Sepsis is a life-threatening systemic-inflammatory
response syndrome triggered by viral, bacterial, fungal or
immunogenic pathogens. This condition initiates a cascade of
reactions culminating in multiorgan dysfunction syndrome. Current
epidemiological data indicate sepsis as the leading cause of
mortality in intensive care units worldwide, with an overall
mortality rate of 25–30% in global cohorts (1–4).
Severe sepsis may induce organ-specific injuries affecting the
kidneys, lungs, brain and heart (5–8).
Notably, sepsis-induced cardiomyopathy (SIC) represents a frequent
complication, occurring in 10–70% of sepsis or septic shock cases
(9). A 2023 cohort study reported
a 20% prevalence of SIC among septic patients, associating with
markedly elevated short-term mortality (10). SIC is defined as an acute,
reversible myocardial depression syndrome during early septic
shock, characterized by infection-driven cardiac dysfunction. While
typically resolving within 7–10 days, SIC may accelerate
cardiovascular collapse (11).
Diagnostic criteria remain non-standardized, but key features
include: i) Left ventricular dilation with normal or reduced
filling pressure; ii) impaired contractility; and iii)
biventricular systolic/diastolic dysfunction manifesting as reduced
ejection fraction (12).
Progressive SIC induces myocardial impairment, as evidenced by
biventricular dilation and decreased left ventricular ejection
fraction (12,13). Notably, myocardial dysfunction
affects 40% of septic patients, with myocardial
dysfunction-associated mortality reaching 70% (14–16),
establishing SIC as a notable threat to patient survival.
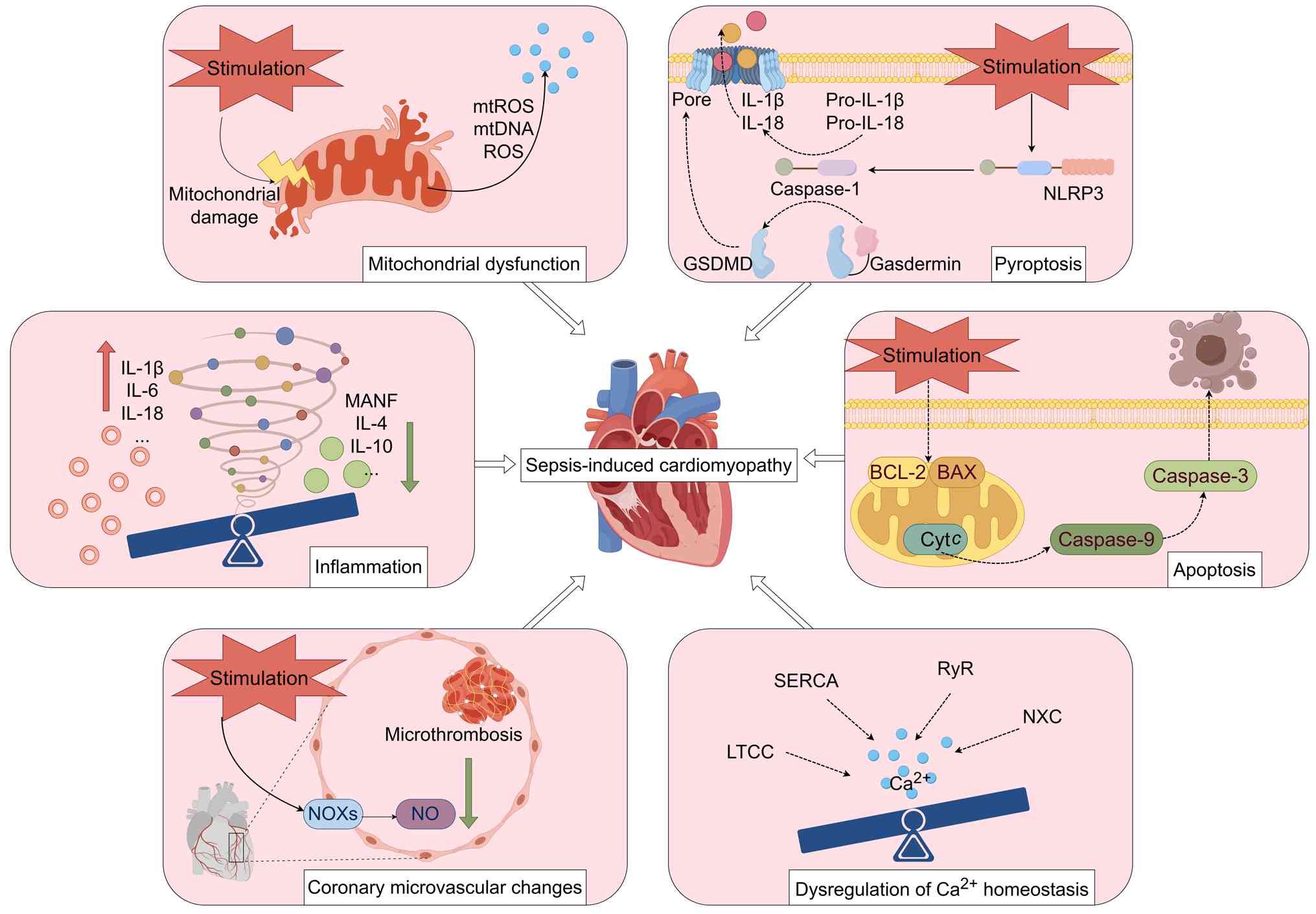
The precise pathogenesis of SIC remains incompletely
elucidated. Current evidence implicates multifactorial mechanisms,
including dysregulated inflammatory responses, programmed cell
death (PCD) mechanisms, such as apoptosis and pyroptosis,
mitochondrial structural or functional impairment, aberrant
calcium-handling protein regulation, endothelial dysfunction and
metabolic disturbances (17–19).
During SIC progression, pathogenic microorganisms and endotoxins
enter systemic circulation, directly activating immune cells. This
triggers excessive cytokine production that amplifies inflammatory
cascades and induces cardiomyocyte cell death. Concurrently,
mitochondrial dysfunction generates an overload of reactive oxygen
species (ROS) and pathological calcium efflux. These interconnected
pathways converge through synergistic amplification, ultimately
impairing myocardial contractility and exacerbating SIC
pathogenesis (Fig. 1).
Previous years have witnessed an intensified
research focus on SIC. Emerging evidence identifies the NOD-like
receptor protein 3 (NLRP3) inflammasome as a notable innate immune
sensor important for maintaining homeostasis, with its
dysregulation implicated in the pathogenesis of diverse chronic
inflammatory and metabolic disorders (20–24).
During SIC development, the NLRP3 inflammasome is activated by
pathogen-associated molecular patterns (PAMPs) and
damage-associated molecular patterns (DAMPs), which triggers
caspase-1-dependent cytokine release, pyroptosis, apoptotic protein
accumulation and mitochondrial injury. These events collectively
drive cardiomyocyte dysfunction and SIC progression (25). Notably, pharmacological inhibition
or genetic downregulation of NLRP3 attenuates SIC-induced cardiac
impairment and improves survival in experimental models (26). Zhu et al (27) demonstrated that the specific NLRP3
inhibitor 5-methoxyindole-3-carboxaldehyde suppresses inflammasome
assembly by disrupting NLRP3-apoptosis-associated speck-like
protein containing a CARD (ASC) interactions. Similarly, MCC950, a
selective small-molecule inhibitor, binds to the NACHT domain of
NLRP3, which prevents the activation of caspase-1 and the resulting
maturation of IL-1β and IL-18, thereby mitigating inflammation and
pyroptosis (28). Although MCC950
demonstrated notable preclinical efficacy, its clinical development
for inflammatory diseases, such as rheumatoid arthritis, was halted
during phase II trials due to off-target liver toxicity related to
carbonic anhydrase inhibition (29). Nevertheless, MCC950 remains a
prototypical and widely used research tool, and its chemical
scaffold continues to inform the design of next-generation NLRP3
inhibitors with improved safety profiles (30). These findings establish NLRP3 as a
central node in the SIC pathological network that coordinates
multiple injury mechanisms, including inflammatory cascades, PCD
and impaired mitophagy. Consequently, NLRP3 represents a promising
therapeutic target for SIC intervention.
Due to the absence of comprehensive reviews
addressing NLRP3 inflammasome signaling in SIC, the present review
provided a systematic analysis of its pathogenic role. The present
review elucidated NLRP3 inflammasome priming and activation
mechanisms, discussing clinical and experimental evidence from
previous studies. Notable emphasis was placed on delineating
molecular pathways through which NLRP3 mediates myocardial injury
in SIC, including inflammatory cascades, pyroptosis, apoptosis and
dysregulated mitophagy. Furthermore, the present review catalogued
chemical compounds and pharmacological agents targeting
NLRP3-associated signaling networks. As such, the present review
consolidated current understanding of the central role of NLRP3 in
SIC pathogenesis and identified promising molecular targets for
therapeutic intervention, thereby informing future research
directions.
NOD-like receptors, a subclass of pattern
recognition receptors, recognize not only PAMPs but also DAMPs such
as adenosine triphosphate (ATP) and mitochondrial ROS (mtROS)
(31). Among the most extensively
studied inflammasomes, the NLRP3 inflammasome comprises three core
components: Pro-caspase-1, ASC and NLRP3 (32). The NLRP3 protein features three
distinct domains: i) A C-terminal leucine-rich repeat domain; ii)
an N-terminal pyrin domain (PYD); and iii) a central NACHT domain
with ATPase activity (33).
Mutations in the NACHT domain impair NLRP3 oligomerization, thereby
reducing caspase-1 activation, IL-1β and IL-18 secretion and
pyroptosis (34). ASC serves as an
important adaptor protein that bridges the PYD of NLRP3 to the CARD
of pro-caspase-1, facilitating inflammasome assembly (34).
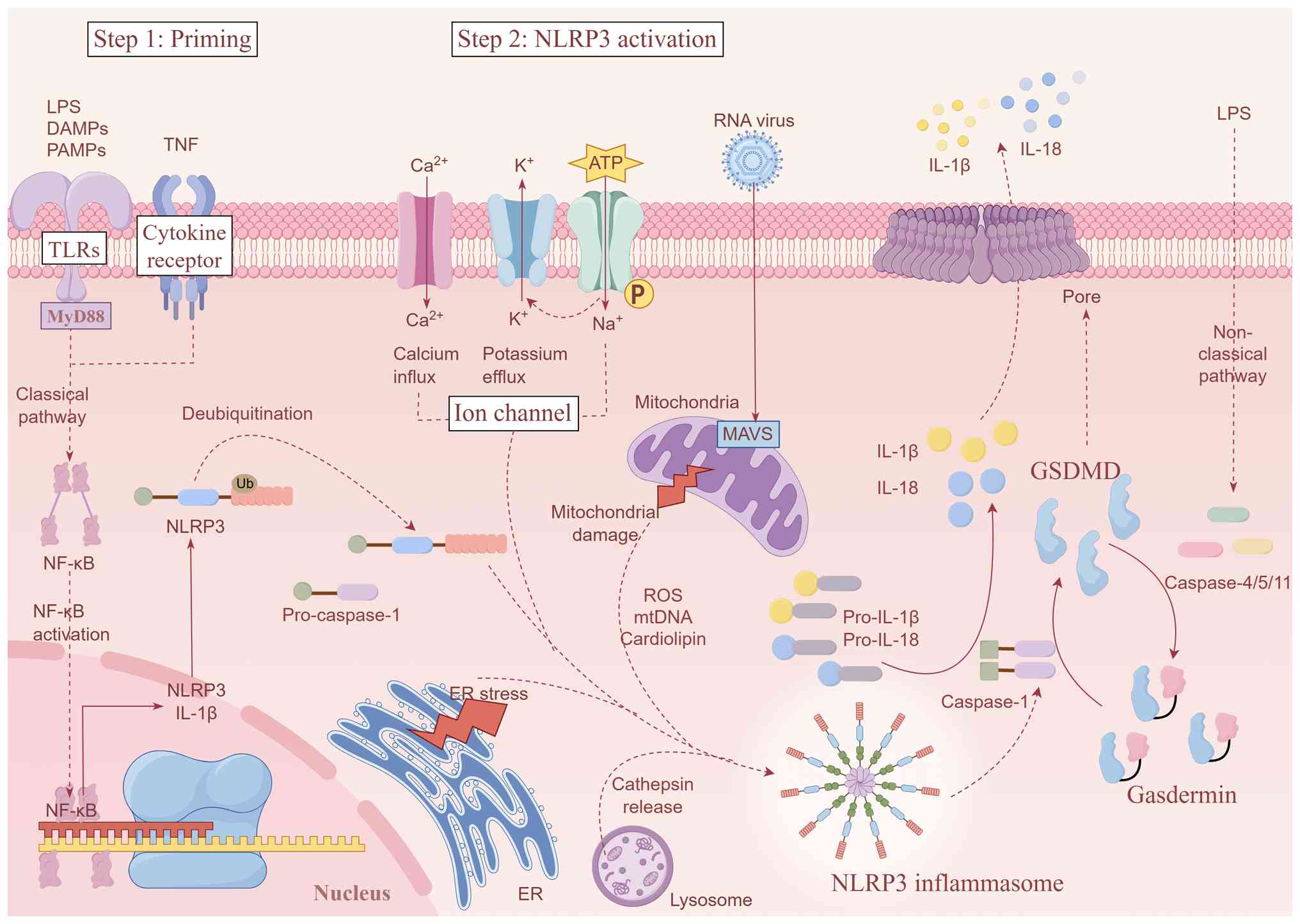
The NLRP3 inflammasome serves as a core regulator of
inflammatory pathways, requiring two distinct signals for canonical
activation: i) A priming signal: Ligands of Toll-like receptors
(TLRs) or cytokine receptors activate the myeloid differentiation
primary response gene 88 (MyD88)/nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB) pathway,
inducing transcriptional upregulation of NLRP3 and the
pro-inflammatory cytokines IL-1β and IL-18 (26); and ii) an activation signal:
Diverse PAMPs, such as bacterial flagellin, lipopolysaccharide
(LPS) and viral RNA, or DAMPs trigger oligomerization of ASC and
pro-caspase-1, culminating in NLRP3 inflammasome complex assembly
(35).
