Introduction
Rheumatoid arthritis (RA) is a chronic autoimmune
disease characterized by immune dysregulation, persistent synovial
inflammation and progressive joint destruction. The pathological
hallmarks of RA include uncontrolled synovial inflammation,
abnormal proliferation of fibroblast-like synoviocytes (FLSs) and
bone erosion. The disease primarily involves small joints, such as
those of the hands and wrists, typically presenting with
symmetrical inflammation and can also manifest with systemic
symptoms, including anemia and fever (1). Despite extensive research, the
molecular mechanisms driving immune-mediated inflammation and bone
damage in RA remain incompletely understood. Epigenetic alterations
may provide a key link between genetic predisposition,
environmental triggers and RA pathogenesis (2,3).
Among epigenetic regulators, microRNAs (miRNAs or miRs), short,
non-coding RNAs that modulate gene expression by binding mRNA and
promoting its degradation or suppressing translation, have
attracted considerable attention (4,5).
miRNAs influence key cellular processes such as differentiation,
proliferation and apoptosis (6,7).
Dysregulation of miRNAs and their downstream
signaling cascades contributes to the disruption of gene networks
that regulate immune responses and skeletal remodeling in RA
(8). Mounting (9,10)
evidence suggests that miRNAs act through dual mechanisms in the
pathophysiology of RA. First, they modulate immune-driven
inflammation. For instance, miR-146a promotes STAT1 activation,
shifting Treg cells toward pro-inflammatory phenotypes (11). Paeonol inhibits TNF-α-induced FLS
proliferation and cytokine release by downregulating miR-155 and
increasing FOXO3 expression (12).
Tocilizumab alters angiogenic pathways by regulating EMMPRIN and
miR-146a-5p (13). Similarly,
miR-155 promotes NF-κB activation via SOCS1 targeting, enhancing
Th17 differentiation and aggravating synovitis (14), while miR-125a-5p decreases
TNF-α-driven osteoclast differentiation and suppresses inflammatory
signaling through the MAPK pathway (15).
Second, miRNAs are implicated in bone metabolism
abnormalities associated with RA. Elevated miR-22-3p improves bone
formation by activating the SOSTDC1-PI3K/AKT pathway (16). The long non-coding RNA MEG3
facilitates osteogenesis in human bone marrow mesenchymal stem
cells by regulating the miR-21-5p/SOD3 pathway (17). Furthermore, miR-21-5p has been
identified as a potential modulator of osteogenesis (18). Although dysregulated miRNAs are
strongly associated with RA-FLS pathogenicity (19), the specific contributions of
individual miRNAs in orchestrating the interplay between synovial
inflammation and bone remodeling remain poorly defined. In
addition, miR-369-3p has been reported as a critical
immunometabolic regulator in inflammatory disorders (20,21);
however, its involvement in RA-related osteoimmunology has not been
explored. It is hypothesized that miR-369-3p functions as a key
epigenetic switch by directly targeting spectrin β,
non-erythrocytic 1 (SPTBN1), a scaffold protein known to inhibit
Wnt/β-catenin signaling through cytoplasmic sequestration of
β-catenin. Aberrant expression of miR-369-3p may suppress SPTBN1,
allowing β-catenin to translocate into the nucleus, where it
amplifies inflammatory cytokine production and promotes
osteoclastogenesis via RANKL activation. This mechanism positions
miR-369-3p at the intersection of synovial inflammation and
subchondral bone erosion, suggesting its potential as a therapeutic
target for interrupting the link between immune dysregulation and
structural joint damage in RA.
A deeper investigation of the molecular pathways
regulating immune inflammation and bone degradation in RA is,
therefore, crucial. Uncovering the role of miR-369-3p may not only
establish it as a biomarker for RA diagnosis but also provide a
rationale for developing miR-369-3p mimics as novel targeted
therapies to alleviate inflammation and prevent joint destruction
by modulating the SPTBN1/Wnt/β-catenin pathway.
Materials and methods
Cell co-cultivation
RA fibroblast-like synoviocytes (RA-FLSs; Hunan
Fenghui Biotechnology Co., Ltd.; cat. no. FHHUM195) were maintained
in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS)
and 1% penicillin-streptomycin (100 U/ml each). Cultures were
incubated at 37°C in a humidified atmosphere containing 5%
CO2 and the medium was refreshed every 72 h. Once cell
confluence reached 80–90%, the cultures were washed twice with PBS
and digested with 0.25% trypsin-EDTA. Trypsinization was monitored
under an inverted microscope to ensure proper detachment, after
which enzymatic activity was neutralized with complete medium.
Cells in the logarithmic growth phase were collected for subsequent
experiments.
For the Transwell co-culture system, RA-FLSs were
seeded into the lower chambers, while peripheral blood mononuclear
cells (PBMCs) were isolated from the peripheral blood of 10
patients (three male, seven female; age, 35–58 years) with RA
(November 2024; approval number: 2023AH-52,) and plated onto the
upper chamber membrane (BIOFIL; TCS-001-006). When cell confluence
reached ~60%, RA-FLSs and PBMCs were harvested separately from the
lower and upper compartments, respectively. Total RNA and protein
were then extracted from each cell population for downstream
reverse transcription-quantitative (RT-q) PCR and western blot
analyses. To establish optimal stimulation conditions, including
concentration and exposure time, RA-FLS viability was assessed
using the CCK-8 assay, with PBMCs derived from RA patients serving
as the stimulating cells.
Cell transfection
Cells were digested with trypsin, seeded at a
density of 5×105 cells per well in five wells of a
six-well plate and cultured at 37°C in 5% CO2 for 24 h
until reaching ~70% confluence. The miR-369-3p sequence:
AAUAAUACAUGGUUGAUCUUU; mimic: Sense, AAUAAUACAUGGUUGAUCUUU and
antisense, AAAGAUCAACCAUGUAUUAUU (both 20 µM; General Biosystems).
A total of 5 µl mimic or negative control (NC; both 20 µM; General
Biosystems) was diluted in 0.25 ml of serum-free medium. Meanwhile,
10 µl of Lipo8000 (Beyotime Institute of Biotechnology) reagent was
diluted separately in 0.5 ml of serum-free medium. The two
solutions were gently vortexed and allowed to stand at room
temperature for 5 min. The transfection reagent was then divided
into two portions, each combined with the mimic solution and
incubated for 20 min to enable complex formation. For the
miR-369-3p inhibitor, (AAAGAUCAACCAUGUAUUAUU; 20 µM; General
Biosystems), and 5 µl was prepared following the same procedure as
the mimic. Transfection was performed at room temperature Prior to
transfection, cells were rinsed 2–3 times with serum-free medium to
remove residual serum. Then, 500 µl of the prepared complexes were
added to each well, gently swirled for 1–2 min and supplemented
with serum-free medium to a final volume of 2 ml. After 4 h of
incubation, the medium was replaced with complete culture medium.
The total transfection duration was 48 h, and subsequent
experiments were performed immediately.
Grouping and model preparation
The RA-FLSs in the logarithmic growth phase were
randomly allocated into six experimental groups using a random
number table to ensure comparable cell density and growth
conditions before treatment. When cultures reached ~60% confluence,
cells were divided into the following groups: Group A (Control):
Normal FLSs co-cultured with PBMCs from healthy donors.
Group B (Model): RA-FLSs co-cultured with PBMCs
derived from RA patients.
Group C (Model + miR-369-3p mimics):
RA-FLSs/RA-PBMCs treated with miR-369-3p mimics.
Group D (Model + mimic-NC): RA-FLSs/RA-PBMCs
treated with mimic negative control.
Group E (Model + miR-369-3p inhibitor):
RA-FLSs/RA-PBMCs treated with miR-369-3p inhibitor; F (Model
+ inhibitor-NC): RA-FLSs/RA-PBMCs treated with inhibitor negative
control.
CCK-8 assay
Cell viability was evaluated by measuring metabolic
activity using the CellTiter-Glo 8 Assay Kit (Promega Corporation;
cat. no. BL1055B). Cells were seeded into 96-well flat-bottom
plates at a density of 5×103 cells per well and allowed
to adhere overnight. Following treatment, 10 µl of CCK-8 reagent
was added directly to each well without removing the culture
medium. Plates were then incubated for 2 h at 37°C under humidified
5% CO2 conditions. Absorbance was measured at 450 nm
using a Rayto RT6100 microplate reader (Rayto Life and Analytical
Sciences Co., Ltd.). All assays were performed in six independent
replicates for each group.
RT-qPCR
Total RNA was isolated from 1×106 cells
using total RNA extraction reagent (ShareBio; cat. no. SB-MR009)
and reverse-transcribed according to the manufacturer's protocol.
RT-qPCR was performed using SYBR Green qPCR Master Mix (iScience;
cat. no. EG20117M) with specific primers on a real-time PCR
detection system. Thermocycling conditions were as follows: initial
denaturation at 95°C for 30 sec; followed by 40 cycles of
denaturation at 95°C for 15 sec and annealing/extension at 60°C for
30 sec. For normalization, U6 small nuclear RNA served as the
internal reference for miR-369-3p, while β-actin was used as the
housekeeping control for mRNAs, including SPTBN1, Wnt and
β-catenin. Relative expression levels of miR-369-3p, SPTBN1, Wnt
and β-catenin were calculated using the 2−ΔΔCq method
(22). Sangon Biotech Co., Ltd.
synthesized primers and their sequences are listed in Table I. Each experiment was conducted
independently three times, with one technical replicate per
group.
 | Table I.List of primer sequences. |
Table I.
