Introduction
The occurrence of renal cell carcinoma (RCC), one of
the most prevalent malignant tumors of the urinary system, is
becoming increasingly common in numerous countries, with high
incidence and mortality rates (1).
RCC encompasses three subtypes [papillary, chromophobe and clear
cell RCC (ccRCC)], with ccRCC being the most prevalent (2). Although early surgical or ablative
interventions can improve patient survival, approximately one-third
of patients develop metastases, and a quarter of them experience
recurrent metastasis following treatment (3). Unlike other types of cancer, ccRCC is
resistant to chemotherapy and radiotherapy. Advancements in
targeted therapies, immunotherapy and combination therapies have
extended the survival of patients with advanced ccRCC to a certain
extent (4). However, not all
patients respond well to these treatments, and the majority of them
will develop resistance due to the immune microenvironment and
immune escape mechanisms of the tumor (5). Patients with early-stage ccRCC have
been shown to benefit from immediate surgical intervention,
although the 5-year survival rate for advanced cases is only 23%
(6). Consequently, there is an
urgent need to deepen the understanding of RCC. Both exploring
novel molecular targets and developing personalized treatment
strategies are crucial for improving survival and treatment
outcomes, especially for patients with advanced disease.
Alternative splicing (AS) is a critical regulatory
mechanism in eukaryotic gene expression, enabling the generation of
protein diversity through spliceosome-mediated exon-intron
rearrangement (7). This process
allows a single gene to produce multiple mRNA variants that encode
distinct proteins, thereby fulfilling vital roles in cellular
development. Dysregulation of AS is a hallmark of tumorigenesis,
contributing to therapeutic resistance (8–10).
Splicing factor 3a subunit 2 (SF3A2), an essential component of the
spliceosome, has emerged as a key regulator in oncogenic processes
and non-neoplastic diseases, functioning through the control of
alternative splicing (AS) networks and protein-protein interactions
(11). In myocardial
ischemia/reperfusion injury, a previous study showed that the
ginsenoside Rb2 mitigated cardiomyocyte damage by inhibiting
p300-mediated SF3A2-K10 acetylation, thereby promoting AS of fascin
actin-bundling protein 1 (FSCN1) and enhancing mitochondrial
respiration (12). These findings
highlighted the role of SF3A2 in cardiovascular pathophysiology,
suggesting that its acetylation status serves as a potential
prognostic biomarker. SF3A2 exhibits context-dependent functions in
various malignancies; for example, in Ewing sarcoma (ES), its
interaction with DExH-box helicase 9 (DHX9) suppresses tumor
progression through splicing modulation (13), whereas in triple-negative breast
cancer (TNBC), SF3A2 overexpression drives cisplatin resistance
through the activation of makorin ring finger protein 1 (MKRN1)
splicing and ubiquitin protein ligase E3 component N-recognin 5
feedback reinforcement (14).
Considered altogether, these dual roles underscore the multifaceted
regulatory potential of SF3A2 across different diseases.
To the best of our knowledge, the role of SF3A2 in
ccRCC progression has not yet been elucidated. The present study
integrated bioinformatics analyses with experimental validation to
characterize the functional involvement of SF3A2 in ccRCC
pathogenesis, thereby providing a basis for exploring SF3A2 as a
potential therapeutic target.
Materials and methods
Open-access data utilization
The present study employed a multimodal analytical
framework to explore the functional dynamics of SF3A2 in ccRCC.
Multi-omics clinical data were sourced from The Cancer Genome Atlas
Kidney Renal Clear Cell Carcinoma (TCGA-KIRC) dataset [n=539 tumor
specimens with RNA-sequencing (RNA-seq) profiles; http://tcga-data.nci.nih.gov/tcga] and the
Genotype-Tissue Expression (GTEx) portal (n=73 normal renal
transcriptomes; http://www.gtexportal.org/). Differential expression
profiling was performed using Gene Expression Profiling Interactive
Analysis 2 (http://gepia2.cancerpku.cn/#index), applying
predefined thresholds (|log2 fold change| >1; false
discovery rate <0.05). Prognostic relevance was assessed using
Kaplan-Meier methodology and log-rank testing for overall survival
(OS). Protein-level validation was achieved by assessing
immunohistochemical data from the Human Protein Atlas repository
(https://www.proteinatlas.org/) (15).
Cell culture systems and viral
transduction
Experimental models included renal tubular
epithelial cells (HK-2) and RCC cell lines (786-O, ACHN, 769-P,
OSRC-2 and A498; all cell lines were purchased from The Cell Bank
of Type Culture Collection of The Chinese Academy of Sciences and
authenticated by short tandem repeat profiling). HK-2 cells were
cultured in DMEM. 786-O, 769-P and OSRC-2 cells were maintained in
RPMI-1640 medium, ACHN cells were grown in MEM, and A498 cells were
cultured in EMEM (or MEM when EMEM was unavailable). All media were
supplemented with 10% fetal bovine serum (cat. no. A5256701; Thermo
Fisher Scientific, Inc.) and 1% penicillin-streptomycin, and all
cells were maintained at 37°C in a humidified incubator containing
5% CO2.
Lentivirus-mediated stable transduction enabled
bidirectional genetic modification: SF3A2 knockdown was achieved
using validated short hairpin (sh)RNAs (shRNAs/shs; sequences of
shRNAs 1–2, respectively: 5′-ACATCAACAAGGACCCGTACT-3′ and
5′-CAAAGTGACCAAGCAGAGAGA-5′). A non-targeting shRNA
(5′-CAACAAGATGAAGAGCACCAA-3′) was used as the negative control. For
overexpression assays, SF3A2 was introduced using an OriGene
LentiORF™ lentiviral expression vector (cat. no. RC201244L4;
Origene Technologies, Inc.), which is based on the
pLenti-CMV-ORF-Puro backbone. The corresponding empty
vector(pLenti-CMV-Puro; cat. no. PS100093V; Origene Technologies,
Inc.) was used as the negative control. Viral particles were
packaged in 293T cells through co-transfection with psPAX2 (cat.
no. #12260; Addgene, Inc.) and pMD2.G (cat. no. #12259; Addgene,
Inc.), and cell supernatant was harvested at 48 h
post-transfection, and subsequently sterile-filtered (0.45-µm
filters). SF3A2 knockdown was performed using pLKO.1-based shRNA
lentiviral plasmids (shRNA-1 and shRNA-2; Shanghai GeneChem Co.,
Ltd.), with a non-targeting pLKO.1 shRNA serving as the control.
Lentiviruses were produced using a third-generation packaging
system in 293T cells (ATCC CRL-3216, obtained from the Cell Bank of
the Chinese Academy of Sciences). 293T cells at 60–70% confluence
were transfected using Lipofectamine® 3000 (Thermo
Fisher Scientific, Inc.) with 4 µg transfer plasmid (pLKO.1-shRNA
or pCMV-SF3A2), 3 µg psPAX2, and 1 µg pMD2.G (total 8 µg DNA per
10-cm dish; 4:3:1 ratio). After 6–8 h at 37°C, the medium was
replaced, and viral supernatants were collected at 48 and 72 h,
filtered through a 0.45-µm membrane, and used immediately or stored
at −80°C. Target cells (786-O, OSRC-2) were infected at 40–50%
confluence with virus supplemented with 8 µg/ml polybrene for 24 h,
followed by medium replacement and recovery for 48–72 h. Stable
knockdown or overexpression lines were established using puromycin
selection (1.5–2.0 µg/ml for selection; 0.5–1.0 µg/ml for
maintenance) for 3–5 days, and cells were expanded for an
additional 3–5 days before downstream experiments. Target cells
were transduced at a multiplicity of infection of 5, followed by
selection with puromycin (2 µg/ml; cat. no. P8833; Sigma-Aldrich;
Merck KGaA) over a 14-day period to establish stable lines.
Finally, the genetic perturbation efficiency was validated using
reverse transcription-quantitative PCR (RT-qPCR) and western blot
analyses, as described subsequently.