During SIC, multiple pathways converge to activate
the NLRP3 inflammasome through distinct mechanisms. Intracellular
K+ depletion constitutes an important upstream event of
NLRP3 inflammasome activation. ATP-mediated purinergic ligand-gated
ion channel 7 receptor activation triggers K+ efflux,
inducing NLRP3 assembly (36). LPS
induces endoplasmic reticulum (ER) stress, promoting
Ca2+ release via inositol 1,4,5-trisphosphate receptors
and subsequent NLRP3 activation (37). SIC causes mitochondrial structural
and functional impairment (38),
increasing mtROS and mitochondrial DNA (mtDNA) release. These
components facilitate NLRP3 oligomerization and exacerbate
myocardial injury, with mitochondrial antiviral-signaling protein
also contributing to inflammasome activation (39). Endocytosed crystalline or
particulate matter, such as silica and cholesterol crystals,
rupture lysosomes, releasing cathepsin B to promote NLRP3
oligomerization (40). Notably,
crosstalk exists between NLRP3 priming and activation mechanisms.
mtROS serves dual roles: As DAMPs activating TLR/MyD88/NF-κB
signaling and as direct promoters of NLRP3 oligomerization
(41). In addition to mtROS, ER
stress and Ca2+ release constitute another crucial
upstream event that orchestrates NLRP3 activation. Specifically, ER
Ca2+ overload induces mitochondrial Ca2+
uptake; this increased mitochondrial Ca2+ load in turn
amplifies mtROS production, thereby establishing a sustained
pathological feedback loop that exacerbates inflammasome activation
(25).
Upon oligomerization and activation, the NLRP3
inflammasome recruits ASC via PYD-PYD interactions. The resulting
complex facilitates pro-caspase-1 autocleavage, generating active
caspase-1 that processes pro-IL-1β and pro-IL-18 into mature
cytokines (42). Concurrently,
activated caspase-1 cleaves gasdermin D (GSDMD), liberating the
GSDMD N-terminal (GSDMD-NT) domain that subsequently translocates
to the plasma membrane, forming pores that mediate the release of
inflammatory mediators, including IL-1β and IL-18, and drive
pyroptosis, a mode of lytic inflammatory cell death (43). In SIC, pyroptosis releases DAMPs,
such as IL-1β, IL-18 and mtDNA, amplifying inflammation and NLRP3
activation through positive feedback. Notably, the inflammatory
milieu and DAMPs (e.g., mtDNA) released during pyroptosis can also
modulate other cell death and clearance pathways, including
apoptosis and mitophagy (44).
This cascade ultimately induces myocardial dysfunction and
structural damage, representing a core pathogenic mechanism in SIC
(45). Pharmacologically, fatty
acid amide hydrolase (FAAH) inhibitors disrupt NLRP3-FAAH
interactions, promoting NLRP3 degradation (46). This evidence indicates that
targeting NLRP3 attenuates inflammatory responses by modulating
interconnected pathways involving inflammation, pyroptosis,
apoptosis and mitophagy. Nevertheless, the precise molecular
targets governing these pathways require further elucidation
(Fig. 2).
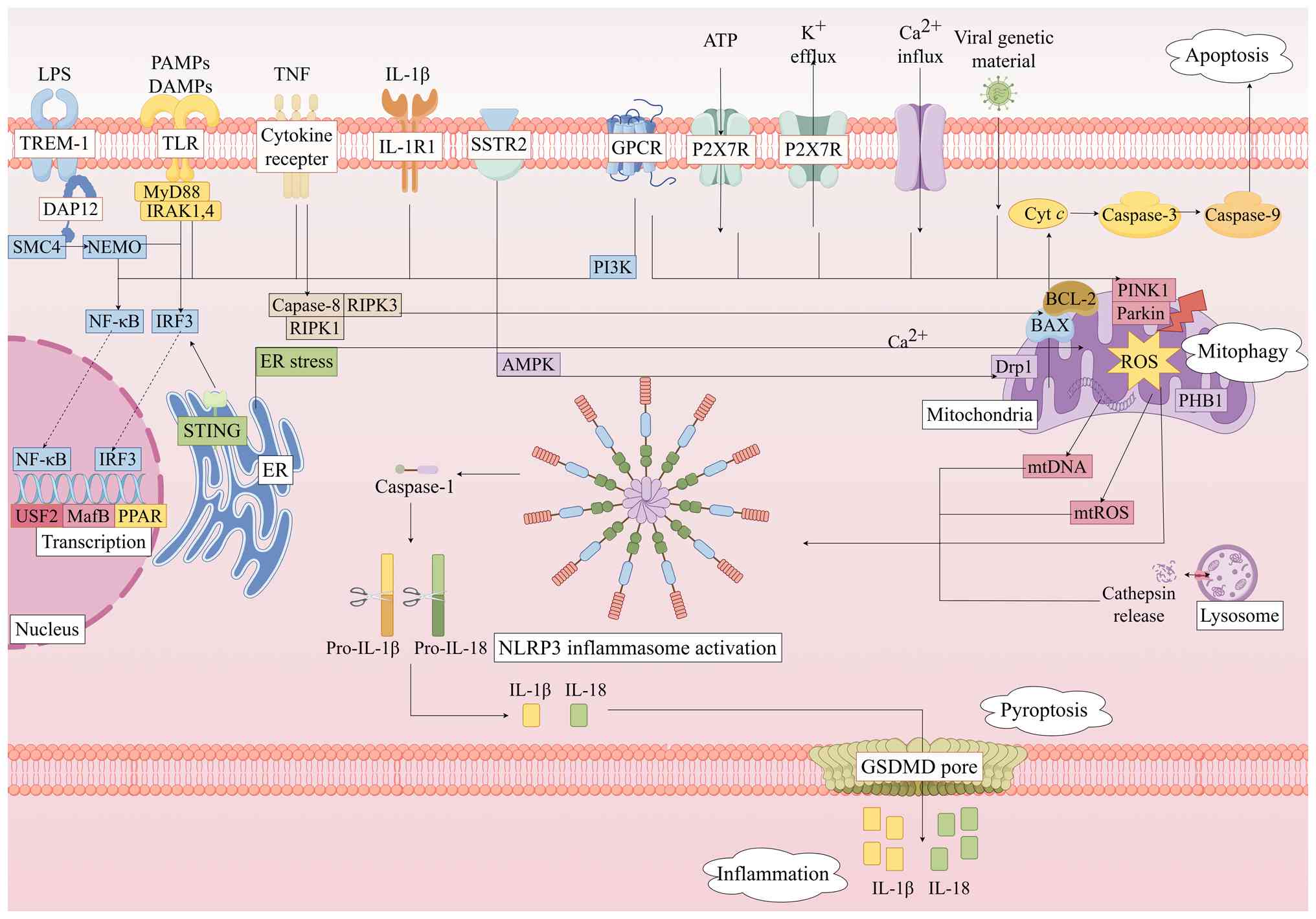
The central pathology of SIC involves a
self-amplifying cascade initiated by NLRP3 inflammasome activation.
Pathogen-derived DAMPs or PAMPs activate the TLR4/NF-κB signaling
pathway, priming NLRP3 expression. This process triggers
mitochondrial dysfunction and impairs mitophagy, leading to the
release of mtROS and mtDNA that directly promote NLRP3
oligomerization. Concurrently, the mitochondrial permeability
transition facilitates the release of cytochrome c (Cyt
c) into the cytoplasm, which is a pivotal event that bridges
mitochondrial damage with downstream programmed cell death (PCD)
pathways (47). Regarding PCD
convergence, these released mitochondrial components, such as Cyt
c, not only initiate intrinsic apoptosis but also contribute to a
complex interplay between apoptosis, pyroptosis, and inflammation
(48). In this context, extrinsic
apoptosis is initiated when TNF-α binds to TNF receptor (TNFR)1,
activating caspase-8, while intrinsic apoptosis occurs as a result
of Cyt c forming apoptosomes to activate caspase-9. Both
pathways converge on caspase-3 of apoptosis-executing factors
(e.g., endonucleases) and other proteins, culminating in apoptosis
(49). In the
pyroptosis-inflammation feedback loop, NLRP3-activated caspase-1
cleaves GSDMD, generating pore-forming GSDMD-NT fragments that
induce pyroptosis in myocardial cells and can further exacerbate
mitochondrial damage (50). This
releases pro-inflammatory cytokines, such as IL-1β and IL-18, which
disseminate systemically to amplify inflammation and generate
secondary DAMPs, thereby reactivating the NLRP3 inflammasome.
The NLRP3 inflammasome exhibits distinct expression
patterns and functional roles across different cardiac cell types,
collectively contributing to the complex pathological network of
SIC. In macrophages, early septic insults induce the expression of
glucocorticoid-induced TNFR-related protein on their surface, which
potentiates NLRP3 inflammasome activation and promotes
pro-inflammatory macrophage polarization by modulating the
post-translational modifications of the NLRP3 inflammasome, thereby
amplifying the inflammatory cascade (51). In cardiomyocytes, NLRP3 activation
directly induces pyroptosis through GSDMD pore formation,
compromising membrane integrity and leading to the release of
intracellular contents. The released inflammatory mediators not
only exacerbate autocrine dysregulation of calcium homeostasis but
also recruit and activate macrophages, perpetuating a
pro-inflammatory microenvironment and impairing cardiomyocyte
contractile function, representing a primary mechanism of acute
cardiac injury in SIC (52).
Furthermore, cardiomyocytes can communicate directly with cardiac
fibroblasts via membrane nanotubes, transmitting inflammasome
activation signals that drive fibroblasts toward a pro-inflammatory
and pro-fibrotic phenotype. This intercellular crosstalk results in
excessive extracellular matrix production, exacerbating acute
injury and laying the foundation for long-term myocardial fibrosis
and diastolic dysfunction in survivors of sepsis (53). In summary, the NLRP3 inflammasome
serves heterogeneous roles across different cardiac cell types,
which collectively constitute the complex pathological network of
SIC. These interconnected mechanisms form a notable cycle:
Mitochondrial damage induces NLRP3 activation, which promotes
inflammation, pyroptosis and apoptosis, resulting in secondary
mitochondrial injury. Targeted disruption of any nodal point in
this cycle represents a promising therapeutic strategy for SIC, as
the overlapping pathways converge on NLRP3 inflammasome activation
to sustain myocardial injury (Fig.
3).