List of primer sequences.
| Gene | Amplicon size
(bp) | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| Hu-β-actin | 96 |
CCCTGGAGAAGAGCTACGAG |
GGAAGGAAGGCTGGAAGAGT |
| Hu-U6 | 94 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| Hu-SPTBN1 | 112 |
CCCTGGAAAGGCTGACTACA |
TCCTCTGAAACCTTCGTGCT |
| Hu-wnt | 154 |
ATCTTCGCTATCACCTCCGC |
GGCCGAAGTCAATGTTGTCG |
| hsa-miR-369-3p |
|
CCGCGCAATACATGGTTG |
AGTGCAGGGTCCGAGGTATT |
| hsa-miR-369-3p
RT |
|
GTCGTATCCAGTGCAGGGTCCGAGGTATTCGCACTGGATACGACAAAGAT |
| Hu-β-catenin | 100 |
ATGTCTGAGGACAAGCCACA |
CAGCAGTCTCATTCCAAGCC |
Western blot analysis
Cell pellets were lysed on ice for 30 min in 100 µl
RIPA buffer (Biosharp Life Sciences; cat. no. BL504A) supplemented
with 1 mM PMSF. Lysates were centrifuged at 13,780 g for 15 min at
4°C and the supernatants were collected as total protein extracts.
The concentration of total protein was determined using the BCA
Protein Assay For SDS-PAGE, proteins were denatured by mixing
samples with 5X loading buffer at a 1:4 ratio, boiled for 10 min
and 30 µg of protein was loaded per well. Gels consisted of a 5%
stacking layer (2 ml) and a 10% resolving layer (5 ml).
Electrophoresis was carried out at 80 V for 30 min (stacking) and
120 V for 1 h (resolving). Proteins were transferred onto PVDF
membranes (MilliporeSigma; cat. no. IPVH00010) that had been
pre-activated in methanol, using a current of 300 mA. Membranes
were blocked with 5% non-fat milk at room temperature for 2 h and
subsequently incubated overnight at 4°C with primary antibodies:
β-actin (1:1,000; Zs-BIO; cat. no. TA-09), SPTBN1 (1:500) (HUABIO,
HA500014), Wnt5 (1:1,000) (Proteintech, 55184-1-AP) and β-catenin
(1:500) (Affinity, AF6266). After washing, membranes were incubated
with HRP-conjugated secondary antibodies (1:10,000) (Zs-BIO,
ZB-2301) for 2 h. Protein bands were visualized using enhanced
chemiluminescence reagent (GLPBIO; cat. no. GK10008). Band
intensities were quantified with ImageJ software (National
Institutes of Health), with β-actin serving as the internal
control. All experiments were performed independently and repeated
three times.
Assay of dual-luciferase reporter
genes
293T cells were seeded into 12-well plates at a
density of 2×105 cells per well 24 h before
transfection. When cell confluence reached 70–80%,
triple-transfection assays were performed with the following
groups: NC-mimics + SPTBN1-wt, miR-369-3p mimics + SPTBN1-wt,
NC-mimics + SPTBN1-Mut and miR-369-3p mimics + SPTBN1-Mut (all
General Biosystems). Transfections were carried out using
Lipofectamine 8000® (Thermo Fisher Scientific, Inc.)
diluted in serum-free DMEM. After 6 h, the transfection medium was
replaced with antibiotic-free DMEM containing 10% FBS and cells
were cultured for another 48 h. Cells were then washed three times
with PBS, lysed on ice for 10 min in 300 µl lysis buffer and
centrifuged at 12,000 × g for 10 min at 4°C. Luciferase activity
was measured using the Dual-Luciferase Reporter Assay Kit (Beyotime
Biotechnology; cat. no. RG027). A 50 µl aliquot of cell lysate was
used to sequentially measure firefly luciferase activity (560 nm)
followed by Renilla luciferase activity (465 nm). All
experiments were performed independently in triplicate.
Flow cytometry detection of cell
cycle
Cells were harvested, washed with pre-chilled PBS
and digested with trypsin. After centrifugation at 382 × g for 5
min at 4°C, the supernatant was reduced to ~50 µl and the pellet
was resuspended in 0.3 ml PBS to obtain a single-cell suspension.
While vortexing, 2.7 ml of ice-cold absolute ethanol was added
dropwise to achieve a final ethanol concentration of 90%. The cells
were fixed at 4°C for 18–24 h or at −20°C for 1 h. Following
fixation, the cells were centrifuged again 382 g, 5 min, 4°C),
washed twice with cold PBS and then resuspended in 0.5 ml of PBS.
The suspension was transferred into a 1.5 ml microcentrifuge tube
and gently dispersed. Next, 100 µl of RNase A reagent
(ready-to-use, provided with the cell cycle detection kit) was
added and samples were incubated at 37°C for 30 min. Cells were
then stained with 400 µl propidium iodide (PI) solution (50 µg/ml,
light-protected, kit-provided) at 4°C for 30–60 min. Within 24 h,
samples were filtered through a 200-mesh strainer and analyzed on a
NovoCyte flow cytometer (Agilent Technologies, Inc.) using
NovoExpress software (version 1.5.6.0) (Agilent Technologies,
Inc.). Each experiment was conducted independently in
triplicate.
Detection of cell proliferation by Edu
staining
Cells were incubated with 10 µM EdU labeling medium
at 37°C for 2 h, followed by fixation, permeabilization and the
Click reaction, as per the manufacturer's instructions (Elabscience
Bionovation Inc.; cat. no. E-CK-A376). After nuclear
counterstaining with 500 µl DAPI working solution at room
temperature for 5–10 min in the dark) and washing (1 ml PBS
containing 3% BSA (Biosharp; cat. no. BS114) per well, washed 3
times for 5 min each), images were captured using an inverted
fluorescence microscope (OLYMPUS; CKX53). Each experiment was
independently repeated three times. EdU-positive cells were
quantified by two blinded observers to minimize bias.
Scratch wound healing assay to detect
cell migration
Horizontal reference lines were drawn on the back of
6-well plates with a marker, spaced 0.5–1 cm apart, with at least
five lines per well. Cells were seeded at a density of
1×106 per well and allowed to reach confluence
overnight. A scratch was made across the cell layer using a 200-µl
pipette tip, perpendicular to the reference lines. Detached cells
were removed by washing three times with PBS and serum-free medium
was added. Plates were then incubated at 37°C in 5% CO2
and wound closure was images captured at 0 and 24 h using an
inverted light microscope (Olympus Corporation; cat. no. CKX53) to
assess migration. Cell migration rate (%) was quantified using
ImageJ software, and the calculation formula was as follows:
[(Wound area at 0 h-Wound area at 12/24 h)/Wound area at 0 h]
×100%. All experiments were independently repeated three times.
Statistical analysis
All statistical analyses were conducted using SPSS
Statistics 26.0 (IBM Corp.), while figures were prepared with
GraphPad Prism 10.1.2 (Dotmatics). Data were expressed as mean ±
standard deviation (SD). Normality of the datasets was assessed
using the Kolmogorov-Smirnov test in SPSS. For normally distributed
data, comparisons between two groups were performed using Student's
t-test, whereas one-way ANOVA was applied for comparisons involving
more than two groups. To minimize type I error from multiple
testing, Tukey's post hoc test was used following ANOVA for
pairwise comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
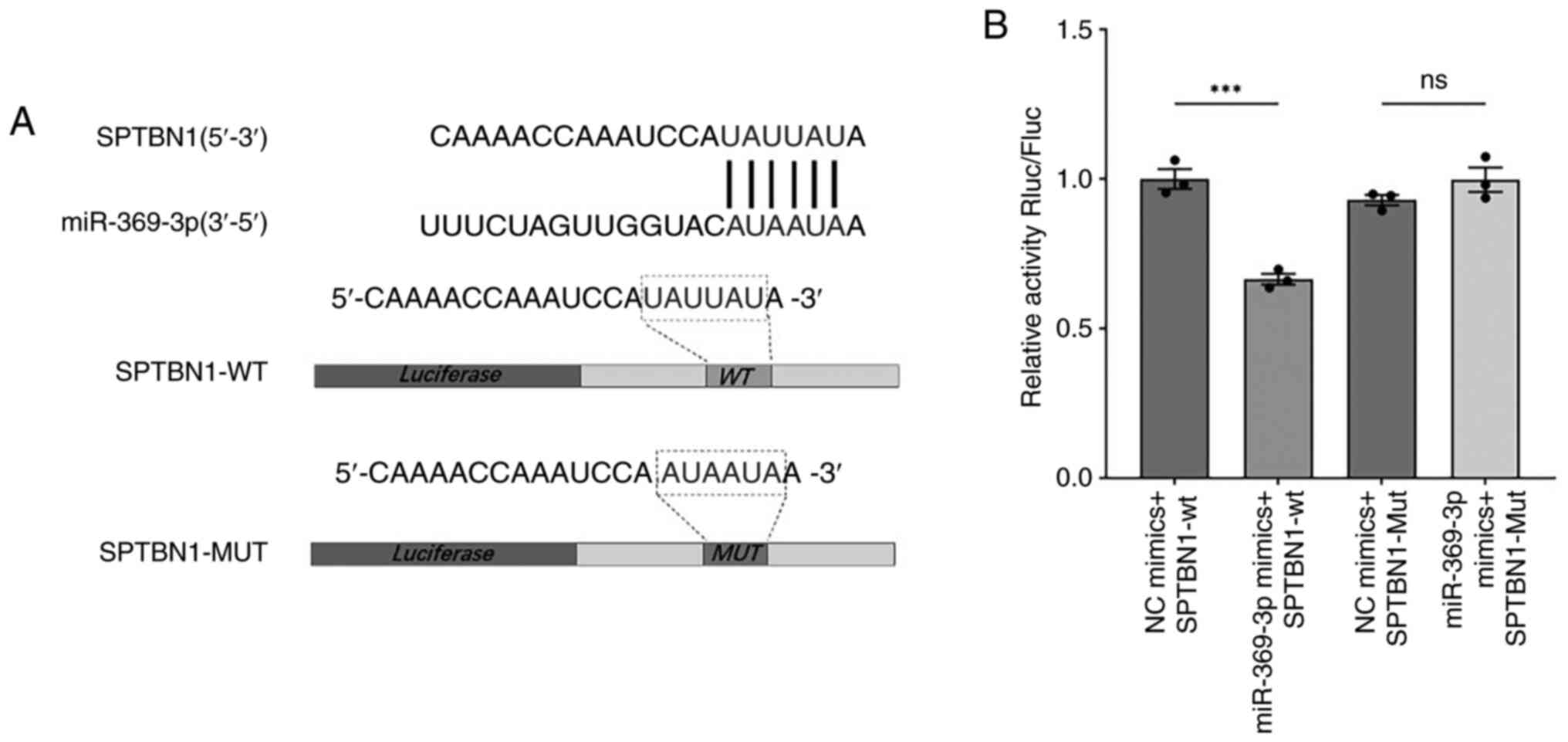
A targeted interaction exists between
miR-369-3p and SPTBN1
From our previous bioinformatics analysis, a
potential binding site between miR-369-3p and the 3′UTR of SPTBN1
was predicted. To validate this interaction, wild-type (WT) and
mutant (Mut) sequences of the predicted binding region were
synthesized and cloned into a dual-luciferase reporter vector.