RT-qPCR analysis
Total RNA was extracted from cellular samples using
TRIzol® reagent (cat. no. 15596026CN; Invitrogen; Thermo
Fisher Scientific, Inc.), and RNA purity was quantified using a
NanoDrop2000™ spectrophotometer (Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. RT) was performed
using the ReverTra Ace® qPCR RT Kit (Toyobo Co., Ltd.)
at 37°C for 15 min (cDNA synthesis), 98°C for 5 min (enzyme
inactivation), hold at 4°C. qPCR was performed using
SYBR® Green qPCR Mix (TransGen Biotech Co., Ltd.) on a
QuantStudio™ real-time PCR system (Thermo Fisher Scientific). The
thermocycling conditions were: 95°C for 30 s (initial
denaturation); 40 cycles of: 95°C for 5 s; 60°C for 30 sec
(annealing/extension); Melt curve analysis: 65–95°C, increment
0.5°C every 5 s; SYBR Green®-based qPCR analysis
(TransGen Biotech Co., Ltd.) was performed using the
2−ΔΔCq method. β-actin acted as the endogenous control
(16).
The forward primer sequence for SF3A2 was
5′-GATTGACTACCCTGAGATCGCC-3′, and the sequence of the reverse
primer was 5′-CTCCCGGTTCCAGTGTGTC-3′. The sequences of the primers
for the reference gene, β-actin, were as follows: Forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT-3′.
Western blot analysis
Protein lysates from cultured cells were prepared
using RIPA buffer with 1% protease inhibitor cocktail (cat. no.
P8340; Sigma-Aldrich; Merck KGaA). Protein concentrations were
determined using a BCA assay (Jiangsu CoWin Biotech Co., Ltd.).
Aliquots (20 µg/lane) were resolved using SDS-PAGE (10% gels; Wuhan
Servicebio Technology Co., Ltd.) and transferred onto 0.45-µm PVDF
membranes. After blocking the membranes with 5% non-fat milk for 1
h at room temperature, washed with TBST containing 0.1% Tween-20,
and incubated overnight at 4°C with the following primary
antibodies: SF3A2 (1:1,000; Abcam, cat. no. ab317408), Actin
(1:5,000; CST Biological Reagents Co., Ltd.; cat. no. 4970),
phosphorylated (p-)AKT (1:2,000; CST Biological Reagents Co., Ltd.
cat. no. 4060) and total AKT (1:2,000; CST Biological Reagents Co.,
Ltd. cat. no. 4691) antibodies. Following washes three times for 10
min each, with TBST. The membrane was incubated with the
HRP-conjugated secondary antibodies (1:5,000, Proteintech, cat. no.
SA00001-1 and SA00001-2) in TBST at room temperature for 1 h,
followed by three washes of 10 min each. prior to ECL detection
(Fdbio Science). Band intensities were semi-quantified using Image
Lab software (version 6.1.0, Bio-Rad Laboratories, Inc.).
Immunohistochemical analysis
For histological examination, tissue samples were
fixed in 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) at
4°C for 24 h. Following fixation, the tissues were dehydrated
through a graded ethanol series, cleared in xylene, and embedded in
paraffin blocks. Serial sections of 5 µm thickness were cut using a
rotary microtome and mounted onto glass slides. Sections were
deparaffinized in xylene and rehydrated through a graded ethanol
series to distilled water. The nuclei were stained with Mayer's
hematoxylin for 8 min at room temperature, followed by rinsing in
running tap water for 10 min. Cytoplasmic counterstaining was
performed by immersing the sections in Eosin Y solution for 2 min
at room temperature. Finally, the stained sections were dehydrated
through a graded alcohol series, cleared in xylene, and
coverslipped with a permanent mounting medium. The stained sections
were examined and imaged using a standard light microscope.
Immunohistochemical analysis was performed on paraffin-embedded
specimens as aforementioned. After antigen retrieval in sodium
citrate buffer (pH 6.0) and peroxidase inactivation with 3%
H2O2, sections were incubated overnight at
4°C with anti-SF3A2 (1:200; Abcam, cat. no. ab317408), anti-PCNA
(1:50; Santa Cruz Biotechnology, cat. no. sc-56), phosphorylated
(p-)AKT (1:100; CST Biological Reagents Co., Ltd., cat. no. 4060).
Following three washes with PBS buffer, sections were then
incubated with an anti-mouse/rabbit-specific protein kit (Envision
Plus, Dako, Carpinteria, CA, USA) at room temperature. The
chromogen used was 3-amino-9-ethylcarbazole (AEC, SK-4205, Vector,
Burlingame, CA, USA). The sections were then counterstained with
Meyer's hematoxylin at room temperature for 30–60 sec. Digital
whole-slide images were generated using a domestic high-throughput
slide scanner (PANOVUE VS200 Panoramic Pathology Scanning
System).
Cell proliferation assay
786-O and OSRC-2 cells were seeded at a density of
500 cells/well. After optimized cell seeding in 96-well plates,
Cell Counting Kit-8 (CCK-8) proliferation assays (cat. no. CK04;
Dojindo Laboratories, Inc.) were performed at 24, 48, 72 and 96 h,
with a 2-h reagent incubation. Absorbance measurements at 450 nm
were subsequently recorded using a BioTek™ Synergy H1 microplate
reader (Agilent Technologies, Inc.), with six technical replicates
per experimental condition.
Colony formation assay
786-O and OSRC-2 cells were seeded at 500 cells per
well in 6-well plates and cultured for 10 days at 37°C in a
humidified incubator containing 5% CO2 with 0.1% DMSO or
SC79 (10 µM). Cells were subsequently fixed with 100% methanol for
15 min at room temperature and stained with 0.1% crystal violet for
15 min. Colonies >50 µm in diameter were counted manually across
three independent biological replicates.
Cell migration and invasion
assays
786-O and OSRC-2 cell migration was assessed using
8-µm Corning® Transwell chambers (Corning, Inc.). Cells
were seeded at 3×104/insert in serum-free medium in the
upper chambers and RPMI-1640 medium containing 20% fetal bovine
serum was added to the lower chambers for 36 h at 37°C. Subsequent
assays were initiated after 36 h, with migrated cells fixed in 4%
paraformaldehyde for 20 min at room temperature and stained with
0.1% crystal violet for 1 h at room temperature. Non-migrated cells
were removed using a cotton swab. For the invasion assays,
Matrigel™-coated membranes (50 µl/insert; BD Biosciences) were
used, the coated chambers were incubated at 37°C for 1 h in a cell
culture incubator to allow the Matrigel to form a thin gel, and
3×104 cells were seeded per insert and incubated for 36
h at 37°C. The culture conditions for the upper and lower chambers
of the invasion assay, as well as the fixation and crystal violet
staining steps were performed as aforementioned. In both assays,
cellular penetration was quantified by counting cells in five
randomly selected fields per replicate at a magnification of light
microscope in ×200.
In vivo animal experiments
Subcutaneous xenograft models were established in
the right axillary subcutaneous region of male BALB/c nude mice
(Beijing Vital River Laboratory Animal Technology Co., Ltd.) by
single axillary implantation of 786-O cells (5×106
cells/mouse in total) with stable SF3A2 knockdown or control
constructs (n=5 mice/group). A total of 10 6-week-old male nude
mice with a body weight of ~20 g were used in total. The 786-O
cells were resuspended in PBS at a density of 5×107
cells/ml. A 100-µl aliquot of the cell suspension was injected
subcutaneously into the ventral region of the right anterior armpit
of nude mice. All animals were housed at a suitable temperature
(22–24°C) and humidity (40–70%) under a 12/12-h light/dark cycle
with unrestricted access to food and water. Tumor growth was
monitored biweekly using digital calipers, and tumor volumes were
calculated according to the formula: (Length ×
width2)/2. After 6 weeks, mice were euthanized by
anesthesia with isoflurane (5%), followed by cervical dislocation.
Death was confirmed both by cessation of respiration and heartbeat,
and by the absence of pedal reflexes, prior to tissue collection.
Excised tumors were fixed in 4% paraformaldehyde for 24 h at 4°C
and were processed for subsequent immunohistochemical analyses. The
animal experiments were performed at The First Affiliated Hospital
of Nanchang University (Nanchang, China), which is a certified
facility authorized to conduct laboratory animal research. All
experiments were performed according to the standard Guidelines for
the Care and Use of Laboratory Animals (17). All procedures adhered to the
guidelines of the Association for Assessment and Accreditation of
Laboratory Animal Care International, and the experimental protocol
was reviewed and approved by the Institutional Animal Care and Use
Committee of The First Affiliated Hospital of Nanchang University
[animal license number: SYXK (Gan) 2021-0003; approval no.
