Introduction
As one of the primary pathological bases of heart
failure (HF), a leading cause of morbidity worldwide with an
estimated prevalence of >64 million, pathological cardiac
hypertrophy is characterized by an absolute increase in ventricular
mass in response to various stressors (1). However, the underlying molecular
mechanism of pathological cardiac remodeling remains largely
unknown; therefore further investigation of novel molecular targets
involved in cardiac hypertrophy is needed.
Iron is an important element for various
biosynthetic processes, including oxygen transport, hemoglobin
synthesis and DNA replication (2).
The presence of iron within cells is particularly important for
maintaining cardiac structure and function. Importantly, iron
overload promotes the formation of reactive oxygen species and
causes lipid peroxidation via the Fenton reaction, triggering an
Fe2+-dependent type of regulated cell death termed
‘ferroptosis’ (3). Research has
demonstrated a strong association between oxidative stress and the
development of cardiac hypertrophy (4). Therefore, targeting ferroptosis may
be a potential therapeutic approach for cardiac hypertrophy. We
previously revealed that an iron-overload environment led to
ferroptosis in myocardial infarction injury (5). Our previous study also demonstrated
that the specific inhibitor of ferroptosis, ferrostatin-1 (Fer-1),
could reduce the progression of cardiac hypertrophy in mice
(6). This evidence supports the
involvement of ferroptosis in cardiac hypertrophy progression;
however, the underlying mechanisms remain ambiguous.
Autophagy is an intracellular catabolic process that
is activated in response to nutrient starvation (7). Russell et al (8) reported that serine/threonine protein
kinase ULK1 (ULK1) induced autophagy by phosphorylating Beclin1 and
activating the lipid kinase PI3K catalytic subunit type 3 (VPS34).
The initiation of autophagy is a complicated process orchestrated
by the ULK1 complex, Beclin1/VPS34 complex and autophagy-related
protein (ATG) 8 machinery (9).
Ferroptosis is a type of autophagy-dependent cell death (10). Ferritin comprises ferritin light
chain and ferritin heavy chain 1 (FTH1) and functions as the major
iron storage protein complex. Nuclear receptor coactivator 4
(NCOA4), a selective cargo receptor, facilitates the degradation of
ferritin in lysosomes, in a process known as ferritinophagy
(11). Notably, the AMP-activated
protein kinase (AMPK)/ULK1 axis has been shown to trigger
ferritinophagy in zinc oxide nanoparticle (ZnONP)-induced
ferroptotic endothelial cell death (12). However, it remains to be fully
elucidated whether ferritinophagy is activated and contributes to
ferroptosis in ULK1-induced cardiac hypertrophy.
In the present study, the role of ULK1 was
investigated in cardiomyocyte ferroptosis and the expression
pattern of the Beclin1/VPS34 complex in cardiomyocyte hypertrophy
was examined. The present study also investigated whether
NCOA4-mediated ferritinophagy was involved in ULK1-induced
cardiomyocyte ferroptosis and sought to determine the effect of the
ULK1 inhibitor SBI-0206965 (SBI) on cardiac hypertrophy and
ferroptosis.
Materials and methods
Reagents
Fer-1 (cat. no. S7243) and 3-methyladenine (3-MA;
cat. no. S2767) were purchased from Selleck Chemicals. The ULK1
inhibitor SBI (cat. no. HY-16966) was purchased from
MedChemExpress. Angiotensin II (Ang II; cat. no. 05-23-0101) was
purchased from Sigma-Aldrich (Merck KGaA). The Ang II AT1-receptor
candesartan (CS; cat. no. HY-B0205) was purchased from
MedChemExpress. Wheat germ agglutinin (WGA; cat. no. 25530) was
purchased from AAT Bioquest, Inc. Lipofectamine® 2000
transfection reagent (cat. no. 11668030) was purchased from Thermo
Fisher Scientific, Inc. The malondialdehyde (MDA; cat. no. S0131),
superoxide dismutase (SOD; cat. no. S0101) and Cell Counting Kit-8
(CCK-8; cat. no. C0037) assay kit were purchased from Beyotime
Biotechnology. The ferrous iron (Fe2+; cat. no. E1046)
assay kit was purchased from Applygen Technologies, Inc. The
Meilunbio® fg super sensitive ECL luminescence reagent
(cat. no. MA0186-1) was purchased from Dalian Meilun Biology
Technology Co., Ltd. The monomeric red fluorescent protein
(mRFP)/monomeric Cherry (mCherry)-green fluorescent protein
(GFP)-microtubule-associated protein 1 light chain 3 (LC3)
adenoviral vectors (cat. no. HB-AP210 000) were purchased from
Hanbio Biotechnology Co., Ltd. ULK1 and Beclin1 overexpression
plasmids were manufactured by Shanghai GeneChem Co., Ltd. The small
interfering RNA (siRNA) sequences against mouse ULK1, NCOA4 or
Beclin1 were generated by Shanghai GenePharma Co., Ltd., and the
siRNA sequences are provided in Table
SI. The primary antibodies used for western blotting and
staining are shown in Tables SII
and SIII.
Animal experiments
Male C57BL/6J mice were obtained from the
Experimental Animal Center of Harbin Medical University [Harbin,
China; experimental animal certification no. SCXK(Hei)2024-002].
The animals were kept under a 12-h light/dark cycle at 22°C with a
humidity of 55–60%, with ad libitum access to food and
water. In total, 72 mice were used, among which 36 mice [n=16 for
the sham group; n=20 for the transverse aortic constriction (TAC)
group] were used to detect alterations in ULK1 expression,
BNP and β-MHC mRNA levels, and autophagy levels after
being subjected to TAC. The other 36 mice were divided into four
groups: i) Sham group (n=6); ii) TAC group (n=10); iii) TAC +
adeno-associated virus 9 (AAV9)-siRNAs targeting ULK1 (siULK1)
group: Mice were injected with AAV9 encoding siULK1 via the tail
vein (n=10); and iv) TAC + AAV9-siNC group: Mice were injected with
AAV9-negative control (NC) siRNA via the tail vein (n=10). Mice in
the TAC groups were subjected to the TAC surgery. The humane
endpoints used to determine when the animals should be sacrificed
to minimize suffering were the inability to maintain normal
activities (n=3; after TAC surgery) or to eat on their own (n=1).
During and after the TAC surgery, 4 mice died within 48 h after the
operation, primarily due to acute cardiac failure or complications
from the surgery itself, which is consistent with the reported
mortality rate for this model (13,14).
The remaining 64 mice were sacrificed at the end of the scheduled
experiment to collect the heart tissue.
A mouse model of cardiac hypertrophy was established
using TAC surgery on male C57BL/6J mice aged 8 weeks (6). Briefly, adult male mice (22±4 g) were
induced and maintained under anesthesia using 3 and 2% isoflurane,
respectively. After successful endotracheal intubation, the cannula
was connected to a rodent ventilator (BL420N; Chengdu Techman
Software Co., Ltd.), the chest was opened and the thoracic aorta
was identified. A 7-0 silk suture was placed around the aorta and
tied to the overlying 28G needle, which was subsequently removed.
Mice in the sham operation group were subjected to the same
surgical procedures as the TAC groups but without constriction.
Finally, the chest was closed and the mice were observed during
recovery from surgery. At the pre-determined time point of 3 weeks,
all mice were sacrificed, and heart tissue samples were isolated
for expression analysis.
In vivo AAV9 infection
On the second day after TAC surgery, mice were
injected with AAV9 encoding siULK1 and NC siRNA via the tail vein
(1.0×1012 viral genomes/ml; 100 µl). AAV9 was diluted in
sterile PBS prior to injection. The sham mice underwent the same
protocol but were injected with 100 µl PBS. The TAC mice underwent
the TAC operation receiving an injection of 100 µl PBS. In this
experiment, mice were divided into 4 groups: i) Sham group; ii) TAC
group; iii) TAC + AAV9-siULK1 group, in which TAC mice were
injected with AAV9-siULK1 for 3 weeks; and iv) TAC + AAV9-siNC
group, containing TAC mice injected with aaV9-nc siRNA for 3
weeks.
Echocardiography
Echocardiography was performed using a
high-frequency ultrasound system (Vevo 2100; VisualSonics, Inc.)
with a 30-MHz linear array transducer (MS400; mouse cardiovascular)
for the assessment of cardiac function. Mice were anesthetized
using 3% isoflurane for induction and maintained under anesthesia
with 2% isoflurane, then placed in a supine position on a heating
pad. The left ventricular area was recorded using M-type
echocardiography. The interventricular septal thickness (IVS) and
left ventricular posterior wall thickness (LVPW) were obtained
using the ultrasound system. LV dimensions were measured at
end-diastole (LVEDd) and end-systole (LVEDs). LV fractional
shortening (FS) was calculated as follows: LVFS
(%)=(LVEDd-LVEDs)/LVEDd ×100%. The LV ejection fraction (EF) was
calculated as follows: LVEF
(%)=(LVEDd3-LVEDs3)/LVEDd3 ×100%.
Offline data analysis was performed in a blinded fashion by an
investigator using a LabChart 8 Reader (ADInstruments, Ltd.) and
Vevo2100 software (VisualSonics, Inc.), ensuring objectivity and
precision in the interpretation of the results.
Determination of the cardiac
index
At the pre-determined time point of 3 weeks, mice
were euthanized by exposure to an overdose of inhaled isoflurane.
Animals were placed in an induction chamber with a high
concentration of 5% isoflurane (vol/vol) in oxygen for 2–3 min
until they lost consciousness, as indicated by the cessation of
purposeful movements and loss of righting reflex. After the
complete cessation of breathing to ensure death, mice were
maintained on 3% isoflurane for an additional 5 min. Death was
confirmed through the absence of a heartbeat, as determined by
cardiac palpation, in conjunction with the absence of respiratory
effort and fixed, dilated pupils. No signs of distress were
observed in any of the animals during the procedure. The hearts
were promptly excised and rinsed with ice cold phosphate buffered
saline (PBS) to eliminate any blood clots. All associated
connective tissues and vessels were meticulously removed. The
hearts were dried with filter paper and weighed to obtain the heart
weight (HW). After removing the atrium and right ventricle, the
left ventricular weight (LVW), inclusive of the ventricular septal
weight, was also determined. Subsequently, the ratios of HW to body
weight (BW) and LVW to BW were calculated. The tibial length (TL)
was measured from the edge of the tibial plateau to the medial
malleolus distance on the right hindlimb. The ratio of HW to TL was
calculated and represented an index of cardiac hypertrophy.
Hematoxylin and eosin (H&E)
staining
The hearts of mice were excised, fixed with 4%
paraformaldehyde (PFA) at room temperature for 48 h, dehydrated in
30% sucrose solution and finally embedded in OCT compound
(Tissue-Tek® O.C.T. Compound 4583; Sakura Finetek USA,
Inc.). Subsequently, the blocks were cut into 5 µm sections for
subsequent experiments. The sections were stained with hematoxylin
solution for 1 min, rinsed in running tap water for 5 min, and
subsequently counterstained with alcoholic eosin solution for 1
min. Finally, the stained sections were dehydrated in increasing
concentrations of ethyl alcohol and cleared in xylene for 2 min.
All procedures were conducted at 37°C. The stained heart was imaged
and histological examination of the myocardium was performed using
a standard light microscope (Olympus Corporation).
Wheat germ agglutinin (WGA)
staining
The hearts of mice were excised, embedded in OCT
compound and sectioned at 5 µm. The heart sections were fixed with
4% formaldehyde for 15 min at room temperature and washed twice
with Hank's Buffer with HEPES. The WGA (cat. no. 25530; AAT
Bioquest, Inc.) was diluted to 5 µg/ml in PBS. The cardiac tissues
were subsequently stained with WGA at 37°C for 20 min and images of
WGA staining were captured using a fluorescence microscope (Olympus
Corporation). The cross-sectional areas of cardiomyocytes were
measured using ImageJ software (version 1.52a; National Institutes
of Health).
Prussian blue staining
The hearts of mice were excised, embedded in OCT
compound and sectioned at 5 µm. The heart sections were fixed with
4% paraformaldehyde for 15 min at room temperature and washed twice
with 1% PBS for 3 min each. Prussian blue staining was performed
using a Prussian Blue and Nuclear Fast Red Staining Kit (C0127S;
Beyotime Biotechnology). Briefly, the sections were incubated in
ferrocyanide acid salt solution at 37°C for 1 h, followed by
rinsing under running tap water at 37°C for 5 min. The sections
were then counterstained with eosin at 37°C for 10 min, rinsed with
distilled water and sealed with neutral resin. The stained heart
was imaged using a fluorescence microscope (Olympus
Corporation).
Electron microscopy
Heart tissue samples (1 mm3) were
primarily fixed with 2.5% glutaraldehyde in 0.1 mol/l sodium
cacodylate buffer at 4°C overnight. After being rinsed three times
with the same buffer, the samples were post-fixed with 1% osmium
tetroxide in cacodylate buffer at 4°C for 2 h in the dark. The
samples were then dehydrated through a graded ethanol series (50,
70, 90 and 100%) for 10 min per step. Subsequently, the samples
were infiltrated with a mixture of propylene oxide and epoxy resin
(SPI-Pon 812R; Structure Probe, Inc.) at 37°C for 2 h, followed by
pure resin overnight at 37°C. Finally, the samples were embedded in
fresh epoxy resin and polymerized at 60°C for 48 h. The samples
were sectioned into 50 nm-thick slices using an ultra-microtome
(Leica EM UC7; Leica Microsystems GmbH), mounted on copper grids,
and doubly stained with 3% uranyl acetate at 37°C for 15 min and
lead citrate at 37°C for 5 min prior to observation under a
transmission electron microscope (Hitachi HT7650; Hitachi
High-Technologies Corporation).
