Introduction
Diabetes mellitus (DM) is a metabolic disease, and
its high incidence has attracted worldwide attention. Type 2 DM
accounts for >95% of all cases of DM (1). DM is a recognized factor that leads
to poor prognosis in patients with acute myocardial infarction
(AMI) (2). Patients with DM have
an increased risk of ischaemic heart disease and an increased
mortality rate due to myocardial infarction (3,4).
Compared with those of non-diabetic patients, the hearts of
patients with DM are more sensitive to reperfusion injury (5). After reperfusion therapy, diabetic
patients often experience more severe reperfusion injury (6). Numerous factors affect the occurrence
and development of diabetic myocardial ischemia-reperfusion (I/R)
injury, including the course of DM, myocardial mitochondrial
dysfunction, cardiac endothelial barrier function and myocardial
microangiopathy (7,8). As the understanding of diabetic
myocardial I/R damage has deepened, researchers have found that the
development of this disease involves multiple factors and multiple
processes, among which inflammation and oxidative stress play key
roles (9). Hyperglycaemia
aggravates inflammation and is among the main factors that increase
diabetic I/R damage (10).
Therefore, reducing myocardial I/R damage in patients with DM has
become a major challenge faced by clinicians (11).
The main obstacle of I/R injury is the positive
feedback loop between inflammation and cardiomyocyte death: The
interaction of leukocytes with cardiomyocytes and endothelial cells
leads to cardiomyocyte death, which activates and attracts
leukocytes and releases cytokines (12). Toll-like receptors (TLRs) are
pattern recognition receptors and play an important role in the
induction of innate immunity and the inflammatory response
(13). TLRs are activated by
adaptor proteins and associated kinases for intracellular signal
transduction. These kinases cause further activation of downstream
kinases, including IκB-α kinase, an inhibitor of NF-κB, leading to
the release of NF-κB, the translocation of NF-κB to the nucleus and
an increase in inflammatory cytokine gene expression, resulting in
a proinflammatory response (14).
At present, an increasing number of studies have shown that TLR2
plays a key role in myocardial I/R (15,16).
A study by Favre et al (17) demonstrated that the myocardial
infarction area in TLR2-knockout mice after 30 min of ischemia and
60 min of reperfusion was reduced compared with that of mice
without TLR2 knockout. Therefore, the present study aimed to
explore whether the TLR2-NF-κB pathway is involved in the process
of myocardial I/R in diabetic mice to further explore feasible
protective measures.
Dexmedetomidine (DEX) is a highly selective α2
adrenergic receptor agonist with a favorable sedative effect that
does not cause respiratory depression (18). Studies have suggested that DEX has
a protective effect against I/R injury (19,20).
Specifically, DEX protects organs during injury, inhibits
proinflammatory signal transduction pathways and reduces cell
death. Several studies have indicated that DEX can treat I/R damage
in the context of DM (21,22). Cheng et al (23) suggested that DEX postprocessing
increased the phosphorylation of GSK-3β in the hearts of diabetic
rats by activating the PI3K/Akt signalling pathway, thereby
inhibiting apoptosis and oxidative stress of the myocardium and
reducing myocardial I/R injury. Li et al (24) demonstrated that DEX reduced
myocardial I/R injury in DM rats, which was associated with the
inhibition of endoplasmic reticulum stress-induced cardiomyocyte
apoptosis. The diabetic myocardial I/R injury process is complex,
and additional mechanisms by which DEX can treat DM myocardial I/R
injury still need to be further studied. At present, to the best of
our knowledge, DEX has not been reported to reduce myocardial I/R
injury in diabetic mice by inhibiting TLR2-mediated
inflammation.
Therefore, the present study aimed to determine the
mechanism by which DEX regulates the TLR2 signalling pathway during
myocardial I/R injury in the context of DM. It was hypothesized
that DEX exerts its cardioprotective effect by downregulating the
TLR2 signalling pathway to inhibit inflammation.
Materials and methods
Animals
A total of 42 male C57BL/6 mice (age, 4 weeks old;
16–18 g) were purchased from Hangzhou Ziyuan Laboratory Animal
Technology Co., Ltd. and raised in a specific pathogen-free
environment at the Animal Experiment Center of Sir Run Run Shaw
Hospital, with free access to water and feed (constant temperature,
24–26°C; constant humidity, 55–60%, 12:12 h day:night cycle). The
animal experimental protocol was approved (approval no.
SRRSH202104015) by and agreed upon by the Experimental Animal
Ethics Committee of Sir Run Run Shaw Hospital (Hangzhou, China).
All animal protocols followed in accordance with the ARRIVE
guidelines.
Establishment of a diabetic mouse
model
Male C57BL/6 mice were given free access to a
standard diet for 7 days and fed a high-fat diet (HFD; containing
60% energy from fat, 20% energy from carbohydrates and 20% energy
from protein) for 4 weeks. After 12 h of fasting, mice received
intraperitoneal injections of streptozotocin (STZ; MilliporeSigma)
dissolved in 0.1 M citrate buffer (pH 4.5) at a dose of 50 mg/kg
body weight for 2 consecutive days. At 3 days and 1 week after STZ
injection, fasting blood glucose was measured through the tail
vein. Mice with a blood glucose level >11.1 mM were defined as
having DM.
Experimental groups
Diabetic mice were randomly assigned to three groups
(n=14 per group): Diabetic sham (SHAM), I/R and DEX-treated I/R
(DEX). Mice in the SHAM and I/R groups received an equal volume of
saline via continuous intravenous infusion. In the DEX group, DEX
(Jiangsu Hengrui Pharmaceutical Co., Ltd.) was dissolved in saline
and administered via continuous intravenous infusion at 6 µg/kg/h,
initiated immediately at the onset of reperfusion and maintained
throughout the 2-h reperfusion period (no loading dose). The
selected dose was based on the clinically relevant maintenance
infusion range of DEX and on body surface area-based interspecies
dose conversion, as previously described (25,26).
A total of seven mice per group were used for infarct size
measurement, and the remaining seven were used for other
experiments.
Establishment of a myocardial I/R
mouse model
The mice continued HFD eating for 4 weeks (until
they grew to 9 weeks old). The mice were intraperitoneally
anaesthetized with sodium pentobarbital (50 mg/kg; i.p.) and
perioperative analgesia was provided with sufentanil (20 µg/kg,
i.v.) (27,28). Anaesthetic and analgesic depth was
continuously assessed during surgery, and additional sufentanil was
administered if nociceptive reflexes or movement responses to
surgical stimulation were observed. After tracheal intubation, an
animal ventilator was used for mechanical ventilation (tidal
volume, 20 ml/kg; frequency, 100 beats/min; HX-300). Body
temperature was continuously monitored using a rectal probe and
maintained at 37.0±0.5°C with a heating pad. Peripheral oxygen
saturation was continuously monitored using a pulse oximeter
throughout the procedure. The right external jugular vein was
opened, and a PE10 catheter was inserted. After the heart was
exposed, the left anterior descending coronary artery was ligated 2
mm below the left auricle with a 6–0 silk suture. A small
polypropylene tube was placed between the ligature and the anterior
descending branch of the left coronary artery. Ischemia was
confirmed by observing the change in colour of the myocardial
tissue in the ischemic area (the normal myocardium was pink, and
the ischemic myocardium was pale). Reperfusion was achieved by
loosening the knot. The artery was occluded for 30 min by
tightening the ligature. After 30 min of ischemia, the ligature was
loosened to allow reperfusion for 2 h. Mice in the SHAM group
underwent the same surgical procedures, apart from tying the 6–0
silk suture. At the end of reperfusion, mice were euthanized by an
overdose of sodium pentobarbital (100 mg/kg; intraperitoneally).
Mortality was confirmed by the absence of respiration and cardiac
activity, after which terminal blood collection and tissue
harvesting were performed.
Infarct size determination
At the end of the experiment, the silk thread was
ligated again. A 2% Evans blue solution (2 ml; Beijing Solarbio
Science & Technology Co., Ltd.) was infused from the catheter.
