The global incidence of cancer continues to rise
annually, remaining a major health challenge worldwide (1). According to the latest estimates from
the International Agency for Research on Cancer, 2022 witnessed
nearly 20 million new cancer cases [including non-melanoma skin
cancers (NMSCs)] and 9.7 million cancer-related deaths (including
NMSCs) (2). Epidemiological
projections indicate that approximately one in five individuals
will develop cancer during their lifetime, with about one in nine
men and one in 12 women succumbing to the disease (3). Current therapeutic strategies for
cancer patients primarily include surgical resection, radiotherapy,
chemotherapy and small-molecule targeted therapies. Unlike
conventional chemotherapy that indiscriminately kills cancer cells,
targeted therapies disrupt specific oncogenic pathways to inhibit
cancer-cell replication and proliferation while minimizing damage
to normal tissues. However, limitations persist, including a
scarcity of actionable therapeutic targets and the emergence of
drug resistance during treatment (4–6).
Consequently, there is an urgent need to identify novel therapeutic
targets and elucidate mechanisms underlying treatment resistance in
oncology research.
Transcription factors are critical regulatory
proteins that govern gene expression by binding to specific DNA
sequences, thereby precisely controlling transcriptional processes
(7). These proteins typically
comprise two functional domains: A DNA-binding domain [e.g., zinc
fingers or helix-loop-helix (HLH) motifs] that recognizes conserved
sequences in promoters or enhancers, and an activation/repression
domain that recruits RNA polymerase or histone-modifying complexes
to initiate or suppress transcription (8). Their spatiotemporal specificity
enables selective activation in distinct cell types or
developmental stages, orchestrating cellular differentiation,
metabolic regulation and stress responses (9). Investigations into transcription
factor networks not only reveal disease mechanisms but also provide
theoretical foundations for targeted therapies. As central hubs of
gene regulatory networks, they hold significant potential in
synthetic biology and cellular reprogramming (10).
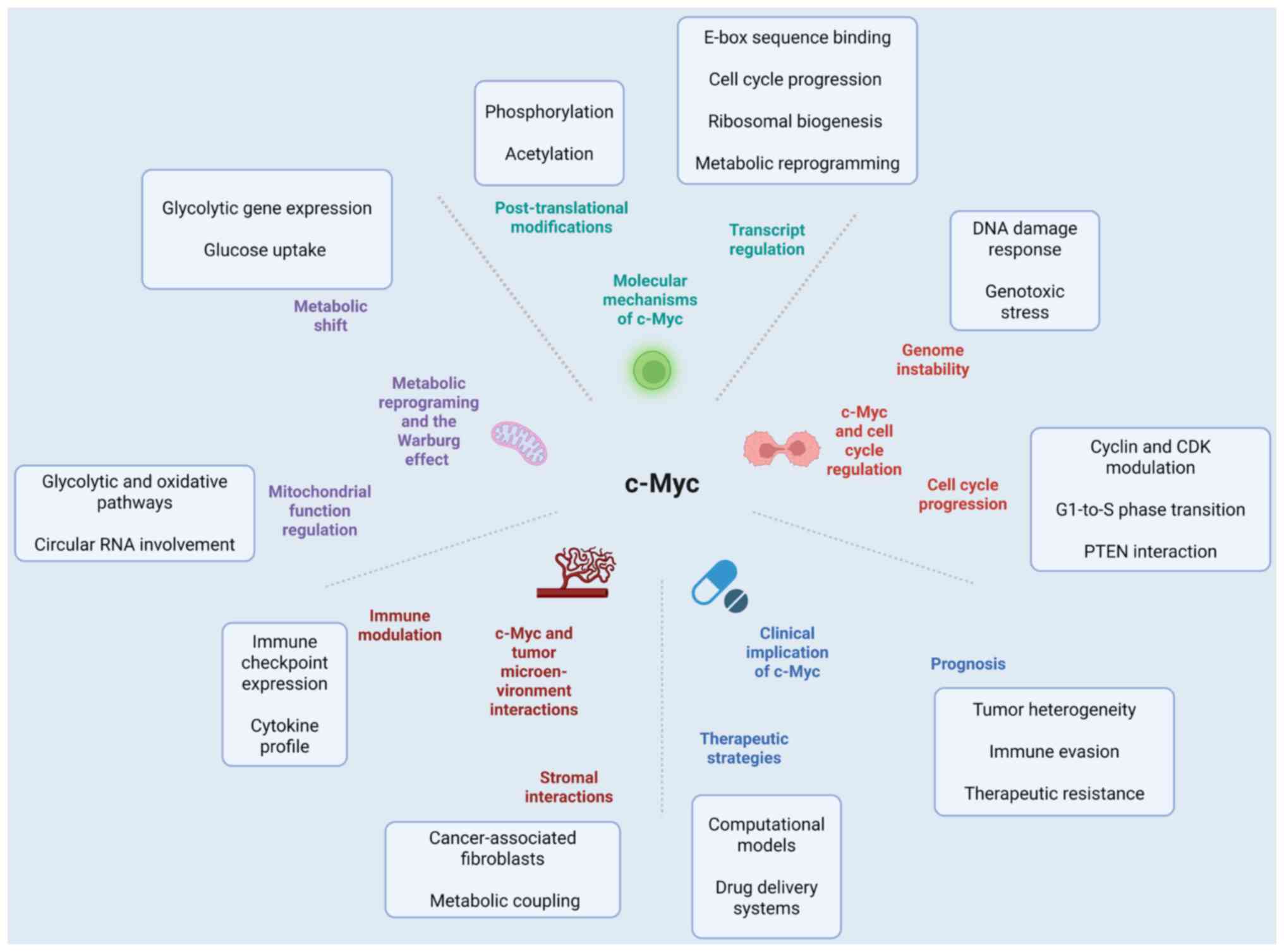
Among these, the myelocytomatosis oncogene (MYC)
family (including c-Myc, N-Myc and L-Myc) represents a pivotal
group of transcription factors regulating cell proliferation and
differentiation. These proteins share a conserved basic HLH leucine
zipper (LZ) domain (11). By
forming heterodimers with MYC associated factor X (MAX) proteins,
they specifically bind E-box sequences (CACGTG) in target gene
promoters, modulating the transcriptional activity of ~15% of human
genes (12). Under physiological
conditions, MYC proteins coordinate cell cycle progression,
metabolic reprogramming (e.g., enhanced glycolysis and
glutaminolysis) and ribosome biogenesis to promote tissue
development and regeneration, while simultaneously suppressing
differentiation signals through antagonism of mother against
decapentaplegic (MAD)/MAX network transcriptional repressor (MNT)
family transcription factors (13).
The pathogenesis and progression of cancer involve a
multi-step, multi-stage process driven by genetic alterations.
Studies have revealed that aberrant activation of MYC family genes
(e.g., through chromosomal translocations, gene amplification or
dysregulation of upstream signaling pathways) is a hallmark of
numerous cancers, with c-Myc overexpression observed in ~70% of
human malignancies (14).
Dysregulated c-Myc expression is closely associated with cancer
initiation and progression (15).