Sequence alignment confirmed complementarity between miR-369-3p and
SPTBN1 (Fig. 1A). The
dual-luciferase reporter assay demonstrated that luciferase
activity in the SPTBN1-WT group was markedly reduced following
transfection with miR-369-3p mimics compared with the NC mimic
group (Fig. 1B).
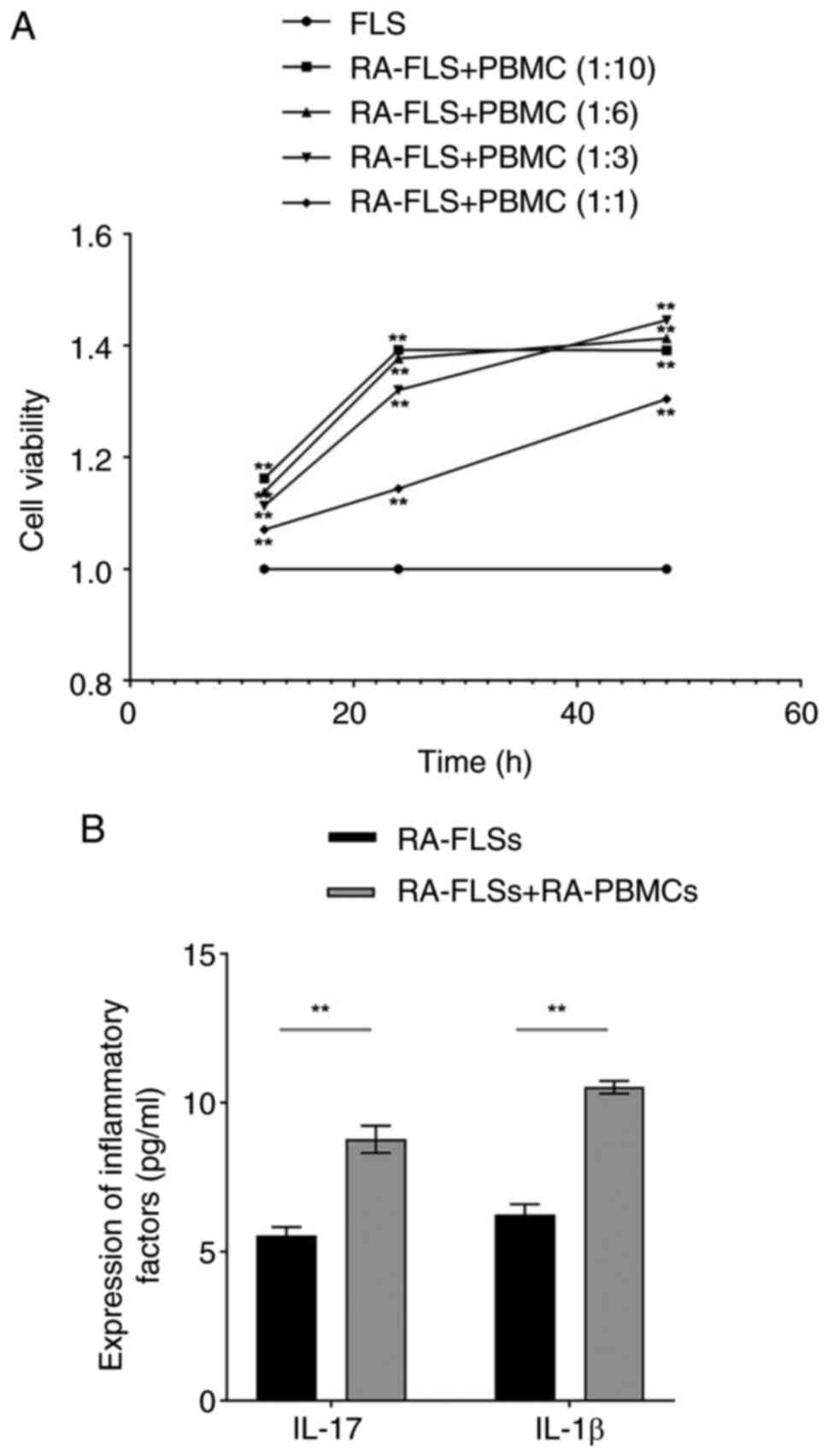
The crosstalk between RA-FLSs and
RA-PBMCs enhances cell viability, determining the optimal
co-culture conditions
To investigate the interaction between RA-FLSs and
RA-PBMCs, a dual-cell co-culture system was developed to mimic the
synovial inflammatory microenvironment of RA. RA-FLSs and RA-PBMCs
were co-cultured at different ratios (1:10, 1:6, 1:3 and 1:1) and
cell viability was measured at 12, 24 and 48 h. CCK-8 analysis
revealed a time-dependent increase in cell viability across all
groups, with the 1:3 ratio showing the greatest viability at 48 h
(Fig. 2A). Accordingly, the
RA-FLSs + RA-PBMCs co-culture model was established at a 1:3 ratio
with 48 h as the optimal activation time for subsequent
experiments. To further assess the inflammatory profile, ELISA was
used to measure cytokine secretion. Compared with RA-FLSs cultured
alone, the co-culture model showed markedly higher levels of IL-17
and IL-1β (Fig. 2B). These
findings indicated that crosstalk between RA-FLSs and RA-PBMCs
promotes cell viability and amplifies inflammatory signaling within
the RA immune microenvironment.
