (30) |
| Ma et al,
2012 | Selenite | Zebrafish | Early
hypomethylation, late hypermethylation | Dysregulated
proliferation-apoptosis homeostasis | Bradycardia,
pericardial edema and reduced cardiac chamber volume with
morphological abnormalities | (39) |
| Li et al,
2009 | Arsenite | Zebrafish | Early
hypomethylation, late hypermethylation | Dysregulated
proliferation-apoptosis homeostasis | Bradycardia and
altered ventricular shape | (42) |
| Jiao et al,
2023 | Cyhexatin | Zebrafish | Global DNA
hypomethylation | Apoptosis,
oxidative stress and endocrine disruption | Abnormal heart rate
and pericardial edema | (44) |
| Chatterjee et
al, 2021 | CMIT/MIT | Zebrafish | Global DNA
hypermethylation | Dysregulated
cardiac developmental gene expression | Abnormal heart rate
and pericardial edema | (48) |
Phthalates, a class of ubiquitous industrial
chemicals, are recognized endocrine-disrupting compounds associated
with potential cardiovascular impairment, reproductive dysfunction
and developmental toxicity (28,29).
Mu et al (30) discovered
through methylated DNA immunoprecipitation sequencing (MeDIP-Seq)
that exposure to di-(2-ethylhexyl) phthalate and di-butyl phthalate
could alter DNA methylation patterns of cardiac development-related
genes, thereby modulating gene expression and subsequently inducing
cardiac developmental defects in zebrafish embryos. This study
observed that reduced DNA methylation at the natriuretic peptide A
(NPPA) and cardiac troponin T2 (cTnT) gene loci was frequently
associated with higher transcriptional activity. By contrast,
increased methylation levels in the T-box transcription factor 5
(TBX5)-b promoter region tended to be associated with decreased
expression, suggesting transcriptional repression. These
methylation-expression relationships support the possibility that
DNA methylation serves as an important epigenetic mechanism
influencing cardiac developmental toxicity following phthalate
exposure.
Particulate matter (PM) represents a heterogeneous
mixture of airborne contaminants comprising chemical components
derived from numerous emission sources, including vehicle
emissions, industrial processes and biomass burning, with PM2.5
specifically referring to fine particulate matter with an
aerodynamic diameter of ≤2.5 µm (31). Accumulating epidemiological
evidence demonstrates that prenatal exposure to particulate matter
is associated with adverse pregnancy outcomes, particularly preterm
delivery, reduced birth weight and impaired neurodevelopment
(32–34). Emerging research highlights the
role of DNA methylation alterations as a key mechanism underlying
PM-induced cardiac developmental toxicity. Genome-wide DNA
methylation profiling revealed that exposure to PM2.5 extracts
induces widespread methylation dysregulation (both hyper- and
hypomethylation) in zebrafish embryonic hearts, leading to
developmental defects (25). This
process is associated with abnormal folate metabolism, suggesting
that PM2.5 may disrupt folate metabolic homeostasis through DNA
methylation modifications, thereby contributing to cardiac
developmental toxicity.
Selenium, while recognized as a key micronutrient
important in maintaining normal physiological functions, such as
development and immune responses, poses marked toxicological risks
due to the narrow therapeutic margin it exhibits between
nutritional adequacy and toxicological thresholds, thereby
warranting serious consideration of environmental exposure hazards
(35,36). As the primary selenium source for
selenoprotein biosynthesis in mammalian cells, selenite has been
demonstrated to induce developmental toxicity and neurobehavioral
abnormalities in zebrafish embryos (37,38).
Ma et al (39) revealed
that selenite induces cardiac developmental toxicity in zebrafish
embryos by disturbing DNA methylation dynamics, manifesting as
aberrant early-stage hypomethylation followed by late-stage
hypermethylation, concomitant with ventricular/atrial morphological
defects and impaired cardiac function. In addition, folate notably
alleviated cardiac defects by rescuing methylation imbalance,
demonstrating that selenite-induced cardiac developmental toxicity
depends on the disruption of DNA methylation dynamics. Similarly,
arsenic is a widely distributed naturally occurring element that
poses hazards to living organisms (40). Particularly in its trivalent form
as arsenite, arsenic has been demonstrated to cause notable harm to
humans through multiple biochemical pathways, including
mitochondrial reactive oxygen species (ROS) generation, due to its
potent carcinogenicity and toxicity (41). A previous study demonstrated that
exposure to 2.0 mM arsenite induced cardiac developmental defects
in zebrafish embryos at 48 h post-fertilization (hpf),
characterized by reduced ventricular volume, morphological
alterations and a markedly increased atrioventricular angle
(42). Concurrent global DNA
hypermethylation was observed, suggesting that this epigenetic
dysregulation may contribute to arsenite-mediated cardiotoxicity.
These findings provide experimental evidence for understanding the
DNA methylation mechanisms underlying the cardiac developmental
toxicity of heavy metals/metalloids.
Cyclohexylltin (CYT), an organotin compound, is
often employed as a broad-spectrum pesticide in modern agricultural
practices for pest management (43). However, its potential cardiotoxic
effects during embryonic development remain to be systematically
elucidated. In a zebrafish model, CYT exposure was shown to induce
genome-wide DNA hypomethylation, characterized by decreased
S-adenosylmethionine (SAM)/S-adenosylhomocysteine (SAH) ratios and
suppressed DNMTs expression, along with cardiac malformations such
as bradycardia and pericardial edema (44). These findings suggest that CYT
disrupts DNA methylation dynamics, potentially resulting in
aberrant transcriptional regulation of genes key to cardiac
development.
5-Chloro-2-methyl-4-isothiazolin-3-one/2-methyl-4-isothiazolin-3-one
(CMIT/MIT) is an isothiazolinone-based biocide that is commonly
formulated in numerous aqueous consumer products, with extensive
applications in disinfectants and cosmetics (45,46).
Previous research has indicated the potential neurotoxic effects of
CMIT/MIT (47), and due to its
extensive commercial applications and consequent exposure risks,
research has shifted focus toward investigating its developmental
toxicity. Chatterjee et al (48) investigated the developmental
toxicity of CMIT/MIT in zebrafish embryos and found that exposure
to this compound markedly upregulated the expression of DNMTs,
including DNMT1, DNMT3a2, DNMT3b1 and DNMT3b4, leading to global
DNA hypermethylation The observed cardiac malformations, including
pericardial edema and bradycardia, were associated with concurrent
epigenetic modifications, indicating that CMIT/MIT-induced
developmental cardiotoxicity may occur through DNMT-dependent
dysregulation of DNA methylation.
Studies utilizing advanced sequencing technologies
have demonstrated that multiple toxicants can disrupt DNA
methylation patterns, inducing gene-specific alterations or
genome-wide epigenetic instability. These modifications impair key
developmental processes such as cardiogenesis, potentially
culminating in developmental cardiotoxicity (25,30).
However, existing research has predominantly identified associative
relationships between DNA methylation changes and developmental
toxicity, without direct experimental validation of causality
between specific epigenetic modifications and toxicological
outcomes. In addition, the mechanistic pathways through which
aberrant DNA methylation influences downstream gene expression to
confer toxicity remain only partially characterized. Despite
intricate crosstalk between DNA methylation and other epigenetic
modifications such as histone modifications and ncRNAs, the precise
regulatory mechanisms governing these interactions remain
incompletely characterized (49,50).
Integrating multi-omics approaches and employing targeted DNA
methylation editing technologies to validate the functional roles
of key methylation sites could advance the understanding of how DNA
methylation influences cardiac developmental toxicity (51).
Histone modification
Histones constitute the key protein subunits that
form octameric nucleosome complexes, creating the structural
scaffold necessary for DNA compaction. These include four core
histone variants (H3, H4, H2A and H2B), which coordinately assemble
to establish the basic repeating unit of chromatin (52). In addition to their structural
function, histones undergo diverse covalent post-translational
modifications, including acetylation, methylation, phosphorylation,
ubiquitination and small ubiquitin-like modifier (SUMO)-ylation,
that serve as key epigenetic regulators modulating transcriptional
activity (53,54). Such modifications frequently target
flexible histone N-terminal domains, whereby genetic activity would
be modulated through the dual mechanisms of directly changing
histone-DNA interaction dynamics or serving as docking sites for
chromatin-associated regulatory proteins (55).
Among the diverse number of histone modifications,
methylation and acetylation constitute two of the most extensively
characterized epigenetic marks. The addition of methyl moieties to
specific lysine or arginine amino acids is enzymatically
facilitated by histone methyltransferases, utilizing
S-adenosylmethionine as the methyl donor molecule (56). Counteracting this process, histone
demethylases catalyze the elimination of these methyl groups
through oxidative reactions. Simultaneously, the acetylation
process introducing acetyl functional groups through histone
acetyltransferases induces chromatin decondensation, creating a
permissive environment for transcriptional machinery (57). This modification is dynamically
reversible through the enzymatic activity of histone deacetylases,
which leads to chromatin compaction associated with gene silencing
(58). These reversible
modifications collectively constitute a sophisticated regulatory
network of histone modifications.
With regard to histone modification
dysregulation-induced cardiac developmental toxicity, existing
evidence has demonstrated that dysregulated histone modifications
can destabilize genomic integrity and dysregulate gene
transcription, ultimately driving multiorgan toxicity
manifestations, particularly hepatotoxicity, neurotoxicity and
reproductive toxicity (59–62).
Furthermore, histone modification-mediated regulatory mechanisms
have become a key focus in developmental toxicity research, as
their dynamic alterations may influence embryonic development and
organogenesis, garnering notable scientific interest (63–65).
The experimental evidence linking toxicant-induced histone
modifications to cardiac developmental toxicity is summarized in
Table II.
| Table II.Histone modification
dysregulation-induced cardiac developmental toxicity. |
Table II.
Histone modification
dysregulation-induced cardiac developmental toxicity.
| First author,
year | Toxicant | Embryo model | Histone
modification | Potential
mechanism | Cardiac toxicity
manifestations | (Refs.) |
|---|
| Zhang et al,
2024 | Fenbuconazole | Zebrafish | H3K9Ac↓
H3K14Ac↓ | Dysregulated
cardiac developmental gene expression | Cardiac arrhythmia
and cardiac morphological defects | (68) |
| Cheng et al,
2016 | Cigarette
smoke | mESCs | Global H3
hypoacetylation | Dysregulated
cardiac developmental gene expression | Decreased cardiac
contraction rate | (75) |
| Wu et al,
2022 | Cadmium | Two-dimensional
cardiac differentiation model and three-dimensional EBs and cardiac
organoid models derived from hESCs | H3K27me3
H3K4me3↓ | Inhibition of
mesoderm formation and suppression of cardiomyocyte differentiation
and cardiac induction | Decreased cardiac
contraction rate | (79) |
Fenbucoonazole (FBZ), a triazole-class fungicide, is
extensively employed in agricultural and horticultural applications
(66). A previous study has
demonstrated that FBZ exposure induces abnormal transcription of
specific genes, thereby compromising cardiac development and
function in larvae (67). However,
the involvement of histone modifications in this process remains
poorly understood. Zhang et al (68) revealed that zebrafish embryos
exposed to FBZ showed markedly decreased acetylation levels of
histone H3K9 and H3K14 (H3K9Ac and H3K14Ac) in the hearts of both
filial (F)0 and F1 generation adult fish. This epigenetic
repression directly downregulated the transcription of core cardiac
developmental genes [GATA binding protein 4 (GATA4), TBX5 and NK2
homeobox 5 (NKX2-5)] and calcium homeostasis-related genes,
resulting in cardiac abnormalities including arrhythmia and
morphological defects. Notably, these adverse effects were
transgenerationally transmitted to the F2 generation. These
findings offer mechanistic insights into histone
acetylation-mediated cardiotoxicity during embryogenesis, while
providing evidence to support transgenerational epigenetic
inheritance.
