Introduction
Cholestasis is characterized by the obstruction of
bile formation, secretion and excretion in hepatocytes or bile duct
epithelial cells, due to various intrahepatic or extrahepatic
causes. This condition prevents bile from entering the duodenum and
results in its direct entry into the bloodstream (1,2). A
previous study indicated that 35% patients newly diagnosed with
chronic liver disease will typically develop cholestasis (3). Cholestasis presents as initial
clinical assessments revealing elevated serum alkaline phosphatase
(ALP) and γ-glutamyl transferase (GGT) activity; however, 56% of
patients do not achieve normal serum biochemical levels of ALP and
GGT upon discharge (4). Without
appropriate treatment, these abnormalities can substantially
increase the risk of hyperbilirubinemia, liver fibrosis, cirrhosis
and liver failure, posing a notable threat to patient survival
(5). Therefore, investigating safe
and effective treatments for cholestasis is of clinical
importance.
Ferroptosis, first identified by Dixon et al
(6) in 2012, is a form of cell
death characterized by iron and reactive oxygen species (ROS)
involvement. Unlike traditional cell death mechanisms, ferroptosis
results from iron-dependent lipid peroxidation and exhibits
distinct ultrastructural features. Elevated lipid peroxidation
levels serve as a key indicator of ferroptosis occurrence (7).
Persistent cholestasis exacerbates damage to bile
duct epithelial cells and hepatocytes, triggering an inflammatory
response (8). Inflammation is
strongly associated with oxidative stress (9,10).
During inflammation, pro-inflammatory factors stimulate phagocytes
and non-phagocytic cells to produce various ROS, such as
O2−, H2O2 and OH−, to
eliminate pathogens (11).
Excessive intracellular ROS then overwhelm glutathione peroxidase 4
(GPX4), disrupting the balance between ROS production and
scavenging, leading to ferroptosis (12). Additionally, inflammatory changes
in hepatocytes, including chronic viral hepatitis, acute hepatitis
and cholestatic hepatitis, can cause intracellular ferritin efflux
(13). Ferritin binds
Fe2+, whereas its depletion increases free
Fe2+ levels. Excess Fe2+ can react with
H2O2 through the Fenton reaction, producing
OH− and hydroxyl radicals with strong oxidizing
properties, thereby promoting ferroptosis (14). Therefore, molecules and signals
involved in iron metabolism and lipid peroxidation are crucial in
the initiation and progression of ferroptosis (15).
The specific link between α-naphthyl isothiocyanate
(ANIT)-induced cholestasis and iron metabolism dysregulation
warrants emphasis. Chronic cholestasis is associated with a
persistent inflammatory response, which can disrupt the hepatic
expression of hepcidin, a master regulator of iron homeostasis
(16,17). Reduced hepcidin levels can lead to
increased iron absorption and mobilization, resulting in hepatic
iron overload (18). This
iron-rich environment, coupled with ANIT-induced oxidative stress,
creates a favorable condition for the Fenton reaction and lipid
peroxidation, thereby priming hepatocytes for ferroptosis (19,20).
Hepatic stellate cells (HSCs) remain quiescent in
healthy liver. By contrast, upon cholestatic liver damage,
persistent intrahepatic inflammation can activate HSCs,
transforming them from vitamin A-storing cells into proliferative,
contractile, inflammatory and chemotactic myofibroblasts that
produce extracellular matrix proteins (21). Ferroptosis in hepatocytes
accelerates this transformation (22). Following ferroptosis activation,
hepatocytes lose their normal functions, weakening their inhibitory
effect on nearby HSCs. The release of free iron ions and ROS from
dead hepatocytes further stimulates HSC activation, perpetuating
ferroptosis. This vicious cycle exacerbates inflammation, enhances
HSC activation and increases extracellular matrix synthesis,
including type IV collagen and laminin, thereby advancing
cholestatic disease progression (23).
Deferoxamine (DFO) is a Food and Drug
Administration-approved iron chelator that has been extensively
applied for treating iron overload disorders such as β-thalassemia
and myelodysplastic syndrome (24). It can effectively reduce free iron
ion levels by chelating them within cells to directly inhibit the
Fenton reaction, thereby preventing cell ferroptosis (25).
Given the intrinsic link between cholestasis and
ferroptosis, it has been hypothesized that hepatocyte ferroptosis
serves as a key driver in the progression of chronic cholestasis.
Therefore, the present study aimed to assess this hypothesis using
the iron chelator DFO to investigate whether the inhibition of
ferroptosis through iron chelation could attenuate the progression
of chronic cholestatic liver injury.
Materials and methods
Animal models and experimental
design
Animal welfare and ethical considerations
All animal experiments were approved by the Animal
Care and Utilization Committee of Shanghai University of
Traditional Chinese Medicine (approval no. PZSHUTCM210715019;
Shanghai, China) and were conducted in strict accordance with the
National Institutes of Health Guide for the Care and Use of
Laboratory Animals (26).
Animal housing
A total of 24 6-week-old male Wistar rats (batch no.
0054518) were purchased from Shanghai SLAC Laboratory Animal Co.,
Ltd. [license no. SCXK (Shanghai) 2017–0005]. The rats weighed
160–180 g upon arrival. They were housed under specific
pathogen-free conditions at 23–24°C with 60±10% humidity, under a
12-h light/12-h dark cycle. After 1 week of acclimation, the rats
were randomly divided into a control group (n=8) and a model group
(n=16).
Dosages
The control group received daily intragastric
administration of an equivalent volume of olive oil (3 ml/kg) and
standard laboratory chow diet. The model group received
intragastric administration of a 2% ANIT (Sigma-Aldrich; Merck
KGaA)-olive oil solution (3 ml/kg) every other day (27) for 12 weeks. From week 9, the model
group was subdivided into the ANIT (n=8) and DFO-treated (n=8)
groups. The DFO-treated group received daily DFO treatment
(MedChemExpress; 100 mg/kg) (28)
for 4 weeks (weeks 9–12). All animals were sacrificed at the end of
week 12 (Fig. 1).
Health monitoring and humane
endpoints
The health and behavior of all animals were
monitored at least twice daily (morning and evening) throughout the
study. Pre-established humane endpoints were strictly adhered to in
order to minimize suffering. Any animal exhibiting one or more of
the following severe clinical signs was sacrificed immediately: i)
>20% loss of initial body weight; ii) prolonged lethargy or
inability to reach food or water; iii) signs of severe pain or
distress (such as persistent vocalization, self-mutilation); iv)
severe dyspnea or cyanosis; v) paralysis or other neurological
deficits that impeded normal mobility. No animals reached these
endpoints prior to the scheduled termination.
Euthanasia, anesthesia and terminal
blood collection
At the end of week 12, all animals were euthanized
for terminal blood collection and tissue harvesting. Euthanasia was
performed by intraperitoneal injection of an overdose of sodium
pentobarbital (150 mg/kg). A total of 6–8 ml of blood was
aseptically collected from the inferior vena cava. Death was
confirmed by the absence of a heartbeat and cessation of
respiration
Analgesia
No additional analgesics were administered during
the chronic model establishment, as the ANIT-induced injury is not
considered to cause notable pain that requires such intervention
and the administration of analgesics could potentially interact
with the study endpoints. Following confirmation of death, liver
tissue was harvested for subsequent analysis.
Biochemical analysis
Detection method of serum liver function and
blood lipids
Blood was collected from the inferior vena cava and
centrifuged (1,000 × g, 20 min and 4°C) to obtain the serum. Serum
alanine aminotransferase (ALT) (cat. no. KF749; Fujifilm Wako Pure
Chemical Corporation), aspartate aminotransferase (AST; cat. no.
KK718; Fujifilm Wako Pure Chemical Corporation), ALP (cat. no.
EH360; Fujifilm Wako Pure Chemical Corporation) and GGT (cat. no.
010260; Yantai Ausbio Biotechnology Co., Ltd.) activities, as well
as total bilirubin (TBIL; cat. no. KK678; Fujifilm Wako Pure
Chemical Corporation), direct bilirubin (DBIL; cat. no. FF870;
Fujifilm Wako Pure Chemical Corporation), total bile acid (TBA;
cat. no. 210428; Yantai Ausbio Biotechnology Co., Ltd.),
triglyceride (TG) (cat. no. L068; Sekisui Medical Co., Ltd.), total
cholesterol (TCH) (cat. no. L979; Sekisui Medical Co., Ltd.) and
low-density lipoprotein (LDL) (cat. no. EG582; Fujifilm Wako Pure
Chemical Corporation) were measured using the automatic biochemical
analyzer (TK-40FR; Canon Medical Systems Corporation).
Detection method of iron ions in serum
and the liver
For serum analysis, 500 µl serum was introduced into
the measurement tube, whereas 500 µl double-distilled water was
placed in the blank tube. A standard application solution (500 µl;
Iron Assay Kit (cat. no. ab83366; Abcam) was added to the standard
tube. Subsequently, 1.5 ml iron chromogenic reagent was added to
each tube according to the manufacturer's instructions and the
tubes were incubated at 100°C for 5 min, before being allowed to
return to room temperature. Subsequently, the samples were
centrifuged at 1,500 × g for 10 min at room temperature. The
optical density (OD) was measured at 520 nm using a microplate
reader (Infinite M200 PRO; Tecan Group, Ltd.) and the total iron
concentration was calculated based on the standard curve and is
expressed as mM. Briefly, the iron content (in nmol) derived from
the standard curve was divided by the sample volume (500 µl) and
multiplied by the sample dilution factor.
Liver tissue samples (10 mg) were homogenized in 90
µl iron assay buffer (cat. no. ab83366; Abcam) and centrifuged at
16,000 × g for 10 min, according to the manufacturer's
instructions. The supernatant was transferred to a 96-well plate.
For standard wells, 100 µl standard diluent was added, whereas
sample wells received 35 µl sample and 65 µl iron assay buffer.
Each well was treated with 5 µl iron reducing agent, followed by
the addition of 100 µl iron probe after a 30-min incubation at
37°C. Following incubation at 37°C for 60 min in the dark, the
absorbance was measured at 520 nm at room temperature. A standard
curve was generated from the OD values of the standard wells and
total iron content in the sample wells was calculated accordingly.