Cell culture and treatment
HL-1 cells were purchased from the Cell Bank of the
Chinese Academy of Medical Sciences (Shanghai, China). All the
cells were suspended in Claycomb medium (cat. no. 51800C;
MilliporeSigma) supplemented with 10% fetal bovine serum (cat. no.
A5669801; Gibco; Thermo Fisher Scientific, Inc.), norepinephrine
and L-glutamine. The HL-1 cell line was authenticated by short
tandem repeat profiling prior to the study and was consistent with
the established reference profile. The cells were confirmed to be
free of mycoplasma contamination and of murine origin without
interspecies contamination. All the cells were cultured at 37°C in
a humidified incubator with 5% CO2. The cells were
harvested after reaching >80% confluency and subsequently
subjected to the specified treatment protocol at 37°C. To establish
an in vitro model of cardiac hypertrophy, the cells were
treated with Ang II (100 nmol/l) for 48 h. To assess the role of
ferroptosis, the cells were treated with Fer-1 (1 µmol/l) for 24 h
prior to Ang II (100 nmol/l) treatment. To assess the role of
autophagy, the cells treated with autophagosome formation inhibitor
3-MA (5 mmol/l) for 24 h prior to Ang II (100 nmol/l) treatment. To
assess the role of ULK1, the cells were treated with SBI-0206965
(SBI; 10 µmol/l) for 24 h prior to Ang II (100 nmol/l) treatment.
To observe the effects of Ang II and its receptor on Beclin1/VPS34
expression levels, the cells were treated with CS (10 µmol/l) for
24 h prior to Ang II (100 nmol/l) treatment. Finally, the Claycomb
medium was added to the cells.
Transfection
HL-1 cells were seeded in 6-well plates before
transfection. Overexpression plasmids for ULK1 and Beclin1 were
manufactured by Shanghai GeneChem Co., Ltd. The overexpression
plasmids for ULK1 and Beclin1 were constructed by cloning the
respective mouse cDNAs into the pcDNA3.1(+) vector backbone
(Shanghai GeneChem Co., Ltd.). This vector contains a CMV promoter
for high-level expression and an ampicillin resistance gene for
selection. For plasmid transfection, when the cell density reached
60%, cells were transfected with 2.5 µg plasmid using Opti-MEM™
(cat. no. 31985070; Thermo Fisher Scientific, Inc.) containing 7.5
µl Lipofectamine® 2000 (cat. no. 11668019; Thermo Fisher
Scientific, Inc.) at 37°C for 24 h. The complex was incubated with
the cells for 24 h under standard conditions (37°C with 5%
CO2) to facilitate transfection and DNA expression.
After 24 h of transfection, the efficiency of plasmid transfection
was determined by western blotting.
For siRNA transfection, when the cell density
reached 60%, cells were transfected with 50 nmol/l siRNA using
Opti-MEM™ medium containing 2 µl Lipofectamine® 2000 at
37°C for 24 h. A single scrambled siRNA was used as the NC for all
siRNA transfections. After 24 h of transfection, the medium
containing Lipofectamine® 2000 was replaced with 10%
fetal bovine serum Claycomb medium. The cells were incubated for 24
h under standard conditions (37°C with 5% CO2) before
subsequent experiments. Finally, the efficiency of siRNA knockdown
was determined by western blotting. The siRNA sequences against
mouse ULK1, NCOA4 and Beclin1, and the negative control sequence
were all generated by Shanghai GenePharma Co., Ltd., and the siRNA
target sequences are shown in Table
SI.
Western blot assay
Total proteins from cardiac tissues and HL-1 cell
samples were extracted with RIPA buffer (cat. no. P0013B; Beyotime
Biotechnology). A BCA Protein Assay Kit (cat. no. P0010; Beyotime
Biotechnology) was used to detect the protein concentration.
Briefly, 30 µg of protein extract was resolved through 10–15% gels,
separated by electrophoresis and subsequently transferred to PVDF
membranes (MilliporeSigma). After blocking the non-specific
antigens in 5% skim milk at 37°C for 1 h, the membranes were
incubated with primary antibodies at 4°C overnight. The primary
antibodies used are listed in Table
SII. Subsequently, HRP-conjugated goat anti-rabbit secondary
antibodies (1:2,000; cat. no. ZB2301; OriGene Technologies, Inc.)
were added for incubation at 37°C for 1 h, and ECL luminescence
reagents were used for immunological detection. Finally, the
signals were detected using the GelView 6000Plus (Guangzhou
Biolight Biotechnology Co., Ltd.). The western blot band densities
were semi-quantified using ImageJ software (version 1.52a; National
Institutes of Health). β-actin served as an internal control.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA from heart tissue and HL-1 cells was
isolated using TRIzol® reagent (Invitrogen; Thermo
Fisher Scientific, Inc.). Subsequently, the mRNA concentration was
assessed using a NanoDrop 2000™ (Thermo Fisher Scientific, Inc.).
Total RNA was reverse transcribed to cDNA using the
PrimeScriptTM RT reagent kit (cat. no. RR037A; Takara
Bio, Inc.) according to the manufacturer's instructions. The
temperature protocol consisted of 37°C for 15 min, followed by 85°C
for 5 sec. qPCR was carried out using TB Green® Premix
Ex Taq™ II (cat. no. RR820A; Takara Bio, Inc.) according to the
manufacturer's instructions. The protocol consisted of 95°C for 30
sec, followed by 40 cycles of 95°C for 5 sec, 60°C for 30 sec and
72°C for 1 min. The primer sequences were as follows: Mouse brain
natriuretic peptide (BNP), forward
5′-TATCTCAAGCTGCTTTGGGCA-3′, reverse 5′-AACAACTTCAGTGCGTTACAGC-3′;
mouse β-myosin heavy chain (β-MHC), forward
5′-CCGAGGTGTGCTCTCCAGAA-3′, reverse 5′-GCTTCATCCACGGCCAATTC-3′;
mouse prostaglandin endoperoxide synthase 2 (Ptgs2), forward
5′-CTGCGCCTTTTCAAGGATGG-3′, reverse 5′-GGGGATACACCTCTCCACCA-3′; and
mouse GAPDH, forward 5′-AAGCTCATTTCCTGGTATGACAA-3′, reverse
5′-CTTACTCCTTGGAGGCCATGT-3′. GAPDH expression served as the
internal control. The levels of detected mRNA were calculated using
the 2−ΔΔCq method (15). RT-qPCR was performed on a TM
Real-Time PCR System (Analytik Jena GmbH) to assess the expression
levels of specific genes.
Immunofluorescence assay
HL-1 cells were seeded in 24-well plates. Following
treatment, the cells were fixed with 4% PFA at 37°C for 15 min,
immersed in 0.1% Triton X-100 for 20 min and blocked in blocking
buffer (P0260; Beyotime Biotechnology) for 1 h at room temperature.
The cells were incubated with primary rabbit anti-4-hydroxynonenal
(anti-4-HNE; 1:100) overnight at 4°C before incubation with
tetramethylrhodamine isothiocyanate (TRITC)-conjugated IgG (H + L)
secondary antibody (1:100; cat. no. AS040; ABclonal Biotech Co.,
Ltd.) at 37°C in the dark for 1 h. The nuclei were stained with
DAPI (1:1,000; cat. no. C1002; Beyotime Biotechnology) at 37°C for
8 min. The colocalization of LC3β (LC3-II) and NCOA4 proteins was
assessed using a fluorescence microscope (Olympus Corporation). The
primary antibodies used for staining are shown in Table SIII.
Measurement of the cell surface
area
The surface area of α-SMA-stained HL-1 cells was
measured after employing Ang II hypertrophic stimuli. HL-1 cells in
20–40 fields were examined in each experiment. Briefly, the cells
were fixed with 4% PFA at 37°C for 15 min, immersed in 0.1% Triton
X-100 for 20 min and blocked in blocking buffer (P0260; Beyotime
Biotechnology) for 1 h at room temperature. Subsequently, the cells
were incubated with anti-α-SMA antibody (1:100; BM4172; Wuhan
Boster Biological Technology, Ltd.) overnight at 4°C before
incubation with TRITC-conjugated IgG (H + L) secondary antibody
(1:100; cat. no. AS040; ABclonal Biotech Co., Ltd.) at room
temperature in the dark for 1 h. The nuclei were stained with DAPI
(1:1,000; C1002; Beyotime Biotechnology) at 37°C for 10 min. Images
were captured using a fluorescence microscope (Olympus
Corporation). The cell surface areas of cardiomyocytes were
measured using ImageJ software (version 1.52a; National Institutes
of Health).
mRFP-GFP-LC3 puncta formation
assays
HL-1 cells were seeded in 24-well plates at a
density of 2×105 cells/well. mRFP-GFP-LC3 adenoviral
vectors were specifically designed to assess the levels of
autophagy flow. The GFP fluorescence signal was quenched under the
decreased pH value condition (pH <5), while the mRFP and mCherry
fluorescence did not change. If green fluorescence and red
fluorescence were colocalized in cells, the autophagosomes were
shown as yellow puncta, known as GFP+/RFP+,
while the autolysosomes were shown as red puncta, demonstrated by
GFP−/RFP+. According to the manufacturer's
instructions, the cells were transfected with an mRFP-GFP-LC3
expression plasmid for 24 h, and the fluorescence intensity was
detected with a fluorescence microscope (Olympus Corporation).
Evaluation of the levels of
Fe2+ content, MDA content and SOD activity
The levels of Fe2+, MDA and SOD in HL-1
cells were determined using their respective assay kits according
to the manufacturer's instructions.
CCK-8 assay
HL-1 cells were seeded into 96-well plates at a
density of 7,000-9,000 cells per well, and the cell viability was
evaluated using a CCK-8 assay kit (C0043; Beyotime Biotechnology).
The cells were treated with 20 µl CCK-8 solution added directly
into the medium (200 µl per well) and incubated at 37°C for 3 h.
The absorbance was measured at 450 nm using an iMarkTM
Microplate Absorbance Reader (1681130; Bio-Rad Laboratories,
Inc.).
Statistical analysis
Each experiment included three replicates. Data are
presented as the mean ± SEM. GraphPad Prism 9.0 (Dotmatics) was
used for data analysis. An unpaired Student's t-test was used for
comparisons between two groups, and differences among groups were
assessed by one-way analysis of variance (ANOVA), followed by
Tukey's multiple comparison post-hoc test. P<0.05 was considered
to indicate a statistically significant difference.
Results
ULK1 expression is elevated in
hypertrophic cardiac tissues and cardiomyocytes
To investigate the role of ULK1 in cardiac
hypertrophy, its expression was first assessed in cardiac tissues
and cardiomyocytes. After TAC surgery, morphometric and
echocardiographic parameters showed an enlarged heart size and
structural abnormalities compared with the sham mice (Fig. 1A). Specifically, the HW/BW ratio in
the TAC group was 20% higher compared with that in the sham group
(Fig. 1B). The LVW/BW ratio in the
TAC group was 37% higher (Fig. 1C)
and the HW/TL ratio in the TAC group was 30% higher compared with
that in the sham group (Fig. 1D).
The TAC mice also showed significantly lower LVEF and FS values,
and significantly higher IVS and LVPW compared with sham mice
(Fig. 1E-H), indicating that TAC
surgery caused more severe impairment of cardiac function.
Furthermore, WGA staining demonstrated that the difference in
cardiomyocyte cross-sectional area was significant, being 80%
higher in the TAC group compared with the sham group (Fig. 1I). Based on these observations, it
can be concluded that TAC surgery induced cardiac hypertrophy and
dysfunction. Accordingly, the mRNA levels of the hypertrophic
biomarkers BNP and β-MHC were also significantly
elevated in the TAC group (Fig. 1J and
K). Additionally, exposure of HL-1 cells to Ang II
significantly increased BNP and β-MHC mRNA expression
(Fig. 1L and M) and significantly
enlarged cell surface areas (Fig. 1N
and O). These results indicated successful establishment of
in vivo and in vitro models.