According to official reports from the U.S. Surgeon
General, tobacco smoke exposure represents a notable environmental
health hazard, accounting for ~480,000 annual mortalities and
constituting one of the most notable pathogenic environmental
factors (69,70). Prenatal exposure to tobacco
pollutants induces embryotoxicity, characterized by growth
restriction, structural anomalies including craniofacial
malformations, congenital heart defects and skeletal abnormalities,
as well as embryonic lethality (71–74).
Cheng et al (75)
demonstrated that both mainstream smoke and sidestream smoke (SS)
exposure during cardiac differentiation of mouse embryonic stem
cells (ESCs) markedly reduced global histone H3 acetylation (H3ac)
levels in developing cardiomyocytes. This study revealed that SS
specifically diminished H3ac enrichment at the promoter region of
GATA4 (a key cardiac transcription factor), leading to its
transcriptional downregulation. The observed epigenetic
modifications suppressed (bone morphogenetic protein (BMP)-SMAD
family member 4 signaling, a key pathway regulating cardiac
morphogenesis. These results demonstrate that dysregulated histone
acetylation constitutes a primary molecular mechanism driving
developmental cardiotoxicity induced by tobacco smoke exposure.
Cadmium (Cd), a highly toxic heavy metal, poses
escalating global health risks due to its environmental persistence
and good aqueous solubility (76).
Studies have demonstrated that Cd exposure exhibits both direct
embryogenic regulatory disruption and association with multiple
pathologies, including developmental cardiotoxicity, establishing
its developmental toxicity as a key concern in mechanistic
toxicology (77,78). Wu et al (79) employed human ESCs (hESCs) in
two-dimensional differentiation models, three-dimensional embryoid
bodies and cardiac organoids as experimental systems. Cd exposure
was found to markedly elevate the levels of repressive histone mark
H3K27me3 while reducing the levels of active histone mark H3K4me3.
This epigenetic dysregulation was associated with the
downregulation of mesoderm markers (including heart and neural
crest derivatives expressed 1 and HOP homeobox) and
cardiac-specific genes (including NKX2-5 and GATA4), ultimately
impairing mesoderm-to-cardiomyocyte differentiation. The
experimental results demonstrate that Cd-induced cardiac
developmental toxicity is regulated by histone methylation,
particularly during mesoderm specification, a developmental phase
exhibiting heightened vulnerability.
Histone ubiquitination regulates diverse
chromatin-associated processes, including transcriptional
regulation, DNA repair and chromatin structure remodeling, with
this regulatory mechanism having attracted growing research
attention in recent years (80–82).
Although direct evidence linking toxicant-induced developmental
cardiotoxicity to histone ubiquitination pathways remains limited,
existing studies have provided a number of theories (83,84).
The cullin-RING E3 ubiquitin ligase (CRL) family has been
demonstrated to regulate cardiac morphogenesis and tissue
maturation through ubiquitin-dependent modifications (83). As a key member of the CRL family,
cullin 4A (CUL4A) serves a key role in heart development by
mediating ubiquitin-dependent degradation of target proteins. A
previous study has shown that CUL4A deficiency induces pericardial
edema, abnormal cardiac looping and pectoral fin defects in
zebrafish embryos, phenotypes associated with impaired
cardiomyocyte proliferation and increased apoptosis (84). These findings suggest that
dysregulation of ubiquitination pathways may contribute to
developmental cardiotoxicity. Given the role of histone
ubiquitination in DNA damage response and cell fate determination,
the present review proposes it may regulate cardiac developmental
toxicity, representing a promising research avenue.
Aberrant histone modifications disrupt cardiac
developmental gene expression through alterations in methylation
and acetylation marks, demonstrating direct mediation of
teratogenic effects. Current research has primarily focused on
histone acetylation and specific methylation events, whereas other
key modifications, including phosphorylation, ubiquitination and
SUMOylation, remain poorly characterized. This knowledge gap limits
comprehensive understanding of the diverse mechanistic
contributions of histone modifications to developmental toxicity
(68,75,79).
While numerous current studies demonstrate associative
relationships between histone modification alterations and toxic
phenotypes, the key molecular mechanisms, particularly how
toxicants precisely regulate histone-modifying enzyme activity
within upstream regulatory networks, remain inadequately elucidated
(68,75,79).
Subsequent investigations should aim to focus on delineating
temporal alterations in histone marks and their interactions with
additional epigenetic regulators to elucidate underlying
pathological mechanisms (55).
NcRNAs
NcRNAs constitute transcriptionally active, yet
untranslated, genetic elements predominant in eukaryotic systems,
with three principal subtypes prevailing, namely long ncRNAs
(lncRNAs), microRNAs (miRNAs) and circular RNAs (circRNAs)
(85,86). Despite their protein-coding
deficiency, these RNA molecules exert comprehensive regulatory
control over important biological processes, including chromatin
remodeling, transcriptional modulation, post-translational
modifications and signal transduction (87,88).
As key regulatory molecules, ncRNAs have been demonstrated to
participate in the mechanisms underlying toxicity induced by
numerous environmental pollutants and pharmaceutical compounds,
such as crude oil and ribavirin (89,90).
With regard to ncRNA dysregulation-induced cardiac
developmental toxicity, emerging scientific evidence has
established ncRNAs as key molecular mediators orchestrating
toxicological responses throughout embryogenesis (91,92).
These discoveries not only offer promising biomarker candidates for
developmental toxicity assessment but also improve the mechanistic
understanding of the epigenetic regulatory circuitry underlying
developmental toxicity. The dysregulation of ncRNA-mediated cardiac
developmental toxicity is summarized in Table III.
| Table III.NcRNAs dysregulation-induced cardiac
developmental toxicity. |
Table III.
NcRNAs dysregulation-induced cardiac
developmental toxicity.
| First author,
year | Toxicant | Embryo model | ncRNAs | Target | Potential
mechanism | Cardiac toxicity
manifestations | (Refs.) |
|---|
| Ye et al,
2020 | Ribavirin | hiPSCs | lncRNA GAS5↑ lncRNA
HBL1↑ | p53 | ROS accumulation
and DNA damage | Pericardial edema,
incomplete cardiac looping and ventricular/atrial enlargement | (96) |
| Xu et al,
2019 | Crude oil | Red drum
(Sciaenops ocellatu) | MiR-18a↓ miR-27b↑
miR-203a↓ | - | Disruption of
cardiac developmental pathways | Pericardial
edema | (101) |
| Magnuson et
al, 2022 | Phenanthrene | Zebrafish | miR-203a↓ | VEGFA | Dysregulated
cardiac developmental gene expression | Abnormal heart rate
and pericardial edema | (110) |
| Guo et al,
2024 | MC-LR | Mice | miR-377-3p | NR6A1 | Macrophage
polarization imbalance | Ventricular
dilation and myocardial thinning | (111) |
LncRNAs represent a distinct class of ncRNAs
characterized by lengths of >200 nucleotides, with certain
transcripts extending to hundreds of kilobases. These RNA molecules
regulate key cellular processes such as proliferation, apoptosis
and cellular migration through multiple molecular mechanisms
(93–95). Notably, dysregulation of lncRNAs
has been implicated in drug-induced developmental toxicity, as
exemplified by antiviral drugs (96). Ribavirin, as a broad-spectrum
antiviral drug, may induce adverse effects in clinical
applications, such as rashes, hemolytic anemia and teratogenicity
(97). However, its potential
cardiotoxicity and underlying mechanisms during cardiac development
remain elusive. Ye et al (96) found that ribavirin upregulates the
expression of lncRNAs growth arrest specific 5 and Heart Brake
lnRNA 1, which inhibits the differentiation of human induced
pluripotent stem cells into cardiomyocytes, triggering DNA damage
and p53 pathway activation. This finding establishes novel
perspectives on the teratogenic mechanisms of ribavirin while
decoding the functional importance of lncRNAs in heart
development.
MiRNAs constitute a class of endogenous small ncRNAs
(~22 nucleotides in length) that post-transcriptionally regulate
gene expression by binding to complementary mRNA sequences,
ultimately inducing mRNA degradation or translational repression
(98). The integration of
high-throughput sequencing technologies with sophisticated
bioinformatics tools has advanced miRNA research, driving
developments in mechanistic studies and translational applications
within this field (99).
Increasing evidence suggests that environmental toxicant exposure
can disrupt normal developmental processes by interfering with
epigenetic regulatory mechanisms, particularly through modulating
miRNA expression profiles. These findings provide novel insights
into toxicological pathway mechanisms (100–102).
Trichloroethylene (TCE), a volatile organic solvent,
is distributed in environmental media including soil, groundwater
and air, posing marked contamination risks (103). A growing body of evidence
indicates that TCE exposure may induce cardiac developmental
abnormalities in humans and other organisms (103–106). Huang et al (91) found that TCE exposure downregulates
miR-133a in zebrafish embryonic hearts, leading to heart
developmental defects by increasing ROS generation and excessive
cell proliferation, yet miR-133a agonists effectively mitigated
this toxic effect. This study demonstrates that miR-133a serves a
key mediating role in TCE-induced cardiac developmental toxicity,
suggesting its potential as a biomarker for cardiotoxicity
assessment.
Crude oil and its chemical components represent
common environmental contaminants in aquatic systems, with their
long-term toxicological impacts remaining an active area of
investigation (107–109). Xu et al (101) employed red drum (Sciaenops
ocellatus) larvae to investigate miRNA-mediated cardiotoxicity
through integrated mRNA-miRNA sequencing analysis. This study
revealed that Deepwater Horizon crude oil exposure altered the
expression of miR-18a, miR-27b and miR-203a and this was
concentration-dependent. Specifically, miR-27b upregulation
promoted physiological/pathological cardiac hypertrophy, while
downregulation of miR-18a and miR-203a impaired heart rate
regulation and cardiac valve morphogenesis, respectively.
Mechanistically, these dysregulated miRNAs disrupted key cardiac
developmental pathways, including calcium signaling and
hypertrophic responses, by modulating their target genes, thereby
providing a novel miRNA-dependent mechanism underlying polycyclic
aromatic hydrocarbon (PAHs)-induced developmental cardiotoxicity in
marine fish. Similarly, another study conducted on larvae mahi-mahi
revealed that exposure to PAHs in crude oil resulted in
dose-dependent upregulation of miR-34b and miR-23b, as well as
downregulation of miR-203a (102). These miRNAs dysregulated cardiac
developmental genes, including components of calcium signaling and
regulators of cardiac hypertrophy, leading to pericardial edema,
bradycardia and other morphological abnormalities. Additionally,
the activation of p53 signaling further exacerbated developmental
cardiac impairments. These findings provide evidence for the
central regulatory role of miRNA-mRNA interactions in crude
oil-induced cardiotoxicity during fish development. The
developmental toxicity mediated by miR-203a in PAHs has garnered
notable research attention. Magnuson et al (110) demonstrated that inhibition of
miR-203a in zebrafish embryos led to heart rate reduction and
pericardial edema, cardiac defects that mimic the developmental
toxicity induced by typical PAHs found in crude oil. This study
elucidated the molecular mechanism wherein miR-203a contributes to
cardiac developmental impairment by regulating key factors such as
VEGFA and fibrosis-associated genes including Krüppel-like factor
4.
