Introduction
The present review aimed to provide a comprehensive
analysis of prolyl hydroxylase domain (PHD) enzymes, detailing
their subtypes, localization, regulatory mechanisms and functional
roles, as well as their involvement in erythropoiesis and the
development of related pharmacological agents. Under hypoxic
conditions, the body initiates a cascade of adaptive biological
responses, numerous of which are mediated by transcriptional
complexes of the hypoxia-inducible factor (HIF) family. The precise
equilibrium among HIF-1α, HIF-2α and HIF-3α is essential for
orchestrating the transcriptional regulation of genes associated
with erythropoiesis, angiogenesis, vascular homeostasis, metabolic
pathways, and cellular proliferation and survival. Simultaneously,
HIF exerts indirect inhibitory effects on the transcription of
genes linked to other biological processes (1,2).
Functionally, the HIF transcription factor exists as a heterodimer
consisting of an oxygen-sensitive α and β subunit. The α subunit
interacts with the nuclear transporter of the constitutively active
aromatic hydrocarbon receptor and its activity is suppressed under
normoxic conditions by the dioxygenase family, which requires
oxygen, Fe, and 2-oxoglutarate (2-OG) for enzymatic function
(3).
In response to hypoxia, HIF-1α levels rise rapidly,
playing a pivotal role in acute hypoxic adaptation, particularly in
regulating erythropoietin (EPO) synthesis, while HIF-2α
predominantly governs long-term hypoxic responses (1,4).
Hydroxylation of two conserved prolyl residues within the HIF-α
subunit by PHD1-3 facilitates its recognition by the von
Hippel-Lindau (VHL) tumor suppressor protein E3 ubiquitin ligase
complex, leading to polyubiquitination and subsequent proteasomal
degradation. In humans, three distinct PHD isoenzymes (PHD1-3) and
an asparaginyl hydroxylase, known as factor-inhibiting HIF (FIH),
have been identified. Hydroxylation of asparagine residues within
the C-terminal transactivation domain (C-TAD) of HIF-α directly
inhibits its interaction with the co-activator p300, thereby
preventing the transcriptional activation of its target genes
(5,6). Moreover, post-translational
modifications such as acetylation and phosphorylation have been
reported to further regulate HIF-α activity, adding additional
layers of complexity to its control mechanisms (6).
PHDs exhibit substrate specificity and differential
tissue distribution, particularly in their hydroxylation of HIF
proline residues. By modulating intracellular metabolism, reactive
oxygen species (ROS) generation, iron (Fe), homeostasis, nitric
oxide (NO) signaling, and redox balance, PHDs serve as critical
regulators of HIF activity, influencing a broad spectrum of
physiological processes under hypoxic conditions. Although
inhibitors targeting the HIF/PHD axis have been successfully
integrated into clinical practice, the development of HIF/PHD
activators or functional restorers remains limited due to
significant technical hurdles. To date, no studies have reported
the successful discovery of HIF/PHD activators, highlighting a
major gap in current research and a potential avenue for future
therapeutic advancements.
Therefore, the present review aimed to
comprehensively summarize the subtypes, localization, regulatory
mechanisms and biological functions of PHDs, along with the
research and development of related pharmacological agents and
their role in erythropoiesis. Activators or functional restorers
targeting the HIF/PHD pathway represent a promising avenue for
therapeutic intervention, particularly in addressing conditions
such as polycythemia and chronic mountain sickness. Advancing
research in this area could pave the way for groundbreaking
developments, offering novel strategies to enhance clinical
management and improve patient outcomes.
PHD subtypes and localization
PHD enzymes function as critical oxygen sensors,
belonging to a family of oxygen-dependent enzymes responsible for
facilitating the proteasomal degradation of HIF-1α under normoxic
conditions (7). The hydroxylation
of HIF-1α is highly dependent on molecular oxygen, making these
enzymes essential regulators of cellular oxygen homeostasis.
Currently, three PHD isoforms, PHD1, PHD2 and PHD3, have been
identified, each exhibiting distinct oxygen-sensing properties
(8). In vitro studies
suggest a hierarchical hydroxylation efficiency among these
isoforms, with PHD2 demonstrating the highest activity, followed by
PHD3 and then PHD1.
PHDs exhibit both substrate specificity and
tissue-specific expression in their hydroxylation of HIFs. PHD2
preferentially hydroxylates HIF-1α over HIF-2α and HIF-3α, whereas
PHD1 and PHD3 display higher efficiency in hydroxylating HIF-2α.
Their expression patterns also vary across tissues: PHD2 is
predominantly found in adipose tissue, PHD1 is highly expressed in
testicular cells, and PHD3 is primarily located in cardiac cells
(8). In terms of subcellular
localization, PHD2 is mainly cytoplasmic, PHD3 is predominantly
nuclear, and PHD1 is distributed in both the cytoplasm and nucleus.
Encoded by the EGLN1 gene, PHD2 serves as the principal oxygen
sensor responsible for modulating HIF-2α activity (9,10).
Among the three PHD isoforms, PHD2 was initially
identified as the most functionally significant hydroxylase in most
cell types (11). Differences in
their intracellular distribution have been elucidated through
studies involving the overexpression of labeled proteins (12,13).
Specifically, fusion protein analyses have revealed that GFP-tagged
EGLN1/HPH-2/PHD2 localizes predominantly to the cytoplasm, whereas
EGLN2/HPH-3/PHD1 is primarily nuclear. Conversely, EGLN3/HPH-1/PHD3
is distributed across both the nucleus and cytoplasm in a
relatively balanced manner (13).
Notably, PHD expression has been implicated in
inflammatory conditions such as ulcerative colitis (UC), a subtype
of inflammatory bowel disease (IBD). In patients with UC, PHD1 and
PHD2 are highly expressed in the lamina propria and colonic
epithelium, while PHD3 is predominantly localized to the vascular
endothelium. Moreover, PHD3 expression is significantly elevated in
inflamed biopsy samples and is positively correlated with
inflammatory cytokines such as IL-8 and TNF-α, as well as the
apoptotic marker caspase-3 (14).
HIF proteins are heterodimeric transcription factors
consisting of a tightly regulated α subunit (HIF-1α, HIF-2α, or
HIF-3α) and a constitutively expressed β subunit. Under normoxic
conditions, HIF-α subunits undergo oxygen-dependent hydroxylation
by PHDs, allowing their recognition by the VHL tumor suppressor
protein, a key component of an E3 ubiquitin ligase complex. This
interaction marks HIF-α for ubiquitination and subsequent
degradation via the proteasome pathway (15,16).
However, under hypoxic conditions, hydroxylation is suppressed,
preventing VHL-mediated degradation and enabling HIF-α to
accumulate and exert its transcriptional effects (12).
Within mammalian cells, the functions of the PHD
protein family (PHD1, PHD2 and PHD3) are diverse, with PHD2
emerging as the predominant oxygen sensor regulating HIF-1α
stability and degradation. In addition to PHDs, FIH-1, an
oxygen-dependent asparaginyl hydroxylase, plays a critical role in
regulating the C-TAD of HIF. FIH-1 provides a direct mechanistic
link between oxygen sensing and HIF-mediated transcription, further
modulating cellular responses to hypoxia (17).
HIF-1α exhibits dynamic nucleocytoplasmic shuttling,
whereas HIF-1β remains permanently localized within the nucleus.
The VHL tumor suppressor protein (pVHL) is distributed across both
the nuclear and cytoplasmic compartments. The nuclear translocation
of PHD1 is mediated by classical nuclear localization signals
(NLS), utilizing the importin α/β receptor pathway. By contrast,
PHD2 nuclear import relies on a non-classical NLS, following an
alternative import pathway, which results in PHD2-mediated
hydroxylation of HIF-1α predominantly occurring within the nucleus.
The nuclear export of PHD2 is facilitated by an N-terminal nuclear
export signal and requires the export receptor chromosome region
maintenance 1. Meanwhile, the nuclear import of PHD3 is regulated
via the importin α/β receptor pathway and also depends on a
non-classical NLS (18).
In the central nervous system, NG2 cells represent a
fourth class of glial cells alongside astrocytes, microglia, and
oligodendrocytes. HIF-2 activation in NG2 cells plays a pivotal
role in promoting neurovascular expansion and remodeling
independently of EPO. The regulatory control of HIF-2 activity in
NG2 cells involves both PHD2 and PHD3. Under conditions where PHD2
and PHD3 are inactive, PHD1 assumes a compensatory role in
regulating the HIF-2 transcriptional response, thereby facilitating
vascular expansion and remodeling. However, the simultaneous
inactivation of PHD1, PHD2 and PHD3 leads to robust HIF-2
activation, further enhancing neurovascular remodeling independent
of EPO signaling (19).
The genetic inactivation of PHD1, PHD2 and PHD3
results in HIF activation, triggering the reprogramming of
myofibroblast-transformed renal EPO-producing cells (MF REPs) to
resume EPO secretion. Specifically, the loss of PHD2 in REPs
restores EPO gene expression in damaged renal tissue, leading to
erythrocytosis. By contrast, the simultaneous deletion of PHD1 and
PHD3 prevents the suppression of EPO expression without inducing
erythrocytosis (20). These
findings underscore the predominant role of PHD2 in EPO regulation
and red blood cell (RBC) production. Given the tissue-specific
regulatory mechanisms of HIF by different PHD isoforms, their
physiological roles vary significantly across organ systems. As
research progresses, pharmacological agents targeting angiogenesis
and EPO synthesis have been integrated into clinical practice for
the treatment of related disorders. Therefore, further
investigation into PHD enzyme functions holds substantial clinical
significance.
The study of PHD enzyme localization primarily
involves the fusion of PHD to the N-terminus of a fluorescent
protein, followed by transient transfection of the fusion construct
into human osteosarcoma (U2OS) cells. Subsequent visualization is
performed using three-dimensional two-photon confocal fluorescence
microscopy (13).
PHDs belong to the family of α-ketoglutarate
(α-KG)-dependent dioxygenases and serve as key regulators of
cellular oxygen sensing and metabolic control. They play a crucial
role in maintaining oxygen homeostasis by mediating the
hydroxylation of target proteins, thereby influencing their
stability and function. A summary of the major PHD isoforms,
subcellular localization, tissue distribution, substrates, and
biological functions is provided in Table I.
 | Table I.Isoforms, subcellular localization,
tissue expression, substrates, and biological functions of
PHDs. |
Table I.
Isoforms, subcellular localization,
tissue expression, substrates, and biological functions of
PHDs.
| Isoforms | Subcellular
localization | Tissue
expression | Substrates | Biological
functions | Disease
associations |
|---|
| PHD1 | Primarily
localized | Highly expressed
in | Substrate
specificity of PHDs depends on isoform | Primarily involved
in | Primarily localized
in the |
| (EGLN2) | in the nucleus
and | skeletal
muscle, | and physiological
context. | fundamental oxygen
sensing | nucleus and
mitochondria, |
|
| mitochondria, | heart, and
kidneys. | Classical
substrates: | and energy
metabolism | associated with
energy |
|
| associated
with |
| – HIF-1α/2α: All
PHD isoforms hydroxylate proline | regulation. | metabolism. |
|
| energy
metabolism. |
| residues in HIF-α
subunits (e.g., Pro402/Pro564 | Closely linked
to |
|
|
|
|
| in HIF-1α),
triggering ubiquitin-mediated | mitochondrial
function |
|
|
|
|
| degradation. | and may modulate
metabolic |
|
|
|
|
| – PHD2 is the
predominant hydroxylase for HIF-1α, | adaptation in
muscle. |
|
|
|
|
| while PHD3 plays a
more prominent role in HIF-2α |
|
|
| PHD2 | Mainly
cytoplasmic | Ubiquitously | regulation. | Hypoxia adaptation:
PHD2 | Mainly cytoplasmic
but |
| (EGLN1) | but can associate
with | expressed, with
high | Non-classical
substrates: | restricts tumor
angiogenesis | can associate with
the |
|
| the plasma
membrane | activity in
vascular | – PHD3 specifically
hydroxylates CRTC2 at | via HIF-1α
degradation. | plasma membrane
or |
|
| or endoplasmic | endothelial cells
and | Pro129/Pro625,
facilitating its nuclear translocation | PHD2
inhibitors | endoplasmic
reticulum, |
|
| reticulum,
dynamically | the tumor
micro- | and activation of
gluconeogenic genes (particularly | (for example,
roxadustat) | dynamically
relocalizing |
|
| relocalizing in
response | environment. | under fasting or
diabetic conditions). | are used to treat
renal | in response to
oxygen |
|
| to oxygen
fluctuations. |
| Other
substrates: | anemia. | fluctuations. |
| PHD3 | Resides in the | Highly expressed
in | – Nuclear factor κB
(NF-κB), RNA polymerase II, | Neuroprotection:
PHD3 | Resides in the
cytoplasm, |
| (EGLN3) | cytoplasm, where
it | the liver,
adipose | among others,
suggesting PHD involvement in | contributes to
ischemic | where it interacts
with |
|
| interacts with
substrates | tissue, and
nervous | inflammation and
transcriptional regulation. | preconditioning in
neurons | substrates such as
CRTC2 |
|
| such as CRTC2
and | system. | The principal
hydroxylase of HIF (HIF-1α), | via HIF-2α
regulation. | and HIF-1α, but
can |
|
| HIF-1α, but
can |
| facilitating its
ubiquitin-mediated degradation, | Metabolic
disorders: The | translocate to the
nucleus |
|
| translocate to
the |
| thereby regulating
hypoxic responses | PHD3-CRTC2 axis
is | under specific
conditions. |
|
| nucleus under
specific |
| (e.g.,
angiogenesis, erythropoiesis). PHD2 knockout | aberrantly
activated in type |
|
|
| conditions. |
| is embryonically
lethal, underscoring its critical role | 2 diabetes, and
PHD3 |
|
|
|
|
| in HIF
regulation. | inhibition has been
shown to |
|
|
|
|
| Beyond its role in
HIF regulation, PHD3 is involved in metabolic control (e.g.,
gluconeogenesis). It hydroxylates CRTC2 (CREB-regulated
transcriptional coactivator), modulating the expression of hepatic
gluconeogenic genes such as PEPCK and G6Pase. | lower blood glucose
levels. |
|
Functions of PHD enzymes
The oxygen-dependent hydroxylation of HIF-α subunits
serves as a key signal for ubiquitination and subsequent
proteasomal degradation, playing a crucial role in regulating HIF
protein abundance and maintaining oxygen homeostasis (21–25).
All three PHD isoforms (PHD1-3) are capable of hydroxylating HIF-α
subunits, although their functional significance varies (12,26).
Among them, genetic studies have highlighted PHD2 as the most
critical regulator of HIF-1α stability. Notably, PHD2 expression is
upregulated under hypoxic conditions, establishing an
HIF-1-mediated autoregulatory feedback loop that fine-tunes
oxygen-dependent responses (11).