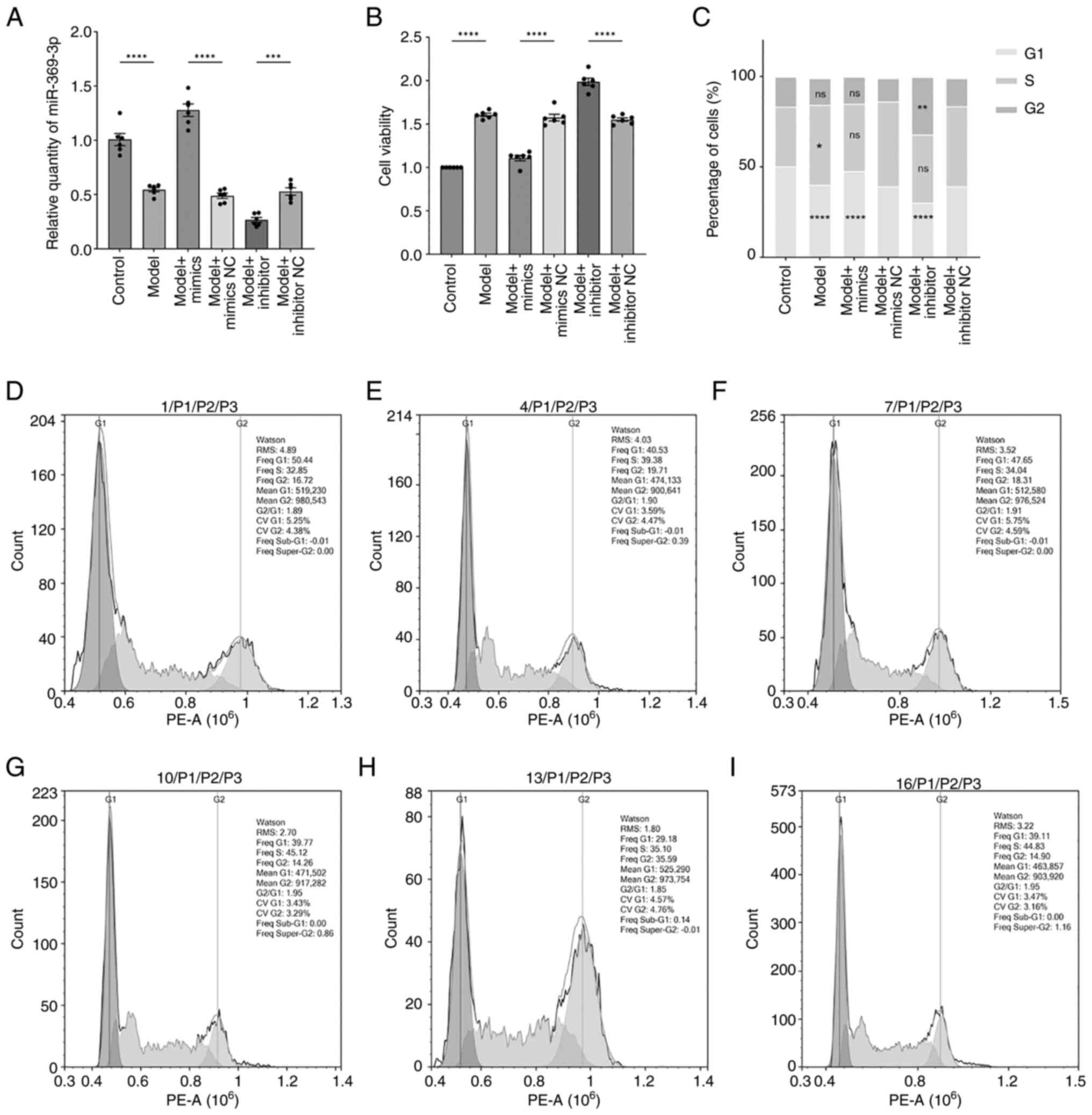
Effect of miR-369-3p modification on
cell viability and cell cycle dynamics
Cells were transfected with miR-369-3p mimics, mimic
negative control (mimic-NC), inhibitors and inhibitor negative
control (inhibitor-NC) (Fig. 3A).
RT-qPCR confirmed altered expression of miR-369-3p across groups.
Compared with the model mimic-NC group, miR-369-3p expression was
markedly upregulated in the model mimic group. At the same time, it
was markedly reduced in the model inhibitor group relative to the
inhibitor-NC group. Model groups displayed lower miR-369-3p
expression compared with control groups.
Cell viability, assessed by CCK-8 assay (Fig. 3B), was markedly higher in model
groups than in controls. However, the introduction of miR-369-3p
mimics suppressed cell viability, whereas the inhibitor group
demonstrated increased proliferation. These results suggested that
upregulation of miR-369-3p inhibits cell proliferation. To further
investigate the role of miR-369-3p in cell proliferation, cell
cycle distribution was analyzed using flow cytometry. The results
were consistent with the CCK-8 findings. Cell cycle analysis
(Fig. 3C-H) showed that RA-FLSs in
the model group had a reduced proportion of cells in the
G1 phase compared with controls. Transfection with
miR-369-3p mimics increased the G1-phase population
relative to mimic-NC, while transfection with the inhibitor
elevated the G2-phase fraction compared with
inhibitor-NC. These findings suggested that inhibiting miR-369-3p
promotes cell cycle progression and accelerates proliferation,
whereas overexpressing miR-369-3p exerts a suppressive effect.
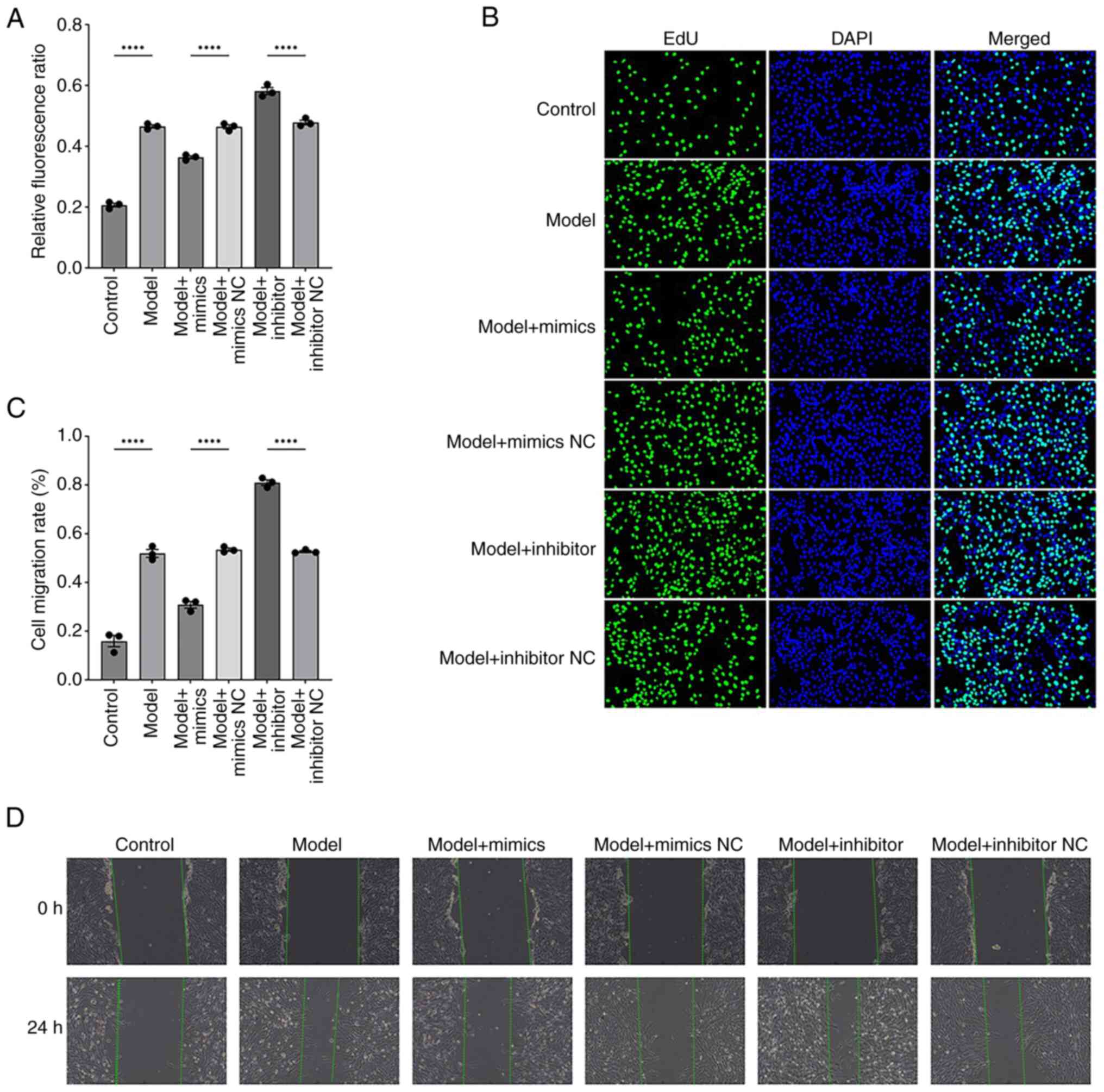
Effects of modifying miR-369-3p on
cellular proliferation and migratory capacity
To further visualize the effect of miR-369-3p on
cell proliferation and migration, EdU and scratch assays were
performed. The EdU assay results were consistent with previous
findings, showing that transfection with miR-369-3p mimics markedly
reduced cell proliferation, whereas the inhibitor group showed the
opposite trend (Fig. 4A and B).
Similarly, scratch assays demonstrated that the mimic group
displayed a significant reduction in migration capacity, while the
inhibitor group showed enhanced migratory activity (Fig. 4C and D).