Research demonstrates that c-Myc regulates >15% of the human
genome and orchestrates transcription mediated by all three RNA
polymerases (I, II and III), directly influencing 2,000-4,000
target genes. This broad regulatory capacity has earned c-Myc the
designation of a ‘master gene regulator’ (16,17).
c-Myc is aberrantly activated in diverse
hematological malignancies, including leukemia (18), lymphoma (19) and multiple myeloma (20), and solid tumors, such as pancreatic
ductal adenocarcinoma (21),
non-small cell lung cancer (NSCLC) (22), SCLC (23), hepatocellular carcinoma (24), prostate cancer (25) and breast cancer (26). Mechanistically, c-Myc drives
tumorigenesis through multiple pathways: Promoting cell
proliferation (27), suppressing
apoptosis (28), reprogramming
metabolism (29), inducing
angiogenesis (30) and modulating
cancer stem cell maintenance (31). Beyond cell-autonomous effects,
c-Myc also remodels the tumor microenvironment (TME) and
facilitates immune evasion (26,32).
These findings collectively position c-Myc as a pivotal therapeutic
target in the era of molecular oncology.
In conclusion, given the direct correlation between
the dysregulation of c-Myc and the development of tumors, this
review summarizes the latest progress in c-Myc research and
systematically examines the three core issues of c-Myc research: i)
c-Myc, as a transcription factor, lacks a typical drug-binding
pocket and is difficult to be directly targeted; furthermore, it
performs essential physiological functions in normal cells and
systemic inhibition is prone to cause toxicity; ii) c-Myc is
embedded in redundant and dynamic transcriptional regulatory
networks, with complex mechanisms, making it difficult to validate
the targets and challenging to meet the requirements of clinical
translation for predictability and controllability; iii) the
function of c-Myc is significantly influenced by tissue type,
genetic background and microenvironment, limiting the applicability
of broad-spectrum intervention strategies. In response to these
issues, four complementary research paths were summarized: i)
Inhibiting the dimerization of c-Myc/Max; ii) targeting key
co-factors such as transcription domain associated protein (TRRAP)
and bromodomain protein 4 (BRD4), to indirectly regulate
c-Myc-dependent transcriptional programs; iii) using degradation
mechanisms to eliminate c-Myc; iv) combining with immunotherapy and
stress pathways. Additionally, in the discussion, the c-Myc
research related to personalized medicine, proteolysis-targeting
chimeras (PROTAC) technology and nano-delivery is outlined,
clarifying the feasible steps for the transformation of basic
discoveries into therapeutic applications, providing a concise and
operational reference for targeting c-Myc.
c-Myc, encoded by the human chromosomal locus 8q24,
is a 439-amino-acid oncoprotein characterized by a C-terminal
DNA-binding domain and an N-terminal transactivation domain (TAD)
(33). The C-terminal region
contains a 100-residue LZ motif that mediates heterodimerization
with its LZ partner MAX, enabling DNA binding to gene promoters
(Fig. 1) (34). As a member of the Myc family, which
also includes N-Myc and L-Myc, c-Myc shares high homology with its
paralogs but exhibits distinct expression patterns (35). While c-Myc is ubiquitously
expressed in proliferating cells and tightly regulated at genetic,
protein and mRNA levels, N-Myc and L-Myc display more restricted
spatiotemporal expression during cellular and tissue development
(36).
The N-terminal TAD (residues 1–143) is an
intrinsically disordered domain critical for c-Myc's
transcriptional activation and biological activity (37). Comprising MB0, MBI and MBII
subdomains, the TAD contains canonical phosphorylation sites (e.g.,
S62 and T58) that regulate c-Myc stability via phosphorylation
cascades (38,39). MBII (residues 129–143), the most
extensively studied subdomain, serves as a hub for key protein
interactions and is indispensable for c-Myc's oncogenic potential
(39). Adjacent to this region,
the MBIIb subdomain (residues 226–270) features a proline (P),
glutamic acid (E), serine (S) and threonine (T)-rich ‘PEST’ motif
involved in stability regulation independent of ubiquitination
(40). The MBIII subdomain
modulates protein stability and enhances cellular transformation,
whereas MBIV exhibits context-dependent variability in
transformation assays (41).
The C-terminal LZ domain (residues 357–439)
facilitates nuclear localization and dimerization with MAX. This
heterodimer binds E-box sequences (CACGTG) in target gene promoters
via disulfide bonds, initiating transcriptional activation
(42,43) (Fig.
1).
Collectively, c-Myc's structural architecture
enables its dual role as a transcriptional activator and repressor,
coordinating critical cellular processes such as transcription
(44), translation (45), chromatin remodeling (46) and proteostasis (47). A study revealed that c-Myc binds
nearly all active promoters and enhancers, regulating genes
essential for cell growth (48).
Under physiological conditions, c-Myc expression is tightly
controlled at transcriptional, post-transcriptional and
post-translational levels. However, dysregulation via chromosomal
translocations, insertional mutagenesis or gene amplification leads
to oncogenic c-Myc accumulation. This drives metabolic
reprogramming to sustain rapid tumor cell proliferation, ultimately
fueling cancer initiation and progression (49,50).
Cancer is a complex and heterogeneous disease driven
by dysregulated oncogene expression, which disrupts the homeostasis
of oncogenic or tumor suppressor signaling pathways (51,52).
Studies have shown that abnormal c-Myc expression, observed in most
malignant tumors, plays a key role in tumorigenesis as it controls
critical cellular processes (53,54).
However, the oncogenic function of c-Myc is not uniform; its
upstream regulatory mechanisms, dominant downstream effector
networks and ultimate clinical significance exhibit profound
‘context dependence’ across different cancer types. This
specificity stems from the unique genetic background,
microenvironment and driving signals of each cancer type, causing
c-Myc to play different roles in tumor progression.
For instance, in breast cancer, the abnormal
activation of c-Myc is often closely related to hormone signaling
and post-transcriptional regulation, with its function strongly
pointing towards metabolic reprogramming, maintenance of stem cell
characteristics and treatment resistance (5). c-Myc enhances VEGF expression by
stimulating the translation of VEGF mRNA, highlighting its role in
mediating the interaction between cancer cells and the TME
(55,56). Elevated estrogen levels are
associated with c-Myc expression, especially in estrogen receptor
(ER)-positive patients, where there is a mutual dependence between
estradiol and c-Myc. c-Myc is considered a classic estrogen-induced
gene in breast cancer cells. Knockdown of insulin-like growth
factor 2 mRNA-binding protein 1 (IGF2BP1) reduces the stability of
c-Myc mRNA, while its ectopic expression enhances the stability of
c-Myc mRNA under normoxic conditions (57). Hypoxia-induced long non-coding RNA
KB-1980E6.3 recruits IGF2BP1 to stabilize c-Myc mRNA, thereby
promoting self-renewal and stemness maintenance in breast cancer
(57). High c-Myc expression
significantly enriches cancer stem cells (CSCs) (58). Diclofenac inhibits the
proliferation of triple-negative breast cancer (TNBC) by
down-regulating c-Myc, reducing glucose uptake and inhibiting
glycolysis (59). c-Myc lies
downstream of the lysine methyltransferase 2D (KMT2D)-histone H3
lysine 4 mono-methylation (H3K4me1)-Y-box binding protein (YBX1)
axis; its expression in TNBC is suppressed upon KMT2D or YBX1
knockdown. c-Myc or SENP1 re-expression rescues the proliferation
and migration in KMT2D/YBX1-deficient TNBC cells (60). Methyltransferase-like protein 3
stabilizes the c-Myc/WD repeat-containing protein 5 (sWDR5)
interaction through a methylation-independent mechanism, enhancing
c-Myc's transcriptional activity on glycolytic genes and promoting
the development of TNBC (61).