Beyond their role in HIF-α hydroxylation, PHDs can
also modulate HIF activity through hydroxylase-independent
pathways. While these enzymes primarily control the stability of
HIF-α proteins, PHD2 and PHD3 themselves are subject to
upregulation via HIF-dependent feedback mechanisms. Functionally,
different PHD isoforms contribute to distinct physiological and
pathological processes, including angiogenesis, erythropoiesis,
tumorigenesis, cell proliferation, differentiation and survival. As
a result, disruptions in PHD expression or function can lead to
diverse biological consequences, reflecting the unique regulatory
roles of each isoform (27). The
selectivity of PHD isoforms toward different HIF-α subunits
exhibits a degree of specificity. PHD2 is preferentially induced by
HIF-1α, whereas PHD3 responds to both HIF-1α and HIF-2α (28). Each PHD isoform regulates the HIF
system in a functionally distinct, non-redundant manner. Under
specific experimental conditions, the relative contribution of each
PHD enzyme depends significantly on its expression levels, with the
hydroxylation rate being directly correlated with the abundance of
active PHD proteins (29). The
oxygen-dependent hydroxylation of specific proline residues in
HIF-1α allows recognition by the VHL tumor suppressor complex, a
key component of the E3 ubiquitin ligase system. This interaction
leads to the polyubiquitination of HIF-1α, triggering its rapid
degradation via the ubiquitin-proteasome pathway (26,30–32).
All three PHD isoforms contribute to HIF regulation,
and their cell-type specificity and inducibility provide HIF with
the flexibility to fine-tune hypoxia responses across different
tissues. The relative specificity of pharmacological PHD inhibition
presents an opportunity for selective modulation of the HIF system,
which holds considerable clinical significance. For instance,
inhibiting PHD2 broadly enhances HIF activation across multiple
cell types, even under normoxic conditions. By contrast, selective
inhibition of PHD3 predominantly amplifies the hypoxic response in
tissues where this enzyme is highly expressed (29). This approach could be leveraged to
activate the HIF system for treating ischemic or hypoxic diseases.
Future research must further elucidate the in vivo
specificity of PHD effects on different HIF-α subunits, as well as
the distinct roles of PHD isoforms in various biological processes.
A deeper understanding of these mechanisms is essential for
minimizing adverse effects associated with PHD inhibitors and
optimizing their therapeutic efficacy.
When PHD function is inhibited, the oxygen-dependent
degradation of VHL and HIFα is disrupted. Similar to PHDs, the
HIF-associated regulatory factor FIH, an asparaginyl hydroxylase,
suppresses the transactivation function of HIFα by hydroxylating
its TAD, thereby inhibiting the p300/CBP signaling pathway, which
plays a crucial role in cell cycle regulation and apoptosis.
Although the pVHL/PHD/HIF axis is one of the most extensively
studied oxygen-sensing mechanisms, numerous aspects remain to be
elucidated (33). The mechanistic
action of the pVHL/PHD/HIF pathway, the alterations in HIF2α-ARNT
dimerization under hypoxia, and the downstream targets of HIF are
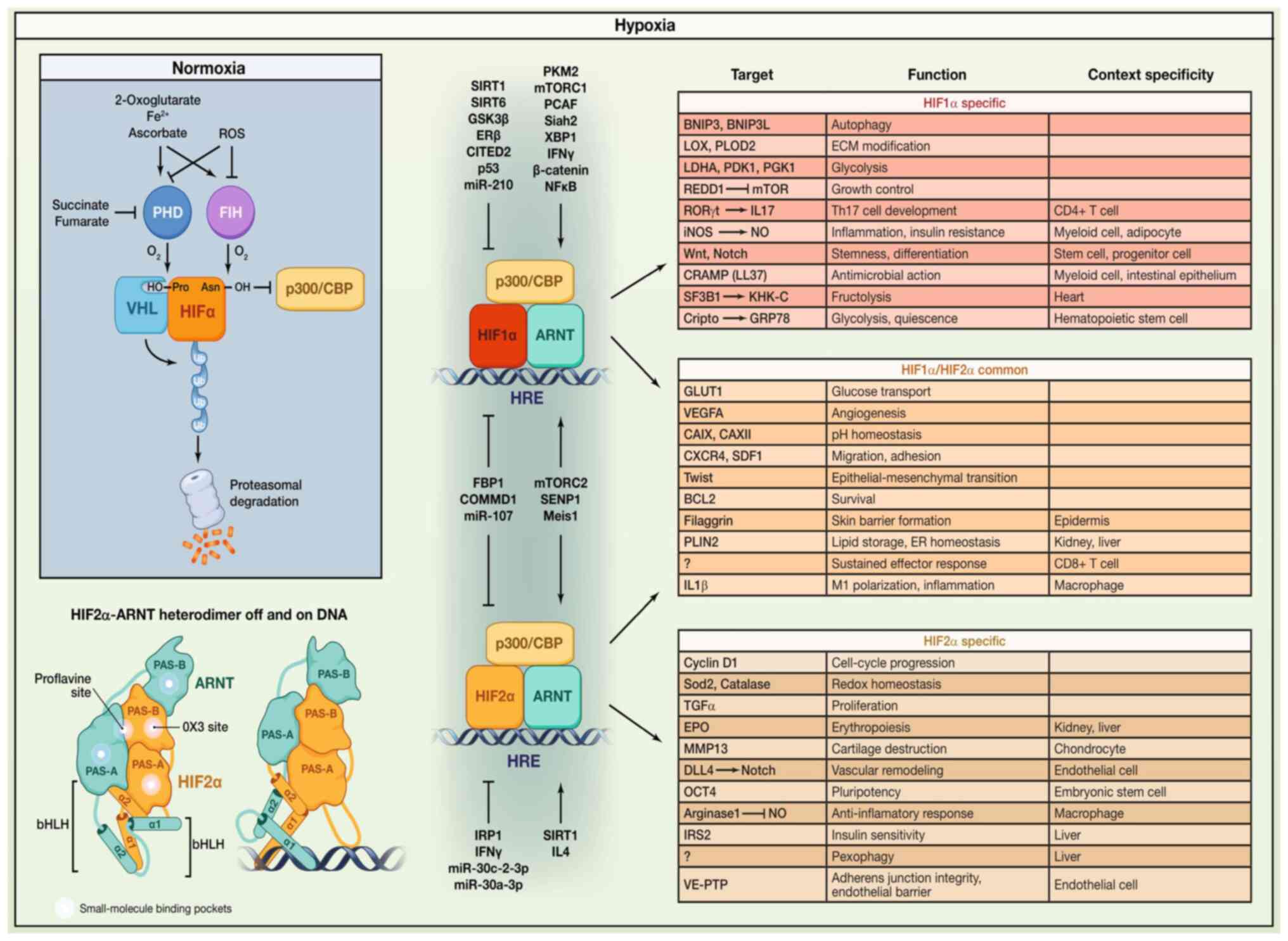
illustrated in Fig. 1.
Regulation of PHD-catalyzed hydroxylation
reactions
PHD1 and PHD2 are large enzymes, each consisting of
>400 amino acids (407 and 426, respectively, in humans), with a
highly conserved hydroxylase domain located in their C-terminal
region, sharing ~55% sequence identity in this region. By contrast,
their N-terminal regions exhibit greater sequence diversity and
comparatively lower activity (26). PHD3, the shortest of the three
isoforms, contains only 239 amino acids in humans. Although it
retains a hydroxylase domain, its N-terminal region is markedly
distinct, comprising only a short, unique segment.
Oxygen dependence
Oxygen serves as a fundamental substrate for
PHD-catalyzed hydroxylation reactions. The Km values for oxygen
across the three PHD isoforms range from 230 to 250 µM, which
slightly exceeds the oxygen concentration in aqueous solutions
equilibrated with ambient indoor air. Since intracellular oxygen
levels are typically lower, this higher Km ensures that hydroxylase
activity remains strictly oxygen-dependent, as long as other
essential substrates and cofactors are adequately available
(34). Under hypoxic conditions
(0.5–2% oxygen), HIF-1α levels increase, a process that requires
functional mitochondria. Notably, inhibition of cytochrome c
oxidase using respiratory chain inhibitors such as NO leads to the
destabilization of HIF-1α, even in low-oxygen environments
(21). This phenomenon is likely
attributed to the rapid mitochondrial oxygen consumption during
oxidative phosphorylation, which significantly reduces cytoplasmic
O2 availability (35).
Intracellular Fe(II)
concentration
PHD enzymes belong to the Fe(II)/2-OG-dependent
dioxygenase family, with PHD2 displaying particularly slow
reactivity with O2, a feature linked to its role in
hypoxia sensing. When PHD2 forms a stable complex with Fe(II) and
2-OG, the water molecules coordinated to Fe(II) remain tightly
bound at the enzyme's active site. Before O2 can bind,
these water molecules must be displaced. A single amino acid
substitution, replacing glutamic acid (D315E) with aspartic acid,
which directly interacts with Fe(II), significantly reduces PHD2's
affinity for Fe(II). However, this modification simultaneously
enhances the reaction rate with O2 by 5-fold, thereby
maintaining PHD2's catalytic efficiency (36).
Competitive inhibition by 2-OG
analogs
2-OG plays a crucial role as an intermediate in the
tricarboxylic acid (TCA) cycle and serves as an essential
co-substrate for PHD activity, aiding in the coordination of Fe(II)
within the catalytic center (26).
The hydroxylation of HIF-α proline residues by Fe(II)- and
2-OG-dependent PHD enzymes is essential for cellular oxygen
sensing. Notably, premature 2-OG binding or prolyl hydroxylation of
HIF-α can drastically reduce the affinity of HIF-α for PHD2, by
nearly 50-fold, thereby inhibiting its interaction with the enzyme.
Consequently, when 2-OG availability is limited, PHD enzymatic
activity decreases, preventing HIF-α degradation and stabilizing
the hypoxic response (37).
Proline substrate specificity
The stabilization of HIF proteins under hypoxic
conditions represents a key adaptive mechanism in oxygen-deprived
environments. The degradation of HIF-α subunits is tightly
regulated by the three PHD isoenzymes (PHD1-3), which catalyze
proline hydroxylation (38). To
date, no proline hydroxylase activity has been detected in non-HIF
proteins or peptides under conditions relevant to HIF-α
hydroxylation (39). Among PHDs,
the C-terminal oxygen-dependent degradation domain (C-ODD) exhibits
higher activity than the N-terminal oxygen-dependent degradation
domain (N-ODD), with the latter being largely inactive in PHD3.
Interestingly, PHD2 demonstrates greater efficiency in
hydroxylating the N-ODD of HIF-2α than that of HIF-1α (40). However, the hydroxylation
efficiency of HIF-2α N-ODD by PHD2 remains lower than that of
HIF-1α N-ODD (40). It has been
suggested that an antiparallel structural motif spanning residues
197 to 380 in PHD2 plays a role in interacting with the ODD domain,
with hydrophobic amino acids contributing to substrate recognition
(41). Furthermore, certain
mutations in PHD2 are associated with altered HIF-2 stability. For
instance, a heterozygous germline mutation at residue H374 leads to
PHD2 destabilization and loss of enzymatic function, ultimately
resulting in upregulated HIF-2α activity (9).
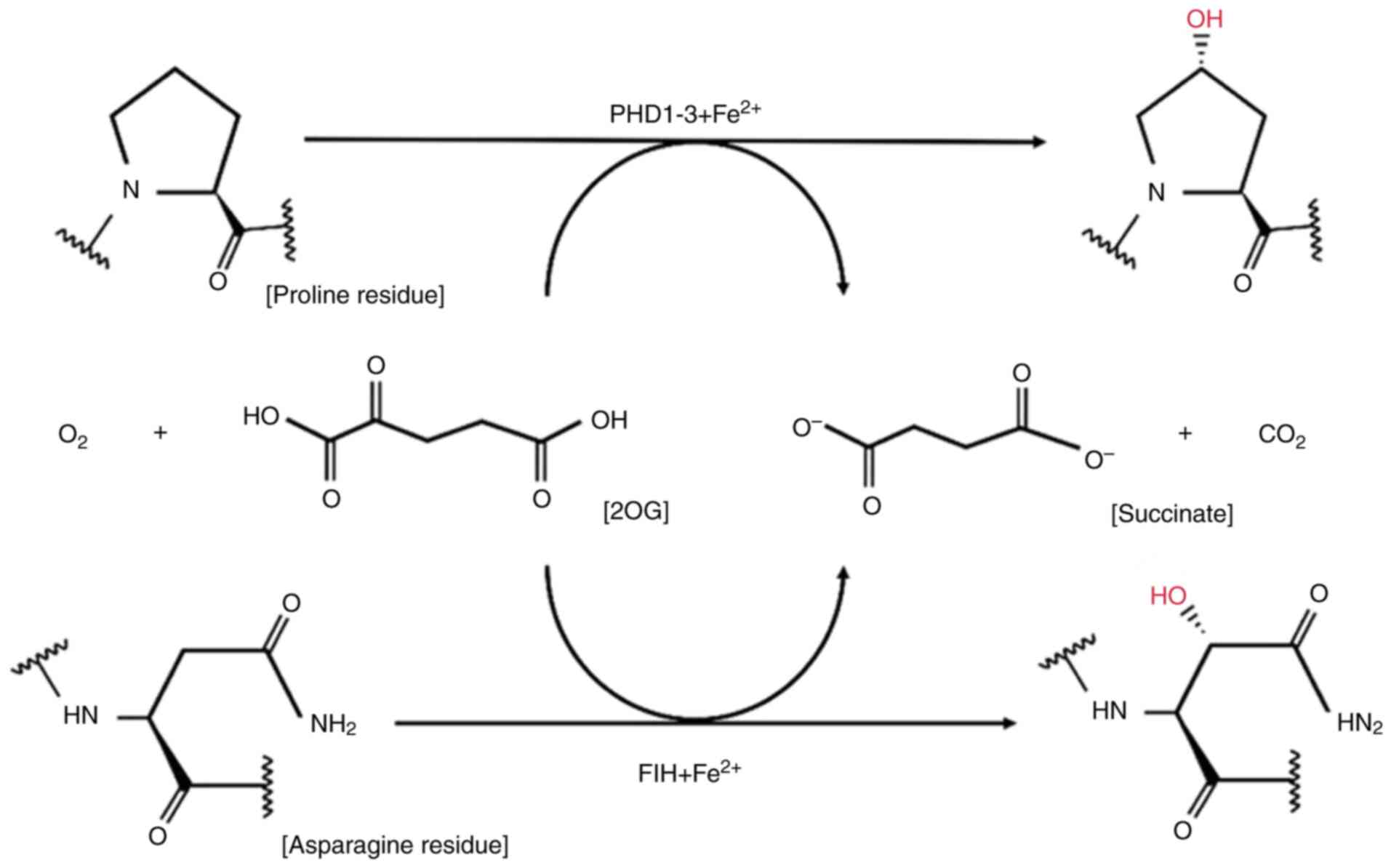
The hydroxylation regulation of PHDs is illustrated
in Fig. 2. Once synthesized, HIF-α
is rapidly degraded if sufficient non-mitochondrial oxygen is
available. HIF PHD1, PHD2 and PHD3 confer oxygen-dependent
proteasomal degradation to the HIF-α subunit. Identifying
alternative targets of HIF hydroxylases is crucial for fully
elucidating the pharmacology of prolyl hydroxylase inhibitors
(PHIs) (42).
