Introduction
Indium, a group IIIA element of the periodic table,
is a rare earth metal characterized by its low resistance, high
thermal conductivity, corrosion resistance, light permeability and
electrical conductivity (1).
Indium is employed in electronics, optoelectronics, defense
aerospace, nuclear power, semiconductor manufacturing and other
industries. Occupational exposure to indium-containing particles
has increased in previous years due to the rising demand for
consumer electronics (2,3). Consequently, an increasing number of
diseases associated with occupational indium exposure have been
reported (4–6). Indium compounds can be ingested or
inhaled into the human body and are excreted through urine. The
biological half-life of indium is ~2 weeks. Acute exposure to
indium can damage the eyes, skin, mucosal epithelium and the
respiratory system (7). Chronic
exposure, particularly through inhalation of a number of
indium-containing compounds, such as indium-tin oxide or indium
oxide, leads to fatal interstitial pneumonia. In animal
experiments, long-term exposure to indium has been shown to affect
the functions of the kidneys, liver, heart, lungs and blood as well
as cause embryotoxicity and fetal malformations (8,9).
Previous studies have also found that continuous exposure to indium
chloride (InCl3) leads to abnormal male reproductive
function. For example, InCl3 impacts male fertility by
causing DNA damage in sperm. In addition, InCl3 also
affects testicular development; however, the underlying molecular
mechanisms remain unclear (10,11).
Anatomically, the testes are divided into numerous
testicular lobules by the invaginating tunica albuginea, each
containing coiled seminiferous tubules for sperm production
(12). The interstitial space
between seminiferous tubules consists of loose connective tissue
containing testicular Leydig cells, which are responsible for
testosterone biosynthesis. In male mammals, ~95% of testosterone in
the body is synthesized and secreted by Leydig cells (13). Therefore, Leydig cells are
important for the development of secondary sexual characteristics
and germ cell differentiation (14). These cells are key in the proper
development of the fetal testis. Damage to Leydig cells impairs
testicular development and in severe cases, leads to male
infertility.
The mechanisms by which indium induces tissue damage
remain largely underexplored. However, numerous studies have
suggested that excessive production of reactive oxygen species
(ROS) serves a central role in this process (15–17).
Excessive ROS induce oxidative stress, resulting in severe DNA
damage. When DNA damage is not adequately repaired, genomic
instability ensues (18). In
response to DNA damage, cells activate DNA damage response (DDR)
pathways to repair the genome. Furthermore, when the extent of
damage exceeds the repair capacity of the cells, cell death is
triggered to eliminate the affected cells.
DDR generally involves the recognition of DNA
lesions and the initiation of signaling cascades that promote
repair and arrest cell cycle progression. DNA damage signaling is
primarily mediated through protein phosphorylation. A total of
three key members of the PI3K-related kinase family,
ataxia-telangiectasia mutated (ATM), ataxia telangiectasia and
Rad3-related (ATR) and DNA-dependent protein kinase (DNA-PK), serve
as principal regulators of the DDR (19). ATM acts as a central kinase
orchestrating the cellular response to double-strand breaks by
regulating DNA repair, checkpoint activation, apoptosis and
senescence (20). Upon
single-strand breaks, ATR is recruited to extended tracts of
single-stranded DNA coated with replication protein A to promote
DNA repair (21). DNA-PK is
composed of a catalytic subunit (DNA-PKcs) and a regulatory
heterodimer (Ku70/Ku80) (22).
When sensing DNA double-strand breaks, Ku70/Ku80 binds to the DNA
ends and recruits DNA-PKcs, forming the active DNA-PK complex. The
primary role of DDR is to repair damaged DNA and halt cell cycle
progression, as if the damage proves irreparable, the DDR machinery
can initiate apoptotic signaling to eliminate the compromised
cells.
ROS also activates autophagy, which promotes either
cell survival by facilitating damaged organelle removal and
metabolic adaptation, or instead contributes to cell death when
excessively activated or when degradation fails (23). Autophagy is a catabolic process and
a key pathway involved in removing damaged organelles, such as
mitochondria (24). Upon autophagy
initiation, the mTOR pathway is inhibited, which leads to the
activation of unc-51-like autophagy activating kinase 1/2
complexes, initiating the autophagy cascade (25). Subsequently, the class III PI3K
complex is activated to promote formation of the phagophore, and
LC3 is cleaved by autophagy-related (ATG)-4 to expose the
C-terminal glycine residue at position 116 (LC3-I). LC3-I is
conjugated to phosphatidylethanolamine forming LC3-II, which is
inserted into both the inner and outer autophagosomal membranes and
thereby facilitates phagophore elongation, cargo sequestration and
closure of the autophagosome (26). Once formed, the autophagosome fuses
with the lysosome, leading to the degradation of sequestered
cytoplasmic components.
The centrosome is the microtubule organization
center of the cell. It is composed of a pair of centrioles and
pericentriolar material which encompasses the surrounding protein
matrix. This pair of centrioles includes a mother centriole and a
daughter centriole (27). During
interphase of the cell cycle, the centrosome orchestrates the
microtubule networks to maintain the shape of the cell and guide
the direction of the cell migration. When the cell enters the
mitotic phase, the duplicated centrosomes move to the opposite
sites of the nucleus and form mitotic spindle poles to promote
chromosomal arrangement on the equatorial plate (28). During cell cycle progression, each
cell contains one centrosome (before duplication) or two
centrosomes (after duplication). However, when cells suffer from
environmental or intercellular stresses, the centrosome undergoes
amplification (cells with >2 centrosomes), thus leading to
aberrant mitotic spindle poles and genomic instability. Therefore,
precise control of the number of centrosomes stabilizes the
inheritance of the cell genome for undisturbed cell proliferation
(29).
The present study aimed to uncover the molecular
mechanism by which InCl3 inhibits testicular cell
proliferation. The present study aimed to elucidate the molecular
mechanisms by which InCl3 inhibits testicular Leydig cell
proliferation, with a particular focus on whether excessive ROS
production, DNA-PK activation, centrosome amplification and
autophagy are involved in this process.
Materials and methods
Cell culture and drug treatment
Mouse progenitor Leydig TM3 cells and tumor Leydig
MA-10 cells were maintained in DMEM-F12 medium (Invitrogen; Thermo
Fisher Scientific, Inc.), supplemented with 10% FBS (Thermo Fisher
Scientific, Inc.), 1% sodium pyruvate (Thermo Fisher Scientific,
Inc.) and 100 IU/ml penicillin-streptomycin (Thermo Fisher
Scientific, Inc.) at 37°C under a humidified atmosphere with 5%
CO2. Leydig cells were exposed to the following drugs
for the durations indicated in the results section:
InCl3 (10, 20, 50, 100 or 200 µM; cat. no. 203440;
Sigma-Aldrich; Merck KGaA), the ROS scavenger N-acetyl-L-cysteine
(NAC; cat. no. A9165; Sigma-Aldrich; Merck KGaA), the DNA-PK
inhibitor vanillin (1 µM; cat.no. V110-4; Sigma-Aldrich; Merck
KGaA), 2′,7′-dichlorofluorescin diacetate (DCFH-DA; cat. no. D6883;
Sigma-Aldrich; Merck KGaA), diphenyleneiodonium (DPI; 10 µM; cat.
no. D2926; Sigma-Aldrich; Merck KGaA), Mito-TEMPO (10 µM; cat. no.
SML0737; Sigma-Aldrich; Merck KGaA) and the autophagy inhibitor
chloroquine (100 µM; cat. no. NBP2-29386; Novus Biologicals;
Bio-Techne).
Small-interfering RNA (siRNA)
transfection
siRNAs against DNA-PKcs (siDNA-PKcs) and ATG7
(siATG7) were purchased from Sigma-Aldrich; Merck KGaA. The
sequences of siRNAs were as follows: siDNA-PKcs, sense,
5′-CCUUCAGUACGAUUAGCGCCC-3′; antisense,
5′-GGGCGCUAAUCGUACUGAAGG-3′; siATG7, sense,
5′-CCGUUCAUUGAUCAAGAACCC-3′; antisense,
5′-GGUUCUUGAUCAAUAUGAACG-3′; and scrambled siRNA, sense,
5′-UCUGAUCGCACGUAUGAUCUU-3′; antisense,
5′-GAUCAUACGUGCGAUCAGAUU-3′.