3-Bromopropionic acid inhibits the growth of TNBC by downregulating
c-Myc, suppressing glycolysis (lactate production, ATP synthesis
and hexokinase activity) and inducing mitochondrial apoptosis
(62,63). Overexpression of protein arginine
methyltransferase 1 (PRMT1) is associated with upregulation of
c-Myc in TNBC, where PRMT1 stabilizes c-Myc to promote tumor
progression and confer resistance to olaparib. Targeting PRMT1 can
enhance the sensitivity to olaparib (64). The above studies demonstrate the
specific role of c-Myc in breast cancer, particularly in the
aggressive subtype TNBC, as a core node of treatment resistance and
a key executor of metabolic reprogramming. By contrast, in
pancreatic cancer, c-Myc more often functions as an integration hub
downstream of oncogenic signaling pathways (such as KRAS), with its
primary role being to drive metabolic reprogramming and construct
an immunosuppressive barrier in response to the nutrient-poor
microenvironment. In pancreatic cancer, c-Myc directly binds to the
fibroblast growth factor binding protein 1 (FGFBP1) promoter to
drive its expression. The F-Box and WD repeat domain containing 7
(Fbw7)/c-Myc axis regulates FGFBP1 levels, and inhibiting c-Myc can
suppress angiogenesis and tumor progression (65). c-Myc cooperates with programmed
cell death 1 (PD-1), and inhibiting c-Myc can enhance PD-1
checkpoint blockade (66). c-Myc
upregulates CD47 transcription, enabling pancreatic cancer cells to
evade immune phagocytosis through the CD47-signal regulatory
protein α (SIRPα) interaction. Blocking CD47 can enhance the
infiltration of CD8+ T cells and macrophages and inhibit tumor
growth (67,68). The mucin 5AC, oligomeric
mucus/gel-forming (MUC5AC)/β-catenin/c-Myc axis promotes
glutamine/glutamate metabolism, pyrimidine biosynthesis and
gemcitabine resistance by upregulating MUC1/hypoxia-inducible
factor (HIF)-1α and glycolysis (69,70).
CD36 inhibits β-catenin/c-Myc signaling by proteasomal degradation
of glypican-4 (GPC4), thereby suppressing glycolysis and tumor
growth. Ectopic GPC4 reactivates β-catenin/c-Myc signaling in
colorectal cancer, suggesting that targeting GPC4 may overcome
treatment resistance and tumor stem cell characteristics (71). Overexpression of c-Myc induces
epithelial-mesenchymal transition in chemotherapy-resistant
cancer-associated fibroblasts and secretion of exosomal microRNA
(miR)-106b, while exosome inhibitors can reverse gemcitabine
resistance (72). Circular RNA
PDK1 acts as a scaffold for the ubiquitin conjugating enzyme E2 O
ubiquitin ligase and forms a ternary complex with bridging
integrator 1 (BIN1) to induce BIN1 degradation, thereby relieving
its inhibition on c-Myc transcriptional activity and promoting
pancreatic cancer growth, metastasis and glycolysis (73). This reveals a specific network of
c-Myc in pancreatic cancer that integrates metabolism, immune
microenvironment and treatment resistance. In other types of
malignant tumor, the regulation and function of c-Myc also have
their unique characteristics. c-Myc transcriptionally activates
colon cancer-associated transcript-1, which in turn enhances c-Myc
expression in cervical cancer through Wnt/β-catenin signaling
(74–76). c-Myc binds to the growth
differentiation factor 15 (GDF-15) promoter to drive its
expression, and GDF-15 indirectly activates c-Myc, which is
associated with the progression and metastasis of cervical cancer
(77–79), suggesting a unique positive
feedback loop in gynecological tumors. In bladder cancer, c-Myc is
a downstream target of the NF-κB pathway, and Rab23 promotes cell
proliferation and invasion by activating NF-κB (80). X-linked Inhibitor of Apoptosis
Protein stabilizes c-Myc by inhibiting glycogen synthase kinase 3β
(GSK-3β)-mediated Thr58 phosphorylation (81). miRNA-451 directly targets the
3′-UTR of c-Myc to inhibit the migration and invasion of bladder
cancer (82), demonstrating the
specificity of regulation at different levels. In hematological
malignancies and certain types of solid tumor, the stability
regulation and direct transcriptional activation mechanisms of
c-Myc are particularly prominent, making its function more akin to
a direct oncogene-driven event. In chronic myeloid leukemia,
overexpression of cancerous inhibitor of protein phosphatase 2A
stabilizes c-Myc by inhibiting protein phosphatase 2A-mediated
dephosphorylation, thereby driving blast crisis (83). Notch homolog 1 (Notch1) directly
transcribes and activates c-Myc to stimulate the proliferation of
leukemia cells (84–86). In SCLC, histone deacetylase (HDAC)7
promotes tumorigenesis by regulating the acetylation and nuclear
transport of β-catenin, upregulating c-Myc and Exportin 1 (XPO1).
XPO1 inhibitors show enhanced efficacy in SCLC models with high
HDAC7 expression (87). Dual mouse
double minute 2 homolog (MDM2)/XPO1 inhibition increases nuclear
p53 levels, suppresses MYC transcription and induces apoptosis in
tumor protein (TP)53 wild-type acute myeloid leukemia (AML), with
cells expressing high levels of c-Myc showing greater sensitivity
(88). These studies indicate that
interventions targeting c-Myc protein stability, transcriptional
complexes or nuclear transport may have specific therapeutic
potential in such cancers.
In summary, c-Myc is not a single-function
pan-cancer driver but a transcriptional regulatory hub that exerts
its effects depending on the specific TME and molecular background.
The downstream biological effects triggered by its abnormal
activation, including metabolic reprogramming, maintenance of stem
cell characteristics, immune evasion and treatment resistance, vary
significantly among different cancer types. This variation stems
from the cancer-type-specific combinations of upstream regulatory
mechanisms (such as hormone signaling, kinase pathways, RNA-binding
proteins or epigenetic modifications) and core effector pathways
(such as glycolysis, Wnt/β-catenin, CD47-SIRPα or NF-κB).
Therefore, systematically dissecting the dominant regulatory nodes
and functional output profiles of c-Myc in various malignant tumors
will help develop more cancer-type-adapted intervention strategies.