The left ventricular risk area was identified from the normal
myocardium by the absence of blue staining. The heart was then
frozen at −20°C for 30 min and cut into 2-mm-thick slices parallel
to the atrioventricular groove. The heart slices were then
incubated with 1% triphenyltetrazolium chloride (MilliporeSigma) in
0.1 M phosphate buffer (pH 7.4) at 37°C for 30 min. The infarcted
tissue in the area at risk (AAR) was stained white, and the
non-infarcted tissue was stained red. After 24 h of fixation with
4% paraformaldehyde, the infarct size was analysed using Adobe
Photoshop CC 2017 (Adobe).
Detection of IL-6, IL-10 and MCP-1 by
ELISA
After 120 min of reperfusion, blood was collected
from the abdominal aorta and centrifuged at ~10,000 × g for 10 min
at 4°C. To assess the early systemic inflammatory response, the
supernatant was collected and serum levels of interleukin-6 (IL-6),
interleukin-10 (IL-10) and monocyte chemoattractant protein-1
(MCP-1) were measured by ELISA using commercial kits: Mouse IL-6
ELISA Kit (Cat. No. F10830), Mouse IL-10 ELISA Kit (Cat. No.
F1087), and Mouse MCP-1 ELISA Kit (Cat. No. F11130) (Shanghai
Xitang Biological Technology Co., Ltd.) according to the
manufacturer's instructions.
Pathological examination
After blood collection, mice were euthanized with a
high dose of anesthetic. Cardiac tissue was fixed in 4%
paraformaldehyde at 4°C overnight, dehydrated, paraffin-embedded,
sectioned to 5-µm thickness and mounted on slides. The
paraffin-embedded sections were stained with H&E according to
the procedure recommended by the supplier (Beyotime Institute of
Biotechnology). Images were observed and acquired using a light
microscope (Nikon Corporation).
Immunohistochemistry assay
Sections were deparaffinized and rehydrated in
xylene and ethanol, followed by antigen retrieval by microwave in
0.1 mol/l citrate buffer, and then placed in 3% hydrogen peroxide
solution (29). After blocking
with 2.5% BSA (MilliporeSigma) at room temperature for 1 h,
sections were incubated with anti-TLR2 (1:200; cat. no. Ab209216;
Abcam) overnight at 4°C, followed by secondary antibody (1:200;
Santa Cruz Biotechnology, Inc.). Stained with 3,3′-diaminobenzidine
tetrahydrochloride (DAB; Beijing Zhongshan Jinqiao Biotechnology
Co., Ltd.), then dehydrated in ethanolic xylene and sealed in
neutral resin. Images were captured under a light microscope (Nikon
Corporation).
Western blot assay
Cardiac tissue was cut into small pieces, placed in
cold RIPA buffer (MilliporeSigma) containing protease inhibitors,
homogenized, centrifuged at ~13,000 × g for 20 min at 4°C. The
total protein concentration was quantified by using a BCA protein
assay kit (Thermo Fisher Scientific, Inc.). Equal amounts of
protein (30 µg/lane) were separated by 10% SDS-polyacrylamide gel
electrophoresis and transferred to PVDF membranes. The membranes
were blocked with 5% skim milk for 2 h, then incubated in primary
antibodies overnight (TLR2 (1:1,000; cat. no. Ab209216; Abcam),
NF-κB P65 (1:1,000; cat. no. Ab7970; Abcam), TNF-α (1:1,000; cat.
no. Ab6671; Abcam), IκB-α (1:1,000, cat. no. 4814; Cell Signaling
Technology, Inc.) and β-actin (1:1,000, cat. no. 4970; Cell
Signaling Technology, Inc.). Bands were detected with a secondary
antibody conjugated to horseradish peroxidase (1:10,000; cat. no.
BL003A; Biosharp Life Sciences) and incubated for 2 h at room
temperature. The signals were visualized by enhanced
chemiluminescence reagents (Thermo Fisher Scientific, Inc.). The
gray value was calculated using ImageJ software (version 1.52k;
National Institutes of Health), and the ratio of the optical
density of each target band to the optical density of the internal
reference protein band was used as the result of statistical
analysis.
Cell culture and H/R cell model
preparation
H9c2 cardiomyocytes, a rat embryonic myocardial cell
line, were purchased from the Cell Bank of the Chinese Academy of
Sciences. Cells were cultured in DMEM supplemented with 10% fetal
bovine serum (FBS; Gibco, Thermo Fisher Scientific, Inc.),
streptomycin and penicillin. All the experimental cells were grown
in a 95% air and 5% CO2 in a 37°C incubator. Cells were
used for experiments when they reach 70–80% confluency according to
the experimental design.
H9c2 cells were adapted to low glucose DMEM and
stimulated with 33 mM high glucose (HG) for 24 h to simulate
hyperglycemia. Cells were exposed to hypoxic conditions (95%
N2 and 5% CO2) for 3 h in medium without
glucose and serum. Following hypoxia, cells were reoxygenated in
high or low glucose medium under normoxic conditions
(reoxygenation) for 6 h to simulate reperfusion. DEX or Pam3CSK4
(PAM; 0.5µM; InvivoGen) was added to the media at the same
time.
Determining the optimal concentration
of DEX and assess whether DEX alleviates hypoxia and reoxygenation
damage in the context of HG
The cells were divided into five groups: The control
group (CON), HG group, oxygen-glucose deprivation and reoxygenation
group (OGD), HG + OGD group and HG + OGD + DEX group. In the HG +
OGD + DEX group, cells were treated with 0.01, 0.1, 1, 10 and 100
µM DEX for 6 h after 3 h of hypoxia.
Effect of DEX on hypoxia and
reoxygenation damage by inhibiting TLR2 expression
Cells were divided into four groups: The HG group,
HG + OGD group, HG + OGD + DEX group and HG + OGD + DEX + PAM
group. After 3 h of hypoxia, 1 µM DEX or 1 µM DEX + 0.5 µM PAM was
added to the reoxygenated medium for incubation for 6 h.
Cell Counting Kit-8 (CCK-8) assay
Cell viability was assessed by CCK-8 assay (Dojindo
Laboratories, Inc.). H9c2 cells were seeded in 96-well plates at a
density of 1×10^4 cells/well and allowed to adhere overnight before
treatment. The cells were processed by modelling procedure.
Subsequently, the cells were treated with 100 µl of CCK-8 solution
at 37°C for 1–3 h. The absorbance at 450 nm was read with a
microplate reader (Bio-Rad Laboratories, Inc.).
Immunofluorescence
H9c2 cells were seeded onto slides and modeled as
aforementioned. Cells were fixed in 4% paraformaldehyde at room
temperature for 15 min, permeabilized with 0.1% Triton X-100 for 15
min and blocked with BSA at room temperature for 30 min. The
samples were incubated overnight at 4°C with primary rabbit
anti-NF-κB p65 antibody (1:100; cat. no. Ab7970; Abcam). DyLight
594-conjugated donkey anti-rabbit IgG (1:200; Jackson
ImmunoResearch Laboratories, Inc.) was used as secondary antibody.
Cells were stained with DAPI (1 µg/ml; Beyotime Institute of
Biotechnology) at room temperature. Immunofluorescence was observed
and captured using a Nikon epifluorescence microscope (Nikon
Corporation).
Western blotting of cellular
proteins
Proteins were isolated from cells, and a large
volume of proteins was loaded, electrophoresed and transferred to
PVDF membranes. The expression of TLR2, NF-κB, TNF-α and β-actin
was detected using the same protocol as aforementioned for the
analysis of mouse myocardial tissue.
Statistical analysis
The data are shown as the mean ± standard deviation.
Differences between groups were determined by one-way analysis of
variance followed by Dunnett's or Tukey's post hoc test. P<0.05
was considered to indicate a statistically significant difference.
GraphPad Prism 7.0 (Dotmatics) and SPSS 21.0 (IBM Corp.) were
applied for data analysis.