As a key metabolic and immunomodulatory organ at the
maternal-fetal interface, the placenta serves indispensable roles
in mediating developmental toxicity through xenobiotic metabolism
and cytokine signaling networks. A study by Guo et al
(111) revealed that the
environmental toxin microcystin-leucine arginine can deliver
miR-377-3p through trophoblast cell-derived extracellular vesicles,
targeting and suppressing the nuclear receptor subfamily 6 group A
member 1 gene in macrophages. This suppression activates the
mammalian target of the mTOR/ribosomal protein S6 kinase β-1/sterol
regulatory element-binding protein signaling pathway, leading to M1
polarization and metabolic disruption in placental macrophages,
ultimately resulting in CHDs in future offspring. To the best of
our knowledge, this study was the first to elucidate the role of
miRNA-mediated placenta-heart axis signaling in environmentally
induced cardiac developmental toxicity, providing novel evidence
for miRNA-regulated transgenerational mechanisms in heart
development.
NcRNAs have been demonstrated to serve as key
regulatory molecules in cardiac developmental toxicity induced by
numerous toxicants, exhibiting marked potential as biomarkers and
therapeutic targets (96,101,110,111). However, current research exhibits
notable limitations. As on the one hand, studies have predominantly
focused on miRNAs, while investigations into the mechanisms of
lncRNAs, circRNAs, small interfering RNAs and Piwi-interacting RNAs
in cardiac developmental toxicity remain markedly insufficient. On
the other hand, the interaction networks and synergistic regulatory
mechanisms among different ncRNA classes are yet to be elucidated,
hindering a comprehensive understanding of ncRNA-mediated
developmental toxicity. Future studies should aim to broaden the
scope of ncRNA research and deepen the exploration of regulatory
relationships among numerous ncRNAs to achieve a more systematic
mechanistic understanding.
m6A methylation
N6-methyladenosine (m6A), initially identified in
eukaryotic messenger RNAs in 1974, represents the methylation of
adenine at the N6 position (112). As the primary internal RNA
modification in eukaryotes, m6A occurs at an average frequency of
1–2 modifications per 1,000 nucleotides (113). Among >170 identified RNA
modifications, m6A is the most abundant and dynamically reversible
epigenetic mark in eukaryotic mRNAs, constituting approximately
one-half all methylated ribonucleotides (114).
The m6A modification system achieves dynamic and
reversible regulation through three functionally distinct classes
of regulatory proteins, namely writers, erasers and readers
(115). Methyltransferases such
as the methyltransferase-like (METTL)-3/METTL14 complex act as
‘writers’, catalyzing m6A RNA modifications (116). In 2011, two demethylases, fat
mass and obesity-associated protein (FTO) and AlkB homolog 5
(ALKBH5), were discovered as ‘erasers’ that remove these methyl
groups (117,118). This finding revealed the dynamic
reversibility of m6A modifications. Notably, both FTO and ALKBH5
belong to the α-ketoglutarate-dependent dioxygenase AlkB homolog
family, highlighting their conserved role in regulating RNA
methylation plasticity (119).
Simultaneously, m6A reader proteins, functioning as recognition
factors, regulate RNA metabolism processes (including splicing,
folding, transport, degradation and translation) through their
specific recognition of m6A-modified sites (114). Together, these core regulatory
components form an integrated m6A modification network, serving not
only as key tools for studying RNA methylation mechanisms but also
emerging as a key research focus in molecular biology due to their
important roles in diverse physiological and pathological
processes.
With regard to m6A methylation dysregulation-induced
cardiac developmental toxicity, the developmental toxicity mediated
by m6A methylation has gradually attracted researchers' attention
(120,121). m6A methylation dynamically
regulates post-transcriptional processes (such as mRNA stability
and translation efficiency) of myocardial development genes.
Dysregulation of this epigenetic mechanism may contribute to
congenital heart malformations. Bisphenol A (BPA) is a
representative industrial chemical used in the production of
polycarbonate plastics and epoxy resins (122). It has been established that BPA
exposure is associated with numerous health disorders, including
metabolic dysregulation, reproductive impairment, cardiovascular
abnormalities, developmental malformations and mammary
tumorigenesis (123). Given the
potential hazards of BPA, bisphenol C (BPC) has been adopted as an
alternative in industrial applications. The presence of BPC is
commonly identified in multiple human biospecimens, particularly
infant urine, indicating a potential exposure hazard for young
children (124). However, whether
BPC exerts toxic effects on embryonic development remains poorly
characterized, highlighting the need for further toxicological
evaluations. Su et al (125) demonstrated that BPC disrupts m6A
modification homeostasis by suppressing the expression of m6A
methyltransferase METTL3. This leads to decreased m6A methylation
levels in mRNAs encoding key cardiac developmental regulators,
including acyl-CoA oxidase 1 involved in fatty acid metabolism and
troponin T2d, key in myocardial contraction. The impaired m6A
modification compromises transcript recognition and stabilization
by m6A reader protein insulin-like growth factor 2 mRNA-binding
protein 2b, ultimately causing structural and functional cardiac
abnormalities in zebrafish. This work reveals environmental
pollutants can interfere with cardiac development by subverting
m6A-mediated epitranscriptomic regulation, identifying novel
molecular targets for evaluating developmental toxicity of
bisphenol analogs.
In addition to the aforementioned DNA methylation
alterations discussed, PM2.5 exposure-induced cardiac developmental
toxicity may also involve dysregulation of m6A methylation. A study
by Ji et al (126) using a
zebrafish larval model revealed that extractable organic matter
from PM2.5 activates the aryl hydrocarbon receptor (AHR), leading
to direct suppression of m6A methyltransferase transcripts METTL14
and METTL3. This resulted in global reduction of cardiac m6A
methylation levels, consequently decreasing m6A modifications while
increasing expression of apoptosis-related genes (TNF receptor
associated factor 4 and BCL2 binding component 3), ultimately
triggering excessive ROS production, apoptosis and cardiac
developmental malformations. These results establish that
AHR-mediated disruption of m6A methylation constitutes a central
mechanism underlying PM2.5-induced cardiac developmental toxicity.
Importantly, supplementation with the methyl donor betaine or
overexpression of METTL14 and METTL3 was found to restore m6A
homeostasis and ameliorate PM2.5-induced cardiotoxicity, thereby
providing key experimental evidence for therapeutic interventions
against environmental pollutant-associated cardiac developmental
defects.
Notable knowledge gaps persist in understanding the
functional mechanisms of m6A RNA methylation during cardiac
developmental toxicity. Although existing studies have established
the critical roles of ‘writers’ such as METTL3/METTL14 in
toxin-induced cardiac malformations, the target-specific
recognition mechanisms of ‘readers’ (such as the insulin-like
growth factor 2 mRNA binding protein family) and the dynamic
regulatory functions of ‘erasers’ (such as FTO/ALKBH5) remain
poorly understood (125,126). Furthermore, the upstream
molecular mechanisms by which environmental pollutants (including
BPC and PM2.5) specifically interfere with m6A-modifying enzyme
activities are yet to be elucidated, although proteomics and
co-immunoprecipitation techniques may help identify their direct
molecular targets. Additionally, the interactions between m6A and
other RNA modifications, such as 5-methylcytosine (m5C) and
N7-methylguanosine (m7G), and their synergistic effects in cardiac
developmental toxicity warrant further investigation (127). Future research should aim to
integrate high-throughput sequencing technologies to
comprehensively delineate the spatiotemporal dynamics of m6A
methylation in cardiac developmental toxicity.
Chromatin accessibility
Chromatin is a dynamic nucleoprotein complex
residing in eukaryotic nuclei, comprising three core components,
namely genomic DNA, histone proteins and non-histone proteins, with
the additional potential association of small RNA molecules
(128). As the organizational
form of eukaryotic genetic material, chromatin facilitates genetic
information storage and transcriptional regulation through its
distinctive three-dimensional architecture, while demonstrating
dynamic spatiotemporal variations in both molecular composition and
structural organization. Chromatin accessibility allows genomic DNA
regions to be recognized and bound by nuclear factors. This
biochemical characteristic is principally governed by local
nucleosome positioning dynamics and competitive binding
interactions among DNA-associated proteins (129–131). Key cellular activities, including
transcription factor-dependent gene regulation, scheduled DNA
replication and repair of genomic damage, all depend on proper
chromatin accessibility (132,133). Research findings demonstrate that
fluctuating chromatin accessibility patterns serve key regulatory
functions across multiple biological pathways, encompassing
senescence, immune system regulation and embryogenesis (134–136). The Assay for
Transposase-Accessible Chromatin using sequencing (ATAC-seq)
enables efficient detection of cell type-specific chromatin
accessibility features and their dynamic alterations under
perturbations or disease conditions, while simultaneously
facilitating genome-wide analysis of transcription factor binding
sites and comparative assessment of chromatin landscapes across
biological contexts (137).
With regard to chromatin accessibility
dysregulation-induced cardiac developmental toxicity, as advances
in epigenetics have occurred, research on the association between
chromatin accessibility and cardiac developmental toxicity is
gradually emerging. A previous study demonstrated that parental
zebrafish exposure to 10 µg/l perfluorobutane sulfonate (PFBS)
disrupts the maternal transmission of transcripts related to
histone-DNA interactions in offspring oocytes, suggesting that this
contaminant may impair transcription factor accessibility to DNA by
altering chromatin compaction (138). Notably, exposure to 100 µg/l PFBS
further results in reduced embryonic heart rates, providing
evidence for PFBS-induced dysregulation of chromatin accessibility
to be associated with cardiac developmental toxicity. Unlike the
mechanistic inference from zebrafish models, experiments in human
stem cells directly demonstrated the chromatin accessibility
alterations that lead to cardiac developmental defects through
ATAC-seq analysis. Liu et al (139) employed ATAC-seq in human induced
pluripotent stem cells and hESCs, demonstrating that
13-cis-retinoic acid (isotretinoin; INN) disrupts cardiac
development by altering chromatin accessibility. This perturbation
leads to aberrant enhancement of transcription factor binding
activities, including hepatocyte nuclear factor 1β, SRY-box
transcription factor 10 and nuclear factor I C. This subsequently
disrupts the normal function of TGF-β and Wnt signaling pathways
and dysregulates the expression of key mesodermal differentiation
genes, such as Eomesodermin, Mix paired-like homeobox 1 and
Dickkopf WNT signaling pathway inhibitor 1. These findings
systematically elucidate the epigenetic mechanisms underlying
INN-induced cardiac developmental toxicity at the human stem cell
level, providing key evidence for drug safety evaluation.
Investigations into toxin-induced perturbations of
cardiac development mediated through chromatin accessibility are
only just emerging. Contemporary research proposes that exposure to
environmental toxicants can disrupt physiological transcription
factor binding to chromatin, potentially inducing aberrant
expression of cardiac developmental gene programs (139). However, these findings only
indicate associations, lacking direct experimental validation
regarding the functionality of open chromatin regions or the
regulatory roles of upstream mechanisms, such as DNA methylation
and histone modifications, in shaping chromatin accessibility.
Furthermore, existing studies predominantly rely on static, in
vitro models at single time-points, failing to capture the
dynamic progression from cardiac mesoderm to cardiomyocyte
differentiation. Limitations in multi-omics approaches [such as
underutilization of WGBS, chromatin immunoprecipitation followed by
sequencing (ChIP-seq)] further hinder systematic dissection of the
epigenetic regulatory network (140). Integrating gene-editing
technologies with model organisms such as zebrafish will enable
more precise elucidation of the mechanisms by which potential
toxicants disrupt cardiac development through chromatin
remodeling.
Epigenetic crosstalk in cardiac
developmental toxicity
Advancements in epigenetic research, particularly
regarding DNA methylation and histone modifications, have
highlighted their intricate interplay and well-coordinated
regulatory networks (141–143).