Despite significant technological advancements,
screening, detecting, and validating alternative functional targets
of PHD and FIH remain challenging. A major limitation is the lack
of high selectivity for PHD isoform-specific inhibitors, which can
lead to a range of adverse effects. The most successful clinical
application to date is in the treatment of anemia associated with
chronic kidney disease (CKD), where PHD inhibitors have been widely
used. However, research on PHD activators or agonists remains in
its early stages.
Biological efficacy of PHDs
Transition metal ions, particularly Fe, play a
pivotal role in the hypoxia-regulated PHD-HIF-EPO signaling axis,
which governs erythropoiesis, angiogenesis, anaerobic metabolism,
adaptation, cell survival and proliferation, thereby maintaining
cellular and systemic homeostasis (43). The regulation of HIF is primarily
mediated by the highly conserved EGLN/PHD family, which
hydroxylates HIF in an oxygen-dependent manner. This hydroxylation
facilitates recognition by VHL family proteins, leading to
ubiquitination and proteasomal degradation of HIF. Interestingly,
PHD3 has been found to enhance HIF signaling through the
hydroxylation of the glycolytic enzyme pyruvate kinase muscle
subtype 2 (PKM2). Beyond its role in the HIF pathway, PHD also
influences synaptic transmission by modulating
α-amino-3-hydroxy-5-methyl-4-isoxazolepropanoic acid (AMPA)
receptor trafficking and regulating transient receptor potential
cation channel member A1 (TRPA1) activity in response to oxygen
levels in sensory neurons. Moreover, PHD activation is modulated by
poly(rC)-binding protein 1 (PCBP1), which functions as an Fe
chaperone, as well as by the (R)-enantiomer of 2-hydroxyglutarate
(2-HG), highlighting the intricate interplay between multiple
regulatory pathways in PHD function (8).
Regulation of lifespan and renal
adaptability
In Caenorhabditis elegans, the activity of
EGL-9, a PHD homolog, is regulated by the hydrogen sulfide-sensing
cysteine synthase-like protein CYSL-1, which, in turn, is modulated
by the acyltransferase RHY-1. Notably, mutations in vhl-1
significantly extend lifespan through a HIF-1-dependent mechanism.
The long-lived phenotype of vhl-1 mutants is suppressed by
mutations in egl-9 and rhy-1, whereas RNA
interference targeting rhy-1 extends the lifespan of
wild-type worms while shortening that of vhl-1 mutants
(44). Additionally, in renal
physiology, the downregulation of PHD by high salt exposure
triggers the activation of peroxisome proliferator-activated
receptor α (PPARα). This PHD/HIF-1α regulatory axis introduces a
novel transcriptional control mechanism, engaging downstream
signaling pathways of NO synthase and heme oxygenase to promote
adaptive renal responses (45).
PHD regulation of EPO production
Renin-expressing cells can be categorized into two
subgroups: Classic glomerular renin-producing cells and mesenchymal
renin-positive cells. The latter population functions as a
reservoir of natural EPO-producing cells, displaying a rapid EPO
response under acute hypoxia via the stabilization of HIF-2.
Interestingly, it is the combined deficiency of PHD2 and PHD3,
rather than PHD2 loss alone, that induces EPO expression in
glomerular renin-positive cells. Sustained HIF-2 activation in
these cells transforms them into EPO-producing cells. The strong
expression of PHD3 in glomerular renin-positive cells serves to
prevent this HIF-2-driven transformation, suggesting that PHD3
plays a crucial role in maintaining the functional stability of the
renin-expressing cell phenotype (43).
Neuroprotection and PHD-mediated
regulation of HIF
In an in vitro hypoxia-ischemia (HI) model
using oxygen-glucose deprivation (OGD) in rat pheochromocytoma
(PCC) (PC-12) cells differentiated with nerve growth factor, EPO
treatment has been found to enhance PHD2 transcription and
translation. This upregulation of PHD2 inhibits HIF-1α expression,
reduces ROS formation, and decreases matrix metalloproteinase-9
(MMP-9) activity, ultimately improving cell survival following
OGD-induced injury. Conversely, silencing PHD2 with small
interfering RNA (siRNA) reverses the neuroprotective effects of
EPO, indicating that PHD2 serves as a key mediator in EPO-induced
HIF-1α suppression and neuroprotection in HI conditions (46).
PHD deficiency and metabolic
consequences
The absence of PHD3 leads to increased blood lipid
levels and elevated hematocrit without affecting atherosclerotic
plaque size in low-density lipoprotein receptor knockout mice
(47). Additionally, studies have
shown that the complete loss of PHD1, PHD2 and PHD3 in the liver
results in severe fatty liver disease and erythrocytosis due to
excessive hepatic EPO production. Mice with hepatocyte-specific
deletion of all three PHD isoforms (PHD1/2/3hKO) exhibit a
1246-fold increase in hepatic EPO expression while renal EPO levels
drop to 6.7% of normal. These mice also develop hematocrit levels
reaching 82.4%, accompanied by severe vascular malformations and
liver steatosis (48).
By contrast, mice with dual liver-specific deletions
of PHD2 and PHD3 (PHD2/3hKO) also show increased hepatic EPO
production and reduced renal EPO expression, but the magnitude of
these changes is significantly lower than in PHD1/2/3hKO mice.
Unlike the triple knockout model, PHD2/3hKO mice maintain normal
hematocrit levels, vascular integrity and hepatic lipid homeostasis
(48). These findings indicate
that overall PHD activity, rather than the function of any single
isoform, plays a dominant role in regulating hepatic EPO production
through the PHD-HIF2α-EPO signaling cascade in vivo
(49).
Impact of PHDs on bone adaptation and
metabolism
The expression levels of negative regulatory
factors, including PHD2, FIH, and histone deacetylase sirtuin-6
(SIRT6), are significantly elevated in the skeletal muscle tissue
of elite athletes, whereas the expression of the hypoxia response
gene pyruvate dehydrogenase kinase 1 (PDK-1) is reduced. This
suggests that exercise-induced training enhances HIF inhibition,
thereby downregulating PDK-1 and contributing to skeletal muscle
adaptation to physical activity (50). In bone cells, the disruption of
PHD2, which is highly expressed in osteoblasts, results in severe
osteoporosis. Notably, treatment with ascorbic acid effectively
suppresses PHD2 expression without affecting PHD1 levels (51). However, when osteoblasts are
treated with PHD inhibitors such as dimethyloxalylglycine (DMOG) or
3,4-dihydroxybenzoate ethyl, the ascorbic acid-mediated modulation
of osteoblast differentiation markers is entirely abolished
(52).
Osteoprotegerin (OPG), a direct target gene of HIF,
plays a crucial role in regulating bone homeostasis. The
inactivation of PHD2 and PHD3 enhances HIF activity, thereby
promoting bone accumulation through the direct modulation of OPG
balance between osteoblasts and osteoclasts. However, the
simultaneous inactivation of all three PHD isoforms (PHD1, PHD2 and
PHD3) leads to excessive angiogenesis-osteogenesis coupling,
resulting in severe erythrocytosis and pathological bone overgrowth
due to extreme HIF activation. Interestingly, the dual knockout of
PHD2 and PHD3 is sufficient to prevent bone loss without
compromising hematopoietic homeostasis in ovariectomized mice
(53).
Additionally, PHD3 expression plays a distinct role
in muscle cell migration. Rhodiola glycoside has been shown to
promote skeletal muscle cell migration and paracrine signaling by
selectively inhibiting the transcription of PHD3 without affecting
PHD1 or PHD2 (54). By contrast,
increased α-KG levels suppress osteoclastogenesis by inhibiting the
NF-κB signaling pathway, which is activated by RANKL, in a
PHD1-dependent manner (55).
Regulation of angiogenesis
PHD enzymes also play a significant role in vascular
remodeling and angiogenesis. The endothelial cell-specific deletion
of PHD2 results in severe pulmonary hypertension characterized by
increased right ventricular systolic pressure and extensive
muscular hypertrophy of the peripheral pulmonary arteries. However,
this phenotype does not involve erythrocytosis. Studies on
endothelial-specific PHD2 mutants with concurrent HIF-1α or HIF-2α
inactivation have demonstrated that pulmonary hypertension is
primarily driven by HIF-2α, rather than HIF-1α. The pathological
effects of HIF-2α in this condition are largely mediated by
upregulated expression of the vasoconstrictor endothelin-1 and
diminished apelin receptor signaling, which normally promotes
vasodilation (56). In HI models,
the expression of PHD proteins exhibits dynamic changes over time.
Following 24 h of HI, PHD3 protein levels, along with HIF-1α
expression, are significantly elevated. However, after 72 h of HI,
PHD3 protein levels decline, while PHD1 and PHD2 levels remain
unchanged throughout the hypoxic period (57).
Role of PHDs in tumor progression and
metabolism
PHD enzymes play a complex and context-dependent
role in cancer, significantly influencing tumor growth and
metabolism (58). The primary
mechanism underlying this effect is the regulation of HIF-1α, which
drives a metabolic shift from oxidative phosphorylation to
anaerobic glycolysis, thereby promoting tumor adaptation to hypoxic
microenvironments. This metabolic reprogramming alters oxidative
stress responses and results in the accumulation of tumor-specific
metabolites that contribute to cancer progression.
In clear cell renal cell carcinoma (ccRCC), the loss
of pVHL leads to the accumulation of HIF-α isoforms and their
downstream target genes. Interestingly, in contrast to most cell
types where PHD3 negatively regulates HIF-1α, ccRCC tumors exhibit
a strong positive correlation between PHD3 and HIF2A mRNA
expression. High PHD3 expression in these tumors sustains elevated
HIF-1α levels and enhances the expression of HIF target genes,
potentially increasing the invasive potential of ccRCC cells
(59).
In non-small cell lung cancer (NSCLC), the
downregulation of PHD1 and PHD2 is associated with tumor initiation
and progression. The loss of these PHD isoforms correlates with
increased activity of downstream HIF pathway genes such as HIF-1α,
PKM2 and PDK1. However, PHD3 does not appear to play a significant
role in NSCLC tumor biology (60).
In colorectal cancer models with varying levels of
drug resistance, silencing PHD1, but not PHD2 or PHD3, prevents p53
activation and impairs DNA repair mechanisms, ultimately leading to
cancer cell death. PHD1 facilitates p53 activation through
hydroxylation-dependent interactions with p38α kinase, enabling
phosphorylation of p53 at serine 15. This modification enhances
p53-mediated nucleotide excision repair by promoting interactions
with the DNA helicase XPB, thereby protecting tumor cells from
chemotherapy-induced apoptosis (61).
Current research suggests that PHD enzymes exhibit
both tumor-promoting and tumor-suppressing functions, depending on
their expression profiles in different cancer types and cellular
contexts. The precise role of each PHD isoform in tumorigenesis
remains an area of active investigation, with implications for the
development of targeted cancer therapies (62).
Regulation in inflammation and glucose
metabolism
PHD1 has been found to be overexpressed in pouchitis
biopsies from patients with ileal pouch-anal anastomosis for UC,
with its expression levels correlating directly with disease
activity. Notably, treatment with the small-molecule PHD inhibitor
DMOG has demonstrated the ability to restore intestinal epithelial
barrier integrity by upregulating the tight junction proteins zona
occludens-1 and claudin-1. Additionally, DMOG alleviates intestinal
epithelial cell apoptosis, thereby mitigating inflammation in the
pouch and improving disease outcomes (63).
In a mouse model of IBD, the expression of PHD1 and
PHD2 progressively increases as the disease advances, whereas PHD3
levels remain unchanged. This suggests that inhibiting all three
PHD subtypes may not be an optimal therapeutic strategy for IBD, as
normal intestinal function appears to rely on the presence of PHD3
(64).
Beyond inflammation, PHD3 also plays a significant
role in glucose metabolism. A sudden loss of PHD3 (also known as
Egln3) in the liver has been shown to enhance insulin sensitivity
by selectively stabilizing HIF-2α. This stabilization promotes the
transcription of insulin receptor substrate 2 (Irs2), which in turn
enhances insulin-stimulated Akt activation. The beneficial
metabolic effects of PHD3 knockout on glucose tolerance and insulin
signaling are entirely dependent on HIF-2α and Irs2, as their
elimination negates these improvements (65).
Additionally, α-KG, a key respiratory substrate, has
been implicated in insulin secretion. Cytoplasmic PHD enzymes
regulate α-KG metabolism, and inhibition of PHDs using ethyl
dihydroxybenzoate (EDHB) significantly suppresses
glucose-stimulated insulin secretion (GSIS) in pancreatic β-cells
(832/13 clone), as well as in rat and human islets. This
suppression occurs due to reduced glucose metabolism, a lower
ATP/ADP ratio, and diminished levels of critical TCA cycle
intermediates, including pyruvate, citrate, fumarate and malate.
Interestingly, silencing PHD1 and PHD3 with siRNA impairs GSIS,
whereas PHD2 knockdown has no impact, suggesting that PHD1 and PHD3
are key regulators of glucose metabolism in pancreatic β-cells
(66).
Regulation of erythropoiesis
A heterozygous PHD2 c.1121A → G (p.H374R) mutation
has been identified in patients with familial polycythemia, though
it is not directly associated with tumor development. However,
individuals with PHD2 mutations linked to polycythemia have been
later diagnosed with recurrent paragangliomas (PGL), suggesting a
potential predisposition. The pathogenic mechanism underlying these
conditions involves dysregulated hypoxia sensitivity, leading to
the stabilization and accumulation of HIF-2α, which creates a
pseudo-hypoxic state. This state, in turn, may contribute to the
development of both polycythemia and hypoxia-associated endocrine
tumors, including PCC and PGL (9).
VHL-associated diseases also result in the
stabilization of HIFs, frequently observed in endocrine tumors such
as PCC and PGL. The aberrant upregulation of HIF not only directly
promotes tumor growth but also reduces apoptosis in endocrine tumor
cells, further exacerbating disease progression. Based on the
classification, localization and functions of PHDs, it is evident
that excessive erythrocytosis is primarily driven by dysregulation
of PHD2, which leads to unchecked HIF-2α accumulation. This, in
turn, results in the overexpression of downstream target genes,
particularly through the hyperactivation of the EPO pathway.