CDYFY-IACUC-202212QR030]. Official protocol approval for the
present study was also obtained from the Ethics Committee of The
First Hospital of Putian City(approval no. 2022-036).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism 9.0 (Dotmatics). All experiments were performed with three
independent biological replicates, parametric data were analyzed
using a paired or unpaired two-tailed Student's t-test (for
two-group comparisons) or one-way ANOVA with Tukey's post hoc test
(for multi-group comparisons). Overall survival analysis was
performed using the TCGA-KIRC cohort. Kaplan-Meier survival curves
were generated using the survival (https://CRAN.R-project.org/package=survival;
version 3.3–1) and survminer (https://CRAN.R-project.org/package=survminer; version
0.4.9) packages in R (version 4.2.1; R Foundation for Statistical
Computing, Vienna, Austria; http://www.r-project.org/). Patients were stratified
into high and low SF3A2 expression groups based on the median
expression value, which was used as the cutoff. Curve fitting was
performed using the survfit function, and statistical significance
between groups was assessed using the log-rank test. Data are
presented as the mean ± SD. P<0.05 was considered to indicate a
statistically significant difference.
Results
High SF3A2 expression predicts poor
prognosis in ccRCC
Integrative analysis of TCGA and GTEx datasets
revealed significant dysregulation of SF3A2 in ccRCC.
Transcriptomic profiling demonstrated marked upregulation of SF3A2
mRNA in tumor tissues compared with normal tissues (P<0.001),
with paired-sample validation confirming tumor-specific
upregulation (Fig. 1A and B). High
SF3A2 expression was associated with poor overall survival in
patients in the TCGA-KIRC dataset (log-rank P=0.001). Kaplan-Meier
curves stratified by median expression are shown in Fig. 1C. Furthermore, immunohistochemical
characterization of normal kidney tissues and ccRCC tissues
revealed a nuclear-enriched localization of SF3A2 protein in ccRCC
specimens from HPA (Fig. 1D).
Further orthogonal validation across five ccRCC cell lines (namely,
the 786-O, 769-P, OSRC-2, A498 and ACHN cell lines) compared with
HK-2 control cells confirmed significant increases in SF3A2
expression at the mRNA levels, with statistical significance
maintained across all cell lines with the exception of ACHN cells
(Fig. 1E).
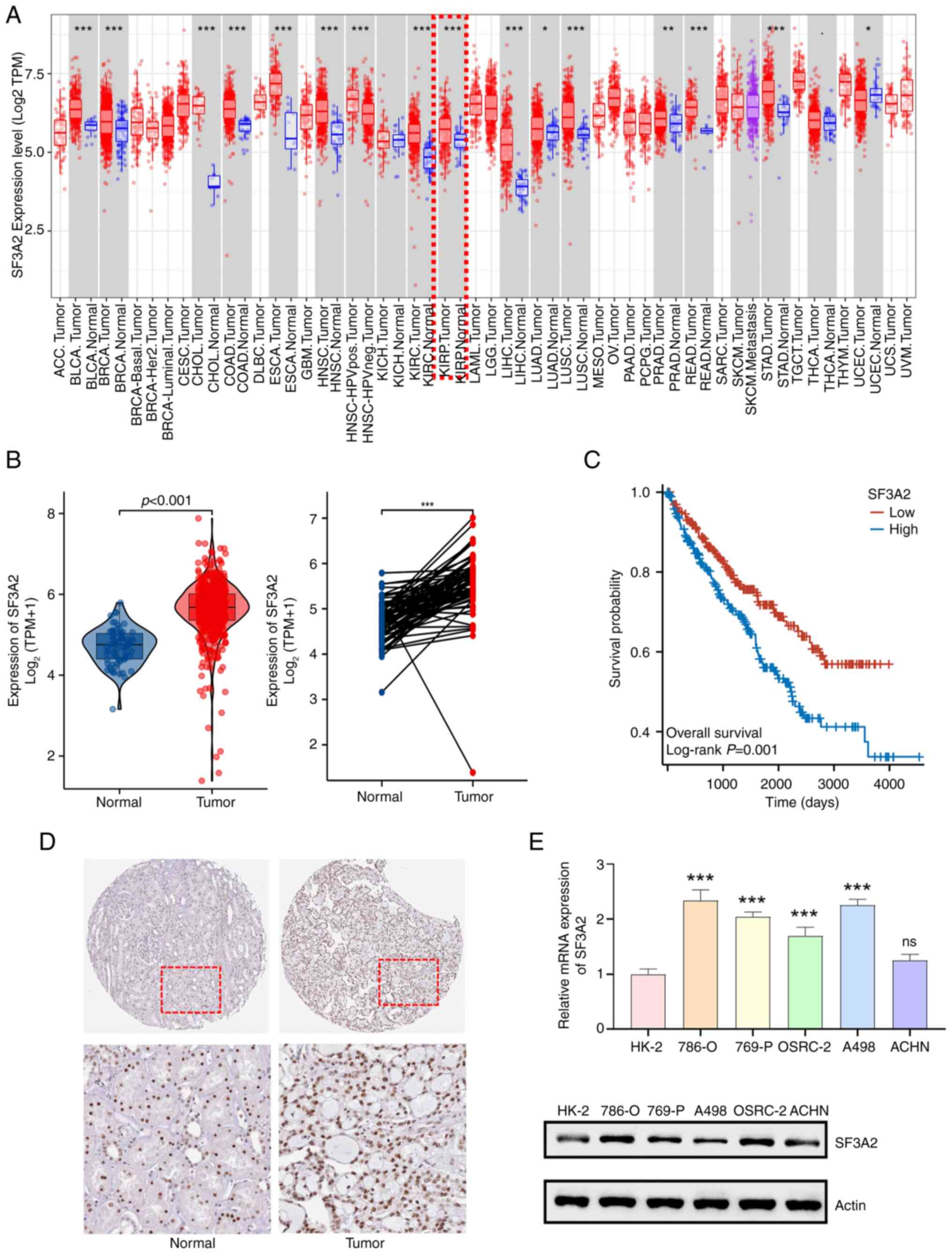 | Figure 1.SF3A2 is highly expressed in ccRCC
and is associated with poorer prognosis. (A) Pan-cancer analysis of
SF3A2 mRNA expression (according to The Cancer Genome
Atlas/Genotype-Tissue Expression databases). ccRCC tissues
(indicated by the red box) exhibited significant upregulation
compared with normal tissues (*P<0.05, **P<0.01
***P<0.001; unpaired t-test). (B) Violin plot comparing SF3A2
mRNA levels in ccRCC tumor tissues vs. normal tissues (unpaired
t-test). Paired analysis confirmed increased SF3A2 expression in
ccRCC tumor tissues compared with matched normal tissues
(***P<0.001; paired t-test). (C) Kaplan-Meier overall survival
curves of patients from The Cancer Genome Atlas Kidney Renal Clear
Cell Carcinoma dataset stratified by SF3A2 expression (median
cut-off). Survival differences were evaluated using the log-rank
test. (D) Immunohistochemical staining, showing weak SF3A2
expression in normal kidney tissues and strong nuclear localization
of SF3A2 in ccRCC tumors. All images were captured at ×200
magnification. (E) Reverse transcription-quantitative PCR analysis
of SF3A2 expression in ccRCC cell lines (786-O, 769-P, OSRC-2, A498
and ACHN) compared with HK-2 normal cells, showing increased SF3A2
mRNA expression in four of the tested cell lines (***P<0.001; ns
in the ACHN cell line; one-way ANOVA). Western blot analysis
confirmed that SF3A2 protein was upregulated in 786-O and OSRC-2
cell lines compared to the HK-2 cell line. ccRCC, clear cell renal
cell carcinoma; ns, not significant; SF3A2, splicing factor 3a
subunit 2; TPM, transcripts per million. |
High SF3A2 expression promotes the
proliferation of ccRCC cells
A stable gene intervention model was subsequently
developed via lentiviral transduction to elucidate the functional
role of SF3A2 in ccRCC. Western blotting and RT-qPCR analyses
confirmed the successful SF3A2 knockdown and overexpression at both
the protein and mRNA levels (Fig. 2A
and B). Functional assays revealed SF3A2 regulated the
proliferation of the cells: CCK-8 assays demonstrated an
attenuation of proliferation of the 786-O and OSRC-2 cells upon
SF3A2 silencing, whereas SF3A2 overexpression led to an enhancement
of the cell replicative potential (Fig. 2C and D). Colony formation assays
further validated these results, showing a significant reduction in
the number of clones formed in the sh-SF3A2 groups compared with
the control, whereas the overexpression group exhibited a
significant enhancement in the numbers of colonies formed (Fig. 2 and F). Taken together, these
findings suggested that SF3A2 may act as an oncogenic driver in
ccRCC, promoting cell proliferation, and thereby establishing its
therapeutic susceptibility for targeted intervention.