Dysregulated inflammation represents a notable
pathophysiological process wherein pro-inflammatory cytokines
activate immune cells and mediate tissue damage, while
anti-inflammatory cytokines counteract these responses. SIC, an
acute cardiac dysfunction syndrome stemming from systemic infection
and inflammation, accelerates cardiovascular collapse by
exacerbating microcirculatory dysfunction and hypoperfusion. Its
pathogenesis involves complex interactions between the immune and
cardiovascular systems, prominently featuring inflammatory
activation (54). Excessive
cytokine production triggers a cytokine storm that amplifies tissue
injury and organ dysfunction (55). During SIC, PAMPs and DAMPs activate
the NLRP3 inflammasome via TLRs (56). This leads to caspase-1-mediated
proteolytic maturation of pro-IL-1β and pro-IL-18. Mature IL-1β and
IL-18 further activate downstream pathways, such as the NF-κB
pathway, inducing secondary inflammatory cytokines, such as TNF-α,
IL-6 and IL-1β, that establish a cytokine storm, ultimately causing
cardiomyocyte dysfunction (57).
Notably, NLRP3-knockout murine SIC models exhibit markedly reduced
expression of IL-1β, IL-6, IL-18 and TNF-α, with concomitant
improvement in cardiac function and attenuated myocardial injury
(58). These findings suggest that
combined targeting of NLRP3-derived IL-1β and IL-18 represents a
novel therapeutic approach for SIC.
Upstream stimulatory factor 2 (USF2), a basic
helix-loop-helix leucine zipper transcription factor, regulates
NLRP3 expression in multiple disease contexts (59,60).
A study reported by Dong et al (61) demonstrated that USF2 silencing
attenuates NLRP3 inflammasome activation in experimental SIC models
by targeting the microRNA (miR/miRNA)-206/Rho-related GTP-binding
protein RhoB/Rho-associated protein kinase signaling axis, thereby
reducing inflammatory responses and cardiac dysfunction.
Conversely, in lupus nephritis, USF2 does not operate through this
miRNA axis but rather directly binds to the NLRP3 promoter to drive
its transcription and exacerbate podocyte injury (60). These findings underscore that the
pathophysiological role of USF2 is not universal but is determined
by specific cellular milieu.
The TLR4/MyD88/NF-κB pathway serves as the dominant
upstream regulator of NLRP3 activation. During SIC, LPS upregulates
NLRP3, IL-18 and IL-1β expression through this pathway,
exacerbating myocardial inflammation. Notably, NF-κB inhibition
with BAY 11-7082, a selective IκBα phosphorylation blocker,
suppresses SIC-induced inflammation in cellular models (62). Myo-inositol oxygenase (MIOX), a key
enzyme in inositol catabolism, functions as both a biomarker and
therapeutic target in renal diseases (63,64).
Notably, MIOX enhances NLRP3 inflammasome activity by inhibiting
its degradation, aggravating infection-induced cardiac inflammation
and dysfunction in SIC models (65).
PCD represents a conserved mechanism for eliminating
damaged cells. Major PCD modalities include apoptosis, autophagy,
ferroptosis, necroptosis and pyroptosis (66). Apoptosis, as a predominant PCD
form, contributes notably to cellular injury during sepsis.
SIC-induced mitochondrial dysfunction alters membrane permeability,
triggering the activation of pro-apoptotic B-cell lymphoma 2
(BCL-2) family proteins, such as BCL-2-associated X protein (BAX),
while suppressing anti-apoptotic members, such as BCL-2. This
facilitates Cyt c release and caspase-3 activation (67). Similarly, TNF-α binding to TNFR1
activates caspase-8, which cleaves and activates the executioner
protein caspase-3. Nuclear translocation of caspase-3 initiates
substrate proteolysis, DNA fragmentation and apoptotic execution
(68). Myocardial apoptosis
severity associates with cardiac damage in sepsis. SIC models
demonstrate impaired cardiac function coincident with the elevated
expression of apoptosis-related proteins, including cleaved
caspase-3 (69). The NLRP3
inflammasome indirectly regulates apoptosis via inflammatory
signaling and cellular stress in SIC. Specifically, TNFR engagement
activates NLRP3 through the receptor-interacting serine/threonine
kinase (RIPK) 1/RIPK3 signaling axis (70), which promotes the release of
pro-inflammatory cytokines, such as IL-1β and IL-18, and induces
mitochondrial damage and ROS production. ROS trigger BAX
oligomerization via compromising mitochondrial outer membrane
integrity by impairing the anti-apoptotic function of BCL-2, which
normally sequesters BAX (71).
This mitochondrial membrane damage facilitates Cyt c release
into the cytoplasm, where it forms apoptosomes that activate
caspase-3 (72). Concurrently,
NLRP3-driven ROS production also activates downstream caspase-8,
which converges with the caspase-3 pathway to induce nuclear
translocation of endonucleases, culminating in DNA fragmentation
and apoptotic cell death (73).
Several upstream regulators have been shown to modulate this
process: Stimulator of interferon genes knockout attenuates
cardiomyocyte injury by suppressing NLRP3 via an interferon
regulatory factor 3-dependent pathway (74); poly(ADP-ribose) polymerase 1
(PARP1) overexpression decreases BCL-2 while elevating BAX and
cleaved caspase-3, a cascade reversed by atractylenolide I
targeting the PARP1/NLRP3 pathway (75); and oxysterols receptor LXR-α
knockout exacerbates apoptosis by enhancing NLRP3 expression
(76).
Pyroptosis represents a pro-inflammatory PCD
distinct from apoptosis, mediated by GSDM family proteins that form
transmembrane pores in the plasma membrane. These pores facilitate
IL-18 and IL-1β release while disrupting ionic homeostasis,
ultimately causing cellular swelling and lysis (77,78).
The NLRP3 inflammasome exemplifies a ‘double-edged sword’ in the
pathogenesis of SIC. As an important component of the innate immune
system, NLRP3 inflammasome activation is necessary for host defense
(79). Moderate pyroptosis, driven
by NLRP3, facilitates the clearance of intracellular pathogens in
SIC by eliminating their replicative niche and exposing them to
extracellular immune surveillance, thereby preserving cardiac
function in the early stages of sepsis (80). Conversely, excessive and
uncontrolled NLRP3 activation triggers an excessive release of
pro-inflammatory cytokines, including IL-1β and IL-18, and
widespread pyroptotic cell death, which exacerbates myocardial
injury and contributes to cardiac dysfunction (81). While inhibition of NLRP3 represents
a promising strategy to mitigate septic cardiomyopathy, complete
and long-term suppression may inadvertently compromise the ability
of the patient to clear infections. Therefore, future therapeutic
paradigms should not aim to abolish NLRP3 activity entirely, but to
precisely modulate it, therefore suppressing its pathological
overactivation while preserving its beneficial role in pathogen
clearance.
Multiple pathways converge on NLRP3 to regulate
pyroptosis: TLR4 activation initiates NLRP3 priming via the
MyD88/NF-κB pathway, while pharmacological inhibition of NF-κB
suppresses the resulting caspase-1 expression and pyroptosis
(62,82). Protective factors such as heat
shock protein 70 (83), apelin via
adenosine 5′-monophosphate-activated protein kinase (AMPK)
signaling (84) and cortistatin
via somatostatin receptor 2 (SSTR2)/AMPK signaling (85) inhibit NLRP3 activation and
downregulate pyroptosis-executing proteins such as GSDMD and
caspase-1. Conversely, phosphatidylinositol-4,5-bisphosphate
3-kinase catalytic subunit γ promotes NLRP3 assembly via G
protein-coupled receptor (GPCR)-induced NF-κB activation,
exacerbating pyroptosis (86,87).
Mitochondria serve as important energy-producing
organelles that regulate cellular functions and maintain
homeostasis, being particularly abundant in cardiomyocytes and
notably vulnerable during sepsis. Autophagy is a lysosomal
degradation process that eliminates damaged macromolecules and
organelles, sustaining intracellular homeostasis (88,89).
Mitophagy specifically targets impaired mitochondria for autophagic
clearance (90), operating
primarily through parkin-dependent and -independent mechanisms. The
PTEN-induced putative kinase 1 (PINK1)/parkin pathway represents
the canonical mitophagy route: Upon mitochondrial damage or
depolarization, PINK1 accumulates on the mitochondrial outer
membrane and recruits Parkin to ubiquitinate damaged mitochondria,
marking them for autophagosomal engulfment (91–94).
Signal transducer and activator of transcription 3 (STAT3), a
cytokine-responsive transcription factor, modulates this process;
STAT3 inhibition in macrophages induces PINK1-dependent mitophagy
(95), which clears dysfunctional
mitochondria, restores mitochondrial membrane potential, suppresses
mtROS release and inactivates the NLRP3 inflammasome (96).
During SIC, the pathogen-activated NLRP3
inflammasome inhibits mitophagy via caspase-1-mediated cleavage of
autophagy-related proteins, such as beclin-1, leading to
mitochondrial structural and functional damage (97). This results in a notable release of
mtROS and mtDNA, which act as DAMPs in a positive feedback loop
that activates the NLRP3 inflammasome, establishing a cycle of
autophagy inhibition and exacerbated inflammation (98). Mitophagy, the selective clearance
of damaged or dysfunctional mitochondria, has been shown to reduce
mtROS levels, suppress NLRP3 inflammasome activation and
consequently mitigate septic cardiomyopathy injury (99). A recent study indicated that
inhibiting pyruvate dehydrogenase kinase suppresses NLRP3
inflammasome activation, reduces downstream caspase-1 cleavage and
IL-1β secretion and diminishes ROS production by enhancing
autophagy (100). Dual
specificity phosphatase 1, a regulator of angiotensin II signaling
(101), has been shown to enhance
FUN14 domain-containing protein 1-dependent mitophagy, thereby
reducing NLRP3 inflammasome formation and the generation of
specific inflammatory cytokines, ultimately attenuating the
inflammatory response (102–104). SSTR2, a GPCR, is an important
functional receptor expressed in cardiomyocytes. The activity of
dynamin-related protein 1 (Drp1), a GTPase primarily regulating
mitophagy, is modulated by phosphorylation (105). During SIC, the neuropeptide
cortistatin can activate SSTR2/Drp1-mediated mitophagy to reduce
ROS production, thereby inhibiting NLRP3 inflammasome activation
and pyroptosis, alleviating SIC-induced myocardial injury (85). Prohibitin 1 (PHB1), a key protein
in the mitochondrial inner membrane, is involved in maintaining
mitochondrial structure, regulating metabolism and inhibiting mtROS
generation. Research demonstrates that PHB1 suppresses mtROS
accumulation by enhancing mitophagy, consequently blocking NLRP3
inflammasome assembly; conversely, PHB1 deficiency exacerbates
mitochondrial damage and inflammation (106). Additionally, autophagy can
directly suppress inflammatory responses by clearing NLRP3
inflammasome components, such as ASC specks, or degrading active
caspase-1 (107,108).
Mitophagy deficiency amplifies mitochondrial danger
signals, including mtROS and mtDNA, which serve as important
triggers and amplifiers for the NLRP3 inflammasome (109). The level of cardiomyocyte
mitophagy is closely associated with the outcome and prognosis of
septic cardiomyopathy; while damaged mitochondria acting as DAMPs
induce NLRP3 inflammasome activation, the activated NLRP3
inflammasome conversely impairs autophagic capacity, establishing a
detrimental positive feedback loop of inflammation and autophagy
imbalance that exacerbates septic cardiomyopathic injury (110). Adaptor protein containing PH
domain, PTB domain and leucine zipper motif 1 (APPL-1), a dynamic
protein of the early endosome, translocates between cellular
organelles under various stress conditions (111). A recent study indicated that
APPL-1 deficiency suppresses mitophagy, consequently triggering
excessive NLRP3 inflammasome activation (112). Additionally, v-maf
musculoaponeurotic fibrosarcoma oncogene homolog B, a protein
belonging to the transcription factor MAF subfamily that is
responsible for binding specific DNA element motifs (113), has been shown to promote NLRP3
inflammasome-mediated mitochondrial damage upon its knockdown in
SIC models, which also display consequent mtROS production and
mtDNA release (114).