Results
DEX protects against I/R-induced
myocardial injury in diabetic mice
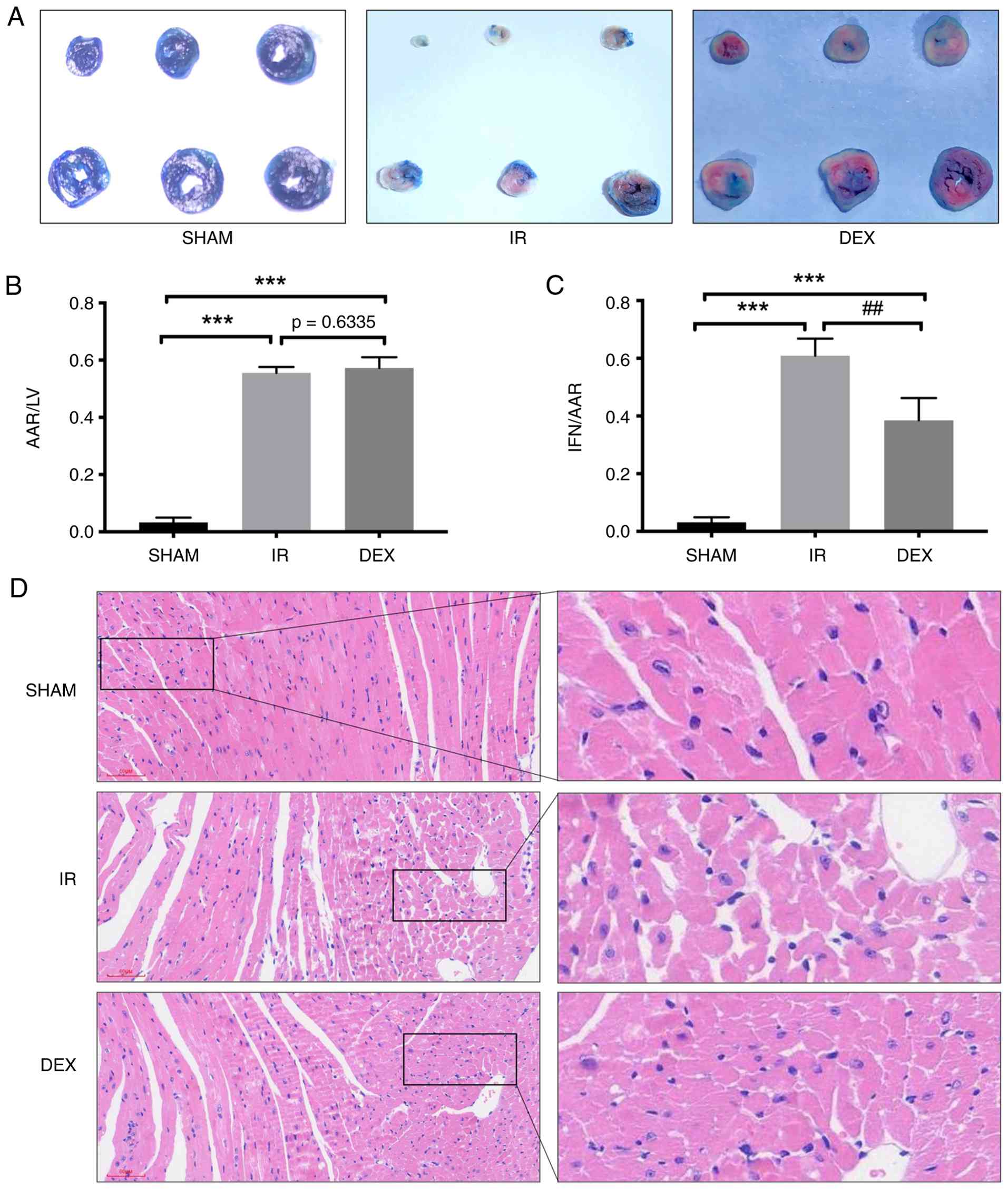
To examine the effects of DEX on I/R-induced
myocardial injury in diabetic mice, myocardial tissue injury in
diabetic mice was detected by Evans blue/TTC double staining and
H&E staining. DEX significantly reduced the area of myocardial
infarction induced by I/R in diabetic mice (Fig. 1A). Evans blue/TTC double staining
demonstrated that the AAR/left ventricular (LV) area was not
different between the DEX group and the I/R group (Fig. 1B). DEX significantly reduced the
ratio of the infarct region/AAR caused by I/R compared with that in
the myocardial I/R group of diabetic mice (Fig. 1C). H&E staining demonstrated
that in the I/R group, myocardial fibres were disordered and broken
and interstitial enlargement, oedema and the necrosis of myocardial
cells were observed, accompanied by round, dissolved or absent
nuclei. These morphological changes were reversed in the DEX group.
H&E staining further revealed that DEX improved the morphology
of cardiomyocytes after I/R (Fig.
1D).
DEX decreases the levels of IL-6 and
MCP-1 and increases the level of IL-10 in diabetic mice
ELISA was used to detect the levels of IL-6, IL-10
and MCP-1 in the serum. DEX reduced the increase in the
inflammatory factors IL-6 and MCP-1 induced by I/R (Fig. 2A and C). Furthermore, DEX
significantly increased the level of the anti-inflammatory factor
IL-10 and inhibited the inflammatory response caused by I/R
(Fig. 2B).
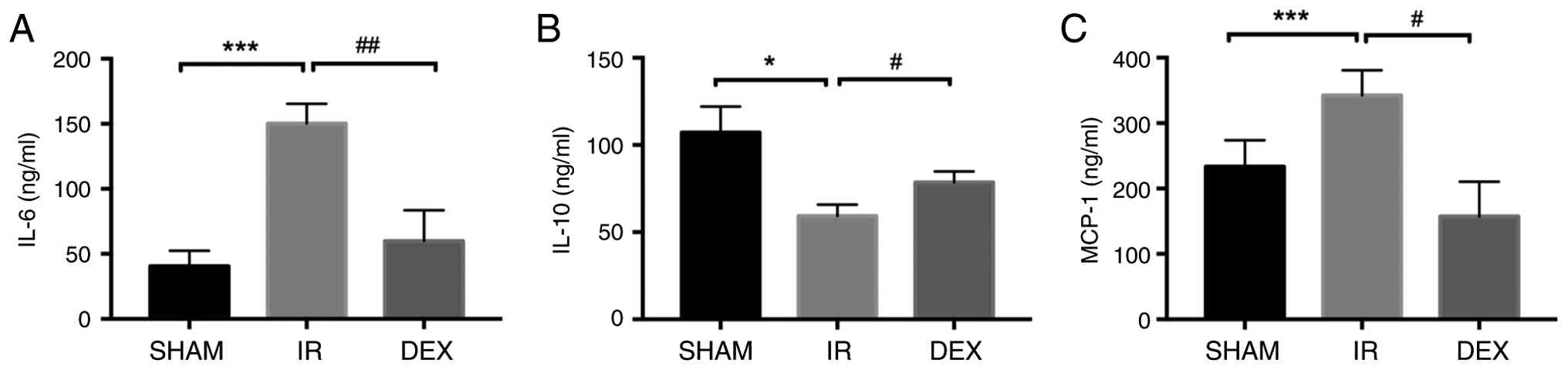 | Figure 2.Effects of DEX on the levels of IL-6,
IL-10 and MCP-1 in diabetic mice. (A-C) Statistical representations
of the serum levels of IL-6, IL-10 and MCP-1, respectively.
Compared with SHAM group, *P<0.05 and ***P<0.001; Compared
with IR group, #P<0.05, ##P<0.01, n=5.