Chuvash polycythemia, a well-characterized genetic
disorder, is associated with a homozygous 598C>T germline
mutation in the VHL gene. This mutation leads to aberrant HIF-1α
upregulation, even under normoxic conditions, driving the excessive
production of EPO and several other hypoxia-responsive genes. The
C>T missense mutation in VHL causes an arginine-to-tryptophan
substitution at residue 200 (Arg200Trp), weakening the interaction
between VHL and HIF-1α. This defective interaction reduces HIF-1α
degradation, thereby leading to the persistent overexpression of
EPO, SLC2A1 (GLUT1, encoding solute carrier family 2), TF (encoding
transferrin), TFRC (encoding transferrin receptors CD71/p90), and
VEGF (encoding vascular endothelial growth factor) (67,68).
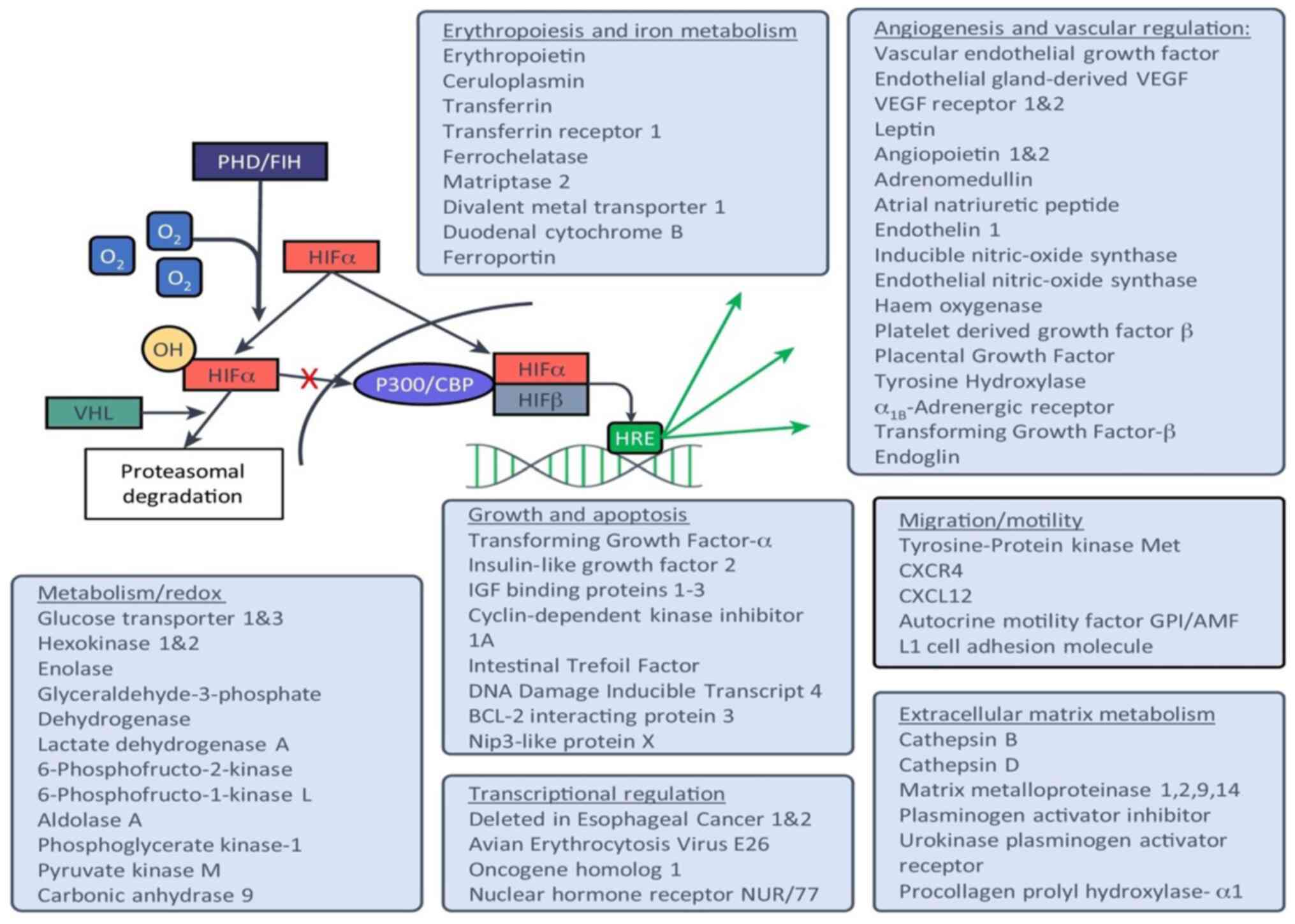
Overall, the HIF/PHD axis plays a complex and
multifaceted role in diverse physiological and pathological
processes (Fig. 3). Significant
progress has been made in the development of PHD inhibitors for
clinical applications (69).
However, different PHD subtypes exert distinct effects across
various biological functions, including angiogenesis,
erythropoiesis, cancer progression, cellular growth,
differentiation and survival. Therefore, further investigations are
required to elucidate the precise impact of PHD enzymes on HIF-α
subtype specificity in in vivo models, as well as their
individual contributions to distinct biological pathways (27).
Pharmaceutical development and therapeutic
applications of PHD inhibitors
The hydroxylation of HIF-α proline residues is
catalyzed by Fe- and 2-OG-dependent dioxygenase enzymes known as
PHDs, whose activity is strictly oxygen-dependent. As a result,
inhibiting PHD function leads to the stabilization of HIF-α,
thereby activating hypoxia-associated signaling pathways even under
normoxic conditions. This mechanism has been leveraged for
therapeutic purposes, with small-molecule PHD inhibitors developed
as clinical treatments for renal anemia. Beyond their application
in anemia management, recent research has highlighted the broader
medical potential of PHD inhibitors in various disease contexts.
Emerging evidence suggests that modulating PHD activity may have
therapeutic implications for ischemic disorders, chronic
inflammatory diseases, metabolic syndromes and even
neurodegenerative conditions. These findings underscore the
expanding role of PHD-targeted therapies and warrant further
investigation into their diverse clinical applications (70).
PHD inhibitors have also been reported in the
treatment of various other diseases (70). There is increasing evidence
supporting the potential of targeting the HIF pathway in acute
myeloid leukemia (AML) therapy. Studies suggest that the selective
PHD2 inhibitor IOX5 exerts its effects by stabilizing HIF-1α,
thereby triggering a cascade of events detrimental to AML cells. By
stabilizing HIF-1α, IOX5 disrupts pro-leukemogenic signaling
pathways, shifts metabolic processes to a state less favorable for
cancer cells and induces apoptosis through BNIP3 upregulation,
highlighting its potential as a non-toxic therapeutic strategy for
AML. Furthermore, when combined with BCL-2 inhibitor venetoclax,
which is already used in clinical settings, IOX5 enhances anti-AML
efficacy. Notably, IOX5 selectively targets PHD without interfering
with other enzymes such as FIH-1, a key feature for minimizing
off-target effects (71).
Roxadustat, another PHD inhibitor, has shown promise
in alleviating anemia in patients with lower-risk myelodysplastic
syndrome, reducing their dependence on RBC transfusions.
Additionally, roxadustat has been identified as a novel therapeutic
candidate for Fe-refractory Fe-deficiency anemia (IRIDA). In mouse
models of IRIDA, it activates the HIF-2α-ferroportin (FPN) axis,
demonstrating significant therapeutic efficacy and clinical
translation potential. Mechanistically, roxadustat stabilizes
HIF-2α in the duodenum, leading to FPN transcriptional activation
and increased intestinal Fe absorption, independent of hepcidin
levels, ultimately ameliorating hepcidin-activated anemia (72).
Moreover, in ischemia/reperfusion injury models,
roxadustat preconditioning has been shown to induce ischemic
tolerance by shifting aerobic respiration to anaerobic metabolism,
thereby maintaining ATP production under hypoxic conditions and
reducing myocardial ischemic damage (73). Additionally, roxadustat activates
the HIF-1α/VEGF/VEGFR2 pathway, promoting angiogenesis, and has
demonstrated therapeutic potential in diabetic wound healing in rat
models (74). Furthermore,
roxadustat induces the accumulation of HIF-1α and WNT7a in the
tibialis anterior muscle, facilitating muscle regeneration
following cyclophosphamide-induced muscle injury and promoting the
formation of significantly larger muscle fibers (75). These findings suggest that
roxadustat could be a promising drug for diabetic wound healing and
the prevention of age-related skeletal muscle atrophy, though
clinical validation is still required.
Given the broad biological functions of HIF,
roxadustat's ability to stabilize HIF has opened new avenues for
treating hypoxia-related diseases, including neuroprotection in
nerve injury (76), oxygen-induced
retinopathy (77), pulmonary
fibrosis (78), acute lung injury
(79), fracture healing (80) and radiation-induced apoptosis
(81).
The therapeutic development of HIF-PHD modulators
has primarily focused on inhibitors designed to elevate hemoglobin
(Hb) levels, particularly for the treatment of renal anemia. To
date, no studies have reported on optimal recovery doses or the
development of PHD activators.
A comprehensive meta-analysis encompassing 30
studies and a total of 13,146 patients has assessed the long-term
efficacy and safety of HIF-PHD inhibitors, including roxadustat,
daprodustat, vadadustat, molidustat, desidustat and enarodustat, in
the management of anemia associated with CKD. The findings
demonstrate that HIF-PHD inhibitors significantly enhance Hb
levels, total Fe-binding capacity, and transferrin concentrations
compared with placebo or erythropoiesis-stimulating agent (ESA)
treatments. Additionally, these inhibitors contribute to a
reduction in cholesterol levels, indicating potential metabolic
benefits.
Regarding safety, patients receiving HIF-PHD
inhibitors exhibit a higher incidence of serious adverse events
compared with those in the placebo group. However, the overall risk
profile is comparable to that observed with ESA therapy. Adverse
effects commonly associated with HIF-PHD inhibitors include
diarrhea, nausea, peripheral edema, hyperkalemia and hypertension,
with a higher likelihood of vomiting, headaches and thrombotic
events when compared with ESA treatment. Despite these risks,
HIF-PHD inhibitors remain effective in elevating Hb levels,
optimizing Fe metabolism, and demonstrating favorable long-term
tolerability in CKD-associated anemia. To mitigate adverse effects
related to excessive Fe utilization, it is recommended that HIF-PHD
inhibitors be administered in conjunction with Fe supplementation
for extended treatment regimens (82).
The research studies on HIF-PHD inhibitors for renal
anemia are as follows [Table
II;(83–94)]: Beyond clinical trials,
investigations have explored the pharmacokinetic interactions of
HIF-PHD inhibitors both in vitro and in healthy volunteers.
In vitro studies have demonstrated that the formation of
chelates between vadadustat and Fe-containing compounds varies
depending on the specific type of Fe reagent in aqueous and
simulated intestinal fluid environments. Notably, when vadadustat
is co-administered with oral Fe supplements, its area under the
plasma concentration-time curve (AUC0-∞) and maximum
plasma concentration (Cmax) are reduced due to
gastrointestinal chelation. This interaction suggests that Fe
supplements may impair vadadustat absorption, necessitating a
dosing interval to optimize its therapeutic efficacy (95).
| Table II.Clinical studies of hypoxia-inducible
factor-PHD inhibitors for renal anemia. |
Table II.
Clinical studies of hypoxia-inducible
factor-PHD inhibitors for renal anemia.