The pathological progression of SIC is driven by the
NLRP3 inflammasome as a central hub, establishing self-amplifying
injury through a triple amplification cascade: i) NLRP3 activation
induces caspase-1-mediated maturation of IL-1β and IL-18,
triggering inflammatory cascades (115); ii) concurrently, caspase-1
cleaves GSDMD to initiate pyroptosis, with resultant pore formation
releasing DAMPs that further activate the TLR4/NLRP3 axis; and iii)
pyroptosis and apoptosis pathways converge at the
caspase-8/BH3-interacting domain death agonist (Bid)/truncated Bid
node, where caspase-8 activation promotes BAX-mediated
mitochondrial outer membrane permeabilization, resulting in
mitochondrial Cyt c release and amplified apoptosis.
Mitochondrial damage releases mtROS and mtDNA that directly
activate NLRP3, while conversely, NLRP3 suppresses
PINK1/parkin-mediated mitophagy via caspase-1 cleavage of beclin-1.
The resulting mitophagy deficiency causes accumulation of damaged
mitochondria and mtROS overproduction, further activating NLRP3 to
establish a cycle of autophagy inhibition and inflammatory
exacerbation (116). Key
molecular targets mediating these interactions include: i)
TNFR-associated factor 2, which bidirectionally regulates the NF-κB
inflammatory pathway and the RIPK1-mediated apoptosis/survival
switch in SIC; and ii) AMPK, an important cellular energy sensor
that concurrently inhibits NLRP3 assembly and enhances
PINK1/parkin-mediated mitophagy during SIC (117). Notably, mtROS released from
damaged mitochondria in SIC cardiomyocytes serve as a common
trigger that interconnect inflammatory activation, BAX
oligomerization and autophagy suppression (118) (Fig.
4).
As aforementioned, pharmacological agents and
compounds targeting the NLRP3 inflammasome and its upstream or
downstream pathways exhibit notable therapeutic potential for
mitigating myocardial injury and treating SIC, potentially forming
the foundation for future therapeutic strategies. The present
section broadly categorizes the mechanisms of action of currently
investigated compounds.
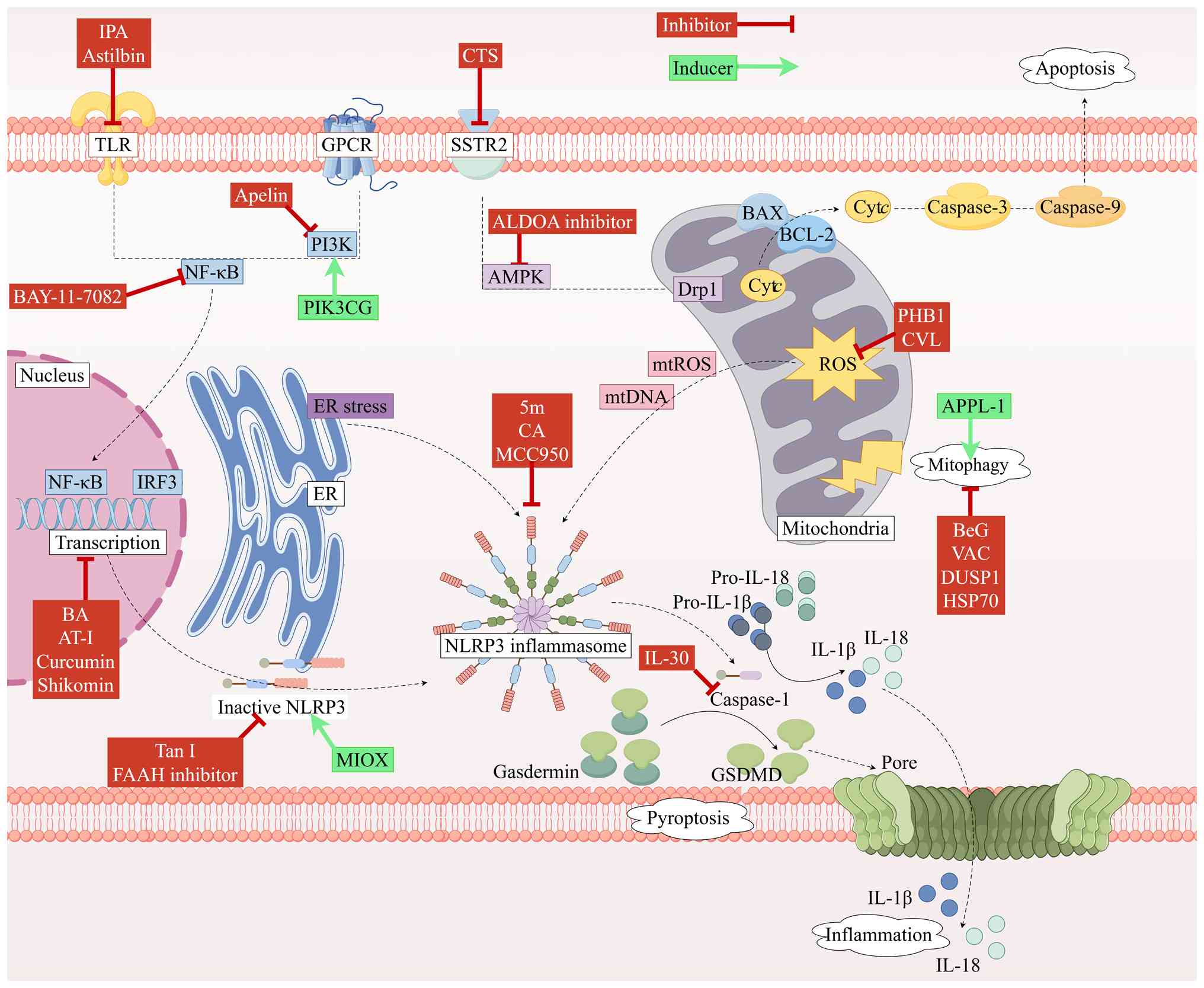
Several pharmacological agents ameliorate
SIC-induced myocardial dysfunction by directly inhibiting NLRP3
inflammasome assembly and activation. Tanshinone I (Tan I), an
active constituent of Salvia miltiorrhiza with established
therapeutic potential across multiple pathologies (119,120), suppresses inflammasome assembly
and downstream caspase-1 activation in SIC model by targeting the
NLRP3 PYD to disrupt its interaction with ASC; this mechanism
positions Tan I and its derivatives as promising next-generation
NLRP3-targeted therapeutics (121). Cinnamyl alcohol, a bioactive
component of cinnamon demonstrating anti-inflammatory and
antioxidant properties (122,123), reduces tissue inflammation and
downregulates inflammatory cytokine expression via NLRP3
inflammasome inhibition, highlighting its therapeutic potential for
sepsis (124). Genetic ablation
of NLRP3 markedly attenuates IL-1β, IL-6, IL-18 and TNF-α
expression while improving cardiac function and mitigating
myocardial injury in SIC model (58). These findings indicate that
combinatorial NLRP3 inflammasome inhibition represents a viable
therapeutic strategy for SIC.
Several agents indirectly mitigate NLRP3
inflammasome-mediated myocardial injury and dysfunction in SIC by
targeting upstream signaling pathways regulating its priming and
activation. NF-κB, a central regulator of NLRP3 priming,
facilitates inflammasome activation via the TLR4/MyD88/NF-κB
pathway (125); a recent study
demonstrated that the NF-κB antagonist BAY 11-7082 reduces
pro-inflammatory cytokine expression, such as that of TNF-α, IL-6
and IL-1β, and suppresses pyroptosis (62). Shikonin, an anti-inflammatory
phytochemical, exerts inhibitory effects on NLRP3
inflammasome-mediated inflammation and pyroptosis via
AMPK/NAD-dependent protein deacetylase sirtuin-1 pathway modulation
when formulated as Zn2+-shikonin nanoparticles (126). Astilbin, a protective flavonoid
(127–131), markedly attenuates inflammatory
marker concentrations and reduces TLR4/NF-κB pathway expression in
septic cardiomyopathy model, indicating that its cardioprotective
mechanism involves TLR4/NF-κB pathway inhibition to suppress NLRP3
inflammasome activation (132).
Brevilin A, an anti-inflammatory sesquiterpene lactone derived from
Centipeda minima, effectively suppresses NF-κB and NLRP3
protein expression in both cellular and animal models of septic
cardiomyopathy, highlighting its promise as a natural therapeutic
for SIC (133).
Indole-3-propionic acid, an anti-inflammatory gut microbiota
metabolite (134–137), inhibits NLRP3 and downstream
caspase-1 expression via TLR4/MyD88/NF-κB pathway suppression in
SIC models (62).
Specific pharmacological agents ameliorate SIC by
inhibiting downstream signaling or effector proteins of the NLRP3
inflammasome. Curcumin, the primary active constituent of turmeric
with demonstrated pleiotropic pharmacological activities (138), reduces NLRP3, ASC and caspase-1
expression in septic animal models when delivered via
arginine-glycine-aspartic acid-anchored liposomal encapsulation,
identifying this as a novel therapeutic strategy for SIC (139). The translational promise of this
strategy is underscored by a phase II randomized controlled trial,
where intervention with nano-curcumin in a cohort of patients with
severe sepsis not only confirmed its safety and feasibility as a
therapeutic but also mechanistically demonstrated a marked
downregulation of NF-κB and NLRP3 mRNA expression in patients
following treatment, thereby providing robust support for its
future application in targeted SIC therapy and prognostic
assessments (140). Furthermore,
IL-30 exhibits anti-inflammatory properties across multiple
pathologies (141–143); a study by Zhao et al
(144) demonstrated that IL-30
treatment effectively suppresses the expression of
pyroptosis-associated proteins, including IL-1β, IL-18, NLRP3 and
GSDMD, in SIC murine models.
Several pharmacological agents mitigate myocardial
injury resulting from SIC by enhancing mitophagy to disrupt the
cycle of NLRP3 inflammasome activation and mitophagy inhibition.
Carvacrol, a monoterpenoid phenol with anti-inflammatory properties
(145), reduces ROS generation
and enhances autophagy to suppress NLRP3 inflammasome formation in
SIC (146). Vaccarin, a bioactive
flavonoid derived from Vaccaria segetalis seeds (147–149), attenuates inflammatory cytokine
expression and modulates mitophagy to confer cardioprotection in
SIC (150). Bergapten, a
bioactive coumarin (151–153), inhibits NLRP3 inflammasome
activation by promoting mitophagy, suggesting therapeutic potential
for SIC (154). Additionally, the
aldolase A inhibitor LYG-202 suppresses activation of the NLRP3
inflammasome and attenuates inflammation in SIC through
AMPK/mitophagy pathway activation (155).
Despite the compelling therapeutic potential of
targeting the NLRP3 inflammasome in SIC, its translation from
promising preclinical data to clinical reality faces numerous
challenges. A critical and expanded analysis of these hurdles,
informed by lessons from broader NLRP3 inhibitor development, is
important for guiding future research.
A defining feature of SIC is its potential
reversibility following sepsis control (161). This characteristic implies a
narrow therapeutic window for NLRP3 inhibition. The
hyperinflammatory phase of early sepsis, when NLRP3 is most active
(162), likely represents the
optimal period for effective intervention to mitigate myocardial
injury and improve patient outcomes. By contrast, administration of
inhibitors during the later immunoparalytic phase may exacerbate
immune imbalance and increase the risk of secondary infections
(163). However, most current
cellular and animal models of NLRP3 inhibition employ prophylactic
or early treatment regimens (164). Therefore, future clinical trials
of NLRP3 inhibitors must carefully define appropriate monitoring
indicators and rigorously evaluate the therapeutic window.
Although several NLRP3 inhibitors have advanced to
clinical trials, current guidelines and clinical experience
emphasize that conventional therapies, including early antibiotic
administration and supportive care, remain the cornerstone of
sepsis management (165). NLRP3
inhibitors are not intended to replace but rather to complement the
existing standard of care, such as antibiotics, fluid resuscitation
and vasopressors. For instance, by mitigating endothelial injury
and capillary leakage (166),
NLRP3 inhibitors may enhance hemodynamic stability achieved through
fluid resuscitation and vasoactive agents. However, potential
antagonistic interactions must be carefully considered in future
clinical use. Certain antibiotics can induce mitochondrial stress
or alter immune cell function, which may indirectly influence NLRP3
inflammasome activation pathways (167). As early and appropriate
antimicrobial therapy is important (168), NLRP3 inhibition must not
interfere with antimicrobial efficacy or the ability of septic
patients to clear bacteria. Furthermore, patients with sepsis
frequently experience acute renal or hepatic dysfunction (169), which can markedly alter the
pharmacokinetics and safety profile of any adjunctive treatment,
including NLRP3 inhibitors. Therefore, integrating NLRP3 inhibitors
into septic care remains challenging and requires rigorous
evaluation in relevant disease-state models. This includes
determining the optimal timing of inhibitor administration relative
to antibiotics, understanding the impact of NLRP3 inhibitors on
hemodynamic management and conducting detailed pharmacokinetic
studies in septic patients exhibiting varying degrees of organ
failure to ensure both the safety and synergistic efficacy of these
inhibitors.