Although direct evidence concerning the crosstalk among epigenetic
modifications in cardiac developmental toxicity remains scarce,
emerging studies provide key mechanistic theories (144–146). Epidemiological data demonstrate a
dose-dependent association between maternal caffeine intake and
adverse pregnancy outcomes, including spontaneous abortion, low
birth weight and congenital cardiac/genital anomalies (147). Experimental investigations
consistently reveal that in utero caffeine exposure disrupts
embryonic cardiac function, leading to developmental cardiotoxicity
(144,145). Of note, Fang et al
(146) demonstrated that
gestational caffeine exposure markedly altered both DNA methylation
patterns and the expression of histone modification regulators in
murine embryonic ventricles, concurrent with the dysregulation of
cardiac-specific miRNAs (miR-208a/b and miR-499). These findings
collectively suggest that synergistic interactions among DNA
methylation, histone modifications and ncRNAs may drive cardiac
maldevelopment through disruption of epigenetic homeostasis.
However, further systems-level investigations integrating
multi-omics profiling and functional validation are key for
delineating precise molecular interactions underpinning this
regulatory hierarchy and establish a comprehensive epigenetic
framework for cardiac developmental toxicity.
Advances in epigenetic research methods and
models for cardiac developmental toxicity
Integrative multi-omics studies: From
single-modification analysis to multi-omics convergence
Advancements in high-throughput sequencing now
permit epigenetic evaluation of cardiac developmental toxicity
through multi-omics integration, overcoming the previous
limitations of single-omics analyses. By contrast with traditional
single-epigenetic-marker detection, multidimensional omics
technologies integrating DNA methylation (WGBS/MeDIP-Seq), histone
modifications (ChIP-Seq), chromatin accessibility (ATAC-Seq), m6A
methylation (methylated RNA immunoprecipitation followed by
sequencing) and ncRNAs [RNA sequencing (RNA-Seq)] enable systematic
elucidation of toxicity-induced epigenetic reprogramming networks
and their regulatory effects on cardiac development (Fig. 2A). For instance, the integrated
multi-omics analysis of RNA-Seq and MeDIP-Seq not only enables
simultaneous detection of DNA methylation modifications and gene
expression changes, but also effectively establishes their causal
associations (30). This
methodology conclusively establishes that toxin-induced cardiac
developmental toxicity primarily arises through DNA
methylation-directed modulation of gene transcription processes.
Furthermore, in combined ChIP-seq and ATAC-seq analyses,
researchers can first map the binding sites of cardiac
developmental transcription factors using ChIP-seq, and then
identify functionally active regions within open chromatin at these
binding sites through ATAC-seq (148). This strategy accurately
identifies core regulatory elements disrupted by toxicants,
demonstrating that cardiac developmental impairment occurs through
direct interference with transcription factor binding and
diminished chromatin accessibility. This comprehensive approach
provides novel mechanistic insights into cardiac developmental
toxicity. Furthermore, it enables the advancement of epigenetic
biomarkers from individual markers to diagnostic signature
profiles, thereby potentially improving both the specificity and
sensitivity of toxicity risk assessment.
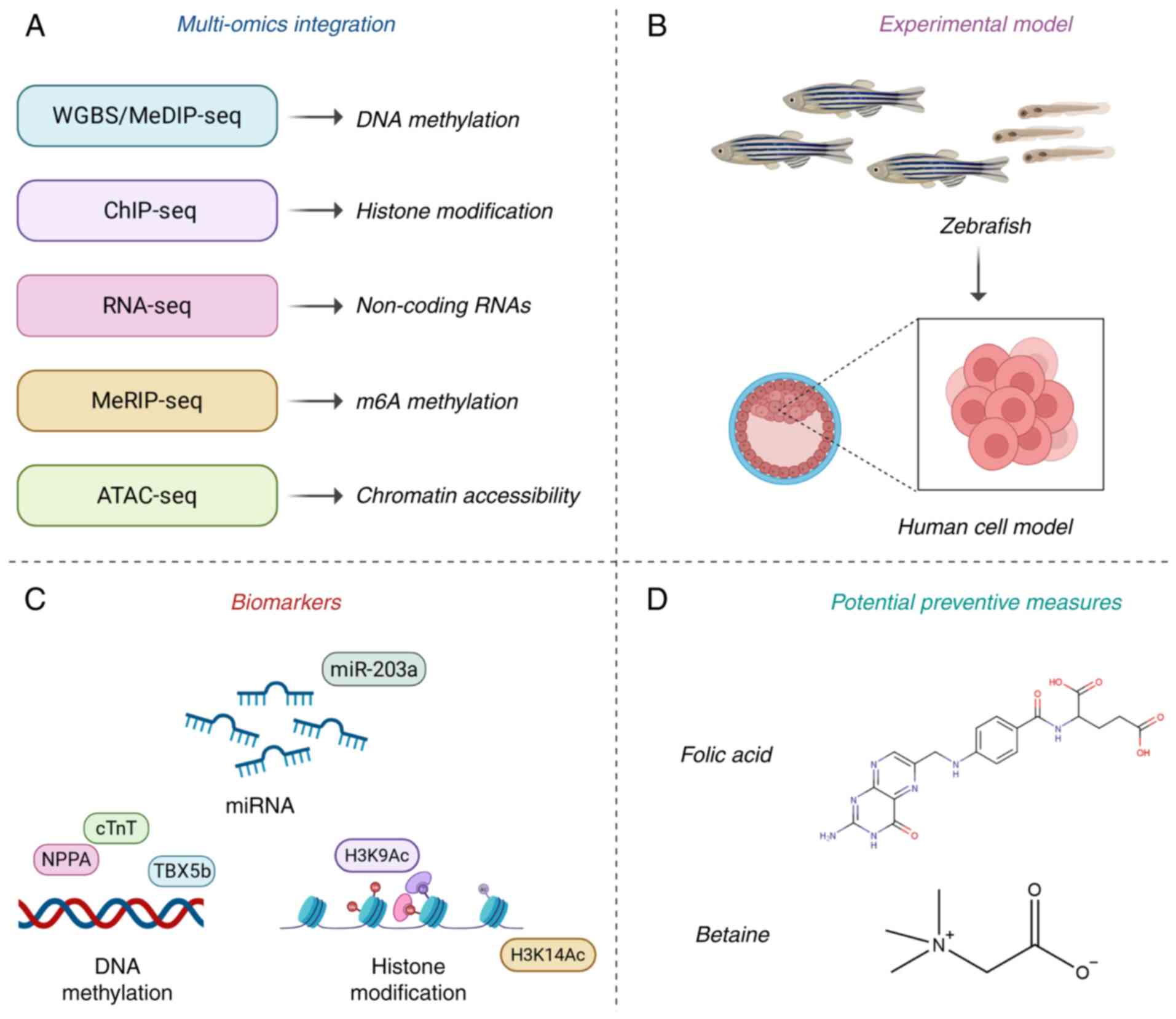 | Figure 2.Integrative application of
multi-omics technologies and models in cardiac developmental
toxicity studies. (A) Multi-omics integration of epigenetic
regulation in cardiac developmental toxicity research. (B) Research
progress in experimental models of cardiac developmental toxicity.
(C) Epigenetic biomarkers identified in cardiac developmental
toxicity research. (D) Preliminary measures for preventing cardiac
developmental toxicity through epigenetic regulation. WGBS,
whole-genome bisulfite sequencing; MeDIP-Seq, methylated DNA
immunoprecipitation sequencing; ChIP-Seq, chromatin
immunoprecipitation followed by sequencing RNA-Seq, RNA sequencing;
MeRIP-Seq, methylated RNA immunoprecipitation followed by
sequencing; ATAC-Seq, Assay for Transposase-Accessible Chromatin
using sequencing; NPPA, natriuretic peptide A; cTnT, cardiac
troponin T2; TBX5b, T-box transcription factor 5b; miR, microRNA;
H3K, histone H3 lysine K. The figure was created using
BioRender.com. |
Cardiac developmental toxicity models:
From zebrafish to human cell-based systems
In cardiac developmental toxicity research,
zebrafish and human cell-based models represent widely utilized
experimental systems (Fig. 2B). In
contemporary developmental biology research, zebrafish serve as a
primary animal model for heart formation studies (149). Notably, the cardiac structure in
zebrafish demonstrates marked conservation with mammalian hearts,
displaying ~70% genetic homology to human cardiac genes (150). Embryonic heart development
follows a well-defined chronological sequence. First, cardiac
progenitor cells begin to differentiate by 5 hpf, forming a linear
heart tube by 16 hpf (151–153). Subsequently, key morphological
changes occur, including cardiac looping into an S-shape (~33 hpf),
initiation of regular contractions (~36 hpf) and formation of
primitive valves (~40 hpf). By 2 days post-fertilization (dpf),
distinct inner and outer curvatures become evident in the
ventricular region (154).
Additionally, zebrafish embryos remain optically transparent until
3–4 dpf, enabling non-invasive real-time visualization and dynamic
monitoring of chemical effects on heart development. These
features, combined with high-throughput screening technologies,
markedly enhance the efficiency of developmental toxicity
assessments. Zebrafish exhibit good reproductive capacity, with
adult individuals capable of year-round cyclical spawning,
producing <200 eggs per female weekly (155). From an ethical standpoint, the
early life stages of zebrafish are considered to experience minimal
pain or distress upon chemical exposure, more closely aligning with
animal welfare guidelines and reducing ethical concerns in
experimental studies (156).
However, this model also presents a number of limitations. Compared
with mammalian models (including mice), zebrafish possess fewer
biological resources such as antibodies and gene-editing tools,
restricting their application in protein biochemistry and related
fields. The cardiac architecture of zebrafish is composed of two
distinct chambers (an atrium and ventricle), differing from the
four-chambered organization typical of mammals. This structural
disparity restricts its effectiveness as a model for investigating
cardiac septation processes or sophisticated hemodynamic parameters
(152). Additionally, zebrafish
embryos primarily absorb test compounds through cutaneous exposure,
differing from common human exposure routes such as oral ingestion
or inhalation (157).
Furthermore, the absence of distinct sex differentiation markers
during early developmental stages hinders the assessment of
sex-specific toxicological effects (158).
Overall cardiac organogenesis relies on animal
models to investigate morphogenesis and systemic integration, yet
early key events (such as stem cell differentiation and cardiac
progenitor fate determination) can be effectively studied using
pluripotent stem cells (PSCs) and in vitro models (159). Through directed differentiation
of ESCs or induced PSCs (iPSCs), researchers can systematically
recapitulate key stages of cardiac development. Specifically,
BMP/Wnt signaling activation initially induces mesoderm formation
and then, under defined growth factors (such as activin A and BMP4)
combined with Wnt pathway inhibition, drives further
differentiation into cardiac progenitor cells, ultimately yielding
functionally contractile cardiomyocytes (159). This process closely mimics the
spatiotemporal features of embryonic heart development, providing
an ideal platform for exploring cardiac developmental mechanisms
and drug toxicity screening (79).
However, this technology exhibits inherent limitations. First, the
differentiation efficiency of ESCs/iPSCs into cardiomyocytes
remains relatively low (typically 1–3%) (159), often resulting in heterogeneous
cellular populations containing undesired non-cardiac lineages that
may compromise experimental reliability (160). Second, the in vitro
culture system cannot fully recapitulate the dynamic regulatory
network governing cardiac development in vivo, thereby
constraining its applicability for comprehensive organ-level
studies. Consequently, this model system may be more suited towards
mechanistic investigations at cellular and molecular levels rather
than integrated organ-scale analyses.
Human cardiac organoids (hCOs) represent a
biomimetic three-dimensional culture model that effectively
recapitulates key aspects of heart development and drug toxicity
responses. These systems are composed of multiple cell types,
including cardiomyocytes, endothelial cells and stromal cells,
establishing a physiologically relevant microenvironment that
surpasses conventional two-dimensional cultures in functional
evaluation (161). hCOs exhibit
mature functional properties, such as spontaneous contractility and
stable electrophysiological signals, enabling simultaneous
assessment of drug effects on cardiomyocyte proliferation and
function. However, it should be noted that hCOs more closely mimic
the postnatal cardiac physiology, limiting their applicability for
toxicity prediction during early embryonic development (162). Additionally, challenges remain,
including prolonged culture duration, technical complexity and
insufficient standardization, which warrant further optimization to
enhance the reproducibility and scalability of the hCO model.