| First author/s,
year | Stage trial | Study design | Object | Medicine | Results | Conclusion | (Refs.) |
|---|
| Kansagra et
al, | Phase I | Randomized, | 100 healthy | ZYAN1 | Maximum
concentration (Cmax) ranged from | Single (10–300 mg)
and | (83) |
| 2018 | clinical | double-blind, | volunteers |
| 566.47±163.03 to
17,858.33±2,899.19 ng/ml. | multiple (100–300
mg) doses |
|
|
| trial | placebo- |
|
| After a single
incremental dose of 10–300 mg | of Zyany1 were safe
and |
|
|
|
| controlled |
|
| The median time
(t/MAX) to Cmax with a | well tolerated in
healthy |
|
|
|
| (evaluation |
|
| 300 mg oral dose of
Zyan1 was ~2.5 h. | volunteers. |
|
|
|
| of safety, |
|
| The mean
Cmax and area (AUC T) area values | The mean C Max
and |
|
|
|
| tolerability, |
|
| under the
concentration-time curve from time | AUCt increased
almost |
|
|
|
| and pharma- |
|
| 0 to time t showed
a dose-proportional increase | proportionally with
the |
|
|
|
| cokinetics |
|
| regardless of
single or multiple dosing. | dose of Zyan
1. |
|
|
|
| after oral |
|
| The average
elimination half-life (t½) is | The average
concentration |
|
|
|
|
administration) |
|
| 6.9–13 h and the
cumulative dose was | of serum EPO showed
a |
|
|
|
|
|
|
| negligible. | dose-response
trend. |
|
|
|
|
|
|
| After a single dose
of Zyany1, the maximum | Zyan 1 once every 2
days |
|
|
|
|
|
|
| mean serum EPO
showed a dose-response | was recommended
for |
|
|
|
|
|
|
| (i.e., 10 and 300
mg Zyany1 doses were | Phase II study
based on |
|
|
|
|
|
|
| 6.6 and 79.9 Miu/l,
respectively), and the | the data of T 1FI
2, |
|
|
|
|
|
|
| average maximum
serum EPO | pharmacodynamic
activity |
|
|
|
|
|
|
| concentration
ranged from 10 to 72 h. | and drug
accumulation. |
|
| Singh et al,
2021 | Phase 3 | Randomized, | 2964 patients | Daprodustat | The mean (±SD)
baseline Hb level was | In CKD patients
with | (84) |
|
| clinical | open-label | with dialysis |
| 10.4±1.0 g per
deciliter overall. The mean | dialysis, the
daprodustat |
|
|
| trial | (1,487 cases
of | CKD patients |
| (±SE) change in the
Hb level from baseline | group is not
inferior to the |
|
|
|
| daprodustat
oral | of Hb levels |
| to weeks 28 through
52 was 0.28±0.02 g | ESAs group in
changes |
|
|
|
|
administration; | ranging from |
| per deciliter in
the daprodustat group and | in Hb levels since
baseline |
|
|
|
| 1,477 cases of | 80 to 115 g/l |
| 0.10±0.02 g per
deciliter in the ESA group | and outcomes of
cardio- |
|
|
|
| ESA) |
|
| [difference, 0.18 g
per deciliter; 95% | vascular adverse
events. |
|
|
|
|
|
|
| confidence interval
(CI), 0.12 to 0.24], |
|
|
|
|
|
|
|
| which met the
prespecified noninferiority |
|
|
|
|
|
|
|
| margin of −0.75 g
per deciliter. |
|
|
|
|
|
|
|
| During a median
follow-up of 2.5 years, a |
|
|
|
|
|
|
|
| major adverse
cardiovascular event occurred |
|
|
|
|
|
|
|
| in 374 of 1487
patients (25.2%) in the |
|
|
|
|
|
|
|
| daprodustat group
and in 394 of 1477 (26.7%) |
|
|
|
|
|
|
|
| in the ESA group
(hazard ratio, 0.93; 95% CI, |
|
|
|
|
|
|
|
| 0.81 to 1.07),
which also met the prespecified |
|
|
|
|
|
|
|
| noninferiority
margin for daprodustat. |
|
|
|
|
|
|
|
| The percentages of
patients with other adverse |
|
|
|
|
|
|
|
| events were similar
in the two groups. |
|
|
| Singh et
al, | Phase 3 | Randomized, | 3872 patients | Daprodustat | The mean (±SE)
change in the Hb level from | In non-dialysis
CKD | (85) |
| 2021 | clinical | open labe | with non- |
| baseline to weeks
28 through 52 was | patients with
anemia, the |
|
|
| trial | (daprodustat: | dialysis CKD |
| 0.74±0.02 g per
deciliter in the daprodustat | daprodustat group
was not |
|
|
|
| darbepoetin | stage 3–5 |
| group and 0.66±0.02
g per deciliter in the | inferior to the
dapepoetin |
|
|
|
| alfa 1:1) |
|
| darbepoetin alfa
group [difference, 0.08 g per | alpha group in
terms of |
|
|
|
|
|
|
| deciliter; 95%
confidence interval (CI), 0.03 to | Hb levels changes
since |
|
|
|
|
|
|
| 0.13], which met
the prespecified noninferiority | baseline and
outcomes of |
|
|
|
|
|
|
| margin of −0.75 g
per deciliter. | cardiovascular
adverse |
|
|
|
|
|
|
| During a median
follow-up of 1.9 years, the | events. |
|
|
|
|
|
|
| first MACE occurred
in 378 of 1937 patients |
|
|
|
|
|
|
|
| (19.5%) in the
daprodustat group and in 371 |
|
|
|
|
|
|
|
| of 1935 patients
(19.2%) in the darbepoetin |
|
|
|
|
|
|
|
| alfa group (hazard
ratio, 1.03; 95% CI, 0.89 to |
|
|
|
|
|
|
|
| 1.19), which met
the prespecified noninferiority |
|
|
|
|
|
|
|
| margin of
1.25. |
|
|
|
|
|
|
|
| The percentages of
patients with adverse events |
|
|
|
|
|
|
|
| were similar in the
two groups. |
|
|
| Fishbane et
al, | Phase 3 | Randomized, | 2133 patients | Roxadustat | Mean (95%
confidence interval) Hb change | Roxadustat
effectively | (86) |
| 2022 | clinical | open label | with dialysis- |
| from baseline was
0.77 (0.69 to 0.85) g/dl with | increased Hb in
patients |
|
|
| trial | (roxadustat: | dependent |
| roxadustat and 0.68
(0.60 to 0.76) g/dl with | with DD-CKD, with
an |
|
|
|
| epoetin alfa
1:1) | (DD) CKD |
| epoetin alfa,
demonstrating noninferiority | AE profile
comparable to |
|
|
|
|
|
|
| [least squares mean
difference (95% CI), | epoetin alfa. |
|
|
|
|
|
|
| 0.09 (0.01 to
0.18); P<0.001]. |
|
|
|
|
|
|
|
| The proportion of
patients experiencing ≥1 AE |
|
|
|
|
|
|
|
| and ≥1 serious AE
was 85.0 and 57.6% with |
|
|
|
|
|
|
|
| roxadustat and 84.5
and 57.5% with epoetin |
|
|
|
|
|
|
|
| alfa,
respectively. |
|
|
| Fishbane et
al, | Phase 3 | Double-blind | CKD stages | Roxadustat | The mean change in
Hb from baseline was | Roxadustat
effectively | (87) |
| 2021 | clinical | randomization | 3–5 of Hb |
| 1.75 g/dl [95%
confidence interval (95% CI), | increased Hb in
patients with |
|
|
| trial | (roxadustat: | <10.0 g/dl |
| 1.68 to 1.81] with
roxadustat vs. 0.40 g/dl |
non-dialysis-dependent CKD |
|
|
|
| placebo 1:1) |
|
| (95% CI, 0.33 to
0.47) with placebo, (P<0.001). | and reduced the
need for |
|
|
|
|
|
|
| Among 411 patients
with baseline elevated | RBC transfusion,
with |
|
|
|
|
|
|
| high-sensitivity
C-reactive protein, the mean | an adverse event
profile |
|
|
|
|
|
|
| change in Hb from
baseline was 1.75 g/dl | comparable to that
of |
|
|
|
|
|
|
| (95% CI, 1.58 to
1.92) with roxadustat vs. | placebo. |
|
|
|
|
|
|
| 0.62 g/dl (95% CI,
0.44 to 0.80) with placebo, |
|
|
|
|
|
|
|
| (P<0.001).
Roxadustat reduced the risk of |
|
|
|
|
|
|
|
| RBC transfusion by
63% (hazard ratio, 0.37; |
|
|
|
|
|
|
|
| 95% CI, 0.30 to
0.44). The most common |
|
|
|
|
|
|
|
| adverse events with
roxadustat and placebo, |
|
|
|
|
|
|
|
| respectively, were
ESKD (21.0% vs. 20.5%), |
|
|
|
|
|
|
|
| urinary tract
infection (12.8% vs. 8.0%), |
|
|
|
|
|
|
|
| pneumonia (11.9%
vs. 9.4%), and hypertension |
|
|
|
|
|
|
|
| (11.5% vs.
9.1%). |
|
|
| Chen et
al, | Phase 3 | Double-blind | 154 patients | Roxadustat | During the primary
analysis period, the mean | The roxadustat
group had | (88) |
| 2019 | clinical | randomization | with non- |
| (±SD) change from
baseline in the Hb level | a higher mean Hb
level |
|
|
| trial | (Roxadustat: | dialysis CKD |
| was an increase of
1.9±1.2 g per deciliter in the | than those in the
placebo |
|
|
|
| placebo= 2:1) | in 29 regions |
| roxadustat group
and a decrease of 0.4±0.8 g | group after 8
weeks. |
|
|
|
|
| of China |
| per deciliter in
the placebo group (P<0.001). | Hyperkalemia and
metabolic |
|
|
|
|
|
|
| The mean reduction
from baseline in the | acidosis occurred
more |
|
|
|
|
|
|
| hepcidin level
(associated with greater Fe | frequently in the
roxadustat |
|
|
|
|
|
|
| availability) was
56.14±63.40 ng per milliliter | group than in the
placebo |
|
|
|
|
|
|
| in the roxadustat
group and 15.10±48.06 ng per | group. The efficacy
of |
|
|
|
|
|
|
| milliliter in the
placebo group. The reduction | roxadustat in Hb
correction |
|
|
|
|
|
|
| from baseline in
the total cholesterol level was | and maintenance
was |
|
|
|
|
|
|
| 40.6 mg per
deciliter in the roxadustat group | maintained during
the |
|
|
|
|
|
|
| and 7.7 mg per
deciliter in the placebo group. | 18-week open-label
period. |
|
| Perkovic et
al, | The | Randomized | 3872 cases | Daprodustat | The median baseline
Hb was 9.9 g/dl, blood | Hb
efficacy,cardiovascular | (89,90) |
| 2022; | American | Open-label
trial | of non- |
| pressure was 135/74
mmHg, and the estimated | (CV) safety and
secondary |
|
| Testi et
al, | CKD | (daprodustat: | dialysis CKD |
| glomerular
filtration rate was 18 ml/min/1.73 m2. | efficacy
outcomes, |
|
| 2023 | anemia | darbepoetin | anemia in |
| Among randomized
patients, 53% were ESA | daprodustat is not
inferior |
|
|
| study | alfa 1: 1) | 38 countries |
| non-users, 57% had
diabetes, and 37% had a | to the control
substance |
|
|
|
|
|
|
| history of CV
disease. At baseline, 61% of | darboetin
alpha. |
|
|
|
|
|
|
| participants were
using renin-angiotensin |
|
|
|
|
|
|
|
| system blockers,
55% were taking statins, |
|
|
|
|
|
|
|
| and 49% were taking
oral iron. |
|
|
|
|
|
|
|
| Baseline
demographics were similar to those |
|
|
|
|
|
|
|
| in other large
non-dialysis anemia trials. |
|
|
| Kurata et
al, | Phase III |
| Non-dialysis- | Roxadustat | Roxadustat
effectively increases and maintains | Roxadustat
effectively | (91) |
| 2022 | clinical |
| dependent |
| Hb levels in both
non-dialysis-dependent and | increases and
maintains |
|
|
| trial |
| and dialysis- |
| dialysis-dependent
CKD patients. Roxadustat | Hb levels.
Roxadustat is an |
|
|
| expert |
| dependent |
| also improved Fe
metabolism and reduced | attractive
alternative |
|
|
| opinion |
| CKD patients |
| intravenous (IV) Fe
requirements. However, | treatment
especially for |
|
|
|
|
|
|
| pooled analyses of
phase 3 studies have | patients with
ESA |
|
|
|
|
|
|
| revealed frequent
thromboembolic events in | hyporesponsive due
to |
|
|
|
|
|
|
| the roxadustat
group, which might be attributed | impaired Fe
utilization. |
|
|
|
|
|
|
| to rapid changes in
Hb and inadequate Fe | So, the appropriate
selection |
|
|
|
|
|
|
| supplementation.
Roxadustat is an attractive | of target patients
and its |
|
|
|
|
|
|
| alternative
treatment especially for patients | proper use are
crucially |
|
|
|
|
|
|
| with ESA
hyporesponsive due to impaired Fe | important. |
|
|
|
|
|
|
| utilization. |
|
|
| Eckardt et
al, | Phase 3 | Random, open- | 3,923 patients | Vadadustat | In the pooled
analysis, the first MACE occurred | vadadustat was
noninferior | (92) |
| 2021 | clinical | label, non- | with |
| in 355 patients
(18.2%) in the vadadustat group | to darbepoetin alfa
with |
|
|
| trial | inferiority | occasional |
| and in 377 patients
(19.3%) in the darbepoetin | respect to
cardiovascular |
|
|
|
| (vadadustat: | or dialysis- |
| alfa group [hazard
ratio, 0.96; 95% confidence | safety and
correction and |
|
|
|
| darbepoetin | dependent |
| interval (CI), 0.83
to 1.11]. The mean | maintenance of
Hb |
|
|
|
| alfa) | (DD-CKD) |
| differences between
the groups in the change |
concentrations. |
|
|
|
|
|
|
| in Hb concentration
were −0.31 g per deciliter |
|
|
|
|
|
|
|
| (95% CI, −0.53 to
−0.10) at weeks 24 to 36 and |
|
|
|
|
|
|
|
| −0.07 g per
deciliter (95% CI, −0.34 to 0.19) |
|
|
|
|
|
|
|
| at weeks 40 to 52
in the incident DD-CKD |
|
|
|
|
|
|
|
| trial and −0.17 g
per deciliter (95% CI, −0.23 |
|
|
|
|
|
|
|
| to −0.10) and −0.18
g per deciliter (95% CI, |
|
|
|
|
|
|
|
| −0.25 to −0.12),
respectively, in the prevalent |
|
|
|
|
|
|
|
| DD-CKD trial. The
incidence of serious adverse |
|
|
|
|
|
|
|
| events in the
vadadustat group was 49.7% in |
|
|
|
|
|
|
|
| the incident DD-CKD
trial and 55.0% in the |
|
|
|
|
|
|
|
| prevalent DD-CKD
trial, and the incidences |
|
|
|
|
|
|
|
| in the darbepoetin
alfa group were 56.5% and |
|
|
|
|
|
|
|
| 58.3%,
respectively. |
|
|
| Provenzano et
al, | Phase 1b | Double-blind | 17 patients | Roxadustat | Maximum plasma
concentration and area | Peak median
endogenous | (93) |
| 2020 | clinical | placebo | with hemo- | (FG-4592) | under the plasma
concentration-time curve | EPO levels were 96
mIU/ml |
|
|
| trial | randomized | dialysis |
| for patients
receiving roxadustat were slightly | and 268 mIU/ml for
the 1- |
|
|
|
| controlled
study | end-stage |
| more than dose
proportional and elimination | and 2-mg/kg
doses, |
|
|
|
| (roxadustat: | renal disease |
| half-life ranged
from 14.7–19.4 h. Roxadustat | respectively,
within |
|
|
|
| Placebo = 3:1) |
|
| was highly protein
bound (99%) in plasma, and | physiologic range
of |
|
|
|
|
|
|
| dialysis
contributed a small fraction of the total | endogenous EPO
responses |
|
|
|
|
|
|
| clearance: only
4.56 and 3.04% of roxadustat | to hypoxia at high
altitude or |
|
|
|
|
|
|
| recovered from the
1 and 2 mg/kg dose groups, | after blood loss.
No serious |
|
|
|
|
|
|
| respectively.
Roxadustat induced transient | adverse events were
reported, |
|
|
|
|
|
|
| elevations of
endogenous EPO that peaked | and there were no
treatment- |
|
|
|
|
|
|
| between 7 and 14 h
after dosing and returned to | or dose-related
trends in |
|
|
|
|
|
|
| baseline by 48 h
after dosing. | adverse event
incidence. |
|
| Akizawa et
al, | Phase 3 | Open-label, | Non-dialysis | roxadustat | Either the
roxadustat or DA comparative group | The roxadustat dose
required | (94) |
| 2021 | clinical | partially | dependent |
| received treatment
(roxadustat, n=131; DA, | to maintain target
Hb in NDD |
|
|
| trial | random | (NDD) |
| n=131). Higher mean
(standard deviation) | patients in Japan
with anemia |
|
|
|
| (roxadustat
and | patients |
| doses of both
roxadustat [63.15 (24.84) mg] | of CKD relative to
DA dose |
|
|
|
| darbepoetin | with CKD |
| and DA [47.33
(29.79) µg] were required in the | may not be impacted
by |
|
|
|
| alfa 1: 1) | anemia |
| highest ESA
resistance index (≥6.8) quartile | low-grade
inflammation. |
|
|
|
|
|
|
| (P=0.003 and
P<0.001, respectively). Patients | Roxadustat may be
beneficial |
|
|
|
|
|
|
| with adequate Fe
repletion had the lowest doses | for
ESA-hyporesponsive |
|
|
|
|
|
|
| for both roxadustat
[45.54 (18.01) mg] and DA | NDD CKD
patients. |
|
|
|
|
|
|
| [28.13 (20.98) µg].