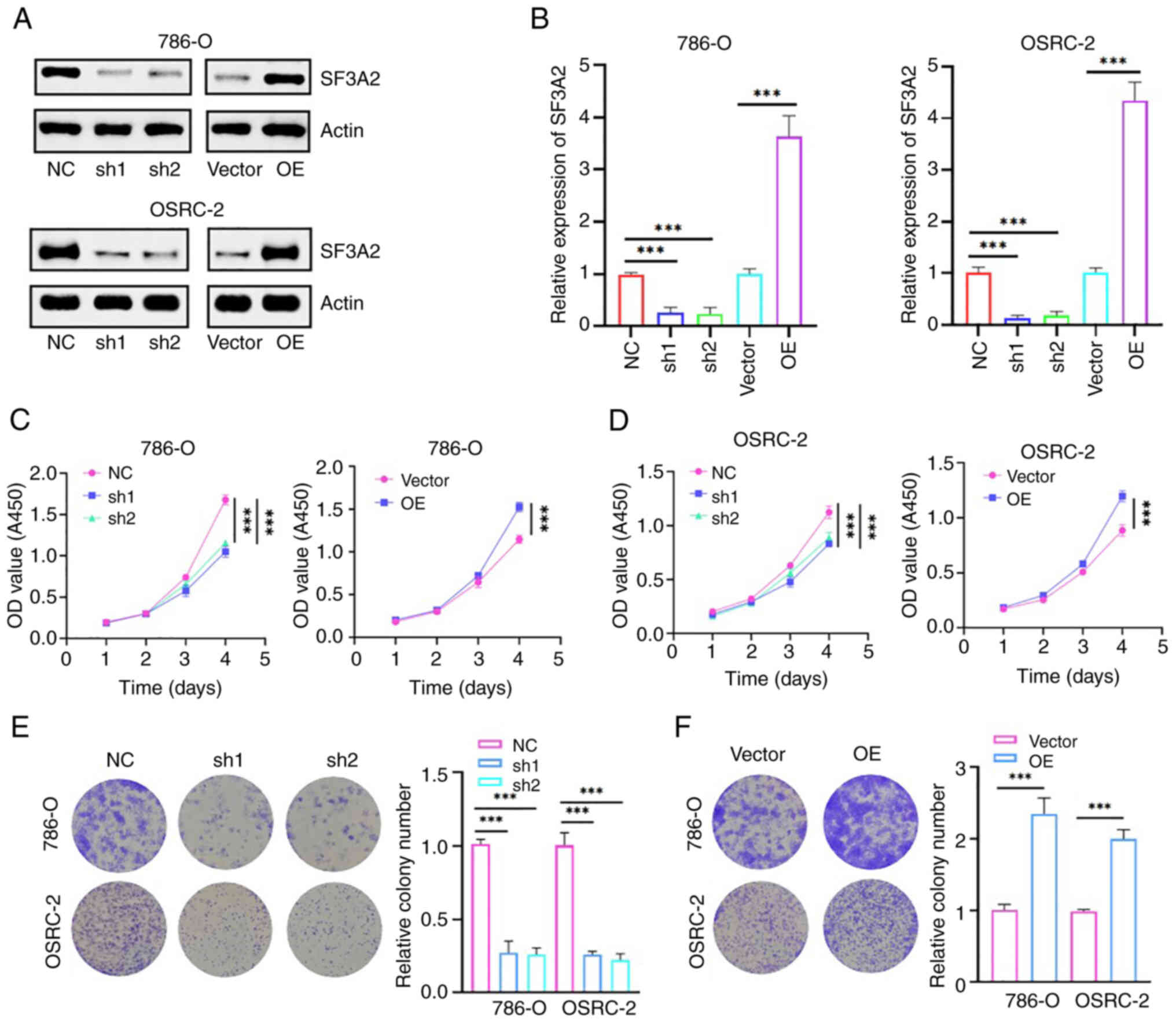 | Figure 2.SF3A2 promotes the proliferation and
colony formation of renal cancer cells. (A) Western blot analysis
of SF3A2 modulation in 786-O and OSRC-2 cells is shown. Effective
knockdown (sh1/sh2) and overexpression of SF3A2 was demonstrated
compared with the controls (NC/Vector). (B) Reverse
transcription-quantitative PCR assays were performed, confirming
SF3A2 transcriptional regulation in 786-O/OSRC-2 cells
(***P<0.001; two-group comparisons were analyzed using unpaired
t-tests, and three-group comparisons were evaluated using one-way
ANOVA). (C) Cell Counting Kit-8 assay, demonstrating
SF3A2-dependent proliferation. (D) sh1/sh2-SF3A2 groups exhibited
suppressed proliferation, whereas the overexpression group
exhibited enhanced proliferation rates (***P<0.001; two-group
comparisons were analyzed using unpaired t-tests, and three-group
comparisons were evaluated using one-way ANOVA). (E) Colony
formation assay showed that SF3A2 expression regulated clonogenic
growth. sh-SF3A2 reduced colony formation. (F) OE promoted
clonogenicity (quantified as relative colony counts; ***P<0.001;
unpaired t-test). NC, negative control; OD, optical density; OE,
overexpression vector; SF3A2, splicing factor 3a subunit 2; sh,
short hairpin RNA. |
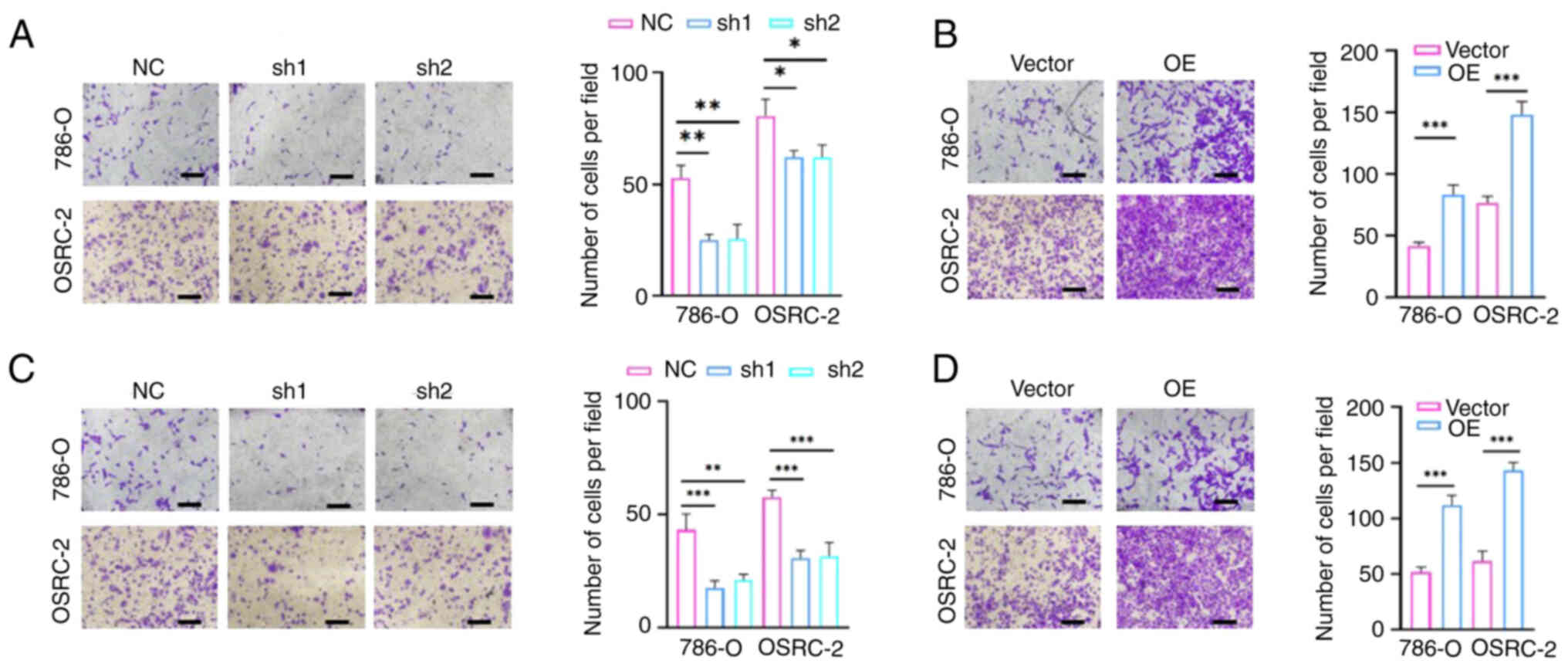
SF3A2 serves an important role in
ccRCC via promotion of the migration and invasion of ccRCC
cells
To assess the regulatory role of SF3A2 in terms of
the invasive phenotype of ccRCC cells, Transwell assays were
performed. SF3A2 knockdown (sh1/sh2-SF3A2) led to a marked
impairment of migration and invasion in the 786-O and OSRC-2 cells,
whereas overexpression of SF3A2 produced an enhancement of these
malignant traits. Quantitative analysis subsequently confirmed a
significant reduction in the numbers of migratory and invasive
cells in the knockdown groups compared with the control group,
whereas the overexpression groups exhibited significantly increased
levels of cell migration and invasion (Fig. 3A-D). Taken together, these results
underscored that SF3A2 acts as a master regulator of ccRCC
metastasis, and its upregulation serves as a driver for tumor
progression by enhancing cell motility. The combined impact on cell
proliferation and invasion further designates SF3A2 as a potential
dual-functional therapeutic target for ccRCC intervention.