To measure Fe2+ content, 100 µl standard diluent and 5
µl iron reducing agent were added to the standard wells, whereas 35
µl sample and 70 µl iron analysis buffer were added to the sample
wells, with all other conditions unchanged.
Detection method of hydroxyproline
(HYP) in the liver
Liver tissue samples (70 mg) were homogenized in 1
ml RIPA buffer (cat. no. SD006; Shanghai Sidings BioTech Co.,
Ltd.). Following hydrolysis at 95°C for 20 min, 10 µl indicator was
added, and the pH was adjusted until the solution turned
yellow-green. The volume of each tube was then increased to 10 ml
with double-distilled water; from this, 4 ml diluted hydrolysate
was combined with 20 mg activated carbon and centrifuged at 1,500 ×
g for 10 min at room temperature. A 1-ml aliquot of the supernatant
was then used as the measurement sample. For controls, 1 ml
double-distilled water was added to the blank tube, with 1 ml
standard solution to the standard tube. Each tube then received 0.5
ml reagents 1, 2 and 3, was mixed thoroughly and was incubated at
60°C for 15 min. After centrifuging at 1,500 × g for 10 min at room
temperature, 200 µl supernatant was taken to measure the OD at 550
nm (cat. no. A030-2; Nanjing Jiancheng Bioengineering
Institute).
Detection method of malondialdehyde
(MDA) in the liver
Liver tissue samples were accurately weighed (50 mg)
and homogenized in 450 µl 0.9% NaCl, followed by centrifugation at
16,000 × g for 10 min at 4°C. The supernatant was collected for
protein concentration analysis using the BCA method. A working
solution was prepared according to the manufacturer's instructions
(cat. no. A003-1; Nanjing Jiancheng Bioengineering Institute), and
4 ml was added to each tube. To the blank tube, 1 ml absolute
ethanol was added, whereas 1 ml standard solution was added to the
standard tube. To the assay tube, 1 ml sample was added. The tubes
were then incubated in a 95°C water bath for 40 min, cooled to room
temperature and centrifuged at 2,000 × g for 10 min at room
temperature. The OD was measured at 532 nm. The MDA content in
liver tissue was calculated and expressed as nmol/mg of protein
(nmol/mg prot) based on the measured OD values and the known
standard concentration (10 nmol/ml), normalized to the protein
concentration of the tissue homogenate (cat. no. A003-1; Nanjing
Jiancheng Bioengineering Institute).
Detection method of glutathione (GSH)
in the liver
Liver GSH content was determined using a commercial
Glutathione Assay kit (cat. no. A006-2-1; Nanjing Jiancheng
Bioengineering Institute). Liver tissues were processed as
aforementioned for the MDA assay. Subsequently, 60 µl reagent 1 was
added to the blank well, whereas 60 µl standard was added to the
standard well and 60 µl sample was added to the assay well; 60 µl
reagent 2 and 15 µl reagent 3 were then added to each well. The
mixtures were allowed to form natural precipitates for 5 min at
37°C, before the OD was measured at 405 nm. The GSH concentration
was calculated according to the manufacturer's instructions.
Detection method of glutathione
peroxidase (GSH-PX) in the liver
GSH-PX activity was determined using a commercial
Glutathione Peroxidase Assay kit (cat. no. A005; Nanjing Jiancheng
Bioengineering Institute). The liver tissues were processed as
aforementioned for the MDA assay. To the non-enzymatic tube, 40 µl
1 mmol/l GSH was added, whereas 20 µl 1 mmol/l GSH and 20 µl
homogenate were added to the enzymatic tube. Following the
enzymatic reaction, 200 µl supernatant was extracted for the
colorimetric reaction. In total, 200 µl GSH standard solvent was
added to the blank tube, 200 µl 20 µmol/l GSH standard solution was
added to the standard tube and 200 µl supernatant was added to both
the non-enzymatic and enzymatic tubes. To each tube, 200 µl reagent
3, 50 µl reagent 4 and 10 µl reagent 5 were added. After allowing
natural sedimentation for 15 min at 37°C, the OD value was measured
at 412 nm.
Histopathological evaluation
Liver tissues were sectioned into 0.5×0.5 cm pieces,
fixed in 4% paraformaldehyde at room temperature for 24–48 h. The
fixed tissue was dehydrated through a graded ethanol series,
cleared in xylene, and embedded in paraffin. Subsequently, 4-µm
thick sections were prepared. H&E staining was performed
according to standard protocols. Sirius red and Prussian blue
staining were performed according to the manufacturers'
instructions, with incubations at room temperature (1 h for Sirius
red and 30 min for Prussian blue). Digital slide scanning and
panoramic imaging of all stained sections were conducted using a
Panoramic Scanning and Analysis System (LEICA SCN 400; Leica
Microsystems GmbH, Germany) equipped with its Leica SCN Slide
Scanner Software, version 2.5; Leica Microsystems). For
quantitative analysis of collagen deposition, the Sirius
red-positive area (representing collagen fibers) was measured in
five randomly selected fields of view/section at 200× magnification
using ImageJ software (version 2.14.0; National Institutes of
Health). The collagen-positive area ratio was calculated as (Sirius
red-positive area/total tissue area) ×100%. Similarly, the Prussian
blue-positive area (representing iron deposition) was quantified
(five random fields at 200× magnification), and the iron
deposition-positive area ratio was calculated.
Ultrastructural pathology
evaluation
Liver tissue was sectioned into 0.1×0.1 cm pieces
and fixed in 2.5% glutaraldehyde at 4°C for 4–6 h. Samples
underwent standard dehydration through a graded ethanol series,
followed by embedding in acetone overnight. Subsequently, 50-nm
ultrathin sections were subjected to double staining with 3% uranyl
acetate and lead citrate at room temperature, each for 15–20 min.
The samples were examined using a transmission electron microscope
(TK-40FR; FEI; Thermo Fisher Scientific, Inc.). Image acquisition
and initial analysis were performed using the microscope's
proprietary software (Tecnai Imaging and Analysis (TIA) Software,
version 4.7; FEI/Thermo Fisher Scientific).
Western blot analysis
Liver tissue lysates were prepared using ice-cold
RIPA Lysis Buffer (cat. no. SD006) supplemented with a protease
inhibitor cocktail (cat. no. SD001; both Shanghai Sidings BioTech
Co., Ltd.). Protein concentrations were measured using a BCA
protein assay kit. Equal protein amounts (20 µg/lane) were
separated by SDS-PAGE on 10%, 7.5%, or 12.5% polyacrylamide gels
and transferred to nitrocellulose membranes The membranes were
blocked at room temperature for 15 min using QuickBlock™ Western
blocking solution (cat. no. P0239; Beyotime Biotechnology). The
membranes were then incubated overnight at 4°C with primary
antibodies as follows: anti-Nrf2 (1:1,000), anti-COX2 (1:1,000),
anti-GPX4 (1:5,000), anti-TFR1 (1:5,000), anti-ASCL4 (1:50,000),
anti-HO-1 (1:50,000), anti-Steap3 (1:10,000), anti-α-SMA (1:2,000),
anti-FPN1 (1:1,000), anti-DMT1 (1:1,000), anti-FTH1 (1:2,000),
anti-XCT (1:2,000), and anti-Keap1 (1:2,000). This was followed by
a 1 h incubation at room temperature with horseradish
peroxidase-conjugated secondary antibodies (goat anti-rabbit IgG,
cat. no. A0208, and goat anti-mouse IgG, cat. no. A0216; Beyotime
Institute of Biotechnology; 1:1,000). β-actin (1:10,000; cat. no.
66009-1; Proteintech) served as an internal control. Antibodies
against nuclear factor erythroid-2-related factor 2 (Nrf2; cat. no.
ab137550), cyclooxygenase 2 (COX2; cat. no. ab52237), GPX4 (cat.
no. ab125066), transferrin receptor 1 (TFR1; cat. no. ab269513),
acyl-CoA synthetase long-chain family member 4 (ASCL4; cat. no.
ab155282), heme oxygenase 1 (HO-1; cat. no. ab68477),
six-transmembrane epithelial antigen of the prostate 3 (Steap3;
cat. no. ab151566) and α-smooth muscle actin (α-SMA; cat. no.
ab7817) were purchased from Abcam. Ferroportin 1 (FPN1; cat. no.
26601-1-AP) and divalent metal transporter 1 (DMT1; cat. no.
20507-1-AP) antibodies were purchased from Proteintech Group, Inc.
Ferritin heavy chain 1 (FTH1; cat. no. PA4412) and
cystine/glutamate transporter (XCT; cat. no. T57046) antibodies
were purchased from Abmart Pharmaceutical Technology Co., Ltd. The
Kelch-like ECH-associated protein 1 (Keap1) antibody (cat. no.
bs-3648R) was purchased from BIOSS. Protein bands were visualized
using Clarity Western ECL Substrate (cat. no. 1705061; Bio-Rad
Laboratories, Inc.). Images of the blots were then captured and the
band intensities were semi-quantified using ImageJ software
(version 1.54p; National Institutes of Health).
Statistical analysis
The sample size (n=8 per group) was determined a
priori using a power analysis conducted with G*Power software
(version 3.1.9.7; gpower.hhu.de/). The effect size was estimated
from preliminary data obtained from a pilot experiment with 6
rats/group, with an α level of 0.05 (two-sided) and a power (1-β)
of 0.8, ensuring adequate statistical power to detect significant
effects. All data are presented as the mean ± standard deviation.
The normality of data distribution was confirmed using the
Shapiro-Wilk test, and the homogeneity of variances was verified
using Levene's test. For data that satisfied both assumptions,
one-way ANOVA was performed, followed by Tukey's honestly
significant difference post hoc test for multiple comparisons. For
data that violated the assumption of normality or homogeneity of
variances, the non-parametric Kruskal-Wallis test was applied,
followed by Dunn's post hoc test for multiple comparisons. All
statistical analyses were performed using SPSS software (version
25.0; IBM Corp.). P<0.05 was considered to indicate a
statistically significant difference.