Valuable lessons can be drawn from the clinical
development of NLRP3-targeting agents in other inflammatory
diseases, offering notable insights for SIC clinical trial design
and risk management (170).
Primarily, the clinical development of pioneering compounds such as
MCC950 and GDC-2394 was halted due to hepatotoxicity, with
MCC950-associated liver injury linked to its off-target inhibition
of carbonic anhydrase 2 (171).
This underscores the necessity for thorough hepatic safety
profiling of any SIC therapeutic candidate. Secondly, while the
anti-IL-1β monoclonal antibody canakinumab has demonstrated
cardiovascular benefits in the Canakinumab Anti-Inflammatory
Thrombosis Outcome Study trial, it concurrently increased the
incidence of fatal infections and hematological toxicities,
highlighting the inherent immunosuppressive risks associated with
broad anti-cytokine therapy in patient populations, such as
patients with SIC (172).
Finally, although glyburide supported the potential of NLRP3
inhibition in preclinical models, its clinical utility was notably
constrained by dose-limiting hypoglycemia and other off-target
effects, serving as a cautionary tale that the therapeutic benefit
of drug repurposing must be carefully weighed against its inherent
polypharmacology (173).
Collectively, these experiences mandate that future SIC clinical
trial designs prioritize hepatic safety, cautiously evaluate the
consequences of immunosuppression and rigorously select therapeutic
agents with high specificity. The compounds reported to target the
NLRP3 inflammasome for the treatment of SIC are summarized in
Table I (27).
Future investigations should prioritize: i)
Cell-specific NLRP3 functions using conditional knockout/knock-in
models; ii) interorganellar communication mechanisms between NLRP3
and ER/golgi networks; iii) rational optimization of natural
compound-derived inhibitors through structure-activity relationship
studies; and iv) clinical translation initiatives. These
initiatives may include: i) Advanced SIC models with improved
clinical relevance; ii) preclinical development of principal
compounds, such as MCC950; iii) predictive biomarker discovery; iv)
novel drug delivery systems, such as nanoparticle encapsulation;
and v) combination therapies with antibiotics, mitochondrial
protectants or anti-apoptotic agents.
The NLRP3 inflammasome has emerged as a central
molecular hub in SIC pathogenesis. By driving cytokine storms,
inducing cardiomyocyte pyroptosis and apoptosis, disrupting
mitochondrial homeostasis and suppressing protective autophagy, the
NLRP3 inflammasome establishes a self-perpetuating cycle of
inflammation and PCD that culminates in myocardial dysfunction.
Pharmacological targeting of NLRP3 signaling pathways represents a
promising therapeutic strategy for SIC. Therefore, further
elucidating the multifaceted roles of NLRP3 in SIC will accelerate
the development of mechanistically-grounded therapeutic
interventions.
Not applicable.
The present work was supported by Beijing Natural Science
Foundation (grant no. 7232126), the Special Scientific Research
Project of Beijing Critical Care Ultrasound Research Association
(grant no. 2023-CCUSG-A-03), the Medical Health Research Project of
Yichang (grant no. A24-2-011) and the Health Promotion
Project-Academic construction project of adsorption
engineering-Scientific research and academic promotion project for
critical and severe diseases (grant no. QS-XFGCJWZZ-0049).
Not applicable.
The original draft of the manuscript and the figures
were produced by YC. ZZ and GZ were responsible for reviewing and
editing the manuscript and supervision of the study. Data
authentication is not applicable. All authors read and approved the
final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Singer M, Deutschman CS, Seymour CW,
Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche
JD, Coopersmith CM, et al: The third international consensus
definitions for sepsis and septic shock (sepsis-3). JAMA.
315:801–810. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bone RC, Balk RA, Cerra FB, Dellinger RP,
Fein AM, Knaus WA, Schein RM and Sibbald WJ: Definitions for sepsis
and organ failure and guidelines for the use of innovative
therapies in sepsis. The ACCP/SCCM consensus conference committee.
American college of chest physicians/society of critical care
medicine. Chest. 101:1644–1655. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cecconi M, Evans L, Levy M and Rhodes A:
Sepsis and septic shock. Lancet. 392:75–87. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gheen N: Sepsis-3 definitions. Ann Emerg
Med. 68:784–785. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gong T, Fu Y, Wang Q, Loughran PA, Li Y,
Billiar TR, Wen Z, Liu Y and Fan J: Decoding the multiple functions
of ZBP1 in the mechanism of sepsis-induced acute lung injury.
Commun Biol. 7:13612024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Hu C, Zhai P, Zhang J, Jiang J, Suo
J, Hu B, Wang J, Weng X, Zhou X, et al: Fibroblastic reticular
cell-derived exosomes are a promising therapeutic approach for
septic acute kidney injury. Kidney Int. 105:508–523. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Song YQ, Lin WJ, Hu HJ, Wu SH, Jing L, Lu
Q and Zhu W: Sodium tanshinone IIA sulfonate attenuates
sepsis-associated brain injury via inhibiting NOD-like receptor
3/caspase-1/gasdermin D-mediated pyroptosis. Int Immunopharmacol.
118:1101112023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xu JQ, Zhang WY, Fu JJ, Fang XZ, Gao CG,
Li C, Yao L, Li QL, Yang XB, Ren LH, et al: Viral sepsis:
Diagnosis, clinical features, pathogenesis, and clinical
considerations. Mil Med Res. 11:782024.PubMed/NCBI
|
|
9
|
Werdan K, Schmidt H, Ebelt H, Zorn-Pauly
K, Koidl B, Hoke RS, Heinroth K and Müller-Werdan U: Impaired
regulation of cardiac function in sepsis, SIRS, and MODS. Can J
Physiol Pharmacol. 87:266–274. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hasegawa D, Ishisaka Y, Maeda T,
Prasitlumkum N, Nishida K, Dugar S and Sato R: Prevalence and
prognosis of sepsis-induced cardiomyopathy: A systematic review and
meta-analysis. J Intensive Care Med. 38:797–808. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Antonucci E, Fiaccadori E, Donadello K,
Taccone FS, Franchi F and Scolletta S: Myocardial depression in
sepsis: From pathogenesis to clinical manifestations and treatment.
J Crit Care. 29:500–511. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Parker MM, Shelhamer JH, Bacharach SL,
Green MV, Natanson C, Frederick TM, Damske BA and Parrillo JE:
Profound but reversible myocardial depression in patients with
septic shock. Ann Intern Med. 100:483–490. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
MacLean LD, Mulligan WG, McLean AP and
Duff JH: Patterns of septic shock in man-a detailed study of 56
patients. Ann Surg. 166:543–562. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Parrillo JE: Pathogenetic mechanisms of
septic shock. N Engl J Med. 328:1471–1477. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Abraham E and Singer M: Mechanisms of
sepsis-induced organ dysfunction. Crit Care Med. 35:2408–2416.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Levy RJ and Deutschman CS: Cytochrome c
oxidase dysfunction in sepsis. Crit Care Med. 35 (9
Suppl):S468–S475. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mebazaa A, De Keulenaer GW, Paqueron X,
Andries LJ, Ratajczak P, Lanone S, Frelin C, Longrois D, Payen D,
Brutsaert DL and Sys SU: Activation of cardiac endothelium as a
compensatory component in endotoxin-induced cardiomyopathy: role of
endothelin, prostaglandins, and nitric oxide. Circulation.
104:3137–3144. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hobai IA, Edgecomb J, LaBarge K and
Colucci WS: Dysregulation of intracellular calcium transporters in
animal models of sepsis-induced cardiomyopathy. Shock. 43:3–15.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Miranda M, Balarini M, Caixeta D and
Bouskela E: Microcirculatory dysfunction in sepsis:
Pathophysiology, clinical monitoring, and potential therapies. Am J
Physiol Heart Circ Physiol. 311:H24–H35. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Henedak NT, El-Abhar HS, Soubh AA and
Abdallah DM: NLRP3 Inflammasome: A central player in renal
pathologies and nephropathy. Life Sci. 351:1228132024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tao S, Fan W, Liu J, Wang T, Zheng H, Qi
G, Chen Y, Zhang H, Guo Z and Zhou F: NLRP3 inflammasome: An
Emerging therapeutic target for Alzheimer's disease. J Alzheimers
Dis. 96:1383–1398. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tengesdal IW, Dinarello CA and Marchetti
C: NLRP3 and cancer: Pathogenesis and therapeutic opportunities.
Pharmacol Ther. 251:1085452023. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sayaf K, Battistella S and Russo FP: NLRP3
inflammasome in acute and chronic liver diseases. Int J Mol Sci.
25:45372024. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, You YK, Guo J, Wang J, Shao B, Li
H, Meng X, Lan HY and Chen H: C-reactive protein promotes diabetic
kidney disease via Smad3-mediated NLRP3 inflammasome activation.
Mol Ther. 33:263–278. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wen Y, Liu Y, Liu W, Liu W, Dong J, Liu Q,
Hao H and Ren H: Research progress on the activation mechanism of
NLRP3 inflammasome in septic cardiomyopathy. Immun Inflamm Dis.
11:e10392023. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Toldo S and Abbate A: The role of the
NLRP3 inflammasome and pyroptosis in cardiovascular diseases. Nat
Rev Cardiol. 21:219–237. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhu W, Bao X, Yang Y, Xing M, Xiong S,
Chen S, Zhong Y, Hu X, Lu Q, Wang K, et al: Peripheral evolution of
tanshinone IIA and cryptotanshinone for discovery of a potent and
specific NLRP3 inflammasome inhibitor. J Med Chem. 68:3460–3479.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zheng Y, Zhang X, Wang Z, Zhang R, Wei H,
Yan X, Jiang X and Yang L: MCC950 as a promising candidate for
blocking NLRP3 inflammasome activation: A review of preclinical
research and future directions. Arch Pharm (Weinheim).
357:e24004592024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li H, Guan Y, Liang B, Ding P, Hou X, Wei
W and Ma Y: Therapeutic potential of MCC950, a specific inhibitor
of NLRP3 inflammasome. Eur J Pharmacol. 928:1750912022. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cabral JE, Wu A, Zhou H, Pham MA, Lin S
and McNulty R: Targeting the NLRP3 inflammasome for inflammatory
disease therapy. Trends Pharmacol Sci. 46:503–519. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Takeuchi O and Akira S: Pattern
recognition receptors and inflammation. Cell. 140:805–820. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y, Ye X, Escames G, Lei W, Zhang X,
Li M, Jing T, Yao Y, Qiu Z, Wang Z, et al: The NLRP3 inflammasome:
Contributions to inflammation-related diseases. Cell Mol Biol Lett.
28:512023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fu J and Wu H: Structural mechanisms of
NLRP3 inflammasome assembly and activation. Annu Rev Immunol.
41:301–316. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu X, Matico RE, Miller R, Chauhan D, Van
Schoubroeck B, Grauwen K, Suarez J, Pietrak B, Haloi N, Yin Y, et
al: Structural basis for the oligomerization-facilitated NLRP3
activation. Nat Commun. 15:11642024. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jo EK, Kim JK, Shin DM and Sasakawa C:
Molecular mechanisms regulating NLRP3 inflammasome activation. Cell
Mol Immunol. 13:148–159. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martínez-García JJ, Martínez-Banaclocha H,
Angosto-Bazarra D, de Torre-Minguela C, Baroja-Mazo A, Alarcón-Vila
C, Martínez-Alarcón L, Amores-Iniesta J, Martín-Sánchez F, Ercole
GA, et al: P2X7 receptor induces mitochondrial failure in monocytes
and compromises NLRP3 inflammasome activation during sepsis. Nat
Commun. 10:27112019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu QR, Yang H, Zhang HD, Cai YJ, Zheng YX,
Fang H, Wang ZF, Kuang SJ, Rao F, Huang HL, et al: IP3R2-mediated
Ca2+ release promotes LPS-induced cardiomyocyte
pyroptosis via the activation of NLRP3/Caspase-1/GSDMD pathway.