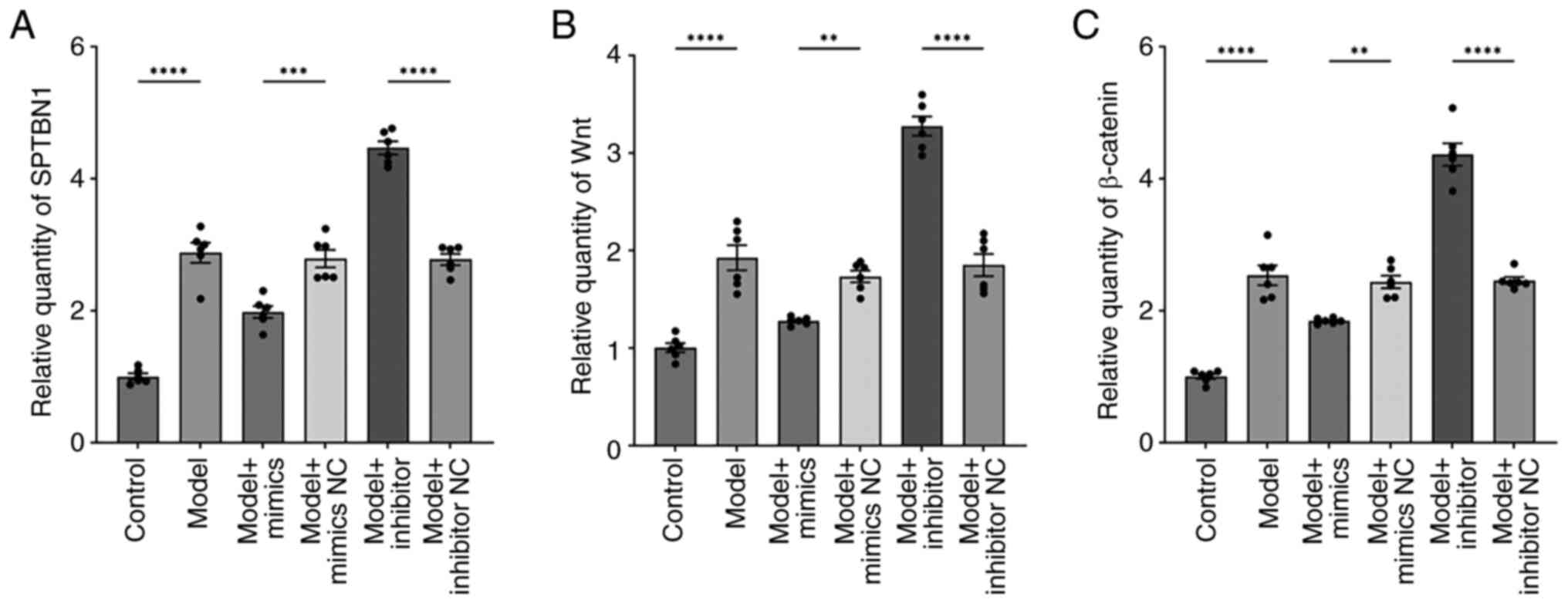
miR-369-3p can regulate SPTBN1 and
Wnt/β-catenin gene expression levels
RT-qPCR (Fig. 5A-C)
revealed that the expression levels of SPTBN1, Wnt and β-catenin
were markedly higher in the model group compared with the control
group. Relative to the model mimics-NC group, these gene
expressions were markedly reduced in the model mimics group. In
comparison, the model inhibitor group showed a pronounced increase
in SPTBN1, Wnt and β-catenin expression compared with the model
inhibitor-NC group. These findings indicated that miR-369-3p
overexpression suppresses, while its inhibition enhances, the
expression of SPTBN1, Wnt and β-catenin in the co-culture
system.
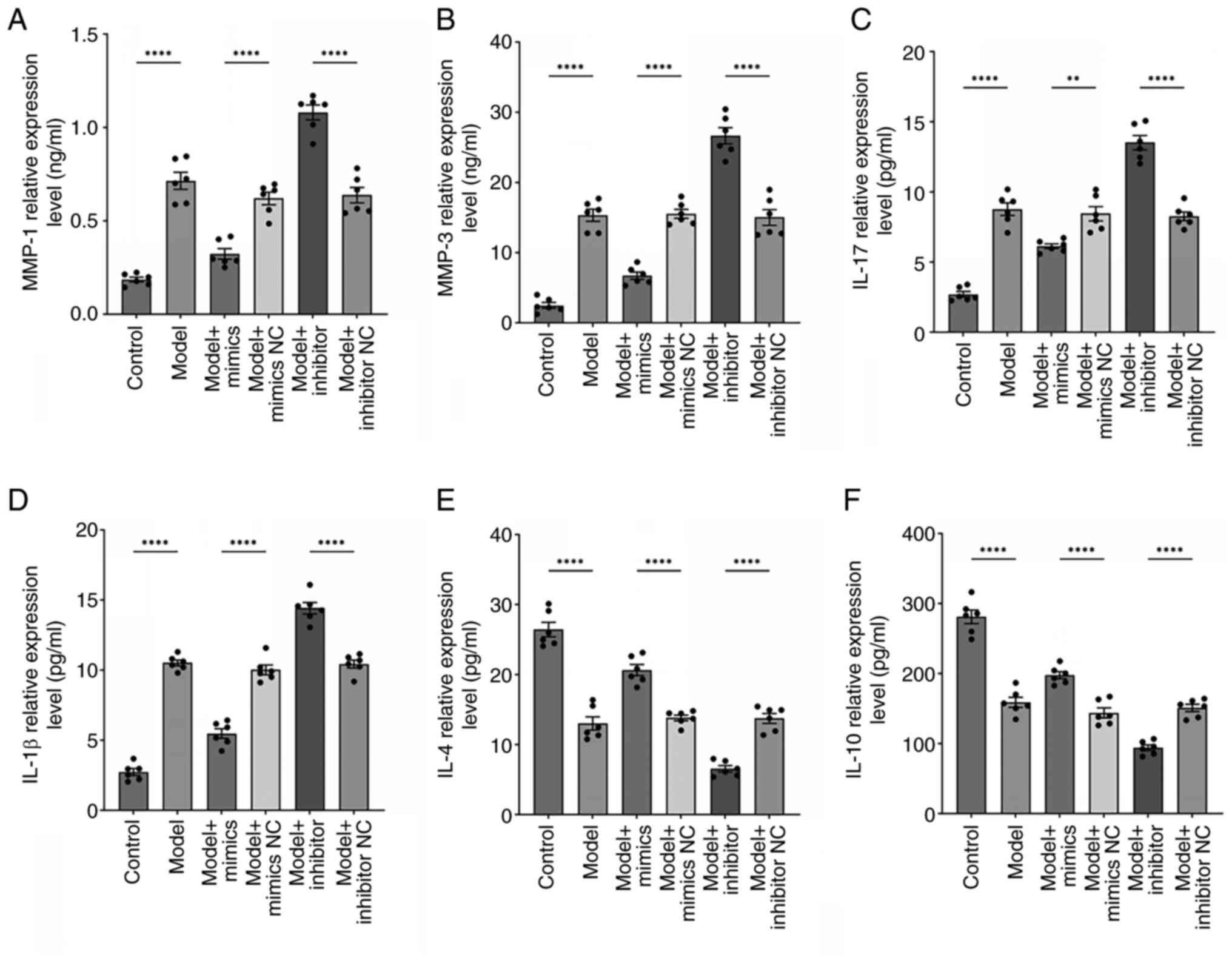
miR-369-3p regulates the
immuno-inflammatory microenvironment in RA
Previous research has highlighted a tightly
regulated pathological network involving FLSs, matrix
metalloproteinases (MMPs) and inflammatory mediators in RA, which
collectively drive synovial inflammation, cartilage destruction and
disease progression (23,24). In the present study, the expression
profiles of two key MMPs (MMP-1 and MMP-3) were investigated and
both pro-inflammatory (IL-17, IL-1β) and anti-inflammatory (IL-4,
IL-10) cytokines were analyzed. ELISA results (Fig. 6A and B) showed that MMP-1 and MMP-3
expression were markedly elevated in the model group compared with
the controls. Transfection with miR-369-3p mimics markedly reduced
MMP-1 and MMP-3 levels relative to the mimic-NC group
(P<0.0001), whereas the inhibitor group showed the opposite
effect compared with its negative control (P<0.0001). Likewise,
model group cells secreted higher levels of IL-17 and IL-1β and
lower levels of IL-4 and IL-10 compared with controls
(P<0.0001). miR-369-3p mimics increased IL-4/IL-10 expression
and suppressed IL-17/IL-1β, while inhibition of miR-369-3p reversed
this pattern (P<0.0001; Fig.
6C-F). These findings suggested that the upregulation of
miR-369-3p mitigates the inflammatory environment in the co-culture
system, whereas its inhibition exacerbates inflammatory
responses.
miR-369-3p ameliorates bone
destruction indicators in RA by regulating the SPTBN1/Wnt/β-catenin
signaling pathway expression
To further investigate the role of miR-369-3p in
regulating bone destruction markers and its involvement in the
SPTBN1/Wnt/β-catenin pathway, bone-related factors were measured
using ELISA and protein expression was evaluated through western
blotting. ELISA results showed that miR-369-3p overexpression
markedly increased osteoprotegerin (OPG) levels, while decreasing
Dickkopf-related protein 1 (DKK1), receptor activator of nuclear
factor κ B (RANK) and RANK ligand (RANKL) expression; however,
miR-369-3p inhibition produced the opposite effect (Fig. 7A-D). Similarly, western blot
analysis (Fig. 7E-H) revealed
higher expression of SPTBN1, Wnt5 and β-catenin proteins in the
model group compared with the control group. In comparison to the
mimic-NC group, cells transfected with miR-369-3p mimics showed
reduced protein expression of SPTBN1, Wnt5 and β-catenin.
Inhibition of miR-369-3p led to increased expression of these
proteins compared with the inhibitor-NC group. These findings
indicated that miR-369-3p overexpression suppresses SPTBN1, Wnt5
and β-catenin protein levels, likely through regulation of the
SPTBN1/Wnt/β-catenin signaling pathway.