For siRNA transfection, Lipofectamine 2000™
(Invitrogen; Thermo Fisher Scientific, Inc.) was mixed with
Opti-MEM (Thermo Fisher Scientific, Inc.), followed by the addition
of 100 nM siRNA. The mixture was then incubated at room temperature
for 25 min before being applied to the cells. Cells were harvested
at room temperature 72 h post-transfection for further
experiments.
Western blotting
Cells were harvested through trypsinization and
lysed on ice for 10 min using the CelLytic™ MT cell lysis reagent
(Sigma-Aldrich; Merck KGaA) supplemented with a commercial protease
inhibitor cocktail (P8340; Sigma-Aldrich; Merck KGaA). The lysates
were centrifuged at 13,000 × g for 10 min at 4°C and the
supernatants were collected. Protein concentrations were determined
using the Bio-Rad Protein Assay Kit (Bio-Rad Laboratories, Inc.).
Equal amounts of protein (50 µg/lane) were mixed with 2X SDS sample
buffer, boiled at 100°C for 10 min and separated by 10% SDS-PAGE at
150 V for 90 min. Proteins were transferred onto PVDF membranes at
20 V and 4°C overnight. Membranes were blocked with 3% BSA in TBST
(containing 0.1% Tween-20) for 1 h at room temperature and
incubated with primary antibodies overnight at 4°C. After washing
with TBST for 30 min, membranes were incubated with HRP-conjugated
secondary antibodies for 1 h at room temperature. Primary (1:7,000)
and secondary (1:7,000) antibody details are provided in Table I. Immunoreactive bands were
visualized using an ECL detection kit (Thermo Fisher Scientific,
Inc.).
 | Table I.Antibodies used for WB and IF. |
Table I.
Antibodies used for WB and IF.
| Antibody | Company | Cat. no. | Application |
|---|
| Actin | MilliporeSigma | MAB1501 | WB |
| GAPDH | GeneTex, Inc. | GTX627408 | WB |
| HSC70 | GeneTex, Inc. | GTX637440 | WB |
| γ-tubulin | GeneTex, Inc. | GTX629704 | IF |
| α-tubulin | GeneTex, Inc. | GTX112141 | WB |
| Cyclin A | Abcam | ab38 | WB |
| Cyclin D | Thermo Fisher
Scientific, Inc. | 2G3G5 | WB |
| Cyclin E | Cell Signaling
Technology, Inc. | 4132 | WB |
| LC3A/B | Cell Signaling
Technology, Inc. | 12741 | WB |
| p-ATM (S1981) | Abcam | Ab81292 | WB |
| ATM | GeneTex, Inc. | GTX70103 | WB |
| p-ATR (S428) | Cell Signaling
Technology, Inc. | 2853 | WB |
| ATR | GeneTex, Inc. | GTX128146 | WB |
| p-DNA-PKcs
(T2609) | GeneTex, Inc. | GTX24194 | WB and IF |
| DNA-PKcs | Santa Cruz
Biotechnology, Inc. | sc-0951 | WB |
| γ-H2AX | Abcam | Ab11175 | IF |
Immunofluorescence microscopy
Following the aforementioned experimental
treatments, cells were fixed and permeabilized with ice-cold
methanol for 5 min. Following permeabilization, cells were
incubated in a blocking buffer containing Triton X-100, Tween-20
and normal goat serum (Thermo Fisher Scientific, Inc.) for 1 h at
room temperature. Primary antibodies were then applied and
incubated overnight at 4°C. The next day, cells were washed three
times with PBS for a total of 30 min and subsequently incubated for
1 h at room temperature with FITC- or Cy3-conjugated goat
anti-mouse or anti-rabbit IgG secondary antibodies (cat. nos.
ab175473 and ab150077; Abcam) in the presence of DAPI for nuclear
staining. Primary (1:200) and secondary (1:500) antibody details
are provided in Table I.
Afterward, cells were washed three additional times with PBS for a
total of 30 min. Coverslips were mounted onto slides using 50%
glycerol in PBS and fluorescence signals were visualized using an
Axio Imager D2 fluorescence microscope (Zeiss AG).
Cell proliferation assay
TM3 cells were cultured at a density of
1×105 cells on culture dishes. After incubation at 37°C
for 24, 48 and 72 h, cells were trypsinized and resuspended with
PBS for counting cell numbers using a hemocytometer under a light
microscope (Zeiss AG). All treatments were performed in triplicate,
with each experiment performed three times.
Flow cytometry
Cell cycle distribution was determined by FACS flow
cytometry. Briefly, TM3 cells were harvested by trypsinization and
resuspended in PBS containing 1 mM EDTA (PBS-E). The cells were
centrifuged at 200 × g for 5 min at 4°C and the pellets were washed
once with PBS-E. After a second centrifugation at 200 × g for 5 min
at 4°C, the cells were resuspended in 0.5 ml PBS-E and fixed by
adding 4.5 ml 70% ice-cold ethanol dropwise with gentle vortexing
at 4°C. Cells were stored at 4°C for 18 h. Before analysis, fixed
cells were washed thoroughly with PBS-E to remove ethanol and then
stained with propidium iodide (SouthernBiotech) for 1 h at room
temperature in the dark. DNA content was measured using a FACScan
flow cytometer (BD Biosciences) and data were analyzed using Kaluza
software version 2.1.3 (Beckman Coulter, Inc.).
5-Ethynyl-2′-deoxyuridine (EdU)
incorporation assay
TM3 cells were seeded on 25×25 mm coverslips in
6-well tissue culture plates and treated with InCl3.
Cell proliferation was assessed using the Click-iT™ EdU Imaging Kit
(Thermo Fisher Scientific, Inc.). Briefly, 1 µl 10 µM EdU working
solution (Component A) was added to the culture medium and cells
were incubated for 1 h at 37°C. After incubation, cells were fixed
and permeabilized with ice-cold methanol for 5 min. The Click-iT™
EdU reaction cocktail was prepared according to the manufacturer's
instructions and added to the cells for 1 h at room temperature in
the dark. Following labeling, cells were washed three times with
PBS for a total of 30 min. EdU signals were visualized using an
Axio Imager D2 fluorescence microscope (Zeiss AG).
Enzyme-linked immunosorbent assay
(ELISA)
Mouse serum testosterone levels were quantified
using a commercially available Mouse T Testosterone ELISA kit (cat.
no. EM1850-HS; Wuhan Fine Biotech Co., Ltd.), according to the
manufacturer's instructions. Briefly, the medium was collected at
room temperature, then centrifuged at 1,000 × g for 20 min at 4°C
to obtain supernatant, which was either assayed immediately or
stored at −80°C until analysis. Standards were prepared by serial
dilution to yield a calibration range of 31.25–2,000 pg/ml. For
each assay, 50 µl samples were added in duplicate to the
T-pre-coated 96-well plate, followed by 50 µl biotin-labeled
detection antibody and incubated for 45 min at 37°C. After three
washes with wash buffer, 100 µl HRP-streptavidin conjugate was
added to each well and incubated for 30 min at 37°C. Plates were
then washed five times, and 90 µl TMB substrate was added and
incubated in the dark at 37°C for 10–20 min, followed by the
addition of 50 µl stop solution to terminate the reaction.
Absorbance was measured at 450 nm using a microplate reader.
Reverse transcription-quantitative
PCR
Total RNA was isolated from TM3 cells treated with
or without InCl3 using TRI Reagent®
(Sigma-Aldrich; Merck KGaA) followed by purification with the
Direct-zol™ RNA Miniprep Kit (Zymo Research Corporation.). After
extraction, RNAs (1 µg) were reverse-transcribed into cDNA using
the SensiFAST™ cDNA Synthesis Kit (Bioline; Meridian Bioscience)
according to the manufacturer's instructions.