DEX, dexmedetomidine. MCP-1, Monocyte Chemoattractant Protein-1;
IR, ischemia-reperfusion. |
DEX reduces the inflammatory response
to myocardial I/R in diabetic mice by inhibiting the TLR2
pathway
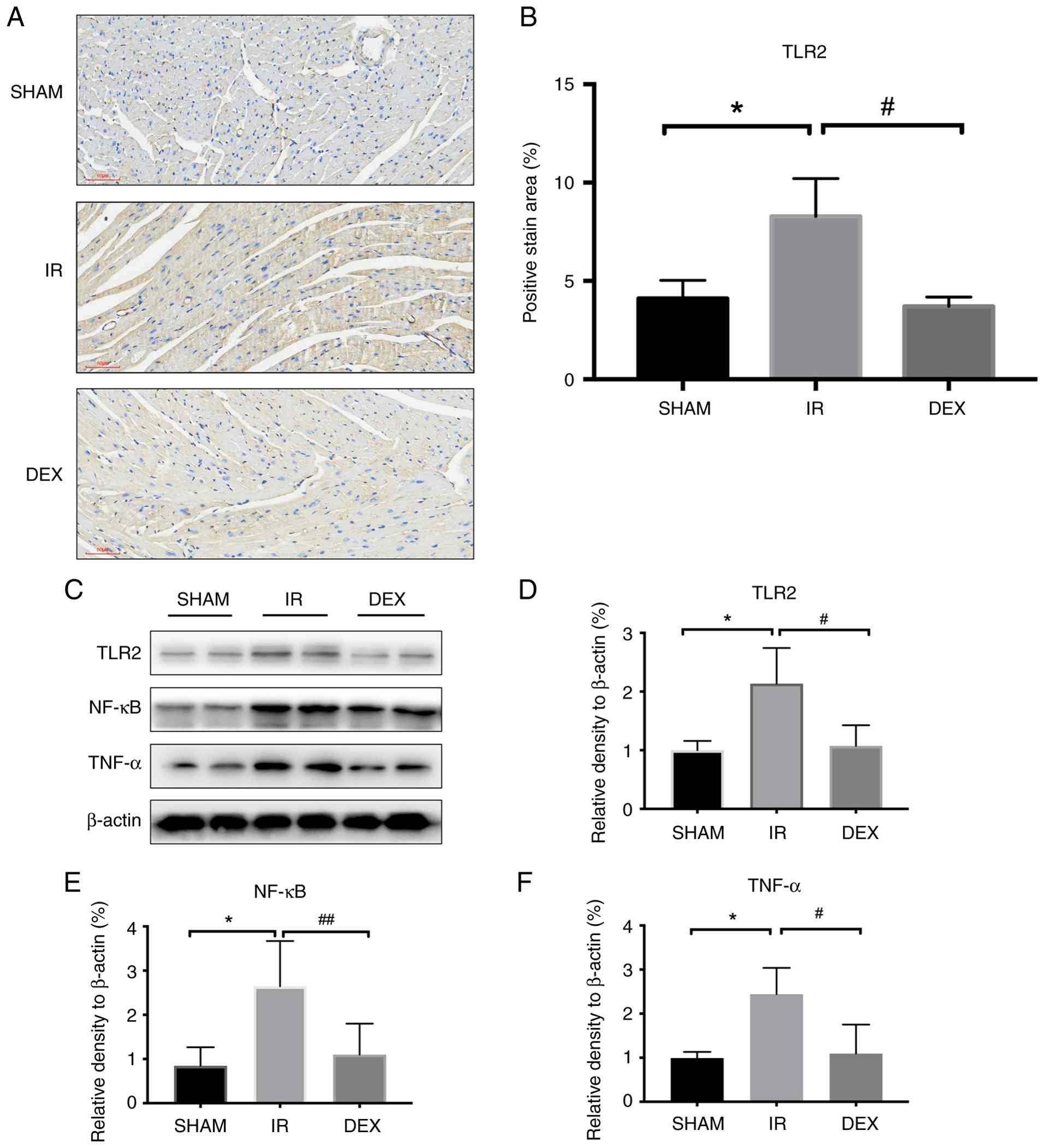
To explore how DEX reduced myocardial I/R injury in
the diabetic mice, immunohistochemical (IHC) staining and western
blot assays of myocardial tissue were performed. Representative
immunohistochemical staining images showed that TLR2 expression was
markedly increased in the I/R group compared with the SHAM group,
whereas DEX treatment reduced TLR2 staining intensity (Fig. 3A). Quantitative analysis further
confirmed that I/R significantly increased the positive staining
area of TLR2, and DEX significantly attenuated this increase
(Fig. 3B). Western blot assays of
the myocardial tissue also demonstrated that I/R increased the
expression of downstream NF-κB and TNF-α. However, DEX inhibited
the expression of NF-κB and TNF-α (Fig. 3C-F).
DEX reduces OGD/R-induced damage to
H9c2 cells under HG conditions
The optimal concentration of DEX was explored for
subsequent experiments by detecting cell viability, and the
experimental results demonstrated that 1 µM DEX was the best
concentration (Fig. 4A). The
expression levels of TLR2, NF-κB and TNF-α in the HG + OGD group of
H9c2 cells were significantly increased compared with those in the
HG group and OGD group. OGD/R in the context of HG aggravated the
expression of inflammatory factors. However, DEX reduced the
expression of TLR2, NF-κB and TNF-α after OGD/R under HG conditions
(Fig. 4B-E).
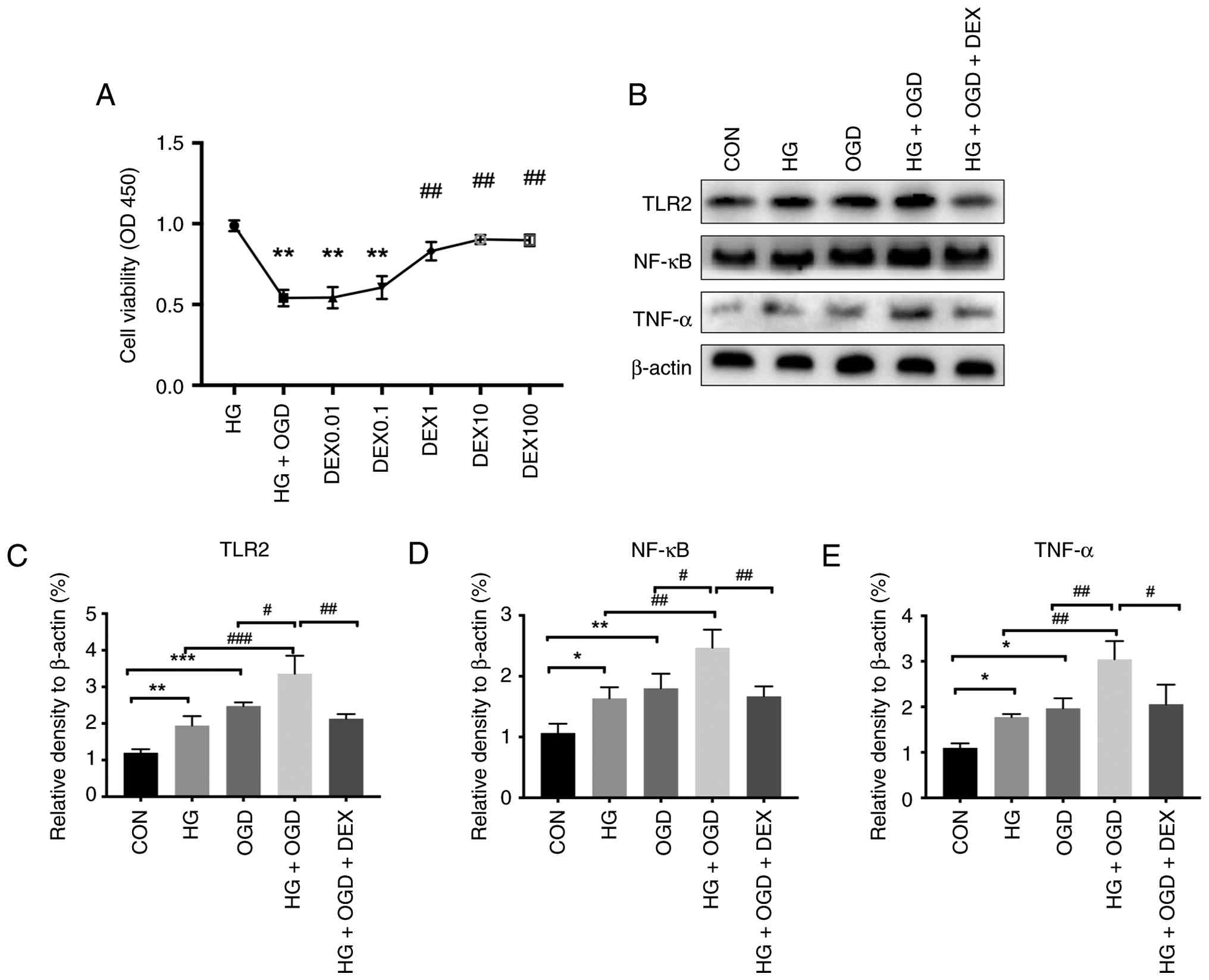 | Figure 4.DEX reduces OGD/R-induced damage to
H9c2 cells under HG conditions. (A) After H9c2 cell modelling, Cell
Counting Kit-8 assays were used to detect cell viability. (B)
Western blot analysis of TLR2, NF-κB, TNF-α and β-actin was carried
out. (C-E) Statistical representations of the relative expression
of TLR2, NF-κB and TNF-α, respectively. Compared with the CON
group, *P<0.05, **P<0.01 and ***P<0.001; compared with the
HG + OGD group, #P<0.05, ##P<0.01 and
###P<0.001; n=5. DEX, dexmedetomidine; OGD/R,
oxygen-glucose deprivation/reoxygenation; HG, high-glucose. |
DEX reduces OGD/R-induced damage to
H9c2 cells under HG conditions through the TLR2-NF-κB pathway
To further determine the molecular mechanism by
which DEX protects against OGD/R-induced damage in the context of
HG, the TLR2 agonist PAM was added along with DEX. DEX reduced the
TLR2, NF-κB and TNF-α expression caused by OGD/R under HG
conditions and increased IκB-α. The use of PAM prevented the effect
of DEX. Therefore, it was hypothesised that DEX inhibits the
inflammatory response by inhibiting the activation of NF-κB
mediated by TLR2 (Fig. 5A-E).