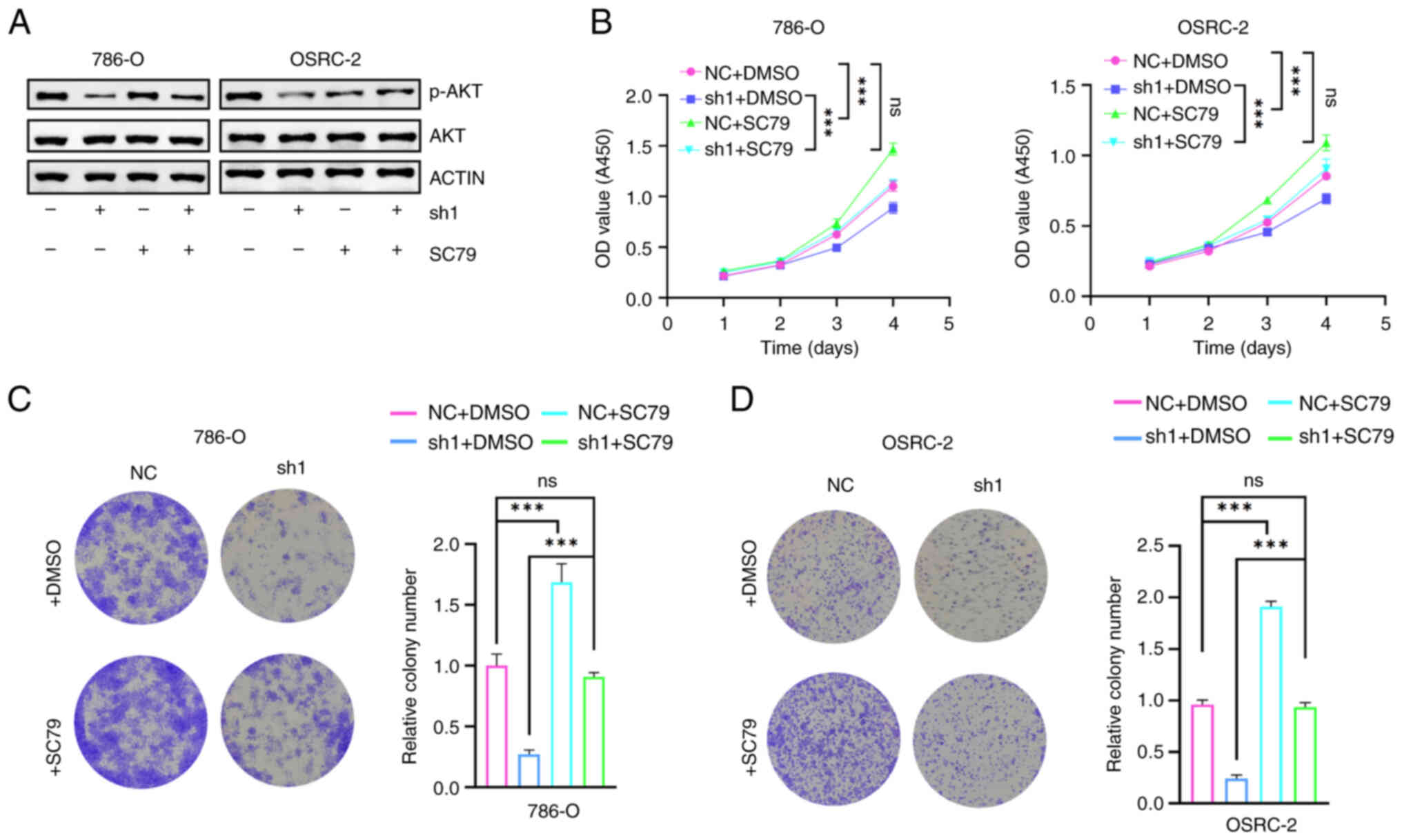
SF3A2 promotes the proliferation of
ccRCC cells by activating the AKT signaling pathway
Subsequently, the activation status of the AKT
signaling axis was systematically analyzed to elucidate the
underlying molecular mechanism via which SF3A2 regulates the
malignant phenotype of ccRCC. Western blotting revealed that SF3A2
knockdown led to a reduction in the level of p-AKT (Fig. 4A). Furthermore, functional CCK-8
assays revealed that SF3A2 silencing inhibited the proliferation of
786-O/OSRC-2 cells, whereas treatment of the cells with SC79
restored their proliferative capacity (Figs. 2C and D and 4B). The results from the colony formation
assays further confirmed this oncogenic dependence: SF3A2 depletion
led to a significant reduction in colony formation, whereas SC79
treatment rescued the clonogenic potential (Figs. 2E, F, 4C and D). Taken together, these findings
established a mechanistic link between SF3A2 and AKT pathway
activation, positioning this axis as a potential therapeutic target
for ccRCC.