Epigenetic discoveries elucidating cardiac
developmental toxicity
Identification of epigenetic
biomarkers in cardiac developmental toxicity
Biomarkers represent measurable biological
parameters that objectively indicate physiological processes,
pathological conditions or pharmacological responses, making them
important tools for disease screening, accurate diagnosis and
reliable prognostic assessment (163). Advances in omics technologies,
particularly high-throughput genomic profiling, have enabled
systematic identification of candidate biomarkers while providing
novel approaches to elucidate disease mechanisms. Epigenetic
markers have gained particular prominence as promising
next-generation biomarkers, given their key role in regulating
disease pathogenesis (7).
In the investigation of cardiac developmental
toxicity, a number of epigenetic biomarkers have been preliminarily
identified as serving as early warning indicators of toxicity
(Fig. 2C). Notably, miR-203a has
been consistently shown to be downregulated during cardiac toxicity
events and zebrafish embryos treated with miR-203a inhibitors
exhibit characteristic cardiac malformations, suggesting its
potential as a biomarker for assessing developmental cardiotoxicity
induced by numerous toxicants (101,102,110). At the DNA methylation level,
hypomethylation of NPPA and cTnT genes, along with hypermethylation
at the TBX5b promoter region, may serve as epigenetic signatures
for phthalate-induced cardiac defects (30). Regarding histone modifications,
reduced H3K9Ac and H3K14Ac levels appear to represent key
epigenetic biomarkers for triazole fungicide (FBZ-induced)
transgenerational cardiotoxicity (68). However, current limitations include
insufficient mechanistic validation of these biomarkers and their
specificity to particular toxicants. This further compromises their
general applicability. Therefore, establishing standardized
epigenetic toxicity assessment systems will markedly facilitate the
identification and application of developmental cardiotoxicity
biomarkers.
Preliminary exploration of potential
preventive measures
Based on current research findings, numerous
candidate drugs have been identified that may exhibit protective
effects against cardiac developmental toxicity (Fig. 2D). Folic acid, a key water-soluble
B vitamin (vitamin B9), cannot be synthesized endogenously and must
be obtained through dietary sources such as leafy green vegetables
and citrus fruits (164). This
biological component has key functions in important cellular
activities such as nucleotide biosynthesis and DNA methylation
processes (165). Research
demonstrates that folic acid, as a vital nutritional factor,
exhibits notable efficacy in preventing cardiac and neurological
developmental defects during embryogenesis (166). Additionally, folic acid has shown
promising potential in exploring epigenetic mechanisms for
preventing cardiac malformations, particularly through DNA
methylation-related pathways. Research has demonstrated that folic
acid supplementation corrects methylation imbalances by restoring
the SAM/SAH ratio and modulating DNMT expression, consequently
ameliorating cardiac malformations (25). This discovery provides a potential
intervention strategy against cardiac developmental toxicity
induced by environmental pollutants such as PM2.5. Similarly,
evidence shows that folate effectively alleviates selenite-induced
genomic methylation disorders and markedly reduces the incidence of
cardiac developmental anomalies, indicating its preventive role
against developmental toxicity through methylation reprogramming
regulation.
Betaine, also known as trimethylglycine, derives its
name from its structural similarity to glycine but with three
additional methyl groups (167).
As a safe and stable natural compound, betaine primarily serves as
a methyl donor in biological systems, participating in
transmethylation reactions and serving a key role in SAM synthesis
(168). SAM, a key cellular
methyl donor, is involved in diverse epigenetic modification
processes, including m6A RNA methylation. By promoting SAM
production, betaine indirectly modulates m6A methylation levels,
thereby influencing gene expression, RNA metabolism, and cellular
functions regulating glucolipid metabolism (168–170). Previous research has demonstrated
that betaine supplementation restores global m6A methylation levels
in the heart, mitigating PM2.5-induced cardiac developmental
toxicity by reducing ROS overproduction, mitochondrial dysfunction
and cell death (126). These
findings provide important evidence for preventing cardiac
developmental toxicity through epigenetic regulatory mechanisms,
although the discussed evidence primarily focuses on DNA
methylation and m6A RNA methylation regulation. Future
investigations should aim to examine more thoroughly the
therapeutic implications of addressing supplementary epigenetic
regulators, such as histone marks, ncRNA-mediated control and DNA
accessibility patterns, in developmental cardiotoxicity contexts.
Currently, the majority of evidence originates from preclinical
studies (cell-based and animal models), whereas clinical validation
in humans remains limited. Unresolved issues, such as optimal
dosage, administration timing and potential adverse effects,
require systematic investigation before this approach can progress
to clinical application.
Conclusion
Environmental pollutants and pharmaceuticals have
become a growing concern owing to their widespread potential to
induce embryonic cardiac developmental toxicity. Epigenetic marks
demonstrate notable dynamic plasticity during embryonic heart
development, with their modification patterns subject to regulation
by numerous environmental factors such as nutritional status,
maternal exposure to environmental contaminants and pharmacological
agents. The present review focuses on epigenetic regulatory
mechanisms and elucidates the molecular pathways through which
specific pollutants and chemicals mediate cardiac developmental
toxicity through epigenetic modifications. In addition, potential
epigenetic biomarkers and preventive pharmaceuticals are
summarized, which may contribute to monitoring embryonic cardiac
developmental toxicity and implementing preventive interventions
(25,68,110,126).
The current understanding of epigenetic mechanisms
underlying cardiac developmental toxicity remains limited, with
numerous key knowledge gaps yet to be resolved. First, the
establishment of causality remains inadequate. The majority of
studies have only observed associations between epigenetic
modifications (such as DNA methylation) and cardiac malformations,
lacking direct experimental evidence demonstrating that specific
modifications drive the toxic phenotypes (42,44,48).
This gap in causality markedly restricts the translational
applicability of existing findings. Second, systematic research
into epigenetic networks is lacking. Although crosstalk exists
among numerous epigenetic modifications, the mechanisms by which
potential toxicants regulate these interactions to induce cardiac
developmental defects remain poorly understood. In addition,
cardiac development is a continuous and precisely timed process,
yet current studies tend to employ static analytical approaches,
examining epigenetic changes at isolated time-points only.
Consequently, they fail to elucidate how pollutant-induced
epigenetic reprogramming dynamically evolves across different
developmental stages (such as cardiac tube formation and
atrioventricular septation) or its stage-dependent impact on
developmental progression. Furthermore, the number of identified
epigenetic biomarkers remains limited and exhibits insufficient
specificity, hindering their utility in clinical diagnosis and
environmental monitoring (30,68,110). Drug development targeting
epigenetic regulation for the prevention of cardiac developmental
defects is notably underdeveloped. Although methyl donors such as
folate and betaine have demonstrated protective effects in a number
of studies, their actions lack specificity and may inadvertently
interfere with normal epigenetic regulation during development
(23,119).
Future research should aim to integrate multi-omics
and epigenomic profiling technologies with CRISPR-based functional
validation to systematically decipher the molecular mechanisms by
which environmental exposures (such as pharmaceuticals and
pollutants) perturb cardiac development through epigenetic
regulation. Additionally, it is important to strengthen the
‘exposure-epigenetics-phenotype’ evidence chain by precisely
defining key time windows and dose-effect relationships. The
development of AI-driven high-throughput biomarker screening
platforms may help identify diagnostic molecular signatures and
facilitate the exploration of targeted epigenetic interventions.
These strategies will enhance early detection and prevention of
embryonic cardiac developmental toxicity, providing a strong
scientific foundation for translational applications.
Acknowledgements
Not applicable.
Funding
The present review was supported by the Talent Development
Program of the Second Affiliated Hospital of Tianjin University of
Traditional Chinese Medicine (grant no. YC-FY202303) and the
National Natural Science Foundation of China (grant no.
8207141582).
Availability of data and materials
Not applicable.
Authors' contributions
ZQ and RC drafted the manuscript and prepared the
figures and tables. RC and DS edited and revised the manuscript.
All the authors read and approved the final version of the
manuscript. Data authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Mahler GJ and Butcher JT: Cardiac
developmental toxicity. Birth Defects Res C Embryo Today.
93:291–297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hoffman JI and Kaplan S: The incidence of
congenital heart disease. J Am Coll Cardiol. 39:1890–1900. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hoffman JI: The global burden of
congenital heart disease. Cardiovasc J Afr. 24:141–145. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hoffman JI: Incidence of congenital heart
disease: II. Prenatal incidence. Pediatr Cardiol. 16:155–165. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sun R, Liu M, Lu L, Zheng Y and Zhang P:
Congenital heart disease: Causes, diagnosis, symptoms, and
treatments. Cell Biochem Biophys. 72:857–860. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jenkins KJ, Correa A, Feinstein JA, Botto
L, Britt AE, Daniels SR, Elixson M, Warnes CA and Webb CL; American
Heart Association Council on Cardiovascular Disease in the Young, :
Noninherited risk factors and congenital cardiovascular defects:
Current knowledge: A scientific statement from the American heart
association council on cardiovascular disease in the young:
Endorsed by the American academy of pediatrics. Circulation.
115:2995–3014. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
de Oliveira DT and Guerra-Sá R: Uncovering
epigenetic landscape: A new path for biomarkers identification and
drug development. Mol Biol Rep. 47:9097–9122. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Maher N, Maiellaro F, Ghanej J, Rasi S,
Moia R and Gaidano G: Unraveling the epigenetic landscape of mature
B cell neoplasia: Mechanisms, biomarkers, and therapeutic
opportunities. Int J Mol Sci. 26:81322025. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Leduque B, Edera A, Vitte C and Quadrana
L: Simultaneous profiling of chromatin accessibility and DNA
methylation in complete plant genomes using long-read sequencing.
Nucleic Acids Res. 52:6285–6297. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi SW and Friso S: Epigenetics: A new
bridge between nutrition and health. Adv Nutr. 1:8–16. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Samanta S, Rajasingh S, Cao T, Dawn B and
Rajasingh J: Epigenetic dysfunctional diseases and therapy for
infection and inflammation. Biochim Biophys Acta Mol Basis Dis.
1863:518–528. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Smith ZD and Meissner A: DNA methylation:
Roles in mammalian development. Nat Rev Genet. 14:204–220. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang H, Lang Z and Zhu JK: Dynamics and
function of DNA methylation in plants. Nat Rev Mol Cell Biol.
19:489–506. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gowher H, Liebert K, Hermann A, Xu G and
Jeltsch A: Mechanism of stimulation of catalytic activity of Dnmt3A
and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol
Chem. 280:13341–13348. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hermann A, Goyal R and Jeltsch A: The
Dnmt1 DNA-(cytosine-C5)-methyltransferase methylates DNA
processively with high preference for hemimethylated target sites.
J Biol Chem. 279:48350–48359. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bansal A and Pinney SE: DNA methylation
and its role in the pathogenesis of diabetes. Pediatr Diabetes.
18:167–177. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bochtler M, Kolano A and Xu GL: DNA
demethylation pathways: Additional players and regulators.
Bioessays. 39:1–13. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Strasenburg W, Borowczak J, Piątkowska D,
Jóźwicki J and Grzanka D: The role of DNA methylation and
demethylation in bladder cancer: A focus on therapeutic strategies.
Front Oncol. 15:15672422025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu F, Li K, Li S, Liu J, Zhang Y, Zhou M,
Zhao H, Chen H, Wu N, Liu Z and Su J: CFEA: A cell-free epigenome
atlas in human diseases. Nucleic Acids Res. 48((D1)): D40–D44.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wu SC and Zhang Y: Active DNA
demethylation: Many roads lead to rome. Nat Rev Mol Cell Biol.