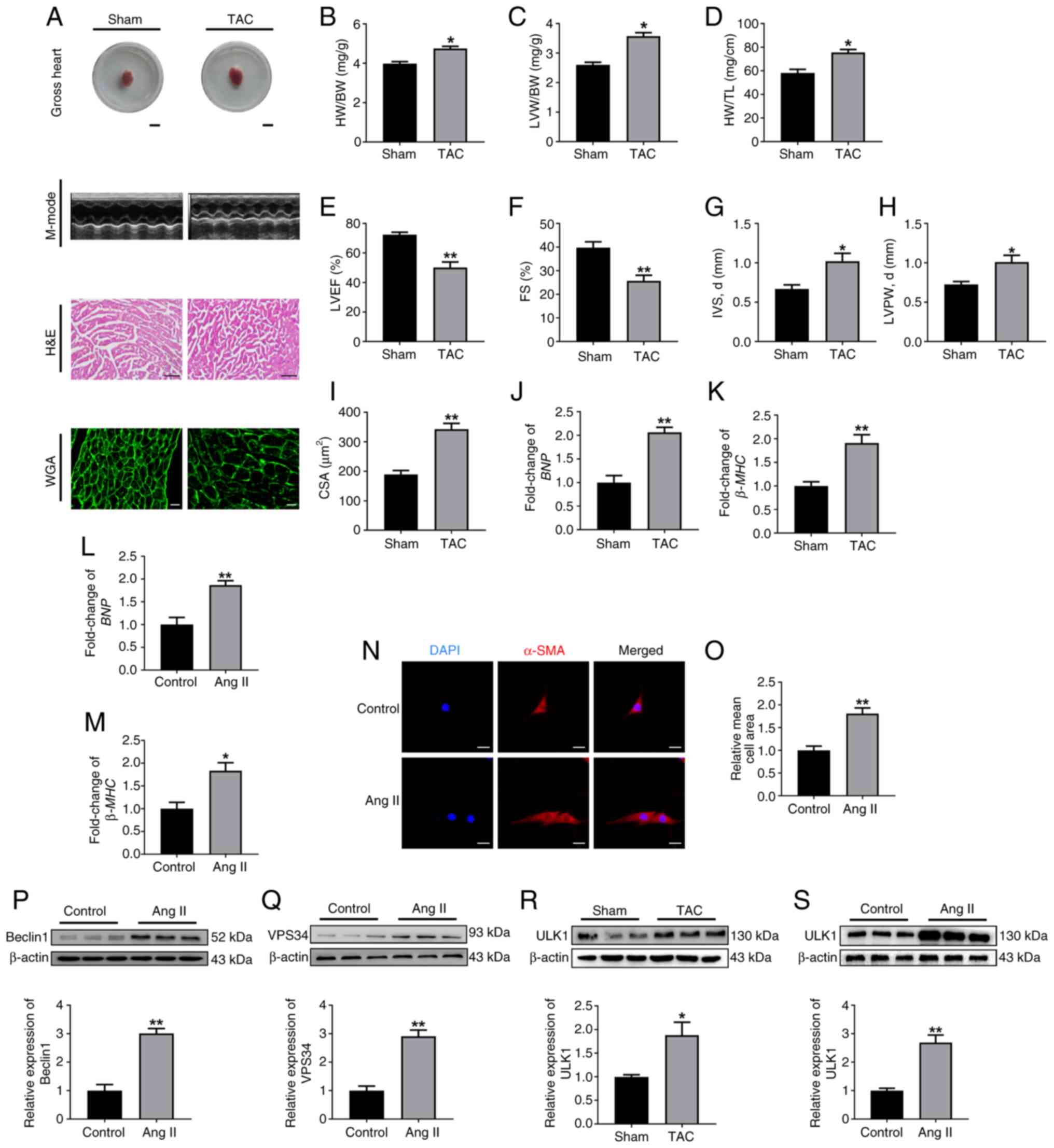 | Figure 1.ULK1 expression is elevated in
hypertrophic cardiac tissues and cardiomyocytes. (A) C57BL/6J mice
at 8-weeks-old were subjected to sham or TAC surgery.
Representative whole heart images (scale bar, 0.5 cm), M-mode
echocardiography, H&E staining (scale bar, 100 µm) and WGA
staining (scale bar, 50 µm) of heart tissue. Assessment of (B)
HW/BW, (C) LVW/BW and (D) HW/TL (n=6). Statistical graphs for (E)
LVEF (%), (F) FS (%), (G) IVS, d (mm) and (H) LVPW, d (mm) (n=6).
(I) Cardiomyocyte cross-sectional area acquired from WGA staining.
Cells were measured from different microscopic fields of 6 samples
in each group. The mRNA levels of (J) BNP and (K)
β-MHC in the hearts from sham or TAC-surgery mice (n=6). The
mRNA levels of (L) BNP and (M) β-MHC in HL-1 cells
treated with Ang II (n=3). (N) Stained cells and (O) quantification
of cell surface area of HL-1 cells treated with Ang II. Scale bar,
50 µm (n=3). Western blot analysis and summarized data
demonstrating (P) Beclin1 and (Q) VPS34 protein expression in HL-1
cells treated with Ang II (n=3). (R) Western blot analysis and
summarized data demonstrating ULK1 protein levels in the hearts
obtained after sham or TAC surgery (n=6). (S) Western blot analysis
and summarized data demonstrating ULK1 protein levels in the
cardiomyocytes stimulated with Ang II (n=6). All data are presented
as mean ± SEM. *P<0.05 and **P<0.01 vs. sham or
control group. ULK1, serine/threonine protein kinase ULK1; TAC,
transverse aortic constriction; HW, heart weight; BW, body weight;
LVW, left ventricular weight; TL, tibia length; LVEF, left
ventricular ejection fraction; FS, fractional shortening; d,
diastole; IVS, interventricular septal thickness; LVPW, left
ventricular posterior wall thickness; CSA, cross-sectional area;
BNP, brain natriuretic peptide; β-MHC, β-myosin heavy
chain; α-SMA, α smooth muscle actin; Ang II, angiotensin II; VPS34,
PI3K catalytic subunit type 3; WGA, wheat germ agglutinin. |
To delve deeper into the signaling pathways
involved, Beclin1 and VPS34 protein expression was assessed in
vitro. The results showed that Ang II augmented the levels of
Beclin1/VPS34 complex in cells (Fig.
1P and Q). Meanwhile, the present study also investigated the
effects of CS on Beclin1/VPS34 activity in vitro. CS, an Ang
II type 1 receptor blocker, is widely used as used as the
first-line drug treatment for hypertension (16,17).
Previous studies have indicated that CS has additional beneficial
effects in diabetes, stroke, dementia and atrial fibrillation
(18). Lebeche et al
(19) have shown that CS abrogates
G protein-coupled receptor agonist-induced MAPK activation and
cardiac myocyte hypertrophy. Subsequently, the effect of CS on
Beclin1/VPS34 activity was investigated in vitro. Firstly,
cells were treated with CS to obverse cell viability for selecting
proper dosage of CS. The results showed that CS prominently
inhibited cell viability in the doses of 40 µmol/l and 80 µmol/l,
and showed no significant influence in other doses by 24 h
treatment compared with the control (0 µmol/l CS) (Fig. S1A). Therefore, the CS
concentrations of 2.5, 5 and 10 µmol/l were chosen for the next
experiment. Cardiomyocytes were pretreated with 100 nmol/l Ang II
prior to administration of different concentrations of CS (2.5, 5
and 10 µmol/l), and notably 10 µmol/l CS treatment significantly
increased cell viability by ~25% (Fig. S1B and C). Subsequently, the
present study further investigated the effects of CS on
Beclin1/VPS34 protein expression (Fig. S1D and E). The results suggested
that the Beclin1/VPS34 complex was upregulated in Ang II-induced
cardiomyocyte hypertrophy and that CS treatment significantly
improved cell viability, decreasing the protein levels of Beclin1
and VPS34.
The present study subsequently assessed the
involvement of ULK1 in cardiac hypertrophy. As shown in Fig. 1R and S, ULK1 expression was
elevated in hypertrophic myocardial tissues and cardiomyocytes.
Collectively, these results suggest that ULK1 may be implicated in
the progression of cardiac hypertrophy.
ULK1 induces ferroptosis in HL-1
cells
To examine the potential role of ULK1 in cardiac
hypertrophy, gain- and loss-of-function studies were performed in
HL-1 cells. The efficiency of ULK1 knockdown is shown in Fig. S2A and the ULK1 overexpression
plasmid efficacy is shown in Fig.
S2F-H. Ang II induced a significant increase in cell surface
area and BNP and β-MHC mRNA levels, both of which
were significantly diminished by ULK1 knockdown in cardiomyocytes
(Fig. S2B-E). Consistently,
overexpression of ULK1 via plasmid transfection in cardiomyocytes
independently elevated the expression of BNP and
β-MHC mRNA and significantly enlarged the surface area of
transfected cells (Fig. S2I-L).
Consistent with the in vivo studies, these data indicated
that ULK1 played a role in stress-induced cardiac hypertrophy.
Previous studies have demonstrated that the
AMPK/ULK1/mTOR axis serves a key role in autophagy and ferroptosis
(20). Therefore, the present
study evaluated the role of ferroptosis in ULK1-knockdown cells by
measuring a number of ferroptotic markers. The results showed that
ULK1 knockdown significantly suppressed cardiomyocyte cell death
compared with the Ang II treatment group (Fig. S3A). As abnormal iron metabolism
can trigger ferroptosis (3), the
present study further tested the levels of Fe2+ in
cardiomyocytes. After 24 h ULK1 knockdown, the levels of
Fe2+ were recovered, which indicated that silencing ULK1
significantly reduced the Ang II-mediated intracellular excess of
Fe2+ (Fig. S3B).
Unlimited lipid peroxidation is a distinguishing feature of
ferroptosis, which leads to the production of active aldehydes such
as MDA and 4-HNE (21). ULK1
knockdown significantly reduced the levels of MDA and markedly
reduced 4-HNE expression (Fig. S3C
and F). Silencing ULK1 also significantly reduced the mRNA
levels of Ptgs2 (Fig.
S3E), which serves as a well-accepted marker of ferroptosis.
Furthermore, the Ang II-induced decrease in SOD was significantly
recovered by ULK1 knockdown (Fig.
S3D). These findings support the hypothesis that ULK1 knockdown
counteracts Ang II-induced ferroptosis in cells.
Subsequently, the ULK1 plasmid was overexpressed in
HL-1 cells. As a result, the viability of ULK1-overexpressing cells
was decreased compared with the control group (Fig. S3G); the levels of Fe2+,
MDA and Ptgs2 mRNA were significantly increased, which was
reflected by a notable increase in 4-HNE, while SOD activity was
significantly inhibited (Fig.
S3H-L). These findings demonstrate that ULK1 overexpression
induces ferroptosis in HL-1 cells.
To ascertain the involvement of ferroptosis in
ULK1-induced cardiomyocyte injury, HL-1 cells were treated with or
without the ferroptosis inhibitor Fer-1 after ULK1 overexpression.
Compared with the ULK1 group, Fer-1 treatment significantly
counteracted increased hypertrophic gene expression and cell
surface area (Fig. 2A-D). Compared
with the control group, ULK1 overexpression induced cellular
damage, and Fer-1 treatment relieved cellular injury caused by ULK1
stimulation (Fig. 2E). The results
of the present study also showed that ULK1 overexpression
significantly induced ferroptosis in cells, which was alleviated by
Fer-1 (Fig. 2F-J). Overall, these
findings demonstrate that Fer-1 reverses ferroptosis in
ULK1-overexpressing cells.
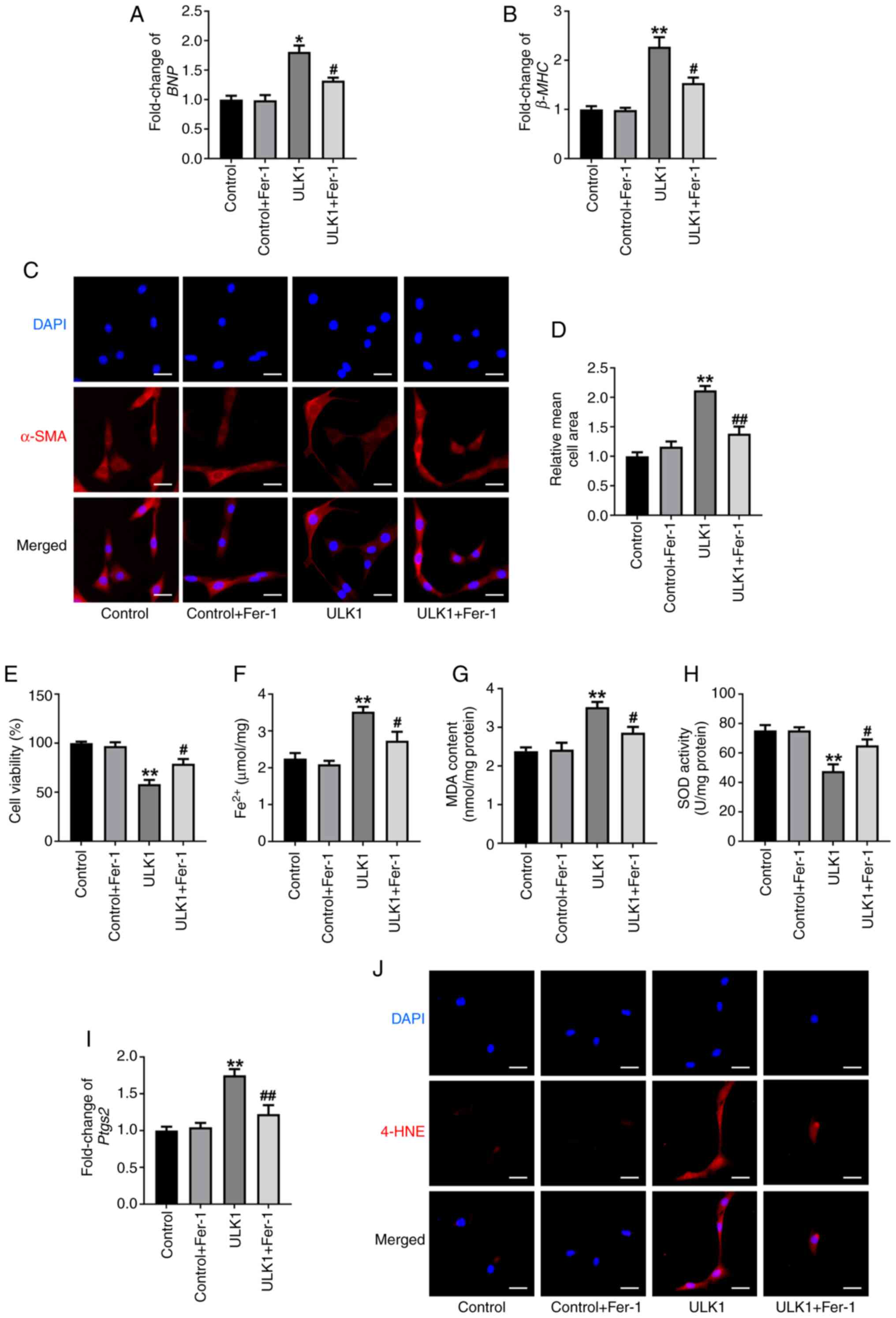 | Figure 2.Fer-1 reverses ferroptosis in
ULK1-overexpressing cells. The mRNA levels of (A) BNP and
(B) β-MHC in HL-1 cells. (C) Images of stained cells and (D)
quantification of cell surface area under the indicated
experimental group. Scale bar, 50 µm. (E) Cell viability of HL-1
cells was assessed using a Cell Counting Kit-8 assay. The levels of
(F) Fe2+, (G) MDA and (H) SOD were determined in the
indicated experimental group. (I) The mRNA levels of Ptgs2.