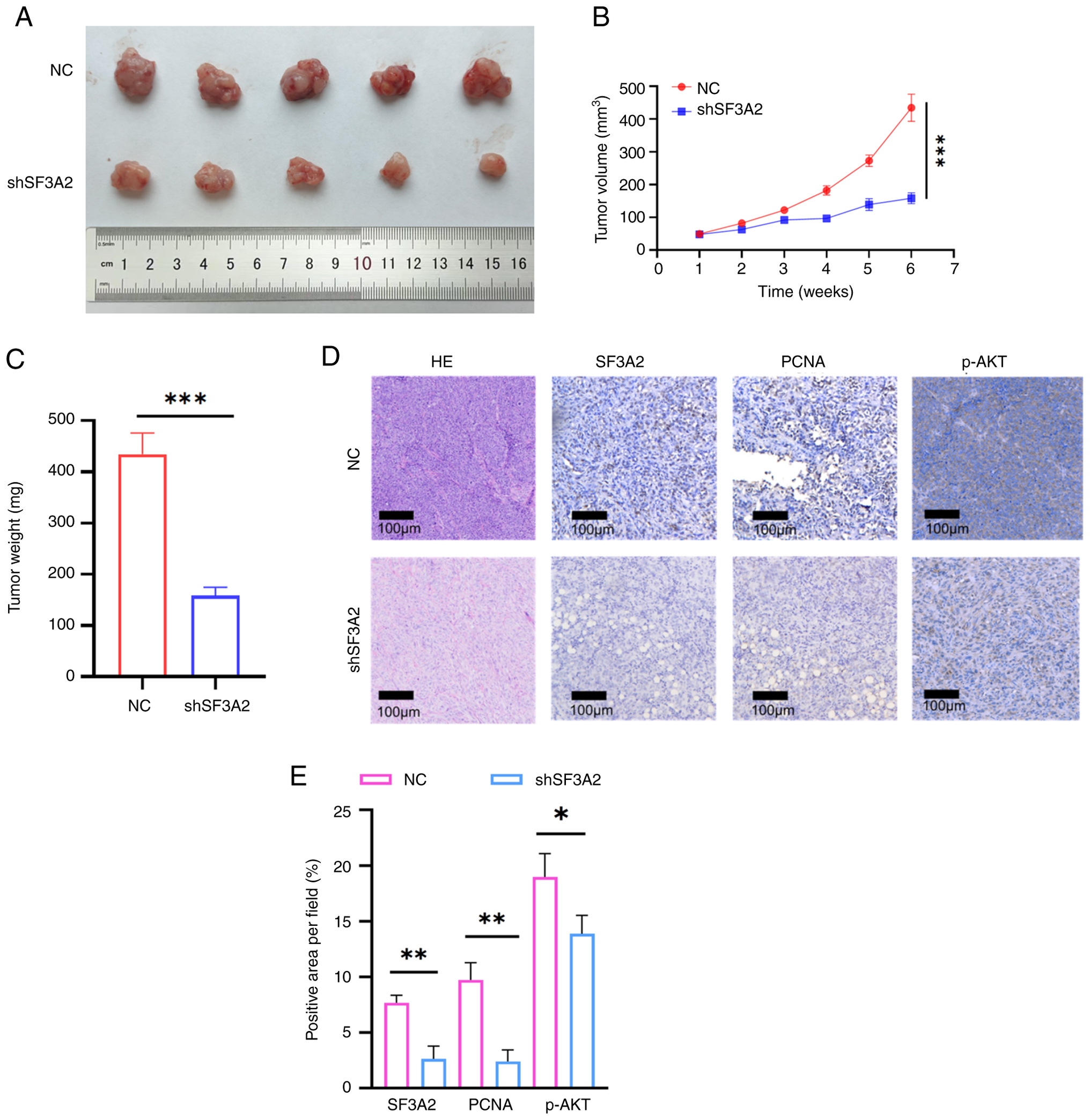
SF3A2 promotes the malignant
biological functions of ccRCC in vivo
To further evaluate the in vivo oncogenic
potential of SF3A2, a ccRCC xenograft tumor model was constructed.
SF3A2 knockdown (shSF3A2) notably inhibited tumorigenic
progression, as evidenced by reduced tumor volume and decreased
final tumor mass (Fig. 5A-C). The
maximum tumor diameter and volume observed in the present study
were 16 mm and 493.96 mm3, respectively. Subsequently,
immunohistochemical analysis revealed a concomitant reduction in
the proportion of proliferating cell nuclear antigen
(PCNA)-positive cells in tumor tissues (P<0.01), linking
SF3A2-driven tumor growth to increased proliferative activity
(Fig. 5D, E). Additionally, p-AKT
staining was also found to be diminished in the SF3A2 knockdown
group (Fig. 5D, E).
Discussion
ccRCC, a highly heterogeneous malignancy, is closely
associated with the dysregulation of critical driver genes.
Increasing evidence has highlighted the role of single-gene
regulatory mechanisms in ccRCC pathogenesis, thereby presenting
potential therapeutic targets for patients with the disease in its
advanced stages (18–20). The present study identified
significantly increased levels of SF3A2 expression in ccRCC tissues
compared with their normal counterparts, and the statistical
significance of these increases was confirmed through both unpaired
and paired analyses. Survival analysis revealed that patients with
high SF3A2 expression had notably shorter OS times compared with
those with low expression. Consistent upregulation at both the
transcriptional and translational levels suggested the involvement
of SF3A2 in ccRCC tumorigenesis, which warranted further
exploration of its molecular functions and the underlying
mechanistic roles in ccRCC pathogenesis.
Located on chromosome 17q21.33, the SF3A2 gene
encodes a core component of the spliceosomal SF3a complex (11). This evolutionarily conserved gene
has been shown to serve a pivotal role in RNA splicing regulation
(21). Mechanistically, SF3A2
directly interacts with NLR family pyrin domain containing 3
(NLRP3), forming indirect associations with neural precursor cell
expressed developmentally downregulated 4 (NEDD4), and establishing
a tripartite regulatory axis. NEDD4 mediates the proteasomal
degradation of NLRP3 through SF3A2 stabilization, with subsequent
NLRP3 depletion impairing the execution of pyroptosis by
suppressing caspase cascade activation (22,23).
Genetic alterations and dysregulated acetylation of SF3A2
compromise its splicing fidelity, leading to pathogenic errors that
disrupt heterodimer assembly in mitochondrial regulators, such as
FSCN1. Post-translational modifications, especially lysine
acetylation, fulfill a crucial role in regulating SF3A2
functionality (12).
The overexpression of SF3A2 has been shown to be
associated with increased radiotherapy sensitivity in cervical
cancer, thereby positioning the splicing factor SF3A2 as a dual
biomarker for predicting therapeutic responses and clinical
outcomes (24,25). Notably, SF3A2 upregulation in TNBC
has been shown to be strongly associated with reduced overall
survival and disease-free survival (14). In the same study, genetic silencing
of SF3A2 was found to potentiate cisplatin chemosensitivity by
suppressing the MKRN1-FAS-associated death domain signaling
pathway, attenuating both exogenous and intrinsic apoptosis, and
impairing the DNA damage response (14). Furthermore, the DHX9-SF3A2 axis has
been shown to drive ES aggressiveness by orchestrating
spliceosome-mediated oncogenic splicing programs (13). In the functional analyses in the
present study, including CCK-8 proliferation assays, colony
formation assays and Transwell cell migration/invasion experiments,
SF3A2 knockdown led to significant impairment of ccRCC cell
proliferation and migration. Conversely, SF3A2 overexpression
markedly accelerated oncogenic progression. These in vitro
findings were validated through animal experiments in vivo,
with constructed xenograft models confirming the tumor-promoting
effect of SF3A2 during ccRCC malignant evolution. Collectively,
this experimental evidence established SF3A2 as a critical
oncogenic driver in ccRCC pathogenesis, underscoring its potential
as both a novel therapeutic target and prognostic biomarker.
Furthermore, the present study aimed to investigate
the underlying mechanisms associated with the role of SF3A2 during
ccRCC malignant evolution. These mechanistic investigations
revealed the involvement of SF3A2 in the AKT signaling axis in
ccRCC pathogenesis. Genetic ablation of SF3A2 specifically reduced
the levels of AKT phosphorylation without affecting total AKT
protein levels. Pharmacological rescue using the AKT pathway
agonist SC79 successfully restored the levels of p-AKT in
SF3A2-depleted cells. Functional assays confirmed that SF3A2
depletion caused impaired cellular proliferation and clonogenic
potential, although these effects were reversed upon treating the
cells with SC79 and inducing SC79-mediated activation of the AKT
pathway. The AKT signaling axis has been shown to be a central
oncogenic driver regulating cellular survival, proliferation and
metabolic homeostasis (26–29).
Reducing AKT pathway stability has also been shown to promote
cuproptosis in oral squamous cell carcinoma cells (30). In another study, zinc finger E-box
binding homeobox 1-induced depletion of the long non-coding RNA
MIR497HG was shown to inhibit PI3K-AKT signaling, whereas PI3K-AKT
inhibition in tamoxifen-resistant breast cancer cells restored
tamoxifen responsiveness (31).