11:607–620. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li X, Cui J, Zhang X, Wu Y, Xu J, Zhao Y,
Hussain S and Chen L: Integrated analysis of methylomic and
transcriptomic profiles in fetal mouse hypothalamus in response to
maternal gestational exposure to arsenic. Ecotoxicol Environ Saf.
299:1184002025. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Demond H, Khan S, Castillo-Fernandez J,
Hanna CW and Kelsey G: Transcriptome and DNA methylation profiling
during the NSN to SN transition in mouse oocytes. BMC Mol Cell
Biol. 26:22025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Jiang Y, Li J, Ren F, Ji C, Aniagu S and
Chen T: PM2.5-induced extensive DNA methylation changes in the
heart of zebrafish embryos and the protective effect of folic acid.
Environ Pollut. 255:1133312019. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Qiu M, Chen J, Liu M, Shi Y, Nie Z, Dong
G, Li X, Chen J, Ou Y and Zhuang J: Reprogramming of DNA
methylation patterns mediates perfluorooctane sulfonate-induced
fetal cardiac dysplasia. Sci Total Environ. 919:1709052024.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cai J, Zhao Y, Liu P, Xia B, Zhu Q, Wang
X, Song Q, Kan H and Zhang Y: Exposure to particulate air pollution
during early pregnancy is associated with placental DNA
methylation. Sci Total Environ. 607–608. 1103–1108. 2017.
|
|
28
|
Heudorf U, Mersch-Sundermann V and Angerer
J: Phthalates: Toxicology and exposure. Int J Hyg Environ Health.
210:623–634. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang Y and Qian H: Phthalates and their
impacts on human health. Healthcare (Basel). 9:6032021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mu X, Chen X, Liu J, Yuan L, Wang D, Qian
L, Qian Y, Shen G, Huang Y, Li X, et al: A multi-omics approach
reveals molecular mechanisms by which phthalates induce cardiac
defects in zebrafish (danio rerio). Environ Pollut. 265((Pt B)):
1138762020. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Thangavel P, Park D and Lee YC: Recent
insights into particulate matter (PM2.5)-mediated
toxicity in humans: An overview. Int J Environ Res Public Health.
19:75112022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cai J, Shen Y, Meng X, Zhao Y, Niu Y, Chen
R, Du W, Quan G, Barnett AL, Jones G, et al: Association of
developmental coordination disorder with early-life exposure to
fine particulate matter in chinese preschoolers. Innovation (Camb).
4:1003472022.PubMed/NCBI
|
|
33
|
Lin CA, Pereira LA, Nishioka DC, Conceição
GM, Braga AL and Saldiva PH: Air pollution and neonatal deaths in
São Paulo, Brazil. Braz J Med Biol Res. 37:765–770. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Gouveia N, Bremner SA and Novaes HM:
Association between ambient air pollution and birth weight in Sao
Paulo, Brazil. J Epidemiol Community Health. 58:11–17. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li L, Zhang B, Li L and Borthwick AGL:
Microbial selenate detoxification linked to elemental sulfur
oxidation: Independent and synergic pathways. J Hazard Mater.
422:1269322022. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lin JH, Liu CC, Liu CY, Hsu TW, Yeh YC,
How CK, Hsu HS and Hung SC: Selenite selectively kills lung
fibroblasts to treat bleomycin-induced pulmonary fibrosis. Redox
Biol. 72:1031482024. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hariharan S, Chauhan S, Marcharla E,
Alphonse CRW, Rajaretinam RK and Ganesan S: Developmental toxicity
and neurobehavioral effects of sodium selenite and selenium
nanoparticles on zebrafish embryos. Aquat Toxicol. 266:1067912024.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Imai T, Kurihara T, Esaki N and Mihara H:
Glutathione contributes to the efflux of selenium from hepatoma
cells. Biosci Biotechnol Biochem. 78:1376–1380. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ma Y, Wu M, Li D, Li XQ, Li P, Zhao J, Luo
MN, Guo CL, Gao XB, Lu CL and Ma X: Embryonic developmental
toxicity of selenite in zebrafish (Danio rerio) and prevention with
folic acid. Food Chem Toxicol. 50:2854–2863. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Ganie SY, Javaid D, Hajam YA and Reshi MS:
Arsenic toxicity: Sources, pathophysiology and mechanism. Toxicol
Res (Camb). 13:tfad1112023. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cantoni O, Zito E, Fiorani M and
Guidarelli A: Arsenite impinges on endoplasmic
reticulum-mitochondria crosstalk to elicit mitochondrial ROS
formation and downstream toxicity. Semin Cancer Biol. 76:132–138.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li D, Lu C, Wang J, Hu W, Cao Z, Sun D,
Xia H and Ma X: Developmental mechanisms of arsenite toxicity in
zebrafish (Danio rerio) embryos. Aquat Toxicol. 91:229–237. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Hrelia P, Vigagni F, Maffei F, Morotti M,
Colacci A, Perocco P, Grilli S and Cantelli-Forti G: Genetic safety
evaluation of pesticides in different short-term tests. Mutat Res.
321:219–228. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Jiao F, Zhao Y, Limbu SM, Kong L, Zhang D,
Liu X, Yang S, Gui W and Rong H: Cyhexatin causes developmental
toxic effects by disrupting endocrine system and inducing
behavioral inhibition, apoptosis and DNA hypomethylation in
zebrafish (Danio rerio) larvae. Chemosphere. 339:1397692023.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kim J and Choi J: Trans- and
multigenerational effects of isothiazolinone biocide CMIT/MIT on
genotoxicity and epigenotoxicity in daphnia magna. Toxics.
11:3882023. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee H, Kim Y, Cho Y, Jeon EJ, Jeong SH,
Lee JH and Kim S: Nociceptive effects and gene alterations of
CMIT/MIT mixture in zebrafish embryos and larvae. J Hazard Mater.
493:1383922025. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Molinari F, Tranchida N, Inferrera F,
Fusco R, Faggio C, Impellitteri F, Cuzzocrea S, Cordaro M and Di
Paola R: Biocide mixture (CMIT/MIT) induces neurotoxicity through
the upregulation of the MAPKs signaling pathways. J Neurophysiol.
134:183–192. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chatterjee N, Lee H, Kim J, Kim D, Lee S
and Choi J: Critical window of exposure of CMIT/MIT with respect to
developmental effects on zebrafish embryos: multi-level endpoint
and proteomics analysis. Environ Pollut. 268((Pt A)): 1157842021.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kausar S, Abbas MN, Gul I, Liu R, Li Q,
Zhao E, Lv M and Cui H: Molecular identification of two DNA
methyltransferase genes and their functional characterization in
the anti-bacterial immunity of antheraea pernyi. Front Immunol.
13:8558882022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Castillo-Aguilera O, Depreux P, Halby L,
Arimondo PB and Goossens L: DNA methylation targeting: The DNMT/HMT
crosstalk challenge. Biomolecules. 7:32017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Liu L, Ni S, Zhang L, Chen Y, Xie M and
Huang X: Molecular insights and clinical implications of DNA
methylation in sepsis-associated acute kidney injury: A narrative
review. BMC Nephrol. 26:2532025. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zhang Y, Zhang Q, Zhang Y and Han J: The
role of histone modification in DNA replication-coupled nucleosome
assembly and cancer. Int J Mol Sci. 24:49392023. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Fagerli E, Escobar I, Ferrier FJ, Jackson
CW, Perez-Lao EJ and Perez-Pinzon MA: Sirtuins and cognition:
implications for learning and memory in neurological disorders.
Front Physiol. 13:9086892022. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang J, Liu H, Liu Y, Luo E and Liu S:
Unlocking the potential of histone modification in regulating bone
metabolism. Biochimie. 227:286–298. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Qi Q, Li L, Liang H and Zeng Y: Role and
research progress of histone modification in cardiovascular
diseases (Review). Exp Ther Med. 30:1322025. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yang Y and Luan Y, Yuan RX and Luan Y:
Histone methylation related therapeutic challenge in cardiovascular
diseases. Front Cardiovasc Med. 8:7100532021. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Mao W, Wang B, Huang R, Sun Z, Yan M and
Dong P: Histone modifications in head and neck squamous cell
carcinoma. Front Oncol. 14:14277252024. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Wang K, Li Y, Qiang T, Chen J and Wang X:
Role of epigenetic regulation in myocardial ischemia/reperfusion
injury. Pharmacol Res. 170:1057432021. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zhang J, Huang H, Ding B, Liu Z, Chen D,
Li S, Shen T and Zhu Q: Histone demethylase KDM4A mediating
macrophage polarization: A potential mechanism of trichloroethylene
induced liver injury. Cell Biol Int. 48:1148–1159. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Kim J and Choi J: Histone
methylation-mediated reproductive toxicity to consumer product
chemicals in caenorhabditis elegans: An epigenetic adverse outcome
pathway (AOP). Environ Sci Technol. 58:19604–19616. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Mehta I, Verma M, Quasmi MN, Kumar D and
Jangra A: Emerging roles of histone modifications in environmental
toxicants-induced neurotoxicity. Toxicology. 515:1541642025.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wang Q, Sun S, Sun G, Han B, Zhang S,
Zheng X and Chen L: Histone modification inhibitors: an emerging
frontier in thyroid cancer therapy. Cell Signal. 131:1117032025.
View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Liu M, Chen B, Pei L, Zhang Q, Zou Y, Xiao
H, Zhou J, Chen L and Wang H: Decreased H3K9ac level of StAR
mediated testicular dysplasia induced by prenatal dexamethasone
exposure in Male offspring rats. Toxicology. 408:1–10. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Zhou J, Liu F, Yu L, Xu D, Li B, Zhang G,
Huang W, Li L, Zhang Y, Zhang W and Wang H: nAChRs-ERK1/2-egr-1
signaling participates in the developmental toxicity of nicotine by
epigenetically down-regulating placental 11β-HSD2. Toxicol Appl
Pharmacol. 344:1–12. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Pei LG, Zhang Q, Yuan C, Liu M, Zou YF, Lv
F, Luo DJ, Zhong S and Wang H: The GC-IGF1 axis-mediated testicular
dysplasia caused by prenatal caffeine exposure. J Endocrinol.
242:M17–M32. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Li Y, Dong F, Liu X, Xu J, Li J, Kong Z,
Chen X and Zheng Y: Environmental behavior of the chiral triazole
fungicide fenbuconazole and its chiral metabolites:
Enantioselective transformation and degradation in soils. Environ
Sci Technol. 46:2675–2683. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wu Y, Yang Q, Chen M, Zhang Y, Zuo Z and
Wang C: Fenbuconazole exposure impacts the development of zebrafish
embryos. Ecotoxicol Environ Saf. 158:293–299. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Zhang Y, Guo J, Tang C, Xu K, Li Z and
Wang C: Early life stage exposure to fenbuconazole causes
multigenerational cardiac developmental defects in zebrafish and
potential reasons. Environ Pollut. 349:1239382024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Madrid JV, Vera-Colón MKM and zur Nieden
NI: Perturbations in osteogenic cell fate following exposure to
constituents present in tobacco: A combinatorial study. Toxics.
11:9982023. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Prasad S, Kaisar MA and Cucullo L:
Unhealthy smokers: Scopes for prophylactic intervention and
clinical treatment. BMC Neurosci. 18:702017. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sabbagh HJ, Baghlaf KK, Jamalellail HMH,
Bakhuraybah AS, AlGhamdi SM, Alharbi OA, AlHarbi KM and Hassan MHA:
Environmental tobacco smoke exposure and non-syndromic orofacial
cleft: Systematic review and meta-analysis. Tob Induc Dis.