For example, targeting the ER-c-Myc axis or the IGF2BP1-mRNA
stability pathway in ER-positive breast cancer; combining
inhibition of CD47 or FGFBP1 downstream of c-Myc in pancreatic
cancer; and using dual MDM2/XPO1 inhibition to induce c-Myc
downregulation in TP53 wild-type AML. Such precision intervention
approaches based on mechanistic heterogeneity will significantly
enhance the clinical feasibility and patient benefit of
c-Myc-targeted therapy.
c-Myc is one of the most frequently dysregulated
transcriptional regulators in tumorigenesis. Its oncogenic effect
stems from the coordinated regulation of multiple malignant
biological processes rather than the activation of a single
pathway. It upregulates cell cycle-related genes by binding to
E-box sequences, driving continuous proliferation and enhancing
ribosome biogenesis and protein translation. To meet the demands of
rapid proliferation, it simultaneously promotes glycolysis and
glutaminolysis to provide energy and biosynthetic precursors. In
the DNA damage response, it exhibits dual regulation: On the one
hand, it exacerbates genomic instability, and on the other hand, it
maintains basic repair capacity to ensure cell survival.
Additionally, it remodels the TME by regulating immune checkpoint
molecules, angiogenic factors and matrix remodeling enzymes. It
also activates stem cell-related genes and inhibits
differentiation, endowing cells with self-renewal, heterogeneity
and treatment resistance. These functions are integrated by c-Myc
and are interdependent, forming a complete oncogenic program
(Fig. 2).
The c-Myc oncoprotein stimulates the cell cycle via
three primary mechanisms: Upregulating cyclins and cyclin-dependent
kinase (CDKs), downregulating CDK inhibitors (p15, p21 and p27),
and accelerating G1-to-S phase transition (89). Dysregulated c-Myc activity drives
uncontrolled proliferation, a hallmark of tumorigenesis (90). c-Myc knockout reduces Cell division
cycle 25A (Cdc25A) expression, diminishes cyclin B1/Cdc2 activity,
enhances radiation-induced G2/M arrest and sensitizes LNCaP cells
to ionizing radiation (91).
Beyond activating cyclins and CDKs, c-Myc promotes cell cycle
progression by impairing ‘braking’ proteins. For example, p27
requires phosphorylation at Thr-187 for recognition and
ubiquitination by the SCF-S-phase kinase associated protein 2
(SKP2) complex (89). c-Myc
facilitates p27 degradation by the following mechanisms: i)
Inducing Skp2 expression (92,93),
ii) activating Cdk2 via cyclin upregulation (94), and iii) activating Cdk1 (89,95).
In lymphoma and osteosarcoma cells, c-Myc inhibition upregulates
p21, inducing G2/M arrest (14,96).
In summary, c-Myc is a central oncogenic
transcription factor regulating cell cycle progression. As a MYC
family member, it forms heterodimers with MAX to bind E-box
sequences (CACGTG) in target gene promoters, directly controlling
~15% of human genes (97). c-Myc
drives G1/S transition by upregulating cyclin D and CDK4/6,
activating E2F transcription and suppressing CDK inhibitors (e.g.,
p21, p27), thereby sustaining proliferative signaling (93,98).
c-Myc influences DNA damage response (DDR)
mechanisms, fostering genomic instability (99). By interacting with repair proteins,
c-Myc modulates cellular responses to genotoxic stress,
underscoring its potential as a therapeutic target for enhancing
DNA-damaging agents (100). c-Myc
binds promoters of DNA double-strand break (DSB) repair genes
[e.g., Nijmegen Breakage Syndrome 1 (NBS1), X-Ray Repair Cross
Complementing 6, Rad5-like protein (Rad5)1, breast cancer type 2
susceptibility protein, Rad50 double-strand break repair protein,
DNA repair and recombination protein Rad54 and DNA-dependent
protein kinase catalytic subunit (DNA-PKcs)], regulating their
expression (100). Inhibiting
c-Myc-mediated DNA repair induces genomic instability and mitotic
catastrophe, sensitizing tumor cells to chemotherapy and radiation
(101). In prostate cancer, c-Myc
knockout suppresses the homologous recombination (HR) [via NBS1,
RAD1 checkpoint DNA exonuclease, structural maintenance of
chromosomes 1A] and non-homologous end joining (NHEJ) [via X-ray
repair cross complementing 6, protein kinase, DNA-activated,
catalytic subunit, polynucleotide kinase 3′-phosphatase] pathways,
enhancing radiosensitivity (101). In embryonal rhabdomyosarcoma,
c-Myc inhibition activates intrinsic apoptosis, exacerbates DSB
damage and impairs DNA-PKc (NHEJ) and RAD51 (HR) recruitment,
increasing radiosensitivity (102). c-Myc also regulates mismatch
repair (MMR) gene expression. MMR corrects base mismatches and
insertion-deletion loops, ensuring replication fidelity (103–105). Silencing MutL homolog 1 (MLH1)
and MutS homolog 2 (MSH2), core MMR proteins, disrupts
post-radiation mismatch correction, sensitizing cells to apoptosis
(106,107). In melanoma, c-Myc downregulation
suppresses MLH1/MSH2 and activates p53-independent apoptosis,
enhancing γ-radiation sensitivity (108). In breast cancer, c-Myc
amplification synergizes with HR deficiency to drive poly
ADP-ribose polymerase inhibitor resistance (109).
Collectively, c-Myc-driven DDR dysregulation
(observed in ~40% of c-Myc-driven tumors) creates ‘synthetic
lethality’ vulnerabilities (e.g., with p53 loss), offering
therapeutic opportunities (e.g., ATR inhibitors) (110,111). These findings highlight c-Myc's
dual role in accelerating tumorigenesis and modulating therapy
sensitivity, emphasizing the need to dissect its regulatory
mechanisms for combination therapy development.
Metabolic reprogramming, particularly the Warburg
effect (aerobic glycolysis), supports biosynthetic demands and
rapid proliferation in cancer (112). c-Myc drives this shift by
enhancing glycolytic gene expression and glucose uptake (113). Non-coding RNAs (e.g., miR-181d)
stabilize c-Myc, further promoting glycolysis and oncogenic
signaling (114). c-Myc also
balances mitochondrial glycolysis and oxidative phosphorylation to
sustain tumor growth (115).
Circular RNA ECE1 regulates the c-Myc/thioredoxin interacting
protein axis, exemplifying its role in metabolic reprogramming
(e.g., in osteosarcoma) (116).
Lactate shuttling between tumor and stromal cells reflects
c-Myc-driven cooperative metabolism within the TME (117). In nasopharyngeal carcinoma, c-Myc
mediates latent membrane protein 1 (LMP1)-induced hexokinase 2
(HK2) upregulation, enhancing glycolysis. HK2 knockdown
radiosensitizes LMP1-overexpressing cells (118). c-Myc also governs glutamine
metabolism by increasing glutamine transporters and glutaminase
expression, promoting glutaminolysis (119,120). Enhanced glutamine metabolism
confers radioresistance via G2/M checkpoint override (121), nucleotide synthesis (122) and redox balance modulation
(123). These findings suggest
targeting c-Myc-mediated metabolic pathways (e.g.,
glucose/glutamine metabolism) may improve radiosensitivity.