Quantitative PCR was performed using FastStart™ SYBR
Green Master Mix (MilliporeSigma) with gene-specific primers at a
final concentration of 0.25 µM. The primer sequences used for mRNA
expression analysis were as follows: Cytochrome P450 family 11
subfamily A member 1 (CYP11A1) forward, 5′-GATCCCGAGGCCCAGCGGTT-3′;
reverse, 5′-AGGGTCATGGAGGTCGTGTCCA-3′; steroidogenic acute
regulatory protein (StAR) forward, 5′-CAGCACTCAGCATGTTCCTCGCT-3′;
reverse, 5′-TCCCCGTTCTCCTGCTGGCTTT-3′; 3-β-hydroxy-δ(5)-steroid dehydrogenase (HSD3B) forward,
5′-TCATTCCCAGGCAGACCATCC-3′; reverse, 5′-CCCTGCAACATCAACTGAGCTG-3′;
and GAPDH forward, 5′-TTTGGCATTGTGGAAGGGCTC-3′; reverse,
5′-CATCACGCCACAGCTTTCCAG-3′.
All reactions were performed in triplicate in a
final reaction volume of 20 µl. The cycling conditions were as
follows: i) Initial denaturation at 95°C for 2 min; ii) 40 cycles
of 95°C for 5 sec; and iii) 60°C for 20 sec. Melt-curve analysis
was conducted to verify amplification specificity. Relative mRNA
expression levels were calculated using the 2−ΔΔCq
method (30) after normalization
to GAPDH as the internal reference.
Statistical analysis
Data are presented as the mean ± SEM. Comparisons
between two groups were performed using unpaired Student's t-tests,
while comparisons among multiple groups were analyzed using one-way
ANOVA tests followed by Tukey's post hoc test for multiple
comparisons. P<0.05 was considered to indicate a statistically
significant difference.
Results
InCl3 inhibits testicular
Leydig cell proliferation
To investigate the effects of InCl3 on
testicular Leydig cell proliferation, mouse progenitor Leydig TM3
cells were treated with a number of concentrations of
InCl3 (10, 20, 50, 100 or 200 µM) for 24, 48 or 72 h,
followed by cell counting. Results demonstrated that the
IC50 value of InCl3 on TM3 cell proliferation
was ~100 µM after 48 h treatment (Fig.
1A). Therefore, this condition was used for the subsequent
experiments. The effect of InCl3 on an additional Leydig
cell line, MA-10, was examined. Treatment with 100 µM
InCl3 inhibited MA-10 cell growth in a time-dependent
manner (Fig. 1B). These findings
indicated that InCl3 suppressed Leydig cell
proliferation.
Subsequently, cell cycle profiles were analyzed
using flow cytometry. The proportion of cells in the
G0/G1 and S-phases was reduced, while those
in the G2/M and polyploidy phases were increased
(Fig. 1C), suggesting that cell
cycle progression was impaired. Given that cell cycle progression
is regulated by cyclin/CDK2 complexes (10), the expression of cyclins and the
activation status of CDK2 were examined. Upon InCl3
treatment, the levels of cyclins D, A and E, as well as the active
form of CDK2 (phosphorylated at Thr161), were reduced (Fig. 1D), supporting the conclusion that
cell cycle progression was inhibited. To further demonstrate this
defect, S-phase entry was assessed using an EdU incorporation
assay. Compared with the control cells, the proportion of
EdU-positive cells was significantly decreased in
InCl3-treated TM3 cells (Fig. 1E and F). Collectively, these
results indicated that InCl3 inhibits the proliferation
of Leydig TM3 cells by impairing cell cycle progression.
Flow cytometry analysis revealed an increased
proportion of polyploid cells, therefore the nuclear morphology was
examined. Following InCl3 treatment, TM3 cells exhibited
irregularly shaped nuclei (Fig.
2A) and a significant increase in nuclear size was also
observed in InCl3-treated cells (Fig. 2B). Additionally, the higher
percentage of cells with micronuclei (small, extranuclear
DNA-containing bodies near the nucleus, as indicated by arrowheads
in Fig. 2C) observed, suggested
that genomic instability had been induced. As genomic instability
is associated with defective mitotic entry, the ability of cells to
enter mitosis was assessed by measuring the mitotic index, defined
as the percentage of cells in mitosis. Cells undergoing mitosis
were identified by DAPI staining, which reveals condensed
chromosomes with increased fluorescence intensity. Findings showed
that the mitotic index was not significantly affected by
InCl3 treatment (Fig.
2D), suggesting that the ability of cells to enter mitosis was
not impaired. Collectively, these findings indicated that
InCl3 treatment induces genomic instability in TM3
cells.
![Genomic instability is observed in
InCl3-treated TM3 cells. InCl3 induces
enlarged nuclei and micronuclei (as indicated by white arrowheads)
in TM3 cells. (A) Nuclear shapes of CTL and
InCl3-treated TM3 cells were analyzed using DAPI
staining. The right panel represents enlarged views of selected
regions. Micronuclei, small DAPI staining near the nucleus, are
indicated by the arrowheads. Mitotic cells were identified and are
marked with red circles. Scale bar, 10 µm. (B) Quantitative results
of the nuclear area shown through DAPI staining. (C) Quantification
of the proportions of cells with micronuclei [as indicated by
selected regions 1 and 2 of (A), right panel]. (D) InCl3
treatment did not affect the mitotic index. The proportions of
mitotic cells (mitotic index) were quantified. (E) InCl3
treatment reduced the levels of testosterone in the culture medium
of TM3 cells. Quantitative results of relative testosterone levels
measured through ELISA in the absence or presence of
InCl3. InCl3 treatment reduced steroidogenic
gene expression. (F) Schematic diagram of testosterone production.
Quantitative results of relative mRNA levels of (G) StAR, (H) HSD3B
and (I) CYP11A1. Results are presented as mean ± SD from ≥3
independent experiments. *P<0.05, **P<0.01 and ***P<0.001.
n.s., not significant; InCl3, indium chloride; StAR,
steroidogenic acute regulatory protein; HSD3B,
3-β-hydroxy-δ(5)-steroid
dehydrogenase; CYP11A1, cytochrome P450 family 11 subfamily A
member 1; E, enlarged nuclei; CTL, control.](/article_images/mmr/33/6/mmr-33-06-13891-g01.jpg) | Figure 2.Genomic instability is observed in
InCl3-treated TM3 cells. InCl3 induces
enlarged nuclei and micronuclei (as indicated by white arrowheads)
in TM3 cells. (A) Nuclear shapes of CTL and
InCl3-treated TM3 cells were analyzed using DAPI
staining. The right panel represents enlarged views of selected
regions. Micronuclei, small DAPI staining near the nucleus, are
indicated by the arrowheads. Mitotic cells were identified and are
marked with red circles. Scale bar, 10 µm. (B) Quantitative results
of the nuclear area shown through DAPI staining. (C) Quantification
of the proportions of cells with micronuclei [as indicated by
selected regions 1 and 2 of (A), right panel]. (D) InCl3
treatment did not affect the mitotic index. The proportions of
mitotic cells (mitotic index) were quantified. (E) InCl3
treatment reduced the levels of testosterone in the culture medium
of TM3 cells. Quantitative results of relative testosterone levels
measured through ELISA in the absence or presence of
InCl3. InCl3 treatment reduced steroidogenic
gene expression. (F) Schematic diagram of testosterone production.
Quantitative results of relative mRNA levels of (G) StAR, (H) HSD3B
and (I) CYP11A1. Results are presented as mean ± SD from ≥3
independent experiments. *P<0.05, **P<0.01 and ***P<0.001.
n.s., not significant; InCl3, indium chloride; StAR,
steroidogenic acute regulatory protein; HSD3B,
3-β-hydroxy-δ(5)-steroid
dehydrogenase; CYP11A1, cytochrome P450 family 11 subfamily A
member 1; E, enlarged nuclei; CTL, control. |
Testicular Leydig cells synthesize testosterone
through the steroidogenic pathway (31). To determine whether
InCl3 influences testosterone production, the levels
were measured following InCl3 treatment. ELISA analysis
revealed a significant reduction in testosterone levels upon
InCl3 treatment (Fig.
2E). Furthermore, the expression levels of steroidogenic genes
StAR and HSD3B, but not CYP11A11, were significantly decreased in
InCl3-treated cells (Fig.