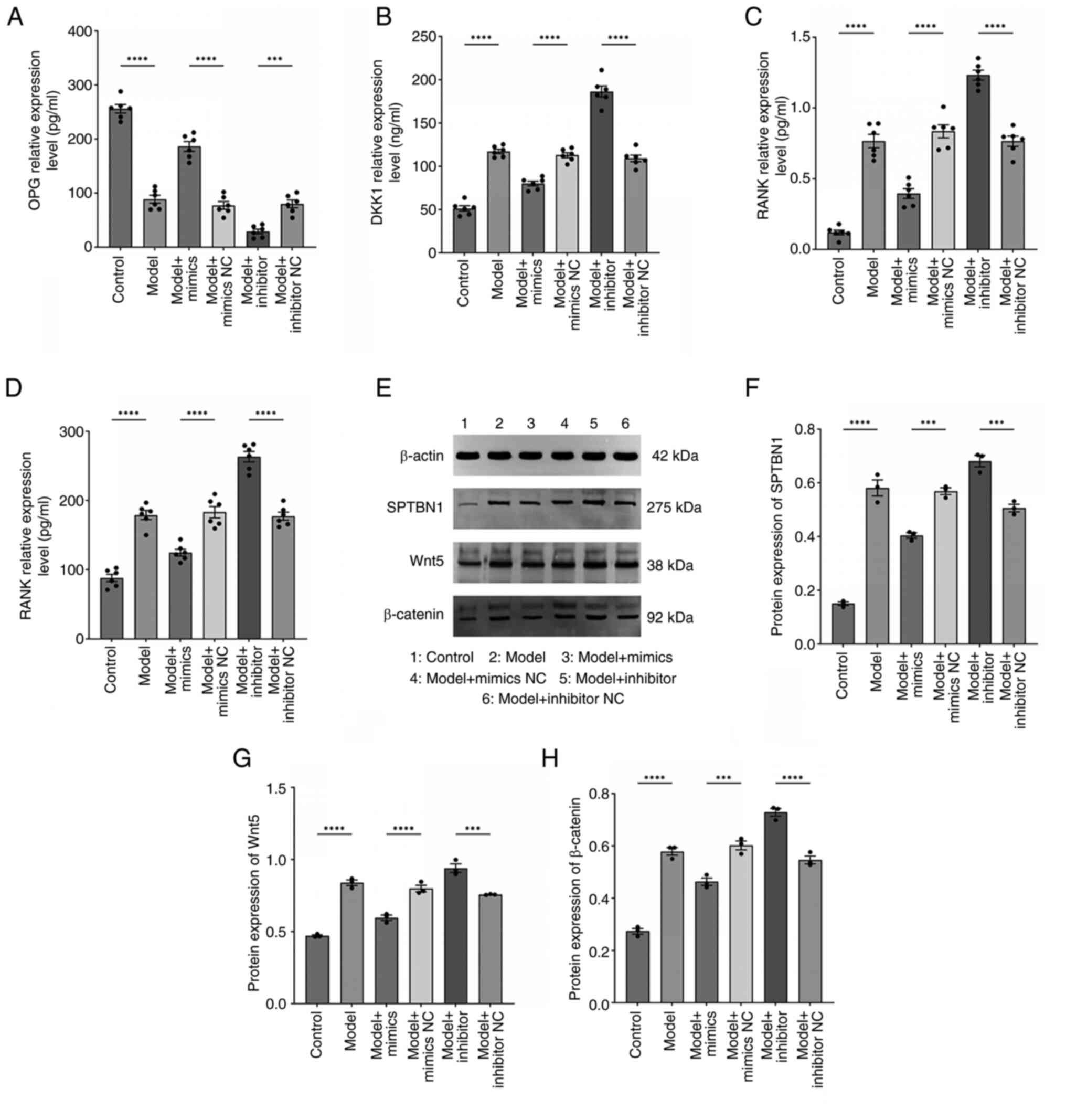 | Figure 7.miR-369-3p regulates RA bone
destruction and the expression of the SPTBN1/Wnt/β-catenin pathway.
ELISA employed for the determination of (A) OPG (pg/ml), (B) DKK1
(ng/ml), (C) RANK (pg/ml) and (D) RANKL (pg/ml) expression in all
cell groups. (n=6). ****P<0.0001. ***P<0.001. (E-H) Western
blot employed for the determination of SPTBN1, Wnt and β-catenin
protein expression in all cell groups (n=3). ****P<0.0001.
***P<0.001. miR, microRNA; RA, rheumatoid arthritis; SPTBN1,
spectrin β, non-erythrocytic 1; OPG, osteoprotegerin; DKK1,
Dickkopf-related protein 1; RANK, receptor activator of nuclear
factor κ B; RANKL, RANK ligand. |
Discussion
The present study began by establishing a co-culture
system of RA-FLSs and RA-PBMCs to simulate the synovial
microenvironment observed in RA more accurately. Comparison of
inflammatory cytokine levels, specifically IL-17 and IL-1β, between
RA-FLSs cultured alone and those in co-culture revealed markedly
higher concentrations in the co-culture group. These findings
suggested that the crosstalk between RA-FLSs and RA-PBMCs activates
inflammatory signaling pathways, amplifying the inflammatory
cascade. This approach more accurately replicates the pathological
interactions between immune cells and stromal cells within RA
synovial tissue. Using CCK-8 assays showed that a co-culture ratio
of 1:3 (RA-FLSs:RA-PBMCs) with a 48-h incubation period provided
optimal stimulation conditions. In parallel, our bioinformatics
analyses predicted potential binding sites for miR-369-3p on
SPTBN1, which were validated through luciferase reporter
assays.
Focusing on the molecular mechanisms underlying
immune inflammation and bone destruction in RA, the present study
revealed that RA-FLSs exhibited decreased miR-369-3p expression,
accompanied by elevated SPTBN1 levels. It was hypothesized that
miR-369-3p directly targets SPTBN1 and regulates the Wnt/β-catenin
signaling pathway, driving RA-FLS proliferation, intensifying
immune-mediated inflammation and contributing to bone damage. By
modulating miR-369-3p expression and assessing its effect on cell
viability, cell cycle progression, proliferation and migration, it
was found that overexpression of miR-369-3p substantially inhibited
the viability, proliferation and migratory activity of RA-FLS.
Simultaneously, the upregulation of miR-369-3p increased the
secretion of anti-inflammatory cytokines while reducing the
production of pro-inflammatory cytokines, MMPs and markers of bone
destruction.
Western blot analyses confirmed that miR-369-3p
modulates immune inflammation and bone destruction through the
SPTBN1/Wnt/β-catenin signaling pathway. To further illustrate the
hierarchical regulatory relationships of this pathway, a schematic
diagram summarizing the entire mechanism is provided in Fig. 8. These findings demonstrated that
miR-369-3p acts as a key epigenetic regulator linking synovial
inflammation to bone erosion through the SPTBN1/Wnt/β-catenin
pathway, highlighting its potential as a therapeutic target for RA.
Furthermore, the miR-369-3p/SPTBN1/Wnt/β-catenin pathway may
constitute a novel post-transcriptional regulatory pathway that
contributes to the epigenetic network governing RA pathogenesis
through miRNA-mediated gene silencing. This pathway may serve a
crucial role in regulating immune inflammation and bone
destruction, underscoring its significance in the molecular
pathogenesis of RA.