High-sensitivity C-reactive |
|
|
|
|
|
|
|
| protein ≥28.57
nmol/l and the estimated |
|
|
|
|
|
|
|
| glomerular
filtration rate <15 ml/min/1.73 m2 |
|
|
|
|
|
|
|
| were associated
with requiring higher DA but |
|
|
|
|
|
|
|
| not roxadustat
doses. |
|
|
Additionally, pharmacokinetic evaluations in healthy
volunteers have assessed the interaction between roxadustat and
warfarin. The combination has been found to be well-tolerated, with
only mild treatment-related adverse events. Importantly,
co-administration does not necessitate warfarin dose adjustments,
indicating that roxadustat does not significantly alter warfarin
metabolism or anticoagulant activity (96).
The potential impact of HIF-PHIs on tumor
development and progression has been a subject of significant
concern, particularly given their close association with VHL
disease. VHL disease is a syndrome characterized by detectable
genetic abnormalities in the VHL gene, frequently associated with
tumor formation (97). HIF
regulates the transcription of numerous target genes, including
VEGF, a key factor that promotes tumor growth. Additionally, two
Phase 3 clinical trials conducted in China have reported a higher
incidence of hyperkalemia in the roxadustat group. However, further
analysis of potassium levels measured by central laboratories does
not confirm an increased risk of hyperkalemia in patients treated
with Roxadustat (88,98).
In the TREAT trial, patients in the darbepoetin alfa
group (target Hb 130 g/l) exhibit a higher risk of fatal or
non-fatal stroke, venous thromboembolism, and arterial
thromboembolism compared with the placebo group (where darbepoetin
alfa is only administered when Hb levels fell below 90 g/l)
(99). Furthermore, in 2019, the
first reported case of roxadustat-induced pulmonary arterial
hypertension is documented (100).
Beyond its role as an HIF prolyl hydroxylase
inhibitor, roxadustat has been suggested to exert HIF-independent
effects, indicating the presence of potential off-target
activities. Notably, it may increase the risk of pulmonary arterial
hypertension and vascular calcification, as well as exacerbate
inflammation and infection (101). Although no significant off-target
effects have been observed in clinical trials or real-world
applications thus far, HIF regulates a vast array of genes and has
pleiotropic effects. Therefore, strict control of HIF activation
levels and duration is essential, and careful consideration of
roxadustat's dosage and treatment regimen is warranted to minimize
unintended side effects resulting from excessive HIF
activation.
PHDs and erythrocytosis
Human survival depends on a continuous and adequate
oxygen supply to meet cellular metabolic demands, primarily through
oxidative phosphorylation, which generates ATP. HIFs play a central
role in regulating gene transcription to maintain oxygen
homeostasis by balancing oxygen supply and demand. The activity of
HIFs is tightly regulated through oxygen-dependent hydroxylation,
primarily mediated by PHD proteins and VHL proteins. Mutations in
VHL, HIF-2α and PHD2 genes can lead to hereditary polycythemia, a
condition characterized by excessive RBC production and PHA due to
aberrantly elevated HIF activity. Furthermore, genetic adaptations
involving variations in PHD2 and HIF-2 contribute to high-altitude
acclimatization by reducing erythropoiesis and pulmonary vascular
reactivity to hypoxia, enabling improved survival in low-oxygen
environments (102).
Population genomic studies have identified EPAS1
(HIF-2α) and EGLN1 (PHD2) as the primary genes responsible for
high-altitude adaptation. These genes exhibit strong associations
with lower Hb levels in the Tibetan population, suggesting an
evolutionary advantage in hypoxic environments (103). PHD2, encoded by EGLN1, functions
as a critical oxygen sensor, facilitating HIF-α hydroxylation and
subsequent degradation under normoxic conditions. In addition to
its catalytic domain, PHD2 contains a highly conserved zinc finger
domain, which independently interacts with HIF-α in vitro. A
C36S/C42S EGLN1 knockout mutation, which disrupts the zinc finger
function, leads to increased EPO gene expression, excessive
erythropoiesis, and an enhanced hypoxic ventilatory response in
vivo. These physiological effects are attributed to
loss-of-function mutations in EGLN1, resulting in prolonged HIF
stabilization and activation (104).
A gain-of-function mutation in EGLN1 (PHD2 D4E:
C127S), along with polymorphisms in EPAS1 (HIF-2α), contributes to
reduced Hb levels among Tibetan highlanders. While the influence of
EGLN1 haplotypes on Hb concentration varies with age in
low-altitude populations, Tibetan highlanders consistently exhibit
lower Hb levels due to adaptation to the EPAS1 rs142764723 C/C
allele. The high-altitude-adapted EGLN1 haplotypes, c.12C>G and
c.380G>C, are unique genetic variations found in Tibetans,
conferring resistance to polycythemia while enabling survival in
chronic hypoxia. Moreover, EPAS1 haplotypes have been incorporated
into the Denisovan genome, suggesting that these genetic
modifications play a critical role in shaping the lower Hb
concentration observed in Tibetans living on the plateau (105).
The EPAS1 variant prevalent in the Tibetan
population has been shown to reduce gene expression in human
umbilical endothelial cells and the placenta. Additionally, mice
with heterozygous EPAS1 knockout exhibit a delayed physiological
response to prolonged hypoxia. This Tibetan EPAS1 variant is
associated not only with lower Hb levels but also with a diminished
pulmonary vasoconstriction response. These findings suggest that
EPAS1 downregulation serves as the molecular basis for Tibetan
adaptation to high-altitude hypoxia, minimizing the risk of chronic
mountain sickness while optimizing oxygen utilization in a
low-oxygen environment (106).
PHD2 serves as a key regulator of HIFs, mediating
their degradation and thereby controlling physiological responses
to hypoxia, including RBC production. The PHD2 p.[Asp4Glu;
Cys127Ser] variant exhibits lower oxygen Km values, which enhances
the degradation of HIFs even under low-oxygen conditions. In
wild-type erythroid progenitor cells, hypoxia typically stimulates
proliferation; however, in patients carrying the EGLN1 c.[12C>G;
380G>C] mutation, progenitor cell proliferation is significantly
suppressed under hypoxic culture conditions. This mutation,
originating from a common haplotype ~8,000 years ago, has been
linked to high-altitude adaptation. Specifically, the EGLN1
c.[12C>G; 380G>C] variant prevents the hypoxia-induced,
HIF-mediated increase in erythropoiesis, thereby acting as a
molecular defense mechanism against high-altitude erythrocytosis in
Tibetans. This genetic adaptation plays a crucial role in
maintaining normal erythrocyte levels in high-altitude populations,
allowing them to thrive in oxygen-deprived environments (107).
Whole-genome sequencing of 10 Tibetan patients with
high-altitude polycythemia (HAPC) has identified two significant
genetic loci associated with erythrocytosis: the egln3/phd3 homolog
(14q13.1, rs1346902) and the PPP1R2P1 (protein phosphatase 1
regulatory inhibitor subunit 2) gene (6p21.32, rs521539). The
egln3/phd3b gene regulates HIF-α stability, while PPP1R2P1 is
involved in ROS homeostasis and oxidative stress regulation
(108).
Furthermore, the zinc finger domain of PHD2 binds
specifically to a Pro-Xaa-Leu-Glu (PXLE) motif found in the
ribosomal chaperone nascent polypeptide complex-α (NACA). This
interaction facilitates the recruitment of PHD2 to the translation
machinery, enabling co-translational hydroxylation of HIF-α.
Notably, this co-translational modification is enhanced by the
presence of a translational pause sequence within HIF-α, which
increases PHD2's efficiency in targeting the protein. Mice carrying
a mutation in Naca that eliminates the PXLE motif exhibit
erythrocytosis, highlighting the functional importance of this
co-translational regulatory mechanism in RBC production (109).
Based on the aforementioned theoretical framework,
HIF stabilization may be detrimental in various disease conditions,
including certain cancers and PHA. Consequently, pharmacological
strategies to inhibit HIFs have been proposed. Early
pharmacological interventions have focused on suppressing HIF
expression or promoting HIF degradation. More recently,
small-molecule inhibitors such as PT2977 have been developed to
block the formation of transcriptionally active HIF-2α/HIF-1β
heterodimers. Depending on the specific HIFα isoform being
targeted, pharmacological interventions can be classified into
non-specific inhibitors targeting both HIF-1α and HIF-2α or
isoform-selective inhibitors specifically targeting either HIF-1α
or HIF-2α.
Non-specific HIF inhibitors include
2-methoxyestradiol, digoxin, and berberine acetate. However, these
compounds have faced significant limitations in clinical
development. 2-Methoxyestradiol, an endogenous estrogen metabolite,
has a limited clinical window; digoxin requires HIF inhibition at
toxic concentrations due to its narrow therapeutic index; and
berberine acetate has potential side effects and a short half-life,
preventing further clinical progress (110).
Selective HIF-1α inhibitors include topotecan
(NSC-609699), PX-478 (S-2-amino-3-[4′-N,N-bis(chloroethyl)amino]
phenylpropionic acid N-oxide dihydrochloride), cyclic peptide
inhibitors (cyc-CLLFVY), and locked nucleic acid antisense
oligonucleotides such as EZN-2968/RO7070179. While these compounds
demonstrate potential therapeutic benefits, further research is
required to establish their safety and efficacy in humans (110).
Selective HIF-2α inhibitors include C76 (methyl
3-{2-[cyano(methylsulfonyl)methylene]hydrazinyl}thiophene-2-carboxylate),
and PT2385, the first clinical HIF-2α inhibitor. Later, the FDA
approved PT2977 (belzutifan, MK-6482) for the treatment of ccRCC in
patients with VHL disease. Additionally, several HIF-2α inhibitors
from Nikang (NKT2152), Novartis (DFF332), and Arcus (AB521) are in
various stages of clinical development (110).
Given that the EPAS1A529V mutation is a key driver
of Pacak-Zhuang syndrome, belzutifan has been investigated as a
potential therapeutic option for a single patient with severe
polycythemia, hypertension and multiple PGL (110). Targeting HIF-2α may offer
therapeutic benefits for PHA, retinal neovascularization and
polycythemia.
While significant advancements have been made in HIF
inhibition, research on enhancing PHD enzyme activity to counteract
HIF accumulation remains scarce. This area presents challenges
similar to small-molecule p53 reactivators, which are further
complicated by the diverse isoforms and subcellular localization of
PHDs. One promising future direction involves RNA-based therapies,
including siRNA and gene editing technologies, alongside the
development of targeted delivery mechanisms. These approaches could
enable precise modulation of HIF activity while minimizing
off-target effects on unrelated tissues or cell types, providing a
novel avenue for HIF activation or inhibition in disease
therapy.
Prospects of PHD-targeted therapies for
high-altitude polycythemia
The link between polycythemia and high-altitude
adaptation was first reported over a century ago, with subsequent
discoveries highlighting the central role of HIF-1 in oxygen
homeostasis. As a heterodimeric transcription factor composed of an
α and β subunit, HIF-1 is primarily regulated by oxygen-dependent
post-translational hydroxylation of its α subunit. This regulatory
mechanism is mediated by non-heme Fe hydroxylases, which
specifically hydroxylate the proline and asparagine residues of
HIF-1 through dioxygenation, utilizing 2-OG as a co-substrate.
Three PHD enzymes and FIH-1 function as essential cellular oxygen
sensors, fine-tuning HIF activity in response to fluctuating oxygen
levels (111).
Genetic studies of high-altitude Tibetans have
identified EPAS1 (HIF-2α), EGLN1 (PHD2), and PPARA as key genes
shaped by natural selection for high-altitude adaptation. Among
these, HIF signaling serves as the primary regulator of
erythropoiesis and other critical physiological functions (112). Hypoxia-induced responses,
spanning cardiovascular, pulmonary and metabolic pathways, provide
crucial insights into disease mechanisms. Understanding these
adaptive pathways may pave the way for targeted therapies for
hypoxia-related conditions, including cardiopulmonary diseases,
neurodegenerative disorders, metabolic syndromes such as diabetes
and obesity, and high-altitude illnesses. To date, no PHD
activators or functional restorers have been developed to enhance
HIF-α degradation, thereby attenuating downstream gene expression
and mitigating associated pathological effects. In Tibetans,
genetic variants of EPAS1 downregulate EPAS1 transcription, serving
as a protective mechanism against erythrocytosis. This natural
adaptation helps maintain relatively low Hb levels and is
associated with reduced pulmonary vasoconstriction responses
(106). Building upon these
genetic insights, strategies aimed at downregulating HIF-α
transcription or enhancing PHD enzymatic activity could provide
therapeutic avenues for HAPC and hypoxia-induced PHA. Such an
approach may help maintain optimal RBC counts, improve
oxygen-carrying capacity, balance erythropoiesis and apoptosis, and
reduce complications arising from increased blood viscosity,
ultimately alleviating patient symptoms and improving
prognosis.
Despite the advancements in developing targeted
inhibitors, the development of PHD activators or functional
restorers remains a significant challenge (113). Although small-molecule p53
reactivators have been explored in clinical trials for restoring
the function of certain p53 mutations, similar approaches have yet
to be translated into clinical applications for hypoxia-related
diseases (114).
Theoretical models suggest that HIF-α inhibitors or
PHD functional restorers could be valuable in treating HAPC by
counteracting excessive erythropoiesis. While extensive research
has been conducted on HIF inhibitors in oncology, no studies have
specifically investigated HIF-α suppression as a therapeutic
strategy for HAPC. Furthermore, efforts to enhance or restore PHD
enzyme function face formidable technological challenges, as
identifying compounds with strong biological efficacy suitable for
clinical development remains difficult.
Additionally, both HIFs and PHDs exhibit
subtype-specific effects in hypoxic and inflammatory environments,
adding another layer of complexity to drug development. However,
promising advancements in targeting hypoxia-driven inflammatory
pathways suggest that novel therapeutic strategies are on the cusp
of entering clinical trials (69).
Acknowledgements
Not applicable.