(J) Immunofluorescence staining for 4-HNE. Scale bar, 50 µm. All
data are presented as mean ± SEM. n=3. *P<0.05 and **P<0.01
vs. control group; #P<0.05 and ##P<0.01
vs. ULK1 group. ULK1, serine/threonine protein kinase ULK1; Fer-1,
ferrostatin-1; BNP, brain natriuretic peptide; β-MHC,
β-myosin heavy chain; α-SMA, α smooth muscle actin; MDA,
malondialdehyde; SOD, superoxide dismutase; Ptgs2,
prostaglandin endoperoxide synthase 2; 4-HNE, 4-hydroxynonenal. |
Autophagy activation is required for
ULK1-induced ferroptosis in HL-1 cells
Subsequently, the autophagy levels of the
hypertrophy models in the present study were investigated. In
vivo, TEM revealed that most mitochondrial membranes exhibited
integrity damage, with loss of mitochondrial cristae structures in
the TAC group compared with the sham group (Fig. S4A). In vitro, LC3
adenovirus infection experiments demonstrated increased autophagic
activity after Ang II stimulation (Fig. S4B). These data indicated that
autophagy was activated in cardiac hypertrophy.
As previous studies have demonstrated that
ferroptosis is dependent on autophagy (22), the present study subsequently
investigated whether autophagy was activated during ULK1-induced
cardiomyocyte ferroptosis. As expected, western blot analysis
showed that, compared with the Ang II treatment group, ULK1
knockdown significantly decreased the protein levels of Beclin1 and
LC3-II, markers of autophagy, and significantly increased that of
the autophagic substrate p62 (Fig.
S5A-C). HL-1 cells were then transfected with mRFP-GFP-LC3
adenoviruses and autophagic flux was observed. The significantly
increased number of autophagosomes and autolysosomes induced by Ang
II stimulation was attenuated after ULK1 knockdown (Fig. S5D and E). By contrast, ULK1
overexpression significantly increased Beclin1 and LC3-II protein
levels, decreased p62 protein levels (Fig. S5F-H) and increased autophagic flux
in cells (Fig. S5I and J). These
results indicated that ULK1 triggered autophagy in HL-1 cells.
Compared with the ULK1 overexpression group,
pretreatment of cells with the autophagy inhibitor 3-MA caused a
significant decrease in hypertrophic markers and cell surface area
following ULK1 treatment (Fig.
S6A-D), suggesting that this autophagy inhibition reduced
ULK1-induced cardiomyocyte hypertrophy. Notably, compared with the
ULK1 overexpression group, Beclin1 and LC3-II expression was
significantly decreased in ULK1 cells when treated with 3-MA
(Fig. 3A and B); however, when
ULK1 overexpressing cells were co-treated with 3-MA, a significant
increase in p62 was observed compared with the ULK1 group (Fig. 3C). Meanwhile, the number of
autophagosomes and autolysosomes were significantly reduced when
cells were co-treated with 3-MA following ULK1 overexpression
compared with the untreated ULK1 group cells (Fig. 3D and E).
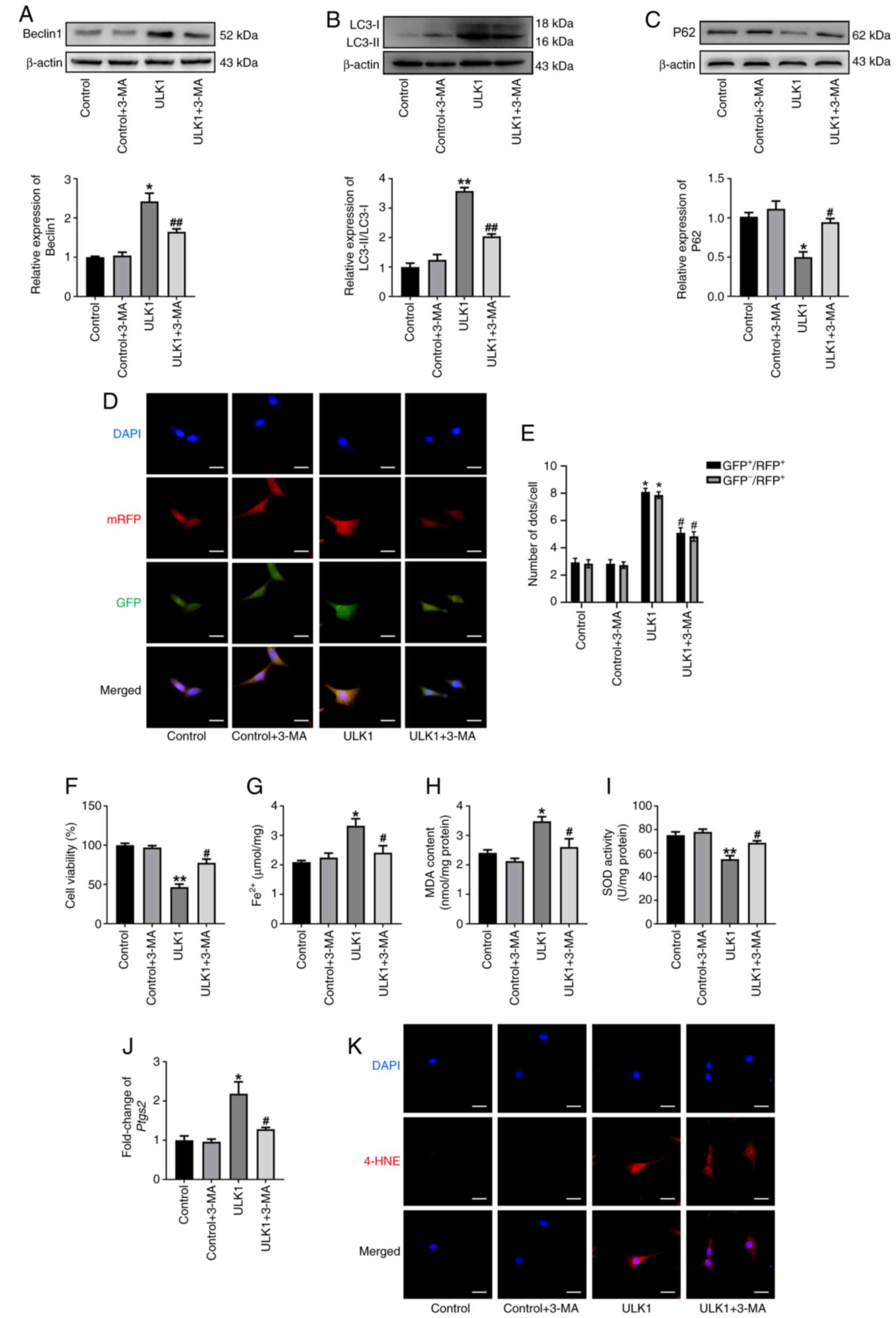 | Figure 3.Autophagy activation is required for
ULK1-induced ferroptosis in HL-1 cells. Western blot analysis and
summarized data demonstrating (A) Beclin1, (B) LC3-II and (C) p62
protein levels in HL-1 cells treated with 3-MA. (D) HL-1 cells were
infected with mRFP-GFP-LC3-labeled adenoviruses and the formation
of autophagosomes (yellow) and autolysosomes (red) were observed in
indicated group. Scale bar, 50 µm. (E) Red and yellow dots/cell in
indicated group. (F) Cell viability of HL-1 cells was detected
using a Cell Counting Kit-8 assay in the 3-MA group. The contents
of (G) Fe2+, (H) MDA and (I) SOD were determined in 3-MA
group. (J) The mRNA level of Ptgs2. (K) Immunofluorescence
staining for 4-HNE in 3-MA group. Scale bar, 50 µm. All data are
presented as mean ± SEM. n=3. *P<0.05 and **P<0.01 vs.
control group; #P<0.05 and ##P<0.01 vs.
ULK1 group. ULK1, serine/threonine protein kinase ULK1; 3-MA,
3-methyladenine; LC3, microtubule-associated protein 1 light chain
3; mRFP, monomeric red fluorescent protein; MDA, malondialdehyde;
SOD, superoxide dismutase; Ptgs2, prostaglandin endoperoxide
synthase 2; 4-HNE, 4-hydroxynonenal. |
The present study then evaluated the relationship
between autophagy and ULK1-induced ferroptosis. Compared with ULK1
overexpression alone, pretreatment with 3-MA significantly
increased cell viability (Fig. 3F)
and blunted ULK1-induced ferroptotic events, as evidenced by
significantly decreased Fe2+ overload (Fig. 3G), MDA production (Fig. 3H), SOD depletion (Fig. 3I), Ptgs2 mRNA (Fig. 3J) and markedly reduced 4-HNE
production (Fig. 3K).
Collectively, these data indicate that activation of autophagy was
required for ULK1-induced ferroptosis of HL-1 cells.
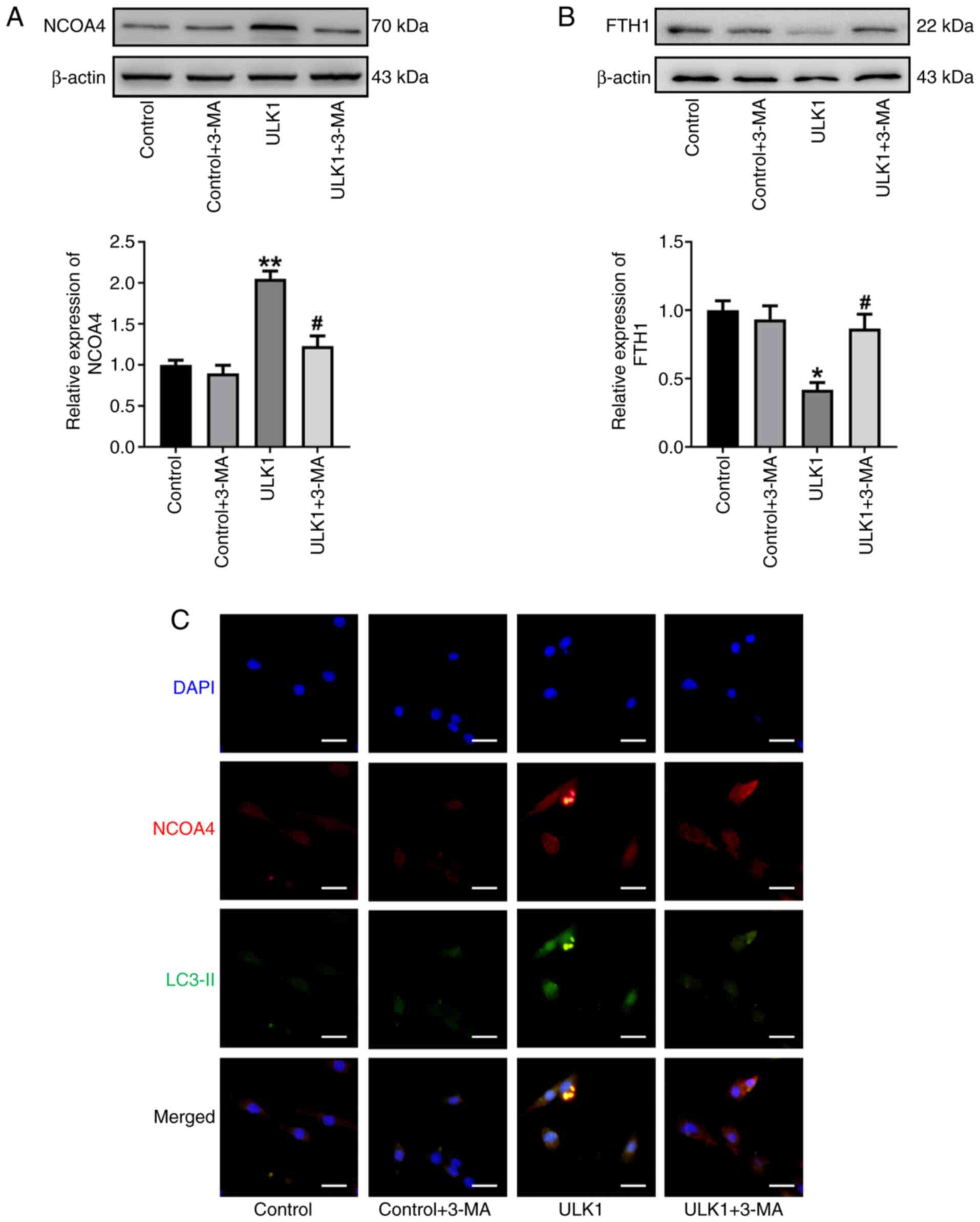
NCOA4-mediated ferritinophagy
contributes to the ferroptosis of HL-1 cells stimulated by
ULK1
NCOA4 is a selective cargo receptor of
ferritinophagy, which is the process of autophagic degradation of
ferritin and subsequent ferroptosis (23). The present study sought to verify
whether NCOA4-mediated ferritinophagy was associated with
ULK1-induced ferroptosis in cells. The western blotting results
revealed that ULK1 knockdown significantly decreased the NCOA4
protein level in Ang II-treated cells, but significantly increased
the FTH1 protein level (Fig. S7A and
B). Furthermore, ferritinophagy was assessed by colocalization
of NCOA4 and LC3-II. Immunofluorescence analysis showed that
silencing ULK1 decreased the colocalization of NCOA4 and LC3-II
compared with the Ang II group (Fig.