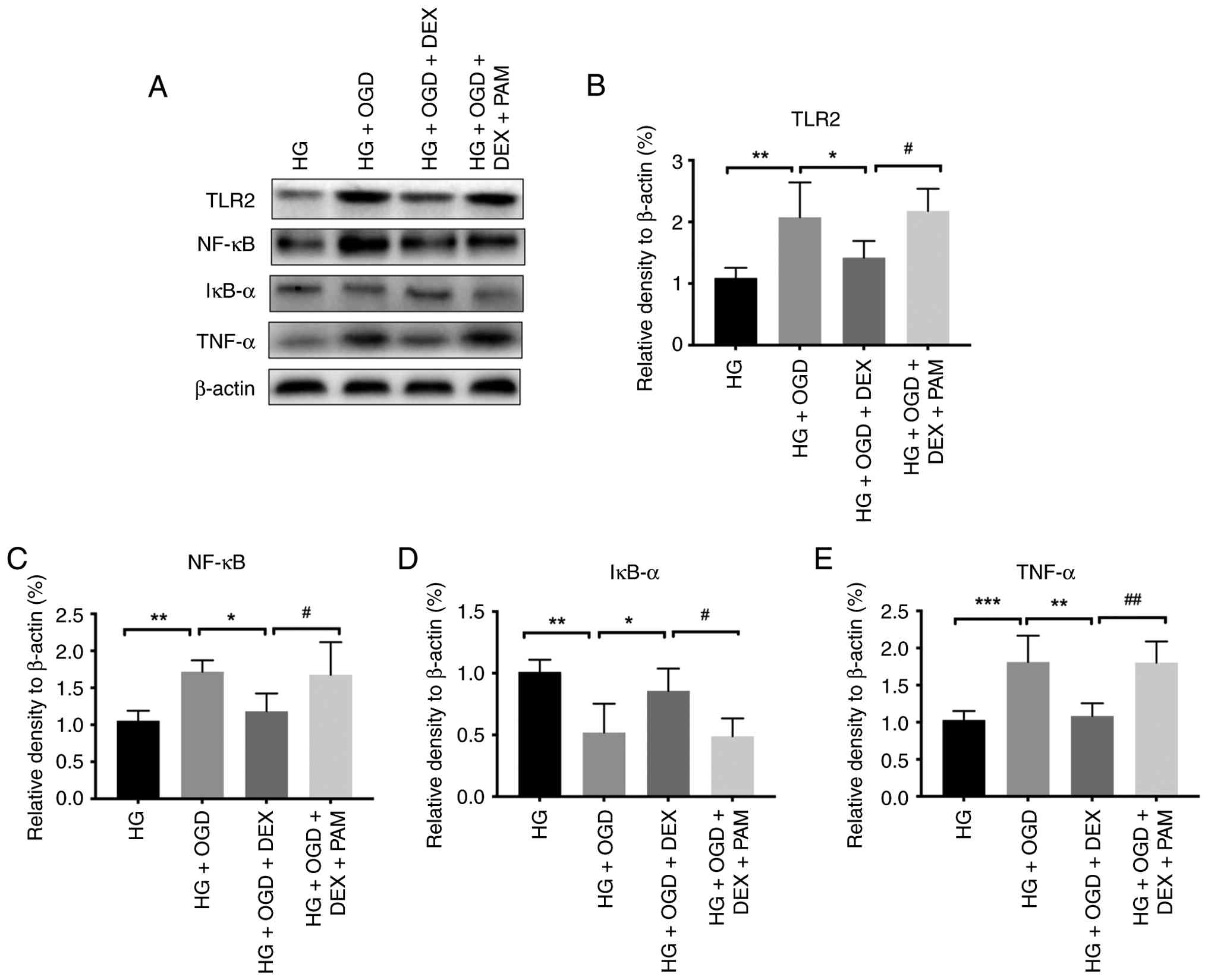 | Figure 5.DEX reduces OGD/R-induced damage to
H9c2 cells under HG conditions through the TLR2-NF-κB pathway. (A)
Western blot analysis of TLR2, NF-κB, TNF-α and β-actin. (B-E)
Statistical representations of the relative expression of TLR2,
NF-κB, IκB-α and TNF-α, respectively. Compared with the HG + OGD
group, *P<0.05, **P<0.01 and ***P<0.001; compared with the
HG + OGD + DEX group, #P<0.05 and
##P<0.01; n=5. DEX, dexmedetomidine; OGD/R,
oxygen-glucose deprivation/reoxygenation; TLR, toll like receptor;
HG, high-glucose; PAM, Pam3CSK4. |
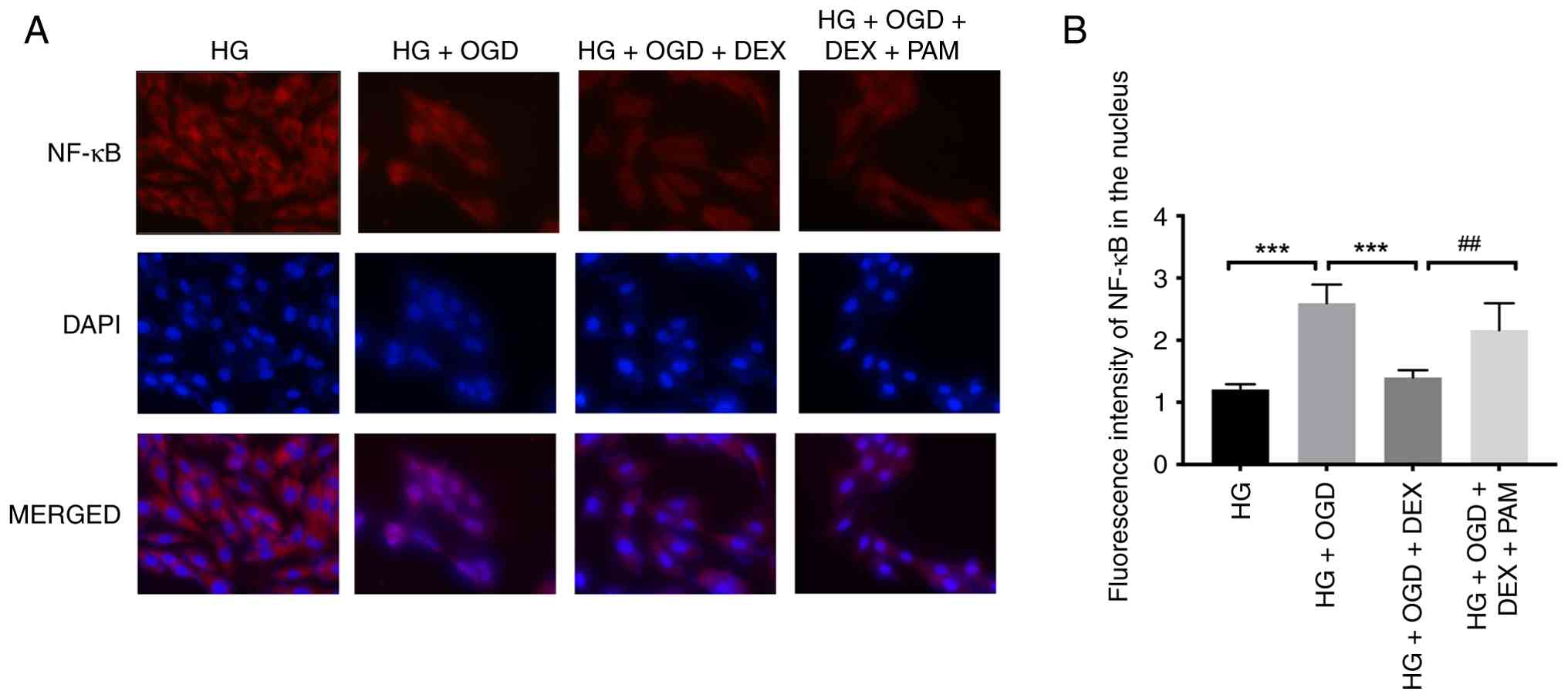
Effect of DEX on the activation of
NF-κB
To explore the effect of DEX on NF-κB downstream of
TLR2, fluorescently labelled NF-κB was used to observe its status.