Funding
The present study search was supported by the Basic Research
Project of Qinghai Provincial Department of Science and Technology
(grant no. 2025-ZJ-730) and the Clinical Research Center for Blood
System Diseases of Qinghai (grant no. 2021-SF-136). Additional
funding was provided by the National Natural Science Foundation of
China (grant no. U24A20749) and the 2022 ‘Kunlun Yingcai’ Advanced
Innovation and Entrepreneurship Top-notch Talents Project in
Qinghai.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YH, ZHZ and YBX drafted the manuscript. WQL, GXH and
KS conducted to the literature search, data organization and edited
content. YBX was responsible for the structure, concept and design
of the article, as well as proofreading the version to be
published. Data authentication is not applicable. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Holmquist-Mengelbier L, Fredlund E,
Lofstedt T, Noguera R, Navarro S, Nilsson H, Pietras A,
Vallon-Christersson J, Borg A, Gradin K, et al: Recruitment of
HIF-1alpha and HIF-2alpha to common target genes is differentially
regulated in neuroblastoma: HIF-2alpha promotes an aggressive
phenotype. Cancer Cell. 10:413–423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xie Y, Shi X, Sheng K, Han G, Li W, Zhao
Q, Jiang B, Feng J, Li J and Gu Y: PI3K/Akt signaling transduction
pathway, erythropoiesis and glycolysis in hypoxia (Review). Mol Med
Rep. 19:783–791. 2019.PubMed/NCBI
|
|
3
|
Stolze IP, Mole DR and Ratcliffe PJ:
Regulation of HIF: Prolyl hydroxylases. Novartis Found Symp.
272:15–25. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ehdaie B and Theodorescu D: Predicting
tumor outcomes in urothelial bladder carcinoma: Turning pathways
into clinical biomarkers of prognosis. Expert Rev Anticancer Ther.
8:1103–1110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pugh CW: Modulation of the hypoxic
response. Adv Exp Med Biol. 903:259–271. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hewitson KS, McNeill LA and Schofield CJ:
Modulating the hypoxia-inducible factor signaling pathway:
Applications from cardiovascular disease to cancer. Curr Pharm Des.
10:821–833. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Thompson CB: Into thin air: How we sense
and respond to hypoxia. Cell. 167:9–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wong BW, Kuchnio A, Bruning U and
Carmeliet P: Emerging novel functions of the oxygen-sensing prolyl
hydroxylase domain enzymes. Trends Biochem Sci. 38:3–11. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ladroue C, Carcenac R, Leporrier M, Gad S,
Le Hello C, Galateau-Salle F, Feunteun J, Pouysségur J, Richard S
and Gardie B: PHD2 mutation and congenital erythrocytosis with
paraganglioma. N Engl J Med. 359:2685–2692. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eltzschig HK, Eckle T and Grenz A: PHD2
mutation and congenital erythrocytosis with paraganglioma. N Engl J
Med. 360:1361–1362. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Berra E, Benizri E, Ginouves A, Volmat V,
Roux D and Pouyssegur J: HIF prolyl-hydroxylase 2 is the key oxygen
sensor setting low steady-state levels of HIF-1alpha in normoxia.
EMBO J. 22:4082–4090. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang J, Zhao Q, Mooney SM and Lee FS:
Sequence determinants in hypoxia-inducible factor-1alpha for
hydroxylation by the prolyl hydroxylases PHD1, PHD2, and PHD3. J
Biol Chem. 277:39792–39800. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Metzen E, Berchner-Pfannschmidt U, Stengel
P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C,
Hellwig-Bürgel T, Jelkmann W, et al: Intracellular localisation of
human HIF-1 alpha hydroxylases: Implications for oxygen sensing. J
Cell Sci. 116:1319–1326. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Van Welden S, Laukens D, Ferdinande L, De
Vos M and Hindryckx P: Differential expression of prolyl
hydroxylase 1 in patients with ulcerative colitis versus patients
with Crohn's disease/infectious colitis and healthy controls. J
Inflamm (Lond). 10:362013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Berra E, Ginouves A and Pouyssegur J: The
hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia
signalling. EMBO Rep. 7:41–45. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaelin WG Jr and Ratcliffe PJ: Oxygen
sensing by metazoans: The central role of the HIF hydroxylase
pathway. Mol Cell. 30:393–402. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lisy K and Peet DJ: Turn me on: Regulating
HIF transcriptional activity. Cell Death Differ. 15:642–649. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Depping R, Jelkmann W and Kosyna FK:
Nuclear-cytoplasmatic shuttling of proteins in control of cellular
oxygen sensing. J Mol Med (Berl). 93:599–608. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Urrutia AA, Guan N, Mesa-Ciller C, Afzal
A, Davidoff O and Haase VH: Inactivation of HIF-prolyl
4-hydroxylases 1, 2 and 3 in NG2-expressing cells induces
HIF2-mediated neurovascular expansion independent of
erythropoietin. Acta Physiol (Oxf). 231:e135472021. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Souma T, Nezu M, Nakano D, Yamazaki S,
Hirano I, Sekine H, Dan T, Takeda K, Fong GH, Nishiyama A, et al:
Erythropoietin synthesis in renal myofibroblasts is restored by
activation of hypoxia signaling. J Am Soc Nephrol. 27:428–438.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pan Y, Mansfield KD, Bertozzi CC, Rudenko
V, Chan DA, Giaccia AJ and Simon MC: Multiple factors affecting
cellular redox status and energy metabolism modulate
hypoxia-inducible factor prolyl hydroxylase activity in vivo and in
vitro. Mol Cell Biol. 27:912–925. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
van der Wel H, Ercan A and West CM: The
Skp1 prolyl hydroxylase from Dictyostelium is related to the
hypoxia-inducible factor-alpha class of animal prolyl
4-hydroxylases. J Biol Chem. 280:14645–14655. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cummins EP, Berra E, Comerford KM,
Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE,
Moynagh P, Pouyssegur J and Taylor CT: Prolyl hydroxylase-1
negatively regulates IkappaB kinase-beta, giving insight into
hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA.
103:18154–18159. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ozer A, Wu LC and Bruick RK: The candidate
tumor suppressor ING4 represses activation of the hypoxia inducible
factor (HIF). Proc Natl Acad Sci USA. 102:7481–7486. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takeda K and Fong GH: Prolyl hydroxylase
domain 2 protein suppresses hypoxia-induced endothelial cell
proliferation. Hypertension. 49:178–184. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Epstein AC, Gleadle JM, McNeill LA,
Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI,
Dhanda A, et al: C. elegans EGL-9 and mammalian homologs define a
family of dioxygenases that regulate HIF by prolyl hydroxylation.
Cell. 107:43–54. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fong GH and Takeda K: Role and regulation
of prolyl hydroxylase domain proteins. Cell Death Differ.
15:635–641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Aprelikova O, Chandramouli GV, Wood M,
Vasselli JR, Riss J, Maranchie JK, Linehan WM and Barrett JC:
Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors.
J Cell Biochem. 92:491–501. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Appelhoff RJ, Tian YM, Raval RR, Turley H,
Harris AL, Pugh CW, Ratcliffe PJ and Gleadle JM: Differential
function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the
regulation of hypoxia-inducible factor. J Biol Chem.
279:38458–38465. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ivan M, Kondo K, Yang H, Kim W, Valiando
J, Ohh M, Salic A, Asara JM, Lane WS and Kaelin WG Jr: HIFalpha
targeted for VHL-mediated destruction by proline hydroxylation:
Implications for O2 sensing. Science. 292:464–468. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Moffitt PE, Wilson GR and Preston RL:
Comparative response of castrate and intact male rats to
diethylstilbestrol. Proc Soc Exp Biol Med. 148:650–652. 1975.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bruick RK and McKnight SL: A conserved
family of prolyl-4-hydroxylases that modify HIF. Science.
294:1337–1340. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lee KE and Simon MC: SnapShot:
Hypoxia-inducible factors. Cell. 163:1288. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hirsila M, Koivunen P, Gunzler V,
Kivirikko KI and Myllyharju J: Characterization of the human prolyl
4-hydroxylases that modify the hypoxia-inducible factor. J Biol
Chem. 278:30772–30780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hagen T, Taylor CT, Lam F and Moncada S:
Redistribution of intracellular oxygen in hypoxia by nitric oxide:
Effect on HIF1alpha. Science. 302:1975–1978. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tarhonskaya H, Chowdhury R, Leung IK, Loik
ND, McCullagh JS, Claridge TD, Schofield CJ and Flashman E:
Investigating the contribution of the active site environment to
the slow reaction of hypoxia-inducible factor prolyl hydroxylase
domain 2 with oxygen. Biochem J. 463:363–372. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Abboud MI, McAllister TE, Leung IKH,
Chowdhury R, Jorgensen C, Domene C, Mecinović J, Lippl K, Hancock
RL, Hopkinson RJ, et al: 2-Oxoglutarate regulates binding of
hydroxylated hypoxia-inducible factor to prolyl hydroxylase domain
2. Chem Commun (Camb). 54:3130–3133. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Osipyants AI, Smirnova NA, Khristichenko
AY, Hushpulian DM, Nikulin SV, Chubar TA, Zakhariants AA, Tishkov
VI, Gazaryan IG and Poloznikov AA: Enzyme-substrate reporters for
evaluation of substrate specificity of HIF prolyl hydroxylase
isoforms. Biochemistry (Mosc). 82:1207–1214. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cockman ME, Lippl K, Tian YM, Pegg HB,
Figg WD Jnr, Abboud MI, Heilig R, Fischer R, Myllyharju J,
Schofield CJ and Ratcliffe PJ: Lack of activity of recombinant HIF
prolyl hydroxylases (PHDs) on reported non-HIF substrates. Elife.
8:e464902019. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gollnick SO, Owczarczak B and Maier P:
Photodynamic therapy and anti-tumor immunity. Lasers Surg Med.
38:509–515. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
McDonough MA, Li V, Flashman E, Chowdhury
R, Mohr C, Liénard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, et
al: Cellular oxygen sensing: Crystal structure of hypoxia-inducible
factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci USA.
103:9814–9819. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Strowitzki MJ, Cummins EP and Taylor CT:
Protein hydroxylation by hypoxia-inducible factor (HIF)
hydroxylases: Unique or ubiquitous? Cells. 8:3842019. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Thevenod F, Schreiber T and Lee WK: Renal
hypoxia-HIF-PHD-EPO signaling in transition metal nephrotoxicity:
Friend or foe? Arch Toxicol. 96:1573–1607. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kruempel JCP, Miller HA, Schaller ML,
Fretz A, Howington M, Sarker M, Huang S and Leiser SF: Hypoxic
response regulators RHY-1 and EGL-9/PHD promote longevity through a
VHL-1-independent transcriptional response. Geroscience.
42:1621–1633. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ozurumba E, Mathew O, Ranganna K, Choi M
and Oyekan A: Regulation of hypoxia inducible factor/prolyl
hydroxylase binding domain proteins 1 by PPARalpha and high salt
diet. J Basic Clin Physiol Pharmacol. 29:165–173. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Souvenir R, Flores JJ, Ostrowski RP,
Manaenko A, Duris K and Tang J: Erythropoietin inhibits HIF-1alpha
expression via upregulation of PHD-2 transcription and translation
in an in vitro model of hypoxia-ischemia. Transl Stroke Res.
5:118–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Demandt JAF, van Kuijk K, Theelen TL,
Marsch E, Heffron SP, Fisher EA, Carmeliet P, Biessen EAL and
Sluimer JC: Whole-body prolyl hydroxylase domain (PHD) 3 deficiency
increased plasma lipids and hematocrit without impacting plaque
size in low-density lipoprotein receptor knockout mice. Front Cell
Dev Biol. 9:6642582021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Duan LJ, Takeda K and Fong GH:
Hematological, hepatic, and retinal phenotypes in mice deficient
for prolyl hydroxylase domain proteins in the liver. Am J Pathol.
184:1240–1250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tojo Y, Sekine H, Hirano I, Pan X, Souma
T, Tsujita T, Kawaguchi S, Takeda N, Takeda K, Fong GH, et al:
Hypoxia signaling cascade for erythropoietin production in
hepatocytes. Mol Cell Biol. 35:2658–2672. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lindholm ME, Fischer H, Poellinger L,
Johnson RS, Gustafsson T, Sundberg CJ and Rundqvist H: Negative
regulation of HIF in skeletal muscle of elite endurance athletes: A
tentative mechanism promoting oxidative metabolism. Am J Physiol
Regul Integr Comp Physiol. 307:R248–R255. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hata M, Yoshitake I, Wakui S, Unosawa S,
Kimura H, Hata H and Shiono M: Long-term patency rate for radial
artery vs. saphenous vein grafts using same-patient materials. Circ
J. 75:1373–1377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Wolf D, Muralidharan A and Mohan S: Role
of prolyl hydroxylase domain proteins in bone metabolism.
Osteoporos Sarcopenia. 8:1–10. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wu C, Rankin EB, Castellini L, Alcudia JF,
LaGory EL, Andersen R, Rhodes SD, Wilson TL, Mohammad KS, Castillo
AB, et al: Oxygen-sensing PHDs regulate bone homeostasis through
the modulation of osteoprotegerin. Genes Dev. 29:817–831. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Zhang J, Kasim V, Xie YD, Huang C,
Sisjayawan J, Ariyanti AD, Yan XS, Wu XY, Liu CP, Yang L, et al:
Inhibition of PHD3 by salidroside promotes neovascularization
through cell-cell communications mediated by muscle-secreted
angiogenic factors. Sci Rep. 7:439352017. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tian J, Bao X, Yang F, Tang X, Jiang Q, Li
Y, Yao K and Yin Y: Elevation of intracellular alpha-ketoglutarate
levels inhibits osteoclastogenesis by suppressing the NF-kappaB
signaling pathway in a PHD1-dependent manner. Nutrients.
15:7012023. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kapitsinou PP, Rajendran G, Astleford L,
Michael M, Schonfeld MP, Fields T, Shay S, French JL, West J and
Haase VH: The endothelial prolyl-4-hydroxylase domain
2/hypoxia-inducible factor 2 axis regulates pulmonary artery
pressure in mice. Mol Cell Biol. 36:1584–1594. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chu HX and Jones NM: Changes in
hypoxia-inducible factor-1 (HIF-1) and regulatory prolyl
hydroxylase (PHD) enzymes following hypoxic-ischemic injury in the
neonatal rat. Neurochem Res. 41:515–522. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Yang M, Su H, Soga T, Kranc KR and Pollard
PJ: Prolyl hydroxylase domain enzymes: Important regulators of
cancer metabolism. Hypoxia (Auckl). 2:127–142. 2014.PubMed/NCBI
|
|
59
|
Miikkulainen P, Hogel H, Seyednasrollah F,
Rantanen K, Elo LL and Jaakkola PM: Hypoxia-inducible factor
(HIF)-prolyl hydroxylase 3 (PHD3) maintains high HIF2A mRNA levels
in clear cell renal cell carcinoma. J Biol Chem. 294:3760–3771.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Koren A, Rijavec M, Krumpestar T, Kern I,
Sadikov A, Čufer T and Korošec P: Gene expression levels of the
prolyl hydroxylase domain proteins PHD1 and PHD2 but not PHD3 are
decreased in primary tumours and correlate with poor prognosis of
patients with surgically resected non-small-cell lung cancer.