S7C). Contrary to the observations of ULK1 silencing
experiments, the levels of ferritinophagy were significantly
aggravated in the ULK1 overexpression group (Fig. S7D and E). The colocalization of
NCOA4 and LC3-II was notably increased after ULK1 overexpression
(Fig. S7F). These data indicated
that NCOA4-mediated ferritinophagy was induced by ULK1
treatment.
To further investigate the role of autophagy in
mediating ferritin degradation, ULK1-overexpressing HL-1 cells were
treated with 3-MA. As shown in Fig. 4A
and B, inhibition of autophagy with 3-MA significantly
decreased NCOA4 expression levels and inhibited FTH1 degradation
compared with untreated ULK1-overexpressing cells. Notably,
immunofluorescence analysis also showed that 3-MA treatment in the
ULK1 group markedly decreased the colocalization of NCOA4 and
LC3-II (Fig. 4C). These results
indicated that this autophagy inhibition significantly reduced
NCOA4-mediated ferritinophagy induced by ULK1 exposure.
Additionally, a rescue experiment was performed to
provide evidence for the important role of ferritinophagy in
ULK1-induced ferroptosis in cells. The efficiency of siRNA-mediated
NCOA4 knockdown was shown in Fig.
S8A. Primarily, NCOA4 knockdown significantly relieved the
hypertrophic events induced by ULK1 overexpression (Fig. S8B-E). As expected, NCOA4 knockdown
significantly alleviated ULK1-induced cell death, iron
accumulation, MDA upregulation, SOD depletion, Ptgs2 mRNA
upregulation and markedly reduced 4-HNE levels (Fig. 5A-F). As shown in Fig. 5G and H, the present study observed
a significant decrease in FTH1 protein in ULK1-induced
cardiomyocytes, which was significantly mitigated by NCOA4
knockdown. These results indicated that NCOA4-mediated
ferritinophagy contributed to the ferroptosis of HL-1 cells
stimulated by ULK1.
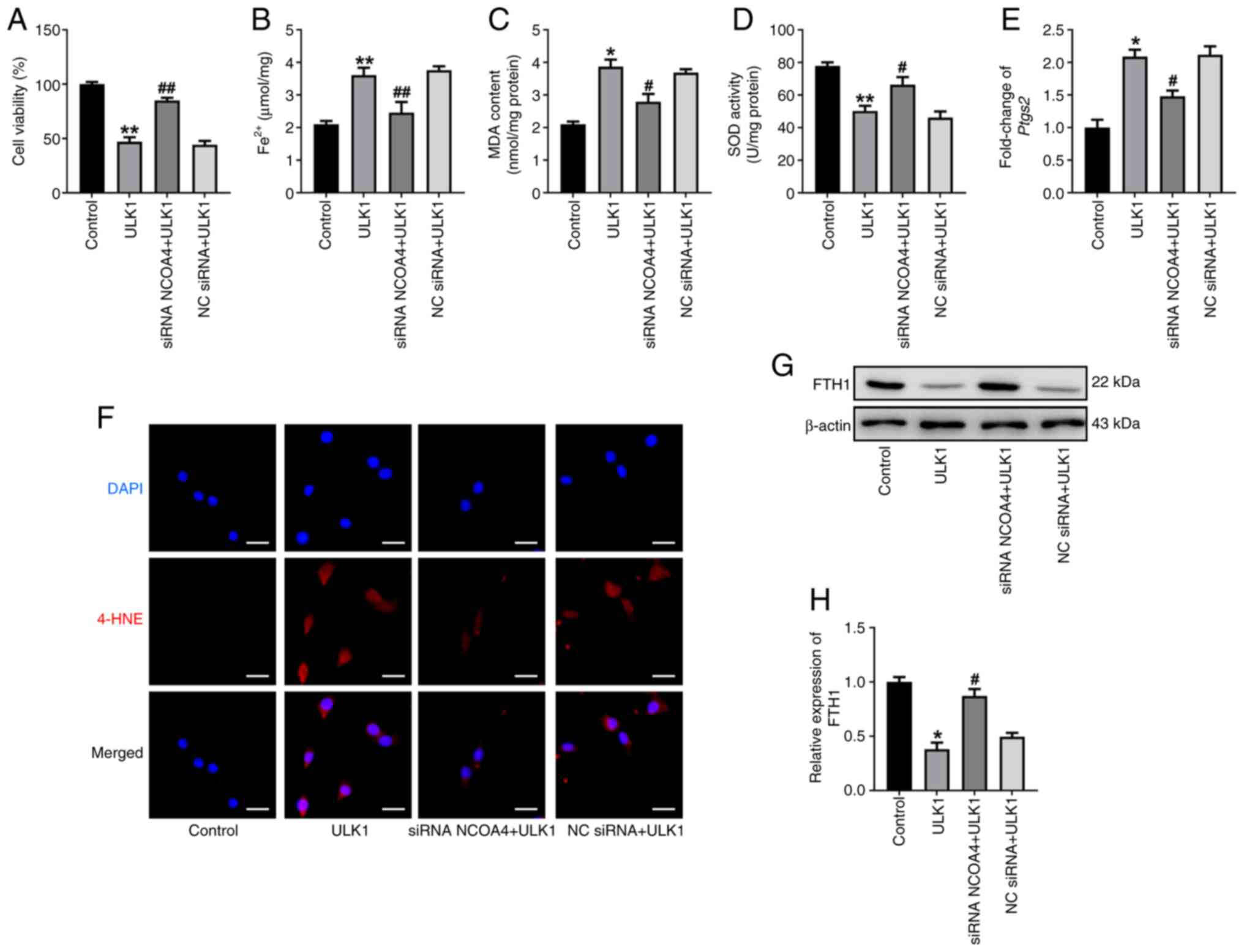 | Figure 5.NCOA4 knockdown alleviates
ULK1-induced ferroptosis events. (A) Cell viability of HL-1 cells
was assessed using a Cell Counting Kit-8 assay in indicated group.
The levels of (B) Fe2+, (C) MDA and (D) SOD were
determined in each indicated group. (E) The mRNA levels of
Ptgs2. (F) Immunofluorescence staining for 4-HNE in
indicated group. Scale bar, 50 µm. (G) Western blot analysis and
(H) summarized data demonstrating FTH1 protein levels in the
indicated groups. All data are presented as mean ± SEM. n=3.
*P<0.05 and **P<0.01 vs. control group; #P<0.05
and ##P<0.01 vs. ULK1 group. ULK1, serine/threonine
protein kinase ULK1; NCOA4, nuclear receptor coactivator 4; MDA,
malondialdehyde; SOD, superoxide dismutase; siRNA, small
interfering RNA; NC, negative control; Ptgs2, prostaglandin
endoperoxide synthase 2; 4-HNE, 4-hydroxynonenal; FTH1, ferritin
heavy chain 1. |
ULK1 activates ferritinophagy
dependent on Beclin1/VPS34 complex in cardiomyocyte
hypertrophy
The present study aimed to identify the regulator of
ferritinophagy activation in response to ULK1-induced cardiomyocyte
hypertrophy. Russell et al (8) found that ULK1 induced autophagy by
phosphorylating Beclin1 and activating VPS34 lipid kinase. The
efficiencies of Beclin1 knockdown and the Beclin1 overexpression
plasmid are shown in Fig. S9A and
F. Beclin1 knockdown significantly decreased the cell surface
area and expression of hypertrophic markers compared with the Ang
II group (Fig. S9B-E), whereas
Beclin1 overexpression significantly induced cardiomyocyte
hypertrophy compared with the control group (Fig. S9G-J). The present study also
determined the ferroptosis levels of Ang II-treated cells in
response to Beclin1 transfections. In response to silencing
Beclin1, ferroptotic events in Ang II-treated cells were
significantly reduced (Fig.
S10A-F); however, the cells showed significant increases in
ferroptotic markers Fe2+, MDA and Ptsg2 following
Beclin1 overexpression, as well as a marked increase in 4-HNE
expression and significant decreases in cell viability and SOD
activity (Fig. S10G-L). These
results indicated that the activation of ferroptosis was involved
in Beclin1-induced cardiomyocyte hypertrophy.
The present study also investigated the role of ULK1
in Beclin1-dependent ferroptosis in cells. Following silencing
Beclin1, ULK1-induced cellular injury and ferroptosis of cells were
significantly inhibited when compared with the ULK1 group (Fig. 6A-F). To further investigate
ferritinophagy in cells, the levels of NCOA4 and FTH1 were
determined by western blotting. Contrasting the significantly
increased expression of FTH1, the levels of NCOA4 were
significantly decreased after Beclin1 knockdown and ULK1
overexpression compared with ULK1 overexpression alone (Fig. 6G and H). Furthermore, the
colocalization between NCOA4 and LC3-II was notably weakened in the
group with silenced Beclin1 and ULK1 overexpression compared with
ULK1 overexpression alone (Fig.
6I). These data indicated that ULK1 activated ferritinophagy,
which was dependent on the Beclin1/VPS34 complex.
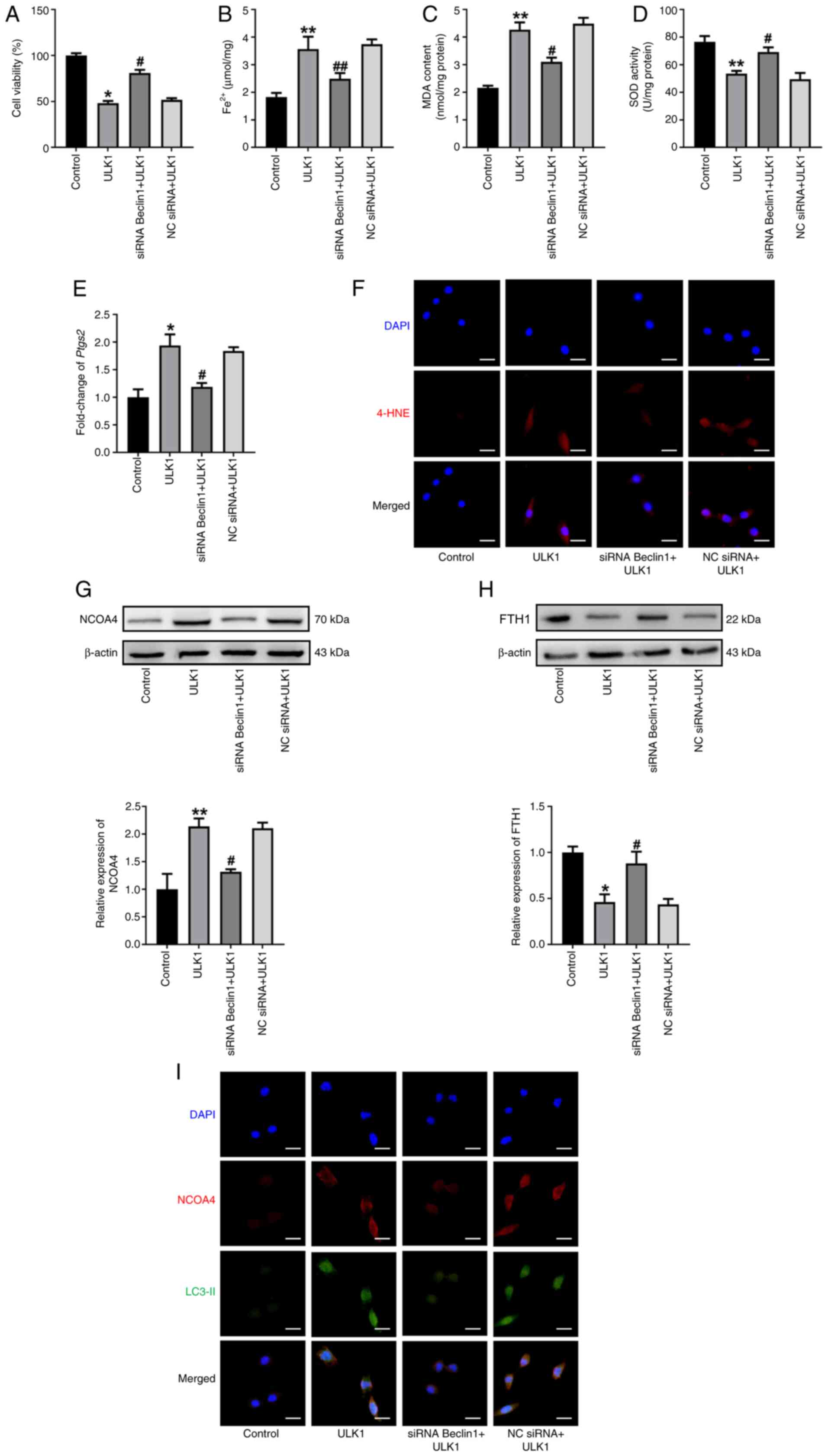 | Figure 6.ULK1 activates ferritinophagy
dependent on Beclin1/VPS34 complex. (A) Cell viability of HL-1
cells was assessed using a Cell Counting Kit-8 assay in the
indicated groups. The levels of (B) Fe2+, (C) MDA and
(D) SOD were determined. (E) The mRNA levels of Ptgs2. (F)
Immunofluorescence staining for 4-HNE in indicated groups. Scale
bar, 50 µm. Western blot analysis and summarized data demonstrating
(G) NCOA4 and (H) FTH1 protein levels in indicated group. (I) The
colocalization of NCOA4 (red) and LC3-II (green) in HL-1 cells was
determined using immunofluorescence staining. Scale bar, 50 µm. All
data are presented as mean ± SEM. n=3. *P<0.05 and **P<0.01
vs. control group; #P<0.05 and ##P<0.01
vs. ULK1 group. ULK1, serine/threonine protein kinase ULK1; VPS34,
PI3K catalytic subunit type 3; siRNA, small interfering RNA; NC,
negative control; MDA, malondialdehyde; SOD, superoxide dismutase;
Ptgs2, prostaglandin endoperoxide synthase 2; 4-HNE,
4-hydroxynonenal; FTH1, ferritin heavy chain 1; NCOA4, nuclear
receptor coactivator 4; LC3, microtubule-associated protein 1 light
chain 3. |
Beclin1/VPS34 complex is associated
with NCOA4-mediated ferritinophagy
Goodwin et al (24) found that ferritinophagy required
ATG9A, VPS34 and Tax1-binding protein 1 (TAX1BP1) to directly bind
to NCOA4. The present study also demonstrated that Beclin1 induced
ferroptosis. To determine whether the Beclin1/VPS34 complex
mediated downstream cellular events, including ferroptosis and
ferritinophagy, a western blot was performed. The results revealed
that, compared with the Beclin1 group, NCOA4 knockdown
significantly inhibited Beclin1-induced ferroptotic events
(Fig. 7A-F), significantly
inhibited NCOA4 expression and significantly promoted FTH1 protein
expression (Fig. 7G and H).