When H9c2 cells underwent OGD/R under HG conditions, NF-κB entered
the nucleus after activation. DEX inhibited the activation of NF-κB
and reduced the inflammatory response. Furthermore, PAM reactivated
the translocation of NF-κB into the nucleus, preventing the effect
of DEX (Fig. 6A and B).
Discussion
The pesent study revealed the protective effect of
DEX against myocardial I/R injury and H9c2 cardiomyocyte OGD/R
injury in diabetic mice under a background of HG. In vivo
experiments revealed that DEX reduced the area of myocardial
infarction caused by I/R injury in diabetic mice, improved the
morphology of cardiomyocytes observed by myocardial H&E
staining, reduced serum inflammatory factor levels, and reduced
myocardial TLR2 and NF-κB protein expression. In cellular
experiments, PAM, an agonist of TLR2, was added. The results
demonstrated that PAM prevented the inhibition of TLR2 by DEX and
promoted the activation of NF-κB to produce inflammatory factors.
In short, DEX reduced diabetic myocardial I/R injury and its
mechanism may be mediated by inhibition of the TLR2-NF-κB
signalling pathway. These results indicate that DEX may have
certain therapeutic value when myocardial I/R injury occurs in
patients with DM.
The risk of heart failure and death after diabetic
myocardial ischemia is 2–3 fold higher when compared with that in
non-diabetic patients (30). A
large number of studies have shown that myocardial I/R injury in
the context of DM is more serious compared with that in
non-diabetic patients (31,32)
and that the protective effects of drugs are reduced (33,34).
The present study established a type 2 DM model in B6 mice through
feeding and STZ induction. Diabetic B6 mice were directly used as a
sham operation control group, and diabetic mice were used to
establish a myocardial infarction model to simulate clinical
myocardial I/R injury. In cellular experiments, CCK-8 and western
blot assays verified that OGD/R damage under a HG background was
more serious when compared with that in the absence of HG.
TLRs are a class of innate immune system receptors
that can bind endogenous ligands derived from a variety of
pathogens and induced by injury (35). TLR2 is used as a microbial sensor
to trigger inflammation and immune responses and induce a variety
of signalling pathways, including the NF-κB signalling pathway,
after ligand binding (36). The
TLR2-NF-κB signalling pathway can induce the expression of
pro-inflammatory mediators and induce an inflammatory response,
leading to an increased cerebral infarction area and aggravated
brain damage (37). Arslan et
al (38) also found that
inhibiting TLR2 can reduce the inflammatory response caused by
myocardial I/R, reduce myocardial infarction and protect heart
function and structure. NF-κB, a key protein downstream of TLR2, is
a dimeric redox-sensitive transcription factor composed of p50 and
p65 (39). It responds quickly to
external changes and plays an important role in I/R injury. Under
normal circumstances, NF-κB and IκB-α in the cell form an inactive
complex. Upon I/R injury, NF-κB is activated through the classic
NF-κB activation pathway: IκB-α is activated by its kinase through
phosphorylation and is degraded, resulting in the disassociation of
NF-κB p65 from the cytoplasmic NF-κB/IκB-α complex (40). Then, NF-κB p65 is activated,
exposing the nuclear localization domain, after which nuclear
translocation occurs and target gene expression is initiated. A
large number of inflammatory factors, such as TNF-α, IL-1β and
IL-6, are released to trigger an inflammatory response and
participate in I/R injury (41).
TNF-α plays an important role in the production and activation of
inflammation. The experimental results revealed that both
myocardial I/R injury and OGD/R injury under HG conditions in
diabetic mice significantly increased the expression of TLR2, NF-κB
and TNF-α. Therefore, the present study used the TLR2-NF-κB pathway
as a target for diabetic myocardial I/R injury.
In vivo and in vitro experiments have
shown that DEX can inhibit the expression of TLR2 and activation of
NF-κB and reduce I/R injury in a HG background. I/R injury is
related to the activation of NF-κB, and NF-κB inhibitors can reduce
I/R injury (42). Activated NF-κB
induces the release of multiple inflammatory mediators, including
pro-IL-1β and IL-18 (43).
Research by Bao et al (44)
demonstrated that DEX can prevent the activation of NF-κB caused by
lipopolysaccharide; reduce the levels of TNF-α, IL-6, IL-1β and
MCP-1; and increase the level of IL-10. As the initiation factor of
the inflammatory cascade, TNF-α promotes the release of a variety
of other inflammatory factors, decreases muscle strength and
further damages myocardial tissue. As an important cytokine, IL-6
participates in the immune and inflammatory responses of the body.
Post-traumatic activation of neutrophils can aggravate the
production of inflammatory mediators, which can effectively predict
the severity of tissue damage. MCP-1 is secreted by cells in
response to pro-inflammatory cytokines. IL-10 can inhibit the
release of inflammatory mediators from monocytes and macrophages.
The research results of the present study show that DEX inhibited
the activation of myocardial NF-κB in diabetic mice, inhibited
secretion of the downstream inflammatory factor TNF-α, decreased
serum levels of the pro-inflammatory factor IL-6 and chemokine
MCP-1, and increased levels of the serum anti-inflammatory factor
IL-10.
To further verify that the role of DEX in diabetic
myocardial I/R injury is mediated through the TLR2-NF-κB pathway,
the TLR2 agonist PAM was used. DEX reduced the expression of TLR2,
inhibited the dissociation of cytoplasmic NF-κB/IκB-α, inhibited
NF-κB nuclear transfer and reduced secretion of the inflammatory
factor TNF-α. PAM prevented the protective effects of DEX. DEX was
identified to reduce isoflurane-induced neurotoxicity by inhibiting
the TLR2/NF-κB signalling pathway (45). Therefore, it was hypothesised that
the protective effects of DEX in the OGD/R model under HG
conditions were mediated through the TLR2-NF-κB pathway.