2F-I). These results indicated that InCl3 not only
suppressed Leydig cell proliferation but also downregulated the
expression of key steroidogenic genes involved in testosterone
biosynthesis.
InCl3 induces centrosome
amplification
It has been established that aberrant mitosis
facilitates genomic instability (22). Therefore, the mitotic apparatus in
InCl3-treated TM3 cells was investigated. Under control
conditions, cells exhibited two spindle poles that aligned
chromosomes at the metaphase plate (Fig. 3A, upper panel; Fig. S1A, left panel). However, upon
InCl3 treatment, numerous spindle poles and misaligned
chromosomes were observed (Figs. 3A
and B and S1A), with this
phenotype also being observed in MA-10 cells (Figs. 3C and D and S1B), suggesting that InCl3
treatment led to aberrant mitosis.
Given that multipolar spindles often result from
centrosome amplification during interphase (32), the number of centrosomes was also
assessed. Under control conditions, cells contain one centrosome
before duplication and two after duplication. By contrast,
InCl3-treated TM3 and MA-10 cell lines exhibited a
marked increase in centrosome amplification, characterized by more
than two discrete γ-tubulin-positive foci per cell instead of the
single pair observed in controls (Figs. 3E-H and S2A and B) at 24, 48 and 72 h, supporting
the hypothesis that InCl3 induces centrosome
amplification. Furthermore, as centrosomes function as the
microtubule-organizing centers that coordinate the microtubule
network, the present study further examined the microtubule
organization. In control cells, a well-organized microtubule
network was observed, whereas in InCl3-treated cells,
microtubules clustered into bundles surrounding each centrosome
(Fig. 3I), suggesting disruption
of the microtubule network. Additionally, analysis of the actin
cytoskeleton revealed that the alignment of actin filaments was
disrupted and actin puncta were present in InCl3-treated
cells (Fig. 3J). Together, these
findings indicated that InCl3 induced centrosome
amplification and disrupted both microtubule networks and actin
filament organization.
InCl3 induces centrosome
amplification via elevating ROS production
InCl3 has been reported to lead to
excessive ROS production in a number of cell types (7,33),
however whether it induces ROS in Leydig cells remains unclear. To
address this question, ROS levels were measured in
InCl3-treated Leydig TM3 cells using a DCFH-DA assay.
InCl3 treatment caused a time-dependent increase in
fluorescence intensity, indicating elevated ROS production in TM3
cells (Fig. 4A).
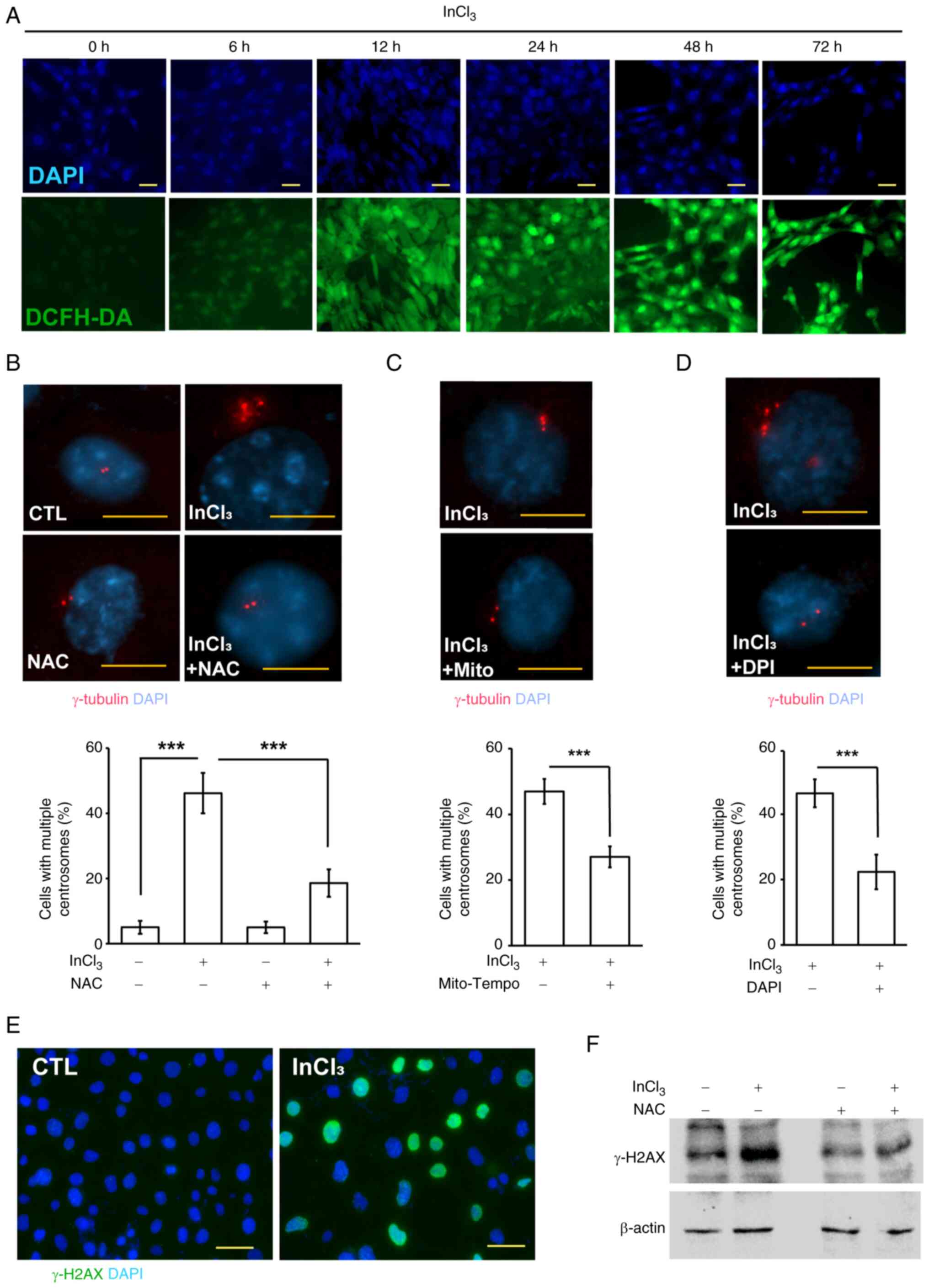 | Figure 4.Treatment of InCl3 induces
ROS production and DNA damage. (A) InCl3 induced ROS
production. Immunofluorescence staining of CTL or
InCl3-treated cells with DCFH-DA (green, ROS-positive
cells) at different time periods. DNA was stained with DAPI. Scale
bars, 20 µm. Inhibition of ROS alleviated InCl3-induced
centrosome amplification. Representative immunofluorescence images
and quantification of TM3 cells with centrosome amplification in
the absence or presence of ROS inhibitors, including (B) NAC, (C)
Mito-Tempo and (D) DPI. Cells were stained with an antibody against
γ-tubulin. DNA was counterstained with DAPI. Scale bars, 10 µm. (E)
InCl3 induced DNA damage. Immunofluorescence staining of
CTL or InCl3-treated cells with an antibody against
γ-H2AX (green). DNA was stained with DAPI. Scale bar, 20 µm. (F)
Inhibition of ROS alleviated InCl3-induced DNA damage.
Extracts of TM3 cells in the absence or presence of ROS inhibitor
(NAC) were analyzed through western blotting with antibodies
against γ-H2AX and actin. Results are presented as mean ± SD from
≥3 independent experiments. ***P<0.001. ROS, reactive oxygen
species; InCl3, indium chloride; NAC,
N-acetyl-L-cysteine; DPI, diphenyleneiodonium; γ-H2AX, γ-H2A
histone family member X; CTL, control. |
Subsequently, the association between ROS and
centrosome amplification was investigated. Notably, treatment with
the ROS scavenger NAC alleviated InCl3-induced
centrosome amplification (Figs. 4B
and S3), suggesting that ROS
generation is required for this phenotype. Two selective inhibitors
were used to further determine whether these ROS originate from
mitochondria or cytosolic NADPH oxidase (NOX) activity,
specifically Mito-TEMPO and DPI. Treatment with Mito-TEMPO, a
mitochondria-targeted scavenger of mitochondrial superoxide,
significantly reduced centrosome amplification (Figs. 4C and S4A). Treatment with DPI, an inhibitor
that primarily suppresses NOX-dependent cytosolic ROS production,
similarly attenuated InCl3-induced centrosome
amplification (Figs. 4D and
S4B). Together, these findings
suggested that InCl3 induced ROS production from both
mitochondrial and NOX-dependent cytosolic sources and that ROS
generation from either pathway contributed to centrosome
amplification.