Cell Death Discov. 10:912024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zeng Y, Cao G, Lin L, Zhang Y, Luo X, Ma
X, Aiyisake A and Cheng Q: Resveratrol attenuates sepsis-induced
cardiomyopathy in rats through anti-ferroptosis via the Sirt1/Nrf2
pathway. J Invest Surg. 36:21575212023. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Liu S, Bi Y, Han T, Li YE, Wang Q, Wu NN,
Xu C, Ge J, Hu R and Zhang Y: The E3 ubiquitin ligase MARCH2
protects against myocardial ischemia-reperfusion injury through
inhibiting pyroptosis via negative regulation of PGAM5/MAVS/NLRP3
axis. Cell Discov. 10:242024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Karasawa T and Takahashi M: The
crystal-induced activation of NLRP3 inflammasomes in
atherosclerosis. Inflamm Regen. 37:182017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ye T, Wang C, Yan J, Qin Z, Qin W, Ma Y,
Wan Q, Lu W, Zhang M, Tay FR, et al: Lysosomal destabilization: A
missing link between pathological calcification and osteoarthritis.
Bioact Mater. 34:37–50. 2023.PubMed/NCBI
|
|
42
|
Du G, Healy LB, David L, Walker C, El-Baba
TJ, Lutomski CA, Goh B, Gu B, Pi X, Devant P, et al: ROS-dependent
S-palmitoylation activates cleaved and intact gasdermin D. Nature.
630:437–446. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang Y, Xu W and Zhou R: NLRP3
inflammasome activation and cell death. Cell Mol Immunol.
18:2114–2127. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Saller BS, Wöhrle S, Fischer L, Dufossez
C, Ingerl IL, Kessler S, Mateo-Tortola M, Gorka O, Lange F, Cheng
Y, et al: Acute suppression of mitochondrial ATP production
prevents apoptosis and provides an essential signal for NLRP3
inflammasome activation. Immunity. 58:90–107.e11. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ting JP, Lovering RC, Alnemri ES, Bertin
J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA,
et al: The NLR gene family: A standard nomenclature. Immunity.
28:285–287. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Zhu Y, Zhang H, Mao H, Zhong S, Huang Y,
Chen S, Yan K, Zhao Z, Hao X, Zhang Y, et al: FAAH served a key
membrane-anchoring and stabilizing role for NLRP3 protein
independently of the endocannabinoid system. Cell Death Differ.
30:168–183. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yu S, Fu J, Wang J, Zhao Y, Liu B, Wei J,
Yan X and Su J: The influence of mitochondrial-DNA-driven
inflammation pathways on macrophage polarization: A new perspective
for targeted immunometabolic therapy in cerebral
ischemia-reperfusion injury. Int J Mol Sci. 23:1352021. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zheng X, Zhong T, Ma Y, Wan X, Qin A, Yao
B, Zou H, Song Y and Yin D: Bnip3 mediates doxorubicin-induced
cardiomyocyte pyroptosis via caspase-3/GSDME. Life Sci.
242:1171862020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Rodrigue-Gervais IG and Saleh M: Caspases
and immunity in a deadly grip. Trends Immunol. 34:41–49. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Miao R, Jiang C, Chang WY, Zhang H, An J,
Ho F, Chen P, Zhang H, Junqueira C, Amgalan D, et al: Gasdermin D
permeabilization of mitochondrial inner and outer membranes
accelerates and enhances pyroptosis. Immunity. 56:2523–2541.e8.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Gritte RB, Souza-Siqueira T, Borges da
Silva E, Dos Santos de Oliveira LC, Cerqueira Borges R, Alves HHO,
Masi LN, Murata GM, Gorjão R, Levada-Pires AC, et al: Evidence for
monocyte reprogramming in a long-term postsepsis study. Crit Care
Explor. 4:e07342022.PubMed/NCBI
|
|
52
|
Jin Y, Fleishman JS, Ma Y, Jing X, Guo Q,
Shang W and Wang H: NLRP3 inflammasome targeting offers a novel
therapeutic paradigm for sepsis-induced myocardial injury. Drug Des
Devel Ther. 19:1025–1041. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Shen J, Wu JM, Hu GM, Li MZ, Cong WW, Feng
YN, Wang SX, Li ZJ, Xu M, Dong ED, et al: Membrane nanotubes
facilitate the propagation of inflammatory injury in the heart upon
overactivation of the β-adrenergic receptor. Cell Death Dis.
11:9582020. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wiersinga WJ, Leopold SJ, Cranendonk DR
and van der Poll T: Host innate immune responses to sepsis.
Virulence. 5:36–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Fajgenbaum DC and June CH: Cytokine storm.
N Engl J Med. 383:2255–2273. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Blevins HM, Xu Y, Biby S and Zhang S: The
NLRP3 inflammasome pathway: A review of mechanisms and inhibitors
for the treatment of inflammatory diseases. Front Aging Neurosci.
14:8790212022. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Napodano C, Carnazzo V, Basile V, Pocino
K, Stefanile A, Gallucci S, Natali P, Basile U and Marino M: NLRP3
inflammasome involvement in heart, liver, and lung diseases-A
lesson from cytokine storm syndrome. Int J Mol Sci. 24:165562023.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Fujimura K, Karasawa T, Komada T, Yamada
N, Mizushina Y, Baatarjav C, Matsumura T, Otsu K, Takeda N,
Mizukami H, et al: NLRP3 inflammasome-driven IL-1β and IL-18
contribute to lipopolysaccharide-induced septic cardiomyopathy. J
Mol Cell Cardiol. 180:58–68. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Sun J, Ge X, Wang Y, Niu L, Tang L and Pan
S: USF2 knockdown downregulates THBS1 to inhibit the TGF-β
signaling pathway and reduce pyroptosis in sepsis-induced acute
kidney injury. Pharmacol Res. 176:1059622022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Xie Y, Li X, Deng W, Nan N, Zou H, Gong L,
Chen M, Yu J, Chen P, Cui D and Zhang F: Knockdown of USF2 inhibits
pyroptosis of podocytes and attenuates kidney injury in lupus
nephritis. J Mol Histol. 54:313–327. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Dong W, Liao R, Weng J, Du X, Chen J, Fang
X, Liu W, Long T, You J, Wang W and Peng X: USF2 activates
RhoB/ROCK pathway by transcriptional inhibition of miR-206 to
promote pyroptosis in septic cardiomyocytes. Mol Cell Biochem.
479:1093–1108. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhang Y, Li S, Fan X and Wu Y:
Pretreatment with indole-3-propionic acid attenuates
lipopolysaccharide-induced cardiac dysfunction and inflammation
through the AhR/NF-κB/NLRP3 pathway. J Inflamm Res. 17:5293–5309.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Konvalinka A: myo-Inositol oxygenase: A
novel kidney-specific biomarker of acute kidney injury? Clin Chem.
60:708–710. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang Y, Lu J, Lin B, Chen J, Lin F, Zheng
Q, Xue X, Wei Y, Chen S and Xu N: Integrated analysis of MIOX gene
in prognosis of clear-cell renal cell carcinoma. Cell Death Dis.
16:3682025. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Zhou W, Yu C and Long Y: Myo-inositol
oxygenase (MIOX) accelerated inflammation in the model of
infection-induced cardiac dysfunction by NLRP3 inflammasome. Immun
Inflamm Dis. 11:e8292023. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Vitale I, Pietrocola F, Guilbaud E,
Aaronson SA, Abrams JM, Adam D, Agostini M, Agostinis P, Alnemri
ES, Altucci L, et al: Apoptotic cell death in disease-current
understanding of the NCCD 2023. Cell Death Differ. 30:1097–1154.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Fu Y, Zhang HJ, Zhou W, Lai ZQ and Dong
YF: The protective effects of sophocarpine on sepsis-induced
cardiomyopathy. Eur J Pharmacol. 950:1757452023. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mahidhara R and Billiar TR: Apoptosis in
sepsis. Crit Care Med. 28 (4 Suppl):N105–N113. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Maluleke TT, Manilall A, Shezi N, Baijnath
S and Millen AME: Acute exposure to LPS induces cardiac dysfunction
via the activation of the NLRP3 inflammasome. Sci Rep.
14:243782024. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Speir M and Lawlor KE: RIP-roaring
inflammation: RIPK1 and RIPK3 driven NLRP3 inflammasome activation
and autoinflammatory disease. Semin Cell Dev Biol. 109:114–124.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Zhou Y, Chai Z, Pandeya A, Yang L, Zhang
Y, Zhang G, Wu C, Li Z and Wei Y: Caspase-11 and NLRP3 exacerbate
systemic Klebsiella infection through reducing mitochondrial ROS
production. Front Immunol. 16:15161202025. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Yang Y, Lei W, Qian L, Zhang S, Yang W, Lu
C, Song Y, Liang Z, Deng C, Chen Y, et al: Activation of NR1H3
signaling pathways by psoralidin attenuates septic myocardial
injury. Free Radic Biol Med. 204:8–19. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Qu M, Wang Y, Qiu Z, Zhu S, Guo K, Chen W,
Miao C and Zhang H: Necroptosis, pyroptosis, ferroptosis in sepsis
and treatment. Shock. 57:161–171. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W
and Tang Q: STING-IRF3 contributes to lipopolysaccharide-induced
cardiac dysfunction, inflammation, apoptosis and pyroptosis by
activating NLRP3. Redox Biol. 24:1012152019. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Wang D, Lin Z, Zhou Y, Su M, Zhang H, Yu L
and Li M: Atractylenolide I ameliorates sepsis-induced
cardiomyocyte injury by inhibiting macrophage polarization through
the modulation of the PARP1/NLRP3 signaling pathway. Tissue Cell.
89:1024242024. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Deng C, Liu Q, Zhao H, Qian L, Lei W, Yang
W, Liang Z, Tian Y, Zhang S, Wang C, et al: Activation of NR1H3
attenuates the severity of septic myocardial injury by inhibiting
NLRP3 inflammasome. Bioeng Transl Med. 8:e105172023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
D'Souza CA and Heitman J: Dismantling the
cryptococcus coat. Trends Microbiol. 9:112–113. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Yu P, Zhang X, Liu N, Tang L, Peng C and
Chen X: Pyroptosis: Mechanisms and diseases. Signal Transduct
Target Ther. 6:1282021. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zheng X, Chen W, Gong F, Chen Y and Chen
E: The role and mechanism of pyroptosis and potential therapeutic
targets in sepsis: A review. Front Immunol. 12:7119392021.
View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Coll RC, Schroder K and Pelegrín P: NLRP3
and pyroptosis blockers for treating inflammatory diseases. Trends
Pharmacol Sci. 43:653–668. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Yarovinsky TO, Su M, Chen C, Xiang Y, Tang
WH and Hwa J: Pyroptosis in cardiovascular diseases: Pumping
gasdermin on the fire. Semin Immunol. 69:1018092023. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Fan Y, Guan B, Xu J, Zhang H, Yi L and
Yang Z: Role of toll-like receptor-mediated pyroptosis in
sepsis-induced cardiomyopathy. Biomed Pharmacother. 167:1154932023.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Song C, Zhang Y, Pei Q, Zheng L, Wang M,
Shi Y, Wu S, Ni W, Fu X, Peng Y, et al: HSP70 alleviates
sepsis-induced cardiomyopathy by attenuating mitochondrial
dysfunction-initiated NLRP3 inflammasome-mediated pyroptosis in
cardiomyocytes. Burns Trauma. 10:tkac0432022. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Cao Z, Li W, Shao Z, Liu X, Zeng Y, Lin P,
Lin C, Zhao Y, Li T, Zhao Z, et al: Apelin ameliorates
sepsis-induced myocardial dysfunction via inhibition of
NLRP3-mediated pyroptosis of cardiomyocytes. Heliyon.
10:e245682024. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Duan F, Li L, Liu S, Tao J, Gu Y, Li H, Yi
X, Gong J, You D, Feng Z, et al: Cortistatin protects against
septic cardiomyopathy by inhibiting cardiomyocyte pyroptosis
through the SSTR2-AMPK-NLRP3 pathway. Int Immunopharmacol.
134:1121862024. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Lu C, Liu J, Escames G, Yang Y, Wu X, Liu
Q, Chen J, Song Y, Wang Z, Deng C, et al: PIK3CG regulates
NLRP3/GSDMD-mediated pyroptosis in septic myocardial injury.