Although DEX is a highly selective α2-adrenergic
receptor (α2-AR) agonist, and its anti-inflammatory actions have
been attributed to α2-AR-mediated neuroimmune modulation (46). The present study suggested that
suppression of the TLR2-NF-κB inflammatory axis plays a key role in
the cardioprotective effects of DEX under hyperglycemic conditions.
Importantly, pharmacological activation of TLR2 with PAM prevented
the inhibitory effects of DEX on NF-κB activation and inflammatory
cytokine production, supporting the notion that TLR2 signaling is a
key downstream determinant of the observed anti-inflammatory
phenotype.
The present study used DEX after myocardial ischemia
for two reasons. First, the time frame of DEX administration in the
present study is clinically relevant. After diabetic patients with
myocardial infarction come to a hospital, DEX is continuously
infused as a sedative or anaesthetic adjuvant during myocardial
reperfusion. DEX can activate central and peripheral presynaptic
membrane α2 receptors, leading to a reduction in the synthesis and
release of norepinephrine, reducing heart rate, improving
myocardial oxygen consumption during ischemia and making it easier
to reach myocardial oxygen during reperfusion (18). Furthermore, DEX balances the supply
and demand of lactic acid and improves lactic acidosis after
myocardial infarction (47).
Perioperative application of low-dose DEX can reduce the use of
opioid analgesics and sedatives in cardiovascular surgery, maintain
haemodynamic stability and reduce myocardial I/R injury (48,49).
DEX also has a partial analgesic effect (50). Second, the inflammatory response
mediated by the TLR2 signalling pathway is significantly
upregulated during myocardial I/R injury. The experimental results
of the present study revealed that DEX treatment can inhibit the
expression of TLR2 and the activation of NF-κB. This reduces the
release of inflammatory factors, reduces the infarct size and
improves the morphology of cardiomyocytes. These effects of DEX are
beneficial to recovery from myocardial I/R injury, indicating its
potential therapeutic prospects in AMI.
In conclusion, the present study found that DEX
protects against myocardial I/R injury in a hyperglycaemic
background by inhibiting the TLR2-NF-κB signalling pathway. The
present study is the first to propose the important role of the
TLR2-NF-κB signalling pathway in the pathogenesis of myocardial I/R
injury under the background of HG, providing a basis for the
clinical application of DEX in patients with diabetic myocardial
I/R injury.
Acknowledgements
The authors would like to thank Professor Xuzhong Xu
(Department of Anesthesiology, The First Affiliated Hospital of
Wenzhou Medical University, Wenzhou, China), for providing
methodological guidance on animal model establishment.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZC participated in conducting the study, data
collection, data analysis and manuscript preparation. LZ was
responsible for study design, conducting the study, data
collection, data analysis and manuscript preparation. GC designed
the study and analyzed data. All authors read and approved the
final manuscript. ZC and LZ confirm the authenticity of all the raw
data.
Ethics approval and consent to
participate
The protocols used for all animal studies were
approved (approval no. SRRSH202104015) by the Experimental Animal
Ethics Committee of Sir Run Run Shaw Hospital (Hangzhou, China) and
Welfare Committee and complied with the NIH guidelines (Guide for
the Care and Use of Laboratory Animals).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Creager MA, Luscher TF, Cosentino F and
Beckman JA: Diabetes and vascular disease: Pathophysiology,
clinical consequences, and medical therapy: Part I. Circulation.
108:1527–1532. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Patel PA, Cubbon RM, Sapsford RJ, Gillott
RG, Grant PJ, Witte KK, Kearney MT and Hall AS: An evaluation of 20
year survival in patients with diabetes mellitus and acute
myocardial infarction. Int J Cardiol. 203:141–144. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Abbasi F, Brown BW Jr, Lamendola C,
McLaughlin T and Reaven GM: Relationship between obesity, insulin
resistance, and coronary heart disease risk. J Am Coll Cardiol.
40:937–943. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brener SJ, Mehran R, Dressler O, Cristea E
and Stone GW: Diabetes mellitus, myocardial reperfusion, and
outcome in patients with acute ST-elevation myocardial infarction
treated with primary angioplasty (from HORIZONS AMI). Am J Cardiol.
109:1111–1116. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Zafrir B, Jaffe R, Rubinshtein R, Karkabi
B, Flugelman MY and Halon DA: Impact of diabetes mellitus on
long-term mortality in patients presenting for coronary
angiography. Am J Cardiol. 119:1141–1145. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Braga JR, Santos IS, Flato UP, Guimaraes
HP and Avezum A: The impact of diabetes mellitus on the mortality
of acute coronary syndromes. Arq Bras Endocrinol Metabol.
51:275–280. 2007.(In Portuguese). View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Doulamis IP, Guariento A, Duignan T,
Orfany A, Kido T, Zurakowski D, Del Nido PJ and McCully JD:
Mitochondrial transplantation for myocardial protection in diabetic
hearts. Eur J Cardiothorac Surg. 57:836–845. 2019. View Article : Google Scholar
|
|
8
|
Qi K, Li X, Geng Y, Cui H, Jin C, Wang P,
Li Y and Yang Y: Tongxinluo attenuates reperfusion injury in
diabetic hearts by angiopoietin-like 4-mediated protection of
endothelial barrier integrity via PPAR-alpha pathway. PLoS One.
13:e01984032018. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Keane KN, Cruzat VF, Carlessi R, de
Bittencourt PI Jr and Newsholme P: Molecular events linking
oxidative stress and inflammation to insulin resistance and β-Cell
dysfunction. Oxid Med Cell Longev. 2015:1816432015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lejay A, Fang F, John R, Van JA, Barr M,
Thaveau F, Chakfe N, Geny B and Scholey JW: Ischemia reperfusion
injury, ischemic conditioning and diabetes mellitus. J Mol Cell
Cardiol. 91:11–22. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xue J, Zhuang J, Wang X, Meng T, Wu J,
Zhang X and Zhang G: Mechanisms and Therapeutic strategies for
myocardial Ischemia-reperfusion injury in diabetic states. ACS
Pharmacol Transl Sci. 7:3691–3717. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Arslan F, de Kleijn DP, Timmers L,
Doevendans PA and Pasterkamp G: Bridging innate immunity and
myocardial ischemia/reperfusion injury: The search for therapeutic
targets. Curr Pharm Des. 14:1205–1216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila toll protein
signals activation of adaptive immunity. Nature. 388:394–397. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liew FY, Xu D, Brint EK and O'Neill LA:
Negative regulation of toll-like receptor-mediated immune
responses. Nat Rev Immunol. 5:446–458. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ha T, Hu Y, Liu L, Lu C, McMullen JR,
Kelley J, Kao RL, Williams DL, Gao X and Li C: TLR2 ligands induce
cardioprotection against ischaemia/reperfusion injury through a
PI3K/Akt-dependent mechanism. Cardiovasc Res. 87:694–703. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liu Y, Li L, Wang Z, Zhang J and Zhou Z:
Myocardial ischemia-reperfusion injury; Molecular mechanisms and
prevention. Microvasc Res. 149:1045652023. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Favre J, Musette P, Douin-Echinard V,
Laude K, Henry JP, Arnal JF, Thuillez C and Richard V: Toll-like
receptors 2-deficient mice are protected against postischemic
coronary endothelial dysfunction. Arterioscler Thromb Vasc Biol.
27:1064–1071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weerink MAS, Struys M, Hannivoort LN,
Barends CRM, Absalom AR and Colin P: Clinical pharmacokinetics and
pharmacodynamics of dexmedetomidine. Clin Pharmacokinet.