InCl3 activates DNA-PK to
induce centrosome amplification
As ROS is known to cause DNA damage (34) and the DDR can induce centrosome
amplification in osteosarcoma cells (22,35,36),
it was hypothesized that InCl3 increases ROS levels,
leading to DNA damage and subsequent centrosome amplification in
testicular Leydig cells. To explore this, DNA damage was assessed
through the detection of γ-H2A histone family member X (γ-H2AX), a
marker of DNA double-strand breaks (37). InCl3 treatment
significantly elevated γ-H2AX levels, as shown by both
immunofluorescence staining and western blotting analysis (Fig. 4E and F), indicating that DNA damage
was induced. Moreover, NAC treatment reduced γ-H2AX levels, further
supporting that ROS mediates DNA damage in InCl3-treated
Leydig TM3 cells. Subsequently, the present study tested whether
ROS contributed to centrosome amplification.
DDR signaling pathways including ATM, ATR and DNA-PK
were investigated. Findings revealed that ATM and ATR were not
activated by InCl3 treatment (Fig. 5A and B). However, DNA-PK was
activated, as evidenced by increased phosphorylation of its
catalytic subunit (DNA-PKcs) following InCl3 treatment
(Fig. 5C). Subsequently, the time
course of DNA-PK activation was investigated. The phosphorylation
of DNA-PKcs increased as early as 12 h after InCl3
treatment, remained elevated up to 48 h and then decreased by 72 h
(Fig. 5D), suggesting transient
DNA-PK activation between 12 and 48 h. Collectively, this suggests
that InCl3 may have activated DNA-PK in Leydig cells.
The present study further investigated whether DNA-PK contributes
to centrosome amplification by treating cells with the selective
DNA-PK inhibitor vanillin (38).
Treatment of cells with vanillin reduced InCl3-induced
centrosome amplification (Figs. 5E and
F and S5A). To further
demonstrate this, DNA-PKcs was depleted using siRNA transfection
(Fig. 5G), which also alleviated
InCl3-induced centrosome amplification (Figs. 5H and I and S5B). Therefore, this indicated that
InCl3 promoted centrosome amplification through DNA-PK
activation in Leydig cells.
 | Figure 5.InCl3 activates DNA-PK to
facilitate centrosome amplification. DNA-PK, but not ATM and ATR,
was activated by InCl3 treatment. Extracts of
InCl3-treated TM3 cells were analyzed using western
blotting with antibodies against (A) ATM, p-ATM, (B) ATR, p-ATR,
(C) DNA-PKcs, p-DNA-PKcs and actin. (D) InCl3 activated
DNA-PK in a time-dependent manner. Inhibition or depletion of
DNA-PK alleviated centrosome amplification. (E) Representative
immunofluorescence images and (F) quantitative results of the
proportions of cells with multiple centrosomes in the absence or
presence of DNA-PK inhibitor vanillin. Cells were stained with an
antibody against γ-tubulin. DNA was counterstained with DAPI. Scale
bar, 10 µm. (H) Representative immunofluorescence images and (I)
quantitative results of the proportions of cells with multiple
centrosomes in the absence or presence of siPKcs. Cells were
stained with an antibody against γ-tubulin. DNA was counterstained
with DAPI. Scale bar, 10 µm. (G) DNA-PKcs was depleted efficiently.
Extracts of cells transfected with siPKcs were examined through
western blotting with antibodies against DNA-PKcs and actin. (J)
p-DNA-PKcs localized to the centrosome upon InCl3
treatment. Immunofluorescence staining of CTL or
InCl3-treated cells with antibodies against γ-tubulin
and p-DNA-PKcs. DNA was stained with DAPI. Scale bar, 10 µm.
Results are presented as mean ± SD from ≥3 independent experiments.
***P<0.001. DNA-PKcs, DNA-dependent protein kinase catalytic
subunit; InCl3, indium chloride; ATM, ataxia
telangiectasia mutated; ATR, ATM and Rad3-related Kinase; p-,
phosphorylated; CTL, control; siPKcs, small interfering RNA against
DNA-PKcs. |
A previous study has shown that phosphorylated
DNA-PKcs localizes to the centrosome, thereby facilitating
centrosome amplification (39). To
determine whether this occurs upon InCl3 treatment, the
subcellular localization of phosphorylated DNA-PKcs was examined.
Under control conditions, phosphorylated DNA-PKcs was barely
detectable. However, after InCl3 treatment, in addition
to its nuclear localization, phosphorylated DNA-PKcs was also
observed at the centrosome (Fig.
5H). Taken together, the present data suggested that
InCl3 induced centrosome amplification by activating
DNA-PK.
InCl3 activates autophagy
to induce centrosome amplification
It has previously been established that DNA damage
can activate autophagy (40). In
addition, autophagy promotes centrosome amplification in
trophoblast cells (41).
Therefore, the present study examined whether InCl3
activates autophagy. LC3 puncta increased markedly in TM3 cells
treated with InCl3 (Fig.
6A), indicating activation of autophagy. To further demonstrate
this finding, LC3 lipidation was examined. InCl3
treatment increased the LC3-II/LC3-I ratio (Fig. 6B, upper panel) and reduced
sequestosome 1/p62 levels (Fig.
6B, lower panel), both of which are consistent with enhanced
autophagic activity. When examining the lysosome, treatment with
InCl3 did not affect the expression of lysosomal
proteins, including lysosome-associated membrane protein 1 (LAMP1)
and cathepsin D (Fig. 6C). The
time course of autophagy induction was next characterized. The
LC3-II/LC3-I ratio increased as early as 12 h after
InCl3 treatment, remained elevated up to 48 h and then
decreased by 72 h (Fig. 6D),
suggesting transient autophagy activation between 12 and 48 h.
Next, the present study investigated whether autophagy was mediated
by DNA-PK activation. Treatment of cells with the DNA-PK inhibitor
vanillin mitigated an increase in the LC3-II/LC3-I ratio (Fig. 6E), suggesting that DNA-PK
activation promoted autophagy under InCl3 treatment.
Furthermore, examination of whether autophagy contributes to
centrosome amplification was conducted. Pharmacological inhibition
of autophagy with chloroquine attenuated InCl3-induced
centrosome amplification (Figs. 6F
and S6A). A genetic approach was
followed to further demonstrate this observation. ATG7 knockdown
with siRNA (Fig. 6G) similarly
reduced InCl3-induced centrosome amplification (Figs. 6H and S6B). Together, these results indicated
that InCl3 activated autophagy, which in turn promoted
centrosome amplification.
 | Figure 6.InCl3 induces autophagy to
induce centrosome amplification. (A) Immunofluorescence staining of
CTL and InCl3-treated cells with antibodies against LC3.
DNA was stained with DAPI. Scale bars, 20 µm. (B) Extracts of
InCl3-treated cells were analyzed using western blotting
with antibodies against LC3, p62, actin and HSC70. (C) Treatment
with InCl3 did not alter the expression of lysosomal
proteins. Extracts of InCl3-treated cells were analyzed
using western blotting with antibodies against LAMP1, cathepsin D
and HSC70. (D) InCl3 induced LC3 lipidation in a
time-dependent manner. Extracts of InCl3-treated cells at different
time periods were analyzed using western blotting with antibodies
against LC3 and actin. (E) Inhibition of DNA-PK alleviated
InCl3-induced autophagy. Extracts of InCl3-treated cells were
analyzed through western blotting with antibodies against LC3 and
actin. Inhibition of autophagy or depletion of ATG7 reduced
centrosome amplification. (F) Quantitative results of TM3 cells
treated with InCl3 in the absence or presence of
chloroquine. (G) ATG7 was depleted in TM3 cells transfected with
siATG7. Extracts of siATG7-transfected cells were analyzed using
western blotting with antibodies against ATG7 and HSC70. (H)
Quantitative results of TM3 cells treated with InCl3 in the absence
or presence of siATG7. Results are presented as the mean ± SD from
≥3 independent experiments. **P<0.01 and ***P<0.001.