21:762023. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kharrazi M, DeLorenze GN, Kaufman FL,
Eskenazi B, Bernert JT Jr, Graham S, Pearl M and Pirkle J:
Environmental tobacco smoke and pregnancy outcome. Epidemiology.
15:660–670. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Deng C, Pu J, Deng Y, Xie L, Yu L, Liu L,
Guo X, Sandin S, Liu H and Dai L: Association between maternal
smoke exposure and congenital heart defects from a case-control
study in China. Sci Rep. 12:149732022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Macdonald-Wallis C, Tobias JH, Davey Smith
G and Lawlor DA: Parental smoking during pregnancy and offspring
bone mass at age 10 years: Findings from a prospective birth
cohort. Osteoporos Int. 22:1809–1819. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cheng W, Zhou R, Feng Y and Wang Y:
Mainstream smoke and sidestream smoke affect the cardiac
differentiation of mouse embryonic stem cells discriminately.
Toxicology. 357–358. 1–10. 2016.PubMed/NCBI
|
|
76
|
Rahbari A, Esmaielpour B, Azarmi R, Fatemi
H, Lajayer HM, Panahirad S, Gohari G and Vita F: Symbiotic fungus
serendipita indica as a natural bioenhancer against cadmium
toxicity in chinese cabbage. Plants (Basel). 14:27732025.PubMed/NCBI
|
|
77
|
Zhu Y, Guan H, Zhu X, Cai J, Jiao X, Shan
J, Li Y, Wu Q and Zhang Z: Astilbin antagonizes developmental
cardiotoxicity after cadmium exposure in chicken embryos by
inhibiting endoplasmic reticulum stress and maintaining calcium
homeostasis. Ecotoxicol Environ Saf. 270:1158472024. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Vasanthakumaran M, Ramesh M, Murugan K,
Hema T, Rajaganesh R and Hwang JS: Developmental toxicity,
biochemical and biomarker in the zebrafish (Danio rerio) embryo
exposed to biosynthesized cadmium oxide nanoparticles. Chemosphere.
369:1438512024. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Wu X, Chen Y, Luz A, Hu G and Tokar EJ:
Cardiac development in the presence of cadmium: An in vitro study
using human embryonic stem cells and cardiac organoids. Environ
Health Perspect. 130:1170022022. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Dasgupta A, Nandi S, Gupta S, Roy S and
Das C: To ub or not to ub: The epic dilemma of histones that
regulate gene expression and epigenetic cross-talk. Biochim Biophys
Acta Gene Regul Mech. 1867:1950332024. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Ghate NB, Nadkarni KS, Barik GK, Tat SS,
Sahay O and Santra MK: Histone ubiquitination: role in genome
integrity and chromatin organization. Biochim Biophys Acta Gene
Regul Mech. 1867:1950442024. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Shu Q, Liu Y and Ai H: The emerging role
of the histone H2AK13/15 ubiquitination: mechanisms of writing,
reading, and erasing in DNA damage repair and disease. Cells.
14:3072025. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Zambrano-Carrasco J, Zou J, Wang W, Sun X,
Li J and Su H: Emerging roles of cullin-RING ubiquitin ligases in
cardiac development. Cells. 13:2352024. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Zhao X, Jiang B, Hu H, Mao F, Mi J, Li Z,
Liu Q, Shao C and Gong Y: Zebrafish cul4a, but not cul4b, modulates
cardiac and forelimb development by upregulating tbx5a expression.
Hum Mol Genet. 24:853–864. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Zhang L, Xu X and Su X: Noncoding RNAs in
cancer immunity: Functions, regulatory mechanisms, and clinical
application. Mol Cancer. 19:482020. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Song Z, Suo C, Liu Y, Jin L, Xie X, Liu J,
Yu B, Wang Y, Zhang Z and Xie D: Comprehensive evaluation of
non-coding RNA-mediated autophagy regulation in myocardial
ischemia-reperfusion injury. Front Pharmacol. 16:15813412025.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Anastasiadou E, Jacob LS and Slack FJ:
Non-coding RNA networks in cancer. Nat Rev Cancer. 18:5–18. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu Y, Yuan J, Zhang Y, Ma T, Ji Q, Tian S
and Liu C: Non-coding RNA as a key regulator and novel target of
apoptosis in diabetic cardiomyopathy: Current status and future
prospects. Cell Signal. 128:1116322025. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Aghaei-Zarch SM, Alipourfard I,
Rasoulzadeh H, Najafi S, Aghaei-Zarch F, Partov S, Movafagh A,
Jahanara A, Toolabi A, Sheikhmohammadi A, et al: Non-coding RNAs:
an emerging player in particulate matter 2.5-mediated toxicity. Int
J Biol Macromol. 235:1237902023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sanjari Nia AH, Reyhani Ardabili M,
Sheikhvand M, Bagheri-Mohammadi S, Niknejad H, Rasoulzadeh H,
Movafagh A, Kharazi Neghad S, Baniasadi M, Ashrafi Asgarabad A, et
al: Non-coding RNAs: A new frontier in benzene-mediated toxicity.
Toxicology. 500:1536602023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Huang Y, Jiang B, Xia Y, Wang J, Ji C,
Tong J, Chen T and Jiang Y: Downregulation of miR-133a contributes
to the cardiac developmental toxicity of trichloroethylene in
zebrafish. Chemosphere. 251:1266102020. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Smirnova L, Block K, Sittka A,
Oelgeschläger M, Seiler AE and Luch A: MicroRNA profiling as tool
for in vitro developmental neurotoxicity testing: The case of
sodium valproate. PLoS One. 9:e988922014. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Xiao H, Zhang F, Zou Y, Li J, Liu Y and
Huang W: The function and mechanism of long non-coding RNA-ATB in
cancers. Front Physiol. 9:3212018. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
D'Angelo E and Agostini M: Long non-coding
RNA and extracellular matrix: The hidden players in cancer-stroma
cross-talk. Noncoding RNA Res. 3:174–177. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Ghorbanzadeh S, Poor-Ghassem N, Afsa M,
Nikbakht M and Malekzadeh K: Long non-coding RNA NR2F2-AS1: Its
expanding oncogenic roles in tumor progression. Hum Cell.
35:1355–1363. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ye D, Bao Z, Yu Y, Han Z, Yu Y, Xu Z, Ma
W, Yuan Y, Zhang L, Xu Y, et al: Inhibition of cardiomyocyte
differentiation of human induced pluripotent stem cells by
ribavirin: Implication for its cardiac developmental toxicity.
Toxicology. 435:1524222020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Ronzoni L, Aghemo A, Rumi MG, Prati G,
Colancecco A, Porretti L, Monico S, Colombo M and Cappellini MD:
Ribavirin suppresses erythroid differentiation and proliferation in
chronic hepatitis C patients. J Viral Hepat. 21:416–423. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Vishnoi A and Rani S: miRNA biogenesis and
regulation of diseases: An updated overview. Methods Mol Biol.
2595:1–12. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Ye J, Xu M, Tian X, Cai S and Zeng S:
Research advances in the detection of miRNA. J Pharm Anal.
9:217–226. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Li W, Mu J, Ni S, Pei W, Wan L, Wu X, Zhu
J, Zhang Z and Li L: Pentachlorophenol exposure delays the recovery
of colitis in association with altered gut microbiota and purine
metabolism. Environ Toxicol. 40:101–110. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Xu EG, Khursigara AJ, Li S, Esbaugh AJ,
Dasgupta S, Volz DC and Schlenk D: mRNA-miRNA-seq reveals
neuro-cardio mechanisms of crude oil toxicity in red drum
(Sciaenops ocellatus). Environ Sci Technol. 53:3296–3305. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Xu EG, Magnuson JT, Diamante G, Mager E,
Pasparakis C, Grosell M, Roberts AP and Schlenk D: Changes in
microRNA-mRNA signatures agree with morphological, physiological,
and behavioral changes in larval mahi-mahi treated with deepwater
horizon oil. Environ Sci Technol. 52:13501–13510. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Deng H, Bai Z, Huangfu C, Wang N, Chen M,
Li G, Huang C, Ao T, Tang X, Xia T, et al: Gremlin1
repression-mediated mitochondrial network hyperfunction contributes
to TCE-induced zebrafish cardiac defects. Cell Commun Signal.
23:3182025. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Drake VJ, Koprowski SL, Lough J, Hu N and
Smith SM: Trichloroethylene exposure during cardiac valvuloseptal
morphogenesis alters cushion formation and cardiac hemodynamics in
the avian embryo. Environ Health Perspect. 114:842–847. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jin H, Ji C, Ren F, Aniagu S, Tong J,
Jiang Y and Chen T: AHR-mediated oxidative stress contributes to
the cardiac developmental toxicity of trichloroethylene in
zebrafish embryos. J Hazard Mater. 385:1215212020. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Johnson PD, Goldberg SJ, Mays MZ and
Dawson BV: Threshold of trichloroethylene contamination in maternal
drinking waters affecting fetal heart development in the rat.
Environ Health Perspect. 111:289–292. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Long SM and Holdway DA: Acute toxicity of
crude and dispersed oil to octopus pallidus (Hoyle, 1885)
hatchlings. Water Res. 36:2769–2776. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Sørhus E, Donald CE, Nakken CL, Perrichon
P, Durif CMF, Shema S, Browman HI, Skiftesvik AB, Lie KK, Rasinger
JD, et al: Co-exposure to UV radiation and crude oil increases
acute embryotoxicity and sublethal malformations in the early life
stages of atlantic haddock (Melanogrammus aeglefinus). Sci Total
Environ. 859:1600802023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Khursigara AJ, Ackerly KL and Esbaugh AJ:
Oil toxicity and implications for environmental tolerance in fish.
Comp Biochem Physiol C Toxicol Pharmacol. 220:52–61. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Magnuson JT, Qian L, McGruer V, Cheng V,
Volz DC and Schlenk D: Relationship between miR-203a inhibition and
oil-induced toxicity in early life stage zebrafish (danio rerio).
Toxicol Rep. 9:373–381. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Guo M, Li X, Choi M, Zhang J, Yan S, Ma D,
Zeng J, Ding W, Wen Y, Li D, et al: Microcystin-LR prenatal
exposure induces coronary heart disease through macrophage
polarization imbalance mediated by trophoblast-derived
extracellular vesicles. Sci Total Environ. 948:1749792024.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Desrosiers R, Friderici K and Rottman F:
Identification of methylated nucleosides in messenger RNA from
Novikoff hepatoma cells. Proc Natl Acad Sci USA. 71:3971–3975.
1974. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
An Y and Duan H: The role of m6A RNA
methylation in cancer metabolism. Mol Cancer. 21:142022. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Qin Y, Li L, Luo E, Hou J, Yan G, Wang D,
Qiao Y and Tang C: Role of m6A RNA methylation in cardiovascular
disease (Review). Int J Mol Med. 46:1958–1972. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Fan Y, Lv X, Chen Z, Peng Y and Zhang M:
m6A methylation: Critical roles in aging and neurological diseases.