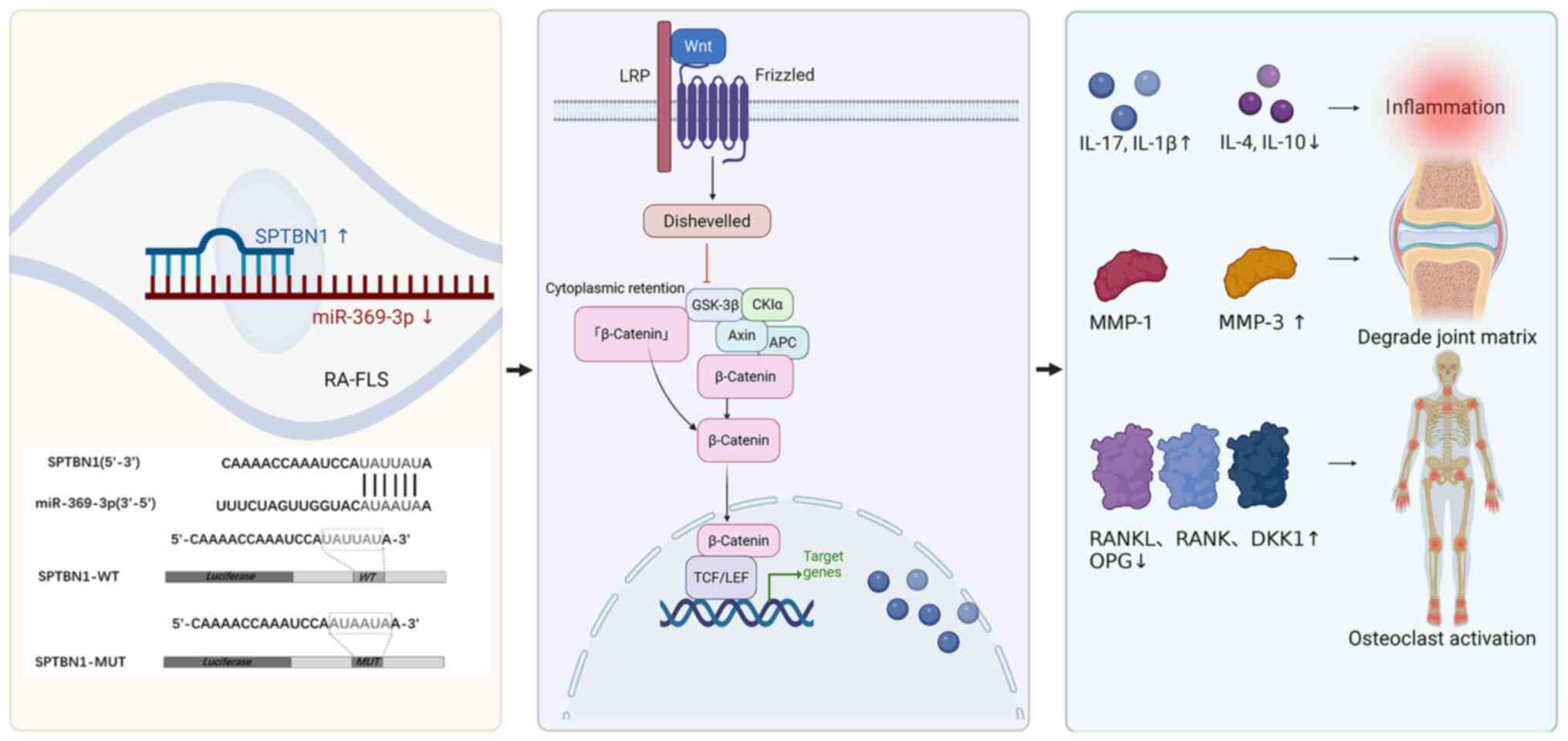 | Figure 8.Schematic diagram of the mechanism
through which miR-369-3p regulates inflammation and bone
destruction via SPTBN1/Wnt/β-catenin pathway. miR, microRNA;
SPTBN1, spectrin β, non-erythrocytic 1; MMP, matrix
metalloproteinases; IL, interleukin; DKK1, Dickkopf-related protein
1; RANK, receptor activator of nuclear factor κ B; RANKL, RANK
ligand; LRP, low-Density Lipoprotein Receptor-Related Protein; TCF,
T-Cell Factor; LEF, Lymphoid Enhancer-Binding Factor. |
The FLSs are a key mesenchymal cell population that
serves as a central mediator of joint damage in RA, primarily due
to their secretion of MMPs, which drive bone resorption (25). In addition, FLS-derived chemokines
recruit leukocytes to the synovium, contributing critically to the
initiation and perpetuation of synovitis. Therefore, targeting FLSs
has emerged as a promising strategy for restoring synovial
homeostasis in RA (26). The
miRNAs, as non-coding RNAs, act as molecular switches in autoimmune
diseases by regulating gene expression through targeted mRNA
degradation or translational inhibition (3,27).
Growing evidence suggests that miRNA-mediated dysregulation of FLS
functions, including proliferation, apoptosis and migration, plays
a pivotal role in RA progression through downstream signaling
pathways (28). Among these,
miR-369-3p has been identified as a pleiotropic regulator of
metabolism and inflammation, demonstrating protective effects in
multiple diseases (21). Its
upregulation suppresses lipopolysaccharide (LPS)-induced
inflammatory responses by downregulating cytokines such as C/EBP-β,
TNF-α and IL-6 and inhibits NLRP3 inflammasome activation through
modulation of deubiquitination pathways (such as Brcc3
downregulation in macrophages), reducing caspase-1-mediated
secretion of pro-inflammatory mediators, including IL-1β and IL-18
(29–31). Due to the key role of these
inflammatory cascades and cytokine networks in RA pathogenesis, the
dynamic expression of miR-369-3p is likely critical for disease
progression. In line with previous studies, the present study
demonstrated downregulation of miR-369-3p in RA-FLSs. Functional
experiments further reveal that modulation of miR-369-3p markedly
reduced SPTBN1 expression and influenced both immune-mediated
inflammation and bone metabolism via the Wnt/β-catenin signaling
pathway.
SPTBN1, which encodes βII-spectrin, is a member of
the spectrin family and serves as an actin-crosslinking
cytoskeletal protein, playing a key role in organizing
transmembrane proteins and cellular organelles (32). Emerging studies have shown that
SPTBN1 modulates disease progression and prognosis by influencing
multiple signaling pathways. For instance, it suppresses primary
osteoporosis by inhibiting osteoblast activity via the Smad3/TGF-β
and STAT1/Cxcl-9 pathways, therefore reducing bone microvascular
blood flow and decreasing VEGF expression (33). In epithelial ovarian cancer, SPTBN1
inhibits tumor progression by regulating the cell cycle through the
suppression of the JAK/STAT3 signaling pathway via SOCS3 (34). Furthermore, SPTBN1 modulates the
Wnt pathway by regulating Kallistatin, a Wnt inhibitor, restraining
the development of hepatocellular carcinoma (35). SPTBN1 has been proposed as a
biomarker to predict therapeutic responses to Wnt/β-catenin pathway
inhibitors.
As a cytoskeletal protein, SPTBN1 has been
recognized for its tumor-suppressive effects in cancer and
osteoporosis, primarily through the inhibition of the Wnt pathway;
however, its role in RA remains largely unexplored. In the present
study, miR-369-3p directly targets SPTBN1, with this miRNA-mediated
regulation affecting β-catenin nuclear translocation. The present
study revealed for the first time, to the best of the authors'
knowledge, that the SPTBN1/Wnt/β-catenin pathway plays a critical
role in RA-related synovial inflammation and bone destruction,
broadening our understanding of Wnt pathway regulation in the
context of autoimmune disease.
Bone destruction in RA results from the synergistic
interplay between synovial inflammation and osteoclast activation,
with the RANKL/RANK/OPG and the Wnt signaling pathway serving as
central regulatory nodes. The present study demonstrated that
miR-369-3p orchestrated the regulation of inflammatory cytokines
and bone metabolism markers through the SPTBN1/Wnt/β-catenin
pathway, highlighting the integrative role of epigenetic factors
within the ‘immunity-bone’ regulatory network. This finding
overcomes the limitations of traditional single-pathway approaches
by revealing the crosstalk between immune regulation and skeletal
homeostasis.
However, the present study has certain limitations.
It relies exclusively on in vitro cell models and lacks
validation in RA animal models or clinical samples. Further
investigations are required to confirm the in vitro
expression patterns and functional relevance of miR-369-3p. In
addition, the upstream regulatory mechanisms of miR-369-3p, such as
transcription factors or inflammatory signals influencing its
promoter, remain unexplored, which limits a comprehensive
understanding of its hierarchical regulation in RA.
Translating miR-369-3p-based therapies into clinical
applications also faces several challenges. First, delivery
efficiency is a major concern: Naked miRNAs are rapidly degraded by
nucleases in vivo and current systemic administration
methods struggle to target RA-affected synovial tissues
effectively, potentially reducing therapeutic impact. Second, miRNA
stability remains an issue, even with delivery vectors. In
addition, the miRNAs may be quickly cleared by the
reticuloendothelial system, necessitating repeated administration
and increasing the risk of off-target effects. Third, the potential
for immune activation cannot be ignored: Exogenous miRNAs or
delivery systems, such as lipid nanoparticles, may provoke innate
immune responses via Toll-like receptors, unintentionally
exacerbating inflammation in RA. Finally, off-target effects are
intrinsic to miRNA therapies because a single miRNA can regulate
multiple mRNAs. For instance, miR-369-3p may influence genes
unrelated to the SPTBN1/Wnt/β-catenin pathway in other tissues,
potentially disrupting normal bone metabolism or immune
homeostasis.
Future studies will aim to validate the therapeutic
efficacy of miR-369-3p mimics in animal models and clinical
cohorts, assessing their capacity to alleviate joint inflammation
and prevent bone erosion. At the same time, efforts will focus on
optimizing delivery strategies, such as the development of
synovial-targeted nanocarriers, to improve the stability and
tissue-specific delivery of miR-369-3p mimics. Furthermore,
bioinformatics-guided target screening, combined with in
vitro validation, will be employed to identify and avoid
off-target genes, while minimizing risks to immune activation.
These approaches aim to improve the safety, specificity and
therapeutic potential of miR-369-3p-based interventions for RA.
Acknowledgements
Not applicable.
Funding
The present study was supported by National Administration of
Traditional Chinese Medicine High-Level Key Discipline Construction
Project-Traditional Chinese Medicine Arthralgia and Bi Syndrome
[project no.: (2023)85], Anhui Provincial Clinical Medical Research
Transformation Special Project (grant no. 202304295107020110) and
the Anhui Province Young Leading Talents Support Program, funded by
the Central Government Development Office [grant no. (2022)4].
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
AW was involved in the study conception and protocol
design, the collection and collation of experimental data; drafted
the initial manuscript and revised the paper. YW was responsible
for experimental design, verification and validation. FL and SD
were responsible for the statistical analysis of experimental data.
ZC and MS participated in the experimental process. AW and YW
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The collection and use of peripheral blood samples
from patients with RA were approved by the Ethics Committee of the
First Affiliated Hospital of Anhui University of Chinese Medicine
(approval no.: 2023AH-52). All patients with RA signed the informed
consent form prior to blood sample collection.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ding Q, Hu W, Wang R, Yang Q, Zhu M, Li M,
Cai J, Rose P, Mao J and Zhu YZ: Signaling pathways in rheumatoid
arthritis: Implications for targeted therapy. Signal Transduct
Target Ther. 8:682023. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kmiołek T and Paradowska-Gorycka A: miRNAs
as biomarkers and possible therapeutic strategies in rheumatoid
arthritis. Cells. 11:4522022. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Payet M, Dargai F, Gasque P and Guillot X:
Epigenetic regulation (including micro-RNAs, DNA methylation and
histone modifications) of rheumatoid arthritis: A systematic
review. Int J Mol Sci. 22:121702021. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nag S, Mitra O, Tripathi G, Samanta S,
Bhattacharya B, Chandane P, Mohanto S, Sundararajan V, Malik S,
Rustagi S, et al: Exploring the theranostic potentials of miRNA and
epigenetic networks in autoimmune diseases: A comprehensive review.