Collectively, these results demonstrated that ULK1 activated
NCOA4-mediated ferritinophagy via the Beclin1/VPS34 complex in
cardiomyocyte hypertrophy.
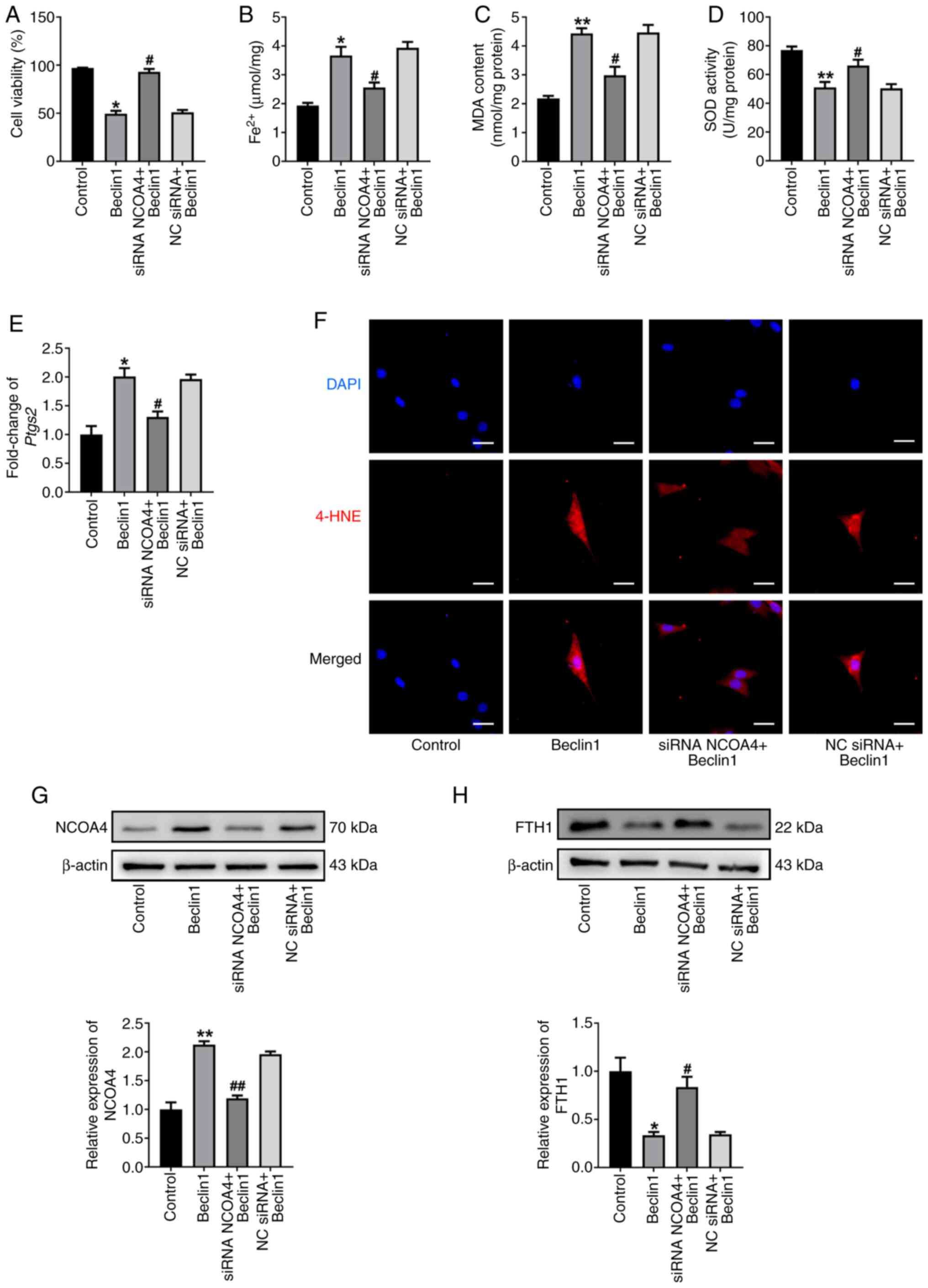 | Figure 7.Beclin1/VPS34 complex is associated
with NCOA4-mediated ferritinophagy. (A) Cell viability of HL-1
cells was assessed using a Cell Counting Kit-8 assay. The levels of
(B) Fe2+, (C) MDA and (D) SOD were determined. (E) The
mRNA levels of Ptgs2. (F) Immunofluorescence staining for
4-HNE in indicated group. Scale bar, 50 µm. Western blot analysis
and summarized data demonstrating (G) NCOA4 and (H) FTH1 protein
levels in the indicated groups. All data are presented as mean ±
SEM. n=3. *P<0.05 and **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. Beclin1 group.
VPS34, PI3K catalytic subunit type 3; NCOA4, nuclear receptor
coactivator 4; siRNA, small interfering RNA; NC, negative control;
MDA, malondialdehyde; SOD, superoxide dismutase; Ptgs2,
prostaglandin endoperoxide synthase 2; 4-HNE, 4-hydroxynonenal;
FTH1, ferritin heavy chain 1. |
Administration of SBI ameliorates
ferritinophagy in ULK1-overexpressing cells
The present study further explored whether targeting
the ULK1/Beclin1 axis could protect against cardiomyocyte
ferroptosis. To this end, SBI, a novel ULK1 inhibitor that can
substantially suppress ULK1 activity (25), was applied. Notably, compared with
the ULK1 group, SBI significantly inhibited ULK1 expression, NCOA4
expression, FTH1 degradation, cell injury and Fe2+
accumulation, and further attenuated ferroptosis progression in
cardiomyocyte hypertrophy (Fig.
8A-G). Notably, histone γ-H2AX expression was markedly
increased upon ULK1 exposure, indicating that ULK1 induced notable
DNA damage, which was reversed by co-treatment with SBI (Fig. 8H). Meanwhile, ULK1 facilitated the
colocalization of NCOA4 and LC3-II, whereas SBI exerted an
inhibitory effect (Fig. 8I). Taken
together, these data demonstrate that the administration of SBI
ameliorated ferritinophagy in ULK1-overexpressing cells.
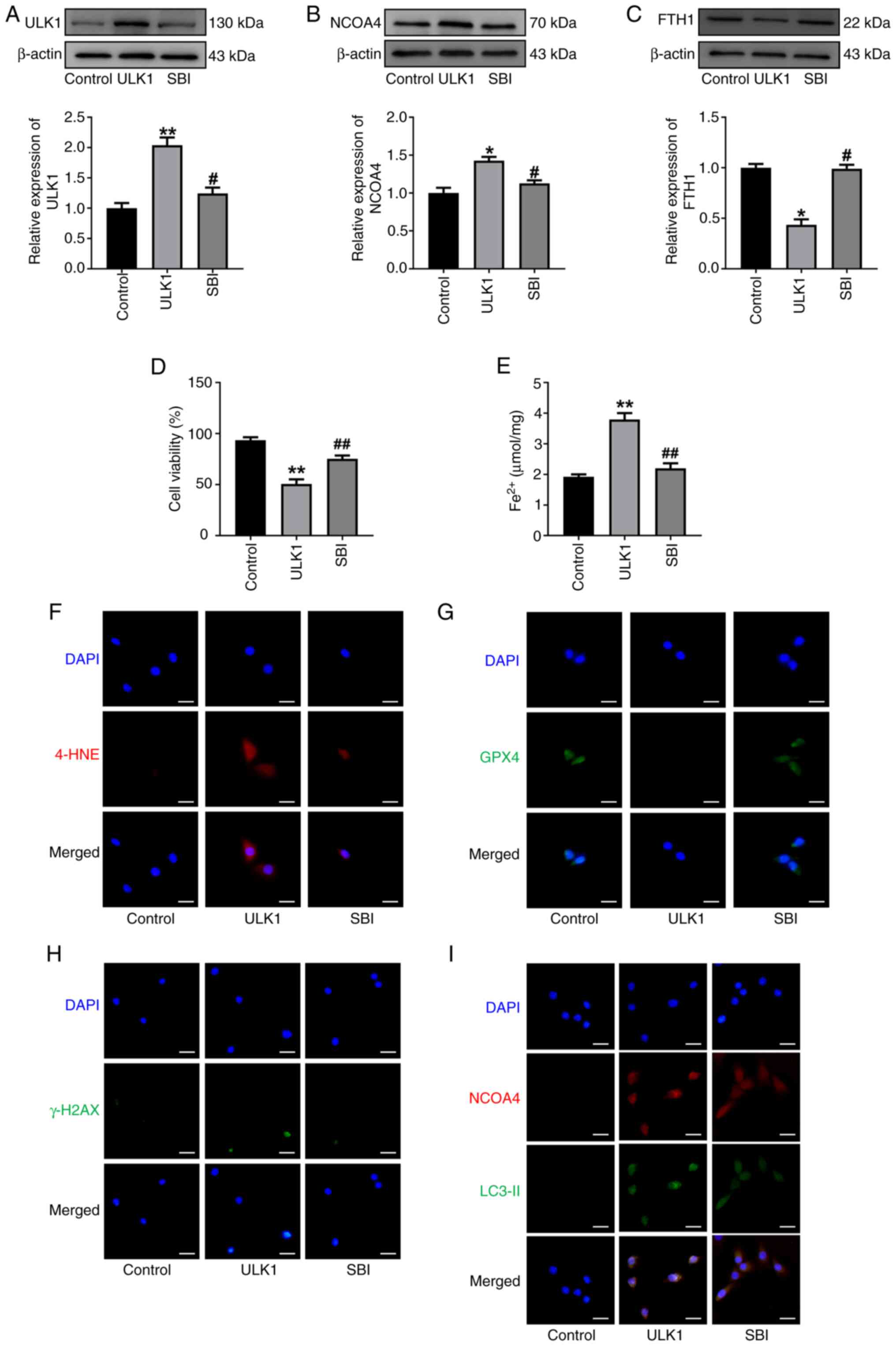 | Figure 8.Administration of SBI-0206965
ameliorates ferritinophagy in ULK1-overexpressing cells. Western
blot analysis and summarized data demonstrating (A) ULK1, (B) NCOA4
and (C) FTH1 protein levels in HL-1 cells treated with SBI-0206965.
(D) Cell viability of HL-1 cells was assessed using a Cell Counting
Kit-8 assay. (E) The levels of Fe2+. (F)
Immunofluorescence staining for 4-HNE. Scale bar, 50 µm. (G)
Immunofluorescence staining for GPX4. Scale bar, 50 µm. (H)
Immunofluorescence staining for γ-H2AX. Scale bar, 50 µm. (I) The
colocalization of NCOA4 (red) and LC3-II (green) in HL-1 cells was
determined by immunofluorescence staining in the indicated groups.
Scale bar, 50 µm. All data are presented as mean ± SEM. n=3.
*P<0.05 and **P<0.01 vs. control group; #P<0.05
and ##P<0.01 vs. ULK1 group. ULK1, serine/threonine
protein kinase ULK1; NCOA4, nuclear receptor coactivator 4; SBI,
SBI-0206965; FTH1, ferritin heavy chain 1; 4-HNE, 4-hydroxynonenal;
GPX4, glutathione peroxidase 4; LC3, microtubule-associated protein
1 light chain 3. |
AAV9-siULK1 reverses
ferritinophagy-related ferroptosis and cardiac hypertrophy
To validate whether inhibition of ULK1 could
attenuate cardiac hypertrophy in a mouse model, the present study
employed an AAV9-mediated loss-of-function approach via tail vein
injection. AAV9 carrying siULK1 was confirmed by high expression
levels in cardiac tissues (Fig.
S11A). Cardiac function analyses demonstrated that AAV9-siULK1
effectively restored LVEF and FS values from significant
TAC-mediated decreases in these parameters (Fig. S11B-D). Additionally, WGA staining
revealed pronounced hypertrophy in mice subjected to TAC surgery,
whereas mild hypertrophy was observed in mice co-treated with
AAV9-siULK1 (Fig. S11E and F).
The mRNA expression of BNP and β-MHC was
significantly increased in TAC surgery mice, which was mediated by
the administration of AAV9-siULK1 (Fig. S11G and H). Notably, the role of
ferroptosis was also evaluated in AAV9-siULK1 treated mice.
Compared with the sham group, TAC treatment resulted in a
significant accumulation of MDA, a decrease in SOD levels and
upregulation of Ptgs2 mRNA levels in cardiac tissues
(Fig. S11I-K). Prussian blue
staining showed that TAC surgery increased iron storage in cardiac
tissues (Fig. S11L). Furthermore,
at the molecular level, the protein levels of ULK1 and NCOA4 were
significantly increased, whereas FTH1 expression was significantly
decreased in the hearts of TAC surgery mice compared with sham mice
(Fig. S11M-O). Concurrently,
AAV9-siULK1 treatment significantly attenuated the aforementioned
TAC-induced effects (Fig.