Front Mol Neurosci. 16:11021472023. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang
L, Jia G, Yu M, Lu Z, Deng X, et al: A METTL3-METTL14 complex
mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem
Biol. 10:93–95. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Zheng G, Dahl JA, Niu Y, Fedorcsak P,
Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al: ALKBH5
is a mammalian RNA demethylase that impacts RNA metabolism and
mouse fertility. Mol Cell. 49:18–29. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang
Y, Yi C, Lindahl T, Pan T, Yang YG and He C: N6-Methyladenosine in
nuclear RNA is a major substrate of the obesity-associated FTO. Nat
Chem Biol. 7:885–887. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zou S, Toh JD, Wong KH, Gao YG, Hong W and
Woon EC: N(6)-Methyladenosine: A conformational marker that
regulates the substrate specificity of human demethylases FTO and
ALKBH5. Sci Rep. 6:256772016. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Yang L, Qi G, Rao W, Cen Y, Chen L, Li W
and Pang Y: Aluminum causes irreversible damage to the development
of hippocampal neurons by regulating m6A RNA methylation. Toxicol
Lett. 399:34–42. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Qian Q, Pu Q, Li X, Liu X, Ni A, Han X,
Wang Z, Wang X, Yan J and Wang H: Acute/chronic triclosan exposure
induces downregulation of m6A-RNA methylation modification via
mettl3 suppression and elicits developmental and immune toxicity to
zebrafish. Chemosphere. 352:1413952024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Gao Y, Li A, Zhang W, Pang S, Liang Y and
Song M: Assessing the toxicity of bisphenol A and its six
alternatives on zebrafish embryo/larvae. Aquat Toxicol.
246:1061542022. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Rezg R, El-Fazaa S, Gharbi N and Mornagui
B: Bisphenol A and human chronic diseases: Current evidences,
possible mechanisms, and future perspectives. Environ Int.
64:83–90. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Liu J, Lin J, Chen J, Maimaitiyiming Y, Su
K, Sun S, Zhan G and Hsu CH: Bisphenol C induces developmental
defects in liver and intestine through mTOR signaling in zebrafish
(Danio rerio). Chemosphere. 322:1381952023. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Su K, Liu J, Chen J, Wu H, Tang W, Sun S,
Lin J, Zhan G and Hsu CH: Bisphenol C induces cardiac developmental
defects by disrupting m6A homeostasis. Environ Sci Technol.
58:17259–17269. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Ji C, Tao Y, Li X, Wang J, Chen J, Aniagu
S, Jiang Y and Chen T: AHR-mediated m6A RNA methylation contributes
to PM2.5-induced cardiac malformations in zebrafish larvae. J
Hazard Mater. 457:1317492023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Wang Y, Zou J and Zhou H: N6-methyladenine
RNA methylation epigenetic modification and diabetic microvascular
complications. Front Endocrinol (Lausanne). 15:14621462024.
View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Chen Y, Liang R, Li Y, Jiang L, Ma D, Luo
Q and Song G: Chromatin accessibility: Biological functions,
molecular mechanisms and therapeutic application. Signal Transduct
Target Ther. 9:3402024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Fyodorov DV, Zhou BR, Skoultchi AI and Bai
Y: Emerging roles of linker histones in regulating chromatin
structure and function. Nat Rev Mol Cell Biol. 19:192–206. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Routh A, Sandin S and Rhodes D: Nucleosome
repeat length and linker histone stoichiometry determine chromatin
fiber structure. Proc Natl Acad Sci USA. 105:8872–8877. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
McBryant SJ, Adams VH and Hansen JC:
Chromatin architectural proteins. Chromosome Res. 14:39–51. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Eustermann S, Patel AB, Hopfner KP, He Y
and Korber P: Energy-driven genome regulation by ATP-dependent
chromatin remodellers. Nat Rev Mol Cell Biol. 25:309–332. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Struhl K: Non-canonical functions of
enhancers: Regulation of RNA polymerase III transcription, DNA
replication, and V(D)J recombination. Trends Genet. 40:471–479.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Wilkinson AL, Zorzan I and Rugg-Gunn PJ:
Epigenetic regulation of early human embryo development. Cell Stem
Cell. 30:1569–1584. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Cao L, Luo Y, Guo X, Liu S, Li S, Li J,
Zhang Z, Zhao Y, Zhang Q, Gao F, et al: SAFA facilitates chromatin
opening of immune genes through interacting with anti-viral host
RNAs. PLoS Pathog. 18:e10105992022. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Lopes-Paciencia S and Ferbeyre G:
Increased chromatin accessibility underpins senescence. FEBS J.
292:4112–4132. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Grandi FC, Modi H, Kampman L and Corces
MR: Chromatin accessibility profiling by ATAC-seq. Nat Protoc.
17:1518–1552. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Tang L, Song S, Hu C, Liu M, Lam PKS, Zhou
B, Lam JCW and Chen L: Parental exposure to perfluorobutane
sulfonate disturbs the transfer of maternal transcripts and
offspring embryonic development in zebrafish. Chemosphere.
256:1271692020. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Liu Q, Van Bortle K, Zhang Y, Zhao MT,
Zhang JZ, Geller BS, Gruber JJ, Jiang C, Wu JC and Snyder MP:
Disruption of mesoderm formation during cardiac differentiation due
to developmental exposure to 13-cis-retinoic acid. Sci Rep.
8:129602018. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Guan G, Abulaiti A, Qu C, Chen CC, Gu Z,
Yang J, Zhang T and Chen X, Zhou Z, Lu F and Chen X: Multi-omics
panoramic analysis of HBV integration, transcriptional regulation,
and epigenetic modifications in PLC/PRF/5 cell line. J Med Virol.
96:e296142024. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Qi C, Jin X, Wang H and Xu D: Crosstalk
between N6-methyladenosine and other epigenetic mechanisms in
central nervous system development and disorders. Biomolecules.
15:10922025. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Tan RZ, Jia J, Li T, Wang L and Kantawong
F: A systematic review of epigenetic interplay in kidney diseases:
Crosstalk between long noncoding RNAs and methylation, acetylation
of chromatin and histone. Biomed Pharmacother. 176:1169222024.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Li W, Luo P, Chen Q, Cheng L, Gan L, Zhang
F, Zhong H, Zheng L and Qian B: Epigenetic modifications in bladder
cancer: Crosstalk between DNA methylation and miRNAs. Front
Immunol. 16:15181442025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Buscariollo DL, Fang X, Greenwood V, Xue
H, Rivkees SA and Wendler CC: Embryonic caffeine exposure acts via
A1 adenosine receptors to alter adult cardiac function and DNA
methylation in mice. PLoS One. 9:e875472014. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Buscariollo DL, Breuer GA, Wendler CC and
Rivkees SA: Caffeine acts via A1 adenosine receptors to disrupt
embryonic cardiac function. PLoS One. 6:e282962011. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Fang X, Mei W, Barbazuk WB, Rivkees SA and
Wendler CC: Caffeine exposure alters cardiac gene expression in
embryonic cardiomyocytes. Am J Physiol Regul Integr Comp Physiol.
307:R1471–1487. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Rohweder R, de Oliveira Schmalfuss T, Dos
Santos Borniger D, Ferreira CZ, Zanardini MK, Lopes GPTF, Barbosa
CP, Moreira TD, Schuler-Faccini L, Sanseverino MTV, et al: Caffeine
intake during pregnancy and adverse outcomes: An integrative
review. Reprod Toxicol. 123:1085182024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Rodriguez-Martinez A, Vuorinen EM,
Shcherban A, Uusi-Mäkelä J, Rajala NKM, Nykter M and Kallioniemi A:
Novel ZNF414 activity characterized by integrative analysis of
ChIP-exo, ATAC-seq and RNA-seq data. Biochim Biophys Acta Gene
Regul Mech. 1865:1948112022. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Eisa-Beygi S, Benslimane FM, El-Rass S,
Prabhudesai S, Abdelrasoul MKA, Simpson PM, Yalcin HC, Burrows PE
and Ramchandran R: Characterization of endothelial cilia
distribution during cerebral-vascular development in zebrafish
(Danio rerio). Arterioscler Thromb Vasc Biol. 38:2806–2818. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhang L and Zhou J: Zebrafish: A smart
tool for heart disease research. J Fish Biol. 105:1487–1500. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Stainier DY, Kontarakis Z and Rossi A:
Making sense of anti-sense data. Dev Cell. 32:7–8. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Poon KL and Brand T: The zebrafish model
system in cardiovascular research: A tiny fish with mighty
prospects. Glob Cardiol Sci Pract. 2013:9–28. 2013.PubMed/NCBI
|
|
153
|
Glickman NS and Yelon D: Cardiac
development in zebrafish: Coordination of form and function. Semin
Cell Dev Biol. 13:507–513. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Salman HE and Yalcin HC: Computational
modeling of blood flow hemodynamics for biomechanical investigation
of cardiac development and disease. J Cardiovasc Dev Dis.
8:142021.PubMed/NCBI
|
|
155
|
Wafer LN, Jensen VB, Whitney JC, Gomez TH,
Flores R and Goodwin BS: Effects of environmental enrichment on the
fertility and fecundity of zebrafish (Danio rerio). J Am Assoc Lab
Anim Sci. 55:291–294. 2016.PubMed/NCBI
|
|
156
|
Ducharme NA, Reif DM, Gustafsson JA and
Bondesson M: Comparison of toxicity values across zebrafish early
life stages and mammalian studies: Implications for chemical
testing. Reprod Toxicol. 55:3–10. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Khabib MNH, Sivasanku Y, Lee HB, Kumar S
and Kue CS: Alternative animal models in predictive toxicology.
Toxicology. 465:1530532022. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Bambino K and Chu J: Zebrafish in
toxicology and environmental health. Curr Top Dev Biol.
124:331–367. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Evans T: Embryonic stem cells as a model
for cardiac development and disease. Drug Discov Today Dis Models.
5:147–155. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Keller G: Embryonic stem cell
differentiation: Emergence of a new era in biology and medicine.
Genes Dev. 19:1129–1155. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Mills RJ, Titmarsh DM, Koenig X, Parker
BL, Ryall JG, Quaife-Ryan GA, Voges HK, Hodson MP, Ferguson C,
Drowley L, et al: Functional screening in human cardiac organoids
reveals a metabolic mechanism for cardiomyocyte cell cycle arrest.
Proc Natl Acad Sci USA. 114:E8372–E8381. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Mills RJ, Parker BL, Quaife-Ryan GA, Voges
HK, Needham EJ, Bornot A, Ding M, Andersson H, Polla M, Elliott DA,
et al: Drug screening in human PSC-cardiac organoids identifies
pro-proliferative compounds acting via the mevalonate pathway. Cell
Stem Cell. 24:895–907.e6. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
163
|
Aronson JK and Ferner RE: Biomarkers-A
general review. Curr Protoc Pharmacol. Mar 17–2017.(Epub ahead of
print). View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Imbard A, Benoist JF and Blom HJ: Neural
tube defects, folic acid and methylation. Int J Environ Res Public
Health. 10:4352–4389. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Movendane Y, Sipalo MG and Chan LCZ:
Advances in folic acid biosensors and their significance in
maternal, perinatal, and paediatric preventive medicine. Biosensors
(Basel). 13:9122023. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Ma Y, Zhang C, Gao XB, Luo HY, Chen Y, Li
HH, Ma X and Lu CL: Folic acid protects against arsenic-mediated
embryo toxicity by up-regulating the expression of Dvr1. Sci Rep.
5:160932015. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Zhao G, He F, Wu C, Li P, Li N, Deng J,
Zhu G, Ren W and Peng Y: Betaine in inflammation: Mechanistic
aspects and applications. Front Immunol. 9:10702018. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Hoffmann L, Brauers G, Gehrmann T,
Häussinger D, Mayatepek E, Schliess F and Schwahn BC: Osmotic
regulation of hepatic betaine metabolism. Am J Physiol Gastrointest
Liver Physiol. 304:G835–G846. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Barak AJ, Beckenhauer HC and Tuma DJ:
Betaine, ethanol, and the liver: A review. Alcohol. 13:395–398.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Chen X, Luo J, Yang L, Guo Y, Fan Y, Liu
J, Sun J, Zhang Y, Jiang Q, Chen T and Xi Q: miR-143-mediated
responses to betaine supplement repress lipogenesis and hepatic
gluconeogenesis by targeting MAT1a and MAPK11. J Agric Food Chem.
70:7981–7992. 2022. View Article : Google Scholar : PubMed/NCBI
|