In summary, c-Myc-driven metabolic reprogramming
(the Warburg effect) is a hallmark of cancer. Deciphering its
molecular underpinnings opens avenues for metabolic intervention
strategies.
c-Myc shapes the TME by regulating immune evasion,
angiogenesis and stromal crosstalk (124,125). It alters immune checkpoint
expression (e.g., PD-L1, CD47) and cytokine profiles, enabling
immune escape. Senescent cells in the TME secrete pro-inflammatory
factors, further promoting tumor growth (26). c-Myc directly binds programmed cell
death ligand 1 (PD-L1) and CD47 promoters to enhance their
transcription (126–128). c-Myc knockout downregulates
PD-L1/CD47, boosting anti-tumor immunity, while PD-L1/CD47
overexpression rescues c-Myc loss-induced growth suppression
(127). c-Myc inhibition reduces
mitochondrial reactive oxygen species (ROS) in hypoxic cells,
diminishing ROS-mediated Fe2+-to-Fe3+
conversion, thereby activating prolyl hydroxylase to degrade HIF-1α
(129). In endometrial cancer,
c-Myc silencing suppresses HIF-1α, enhancing radiosensitivity.
HIF-1α overexpression reverses this effect, implicating c-Myc in
radioresistance via HIF-1α stabilization (130).
These findings position c-Myc as a master regulator
of both intrinsic tumor proliferation and TME remodeling. Targeting
the c-Myc-TME axis (e.g., combined with anti-angiogenics or immune
checkpoint inhibitors) may enhance therapeutic efficacy, though its
dynamic complexity warrants further exploration for precision
strategies.
CSCs, with self-renewal and tumor-initiating
capacities, drive cancer progression (131). c-Myc maintains CSC stemness by
directly activating pluripotency genes [Nanog homeobox (NANOG),
octamer-binding transcription factor 4, SRY-box transcription
factor 2(SOX2)] and modulating the Wnt/β-catenin, Notch and Hippo
pathways, while suppressing differentiation signals (e.g., miR-34a)
(132–137). The arginyl-tRNA
synthetase-mitotic arrest deficient 1 like 1 fusion gene promotes
nasopharyngeal carcinogenesis and chemoradioresistance by
activating the Far upstream element binding protein 1/c-Myc axis,
inducing CSC-like properties (138). In T-cell acute lymphoblastic
lymphoma, c-Myc binds the HIF-2α promoter, sustaining CSC
self-renewal via Nanog and SOX2 (139). Triptolide (C1572), a natural
compound, selectively depletes therapy-resistant CSCs in TNBC by
degrading c-Myc via the proteasome. c-Myc knockout mimics this
effect, inducing CSC senescence (140). In conclusion, c-Myc also drives
CSC plasticity through epigenetic remodeling (e.g., DNA
demethylation), conferring adaptive advantages (141). As a central node in CSC
regulatory networks, c-Myc represents a promising target for
eradicating tumor recurrence.
Overall, c-Myc-driven tumorigenesis is a highly
integrated biological process. The cell cycle progression,
metabolic reprogramming, DNA damage response, TME remodeling and
stem cell characteristics it regulates are not independent of each
other but form a functionally interconnected and
multi-feedback-regulated network. In this network, continuous
proliferation is the core phenotype, and metabolic reprogramming
and adaptive regulation of the DNA damage response jointly support
the maintenance of this phenotype. The biosynthetic demands
triggered by c-Myc's acceleration of the cell cycle are met by its
simultaneous activation of glycolysis and glutaminolysis programs;
the corresponding metabolites (such as acetyl-CoA and
α-ketoglutarate) can serve as substrates for epigenetic
modifications, feeding back to enhance c-Myc's transcriptional
activity and the stemness features it induces. Meanwhile, the
replication stress and ROS accumulation caused by rapid
proliferation and metabolic activities trigger genomic instability.
c-Myc selectively regulates homologous recombination, mismatch
repair and other pathways, allowing for limited genetic variation
accumulation under the premise of ensuring basic cell survival,
thereby influencing the evolutionary trajectory of tumors. c-Myc
also shapes an immunosuppressive microenvironment by upregulating
immune checkpoint molecules and angiogenic factors, providing
protection for the above-mentioned intrinsic oncogenic programs;
its activation of stem cell-related genes helps maintain tumor
heterogeneity, invasion potential and treatment resistance.
Therefore, c-Myc is a core regulatory hub coordinating multiple
oncogenic functions. This systemic feature suggests that targeting
a single downstream effector molecule is susceptible to network
compensation mechanisms; in contrast, directly interfering with
c-Myc protein itself (such as through protein degradation or
transcriptional inhibition), or jointly blocking multiple key
functional outputs (such as metabolic intervention combined with
immune checkpoint blockade), may more effectively disrupt the
oncogenic steady state it drives (Fig.
2).
As a transcription factor, c-Myc plays a central
role in cell proliferation, differentiation and metabolism by
regulating downstream gene expression. Its aberrant activation is
implicated in >70% of human cancers (44). However, c-Myc has long been
considered an ‘undruggable’ target due to the lack of a
well-defined binding pocket, its reliance on protein-protein
interactions, and its nuclear localization, which complicates
therapeutic targeting. Additionally, c-Myc is frequently activated
via amplification rather than mutation in most tumors, and its
inhibition may disrupt normal cellular functions, leading to severe
toxicity (13,54). Recent advances in protein
engineering, RNA technology and artificial intelligence have
revitalized efforts to target MYC, with multiple inhibitors now in
clinical trials, offering new hope for cancer therapy (36). Below, current c-Myc-targeted
strategies and clinical progress are discussed.
As a typical intrinsically disordered protein, c-Myc
exhibits high dynamics and lacks a stable tertiary structure.
Coupled with its strict nuclear localization property, this poses
significant scientific challenges for targeted intervention: The
extended and conformationally diverse protein surface makes it
difficult to form deep and clear small molecule binding sites,
severely limiting the design of traditional inhibitors;
furthermore, the nuclear membrane barrier further restricts the
effective delivery of candidate drugs to the target site.
Therefore, current research and development strategies have
systematically shifted towards an indirect intervention mode based
on mechanistic understanding, aiming to dismantle the oncogenic
function of c-Myc from multiple dimensions. This transformation
needs to be understood in the broader context of the evolution of
the target biology paradigm-compared to traditional kinase targets,
c-Myc lacks a clear active groove at the structural level, and its
protein-protein interaction interface exhibits a dynamic flat
characteristic, making it difficult to provide high-affinity
small-molecule binding sites; in terms of the mechanism, it does
not rely on catalytic activity, so the ‘enzyme activity inhibition’
strategy (such as blocking ATP binding) cannot be adopted, and
instead, it must interfere with its dynamic assembly processes such
as dimerization, DNA binding and cofactor recruitment; in terms of
design thinking, the ‘occupation-driven’ classic model is largely
ineffective, breakthroughs are concentrated on ‘function-driven’
(such as blocking MYC/MAX) or ‘elimination-driven’ (such as PROTAC
degradation, RNA interference) approaches, with the core logic
shifting from ‘inhibition’ to ‘elimination’; in the clinical
transformation aspect, although there are a large number of kinase
inhibitors approved for market, direct small molecule inhibitors
for c-Myc have not yet achieved success, highlighting the necessity
and challenge of the paradigm shift. This comparison aims to
illustrate that the latest progress in the c-Myc field (such as
degraders and RNA therapies) not only represents technological
iteration but also represents a fundamental evolution of the drug
design concept for ‘undruggable’ targets.