Mechanistically, the AKT-mediated phosphorylation of its canonical
substrate, GSK3β, causes the inactivation of this multifunctional
kinase, which thereby modulates numerous downstream effectors
(32–35). In ccRCC, dysregulation of this
pathway has been shown to be critical for tumor initiation and
progression, and therapeutic resistance (36–38).
Furthermore, S-palmitoylation of acylglycerol kinase (AGK) mediated
by the palmitoyltransferase ZDHHC2AGK has been shown to promote AGK
translocation to the plasma membrane, which activated the
PI3K-AKT-mTOR signaling pathway in ccRCC, thereby modulating
sensitivity to the tyrosine kinase inhibitor (TKI) sunitinib
(39). Furthermore, Liu et
al showed that ovarian tumor domain-containing deubiquitinase 1
inhibits the AKT pathway in renal cancer cells, which thereby
suppresses tumor growth and modulates resistance to TKIs (40). Additionally, the zinc finger
protein ZIC2 has been shown to positively regulate ubiquitin
conjugating enzyme E2 C, thereby activating the AKT pathway, and
consequently promoting tumor cell proliferation, invasion and
migration, and G2/M phase blockade, ultimately enhancing
tumor formation and lung metastasis (41). Considered altogether, these studies
have collectively reinforced the hypothesis that SF3A2 may exert a
pro-oncogenic role through the AKT signaling pathway in ccRCC.
Although SF3A2 has been traditionally recognized as
a core component of the spliceosomal SF3a complex, the present
study primarily focused on its role in downstream oncogenic
signaling via the AKT signaling pathway. Recent studies have
demonstrated that SF3A2 is able to influence splicing events in key
regulators, thereby contributing to chemoresistance and aggressive
tumor phenotypes (13,14). The nuclear localization of SF3A2
may facilitate the regulation of splicing networks that indirectly
activate either the AKT signaling axis or other oncogenic pathways.
Due to funding and time limitations, large-scale RNA-seq or
splicing microarray analyses were not performed in the present
study; however, the absence of these analyses is acknowledged as
one of its limitations, and future work will aim to systematically
establish the profile of SF3A2-dependent splicing events and their
association with AKT pathway regulation. Additionally, the
nuclear-enriched localization of splicing factors, including SF3A2,
has been shown to serve a unique role in coordinating
transcriptional and post-transcriptional processes that drive
malignant transformation (11).
In spite of this limitation, the present
investigation systematically characterized the oncogenic function
of SF3A2 in ccRCC through in functional validation (both in
vitro and in vivo) and a mechanistic exploration of its
signaling axis. Although the present findings have highlighted the
pathological relevance of SF3A2, there are methodological
constraints that still need to be addressed. First, validation
across expanded clinical cohorts is required to confirm expression
patterns. Secondly, a deeper mechanistic interrogation of
SF3A2-mediated spliceosomal regulation and its impact on AKT
signaling will be required to further delineate its molecular role
in ccRCC. Lastly, given that the present study focused on the
genetic modulation of SF3A2, future studies will need to assess the
potential of small-molecule inhibitors, such as E7107, in order to
validate SF3A2 as a druggable target in ccRCC.
In conclusion, the present study identified SF3A2
upregulation as a clinically significant prognostic marker in
ccRCC, which was associated with poor clinical outcomes and
accelerated tumor progression. Mechanistically, SF3A2 was shown to
promote tumor growth through activation of the AKT signaling
pathway. Collectively, these findings have positioned SF3A2 as a
dual-functional biomarker with translational relevance, offering
both prognostic stratification and actionable therapeutic targeting
opportunities in ccRCC management.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Fujian Provincial Natural
Science Foundation of China (grant no. 2023J011724).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JX designed the study, analyzed the data and wrote
the manuscript. RC collected the data and prepared the figures. JX
and RC both contributed to the data analysis, conceived the study,
and critically revised the manuscript. JX and RC confirm the
authenticity of all the raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of The First Affiliated
Hospital of Nanchang University (Nanchang, Jiangxi, China)
(approval no. CDYFY-IACUC-202212QR030) and were conducted in
accordance with the Guidelines for the Care and Use of Laboratory
Animals. The present study involved animal experiments only. The
research protocol was administratively reviewed and approved by The
First Hospital of Putian City (approval no. 2022-036), which served
as the primary institutional oversight body for this project.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
AS
|
alternative splicing
|
|
CCK-8
|
Cell Counting Kit-8
|
|
RCC
|
renal cell carcinoma
|
|
ccRCC
|
clear cell RCC
|
|
DHX9
|
DExH-box helicase 9
|
|
ES
|
Ewing sarcoma
|
|
FSCN1
|
fascin actin-bundling protein 1
|
|
GTEx
|
Genotype-Tissue Expression
|
|
MKRN1
|
makorin ring finger protein 1
|
|
NEDD4
|
neural precursor cell expressed
developmentally downregulated 4
|
|
NLRP3
|
NLR family pyrin domain containing
3
|
|
RNA-seq
|
RNA-sequencing
|
|
SF3A2
|
splicing factor 3a subunit 2
|
|
TCGA
|
The Cancer Genome Atlas
|
|
TNBC
|
triple-negative breast cancer
|
|
TKI
|
tyrosine kinase inhibitor
|
|
RT-qPCR
|
reverse transcription-quantitative
PCR
|
References
|
1
|
Rose TL and Kim WY: Renal cell carcinoma:
A review. JAMA. 332:1001–1010. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Barata P, Gulati S, Elliott A, Hammers HJ,
Burgess E, Gartrell BA, Darabi S, Bilen MA, Basu A, Geynisman DM,
et al: Renal cell carcinoma histologic subtypes exhibit distinct
transcriptional profiles. J Clin Invest. 134:e1789152024.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Young M, Jackson-Spence F, Beltran L, Day
E, Suarez C, Bex A, Powles T and Szabados B: Renal cell carcinoma.
Lancet. 404:476–491. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Atkins MB and Tannir NM: Current and
emerging therapies for first-line treatment of metastatic clear
cell renal cell carcinoma. Cancer Treat Rev. 70:127–137. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Powles T, Albiges L, Bex A, Comperat E,
Grunwald V, Kanesvaran R, Kitamura H, McKay R, Porta C, Procopio G,
et al: Renal cell carcinoma: ESMO clinical practice guideline for
diagnosis, treatment and follow-up. Ann Oncol. 35:692–706. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bedke J, Ghanem YA, Albiges L, Bonn S,
Campi R, Capitanio U, Dabestani S, Hora M, Klatte T, Kuusk T, et
al: Updated European association of urology guidelines on the use
of adjuvant immune checkpoint inhibitors and subsequent therapy for
renal cell carcinoma. Eur Urol. 87:491–496. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Marasco LE and Kornblihtt AR: The
physiology of alternative splicing. Nat Rev Mol Cell Biol.
24:242–254. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bradley RK and Anczukow O: RNA splicing
dysregulation and the hallmarks of cancer. Nat Rev Cancer.
23:135–155. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bian Z, Yang F, Xu P, Gao G, Yang C, Cao
Y, Yao S, Wang X, Yin Y, Fei B and Huang Z: LINC01852 inhibits the
tumorigenesis and chemoresistance in colorectal cancer by
suppressing SRSF5-mediated alternative splicing of PKM. Mol Cancer.
23:232024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Meng K, Li Y, Yuan X, Shen HM, Hu LL, Liu
D, Shi F, Zheng D, Shi X, Wen N, et al: The cryptic lncRNA-encoded
microprotein TPM3P9 drives oncogenic RNA splicing and
tumorigenesis. Signal Transduct Target Ther. 10:432025. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Zhan X, Bian T, Yang F, Li P, Lu
Y, Xing Z, Fan R, Zhang QC and Shi Y: Structural insights into
branch site proofreading by human spliceosome. Nat Struct Mol Biol.