Results
Liver function of rats with chronic
cholestasis is impaired and histopathological examination of liver
tissue reveals inflammation
ANIT-induced cholestasis led to significant
increases in serum ALT, AST, ALP and GGT activities, and TBIL,
DBIL, TBA, TCH and LDL levels, whereas TG levels were significantly
decreased compared with those in the control group (Fig. 2A-J). DFO significantly restored
serum ALT, ALP and GGT activities, and TBIL, DBIL, TBA, TCH, LDL
and TG levels, compared with those in the ANIT group. H&E
staining revealed bile duct hyperplasia and inflammatory cell
infiltration in the portal area of rats in the ANIT group, which
were markedly inhibited by DFO treatment (Fig. 2K).
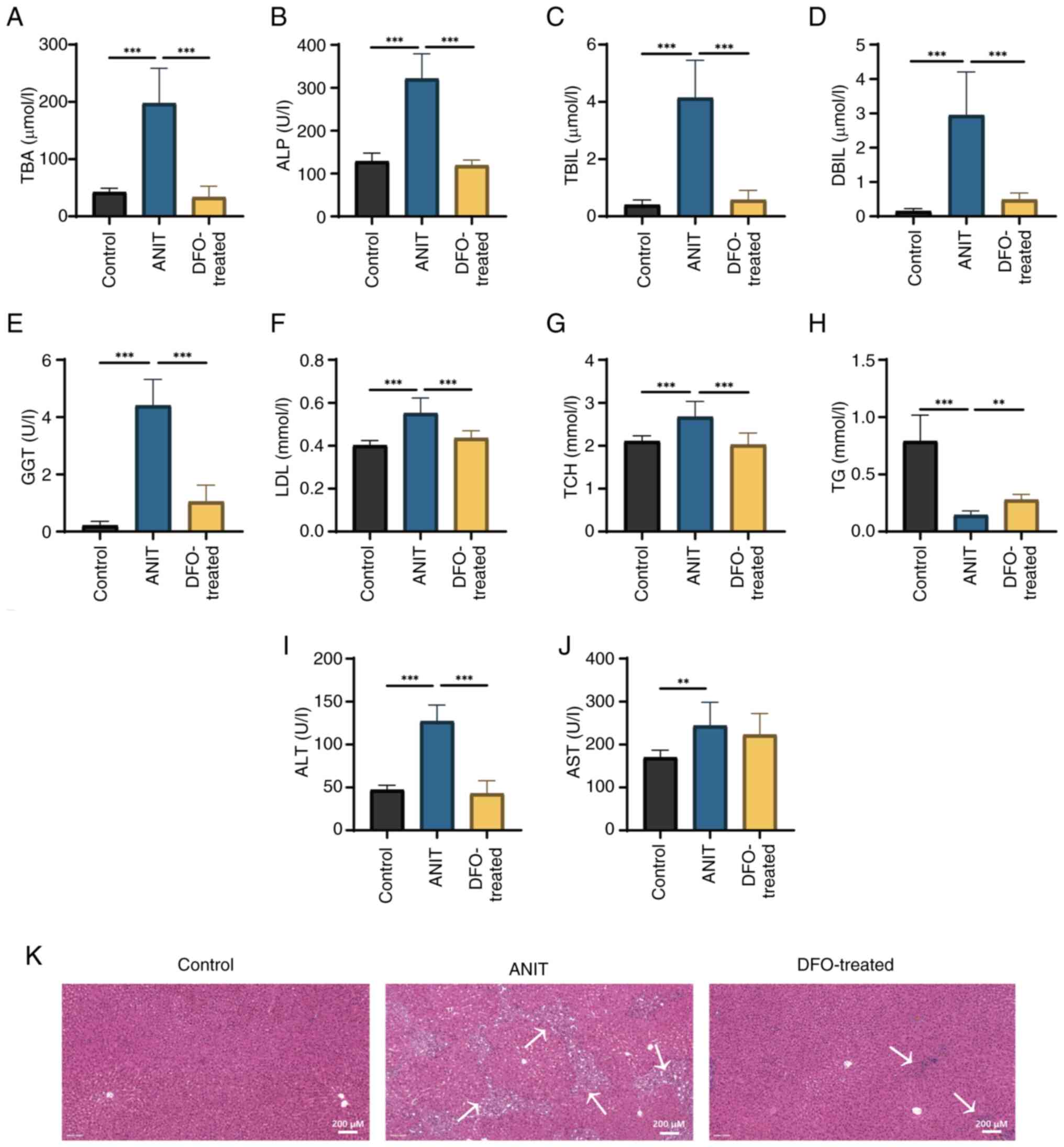 | Figure 2.Liver function of rats with chronic
cholestasis is impaired and histopathological analysis of liver
tissue reveals inflammation. The control group was fed a chow diet,
and the ANIT and DFO-treated groups were intragastrically
administered ANIT olive oil solution with or without DFO
(n=8/group). (A) TBA, (B) ALP, (C) TBIL, (D) DBIL, (E) GGT, (F)
LDL, (G) TCH, (H) TG, (I) ALT and (J) AST were measured.
**P<0.01, ***P<0.001. (K) Hematoxylin and eosin staining
(scale bar, 200 µm). Arrows indicate bile duct hyperplasia and
inflammatory cell infiltration in the portal area. ANIT, α-naphthyl
isothiocyanate; DFO, deferoxamine; TBA, total bile acid; ALP,
alkaline phosphatase; TBIL, total bilirubin; DBIL, direct
bilirubin; GGT, γ-glutamyl transferase; LDL, low-density
lipoprotein; TCH, total cholesterol; TG, triglyceride; ALT, alanine
aminotransferase; AST, aspartate aminotransferase. |
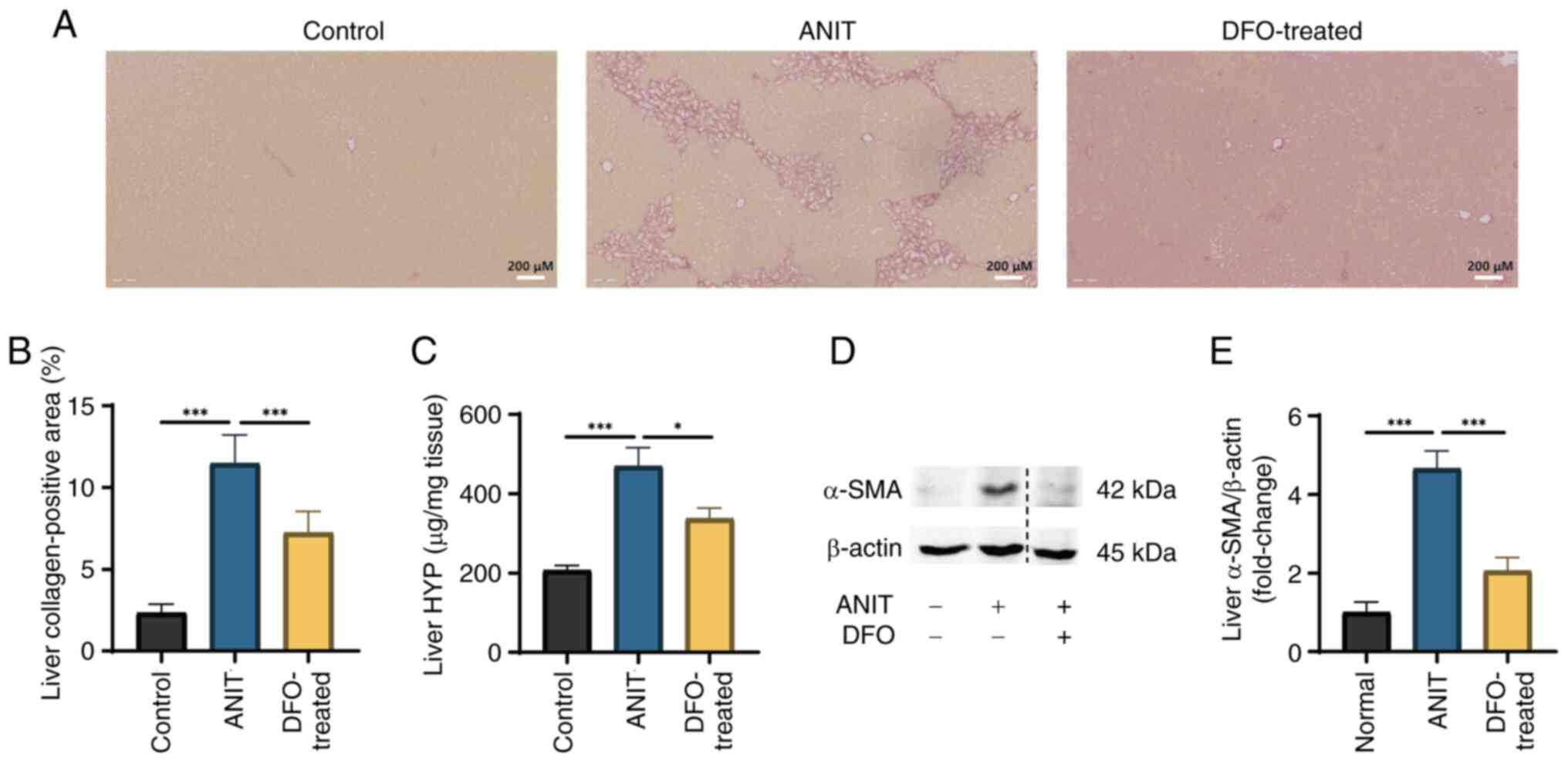
Intrahepatic collagen fiber deposition
is increased in rats with chronic cholestasis
Sirius red staining revealed significant collagen
fiber deposition at the periphery of rat hepatic lobules following
ANIT-induced cholestasis (Fig. 3A and
B). DFO treatment, by reducing iron overload, was shown to
markedly improve ductular function and inflammatory cell
infiltration (Fig. 2K, while
significantly reducing intrahepatic collagen fiber deposition; as
evidenced by a decreased Sirius red-positive staining area ratio
(Fig. 3A and B). HYP levels were
significantly elevated in samples from rats with ANIT-induced
cholestasis compared with those in the control group, whereas DFO
treatment substantially restored HYP levels (Fig. 3C). Furthermore, liver α-SMA protein
expression was significantly increased in the ANIT group, whereas
it was significantly reversed by DFO treatment (Fig. 3D and E).