At the level of transcriptional complexes, c-Myc
must form a heterodimer with MAX to specifically recognize and bind
to the E-box sequence (5′-CACGTG-3′) in the promoter region of
target genes, thereby initiating the expression of downstream
oncogenes (142,143). Thus, targeting the
protein-protein interaction interface or interfering with its DNA
binding ability has become a key path to block oncogenic signaling,
and such strategies (such as OMO-103) have been proven not only to
inhibit tumor cell proliferation but also to down-regulate the
expression of c-Myc-driven immunosuppressive molecules (such as
PD-L1), potentially reversing the immune microenvironment (143). At the level of protein
homeostasis regulation, c-Myc has an extremely short half-life
(~20–30 min), and its ubiquitin-proteasome-dependent degradation is
precisely regulated by a phosphorylation cascade-phosphorylation of
serine 62 (S62) mediated by ERK, CDK or JNK can enhance its
stability, while subsequent phosphorylation of threonine 58 (T58)
by GSK-3β triggers recognition by E3 ubiquitin ligases such as
FBW7, ultimately leading to proteasomal degradation (144). Based on this, small molecule
compounds (such as 361 and 975) can accelerate the clearance of
c-Myc by promoting T58 phosphorylation or inhibiting S62
modification (66), and this
clearance at the protein level is thought to simultaneously relieve
c-Myc's transcriptional drive on multiple immune checkpoints. More
advanced strategies rely on PROTAC technology to achieve controlled
degradation: Currently, they mainly fall into three categories-the
first is ‘indirect targeting’, which targets key transcriptional
co-factors of c-Myc, such as bromodomain and extra terminal domain
(BET) family protein BRD4 (which recruits c-Myc to chromatin by
recognizing histone acetylation marks and is a core co-factor for
c-Myc transcriptional activation). Several BRD4-PROTACs based on
JQ1 or OTX015 (such as ARV-825) have entered preclinical studies
and have been shown to significantly downregulate c-Myc expression
through efficient degradation of BRD4, potentially relieving
multiple inhibitions on the immune system; the second is ‘direct
targeting’-molecules such as AU-15330 developed by Aurigene aim to
bind to the c-Myc/Max dimer interface and recruit mental
retardation E3 ligase, which has been confirmed in preclinical
models to induce endogenous c-Myc degradation and is currently in
the in-depth validation stage, with its potential for combination
with immunotherapy being highly anticipated; the third is an
exploratory ‘RNA-PROTAC fusion strategy’, which involves designing
small-molecule ligands that specifically recognize specific
secondary structures of c-Myc mRNA (such as G-quadruplexes in the
5′UTR region) as ‘warheads’, and then coupling them with E3 ligase
ligands to construct bifunctional molecules. This strategy is still
in the early stage of concept validation (145,146). At the level of transcriptional
regulation, given that the c-Myc promoter is rich in
super-enhancers and highly dependent on epigenetic cooperation,
targeting the upstream transcriptional machinery is also an
effective alternative approach: BET bromodomain inhibitors (such as
JQ1 and OTX015) weaken the anchoring of BRD4 at the c-Myc promoter
and its recruitment of the transcriptional elongation complex by
blocking BRD4′s recognition of acetylated histones (147–149). These inhibitors have shown
potential for synergy with anti-PD-1/PD-L1 therapies in preclinical
studies; CDK7/9 inhibitors (such as KB-0742) directly inhibit the
phosphorylation of RNA polymerase II and transcriptional
elongation, thereby globally suppressing c-Myc mRNA synthesis
(150), and may also sensitize to
immunotherapy by reducing the expression of immune checkpoint
molecules (150). In terms of
RNA-level intervention, RNA interference technology also shows
significant value: Small interfering RNA (siRNA) requires a
delivery system (such as lipid nanoparticles or GalNAc conjugation)
to enter cells. The representative drug DCR-MYC (developed by
Dicerna, using the proprietary GalXC™ delivery
technology) is an intravenous siRNA designed to specifically
silence MYC mRNA; antisense oligonucleotides (ASO) are
single-stranded DNA/RNA molecules that mainly inhibit translation
through RNase H-mediated mRNA degradation or steric hindrance.
Although AZD4785 (developed by Ionis/AstraZeneca, targeting KRAS
mRNA) does not directly act on c-Myc, its clinical success strongly
validates the feasibility of ASO technology in targeting
‘undruggable’ oncogenic transcripts. ASOs targeting c-Myc have
demonstrated efficacy in preclinical models and silencing c-Myc can
reshape the tumor immune microenvironment, but still faces
challenges in in vivo delivery efficiency and nucleic acid
stability; short hairpin RNA is usually delivered by viral vectors
(such as lentivirus or adeno-associated virus) and processed into
siRNA in cells. Currently, it is more commonly used in gene therapy
research and cell therapy (for example, in the modification of
chimeric antigen receptor-T cells to knockdown c-Myc to enhance
their in vivo persistence). At the level of stress pathway
synergy, overexpression of c-Myc can lead to a sharp increase in
protein synthesis load, imbalance in ribosome biogenesis and
intensified endoplasmic reticulum stress, making tumor cells
specifically dependent on pathways such as the unfolded protein
response (UPR). Therefore, combined targeting of key nodes in the
UPR (such as inositol-requiring enzyme 1 or protein kinase R-like
endoplasmic reticulum kinase) can significantly amplify the
apoptotic signals induced by c-Myc inhibition, achieving a
synergistic anti-tumor effect (151,152).
In summary, although the above strategies take
different paths, they are complementary and work in concert,
collectively forming a multi-level targeted network covering
transcription, translation, protein homeostasis and stress
response, systematically dismantling the oncogenic program driven
by c-Myc. This provides a solid, diverse and translational
scientific basis for breaking through the long-held perception of
c-Myc as ‘undruggable’. It is particularly worth emphasizing that
the combination of these c-Myc-targeting strategies with immune
checkpoint inhibitors is emerging as a highly promising new
direction for overcoming immune therapy resistance and converting
‘cold tumors’ into ‘hot tumors’. The exploration in this area is
ongoing. Collectively, these multi-pronged strategies provide
innovative avenues to overcome c-Myc's ‘undruggability’.
At present, the clinical intervention strategies
targeting c-Myc mainly include direct inhibition, functional
interference, epigenetic regulation, protein degradation and
indirect transcriptional inhibition (Table I). These explorations not only
reflect the continuous efforts to target this core oncogene but
also systematically reveal the multiple challenges it faces in
terms of biological regulatory complexity and drug development
feasibility. A careful analysis of the clinical outcomes of each
strategy provides an important basis for subsequent rational drug
design (153).