31:835–845. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang Q, Yao Y, Wang Y, Li J, Chen J, Wu
M, Guo C, Lou J, Yang W, Zhao L, et al: Ginsenoside Rb2 inhibits
p300-mediated SF3A2 acetylation at lysine 10 to promote Fscn1
alternative splicing against myocardial ischemic/reperfusion
injury. J Adv Res. 65:365–379. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Frezza V, Chellini L, Riccioni V,
Bonvissuto D, Palombo R and Paronetto MP: DHX9 helicase impacts on
splicing decisions by modulating U2 snRNP recruitment in Ewing
sarcoma cells. Nucleic Acids Res. 53:gkaf0682025. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deng L, Liao L, Zhang YL, Yang SY, Hu SY,
Andriani L, Ling YX, Ma XY, Zhang FL, Shao ZM and Li DQ: SF3A2
promotes progression and cisplatin resistance in triple-negative
breast cancer via alternative splicing of MKRN1. Sci Adv.
10:eadj40092024. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Karlsson M, Zhang C, Mear L, Zhong W,
Digre A, Katona B, Sjostedt E, Butler L, Odeberg J, Dusart P, et
al: A single-cell type transcriptomics map of human tissues. Sci
Adv. 7:eabh21692021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Council N.R, . Guide for the Care and Use
of Laboratory Animals. National Academies Press; Washington, DC:
2010, PubMed/NCBI
|
|
18
|
Miao D, Wang Q, Shi J, Lv Q, Tan D, Zhao
C, Xiong Z and Zhang X: N6-methyladenosine-modified DBT alleviates
lipid accumulation and inhibits tumor progression in clear cell
renal cell carcinoma through the ANXA2/YAP axis-regulated Hippo
pathway. Cancer Commun (Lond). 43:480–502. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Xu Y, Li L, Yang W, Zhang K, Zhang Z, Yu
C, Qiu J, Cai L, Gong Y, Zhang Z, et al: TRAF2 promotes
M2-polarized tumor-associated macrophage infiltration, angiogenesis
and cancer progression by inhibiting autophagy in clear cell renal
cell carcinoma. J Exp Clin Cancer Res. 42:1592023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang B, Ren J, Ma Q, Yang F, Pan X, Zhang
Y, Liu Y, Wang C, Zhang D, Wei L, et al: A novel peptide
PDHK1-241aa encoded by circPDHK1 promotes ccRCC progression via
interacting with PPP1CA to inhibit AKT dephosphorylation and
activate the AKT-mTOR signaling pathway. Mol Cancer. 23:342024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pellacani C, Bucciarelli E, Renda F,
Hayward D, Palena A, Chen J, Bonaccorsi S, Wakefield JG, Gatti M
and Somma MP: Splicing factors Sf3A2 and Prp31 have direct roles in
mitotic chromosome segregation. Elife. 7:e403252018. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sun W, Lu H, Cui S, Zhao S, Yu H, Song H,
Ruan Q, Zhang Y, Chu Y and Dong S: NEDD4 ameliorates myocardial
reperfusion injury by preventing macrophages pyroptosis. Cell
Commun Signal. 21:292023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hirokawa M, Morita H, Tajima T, Takahashi
A, Ashikawa K, Miya F, Shigemizu D, Ozaki K, Sakata Y, Nakatani D,
et al: A genome-wide association study identifies PLCL2 and
AP3D1-DOT1L-SF3A2 as new susceptibility loci for myocardial
infarction in Japanese. Eur J Hum Genet. 23:374–380. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Ouyang Y, Cao X and Cai Q:
Identifying hub genes for chemo-radiotherapy sensitivity in
cervical cancer: A bi-dataset in silico analysis. Discov Oncol.
15:4342024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Long G and Li Z, Gao Y, Zhang X, Cheng X,
Daniel IE, Zhang L, Wang D and Li Z: Ferroptosis-related
alternative splicing signatures as potential biomarkers for
predicting prognosis and therapy response in gastric cancer.
Heliyon. 10:e343812024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Song M, Bode AM, Dong Z and Lee MH: AKT as
a therapeutic target for cancer. Cancer Res. 79:1019–1031. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhang HL, Hu BX, Ye ZP, Li ZL, Liu S,
Zhong WQ, Du T, Yang D, Mai J, Li LC, et al: TRPML1 triggers
ferroptosis defense and is a potential therapeutic target in
AKT-hyperactivated cancer. Sci Transl Med. 16:eadk03302024.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martin F, Alcon C, Marin E,
Morales-Sanchez P, Manzano-Munoz A, Diaz S, Garcia M, Samitier J,
Lu A, Villanueva A, et al: Novel selective strategies targeting the
BCL-2 family to enhance clinical efficacy in ALK-rearranged
non-small cell lung cancer. Cell Death Dis. 16:1942025. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yu L, Wei J and Liu P: Attacking the
PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment
in human cancer. Semin Cancer Biol. 85:69–94. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yu W, Yin S, Tang H, Li H, Zhang Z and
Yang K: PER2 interaction with HSP70 promotes cuproptosis in oral
squamous carcinoma cells by decreasing AKT stability. Cell Death
Dis. 16:1922025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian Y, Chen ZH, Wu P, Zhang D, Ma Y, Liu
XF, Wang X, Ding D, Cao XC and Yu Y: MIR497HG-Derived miR-195 and
miR-497 mediate tamoxifen resistance via PI3K/AKT signaling in
breast cancer. Adv Sci (Weinh). 10:e22048192023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Huang J, Wang J, Xia S, Ran H, Gao
L, Feng C, Gui L, Zhou Z and Yuan J: Human umbilical cord-derived
mesenchymal stem cell transplantation supplemented with curcumin
improves the outcomes of ischemic stroke via AKT/GSK-3β/β-TrCP/Nrf2
axis. J Neuroinflammation. 20:492023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu J, Li SM, Tang YJ, Cao JL, Hou WS,
Wang AQ, Wang C and Jin CH: Jaceosidin induces apoptosis and
inhibits migration in AGS gastric cancer cells by regulating
ROS-mediated signaling pathways. Redox Rep. 29:23133662024.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Liu X, Song J, Zhang H, Liu X, Zuo F, Zhao
Y, Zhao Y, Yin X, Guo X, Wu X, et al: Immune checkpoint
HLA-E:CD94-NKG2A mediates evasion of circulating tumor cells from
NK cell surveillance. Cancer Cell. 41:272–287. e92023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jiang Q, Zheng N, Bu L, Zhang X, Zhang X,
Wu Y, Su Y, Wang L, Zhang X, Ren S, et al: SPOP-mediated
ubiquitination and degradation of PDK1 suppresses AKT kinase
activity and oncogenic functions. Mol Cancer. 20:1002021.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang Z, Zhang Y and Zhang R: P4HA3
promotes clear cell renal cell carcinoma progression via the
PI3K/AKT/GSK3β pathway. Med Oncol. 40:702023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Mao H, Zhao Y, Lei L, Hu Y, Zhu H, Wang R,
Ni D, Liu J, Xu L, Xia H, et al: Selenoprotein S regulates
tumorigenesis of clear cell renal cell carcinoma through
AKT/GSK3β/NF-ĸB signaling pathway. Gene. 832:1465592022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhou Z, Li Y, Chai Y, Zhang Y and Yan P:
Analysis of mRNA pentatricopeptide repeat domain 1 as a prospective
oncogene in clear cell renal cell carcinoma that accelerates tumor
cells proliferation and invasion via the Akt/GSK3β/β-catenin
pathway. Discov Oncol. 16:222025. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Sun Y, Zhu L, Liu P, Zhang H, Guo F and
Jin X: ZDHHC2-Mediated AGK palmitoylation activates AKT-mTOR
signaling to reduce sunitinib sensitivity in renal cell carcinoma.
Cancer Res. 83:2034–2051. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu W, Yan B, Yu H, Ren J, Peng M, Zhu L,
Wang Y, Jin X and Yi L: OTUD1 stabilizes PTEN to inhibit the
PI3K/AKT and TNF-alpha/NF-kappaB signaling pathways and sensitize
ccRCC to TKIs. Int J Biol Sci. 18:1401–1414. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lv Z, Wang M, Hou H, Tang G, Xu H, Wang X,
Li Y, Wang J and Liu M: FOXM1-regulated ZIC2 promotes the malignant
phenotype of renal clear cell carcinoma by activating UBE2C/mTOR
signaling pathway. Int J Biol Sci. 19:3293–3306. 2023. View Article : Google Scholar : PubMed/NCBI
|