S11A-O). Overall, these in vivo data suggest that
ferroptosis was activated in TAC-induced mice with cardiac injury,
while AAV9-siULK1 treatment suppressed ferroptosis.
Discussion
Cardiac hypertrophy initially functions as a
compensatory response to various stressors, but sustained
hypertrophy has emerged as a prominent predictor of the development
of HF (26). Several
disease-related conditions activate endocrine, paracrine and
autocrine regulatory circuits, which directly influence
cardiomyocyte hypertrophy via serine/threonine kinases and receptor
tyrosine kinases (27). Our
previous studies have focused on the function of ULK1 in the
autophagy pathway in cardiac hypertrophy (28). The present study aimed to
investigate the non-autophagy role of ULK1 under stressful
conditions. In the present study, ULK1 expression was found to be
increased in hypertrophic cardiac tissues and cardiomyocytes. ULK1
overexpression induced iron accumulation, lipid peroxidation and
ferroptotic cell death in cardiomyocytes, which were suppressed by
ULK1 knockdown. Mechanistically, the present study demonstrated
that ULK1 activated NCOA4-mediated ferritinophagy via the
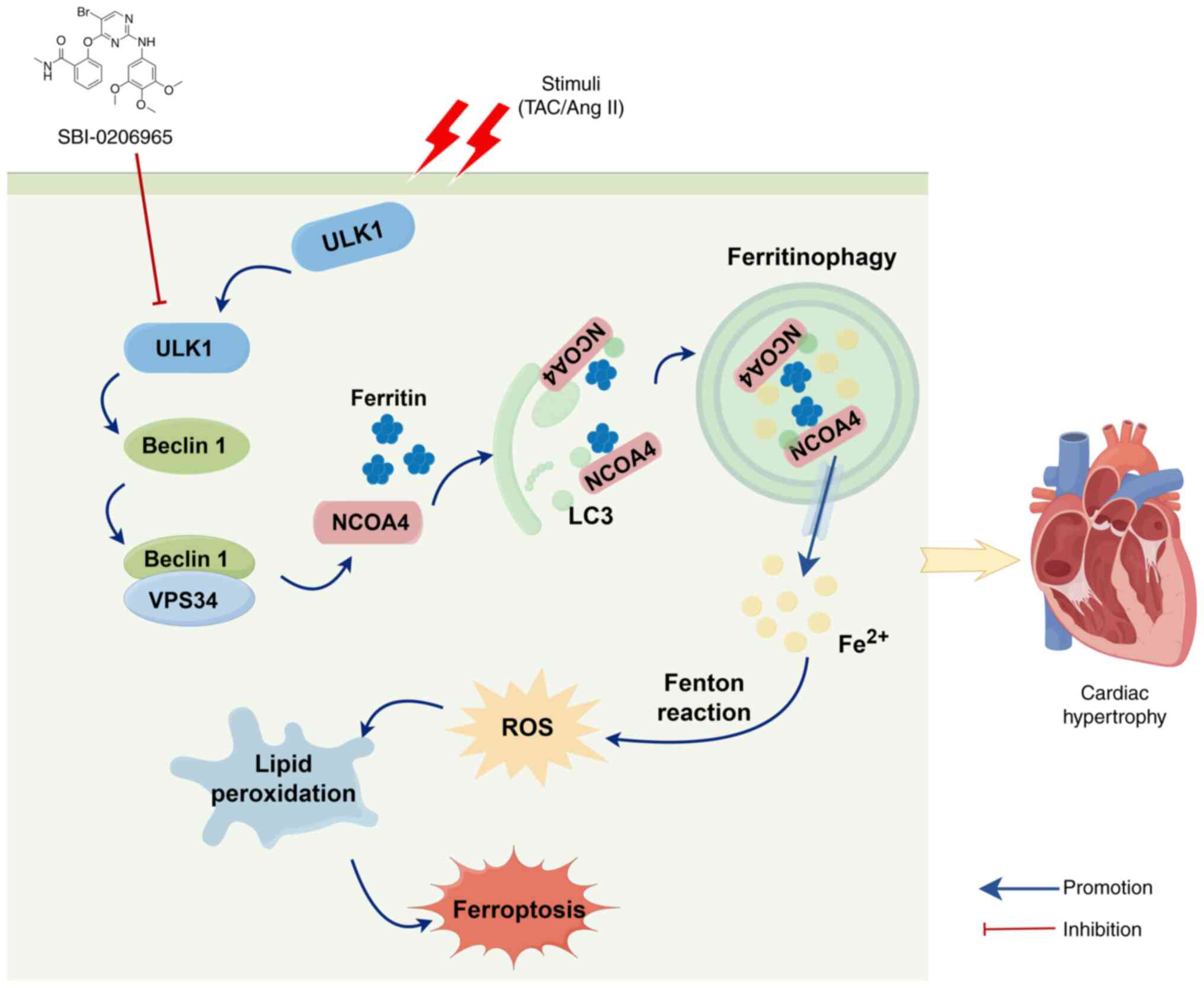
Beclin1/VPS34 complex in cardiomyocyte hypertrophy (Fig. 9). Notably, the ULK1 inhibitor SBI
ameliorated NCOA4-mediated ferritinophagy in cardiomyocyte
hypertrophy. These findings indicate that ULK1 is involved in the
regulation of intracellular iron metabolism and ferroptosis,
highlighting its potential as a therapeutic target for the
treatment of cardiac hypertrophy.
In the present study, ferroptosis represents a novel
mechanism underlying cardiomyocyte dysfunction induced by ULK1
exposure. Since ferroptosis was first reported in 2012, there has
been a notable growth in research on cardiovascular diseases,
including atherosclerosis, myocardial infarction, myocardial
ischemia-reperfusion injury, HF, cardiac hypertrophy,
cardiomyopathy and abdominal aortic aneurysm (29–31).
Ferroptosis is characterized by the iron-dependent accumulation of
lipid peroxidation, causing non-apoptotic cell death (32). In the present study, ULK1-treated
HL-1 cells exhibited iron overload, lipid peroxidation,
demonstrated by both MDA and 4-HNE, and SOD deprivation, which were
alleviated by the ferroptosis inhibitor Fer-1. These findings
suggest that ferroptosis represents an important contributor to
ULK1-induced HL-1 cell death.
The process of ferritinophagy was defined by Mancias
et al (33) in 2014. The
study used quantitative proteomics to identify NCOA4 as a selective
cargo receptor, which mediates ferritin degradation in
autophagosomes and leads to the release of Fe2+.
NCOA4-mediated ferritinophagy has been implicated in the
progression of cardiovascular diseases (34). For instance, NCOA4 has been
reported to be involved in pressure overload-induced cardiac
remodeling through ferritinophagy regulation (35). The apelin-13/apelin receptor system
has also been shown to promote ferritinophagy and contribute to
cardiac hypertrophy (36).
Furthermore, rats with myocardial ischemia-reperfusion injury have
been found to present with enhanced NCOA4-mediated ferritinophagy,
ferroptosis and deteriorated cardiac function, all of which could
be reversed by the administration of baicalin (37). However, to the best of our
knowledge, the role of NCOA4-mediated ferritinophagy in
ULK1-induced cardiomyocyte hypertrophy has not been reported
previously.
Autophagy is a lysosomal-dependent degradation
pathway that is a key regulator of cellular homeostasis (38). In ULK1-mediated classical
autophagy, the ULK1 complex, comprising ULK1, ATG13, RB1-inducible
coiled-coil protein and ATG101, serves an important role in
initiating the autophagic process (39). The present results provide evidence
that ULK1 activation induces autophagy and that ferroptosis is a
form of autophagic cell death. The results of the present study
demonstrated that inhibition of autophagy using 3-MA led to a
significant reduction in cell death, lipid peroxidation and SOD
depletion following ULK1 induction. These results indicate that
autophagy activation is required for ULK1-induced ferroptosis of
HL-1 cells.
Correspondingly, Qin et al (12) found that AMPK/ULK1 axis-mediated
autophagy activation was associated with ZnONP-induced ferroptosis
of human umbilical vein endothelial cells. Ferritinophagy, as a new
type of autophagy, is closely associated with various physiological
and pathophysiological processes. It is tightly regulated by an
iron-dependent protein network that coordinates intracellular
Fe2+ homeostasis and its associated physiological
functions (11). The present study
provided evidence that NCOA4-mediated ferritinophagy is an
important mechanism for ULK1-induced ferroptosis in cardiomyocytes.
Supplementation with the autophagy inhibitor 3-MA decreased NCOA4
expression and FTH1 degradation. Meanwhile, colocalization between
NCOA4 and LC3-II were also inhibited, indicating a specific role of
NCOA4-mediated ferritinophagy during ULK1 treatment. Furthermore,
NCOA4 knockdown significantly reduced iron accumulation and lipid
peroxidation and recovered cell viability in response to ULK1
overexpression, further supporting the notion that ULK1 induces
ferroptosis in a manner dependent on NCOA4-mediated ferritinophagy.
The present study expanded the known functions of ferritinophagy to
include its involvement in ULK1-mediated cardiomyocyte injury.
The present study also explored the possible
mechanisms of ferritinophagy activation in response to ULK1
overexpression. ULK1 is best known for its evolutionarily conserved
role in the autophagy pathway, and the ULK1 and Beclin1/VPS34
complexes are key signaling complexes required for autophagosome
formation (40). In recent years,
ULK1 has been shown to have additional functions beyond autophagy,
including protein trafficking and signaling events that affect
cellular homeostasis and cell fate (41). ULK1 mediates autophagy by
phosphorylating Beclin1 and activating VPS34 lipid kinase (42). However, the role of the
Beclin1/VPS34 complex in ferroptosis and ferritinophagy in
ULK1-induced cardiomyocyte injury has, to the best of our
knowledge, not been clarified. The present study demonstrated that
the Beclin1/VPS34 complex was upregulated in hypertrophic cells.
Notably, the results of the present study demonstrated that Beclin1
was linked with ferritinophagy and ferroptosis by regulating NCOA4
and FTH1 activity. Beclin1 knockdown significantly decreased NCOA4
protein levels and increased FTH1 protein levels to protect HL-1
cells from ferritinophagy-mediated ferroptosis. The roles of the
Beclin1/VPS34 complex in ULK1-induced ferroptosis require further
investigation.
Ferritinophagy requires direct binding of ATG9A,
VPS34 and TAX1BP1 to NCOA4 (24).
The present study observed that NCOA4 deficiency notably inhibited
Beclin1-induced ferroptosis events and reduced the colocalization
of NCOA4 and LC3-II, suggesting that Beclin1 activates ferroptosis
in a manner dependent on NCOA4-mediated ferritinophagy. Several
other mechanisms are involved in Beclin1-induced ferroptosis. It
has been reported that phosphorylated AMPK could induce
Beclin1-cystine/glutamate transporter complex formation, which
causes ferroptosis via blocking the cystine/glutamate transporter
(43). The present study
demonstrated that ferroptosis, ferritinophagy and cardiomyocyte
injury were closely interconnected, with Beclin1 potentially
serving as a central mediator in this relationship. To some extent,
the mechanism of activation of the Beclin1/VPS34 complex and
ferritinophagy is just the tip of the iceberg.
The present study also reported the effects of ULK1
inhibitors in ferritinophagy-related ferroptosis. A previous study
reported that SBI has been shown to reverse the induction of
autophagy and autophagic flux in cardiomyocyte hypertrophic injury
(44). In the present study,
inhibition of the ULK1/Beclin1 axis was found to suppress ferritin
degradation and ultimately alleviate cardiomyocyte ferroptosis.
AAV9-siULK1 attenuated cardiac hypertrophy and suppressed
ferritinophagy-related ferroptosis. This suppression was evidenced
by changes in direct ferroptosis markers, including decreased MDA
content, increased SOD activity and reduced Ptgs2 mRNA
levels. A reduction in Prussian blue-positive iron deposits was
also noted, consistent with decreased iron storage
(Fe3+); however, this method does not specifically
detect the causative redox-active Fe2+ pool. These
findings support the important role of the ULK1/Beclin1/NCOA4 axis
in cardiomyocyte ferroptosis. However, further studies are needed
to demonstrate the clinical potential of SBI.
The present study had some limitations. While the
genetic knockdown approaches both in vitro and in
vivo robustly established ULK1 as a critical upstream regulator
of ferritinophagy, the precise molecular mechanisms downstream of
ULK1 remain only partially understood. We hypothesized that ULK1
acts through the Beclin1/VPS34 complex; however, direct evidence
for a functional interaction, such as the exact phosphorylation
sites at which ULK1 modifies Beclin1, was not observed.
Furthermore, the specific mechanism by which the Beclin1/VPS34
complex promotes NCOA4-mediated ferritinophagy remains unclear.
Future studies utilizing co-immunoprecipitation, glutathione
S-transferase pull down and co-localization analyses will be
essential to establish these direct molecular links and fully
validate the proposed signaling pathway.
In summary, the present study reported that ULK1
induced ferroptosis of HL-1 cells, which was associated with the
activation of autophagy, especially the autophagic degradation of
ferritin-ferritinophagy. Specifically, ULK1 activated
NCOA4-mediated ferritinophagy dependent on the Beclin1/VPS34
complex. Furthermore, SBI, an inhibitor of ULK1/2, inhibited
ferroptosis by suppressing the Beclin1/VPS34 complex/NCOA4 axis.
Collectively, the present study provided evidence to suggest that
ferritinophagy-mediated ferroptosis is a novel form of cell death
induced by ULK1 in vitro in cardiac hypertrophy.
Supplementary Material
Supporting Data
Supporting Data
Acknowledgements
Not applicable.