G-quadruplex stabilizers such as CX-3543 can inhibit
c-Myc transcription by stabilizing specific DNA secondary
structures in the c-Myc promoter region and preventing
transcription factor binding. Although CX-3543 showed promising
antitumor activity in early clinical trials in neuroendocrine
tumors, poor pharmacokinetic properties, unacceptable dose-limiting
toxicity and unsatisfactory clinical efficacy led to the
termination of clinical trials. Its research and development work
was terminated. The specific reasons are as follows: i) The
pharmacokinetic profile of CX-3543 was a fatal flaw in its clinical
development. Studies have shown that the drug has a high systemic
clearance (rapid elimination from the body), making it difficult to
maintain blood drug concentrations within the effective therapeutic
window. In order to achieve the target inhibition effect, patients
need frequent intravenous injection (such as once a day or multiple
times a week), which not only seriously affects patient compliance,
but also increases the risk of complications such as infection and
phlebitis. ii) In the phase II study, CX-3543 showed severe retinal
toxicity (e.g., retinal pigment epithelial cell damage, visual
field defects), and the incidence of toxicity increased with
increasing doses. This toxicity was ‘dose-limiting’ (i.e., could
not be avoided by dose adjustment) and could not be mitigated by
dose reductions, ultimately preventing treatment continuation.
Retinal toxicity is an important safety hazard of anticancer drugs,
especially for patients with long-term use, which may lead to
permanent vision impairment. Therefore, regulatory agencies have a
low tolerance for retinal toxicity. iii) Although CX-3543 showed
inhibition of c-Myc pathway and ribosome reconstitution in multiple
cell models (e.g., tumor cell lines and animal transplanted
tumors), the objective response rate of CX-3543 in phase II
clinical trials (for a variety of solid tumors, e.g.,
neuroendocrine tumors and lymphomas) was low (well below the
prespecified response threshold). This impractical dosing regimen,
combined with the observed dose-limiting retinal toxicity in
patients, resulted in an unacceptable benefit-risk ratio that did
not meet regulatory criteria for further clinical advancement
(ClinicalTrials.gov no, NCT00780663) (153–156). This finding suggests that the
discontinuation of CX-3543 was not due to a single factor but
rather to a combination of pharmacokinetic, safety and efficacy
failures. Therefore, the development and use of c-Myc inhibitors
should be further explored. At the same time, the specific
recognition and effective targeting of nuclear DNA structures by
small molecules still face significant limitations in
pharmacokinetics and delivery efficiency. Antisense
oligonucleotides (such as INX-3280) degrade c-Myc mRNA through an
RNase H-dependent pathway. Although the proof-of-concept study in
AML was discontinued, it confirmed the feasibility of RNA-level
targeting (157,158); current research and development
efforts are focused on improving the tumor-targeting delivery
efficiency, serum stability and cellular uptake of nucleic acid
drugs. OMO-103 (Omomyc) is a small protein that can penetrate the
cell membrane and inhibit the formation of c-Myc/MAX heterodimers
by competitively binding to MAX. It has shown good safety in phase
I/II trials for metastatic pancreatic ductal adenocarcinoma and
osteosarcoma, and can reduce c-Myc activity in tumor tissues,
inhibiting proliferation and migration (159,160). Its potential synergistic value
lies in the possible alleviation of the c-Myc-driven
immunosuppressive microenvironment, but attention should be paid to
the immunogenicity risk of peptide drugs, the difficulty of
large-scale production processes, and the tissue distribution and
target occupancy in solid tumors after systemic administration.
OTX-2002 is an mRNA-based lipid nanoparticle delivery system
designed to induce epigenetic silencing at the c-Myc gene locus. In
early clinical trials for hepatocellular carcinoma (NCT05497453),
it was observed that c-Myc expression was downregulated and tumor
growth was inhibited (161); its
long-term application requires further assessment of off-target
epigenetic editing risks, organ selectivity of the nanoparticle
carrier and potential immune activation effects. WBC100 promotes
the proteasomal degradation of c-Myc by interfering with its
nuclear localization signal and has shown acceptable safety and
target inhibition activity in phase I trials for c-Myc-positive
advanced solid tumors (162); its
clinical translation potential depends on the depth, duration and
tolerance of the therapeutic effect, and related mechanism studies
will also help guide the development of more precise protein
degradation strategies. BET inhibitors (such as JQ1) indirectly
inhibit c-Myc transcription by blocking the binding of coactivators
such as BRD4 to acetylated histones. Early clinical data show that
they have broad-spectrum anti-tumor activity (147,149); however, the first-generation
compounds are limited by hematological toxicity (such as
thrombocytopenia) due to insufficient target selectivity, which has
driven the exploration of highly selective second-generation
inhibitors and combination therapy regimens.
In summary, the clinical practice of c-Myc targeted
therapy has been continuously accumulating key experiences:
Terminated trials have revealed critical bottlenecks in aspects
such as target biology, drug delivery and patient selection, while
strategies in the clinical development stage need further
validation in terms of delivery efficiency, resistance control and
long-term safety. Future research should focus on deepening the
understanding of translational mechanisms, optimizing subject
selection based on molecular typing, prospectively designing
combined intervention plans and developing new delivery
technologies with greater tissue selectivity to effectively advance
c-Myc as a druggable and manageable clinical target. Furthermore,
these clinical advancements highlight the potential of c-Myc in the
treatment of various cancers. Ongoing research and technological
innovations are expected to develop more effective c-Myc targeted
drugs, bringing new hope to patients and enriching the scientific
basis of precision oncology. It is particularly worth emphasizing
that the success of future c-Myc targeted therapies is likely not
only due to their direct anti-proliferative effects but also to the
synergistic sensitization effects produced by combined strategies
such as immunotherapy, opening up new avenues for conquering
refractory solid tumors by reshaping the immune
microenvironment.
Overall, the successful paradigm of traditional
targeted therapy is mainly based on enzyme targets such as kinases
with well-defined catalytic domains. These targets typically have
stable three-dimensional conformations and deep, conserved
ligand-binding pockets, and their functions are highly dependent on
enzymatic activities that can be competitively inhibited by small
molecules (such as ATP binding), thus supporting rational drug
design based on X-ray crystallography or cryo-electron microscopy
structures and successfully driving the approval of multiple kinase
inhibitors for clinical use. In contrast, c-Myc, as a typical
transcription factor, presents fundamentally different scientific
challenges for targeting. There are systematic differences between
kinases and c-Myc in terms of structural characteristics,
functional realization mechanisms and intervention logics: Kinases
have rigid, structured active centers, while c-Myc (especially its
N-terminal transactivation domain) is an intrinsically disordered
protein, lacking a persistent, recognizable hydrophobic binding
interface; the oncogenic effect of kinases directly stems from
their catalytic activity, and inhibiting this activity can
effectively interrupt downstream signal transduction, while the
functional realization of c-Myc depends on multiple dynamic
processes-including forming heterodimers with MAX, specifically
binding to E-box sequences (CACGTG) on DNA, and assembling
functional transcriptional complexes with co-regulators such as
TRRAP and WDR5, and its oncogenicity is the result of a combination
of increased protein expression levels, enhanced complex stability
and amplified transcriptional output; therefore, the
‘occupation-driven’ inhibition strategy for kinases is not
applicable to c-Myc. Current promising intervention approaches
focus on event-driven strategies, such as recruiting E3 ubiquitin
ligases to c-Myc protein or its key co-factors (such as BRD4, WDR5)
through PROTAC technology to induce ubiquitination modification and
proteasomal degradation; or using RNA interference (siRNA, ASO),
targeted protein degradation or transcriptional inhibition to
reduce its functional abundance at the mRNA or protein level. These
methods do not rely on the recognition of traditional binding
pockets but rather weaken the oncogenic function output of c-Myc by
regulating protein homeostasis or gene expression. This shift from
‘inhibiting activity’ to ‘regulating abundance and assembly’
reflects that drug development targeting transcription factor-like
targets is gradually moving towards a new stage with clearer
mechanisms and more feasible pathways, and also provides a
methodological framework for addressing other ‘undruggable’
targets.