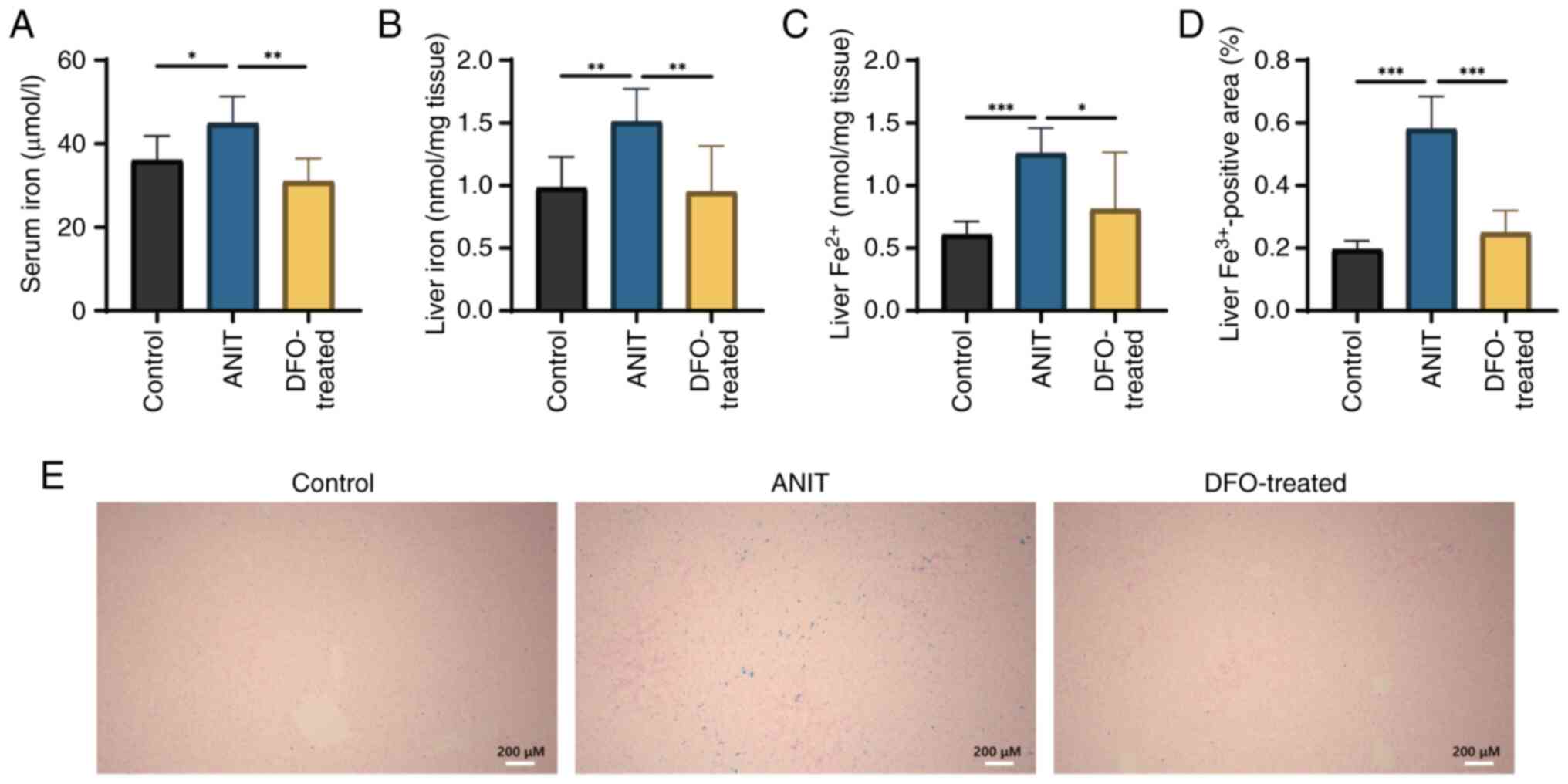
Free iron ions in serum and liver are
aberrantly increased in rats with chronic cholestasis
Serum total iron, liver total iron and
Fe2+ content were significantly elevated in rats with
ANIT-induced cholestasis compared with those in the control group;
however, DFO treatment significantly decreased these levels
compared with those in the ANIT group (Fig. 4A-C). Furthermore, DFO treatment
significantly reduced hepatic iron deposition, as evidenced by the
decreased positive Prussian blue staining area ratio (Fig. 4D). Prussian blue staining revealed
extensive blue staining in the liver of rats with chronic
cholestasis, indicating increased iron deposition (Fig. 4E).
Lipid peroxidation and ferroptosis
marker levels are increased in the livers of rats with and the
liver ultrastructure is affected in a DFO-reversible manner
In rats with ANIT-induced cholestasis, liver MDA
levels were significantly increased, whereas GSH content and GSH-PX
activity were significantly decreased, compared with those in the
control group (Fig. 5A-C). DFO
treatment, through its iron-chelating action, notably restored
liver GSH-PX activity and MDA and GSH levels compared with those in
the ANIT group. Furthermore, the protein expression levels of ASCL4
and COX2 were significantly elevated in the liver of rats with
ANIT-induced cholestasis compared with those in the control group
(Fig. 5D-F). By contrast, DFO
treatment significantly reversed the aberrant expression of these
aforementioned ferroptosis markers compared with those in the ANIT
group. Transmission electron microscopy revealed that hepatocytes
from rats with chronic cholestasis exhibited intact nuclei but
characteristic mitochondrial abnormalities, including reduced size,
blurred and fused cristae, and increased bilayer membrane density
(Fig. 5G). By contrast,
DFO-treated rats exhibited intact hepatocyte nuclei and
morphologically normal mitochondrial cristae. Furthermore, the
mitochondrial size in the DFO group appeared increased compared
with that in the ANIT group, indicating a partial restoration of
mitochondrial morphology towards a normal state.
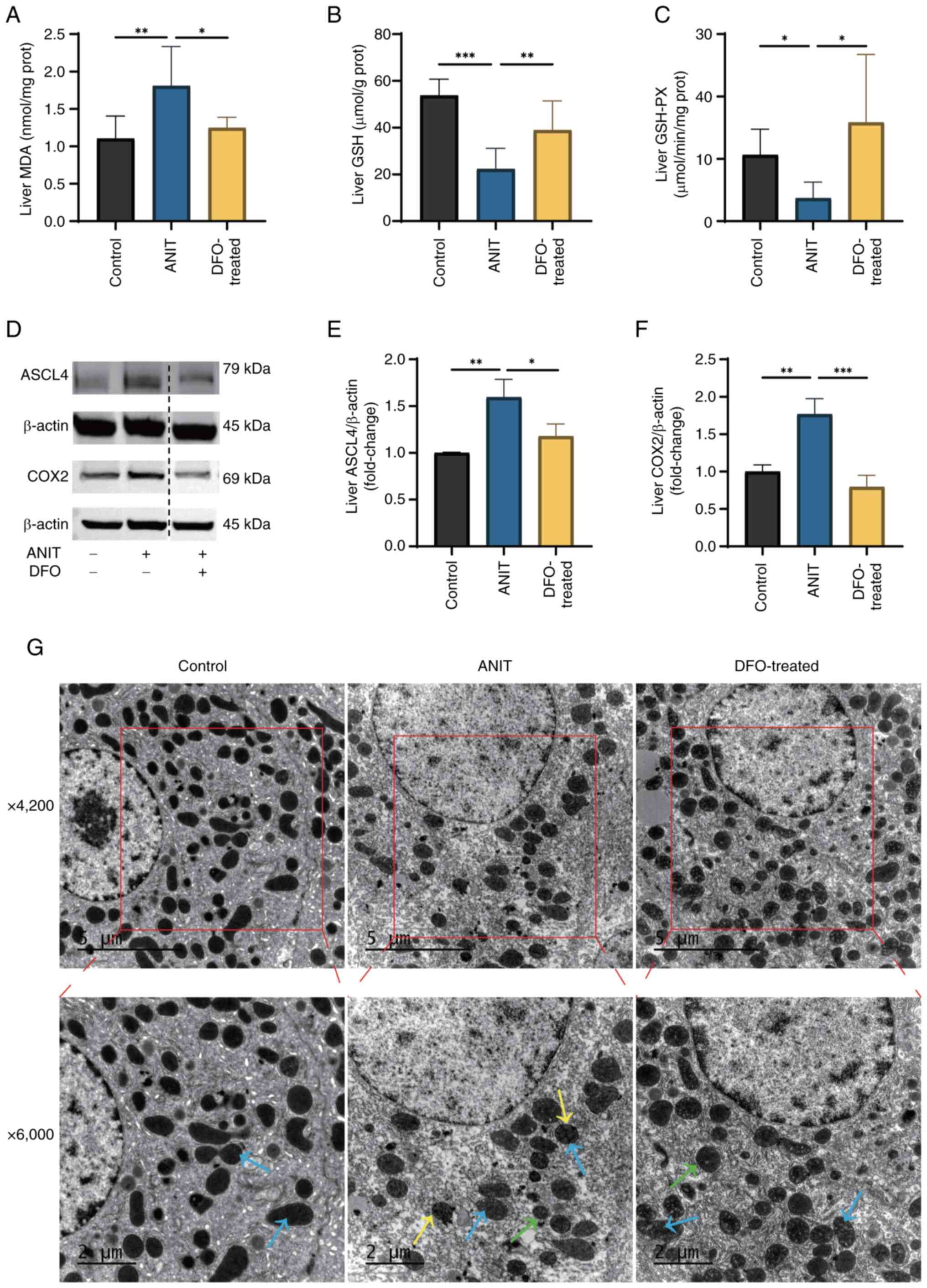 | Figure 5.Levels of lipid peroxidation and
ferroptosis markers are increased in the liver of rats with chronic
cholestasis and the liver ultrastructure is affected in a
DFO-reversible manner. Control group was fed a chow diet, whereas
ANIT and DFO-treated groups were intragastrically administered ANIT
olive oil solution with or without DFO (n=8/group). Levels of (A)
MDA, (B) GSH and (C) GSH-PX were detected. (D) Changes in
ferroptosis marker protein expression. The blots shown are
representative of three independent experiments. A dotted line
indicates that the lanes were non-adjacent on the original gel. All
target protein bands and their corresponding loading control bands
shown side-by-side were derived from the same membrane. Relative
expression levels of (E) ASCL4 and (F) COX2. Data are presented as
the mean ± SD. P-values were determined by one-way ANOVA.