Funding
The present work was supported by Heilongjiang Provincial
Natural Science Foundation (grant no. LH2024H034), the Fundamental
Research Funds for the Provincial Universities (grant no.
JFYQPY202401) and the Postgraduate Research & Practice
Innovation Program of Harbin Medical University (grant no.
YJSCX2023-100HYD).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
QZ, MZ and HS conceived and designed the
experiments. QZ, MZ and YL performed the experiments. QZ, MZ and PS
revised the manuscript. YL, PS, HQ and MJ analyzed and interpreted
the data. YC interpreted data. QZ and MZ wrote the manuscript. QZ,
MZ and YC critically revised the manuscript for important
intellectual content. QZ and HS confirm the authenticity of all the
raw data. All authors read and approved the final version of the
manuscript.
Ethics approval and consent to
participate
The animal study protocol was reviewed and approved
by the Experimental Animal Ethics Committee of Harbin Medical
University-Daqing (approval no. HMUDQ20250331001). All methods were
performed in accordance with the relevant guidelines and
regulations.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Authors' information
Hongli Sun, sunhongli@hmudq.edu.cn; Qianhui
Zhang, zhangqianhui@hmudq.edu.cn;
Meitian Zhang, 651861365@qq.com; Yongsheng Liu,
lyongsheng2018@163.com;
Pilong Shi, 283073952@qq.com;
Hanping Qi, 445817291@qq.com;
Man Jiang, 1667447336@qq.com; Yonggang Cao,
437343482@qq.com.
Glossary
Abbreviations
Abbreviations:
|
Ang II
|
angiotensin II
|
|
BNP
|
brain natriuretic peptide
|
|
BW
|
body weight
|
|
CCK-8
|
Cell Counting Kit-8
|
|
LVEF
|
left ventricular ejection
fraction
|
|
FS
|
fractional shortening
|
|
Fer-1
|
ferrostatin-1
|
|
FTH1
|
ferritin heavy chain 1
|
|
H&E
|
hematoxylin and eosin
|
|
4-HNE
|
4-hydroxynonenal
|
|
HW
|
heart weight
|
|
GFP
|
green fluorescent protein
|
|
LVW
|
left ventricular weight
|
|
3-MA
|
3-methyladenine
|
|
β-MHC
|
β-myosin heavy chain
|
|
MDA
|
malondialdehyde
|
|
PFA
|
paraformaldehyde
|
|
SOD
|
superoxide dismutase
|
|
TAC
|
transverse aortic constriction
|
|
TL
|
tibial length
|
|
ULK1
|
serine/threonine protein kinase
ULK1
|
|
WGA
|
wheat germ agglutinin
|
References
|
1
|
Martin SS, Aday AW, Allen NB, Almarzooq
ZI, Anderson CAM, Arora P, Avery CL, Baker-Smith CM, Bansal N,
Beaton AZ, et al: 2025 Heart disease and stroke statistics: A
report of US and global data from the American heart association.
Circulation. 151:e41–e660. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dev S and Babitt JL: Overview of iron
metabolism in health and disease. Hemodial Int. 21:S6–S20. 2017.
View Article : Google Scholar
|
|
3
|
Chen X, Yu C, Kang R and Tang D: Iron
metabolism in ferroptosis. Front Cell Devel Biol. 8:5902262020.
View Article : Google Scholar
|
|
4
|
Wu R, Wyatt E, Chawla K, Tran M, Ghanefar
M, Laakso M, Epting CL and Ardehali H: Hexokinase II knockdown
results in exaggerated cardiac hypertrophy via increased ROS
production. EMBO Mol Med. 4:633–646. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fan K, Huang W, Qi H, Song C, He C, Liu Y,
Zhang Q, Wang L and Sun H: The Egr-1/miR-15a-5p/GPX4 axis regulates
ferroptosis in acute myocardial infarction. Eur J Pharmacol.
909:1744032021. View Article : Google Scholar
|
|
6
|
Zhang Q, Song C, Zhang M, Song C, He C,
Liu Y, Zhang Q, Wang L and Sun H: Super-enhancer-driven lncRNA
Snhg7 aggravates cardiac hypertrophy via Tbx5/GLS2/ferroptosis
axis. Eur J Pharmacol. 953:1758222023. View Article : Google Scholar
|
|
7
|
Ichimiya T, Yamakawa T, Hirano T, Yokoyama
Y, Hayashi Y, Hirayama D, Wagatsuma K, Itoi T and Nakase H:
Autophagy and autophagy-related diseases: A review. Int J Mol Sci.
21:89742020. View Article : Google Scholar
|
|
8
|
Russell RC, Tian Y, Yuan H, Park HW, Chang
YY, Kim J, Kim H, Neufeld TP, Dillin A and Guan KL: ULK1 induces
autophagy by phosphorylating Beclin-1 and activating VPS34 lipid
kinase. Nat Cell Biol. 15:741–750. 2013. View Article : Google Scholar
|
|
9
|
Lin MG and Hurley JH: Structure and
function of the ULK1 complex in autophagy. Curr Opin Cell Biol.
39:61–68. 2016. View Article : Google Scholar
|
|
10
|
Pierzynowska K, Rintz E, Gaffke L and
Węgrzyn G: Ferroptosis and its modulation by autophagy in light of
the pathogenesis of lysosomal storage diseases. Cells. 10:3652021.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Santana-Codina N and Mancias JD: The role
of NCOA4-mediated ferritinophagy in health and disease.
Pharmaceuticals (Basel). 11:1142018. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Qin X, Zhang J, Wang B, Xu G, Yang X, Zou
Z and Yu C: Ferritinophagy is involved in the zinc oxide
nanoparticles-induced ferroptosis of vascular endothelial cells.
Autophagy. 17:4266–4285. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nakao Y, Aono J, Hamaguchi M, Takahashi K,
Sakaue T, Inoue K, Ikeda S and Yamaguchi O: O-ring-induced
transverse aortic constriction (OTAC) is a new simple method to
develop cardiac hypertrophy and heart failure in mice. Sci Rep.
12:852022. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Merino D, Gil A, Gómez J, Ruiz L, Llano M,
García R, Hurlé MA and Nistal JF: Experimental modelling of cardiac
pressure overload hypertrophy: Modified technique for precise,
reproducible, safe and easy aortic arch banding-debanding in mice.
Sci Rep. 8:31672018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Martinez VR, Martins Lima A, Stergiopulos
N, Velez Rueda JO, Islas MS, Griera M, Calleros L, Rodriguez Puyol
M, Jaquenod de Giusti C, Portiansky EL, et al: Effect of the
structural modification of candesartan with zinc on hypertension
and left ventricular hypertrophy. Eur J Pharmacol. 946:1756542023.
View Article : Google Scholar
|
|
17
|
Wu D, Tang X, Ding L, Cui J, Wang P, Du X,
Yin J, Wang W, Chen Y and Zhang T: Candesartan attenuates
hypertension-associated pathophysiological alterations in the gut.
Biomed Pharmacother. 116:1090402019. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mahmoud DM, Ali MRA, Aldosari BN, Zaki RM,
Afzal O, Tulbah AS, Naguib DM, Zanaty MI, Attia ME, Abo El-Ela FI
and Fouad AG: Functional candesartan loaded lipid nanoparticles for
the control of diabetes-associated stroke: In vitro and in vivo
studies. Int J Pharm X. 7:1002272024.PubMed/NCBI
|
|
19
|
Lebeche D, Zhao Bin K and Hajjar R:
Candesartan abrogates G protein-coupled receptors agonist-induced
MAPK activation and cardiac myocyte hypertrophy. J
Renin-Angiotensin-Aldosterone Syst. 2:S154–S161. 2001. View Article : Google Scholar
|
|
20
|
Shang L and Wang X: AMPK and mTOR
coordinate the regulation of Ulk1 and mammalian autophagy
initiation. Autophagy. 7:924–926. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Endale HT, Tesfaye W and Mengstie TA: ROS
induced lipid peroxidation and their role in ferroptosis. Fron Cell
Dev Biol. 11:12260442023. View Article : Google Scholar
|
|
22
|
Kang R and Tang D: Autophagy and
ferroptosis-What's the connection? Curr Pathobiol Rep. 5:153–159.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Santana-Codina N, Gikandi A and Mancias
JD: The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv
Exp Med Biol. 1301:41–57. 2021. View Article : Google Scholar
|
|
24
|
Goodwin JM, Dowdle WE, DeJesus R, Wang Z,
Bergman P, Kobylarz M, Lindeman A, Xavier RJ, McAllister G, Nyfeler
B, et al: Autophagy-independent lysosomal targeting regulated by
ULK1/2-FIP200 and ATG9. Cell Rep. 20:2341–2356. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ahwazi D, Neopane K, Markby GR, Kopietz F,
Ovens AJ, Dall M, Hassing AS, Gräsle P, Alshuweishi Y, Treebak JT,
et al: Investigation of the specificity and mechanism of action of
the ULK1/AMPK inhibitor SBI-0206965. Biochem J. 478:2977–2997.
2021. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Caturano A, Vetrano E, Galiero R,
Salvatore T, Docimo G, Epifani R, Alfano M, Sardu C, Marfella R,
Rinaldi L, et al: Cardiac hypertrophy: From pathophysiological
mechanisms to heart failure development. Rev Cardiovasc Med.
23:1652022. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chukwuma Sr D: Exploring the complex
interplay in the regulation of cardiac pathophysiological functions
by protein kinases and phosphatases. MOJ Biol Med. 6:165–170. 2021.
View Article : Google Scholar
|
|
28
|
Song C, Qi H, Liu Y, Chen Y, Shi P, Zhang
S, Ren J, Wang L, Cao Y and Sun H: Inhibition of lncRNA Gm15834
attenuates autophagy-mediated myocardial hypertrophy via the
miR-30b-3p/ULK1 axis in mice. Mol Ther. 29:1120–1137. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fatima S, Zhou H, Chen Y and Liu Q: Role
of ferroptosis in the pathogenesis of heart disease. Front Physiol.
15:14506562024. View Article : Google Scholar
|
|
30
|
Zhang K, Tian XM, Li W and Hao LY:
Ferroptosis in cardiac hypertrophy and heart failure. Biomed
Pharmacother. 168:1157652023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang Y, Xin L, Xiang M, Shang C, Wang Y,
Wang Y, Cui X and Lu Y: The molecular mechanisms of ferroptosis and
its role in cardiovascular disease. Biomed Pharmacother.
145:1124232022. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ma TL, Chen JX, Zhu P, Zhang CB, Zhou Y
and Duan JX: Focus on ferroptosis regulation: Exploring novel
mechanisms and applications of ferroptosis regulator. Life Sci.
307:1208682022. View Article : Google Scholar
|
|
33
|
Mancias JD, Wang X, Gygi SP, Harper JW and
Kimmelman AC: Quantitative proteomics identifies NCOA4 as the cargo
receptor mediating ferritinophagy. Nature. 509:105–109. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Qin Y, Qiao Y, Wang D, Tang C and Yan G:
Ferritinophagy and ferroptosis in cardiovascular disease:
Mechanisms and potential applications. Biomed Pharmacother.
141:1118722021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ito J, Omiya S, Rusu MC, Ueda H, Murakawa
T, Tanada Y, Abe H, Nakahara K, Asahi M, Taneike M, et al: Iron
derived from autophagy-mediated ferritin degradation induces
cardiomyocyte death and heart failure in mice. Elife.
10:e621742021. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tang M, Huang Z, Luo X, Liu M, Wang L, Qi
Z, Huang S, Zhong J, Chen JX, Li L, et al: Ferritinophagy
activation and sideroflexin1-dependent mitochondria iron overload
is involved in apelin-13-induced cardiomyocytes hypertrophy. Free
Radic Biol Med. 134:445–457. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fan Z, Cai L, Wang S, Wang J and Chen B:
Baicalin prevents myocardial ischemia/reperfusion injury through
inhibiting ACSL4 mediated ferroptosis. Front Pharmacol.
12:6289882021. View Article : Google Scholar
|
|
38
|
Ktistakis NT and Tooze SA: Digesting the
expanding mechanisms of autophagy. Trends Cell Biol. 26:624–635.
2016. View Article : Google Scholar
|
|
39
|
Hama Y, Fujioka Y, Yamamoto H, Mizushima N
and Noda NN: The triad interaction of ULK1, ATG13, and FIP200 is
required for ULK complex formation and autophagy. Elife.
13:RP1015312025. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wirth M, Joachim J and Tooze SA:
Autophagosome formation-the role of ULK1 and Beclin1-PI3KC3
complexes in setting the stage. Semin Cancer Biol. 23:301–309.
2013. View Article : Google Scholar
|
|
41
|
Pareek G and Kundu M: Physiological
functions of ULK1/2. J Mol Biol. 436:1684722024. View Article : Google Scholar
|
|
42
|
Jia M, Yue X, Sun W, Zhou Q, Chang C, Gong
W, Feng J, Li X, Zhan R, Mo K, et al: ULK1-mediated metabolic
reprogramming regulates Vps34 lipid kinase activity by its
lactylation. Sci Adv. 9:eadg49932023. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Yan K, Hou T, Zhu L, Ci X and Peng L:
PM2.5 inhibits system Xc- activity to induce ferroptosis by
activating the AMPK-Beclin1 pathway in acute lung injury.
Ecotoxicol Environ Saf. 245:1140832022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li RL, Wu SS, Wu Y, Wang XX, Chen HY, Xin
JJ, Li H, Lan J, Xue KY, Li X, et al: Irisin alleviates pressure
overload-induced cardiac hypertrophy by inducing protective
autophagy via mTOR-independent activation of the AMPK-ULK1 pathway.
J Mol Cell Cardiol. 121:242–255. 2018. View Article : Google Scholar
|