InCl3, indium chloride; HSC70, heat shock cognate
protein 70; ATG7, autophagy related 7; siATG7, small-interfering
RNA against ATG7; DNA-PK, DNA-dependent protein kinase; CTL,
control; LAMP1, lysosome-associated membrane protein 1. |
Discussion
Consistent with previous reports that indium
compounds impair testicular function and sperm quality (10,11),
the present study demonstrated that InCl3 inhibits the
proliferation of testicular Leydig cells. InCl3 markedly
reduced Leydig cell proliferation and caused marked genomic
instability. Mechanistically, InCl3 increased
intracellular ROS levels, leading to extensive DNA damage, as
evidenced by elevated γ-H2AX levels. ROS-mediated DNA damage either
promotes DNA-PK activation or autophagy induction to centrosome
amplification. These amplified centrosomes became multiple mitotic
spindle poles, thereby causing aberrant mitosis and genomic
instability. In alignment with prior studies linking oxidative
stress and abnormal centrosome duplication in human HCT116 colon
cancer and rat IEC-6 normal small intestine cell lines (42,43),
the present findings provide new insights detailing the mechanisms
by which InCl3 disrupts Leydig cell proliferation and
potentially contributes to male reproductive toxicity.
Human epidemiological data directly associating
occupational indium exposure with male reproductive toxicity remain
very limited. The majority of available information comes from
animal experiments and indirect occupational health observations.
Current biomonitoring studies in indium-exposed workers have
focused mainly on lung, kidney and liver toxicity and have not
systematically evaluated reproductive outcomes (44). By contrast, rodent studies have
consistently showed that indium compounds may cause marked
testicular injury, including reduced testis weight, vacuolar
degeneration, germ-cell loss and abnormal sperm morphology
(7). The severity of these effects
is associated with serum indium concentrations (10). For example, repeated intratracheal
administration of indium compounds in hamsters has resulted in
marked reductions in testis and epididymis weights and severe
long-lasting testicular lesions (45). To the best of our knowledge, at
present, no comparable human data exist regarding testis size or
structural damage among indium-exposed workers. Therefore, concerns
regarding male reproductive toxicity have been based primarily on
animal findings. Although population studies of male infertility in
industrial settings suggest that occupational and environmental
exposures may contribute to semen abnormalities (9,11,45),
a causal association with indium exposure has not been established.
Overall, notable animal evidence and general occupational
infertility data highlight a potential reproductive risk associated
with indium exposure and underscore the need for dedicated human
studies in exposed populations.
Although the IC50 value of 100 µM used in
the present study exceeds the typical levels of environmental or
occupational indium exposure, such concentrations are commonly
employed in in vitro models to elicit detectable cellular
responses, due to the absence of systemic metabolism, distribution
and clearance mechanisms. A previous study has reported that indium
levels in the blood of exposed workers can reach a high ng/ml value
(~10−8 M) (46), while
a further study has demonstrated that indium can accumulate in the
lungs and enter systemic circulation, raising concerns about
long-term low-dose exposure (47).
Furthermore, in vivo studies have indicated that indium
compounds can induce testicular toxicity. For example, testicular
damage and reduced testosterone levels in indium-exposed mice
(48). In addition, similar toxic
effects were observed in rats following inhalation exposure
(49). These findings collectively
support the biological plausibility of the present in vitro
results and highlight the potential reproductive risks associated
with indium exposure. Therefore, while the concentration used in
the present assays may not directly reflect physiological levels,
it serves as a useful model for elucidating the underlying cellular
mechanisms involved in indium-induced testicular toxicity.
Furthermore, it should be noted that the concentration used in the
present study (100 µM) is higher than physiologically relevant
levels and thus represents a limitation of the present study.
However, such non-physiological doses are often necessary in
vitro to reveal potential cellular mechanisms that may not be
detectable under lower exposure conditions.
Aberrant copy numbers of centrosomes drive mitotic
errors and chromosomal instability. Under normal conditions,
centrosome duplication is tightly regulated to ensure the formation
of a bipolar spindle that accurately segregates chromosomes during
mitosis. However, when centrosome amplification occurs, as observed
in InCl3-treated Leydig cells, multipolar mitotic
spindles are observed, thus leading to chromosome mis-segregation,
aneuploidy and genomic instability. Genomic instability imposes a
number of consequences on cell physiology. For example, aneuploid
or polyploid cells often experience cell cycle arrest, senescence
or apoptosis due to activation of the p53-dependent surveillance
mechanisms. This aligns with the present observation regarding
impaired Leydig cell proliferation following InCl3
exposure. In addition, cells that escape checkpoint control may
propagate with abnormal karyotypes, leading to further genetic
imbalances and functional decline. Within the context of Leydig
cells, which are terminally differentiated endocrine cells
responsible for testosterone biosynthesis, persistent genomic
instability may compromise the expression of StAR or CYP11A1
enzymes, thereby reducing androgen production. Chronic exposure to
InCl3, by continuously inducing centrosome amplification
and mitotic defects, gradually depletes the Leydig cell population
or renders them functionally incompetent. This, in turn, would
disrupt the paracrine signaling network between Leydig and Sertoli
cells, leading to defective germ cell maturation and reduced sperm
quality.
Genomic instability in somatic testicular cells,
including Leydig cells, has been associated with long-term
reproductive pathologies. In rodent models, centrosome
amplification in the testis is associated with testicular atrophy,
decreased sperm count and increased sperm DNA fragmentation, all of
which contribute to subfertility or infertility. Given that Leydig
cells are relatively quiescent under physiological conditions, the
induction of centrosome overduplication and consequent mitotic
stress represents a novel mechanism by which InCl3
exerts cumulative damage over time, even at sublethal doses. Taken
together, the present study underscores that centrosome
amplification is not only a transient cellular aberration but a key
event that associates oxidative stress and DNA damage with
endocrine dysfunction and reproductive failure. In the broader
context of occupational and environmental health, prolonged indium
exposure may pose a marked threat to male fertility by targeting
Leydig cells, disrupting hormone production and indirectly
impairing spermatogenesis. Future studies should aim to investigate
whether antioxidant therapies or inhibitors of centrosome
overduplication could mitigate these deleterious effects and
preserve testicular function in individuals exposed to indium
compounds.
The present study demonstrated that DNA-PK, but not
ATM or ATR, was selectively activated following InCl3
exposure. This was a key observation as ATM and ATR are considered
the canonical kinases in the DNA damage response, with ATM
primarily responding to double-strand breaks and ATR responding to
replication stress. By contrast, DNA-PK serves a role in
non-homologous end joining, directly binding to DNA ends through
the Ku70/Ku80 heterodimer and facilitating DNA repair. The present
findings indicate that DNA-PK was the primary activated DDR kinase
in Leydig cells upon InCl3 treatment. Notably, DNA-PK
phosphorylation was observed not only in the nucleus but also at
the centrosome, indicating a non-canonical role in regulating
centrosome dynamics. This dual localization highlights the emerging
concept that DNA-PK exhibits cytoplasmic functions beyond genome
maintenance, particularly in modulating centrosome homeostasis.
Furthermore, phosphorylated DNA-PKcs has been previously detected
at centrosomes under genotoxic stress (50), where it interacts with key
centrosomal proteins such as γ-tubulin, pericentrin and nuclear
mitotic apparatus, thereby promoting overduplication and multipolar
spindle formation.