*P<0.05, **P<0.01, ***P<0.001. (G) Transmission electron
microscope sections, blue arrows indicate mitochondrial cristae,
yellow arrows indicate outer mitochondrial membrane and green
arrows point to mitochondria (scale bars, 5 and 2 µ m). DFO,
deferoxamine; ANIT, α-naphthyl isothiocyanate; MDA,
malondialdehyde; GSH, glutathione; GSH-PX, glutathione peroxidase;
ASCL4, acyl-CoA synthetase long-chain family member 4; COX2,
cyclooxygenase 2. |
Abnormal expression of antioxidant and
iron metabolism-related proteins in liver samples of rats with
chronic cholestasis
In rats with ANIT-induced cholestasis, liver protein
expression levels of Nrf2, XCT and GPX4 were significantly reduced,
whereas those of Keap1 and HO-1 were increased, compared with those
in rats in the control group (Fig.
6A-F). DFO treatment notably increased the expression of Nrf2,
XCT and GPX4, decreased Keap1 expression and further enhanced HO-1
expression. Similarly, the protein expression levels of liver FPN1
and FTH1 were significantly decreased, whereas those of TFR1, DMT1
and Steap3 were elevated in cholestasis-affected rats (Fig. 6G-L). DFO treatment effectively
normalized these disruptions in the expression of iron
metabolism-related proteins.
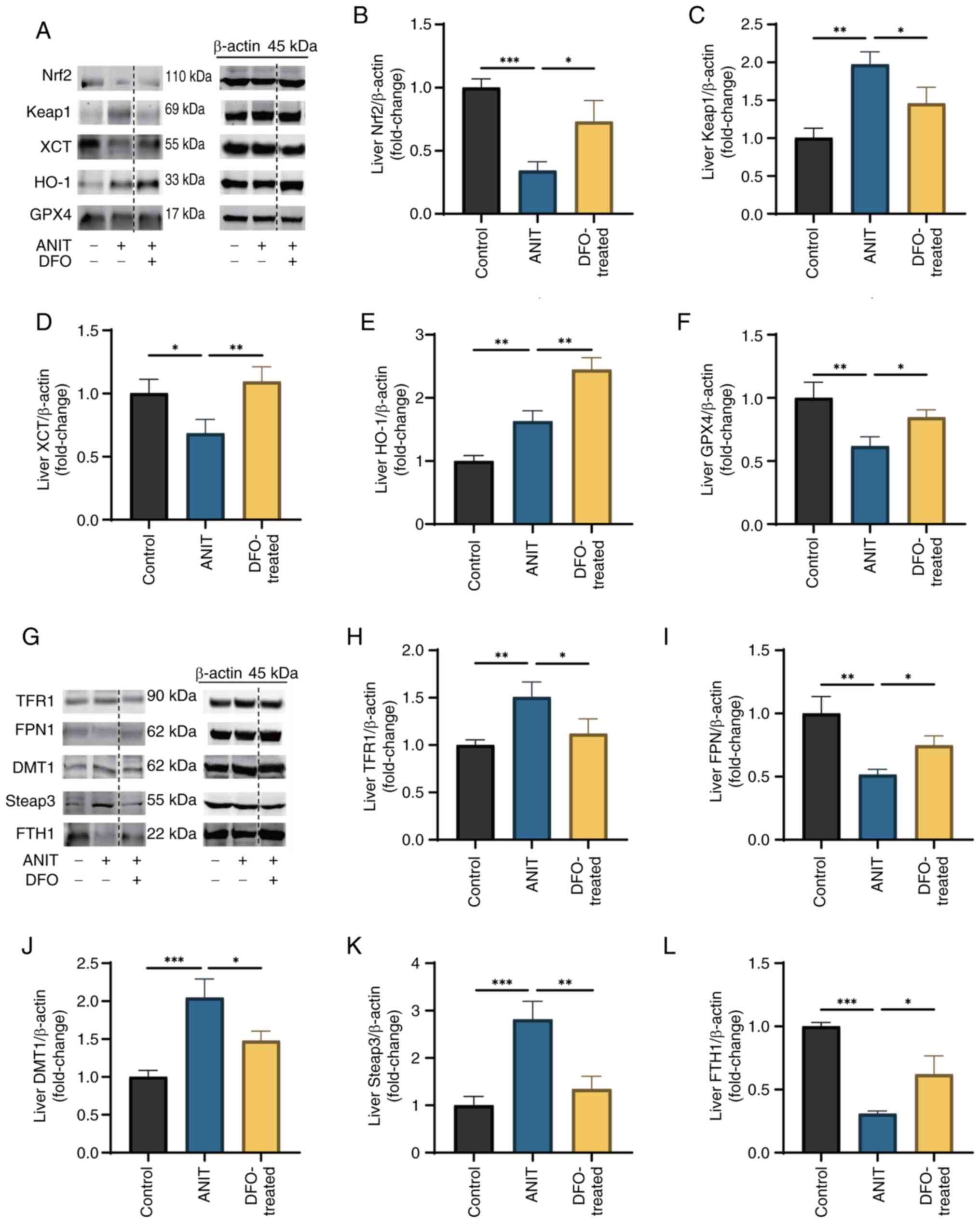 | Figure 6.Abnormal expression of antioxidant
and iron metabolism-related proteins in liver samples of rats with
chronic cholestasis. Control group was fed a chow diet, whereas the
ANIT and DFO-treated groups were intragastrically administered ANIT
olive oil solution with or without DFO (n=8/group). (A) Changes in
ferroptosis antioxidant-related protein expression. The blots shown
are representative of three independent experiments. A dotted line
indicates that the lanes were non-adjacent on the original gel. All
target protein bands and their corresponding loading control bands
shown side-by-side were derived from the same membrane. Relative
expression levels of (B) Nrf2, (C) Keap1, (D) XCT, (E) HO-1 and (F)
GPX4. (G) Changes in iron metabolism-related protein expression.
The blots shown are representative of three independent
experiments. A dotted line indicates that the lanes were
non-adjacent on the original gel. All target protein bands and
their corresponding loading control bands shown side-by-side were
derived from the same membrane. Relative expression levels of (H)
TFR1, (I) FPN1, (J) DMT1, (K) Steap3 and (L) FTH1. Data are
presented as the mean ± SD. P-values were determined by one-way
ANOVA. *P<0.05, **P<0.01, ***P<0.001. ANIT, α-naphthyl
isothiocyanate; DFO, deferoxamine; Nrf2, nuclear factor
erythroid-2-related factor 2; Keap1, Kelch-like ECH-associated
protein 1, XCT, cystine/glutamate transporter; HO-1, heme oxygenase
1; GPX4, glutathione peroxidase 4; TFR1, transferrin receptor 1;
FPN1, ferroportin 1; DMT1, divalent metal transporter 1; Steap3,
six-transmembrane epithelial antigen of the prostate 3; FTH1,
ferritin heavy chain 1. |
Discussion
Cholestasis is a pathological condition that
suppresses bile formation and excretion, which is influenced by
various factors affecting the liver (29). During cholestatic liver disease
progression, sustained cholestasis can trigger inflammation in bile
duct cells and hepatocytes (30).
This inflammatory response can then prompt immune cells to generate
abundant ROS in the presence of proinflammatory factors to combat
pathogens. However, failure to promptly eliminate the excessive ROS
results in their accumulation, serving as a pathogenic stimulus to
culminate in hepatocyte cell death (12).
A hallmark of ferroptosis is the decreased uptake of
intracellular cystine and synthesis of cysteine, which limits GSH
biosynthesis and reduces GSH-PX activity (31). GSH and GSH-PX scavenge free
radicals and ROS-induced lipid peroxides in vivo, thereby
mitigating oxidative damage within the organism (32,33).
MDA is a primary toxic byproduct of lipid peroxidation, serving as
an indicator of tissue peroxidative damage (34). Serum and liver tissue total iron,
in addition to Fe2+ and Fe3+ levels, indicate
potential iron overload in the organism. Excess free
Fe2+ reacts with intracellular
H2O2 through the Fenton reaction, producing
Fe3+ and potent oxidants. These oxidants damage cell
membranes and facilitate cellular ferroptosis. Strong oxidants can
damage cell membranes and induce ferroptosis (14). As described in previous studies
(13,35), transmission electron microscopy
hallmarks of ferroptosis include hepatocyte mitochondria with
reduced size, increased bilayer density, ruptured and crumpled
outer membranes, and diminished or absent cristae, whilst the
nuclei remain intact. Consistent with these established features,
our ultrastructural analysis revealed characteristic mitochondrial
abnormalities in cholestatic rats, including reduced size, blurred
and fused cristae, and increased bilayer membrane density),
supporting the occurrence of ferroptosis in our model. ASCL4 and
COX2 are key proteins involved in detecting ferroptosis. ASCL4
catalyzes the formation of polyunsaturated fatty acid-acyl coenzyme
A derivatives (36), which bind to
phospholipids in cell membranes through the action of
lysophosphatidylcholine acyltransferase 3, increasing membrane
susceptibility to oxidation and lipid peroxide-induced damage
(37,38). By contrast, COX2 enhances
nicotinamide adenine dinucleotide phosphate oxidase 1-mediated
lipid peroxidation to elevate intracellular ROS levels (39).