Cancers (Basel). 13:23092021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Deschoemaeker S, Di Conza G, Lilla S,
Martín-Pérez R, Mennerich D, Boon L, Hendrikx S, Maddocks OD, Marx
C, Radhakrishnan P, et al: PHD1 regulates p53-mediated colorectal
cancer chemoresistance. EMBO Mol Med. 7:1350–1365. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Jokilehto T and Jaakkola PM: The role of
HIF prolyl hydroxylases in tumour growth. J Cell Mol Med.
14:758–770. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Harnoss JM, Gebhardt JM, Radhakrishnan P,
Leowardi C, Burmeister J, Halligan DN, Yuan S, Kennel KB,
Strowitzki MJ, Schaible A, et al: Prolyl hydroxylase inhibition
mitigates pouchitis. Inflamm Bowel Dis. 26:192–205. 2020.PubMed/NCBI
|
|
64
|
Bakshi HA, Mishra V, Satija S, Mehta M,
Hakkim FL, Kesharwani P, Dua K, Chellappan DK, Charbe NB,
Shrivastava G, et al: Dynamics of prolyl hydroxylases levels during
disease progression in experimental colitis. Inflammation.
42:2032–2036. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Taniguchi CM, Finger EC, Krieg AJ, Wu C,
Diep AN, LaGory EL, Wei K, McGinnis LM, Yuan J, Kuo CJ and Giaccia
AJ: Cross-talk between hypoxia and insulin signaling through Phd3
regulates hepatic glucose and lipid metabolism and ameliorates
diabetes. Nat Med. 19:1325–1330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Huang M, Paglialunga S, Wong JM, Hoang M,
Pillai R and Joseph JW: Role of prolyl hydroxylase domain proteins
in the regulation of insulin secretion. Physiol Rep. 4:e127222016.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hickey MM, Lam JC, Bezman NA, Rathmell WK
and Simon MC: von Hippel-Lindau mutation in mice recapitulates
Chuvash polycythemia via hypoxia-inducible factor-2alpha signaling
and splenic erythropoiesis. J Clin Invest. 117:3879–3889.
2007.PubMed/NCBI
|
|
68
|
Gordeuk VR and Prchal JT: Vascular
complications in Chuvash polycythemia. Semin Thromb Hemost.
32:289–294. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Watts ER and Walmsley SR: Inflammation and
hypoxia: HIF and PHD isoform selectivity. Trends Mol Med. 25:33–46.
2019. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Minamishima YA: Hypoxic response as a
therapeutic target of human diseases. Nihon Yakurigaku Zasshi.
155:40–45. 2020.(In Japanese). View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Lawson H, Holt-Martyn JP, Dembitz V,
Kabayama Y, Wang LM, Bellani A, Atwal S, Saffoon N, Durko J, van de
Lagemaat LN, et al: The selective prolyl hydroxylase inhibitor IOX5
stabilizes HIF-1alpha and compromises development and progression
of acute myeloid leukemia. Nat Cancer. 5:916–937. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yu Y, Su Y, Yang S, Liu Y, Lin Z, Das NK,
Wu Q, Zhou J, Sun S, Li X, et al: Activation of Intestinal
HIF2alpha ameliorates iron-refractory anemia. Adv Sci (Weinh).
11:e23070222024. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Locatelli F, Del Vecchio L, De Nicola L
and Minutolo R: Are all erythropoiesis-stimulating agents created
equal? Nephrol Dial Transplant. 36:1369–1377. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Zhu Y, Wang Y, Jia Y, Xu J and Chai Y:
Roxadustat promotes angiogenesis through HIF-1alpha/VEGF/VEGFR2
signaling and accelerates cutaneous wound healing in diabetic rats.
Wound Repair Regen. 27:324–334. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Cirillo F, Resmini G, Angelino E, Ferrara
M, Tarantino A, Piccoli M, Rota P, Ghiroldi A, Monasky MM, Ciconte
G, et al: HIF-1alpha directly controls WNT7A expression during
myogenesis. Front Cell Dev Biol. 8:5935082020. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li X, Cui XX, Chen YJ, Wu TT, Xu H, Yin H
and Wu YC: Therapeutic potential of a prolyl hydroxylase inhibitor
FG-4592 for Parkinson's diseases in vitro and in vivo: Regulation
of redox biology and mitochondrial function. Front Aging Neurosci.
10:1212018. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Singh C, Hoppe G, Tran V, McCollum L,
Bolok Y, Song W, Sharma A, Brunengraber H and Sears JE: Serine and
1-carbon metabolism are required for HIF-mediated protection
against retinopathy of prematurity. JCI Insight. 4:e1293982019.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Schley G, Klanke B, Kalucka J, Schatz V,
Daniel C, Mayer M, Goppelt-Struebe M, Herrmann M, Thorsteinsdottir
M, Palsson R, et al: Mononuclear phagocytes orchestrate prolyl
hydroxylase inhibition-mediated renoprotection in chronic
tubulointerstitial nephritis. Kidney Int. 96:378–396. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Han F, Wu G, Han S, Li Z, Jia Y, Bai L, Li
X, Wang K, Yang F, Zhang J, et al: Hypoxia-inducible factor
prolyl-hydroxylase inhibitor roxadustat (FG-4592) alleviates
sepsis-induced acute lung injury. Respir Physiol Neurobiol.
281:1035062020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Chen C, Yan S, Qiu S, Geng Z and Wang Z:
HIF/Ca(2+)/NO/ROS is critical in roxadustat treating bone fracture
by stimulating the proliferation and migration of BMSCs. Life Sci.
264:1186842021. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Zhang P, Du J, Zhao H, Cheng Y, Dong S,
Yang Y, Li B, Gao F, Sun X, Cai J and Liu C: Radioprotective
effects of roxadustat (FG-4592) in haematopoietic system. J Cell
Mol Med. 23:349–356. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Chen H, Cheng Q, Wang J, Zhao X and Zhu S:
Long-term efficacy and safety of hypoxia-inducible factor prolyl
hydroxylase inhibitors in anaemia of chronic kidney disease: A
meta-analysis including 13,146 patients. J Clin Pharm Ther.
46:999–1009. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Kansagra KA, Parmar D, Jani RH, Srinivas
NR, Lickliter J, Patel HV, Parikh DP, Heading H, Patel HB, Gupta
RJ, et al: Phase I clinical study of ZYAN1, A novel
prolyl-hydroxylase (PHD) inhibitor to evaluate the safety,
tolerability, and pharmacokinetics following oral administration in
healthy volunteers. Clin Pharmacokinet. 57:87–102. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Singh AK, Carroll K, Perkovic V, Solomon
S, Jha V, Johansen KL, Lopes RD, Macdougall IC, Obrador GT, Waikar
SS, et al: Daprodustat for the treatment of anemia in patients
undergoing dialysis. N Engl J Med. 385:2325–2335. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Singh AK, Carroll K, McMurray JJV, Solomon
S, Jha V, Johansen KL, Lopes RD, Macdougall IC, Obrador GT, Waikar
SS, et al: Daprodustat for the treatment of anemia in patients not
undergoing dialysis. N Engl J Med. 385:2313–2324. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Fishbane S, Pollock CA, El-Shahawy M,
Escudero ET, Rastogi A, Van BP, Frison L, Houser M, Pola M, Little
DJ, et al: Roxadustat versus epoetin alfa for treating anemia in
patients with chronic kidney disease on dialysis: Results from the
randomized phase 3 ROCKIES study. J Am Soc Nephrol. 33:850–866.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Fishbane S, El-Shahawy MA, Pecoits-Filho
R, Escudero ET, Rastogi A, Van BP, Frison L, Houser M, Pola M,
Little DJ, et al: Roxadustat for treating anemia in patients with
CKD not on dialysis: Results from a randomized phase 3 study. J Am
Soc Nephrol. 32:737–755. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Chen N, Hao C, Peng X, Lin H, Yin A, Hao
L, Tao Y, Liang X, Liu Z, Xing C, et al: Roxadustat for anemia in
patients with kidney disease not receiving dialysis. N Engl J Med.
381:1001–1010. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Perkovic V, Blackorby A, Cizman B, Carroll
K, Cobitz AR, Davies R, DiMino TL, Jha V, Johansen KL, Lopes RD, et
al: The ASCEND-ND trial: Study design and participant
characteristics. Nephrol Dial Transplant. 37:2157–2170. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Testi I, Calcagni A, Barton K, Gooch J and
Petrushkin H: Hypotony in uveitis: An overview of medical and
surgical management. Br J Ophthalmol. 107:1765–1770. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Kurata Y, Tanaka T and Nangaku M: An
evaluation of roxadustat for the treatment of anemia associated
with chronic kidney disease. Expert Opin Pharmacother. 23:19–28.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Eckardt KU, Agarwal R, Aswad A, Awad A,
Block GA, Bacci MR, Farag YMK, Fishbane S, Hubert H, Jardine A, et
al: Safety and efficacy of vadadustat for anemia in patients
undergoing dialysis. N Engl J Med. 384:1601–1612. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Provenzano R, Tumlin J, Zabaneh R, Chou J,
Hemmerich S, Neff TB and Yu KP: Oral hypoxia-inducible factor
prolyl hydroxylase inhibitor roxadustat (FG-4592) for treatment of
anemia in chronic kidney disease: A placebo-controlled study of
pharmacokinetic and pharmacodynamic profiles in hemodialysis
patients. J Clin Pharmacol. 60:1432–1440. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Akizawa T, Tanaka-Amino K, Otsuka T and
Yamaguchi Y: Factors affecting doses of roxadustat versus
darbepoetin alfa for anemia in nondialysis patients. Am J Nephrol.
52:702–713. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Kokado Y, Kawai K, Nanjo T, Kinoshita S
and Kondo K: In vitro and clinical pharmacokinetic studies of the
effects of iron-containing agents on vadadustat, an oral
hypoxia-inducible factor-prolyl hydroxylase inhibitor. Clin Ther.
43:1408–1418. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Hansen MB, Kondziella D, Danielsen ER,
Larsen VA, Jansen EC and Hyldegaard O: Cerebral proton magnetic
resonance spectroscopy demonstrates reversibility of
N-acetylaspartate/creatine in gray matter after delayed
encephalopathy due to carbon monoxide intoxication: A case report.
J Med Case Rep. 8:2112014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Gossage L, Eisen T and Maher ER: VHL, the
story of a tumour suppressor gene. Nat Rev Cancer. 15:55–64. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Chen N, Hao C, Liu BC, Lin H, Wang C, Xing
C, Liang X, Jiang G, Liu Z, Li X, et al: Roxadustat treatment for
anemia in patients undergoing long-term dialysis. N Engl J Med.
381:1011–1022. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Pfeffer MA, Burdmann EA, Chen CY, Cooper
ME, de Zeeuw D, Eckardt KU, Feyzi JM, Ivanovich P, Kewalramani R,
Levey AS, et al: A trial of darbepoetin alfa in type 2 diabetes and
chronic kidney disease. N Engl J Med. 361:2019–2032. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Cygulska K, Wejner-Mik P, Plewka M, Figiel
L, Chrzanowski L and Kasprzak JD: Roxadustat: Another drug that
causes pulmonary hypertension? Report of first human case. Pol Arch
Intern Med. 129:344–345. 2019.PubMed/NCBI
|
|
101
|
Zhu X, Jiang L, Wei X, Long M and Du Y:
Roxadustat: Not just for anemia. Front Pharmacol. 13:9717952022.
View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Semenza GL: The genomics and genetics of
oxygen homeostasis. Annu Rev Genomics Hum Genet. 21:183–204. 2020.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Wu TY, Liu FY, Ouzhou L, Cui CY, Qi XB and
Su B: A genetic adaptive pattern-low hemoglobin concentration in
the Himalayan highlanders. Zhongguo Ying Yong Sheng Li Xue Za Zhi.
29:481–493. 2013.PubMed/NCBI
|
|
104
|
Arsenault PR, Song D, Chung YJ, Khurana TS
and Lee FS: The zinc finger of prolyl hydroxylase domain protein 2
is essential for efficient hydroxylation of hypoxia-inducible
factor alpha. Mol Cell Biol. 36:2328–2343. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Tashi T, Reading NS, Wuren T, Zhang X,
Moore LG, Hu H, Tang F, Shestakova A, Lorenzo F, Burjanivova T, et
al: Gain-of-function EGLN1 prolyl hydroxylase (PHD2 D4E:C127S) in
combination with EPAS1 (HIF-2alpha) polymorphism lowers hemoglobin
concentration in Tibetan highlanders. J Mol Med (Berl). 95:665–670.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Peng Y, Cui C, He Y, Ouzhuluobu Zhang H,
Yang D, Zhang Q, Bianbazhuoma Yang L, He Y, et al: Down-regulation
of EPAS1 transcription and genetic adaptation of tibetans to
high-altitude hypoxia. Mol Biol Evol. 34:818–830. 2017.PubMed/NCBI
|
|
107
|
Lorenzo FR, Huff C, Myllymaki M, Olenchock
B, Swierczek S, Tashi T, Gordeuk V, Wuren T, Ri-Li G, McClain DA,
et al: A genetic mechanism for Tibetan high-altitude adaptation.
Nat Genet. 46:951–956. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Gesang L, Gusang L, Dawa C, Gesang G and
Li K: Whole-Genome sequencing identifies the egl nine homologue 3
(egln3/phd3) and protein phosphatase 1 regulatory inhibitor subunit
2 (PPP1R2P1) associated with high-altitude polycythemia in Tibetans
at high altitude. Dis Markers. 2019:59464612019. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Song D, Peng K, Palmer BE and Lee FS: The
ribosomal chaperone NACA recruits PHD2 to cotranslationally modify
HIF-α. EMBO J. 41:e1120592022. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Yuan X, Ruan W, Bobrow B, Carmeliet P and
Eltzschig HK: Targeting hypoxia-inducible factors: Therapeutic
opportunities and challenges. Nat Rev Drug Discov. 23:175–200.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Stockmann C and Fandrey J: Hypoxia-induced
erythropoietin production: A paradigm for oxygen-regulated gene
expression. Clin Exp Pharmacol Physiol. 33:968–979. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Simonson TS, McClain DA, Jorde LB and
Prchal JT: Genetic determinants of Tibetan high-altitude
adaptation. Hum Genet. 131:527–533. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Hassin O and Oren M: Drugging p53 in
cancer: One protein, many targets. Nat Rev Drug Discov. 22:127–144.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Zheng J, Deng Y, Huang B and Chen X:
Efficacy and safety of immune checkpoint inhibitors combined with
chemotherapy as first-line treatment for extensive-stage small cell
lung cancer: a meta-analysis based on mixed-effect models. Front
Med (Lausanne). 10:11989502023. View Article : Google Scholar : PubMed/NCBI
|