Immun Inflamm Dis. 11:e11212023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yao Q, Chen Y and Zhou X: The roles of
microRNAs in epigenetic regulation. Curr Opin Chem Biol. 51:11–17.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Diener C, Keller A and Meese E: The
miRNA-target interactions: An underestimated intricacy. Nucleic
Acids Res. 52:1544–1557. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Sell MC, Ramlogan-Steel CA, Steel JC and
Dhungel BP: MicroRNAs in cancer metastasis: Biological and
therapeutic implications. Expert Rev Mol Med. 25:e142023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gao B, Sun G, Wang Y, Geng Y, Zhou L and
Chen X: microRNA-23 inhibits inflammation to alleviate rheumatoid
arthritis via regulating CXCL12. Exp Ther Med. 21:4592021.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bao X, Ma L and He C: MicroRNA-23a-5p
regulates cell proliferation, migration and inflammation of
TNF-α-stimulated human fibroblast-like MH7A synoviocytes by
targeting TLR4 in rheumatoid arthritis. Exp Ther Med. 21:4792021.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang X, Liu D, Cui G and Shen H:
Circ_0088036 mediated progression and inflammation in
fibroblast-like synoviocytes of rheumatoid arthritis by
miR-1263/REL-activated NF-κB pathway. Transpl Immunol.
73:1016042022. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou Q, Haupt S, Kreuzer JT, Hammitzsch A,
Proft F, Neumann C, Leipe J, Witt M, Schulze-Koops H and Skapenko
A: Decreased expression of miR-146a and miR-155 contributes to an
abnormal treg phenotype in patients with rheumatoid arthritis. Ann
Rheum Dis. 74:1265–1274. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu N, Feng X, Wang W, Zhao X and Li X:
Paeonol protects against TNF-α-induced proliferation and cytokine
release of rheumatoid arthritis fibroblast-like synoviocytes by
upregulating FOXO3 through inhibition of miR-155 expression.
Inflamm Res. 66:603–610. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zisman D, Safieh M, Simanovich E, Feld J,
Kinarty A, Zisman L, Gazitt T, Haddad A, Elias M, Rosner I, et al:
Tocilizumab (TCZ) decreases angiogenesis in rheumatoid arthritis
through its regulatory effect on miR-146a-5p and EMMPRIN/CD147.
Front Immunol. 12:7395922021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kmiołek T, Rzeszotarska E, Wajda A,
Walczuk E, Kuca-Warnawin E, Romanowska-Próchnicka K, Stypinska B,
Majewski D, Jagodzinski PP, Pawlik A and Paradowska-Gorycka A: The
interplay between transcriptional factors and MicroRNAs as an
important factor for Th17/Treg balance in RA patients. Int J Mol
Sci. 21:71692020. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Safari F, Damavandi E, Rostamian AR,
Movassaghi S, Imani-Saber Z, Saffari M, Kabuli M and Ghadami M:
Plasma levels of MicroRNA-146a-5p, MicroRNA-24-3p, and
MicroRNA-125a-5p as potential diagnostic biomarkers for rheumatoid
arthris. Iran J Allergy Asthma Immunol. 20:326–337. 2021.PubMed/NCBI
|
|
16
|
Wang C, Wang X, Cheng H and Fang J:
MiR-22-3p facilitates bone marrow mesenchymal stem cell
osteogenesis and fracture healing through the SOSTDC1-PI3K/AKT
pathway. Int J Exp Path. 105:52–63. 2024. View Article : Google Scholar
|
|
17
|
Wang S, Xiong G, Ning R, Pan Z, Xu M, Zha
Z and Liu N: LncRNA MEG3 promotes osteogenesis of hBMSCs by
regulating miR-21-5p/SOD3 axis. Acta Biochim Pol. 69:71–77.
2022.PubMed/NCBI
|
|
18
|
Lian F, Zhao C, Qu J, Lian Y, Cui Y, Shan
L and Yan J: Icariin attenuates titanium particle-induced
inhibition of osteogenic differentiation and matrix mineralization
via miR-21-5p. Cell Biol Int. 42:931–939. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chang C, Xu L, Zhang R, Jin Y, Jiang P,
Wei K, Xu L, Shi Y, Zhao J, Xiong M, et al: MicroRNA-Mediated
epigenetic regulation of rheumatoid arthritis susceptibility and
pathogenesis. Front Immunol. 13:8388842022. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Scalavino V, Piccinno E, Labarile N,
Armentano R, Giannelli G and Serino G: Anti-inflammatory effects of
miR-369-3p via PDE4B in intestinal inflammatory response. Int J Mol
Sci. 25:84632024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Rawal S, Randhawa V, Rizvi SHM, Sachan M,
Wara AK, Pérez-Cremades D, Weisbrod RM, Hamburg NM and Feinberg MW:
miR-369-3p ameliorates diabetes-associated atherosclerosis by
regulating macrophage succinate-GPR91 signalling. Cardiovasc Res.
120:1693–1712. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mahmoud DE, Kaabachi W, Sassi N, Tarhouni
L, Rekik S, Jemmali S, Sehli H, Kallel-Sellami M, Cheour E and
Laadhar L: The synovial fluid fibroblast-like synoviocyte: A
long-neglected piece in the puzzle of rheumatoid arthritis
pathogenesis. Front Immunol. 13:9424172022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Xin PL, Jie LF, Cheng Q, Bin DY and Dan
CW: Pathogenesis and function of interleukin-35 in rheumatoid
arthritis. Front Pharmacol. 12:6551142021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Waltereit-Kracke V, Wehmeyer C, Beckmann
D, Werbenko E, Reinhardt J, Geers F, Dienstbier M, Fennen M,
Intemann J, Paruzel P, et al: Deletion of activin a in mesenchymal
but not myeloid cells ameliorates disease severity in experimental
arthritis. Ann Rheum Dis. 81:1106–1118. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Nygaard G and Firestein GS: Restoring
synovial homeostasis in rheumatoid arthritis by targeting
fibroblast-like synoviocytes. Nat Rev Rheumatol. 16:316–333. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Mirzaei R, Zamani F, Hajibaba M,
Rasouli-Saravani A, Noroozbeygi M, Gorgani M, Hosseini-Fard SR,
Jalalifar S, Ajdarkosh H, Abedi SH, et al: The pathogenic,
therapeutic and diagnostic role of exosomal microRNA in the
autoimmune diseases. J Neuroimmunol. 358:5776402021. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Peng Y, Zhang M and Hu J: Non-coding RNAs
involved in fibroblast-like synoviocyte functioning in arthritis
rheumatoid: From pathogenesis to therapy. Cytokine. 173:1564182024.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Scalavino V, Liso M, Cavalcanti E, Gigante
I, Lippolis A, Mastronardi M, Chieppa M and Serino G: miR-369-3p
modulates inducible nitric oxide synthase and is involved in
regulation of chronic inflammatory response. Sci Rep. 10:159422020.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Galleggiante V, De Santis S, Liso M, Verna
G, Sommella E, Mastronardi M, Campiglia P, Chieppa M and Serino G:
Quercetin-induced miR-369-3p suppresses chronic inflammatory
response targeting C/EBP-β. Mol Nutr Food Res. 63:e18013902019.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Scalavino V, Piccinno E, Valentini AM,
Schena N, Armentano R, Giannelli G and Serino G: miR-369-3p
modulates intestinal inflammatory response via BRCC3/NLRP3
inflammasome axis. Cells. 12:21842023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Susuki K, Zollinger DR, Chang KJ, Zhang C,
Huang CY, Tsai CR, Galiano MR, Liu Y, Benusa SD, Yermakov LM, et
al: Glial βII spectrin contributes to paranode formation and
maintenance. J Neurosci. 38:6063–6075. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Xu X, Yang J, Ye Y, Chen G, Zhang Y, Wu H,
Song Y, Feng M, Feng X, Chen X, et al: SPTBN1 prevents primary
osteoporosis by modulating osteoblasts proliferation and
differentiation and blood vessels formation in bone. Front Cell Dev
Biol. 9:6537242021. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen M, Zeng J, Chen S, Li J, Wu H, Dong
X, Lei Y, Zhi X and Yao L: SPTBN1 suppresses the progression of
epithelial ovarian cancer via SOCS3-mediated blockade of the
JAK/STAT3 signaling pathway. Aging (Albany NY). 12:10896–10911.
2020. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhi X, Lin L, Yang S, Bhuvaneshwar K, Wang
H, Gusev Y, Lee MH, Kallakury B, Shivapurkar N, Cahn K, et al:
βII-Spectrin (SPTBN1) suppresses progression of hepatocellular
carcinoma and Wnt signaling by regulation of Wnt inhibitor
kallistatin. Hepatology. 61:598–612. 2015. View Article : Google Scholar : PubMed/NCBI
|