In the present study, rats with chronic cholestasis
exhibited significantly reduced GSH content and GSH-PX activity,
alongside a marked increase in MDA content in liver tissue,
indicating a severe deficiency in antioxidant capacity and
substantial lipid peroxidation damage due to lipid peroxide
accumulation. Total serum iron, total iron and Fe2+
levels in liver tissue were significantly elevated in these model
rats. Prussian blue staining revealed extensive blue staining in
liver tissue, signifying abnormal iron accumulation, particularly
Fe3+, in cholestatic rats. Transmission electron
microscopy also demonstrated indistinct hepatocyte boundaries,
intact nuclei, reduced mitochondrial size, blurred and fused
mitochondrial cristae, with increased bilayer membrane density.
These ultrastructural and morphological alterations in hepatocytes
align with the characteristics of ferroptosis. Western blotting
revealed markedly increased expression levels of ASCL4 and COX2 in
the liver tissue of model rats, signifying a substantial rise in
lipid peroxide levels and the occurrence of ferroptosis. These
findings suggest that ferroptosis of hepatocytes may occur during
ANIT-induced chronic cholestasis in rats.
Although the present study was conducted in an
animal model, the relevance of ferroptosis to human cholestatic
liver diseases has been recognized. Previous studies have reported
iron accumulation and dysregulation of ferroptosis-related genes
(such as GPX4 and SLC7A11) in liver tissues from patients with
intrahepatic cholestasis of pregnancy or cholestatic drug-induced
liver injury (40,41). This converging evidence suggests
that the ferroptotic pathway identified in the present study may
have translational importance in human cholestatic conditions.
In the ferroptosis antioxidant pathway, System Xc-
is comprised of two proteins, XCT and 4F2 cell-surface antigen
heavy chain, facilitating a 1:1 exchange of extracellular cystine
for intracellular glutamate (42).
Cystine serves as a vital precursor for intracellular cysteine
synthesis, positioning XCT as a key component in this pathway. GPX4
is a selenoprotein that reduces lipid peroxides to lipid alcohols,
thereby preventing ferroptosis by scavenging excess intracellular
lipid peroxides. This enzyme is the most potent antioxidant enzyme
in the GPX family, which serves as a pivotal target within the
antioxidant pathway (43). The
Keap1/Nrf2/HO-1 axis constitutes a key intracellular antioxidant
pathway (44). Keap1 inhibits Nrf2
by retaining it in the cytoplasm (45), thereby preventing its nuclear
translocation and subsequent antioxidant activity (46). Nrf2 is a central component in the
antioxidant system, regulating the upstream factor HO-1 (47), which enhances the expression of
both HO-1 and GPX4 (48). HO-1
exhibits antioxidant and anti-inflammatory properties, mitigating
mitochondrial and lipid peroxidation damage (49).
In the present study, Keap1 protein expression was
elevated, whereas XCT, GPX4 and Nrf2 protein expression levels were
reduced, in the liver tissues of rats following ANIT-induced
chronic cholestasis. This indicated that the System Xc- transporter
was inhibited during cholestasis, resulting in cysteine deficiency
and limited GPX4 synthesis, which failed to efficiently scavenge
excessive ROS. The Keap1/Nrf2/HO-1 pathway was suppressed,
impairing antioxidant activity. The observed upregulation of HO-1
in the present model appeared paradoxical to the overall
suppression of the Nrf2 axis. However, this can be interpreted as a
compensatory stress response to severe oxidative injury. The
initial induction of HO-1 may represent the attempt of the liver to
mitigate damage. In the face of sustained cholestatic insult, this
compensatory mechanism ultimately proved insufficient to prevent
the progression of ferroptosis, as evidenced by the downregulation
of other key antioxidants, such as GPX4 (50,51).
Consequently, the antioxidant capacity of the body was diminished
and excessive intracellular lipid peroxides were not effectively
eliminated, leading to ferroptosis.
In the iron metabolism pathway, TFR1 on the cell
membrane facilitates the intracellular transport of iron ions by
binding to the extracellular Tf-Fe complex (52). Steap3 reduces transferrin-bound
Fe3+ to Fe2+ (53), which is then translocated into the
intracytoplasmic labile iron pool (LIP) through DMT1. DMT1 is
crucial for the intracellular transport of ferric ions and serves
as an essential metal ion transporter in the iron metabolism
pathway (54). Cytoplasmic
ferritin as a multimer can decrease intracellular free iron ion
levels by binding to free iron ions in the LIP. FPN is the sole
protein facilitating iron efflux in mammals, enabling the transfer
of iron ions out of cells and thereby reducing intracellular free
iron ion concentrations (55). The
mechanism of iron-induced cell death is illustrated in Fig. 7.
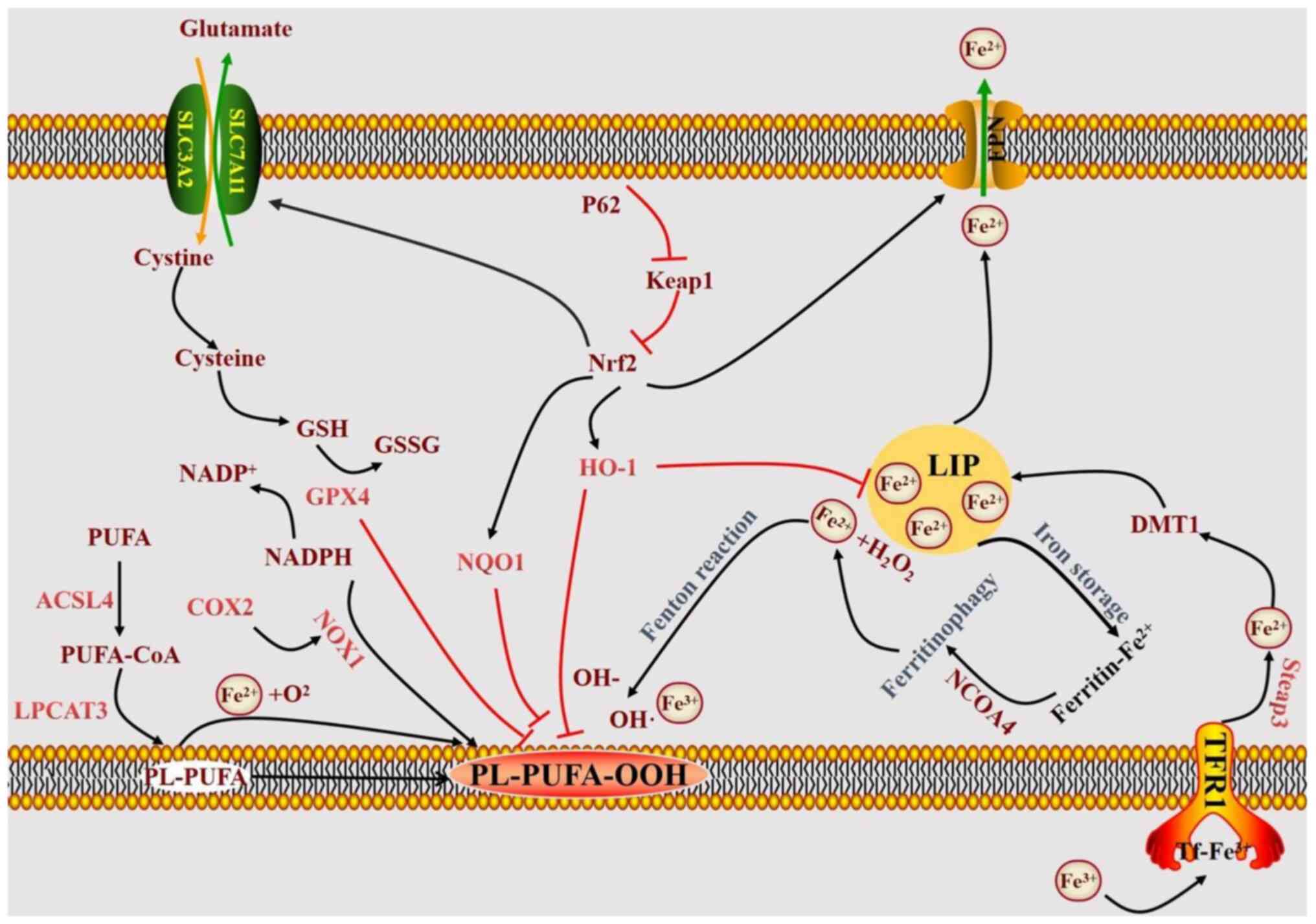 | Figure 7.Diagram of the mechanism of
ferroptosis. ASCL4, acyl-CoA synthetase long-chain family member 4;
COX2, cyclooxygenase 2; DMT1, divalent metal transporter 1; FPN,
ferroportin; GSSG, oxidized glutathione; GPX4, glutathione
peroxidase 4; GSH, glutathione; HO-1, heme oxygenase 1; Keap1,
Kelch-like ECH-associated protein 1; LIP, labile iron pool; LPCAT3,
lysophosphatidylcholine acyltransferase 3; NCOA4, nuclear receptor
coactivator 4; NOX1, nicotinamide adenine dinucleotide phosphate
oxidase 1; NQO1, NAD(P)H quinone dehydrogenase 1; Nrf2, nuclear
factor erythroid-2-related factor 2; PL-PUFA-OOH,
phospholipid-polyunsaturated fatty acid hydroperoxide; SLC7A11,
solute carrier family 7 member 11; Steap3, six-transmembrane
epithelial antigen of the prostate 3; TFR1, transferrin receptor
1. |
The present study revealed that in liver tissues of
rats after ANIT-induced chronic cholestasis, TFR1, Steap3 and DMT1
protein expression increased, whereas FTH1 and FPN1 expression
decreased. This suggests that cholestasis can enhance cellular iron
ion uptake, reduce ferritin storage function, diminish iron
exclusion capacity, elevate intracellular free iron levels,
intensify the Fenton reaction and promote excessive ROS, leading to
lipid peroxidation and ferroptosis in hepatocytes.
In summary, these aforementioned observations
suggested that decreased antioxidant and iron metabolism in
hepatocytes, resulting in iron-induced cell death, is a key
pathological mechanism in chronic cholestasis.