Inflammation. 46:2416–2432. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Liu Q, Dong Y, Escames G, Wu X, Ren J,
Yang W, Zhang S, Zhu Y, Tian Y, Acuña-Castroviejo D and Yang Y:
Identification of PIK3CG as a hub in septic myocardial injury using
network pharmacology and weighted gene co-expression network
analysis. Bioeng Transl Med. 8:e103842022. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Liu S, Yao S, Yang H, Liu S and Wang Y:
Autophagy: Regulator of cell death. Cell Death Dis. 14:6482023.
View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Ren C, Zhang H, Wu TT and Yao YM:
Autophagy: A potential therapeutic target for reversing
sepsis-induced immunosuppression. Front Immunol. 8:18322017.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Wang S, Long H, Hou L, Feng B, Ma Z, Wu Y,
Zeng Y, Cai J, Zhang DW and Zhao G: The mitophagy pathway and its
implications in human diseases. Signal Transduct Target Ther.
8:3042023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Shan X, Tao W, Li J, Tao W, Li D, Zhou L,
Yang X, Dong C, Huang S, Chu X and Zhang C: Kai-Xin-San ameliorates
Alzheimer's disease-related neuropathology and cognitive impairment
in APP/PS1 mice via the mitochondrial autophagy-NLRP3 inflammasome
pathway. J Ethnopharmacol. 329:1181452024. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Lei X, Wang J, Zhang F, Tang X, He F,
Cheng S, Zou F and Yan W: Micheliolide ameliorates
lipopolysaccharide-induced acute kidney injury through suppression
of NLRP3 activation by promoting mitophagy via Nrf2/PINK1/Parkin
axis. Int Immunopharmacol. 138:1125272024. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Zhou F, Lian W, Yuan X, Wang Z, Xia C, Yan
Y, Wang W, Tong Z, Cheng Y, Xu J, et al: Cornuside alleviates
cognitive impairments induced by Aβ1-42 through
attenuating NLRP3-mediated neurotoxicity by promoting mitophagy.
Alzheimers Res Ther. 17:472025. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Ajoolabady A, Chiong M, Lavandero S,
Klionsky DJ and Ren J: Mitophagy in cardiovascular diseases:
Molecular mechanisms, pathogenesis, and treatment. Trends Mol Med.
28:836–849. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Zhu L, Wang Z, Sun X, Yu J, Li T, Zhao H,
Ji Y, Peng B and Du M: STAT3/Mitophagy axis coordinates macrophage
NLRP3 inflammasome activation and inflammatory bone loss. J Bone
Miner Res. 38:335–353. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Luo L, Wang F, Xu X, Ma M, Kuang G, Zhang
Y, Wang D, Li W, Zhang N and Zhao K: STAT3 promotes NLRP3
inflammasome activation by mediating NLRP3 mitochondrial
translocation. Exp Mol Med. 56:1980–1990. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Nedel W, Deutschendorf C and Portela LVC:
Sepsis-induced mitochondrial dysfunction: A narrative review. World
J Crit Care Med. 12:139–152. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Jing J, Yang F, Wang K, Cui M, Kong N,
Wang S, Qiao X, Kong F, Zhao D, Ji J, et al: UFMylation of NLRP3
prevents its autophagic degradation and facilitates inflammasome
activation. Adv Sci (Weinh). 12:e24067862025. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Yang S, Huang G and Ting JP: Mitochondria
and NLRP3: To die or inflame. Immunity. 58:5–7. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Meyers AK, Wang Z, Han W, Zhao Q, Zabalawi
M, Duan L, Liu J, Zhang Q, Manne RK, Lorenzo F, et al: Pyruvate
dehydrogenase kinase supports macrophage NLRP3 inflammasome
activation during acute inflammation. Cell Rep. 42:1119412023.
View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Thorburn J, Xu S and Thorburn A: MAP
kinase- and Rho-dependent signals interact to regulate gene
expression but not actin morphology in cardiac muscle cells. EMBO
J. 16:1888–1900. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Tan Y, Zhang Y, He J, Wu F, Wu D, Shi N,
Liu W, Li Z, Liu W, Zhou H and Chen W: Dual specificity phosphatase
1 attenuates inflammation-induced cardiomyopathy by improving
mitophagy and mitochondrial metabolism. Mol Metab. 64:1015672022.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Li FJ, Hu H, Wu L, Luo B, Zhou Y, Ren J,
Lin J, Reiter RJ, Wang S, Dong M, et al: Ablation of mitophagy
receptor FUNDC1 accentuates septic cardiomyopathy through
ACSL4-dependent regulation of ferroptosis and mitochondrial
integrity. Free Radic Biol Med. 225:75–86. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Nie J and Qiu H: DUSP1 mitigates
MSU-induced immune response in gouty arthritis reinforcing
autophagy. Front Biosci (Landmark Ed). 29:2222024. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Jiang H, Chen F, Song D, Zhou X, Ren L and
Zeng M: Dynamin-related protein 1 is involved in mitochondrial
damage, defective mitophagy, and NLRP3 inflammasome activation
induced by MSU crystals. Oxid Med Cell Longev. 2022:50644942022.
View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Chen S, Ma J, Yin P and Liang F: The
landscape of mitophagy in sepsis reveals PHB1 as an NLRP3
inflammasome inhibitor. Front Immunol. 14:11884822023. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Song D, Tao W, Liu F, Wu X, Bi H, Shu J,
Wang D and Li X: Lipopolysaccharide promotes NLRP3 inflammasome
activation by inhibiting TFEB-mediated autophagy in NRK-52E cells.
Mol Immunol. 163:127–135. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Zhang R, Guan S, Meng Z, Zhang D and Lu J:
Ginsenoside Rb1 alleviates 3-MCPD-induced renal cell pyroptosis by
activating mitophagy. Food Chem Toxicol. 186:1145222024. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Hu D, Sheeja Prabhakaran H, Zhang YY, Luo
G, He W and Liou YC: Mitochondrial dysfunction in sepsis:
Mechanisms and therapeutic perspectives. Crit Care. 28:2922024.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Silva RCMC: Mitochondria, autophagy and
inflammation: Interconnected in aging. Cell Biochem Biophys.
82:411–426. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Miaczynska M, Christoforidis S, Giner A,
Shevchenko A, Uttenweiler-Joseph S, Habermann B, Wilm M, Parton RG
and Zerial M: APPL proteins link Rab5 to nuclear signal
transduction via an endosomal compartment. Cell. 116:445–456. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Wu KKL and Cheng KKY: A new role of the
early endosome in restricting NLRP3 inflammasome via mitophagy.
Autophagy. 18:1475–1477. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Yu WM, Appler JM, Kim YH, Nishitani AM,
Holt JR and Goodrich LV: A Gata3-Mafb transcriptional network
directs post-synaptic differentiation in synapses specialized for
hearing. Elife. 2:e013412013. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Cui H, Banerjee S, Xie N, Dey T, Liu RM,
Sanders YY and Liu G: MafB regulates NLRP3 inflammasome activation
by sustaining p62 expression in macrophages. Commun Biol.
6:10472023. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Wang J, Wu M, Magupalli VG, Dahlberg PD,
Wu H and Jensen GJ: Human NLRP3 inflammasome activation leads to
formation of condensate at the microtubule organizing center.
bioRxiv [Preprint]. 2024.09.12.612739. 2024.
|
|
116
|
Chen X, Yuan T, Zheng D, Li F, Xu H, Ye M,
Liu S and Li J: Cardiomyocyte mitochondrial mono-ADP-ribosylation
dictates cardiac tolerance to sepsis by configuring bioenergetic
reserve in male mice. Nat Commun. 16:81192025. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Wan C and Wang Y: Integrated multi-omics
of mitophagy-related molecular subtype characterization and
biomarker identification in sepsis. Sci Rep. 16:7012025. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Mohd S, Sharma V, Harish V, Kumar R and
Pilli G: Exploring thiazolidinedione-naphthalene analogues as
potential antidiabetic agents: Design, synthesis, molecular docking
and in-vitro evaluation. Cell Biochem Biophys. 83:2213–2226. 2025.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Hu C, He X, Zhang H, Hu X, Liao L, Cai M,
Lin Z, Xiang J, Jia X, Lu G, et al: Tanshinone I limits
inflammasome activation of macrophage via docking into Syk to
alleviate DSS-induced colitis in mice. Mol Immunol. 173:88–98.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Liang D, Tang S, Liu L, Zhao M, Ma X, Zhao
Y, Shen C, Liu Q, Tang J, Zeng J and Chen N: Tanshinone I
attenuates gastric precancerous lesions by inhibiting epithelial
mesenchymal transition through the p38/STAT3 pathway. Int
Immunopharmacol. 124:1109022023. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Zhao J, Liu H, Hong Z, Luo W, Mu W, Hou X,
Xu G, Fang Z, Ren L, Liu T, et al: Tanshinone I specifically
suppresses NLRP3 inflammasome activation by disrupting the
association of NLRP3 and ASC. Mol Med. 29:842023. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Dai Y, Zhang X, Xu Y, Wu Y and Yang L: The
protective effects of cinnamyl alcohol against hepatic steatosis,
oxidative and inflammatory stress in nonalcoholic fatty liver
disease induced by childhood obesity. Immunol Invest. 52:1008–1022.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Yoshizaki K, Frias DP, Maier K, Smelan J,
Correia AT, Oliveira LMDS, Amato-Lourenço LF, Santillo BT, Prado
CM, Oshiro TM, et al: Exposure of cinnamyl alcohol in co-culture of
BEAS-2B and dendritic cells: Interaction between CYP450 and
cytokines. J Appl Toxicol. 44:1317–1328. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zou L, Li C, Chen X, Yu F, Huang Q, Chen
L, Wu W and Liu Q: The anti-inflammatory effects of cinnamyl
alcohol on sepsis-induced mice via the NLRP3 inflammasome pathway.
Ann Transl Med. 10:482022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Cheng Z, Huang M, Li W, Hou L, Jin L, Fan
Q, Zhang L, Li C, Zeng L, Yang C, et al: HECTD3 inhibits NLRP3
inflammasome assembly and activation by blocking NLRP3-NEK7
interaction. Cell Death Dis. 15:862024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Guo J, Miao Y, Nie F, Gao F, Li H, Wang Y,
Liu Q, Zhang T, Yang X, Liu L, et al: Zn-Shik-PEG nanoparticles
alleviate inflammation and multi-organ damage in sepsis. J
Nanobiotechnology. 21:4482023. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Ding Y, Liu L, Wu Y, Wang Y and Zhao R:
Optimization of the transdermal delivery system in astilbin
microemulsion with improved stability and anti-psoriatic activity.
Curr Drug Deliv. 20:281–291. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Li C, Huang Y, Wu C, Qiu Y, Zhang L, Xu J,
Zheng J, Zhang X, Li F and Xia D: Astilbin inhibited neutrophil
extracellular traps in gouty arthritis through suppression of
purinergic P2Y6 receptor. Phytomedicine. 130:1557542024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Geng X, Fu Z, Geng G, Chi K, Liu C, Hong
H, Cai G, Chen X and Hong Q: Astilbin improves the therapeutic
effects of mesenchymal stem cells in AKI-CKD mice by regulating
macrophage polarization through PTGS2-mediated pathway. Stem Cell
Res Ther. 15:4272024. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Dou JY, Zhou MJ, Xuan MY, Guo J, Liu SH,
Lian LH, Cui ZY, Nan JX and Wu YL: Astilbin alleviates hepatic
fibrosis through PXR-PINK1/Parkin pathway: A new strategy by
regulating hepatic stellate cells-macrophage crosstalk.
Phytomedicine. 135:1561442024. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Yang D and Zhang QF: The natural source,
physicochemical properties, biological activities and metabolism of
astilbin. Crit Rev Food Sci Nutr. 63:9506–9518. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Fang Z, Wang G, Huang R, Liu C,
Yushanjiang F, Mao T and Li J: Astilbin protects from
sepsis-induced cardiac injury through the NRF2/HO-1 and TLR4/NF-κB
pathway. Phytother Res. 38:1044–1058. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Liu YF, Li WQ, Hu ND, Ai B, Xia HX, Guo X,
Chen Z and Xia H: Brevilin A ameliorates sepsis-induced
cardiomyopathy through inhibiting NLRP3 inflammation. Ann Med Surg
(Lond). 85:5952–5962. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Fang H, Wang Y, Deng J, Zhang H, Wu Q, He
L, Xu J, Shao X, Ouyang X, He Z, et al: Sepsis-induced gut
dysbiosis mediates the susceptibility to sepsis-associated
encephalopathy in mice. mSystems. 7:e01399212022. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Zhuang H, Ren X, Jiang F and Zhou P:
Indole-3-propionic acid alleviates chondrocytes inflammation and
osteoarthritis via the AhR/NF-κB axis. Mol Med. 29:172023.