The present study observed that pharmacological
inhibition or siRNA-mediated depletion of DNA-PKcs significantly
alleviated InCl3-induced centrosome amplification,
suggesting that aberrant activation of DNA-PKcs in the centrosome
promotes centrosome amplification. DNA-PK activation at the
centrosome drives centrosome amplification through a number of
pathways. One possibility is that DNA-PK phosphorylates centrosome
cycle regulators, such as polo-like kinase 4 (PLK4), Aurora A or
NIMA-related kinase 2, which are involved in centriole biogenesis
and separation (51–53). Alternatively, DNA-PK may contribute
to cytoskeletal remodeling by modifying microtubule dynamics
(54), indirectly altering
centrosome positioning and spindle assembly. Within the context of
environmental toxicology, the present study highlights that DNA-PK
activation is a key molecular mediator of indium-induced
reproductive toxicity, making it a potential therapeutic
target.
Although DNA-PKcs has been implicated in regulating
centrosome amplification, whether it directly phosphorylates
centrosomal proteins such as PLK4 or centrosomal protein 192 kDa
(CEP192) remains unclear. Due to the unavailability of
phospho-specific antibodies against PLK4 and CEP192 and the low
efficiency of immunoprecipitation using anti-PLK4 and anti-CEP192
antibodies, the present study was unable to determine their
phosphorylation status. Future studies employing improved detection
methods or phospho-proteomic approaches are required to clarify
whether DNA-PKcs directly phosphorylates these proteins.
The present study demonstrated that autophagy
contributes to InCl3-induced centrosome amplification.
Autophagy activation was observed following InCl3
treatment, as evidenced by increased LC3 puncta and conversion of
LC3-I to LC3-II. Blocking autophagy with chloroquine significantly
reduced centrosome amplification, suggesting that autophagy
provides a permissive environment for aberrant centrosome
duplication. This finding aligns with previous studies
demonstrating that autophagy can promote centrosome amplification
in trophoblast and cancer cells (28,34,41).
Therefore, ROS-induced autophagy may act synergistically with
DNA-PK activation to disrupt centrosome homeostasis.
Collectively, the present findings establish a
mechanistic cascade whereby InCl3 generates ROS, which
induces DNA damage and activates DNA-PK. Activated DNA-PK
accumulates at centrosomes and triggers their amplification.
Concurrently, ROS also stimulates autophagy, which further
facilitates centrosome amplification. This dual pathway ultimately
disrupts mitotic integrity and suppresses Leydig cell
proliferation. These findings not only contribute to the current
understanding of indium-associated reproductive toxicity but also
present novel mechanistic associations between oxidative stress,
DNA damage response, autophagy and centrosome homeostasis.
Supplementary Material
Supporting Data
Acknowledgements
The authors would like to thank Professor Wen-Tai
Chiu (Bioimaging Core Facility of the National Core Facility for
Biopharmaceuticals, Ministry of Science and Technology, Tainan,
Taiwan) for their technical support.
Funding
The present study was supported by grants from The National
Science and Technology Council of Taiwan (grant no.
NSTC-113-2314-B-024-001), The National Cheng Kung University
Hospital (grant no. NCKUH-11402027) and The An Nan Hospital, China
Medical University (grant no. ANHRF113-36).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YNT, CYW, PJS and KCW conceptualized the present
study. YNT, CYW, MYK and RCL devised the methodology. PJS and KCW
conducted the investigation. RCL and MYK were responsible for
software and formal analysis. YNT, CYW and RCL wrote, reviewed and
edited the original manuscript. YNT, CYW, PJS and KCW provided
supervision and acquired funding for the present study. YNT and CYW
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
InCl3
|
indium chloride
|
|
ATM
|
ataxia telangiectasia mutated
|
|
ATR
|
ATM and Rad3-related kinase
|
|
DNA-PK
|
DNA-dependent protein kinase
|
|
DNA-PKcs
|
DNA-PK catalytic subunit
|
|
NAC
|
N-acetyl-L-cysteine
|
|
DCFH-DA
|
2′,7′-dichlorofluorescin
diacetate
|
|
ROS
|
reactive oxygen species
|
|
DDR
|
DNA damage response
|
|
DPI
|
diphenyleneiodonium
|
|
siRNA
|
small-interfering RNA
|
|
γ-H2AX
|
γ-H2A histone family member X
|
|
ATG7
|
autophagy related 7
|
References
|
1
|
Scansetti G: Exposure to metals that have
recently come into use. Sci Total Environ. 120:85–91. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Choi KM and An HC: Characterization and
exposure measurement for indium oxide nanofibers generated as
byproducts in the LED manufacturing environment. J Occup Environ
Hyg. 13:D23–D30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cummings KJ, Virji MA, Park JY, Stanton
ML, Edwards NT, Trapnell BC, Carey B, Stefaniak AB and Kreiss K:
Respirable indium exposures, plasma indium, and respiratory health
among indium-tin oxide (ITO) workers. Am J Ind Med. 59:522–531.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sato A, Saito K, Abe K, Sugimoto K, Nagao
T, Sukeda A and Yunaiyama D: Indium chloride bone marrow
scintigraphy for hepatic myelolipoma: A case report. World J Clin
Cases. 11:4377–4383. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Eskandari A, Glerum DM and Tsui TY:
Influence of Indium (III) chloride on human dermal fibroblast cell
adhesion on tantalum/silicon oxide Nano-composites. Materials
(Basel). 15:35772022. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kimura E, Ahmed S, Chen H and Hiraku Y:
Epithelial-mesenchymal transition in human alveolar cells exposed
to indium chloride. J Appl Toxicol. 45:2353–2362. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tsai PK, Wu SW, Chiang CY, Lee MW, Chen
HY, Chen WY, Chen CJ, Yang SF, Yeh CB and Kuan YH: Evaluation of
cytotoxicity, apoptosis, and genotoxicity induced by indium
chloride in macrophages through mitochondrial dysfunction and
reactive oxygen species generation. Ecotoxicol Environ Saf.
193:1103482020. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Manno RA, Grassetti A, Oberto G, Nyska A
and Ramot Y: The minipig as a new model for the evaluation of
doxorubicin-induced chronic toxicity. J Appl Toxicol. 36:1060–1072.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bomhard EM: The toxicology of indium
oxide. Environ Toxicol Pharmacol. 58:250–258. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lee KH, Chen HP, Leung CM, Chen HL, Tsai
SS and Hsu PC: Effects of indium chloride exposure on sperm
morphology and DNA integrity in rats. J Food Drug Anal. 23:152–160.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee KH, Chen HL, Leung CM, Chen HP and Hsu
PC: Indium acetate toxicity in male reproductive system in rats.
Environ Toxicol. 31:68–76. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Teves ME and Roldan ERS: Sperm bauplan and
function and underlying processes of sperm formation and selection.
Physiol Rev. 102:7–60. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen HB, Pineda Garcia JC, Arizono S,
Takeda T, Li RS, Hattori Y, Sano H, Miyauchi Y, Hirota Y, Tanaka Y
and Ishii Y: DAPL1 is a novel regulator of testosterone production
in Leydig cells of mouse testis. Sci Rep. 11:185322021. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walker WH: Testosterone signaling and the
regulation of spermatogenesis. Spermatogenesis. 1:116–120. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Collin F: Chemical basis of reactive
oxygen species reactivity and involvement in neurodegenerative
diseases. Int J Mol Sci. 20:24072019. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gottschling BC, Maronpot RR, Hailey JR,
Peddada S, Moomaw CR, Klaunig JE and Nyska A: The role of oxidative
stress in indium phosphide-induced lung carcinogenesis in rats.
Toxicol Sci. 64:28–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Brun NR, Christen V, Furrer G and Fent K:
Indium and indium tin oxide induce endoplasmic reticulum stress and
oxidative stress in zebrafish (Danio rerio). Environ Sci Technol.