DFO is a potent iron chelator that is extensively
used in clinical settings to treat iron overload disorders such as
β-thalassemia and myelodysplastic syndrome (24). In the present study, the protective
effect of DFO was shown to be primarily mediated through its
capacity to chelate iron. DFO treatment significantly reduced serum
and hepatic total iron levels as well as Fe2+ content,
and markedly decreased hepatic iron deposition. Furthermore, DFO
alleviated indices of iron-dependent oxidative damage, including
lowering lipid peroxidation (MDA) and restoring antioxidant
capacity (GSH and GSH-PX). In addition, DFO normalized the
expression of key proteins involved in iron metabolism (TFR1, FPN1,
etc., Fig. 6G-L) and ferroptosis
(ASCL4, COX2). By reducing intracellular and systemic iron load,
DFO can limit the substrates available for the Fenton reaction
(56), thereby decreasing the
generation of hydroxyl radicals and subsequent lipid peroxidation
(57,58). This reduction in iron-driven
oxidative stress ultimately mitigates the process of ferroptosis
(59).
The present study investigated the role of the
ferroptosis inhibitor DFO in addressing ANIT-induced chronic
cholestasis in rats, aiming to test the hypothesis that
hepatocellular ferroptosis is a key pathogenic mechanism in chronic
cholestasis and that targeting it may be therapeutic. The findings
indicated that DFO may markedly ameliorate cholestasis.
Biochemically, DFO reduced serum levels of TBA, TBIL, DBIL, LDL and
TCH, decreased GGT, ALP and ALT activities, increased serum TG
levels and mitigated hepatocyte dysfunction in cholestatic rats. In
hepatic tissues, DFO enhanced GSH content and GSH-PX activity,
boosted antioxidant capacity, lowered MDA content, reduced lipid
peroxidation and decreased HYP content in the liver of model rats.
Biochemical analysis demonstrated that DFO effectively reduced
serum total iron, liver tissue total iron and Fe2+
levels in model rats. Hepatic histopathology revealed that DFO
decreased small bile duct proliferation in the hepatic portal
areas, reduced inflammatory cell infiltration and diminished
collagen fiber deposition and Fe3+ accumulation.
Transmission electron microscopy indicated that hepatocyte nuclei
in the DFO group remained intact, with normal mitochondrial
morphology, size and clear cristae. Western blotting showed that
DFO downregulated the protein expression of α-SMA, a classical
marker of activated HSCs, in the rat liver tissue, indicating the
inhibition of HSC activation. This contributed to the deceleration
of hepatic fibrosis progression in cholestasis. DFO downregulated
ASCL4 and COX2 protein expression in liver tissues, and inhibited
ANIT-induced ferroptosis in chronically cholestatic rats. After
intervention, DFO reduced Keap1, TFR1, DMT1 and Steap3 protein
levels, whereas it upregulated Nrf2, XCT, HO-1, GPX4, FPN1 and FTH1
in liver tissues. This suggested that DFO can mitigate ferroptosis
by enhancing hepatocyte iron storage and excretion, reducing
extracellular ferric iron translocation and decreasing free ferric
iron content, thereby boosting antioxidant capacity and preventing
ferroptosis.
However, it is important to acknowledge that DFO is
a pleiotropic agent with biological effects beyond iron chelation.
These include intrinsic antioxidant properties and potential
modulatory effects on non-parenchymal cells, such as HSCs (60,61).
While the present data strongly supported iron chelation as the
primary mechanism, the contribution of these additional pleiotropic
effects to the overall hepatoprotection observed cannot be ruled
out.
The present study has several limitations. The
findings are based on a single animal model of cholestasis.
Validation in additional models would strengthen the
generalizability of the conclusions. In addition, the use of DFO,
though effective, prevents the unequivocal attribution of all
protective effects solely to the reduction of ferroptosis through
iron chelation, due to its pleiotropic nature. Future studies using
more specific ferroptosis inhibitors (such as ferrostatin-1)
(62) or genetic approaches to
manipulate key ferroptosis regulators (including GPX4) would be
invaluable. The lack of in vitro data also limited the
mechanistic depth. Establishing in vitro models of bile
acid-induced hepatocyte injury will be essential for dissecting the
precise molecular sequence of events.
In conclusion, the present findings demonstrated
that ferroptosis, driven by the concurrent disruption of hepatocyte
antioxidant and iron metabolism capacities, is a key pathological
mechanism in chronic cholestasis. The iron chelator DFO alleviated
cholestatic liver injury, an effect likely mediated by the
reduction of iron overload and the subsequent ferroptotic process.
The present study highlighted the ferroptosis pathway as a
promising therapeutic target for cholestatic liver diseases.
Acknowledgements
Not applicable.
Funding
The present study was funded by the National Natural Science
Foundation of China (grant no. 81774196), the Natural Science
Foundation of Shanghai Municipality of China (grant no.
22ZR1459200) and the Luzhou Science and Technology Innovation City
Development Fund of China (grant no. 2024RQN226).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
XW designed the present study. ZG performed
experiments. JW performed data interpretation. YW performed data
analysis. XL, YX, LQ, JL and PL assisted in collecting the samples.
All authors read and approved the final manuscript. XW and ZG
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The animal protocol was approved by the Animal Care
and Utilization Committee of Shanghai University of Traditional
Chinese Medicine (Ethics Review Number: PZSHUTCM210715019) and was
conducted following the Guide for the Care and Use of Laboratory
Animals (26).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Karpen SJ, Kelly D, Mack C and Stein P:
Ileal bile acid transporter inhibition as an anticholestatic
therapeutic target in biliary atresia and other cholestatic
disorders. Hepatol Int. 14:677–689. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Pollock G and Minuk GY: Diagnostic
considerations for cholestatic liver disease. J Gastroenterol
Hepatol. 32:1303–1309. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bortolini M, Almasio P, Bray G, Budillon
G, Coltorti M, Frezza M, Okolicsanyi L, Salvagnini M and Williams
R: Multicentre survey of the prevalence of intrahepatic cholestasis
in 2520 consecutive patients with newly diagnosed chronic liver
disease. Drug Invest. 4 (Suppl 4):S83–S89. 1992. View Article : Google Scholar
|
|
4
|
Xie W, Cao Y, Xu M, Wang J, Zhou C, Yang
X, Geng X, Zhang W, Li N and Cheng J: Prognostic significance of
elevated cholestatic enzymes for fibrosis and hepatocellular
carcinoma in hospital discharged chronic viral hepatitis patients.
Sci Rep. 7:102892017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ibrahim SH, Kamath BM, Loomes KM and
Karpen SJ: Cholestatic liver diseases of genetic etiology: Advances
and controversies. Hepatology. 75:1627–1646. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bayir H, Dixon SJ, Tyurina YY, Kellum JA
and Kagan VE: Ferroptotic mechanisms and therapeutic targeting of
iron metabolism and lipid peroxidation in the kidney. Nat Rev
Nephrol. 19:315–336. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang W: Iron turns to wild when the
transferrin is away. Blood. 136:649–650. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Buckley CD, Barone F, Nayar S, Benezech C
and Caamano J: Stromal cells in chronic inflammation and tertiary
lymphoid organ formation. Annu Rev Immunol. 33:715–745. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forrester SJ, Kikuchi DS, Hernandes MS, Xu
Q and Griendling KK: Reactive oxygen species in metabolic and
inflammatory signaling. Circ Res. 122:877–902. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang WB, Yang F, Wang Y, Jiao FZ, Zhang
HY, Wang LW and Gong ZJ: Inhibition of HDAC6 attenuates LPS-induced
inflammation in macrophages by regulating oxidative stress and
suppressing the TLR4-MAPK/NF-ĸB pathways. Biomed Pharmacother.
117:1091662019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun
X, Kang R and Tang D: Ferroptosis: Process and function. Cell Death
Differ. 23:369–379. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Han C, Liu Y, Dai R, Ismail N, Su W and Li
B: Ferroptosis and its potential role in human diseases. Front
Pharmacol. 11:2392020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang Y and Tang M: PM2.5 induces
ferroptosis in human endothelial cells through iron overload and
redox imbalance. Environ Pollut. 254:1129372019. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jiang X, Stockwell BR and Conrad M:
Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol
Cell Biol. 22:266–282. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lyberopoulou A, Chachami G, Gatselis NK,
Kyratzopoulou E, Saitis A, Gabeta S, Eliades P, Paraskeva E, Zachou
K, Koukoulis GK, et al: Low serum hepcidin in patients with
autoimmune liver diseases. PLoS One. 10:e01354862015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marques O, Horvat NK, Zechner L, Colucci
S, Sparla R, Zimmermann S, Neufeldt CJ, Altamura S, Qiu R, Mudder
K, et al: Inflammation-driven NF-kappab signaling represses
ferroportin transcription in macrophages via HDAC1 and HDAC3.
Blood. 145:866–880. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xie X, Chang L, Zhu X, Gong F, Che L,
Zhang R, Wang L, Gong C, Fang C, Yao C, et al: Rubiadin mediates
the upregulation of hepatic hepcidin and alleviates iron overload
via BMP6/SMAD1/5/9-signaling pathway. Int J Mol Sci. 26:13852025.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Delesderrier E, Monteiro JDC, Freitas S,
Pinheiro IC, Batista MS and Citelli M: Can iron and polyunsaturated
fatty acid supplementation induce ferroptosis? Cell Physiol
Biochem. 57:24–41. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu C, Liu Z, Dong Z, Liu S, Kan H and
Zhang S: Multifaceted interplays between the essential players and
lipid peroxidation in ferroptosis. J Genet Genomics. 52:1071–1081.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yu Y, Jiang L, Wang H, Shen Z, Cheng Q,
Zhang P, Wang J, Wu Q, Fang X, Duan L, et al: Hepatic transferrin
plays a role in systemic iron homeostasis and liver ferroptosis.