56:893–913. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yuan H, Guo J, Wang C and Zhang C:
Alleviation effects of dexmedetomidine on myocardial
ischemia/reperfusion injury through fatty acid metabolism pathway
via Elovl6. Int Immunopharmacol. 138:1125882024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Sun Z, Tianyun Z, Shaojun L, Ying G, Joe M
and Hao W: Dexmedetomidine attenuates spinal cord
ischemia-reperfusion injury through both anti-inflammation and
anti-apoptosis mechanisms in rabbits. J Transl Med. 16:2092018.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Zhang B, Li G, Zhao H, Tian X, Yu
J, Yin Y and Meng C: Dexmedetomidine alleviates lung oxidative
stress injury induced by ischemia-reperfusion in diabetic rats via
the Nrf2-sulfiredoxin1 pathway. Biomed Res Int. 2022:55847332022.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen L, Cao J, Cao D, Wang M, Xiang H,
Yang Y, Ying T and Cong H: Protective effect of dexmedetomidine
against diabetic hyperglycemia-exacerbated cerebral
ischemia/reperfusion injury: An in vivo and in vitro study. Life
Sci. 235:1165532019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng X, Hu J, Wang Y, Ye H, Li X, Gao Q
and Li Z: Effects of dexmedetomidine postconditioning on myocardial
ischemia/reperfusion injury in diabetic rats: Role of the
PI3K/Akt-Dependent signaling pathway. J Diabetes Res.
2018:30719592018. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li J, Zhao Y, Zhou N, Li L and Li K:
Dexmedetomidine attenuates myocardial ischemia-reperfusion injury
in diabetes mellitus by inhibiting endoplasmic reticulum stress. J
Diabetes Res. 2019:78693182019. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Dexmedetomidine Hydrochloride Injection
[package insert]. WG Critical Care, LLC; Paramus, NJ: 2021
|
|
26
|
U.S. Food and Drug Administration:
Guidance for Industry: Estimating the maximum safe starting dose in
initial clinical trials for therapeutics in adult healthy
volunteers. Center for Drug Evaluation and Research (CDER);
Rockville, MD: 2005
|
|
27
|
Flecknell P: Analgesia and Post-Operative
Care. Laboratory Animal Anaesthesia. 4th edition. Flecknell P:
Academic Press; Oxford: pp. 141–192. 2015
|
|
28
|
Niemegeers CJ, Schellekens KH, Van Bever
WF and Janssen PA: Sufentanil, a very potent and extremely safe
intravenous morphine-like compound in mice, rats and dogs.
Arzneimittelforschung. 26:1551–1556. 1976.PubMed/NCBI
|
|
29
|
Katoh K: Microwave-assisted tissue
preparation for rapid fixation, decalcification, antigen retrieval,
cryosectioning, and immunostaining. Int J Cell Biol.
2016:70769102016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Matheus AS, Tannus LR, Cobas RA, Palma CC,
Negrato CA and Gomes MB: Impact of diabetes on cardiovascular
disease: An update. Int J Hypertens. 2013:6537892013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li M, Wang J, Ding S, Ding B, Oketunbi TJ,
Song X, Li Y, Niu Q, Shi X, Gao D, et al: Cardiac magnetic
resonance imaging-derived pathophysiology and prognosis of diabetes
mellitus with acute myocardial infarction after revascularization:
A prospective cohort study. Ann Med. 56:23997512024. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gan L, Xie D, Liu J, Lau WB, Christopher
TA, Lopez B, Zhang L, Gao E, Koch W, Ma XL and Wang Y: Small
extracellular microvesicles mediated pathological communications
between dysfunctional adipocytes and cardiomyocytes as a novel
mechanisms exacerbating Ischemia/reperfusion injury in diabetic
mice. Circulation. 141:968–983. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ferdinandy P, Andreadou I, Baxter GF,
Botker HE, Davidson SM, Dobrev D, Gersh BJ, Heusch G, Lecour S,
Ruiz-Meana M, et al: Interaction of cardiovascular nonmodifiable
risk factors, comorbidities and comedications with
Ischemia/Reperfusion injury and cardioprotection by pharmacological
treatments and ischemic conditioning. Pharmacol Rev. 75:159–216.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Russo I, Penna C, Musso T, Popara J,
Alloatti G, Cavalot F and Pagliaro P: Platelets, diabetes and
myocardial ischemia/reperfusion injury. Cardiovasc Diabetol.
16:712017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Brown J, Wang H, Hajishengallis GN and
Martin M: TLR-signaling networks: An integration of adaptor
molecules, kinases, and cross-talk. J Dent Res. 90:417–427. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tye H, Kennedy CL, Najdovska M, McLeod L,
McCormack W, Hughes N, Dev A, Sievert W, Ooi CH, Ishikawa TO, et
al: STAT3-driven upregulation of TLR2 promotes gastric
tumorigenesis independent of tumor inflammation. Cancer Cell.
22:466–478. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Li HY, Yuan ZY, Wang YG, Wan HJ, Hu J,
Chai YS, Lei F, Xing DM and Du LJ: Role of baicalin in regulating
toll-like receptor 2/4 after ischemic neuronal injury. Chin Med J
(Engl). 125:1586–1593. 2012.PubMed/NCBI
|
|
38
|
Arslan F, Smeets MB, O'Neill LAJ, Keogh B,
McGuirk P, Timmers L, Tersteeg C, Hoefer IE, Doevendans PA,
Pasterkamp G, et al: Myocardial ischemia/reperfusion injury is
mediated by leukocytic toll-like receptor-2 and reduced by systemic
administration of a novel Anti-Toll-Like Receptor-2 antibody.
Circulation. 121:80–90. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhang T, Ma C, Zhang Z, Zhang H and Hu H:
NF-κB signaling in inflammation and cancer. MedComm (2020).
2:618–653. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hayden MS and Ghosh S: NF-κB, the first
quarter-century: Remarkable progress and outstanding questions.
Genes Dev. 26:203–234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Eltzschig HK and Eckle T: Ischemia and
reperfusion-from mechanism to translation. Nat Med. 17:1391–1401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Dong P, Liu K and Han H: The role of
NF-kappaB in myocardial Ischemia/reperfusion injury. Curr Protein
Pept Sci. 23:535–547. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Timmers L, Sluijter JP, van Keulen JK,
Hoefer IE, Nederhoff MG, Goumans MJ, Doevendans PA, van Echteld CJ,
Joles JA, Quax PH, et al: Toll-like receptor 4 mediates maladaptive
left ventricular remodeling and impairs cardiac function after
myocardial infarction. Circ Res. 102:257–264. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Bao Y, Zhu Y, He G, Ni H, Liu C, Ma L,
Zhang L and Shi D: Dexmedetomidine attenuates neuroinflammation in
LPS-Stimulated BV2 microglia cells through upregulation of miR-340.
Drug Des Devel Ther. 13:3465–3475. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Pang X, Zhang P, Zhou Y, Zhao J and Liu H:
Dexmedetomidine pretreatment attenuates isoflurane-induced
neurotoxicity via inhibiting the TLR2/NF-κB signaling pathway in
neonatal rats. Exp Mol Pathol. 112:1043282020. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Xiang H, Hu B, Li Z and Li J:
Dexmedetomidine controls systemic cytokine levels through the
cholinergic anti-inflammatory pathway. Inflammation. 37:1763–1770.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Du J, Xu Z, Zhen J, Liu J, Yang D, Zheng
EL and Leng JY: Dexmedetomidine attenuates myocardial
ischemia/reperfusion injury through regulating lactate signaling
cascade in mice. Eur Rev Med Pharmacol Sci. 23:3527–3532.
2019.PubMed/NCBI
|
|
48
|
Cheung CW, Qiu Q, Liu J, Chu KM and Irwin
MG: Intranasal dexmedetomidine in combination with
patient-controlled sedation during upper gastrointestinal
endoscopy: A randomised trial. Acta Anaesthesiol Scand. 59:215–223.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cheung CW, Ng KF, Liu J, Yuen MY, Ho MH
and Irwin MG: Analgesic and sedative effects of intranasal
dexmedetomidine in third molar surgery under local anaesthesia. Br
J Anaesth. 107:430–437. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Valverde A and Skelding AM: Alternatives
to opioid analgesia in small animal anesthesia: Alpha-2 agonists.
Vet Clin North Am Small Anim Pract. 49:1013–1027. 2019. View Article : Google Scholar : PubMed/NCBI
|