View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Heumel S, de Rezende Rodovalho V, Urien C,
Specque F, Brito Rodrigues P, Robil C, Delval L, Sencio V, Descat
A, Deruyter L, et al: Shotgun metagenomics and systemic targeted
metabolomics highlight indole-3-propionic acid as a protective gut
microbial metabolite against influenza infection. Gut Microbes.
16:23250672024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Ilha M, Sehgal R, Matilainen J, Rilla K,
Kaminska D, Gandhi S, Männistö V, Ling C, Romeo S, Pajukanta P, et
al: Indole-3-propionic acid promotes hepatic stellate cells
inactivation. J Transl Med. 23:2532025. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Rapti E, Adamantidi T, Efthymiopoulos T,
Kyzas GZ and Tsoupras A: Potential applications of the
anti-inflammatory, antithrombotic and antioxidant health-promoting
properties of curcumin: A critical review. Nutraceuticals.
4:562–595. 2024. View Article : Google Scholar
|
|
139
|
Shi Y, Wu Q, Lu Y, Meng LP, Xu XL, Wang XJ
and Chen W: Arginine-glycine-aspartic acid-anchored curcumin-based
nanotherapeutics inhibit pyroptosis-induced cytokine release
syndrome for in vivo and in vitro sepsis applications. Curr Pharm
Des. 29:283–294. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Karimi A, Pourreza S, Vajdi M, Mahmoodpoor
A, Sanaie S, Karimi M and Tarighat-Esfanjani A: Evaluating the
effects of curcumin nanomicelles on clinical outcome and cellular
immune responses in critically ill sepsis patients: A randomized,
double-blind, and placebo-controlled trial. Front Nutr.
9:10378612022. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Kim D, Kim S, Kang MS, Yin Z and Min B:
Cell type specific IL-27p28 (IL-30) deletion in mice uncovers an
unexpected regulatory function of IL-30 in autoimmune inflammation.
Sci Rep. 13:18122023. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Sung M, Lim S, Park S, Choi Y and Kim S:
Anti-inflammatory effects of phytosphingosine-regulated cytokines
and NF-kB and MAPK mechanism. Cell Mol Biol (Noisy-le-grand).
70:22–30. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Shehata RR, Atta SA, Fatma AS, Aml RA and
Gomaa AS: Association of serum IL-30 and soluble GP130 with the
risk of psoriasis vulgaris. Egypt J Immunol. 31:61–70. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhao M, Zheng Z, Zhang P, Xu Y, Zhang J,
Peng S, Liu J, Pan W, Yin Z, Xu S, et al: IL-30 protects against
sepsis-induced myocardial dysfunction by inhibiting
pro-inflammatory macrophage polarization and pyroptosis. iScience.
26:1075442023. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Arruri VK, Gundu C, Kalvala AK, Sherkhane
B, Khatri DK and Singh SB: Carvacrol abates NLRP3 inflammasome
activation by augmenting Keap1/Nrf-2/p62 directed autophagy and
mitochondrial quality control in neuropathic pain. Nutr Neurosci.
25:1731–1746. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Joshi S, Kundu S, Priya VV, Kulhari U,
Mugale MN and Sahu BD: Anti-inflammatory activity of carvacrol
protects the heart from lipopolysaccharide-induced cardiac
dysfunction by inhibiting pyroptosis via NLRP3/caspase1/gasdermin D
signaling axis. Life Sci. 324:1217432023. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Wu T, Ma W, Lu W, Huangshen Z, Chen S,
Yang Q, Li C, Li Z, Li N, Feng X, et al: Vaccarin alleviates
cisplatin-induced acute kidney injury via decreasing NOX4-derived
ROS. Heliyon. 9:e212312023. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Zhu X, Meng X, Du X, Zhao C, Ma X, Wen Y,
Zhang S, Hou B, Cai W, Du B, et al: Vaccarin suppresses diabetic
nephropathy through inhibiting the EGFR/ERK1/2 signaling pathway.
Acta Biochim Biophys Sin (Shanghai). 56:1860–1874. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Fan Q, Liu D, Chu C, Wang Y, Liu M, Liu Y,
Huang Y, Zhang J and Wen J: Vaccarin alleviates renal
ischemia-reperfusion injury by inhibiting inflammation and
ferroptosis. Int Immunopharmacol. 153:1144632025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Zhu XX, Meng XY, Zhang AY, Zhao CY, Chang
C, Chen TX, Huang YB, Xu JP, Fu X, Cai WW, et al: Vaccarin
alleviates septic cardiomyopathy by potentiating NLRP3
palmitoylation and inactivation. Phytomedicine. 131:1557712024.
View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Liao R, Sun ZC, Wang L, Xian C, Lin R,
Zhuo G, Wang H, Fang Y, Liu Y, Yang R, et al: Inhalable and
bioactive lipid-nanomedicine based on bergapten for targeted acute
lung injury therapy via orchestrating macrophage polarization.
Bioact Mater. 43:406–422. 2024.PubMed/NCBI
|
|
152
|
Jiang Y, Nguyen TV, Jin J, Yu ZN, Song CH
and Chai OH: Bergapten ameliorates combined allergic rhinitis and
asthma syndrome after PM2.5 exposure by balancing Treg/Th17
expression and suppressing STAT3 and MAPK activation in a mouse
model. Biomed Pharmacother. 164:1149592023. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Zhu S, Cheng L, Chen T, Liu X, Zhang C,
Aji A, Guo W, Zhu J, Chu Y, Guo D and Li F: Bergapten ameliorates
psoriatic skin lesions and IL-17A-induced activation of the NF-κB
signaling pathway via the downregulation of CYP1B1. Phytother Res.
39:661–675. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Luo T, Jia X, Feng WD, Wang JY, Xie F,
Kong LD, Wang XJ, Lian R, Liu X, Chu YJ, et al: Bergapten inhibits
NLRP3 inflammasome activation and pyroptosis via promoting
mitophagy. Acta Pharmacol Sin. 44:1867–1878. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Bai D, Du J, Bu X, Cao W, Sun T, Zhao J,
Zhao Y and Lu N: ALDOA maintains NLRP3 inflammasome activation by
controlling AMPK activation. Autophagy. 18:1673–1693. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Honda TSB, Ku J and Anders HJ: Cell
type-specific roles of NLRP3, inflammasome-dependent and
-independent, in host defense, sterile necroinflammation, tissue
repair, and fibrosis. Front Immunol. 14:12142892023. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Yu Y, Yu S, Lu Z, Qiang L, Zhong Y, Ge P,
Lei Z, Qiu C, Fang Y, Zhang X, et al: Pathogenic phosphorylation of
linear ubiquitin machinery causes inflammasome sensor degradation.
Cell Rep. 44:1162862025. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Liang S, Zhou J, Cao C, Liu Y, Ming S, Liu
X, Shang Y, Lao J, Peng Q, Yang J and Wu M: GITR exacerbates
lysophosphatidylcholine-induced macrophage pyroptosis in sepsis via
posttranslational regulation of NLRP3. Cell Mol Immunol.
21:674–688. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Cui LG, Zhai MM, Yin JJ, Wang ZM, Wang SH,
Zhou YJ, Li PP, Wang Y, Xia L, Wang P, et al: Targeting the
ALKBH5-NLRP3 positive feedback loop alleviates cardiomyocyte
pyroptosis after myocardial infarction. Eur J Pharmacol.
989:1772472025. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Chen L, Zhang W, Chen H, Zhang Y, Guo B,
Yang L, Yin C, Zuo Q, Ren L, Bai L, et al: HDAC3 activates
endothelial NLRP3 inflammasome and promotes atherosclerosis via
inhibiting the acetylation of specificity protein 1. Cell Death
Differ. Nov 26–2025.(Epub ahead of print). View Article : Google Scholar
|
|
161
|
Liu S, Wu Z, Su Y and Qiu F: Successful
treatment of sepsis-induced cardiomyopathy with 36 h refractory
ventricular fibrillation: A case report. Heliyon. 10:e350842024.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Borkowski P, Borkowski M, Borkowska N,
Modak V, Nazarenko N, Mangeshkar S, Osabutey A, Maliha M, Chowdhury
I, Batikyan A, et al: The Complexities of sepsis-induced
cardiomyopathy: A clinical case and review of inflammatory pathways
and potential therapeutic targets. Cureus. 16:e751732024.PubMed/NCBI
|
|
163
|
Silva EE, Skon-Hegg C, Badovinac VP and
Griffith TS: The calm after the storm: Implications of sepsis
immunoparalysis on host immunity. J Immunol. 211:711–719. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
164
|
Yu X, Song Y, Dong T, Ouyang W, Shao L,
Quan C, Lee KE, Tan T, Tsung A, Kurabayashi K, et al: Loss of PADI2
and PADI4 ameliorates sepsis-induced acute lung injury by
suppressing NLRP3+ macrophages. JCI Insight. 9:e1816862024.
View Article : Google Scholar : PubMed/NCBI
|
|
165
|
Evans L, Rhodes A, Alhazzani W, Antonelli
M, Coopersmith CM, French C, Machado FR, Mcintyre L, Ostermann M,
Prescott HC, et al: Surviving sepsis campaign: International
guidelines for management of sepsis and septic shock 2021.
Intensive Care Med. 47:1181–1247. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
166
|
Duan Y, Li Q, Wu J, Zhou C, Liu X, Yue J,
Chen X, Liu J, Zhang Q, Zhang Y and Zhang L: A detrimental role of
endothelial S1PR2 in cardiac ischemia-reperfusion injury via
modulating mitochondrial dysfunction, NLRP3 inflammasome
activation, and pyroptosis. Redox Biol. 75:1032442024. View Article : Google Scholar : PubMed/NCBI
|
|
167
|
Liu S, Tan M, Cai J, Li C, Yang M, Sun X
and He B: Ribosome-targeting antibiotic control NLRP3-mediated
inflammation by inhibiting mitochondrial DNA synthesis. Free Radic
Biol Med. 210:75–84. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
168
|
Licini C, Morroni G, Lucarini G, Vitto
VAM, Orlando F, Missiroli S, D'Achille G, Perrone M, Spadoni T,
Graciotti L, et al: ER-mitochondria association negatively affects
wound healing by regulating NLRP3 activation. Cell Death Dis.
15:4072024. View Article : Google Scholar : PubMed/NCBI
|
|
169
|
Zhao PY, Yao RQ, Ren C, Li SY, Li YX, Zhu
SY, Yao YM and Du XH: De ritis ratio as a significant prognostic
factor in patients with sepsis: A retrospective analysis. J Surg
Res. 264:375–385. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
170
|
Vande Walle L and Lamkanfi M: Drugging the
NLRP3 inflammasome: From signalling mechanisms to therapeutic
targets. Nat Rev Drug Discov. 23:43–66. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
171
|
Tang F, Kunder R, Chu T, Hains A, Nguyen
A, McBride JM, Zhong Y, Santagostino S, Wilson M, Trenchak A, et
al: First-in-human phase 1 trial evaluating safety,
pharmacokinetics, and pharmacodynamics of NLRP3 inflammasome
inhibitor, GDC-2394, in healthy volunteers. Clin Transl Sci.
16:1653–1666. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
172
|
Ridker PM, MacFadyen JG, Thuren T, Everett
BM, Libby P and Glynn RJ; CANTOS Trial Group, : Effect of
interleukin-1β inhibition with canakinumab on incident lung cancer
in patients with atherosclerosis: exploratory results from a
randomised, double-blind, placebo-controlled trial. Lancet.
390:1833–1842. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
173
|
Saresella M, Zoia CP, La Rosa F, Bazzini
C, Sala G, Grassenis E, Marventano I, Hernis A, Piancone F, Conti
E, et al: Glibenclamide-loaded engineered nanovectors (GNVs)
modulate autophagy and NLRP3-inflammasome activation.
Pharmaceuticals (Basel). 16:17252023. View Article : Google Scholar : PubMed/NCBI
|