Blood. 136:726–739. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang Q, Qu Y, Zhang Q, Li F, Li B, Li Z,
Dong Y, Lu L and Cai X: Exosomes derived from hepatitis b
virus-infected hepatocytes promote liver fibrosis via mir-222/TFRC
axis. Cell Biol Toxicol. 39:467–481. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang L, Xie P, Wu J, Yu J, Li X, Ma H, Yu
T, Wang H, Ye J, Wang J and Zheng H: Deferoxamine treatment
combined with sevoflurane postconditioning attenuates myocardial
ischemia-reperfusion injury by restoring HIF-1/BNIP3-mediated
mitochondrial autophagy in GK rats. Front Pharmacol. 11:62020.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Duscher D, Neofytou E, Wong VW, Maan ZN,
Rennert RC, Inayathullah M, Januszyk M, Rodrigues M, Malkovskiy AV,
Whitmore AJ, et al: Transdermal deferoxamine prevents
pressure-induced diabetic ulcers. Proc Natl Acad Sci USA.
112:94–99. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
National RCUC, . Guide for the Care and
Use of Laboratory Animals. National Academies Press; Washington,
DC: 2011
|
|
27
|
Liu X, Wang J, Li M, Qiu J, Li X, Qi L,
Liu J, Liu P, Xie G and Wang X: Farnesoid × receptor is an
important target for the treatment of disorders of bile acid and
fatty acid metabolism in mice with nonalcoholic fatty liver disease
combined with cholestasis. J Gastroenterol Hepatol. 38:1438–1446.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang H, Jiang C, Yang Y, Li J, Wang Y,
Wang C and Gao Y: Resveratrol ameliorates iron overload induced
liver fibrosis in mice by regulating iron homeostasis. PeerJ.
10:e135922022. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiang D, Liu Y, Zu Y, Yang J, He W, Zhang
C and Liu D: Calculus bovis sativus alleviates estrogen
cholestasis-induced gut and liver injury in rats by regulating
inflammation, oxidative stress, apoptosis, and bile acid profiles.
J Ethnopharmacol. 302:1158542023. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Zhuang Y, Ortega-Ribera M, Thevkar Nagesh
P, Joshi R, Huang H, Wang Y, Zivny A, Mehta J, Parikh SM and Szabo
G: Bile acid-induced IRF3 phosphorylation mediates cell death,
inflammatory responses, and fibrosis in cholestasis-induced liver
and kidney injury via regulation of ZBP1. Hepatology. 79:752–767.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang WS and Stockwell BR: Ferroptosis:
Death by lipid peroxidation. Trends Cell Biol. 26:165–176. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Alajbeg IZ, Lapic I, Rogic D, Vuletic L,
Andabak Rogulj A, Illes D, Knezović Zlatarić D, Badel T, Vrbanovic
E and Alajbeg I: Within-subject reliability and between-subject
variability of oxidative stress markers in saliva of healthy
subjects: A longitudinal pilot study. Dis Markers.
2017:26974642017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hakkoymaz H, Nazik S, Seyithanoglu M,
Guler O, Sahin AR, Cengiz E and Yazar FM: The value of
ischemia-modified albumin and oxidative stress markers in the
diagnosis of acute appendicitis in adults. Am J Emerg Med.
37:2097–2101. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Tangvarasittichai S: Oxidative stress,
insulin resistance, dyslipidemia and type 2 diabetes mellitus.
World J Diabetes. 6:456–480. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Moore DD, Kato S, Xie W, Mangelsdorf DJ,
Schmidt DR, Xiao R and Kliewer SA: International union of
pharmacology. LXII. The NR1h and NR1i receptors: Constitutive
androstane receptor, pregnene × receptor, farnesoid × receptor
alpha, farnesoid × receptor beta, liver × receptor alpha, liver ×
receptor beta, and vitamin d receptor. Pharmacol Rev. 58:742–759.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Doll S, Proneth B, Tyurina YY, Panzilius
E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A,
et al: ACSL4 dictates ferroptosis sensitivity by shaping cellular
lipid composition. Nat Chem Biol. 13:91–98. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Daiber A, Di Lisa F, Oelze M,
Kroller-Schon S, Steven S, Schulz E and Munzel T: Crosstalk of
mitochondria with NADPH oxidase via reactive oxygen and nitrogen
species signalling and its role for vascular function. Br J
Pharmacol. 174:1670–1689. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
You Y, Qian Z, Jiang Y, Chen L, Wu D, Liu
L, Zhang F, Ning X, Zhang Y and Xiao J: Insights into the
pathogenesis of gestational and hepatic diseases: The impact of
ferroptosis. Front Cell Dev Biol. 12:14828382024. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Teschke R: Treatment of drug-induced liver
injury. Biomedicines. 11:152022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lewerenz J, Hewett SJ, Huang Y, Lambros M,
Gout PW, Kalivas PW, Massie A, Smolders I, Methner A, Pergande M,
et al: The cystine/glutamate antiporter system x(c)(−) in health
and disease: From molecular mechanisms to novel therapeutic
opportunities. Antioxid Redox Signal. 18:522–555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kan X, Yin Y, Song C, Tan L, Qiu X, Liao
Y, Liu W, Meng S, Sun Y and Ding C: Newcastle-disease-virus-induced
ferroptosis through nutrient deprivation and ferritinophagy in
tumor cells. iScience. 24:1028372021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R
and Tang D: Activation of the p62-keap1-NRF2 pathway protects
against ferroptosis in hepatocellular carcinoma cells. Hepatology.
63:173–184. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yoo S, Kim M, Bae JY, Lee SA and Koh G:
Bardoxolone methyl inhibits ferroptosis through the Keap1-Nrf2
pathway in renal tubular epithelial cells. Mol Med Rep. 32:2672025.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ichimura Y, Waguri S, Sou YS, Kageyama S,
Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, et
al: Phosphorylation of p62 activates the Keap1-Nrf2 pathway during
selective autophagy. Mol Cell. 51:618–631. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chen Y, Ma L, Yan Y, Wang X, Cao L, Li Y
and Li M: Ophiopogon japonicus polysaccharide reduces
doxorubicin-induced myocardial ferroptosis injury by activating
Nrf2/GPX4 signaling and alleviating iron accumulation. Mol Med Rep.
31:362025. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Xiao P, Huang H, Zhao H, Liu R, Sun Z, Liu
Y, Chen N and Zhang Z: Edaravone dexborneol protects against
cerebral ischemia/reperfusion-induced blood-brain barrier damage by
inhibiting ferroptosis via activation of nrf-2/HO-1/GPX4 signaling.
Free Radic Biol Med. 217:116–125. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hu T, Wei G, Xi M, Yan J, Wu X, Wang Y,
Zhu Y, Wang C and Wen A: Synergistic cardioprotective effects of
danshensu and hydroxysafflor yellow a against myocardial
ischemia-reperfusion injury are mediated through the Akt/Nrf2/HO-1
pathway. Int J Mol Med. 38:83–94. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lin Q, Li S, Jin H, Cai H, Zhu X, Yang Y,
Wu J, Qi C, Shao X, Li J, et al: Mitophagy alleviates
cisplatin-induced renal tubular epithelial cell ferroptosis through
ROS/HO-1/GPX4 axis. Int J Biol Sci. 19:1192–1210. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Choi AM and Alam J: Heme oxygenase-1:
Function, regulation, and implication of a novel stress-inducible
protein in oxidant-induced lung injury. Am J Respir Cell Mol Biol.
15:9–19. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fisher AL, Wang CY, Xu Y, Joachim K, Xiao
X, Phillips S, Moschetta GA, Alfaro-Magallanes VM and Babitt JL:
Functional role of endothelial transferrin receptor 1 in iron
sensing and homeostasis. Am J Hematol. 97:1548–1559. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Deng P, Li J, Lu Y, Hao R, He M, Li M, Tan
M, Gao P, Wang L, Hong H, et al: Chronic cadmium exposure triggered
ferroptosis by perturbing the STEAP3-mediated glutathione redox
balance linked to altered metabolomic signatures in humans. Sci
Total Environ. 905:1670392023. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Gao X, Hu W, Qian D, Bai X, He H, Li L and
Sun S: The mechanisms of ferroptosis under hypoxia. Cell Mol
Neurobiol. 43:3329–3341. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Nairz M, Fritsche G, Brunner P, Talasz H,
Hantke K and Weiss G: Interferon-gamma limits the availability of
iron for intramacrophage salmonella typhimurium. Eur J Immunol.
38:1923–1936. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Ge W, Jie J, Yao J, Li W, Cheng Y and Lu
W: Advanced glycation end products promote osteoporosis by inducing
ferroptosis in osteoblasts. Mol Med Rep. 25:1402022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Chen GH, Song CC, Pantopoulos K, Wei XL,
Zheng H and Luo Z: Mitochondrial oxidative stress mediated
Fe-induced ferroptosis via the NRF2-ARE pathway. Free Radic Biol
Med. 180:95–107. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Holden P and Nair LS: Deferoxamine: An
angiogenic and antioxidant molecule for tissue regeneration. Tissue
Eng Part B Rev. 25:461–470. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Zeng Z, Huang H, Zhang J, Liu Y, Zhong W,
Chen W, Lu Y, Qiao Y, Zhao H, Meng X, et al: HDM induce airway
epithelial cell ferroptosis and promote inflammation by activating
ferritinophagy in asthma. FASEB J. 36:e223592022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Mohammed A, Abd Al Haleem EN, El-Bakly WM
and El-Demerdash E: Deferoxamine alleviates liver fibrosis induced
by CCl4 in rats. Clin Exp Pharmacol Physiol. 43:760–768. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Liu MX, Gu YY, Nie WY, Zhu XM, Qi MJ, Zhao
RM, Zhu WZ and Zhang XL: Formononetin induces ferroptosis in
activated hepatic stellate cells to attenuate liver fibrosis by
targeting NADPH oxidase 4. Phytother Res. 38:5988–6003. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xie J, Ye Z, Li L, Xia Y, Yuan R, Ruan Y
and Zhou X: Ferrostatin-1 alleviates oxalate-induced renal tubular
epithelial cell injury, fibrosis and calcium oxalate stone
formation by inhibiting ferroptosis. Mol Med Rep. 26:2562022.
View Article : Google Scholar : PubMed/NCBI
|