48:11679–11687. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cheng SM, Shieh MC, Lin TY and Cheung CHA:
The ‘Dark Side’ of autophagy on the maintenance of genome
stability: Does it really exist during excessive activation? J Cell
Physiol. 237:178–188. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Blackford AN and Jackson SP: ATM, ATR, and
DNA-PK: The Trinity at the Heart of the DNA damage response. Mol
Cell. 66:801–817. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shiloh Y and Ziv Y: The ATM protein
kinase: Regulating the cellular response to genotoxic stress, and
more. Nat Rev Mol Cell Biol. 14:197–210. 2013. View Article : Google Scholar
|
|
21
|
Fokas E, Prevo R, Hammond EM, Brunner TB,
McKenna WG and Muschel RJ: Targeting ATR in DNA damage response and
cancer therapeutics. Cancer Treat Rev. 40:109–117. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang CY, Huang EY, Huang SC and Chung BC:
DNA-PK/Chk2 induces centrosome amplification during prolonged
replication stress. Oncogene. 34:1263–1269. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Filomeni G, De Zio D and Cecconi F:
Oxidative stress and autophagy: The clash between damage and
metabolic needs. Cell Death Differ. 22:377–388. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Codogno P and Meijer AJ: Autophagy and
signaling: Their role in cell survival and cell death. Cell Death
Differ. 12 (Suppl 2):S1509–S1518. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ichimura Y, Kirisako T, Takao T, Satomi Y,
Shimonishi Y, Ishihara N, Mizushima N, Tanida I, Kominami E, Ohsumi
M, et al: A ubiquitin-like system mediates protein lipidation.
Nature. 408:488–492. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang CY, Kao YH, Lai PY, Chen WY and Chung
BC: Steroidogenic factor 1 (NR5A1) maintains centrosome homeostasis
in steroidogenic cells by restricting centrosomal DNA-dependent
protein kinase activation. Mol Cell Biol. 33:476–484. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Teng YN, Chang HC, Chao YY, Cheng HL, Lien
WC and Wang CY: Etoposide triggers cellular senescence by inducing
multiple centrosomes and primary cilia in adrenocortical tumor
cells. Cells. 10:14662021. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lai PY, Wang CY, Chen WY, Kao YH, Tsai HM,
Tachibana T, Chang WC and Chung BC: Steroidogenic Factor 1 (NR5A1)
resides in centrosomes and maintains genomic stability by
controlling centrosome homeostasis. Cell Death Differ.
18:1836–1844. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tsai YC, Kuo TN, Chao YY, Lee PR, Lin RC,
Xiao XY, Huang BM and Wang CY: PDGF-AA activates AKT and ERK
signaling for testicular interstitial Leydig cell growth via
primary cilia. J Cell Biochem. 124:89–102. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang CY, Hong YH, Syu JS, Tsai YC, Liu XY,
Chen TY, Su YM, Kuo PL, Lin YM and Teng YN: LRWD1 regulates
microtubule nucleation and proper cell cycle progression in the
human testicular embryonic carcinoma cells. J Cell Biochem.
119:314–326. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lin RH, Yang ML, Li YC, Chang HM and Kuan
YH: Indium chloride-induced micronuclei via reactive oxygen species
in Chinese hamster lung fibroblast V79 cells. Environ Toxicol.
28:595–600. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin RC, Chao YY, Lien WC, Chang HC, Tsai
SW and Wang CY: Polo-like kinase 1 selective inhibitor BI2536
(dihydropteridinone) disrupts centrosome homeostasis via ATM-ERK
cascade in adrenocortical carcinoma. Oncol Rep. 50:1672023.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Tsai YC, Kuo TN, Chao YY, Lin RC, Chien
HH, Peng IT, Tsai YF, Su PJ and Wang CY: Pericentriolar material 1
aggregation maintains cell survival upon prolonged replication
stress. Arch Biochem Biophys. 768:1103832025. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang CY, Tsai SW, Chien HH, Chen TY, Sheu
SY, So EC and Huang BM: Cordycepin inhibits human gestational
choriocarcinoma cell growth by disrupting centrosome homeostasis.
Drug Des Devel Ther. 14:2987–3000. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chao YY, Huang BM, Peng IC, Lee PR, Lai
YS, Chiu WT, Lin YS, Lin SC, Chang JH, Chen PS, et al: ATM- and
ATR-induced primary ciliogenesis promotes cisplatin resistance in
pancreatic ductal adenocarcinoma. J Cell Physiol. 237:4487–4503.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen TY, Huang BM, Tang TK, Chao YY, Xiao
XY, Lee PR, Yang LY and Wang CY: Genotoxic stress-activated
DNA-PK-p53 cascade and autophagy cooperatively induce ciliogenesis
to maintain the DNA damage response. Cell Death Differ.
28:1865–1879. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang CY, Lai PY, Chen TY and Chung BC:
NR5A1 prevents centriole splitting by inhibiting centrosomal DNA-PK
activation and beta-catenin accumulation. Cell Commun Signal.
12:552014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lien WC, Chen TY, Sheu SY, Lin TC, Kang
FC, Yu CH, Kuan TS, Huang BM and Wang CY: 7-hydroxy-staurosporine,
UCN-01, induces DNA damage response, and autophagy in human
osteosarcoma U2-OS cells. J Cell Biochem. 119:4729–4741. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsai YC, Kuo TN, Lin RC, Tsai HL, Chao YY,
Lee PR, Su PJ and Wang CY: MicroRNA-155-5p inhibits trophoblast
cell proliferation and invasion by disrupting centrosomal function.
Mol Med Rep. 29:852024. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bian XK, Guo JL, Xu SX, Han YW, Lee SC and
Zhao JZ: Hexavalent chromium induces centrosome amplification
through ROS-ATF6-PLK4 pathway in colon cancer cells. Cell Biol Int.
46:1128–1136. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Zhang RK, Wang P, Lu YC, Lang L, Wang L
and Lee SC: Cadmium induces cell centrosome amplification via
reactive oxygen species as well as endoplasmic reticulum stress
pathway. J Cell Physiol. 234:18230–18248. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hoet P, De Graef E, Swennen B, Seminck T,
Yakoub Y, Deumer G, Haufroid V and Lison D: Occupational exposure
to indium: What does biomonitoring tell us? Toxicol Lett.
213:122–128. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tanaka A: Health effects of indium
compounds in animal experiments. J Occup Health. 67:uiaf0072025.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Choi S, Won YL, Kim D, Yi GY, Park JS and
Kim EA: Subclinical interstitial lung damage in workers exposed to
indium compounds. Ann Occup Environ Med. 25:242013. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Ding CG, Wang HQ, Song HB, Li ZH, Li XP,
Ye SS, Zhang FG, Cui SW, Yan HF and Li T: Occupational exposure to
indium of indium smelter workers. Biomed Environ Sci. 29:379–384.
2016.PubMed/NCBI
|
|
48
|
Nagano K, Nishizawa T, Umeda Y, Kasai T,
Noguchi T, Gotoh K, Ikawa N, Eitaki Y, Kawasumi Y, Yamauchi T, et
al: Inhalation carcinogenicity and chronic toxicity of indium-tin
oxide in rats and mice. J Occup Health. 53:175–187. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Maghraoui S, Florea A, Ayadi A, Matei H
and Tekaya L: Changes in organ weight, sperm quality and
testosterone levels after aluminum (Al) and indium (In)
administration to Wistar rats. Biol Trace Elem Res. 201:766–775.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Zhang S, Hemmerich P and Grosse F:
Centrosomal localization of DNA damage checkpoint proteins. J Cell
Biochem. 101:451–465. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhou K, He Y, Lin X, Zhou H, Xu X and Xu
J: KIFC1 depends on TRIM37-mediated ubiquitination of PLK4 to
promote centrosome amplification in endometrial cancer. Cell Death
Discov. 10:4192024. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Qi H, Kikuchi M, Yoshino Y, Fang Z, Ohashi
K, Gotoh T, Ideta R, Ui A, Endo S, Otsuka K, et al: BRCA1
transports the DNA damage signal for CDDP-induced centrosome
amplification through the centrosomal Aurora A. Cancer Sci.
113:4230–4243. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bobbitt JR, Cuellar-Vite L, Weber-Bonk KL,
Yancey MR, Majmudar PR and Keri RA: Targeting the mitotic kinase
NEK2 enhances CDK4/6 inhibitor efficacy by potentiating genome
instability. J Biol Chem. 301:1081962025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ma S, Rong Z, Liu C, Qin X, Zhang X and
Chen Q: DNA damage promotes microtubule dynamics through a
DNA-PK-AKT axis for enhanced repair. J Cell Biol.
220:e2019110252021. View Article : Google Scholar : PubMed/NCBI
|
