c-Myc, as a proto-oncogenic transcription factor,
regulates biological processes such as cell proliferation,
differentiation and metabolism, and its aberrant activation is
implicated in >70% of human cancers. However, several challenges
persist in c-Myc research: First, developing targeted therapeutic
strategies for c-Myc faces significant hurdles due to its
ubiquitous expression in both normal and cancer cells, and direct
inhibition strategies have yet to achieve substantial clinical
success. Second, while c-Myc expression and activity are regulated
by multiple signaling pathways, its precise regulatory network
remains incompletely understood, necessitating further elucidation
to inform effective therapeutic development. Finally, the
context-dependent roles of c-Myc across cancer types-such as
promoting proliferation in certain malignancies while suppressing
differentiation in others- add complexity to its study. Although
the discovery of c-Myc has opened new avenues in cancer research,
its intricate regulatory mechanisms and pleiotropic functions pose
ongoing challenges. Future studies must prioritize unraveling
c-Myc's molecular mechanisms and devising precise therapeutic
strategies to overcome its limitations in cancer treatment.
Despite extensive research into c-Myc's role in
cancer, its complex regulatory mechanisms and potential therapeutic
targets require further exploration. Future directions may include:
i) Targeted therapy: Developing small-molecule drugs to directly or
indirectly inhibit c-Myc function, such as by disrupting c-Myc-Max
complex formation or blocking its DNA binding; ii) transcriptional
co-factor studies: Delving into c-Myc's interactions with
transcriptional co-factors to clarify its gene regulatory
mechanisms; iii) isoform-specific functions: Investigating
functional differences among c-Myc isoforms and their
cancer-specific roles; and iv) immunotherapy integration: Exploring
c-Myc's role in tumor immune evasion and its potential synergy with
immune checkpoint inhibitors. In summary, as a master regulator of
cell proliferation and cancer progression, c-Myc's structural and
functional features offer critical research avenues. Future efforts
should focus on refining targeted therapies and dissecting its
roles in the TME and immune evasion to advance cancer
treatment.
To date, personalized medicine has emerged as a key
focus, as genomic technologies enable tailoring therapies to
individual tumor genetic profiles. This approach enhances treatment
precision while minimizing adverse effects. Advances in drug
delivery systems, particularly nanotechnology-based platforms,
improve c-Myc targeting and therapeutic specificity. Novel
inhibitors, including peptide-based drugs and PROTACs, represent
significant progress in overcoming c-Myc's targeting
challenges.
In conclusion, c-Myc is a multifunctional protein
with vital biological roles, yet the relationship between its
structure and function demands deeper investigation. A
comprehensive understanding of its structure, function and
interactions will illuminate c-Myc's central role in cellular
biology and inspire innovative therapeutic strategies for related
diseases.
Not applicable.
This work was supported by a program for the grants from the
Scientific Research Project of Education Department of Yunnan
Province (grant no. 2023Y0787).
Not applicable.
MY and YT were involved in the conception and design
of the study. MY, YT, JL, JZ, LZ, XZ, SY and YS wrote the first
draft of the review, while XZ, SY and YS collected the information
needed for the review, including references and images. YT revised
the manuscript. Data authentication is not applicable. All authors
read and approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011.PubMed/NCBI
|
|
2
|
Bray F, Laversanne M, Sung H, Ferlay J,
Siegel RL, Soerjomataram I and Jemal A: Global cancer statistics
2022: GLOBOCAN estimates of incidence and mortality worldwide for
36 cancers in 185 countries. CA Cancer J Clin. 74:229–263.
2024.PubMed/NCBI
|
|
3
|
Xia C, Dong X, Li H, Cao M, Sun D, He S,
Yang F, Yan X, Zhang S, Li N and Chen W: Cancer statistics in China
and United States, 2022: Profiles, trends, and determinants. Chin
Med J (Engl). 135:584–590. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Cai M, Song XL, Li XA, Chen M, Guo J, Yang
DH, Chen Z and Zhao SC: Current therapy and drug resistance in
metastatic castration-resistant prostate cancer. Drug Resist Updat.
68:1009622023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Xiao Y, Liu P, Wei J, Zhang X, Guo J and
Lin Y: Recent progress in targeted therapy for non-small cell lung
cancer. Front Pharmacol. 14:11255472023. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lev S: Targeted therapy and drug
resistance in triple-negative breast cancer: The EGFR axis. Biochem
Soc Trans. 48:657–665. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wagh K, Stavreva DA, Upadhyaya A and Hager
GL: Transcription factor dynamics: One molecule at a time. Annu Rev
Cell Dev Biol. 39:277–305. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mann R and Notani D: Transcription factor
condensates and signaling driven transcription. Nucleus.
14:22057582023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mazzocca M, Colombo E, Callegari A and
Mazza D: Transcription factor binding kinetics and transcriptional
bursting: What do we really know? Curr Opin Struct Biol.
71:239–248. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Krebs AR: Studying transcription factor
function in the genome at molecular resolution. Trends Genet.
37:798–806. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
García-Gutiérrez L, Delgado MD and León J:
MYC oncogene contributions to release of cell cycle brakes. Genes
(Basel). 10:2442019. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nair SK and Burley SK: X-ray structures of
Myc-Max and Mad-Max recognizing DNA. Molecular bases of regulation
by proto-oncogenic transcription factors. Cell. 112:193–205. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Llombart V and Mansour MR: Therapeutic
targeting of ‘undruggable’ MYC. EBioMedicine. 75:1037562022.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang Y, Xue K, Li Z, Zheng W, Dong W, Song
J, Sun S, Ma T and Li W: [Corrigendum] c-Myc regulates the
CDK1/cyclin B1 dependent-G2/M cell cycle progression by histone H4
acetylation in Raji cells. Int J Mol Med. 44:19882019.PubMed/NCBI
|
|
15
|
Carabet LA, Rennie PS and Cherkasov A:
Therapeutic Inhibition of Myc in cancer. Structural bases and
computer-aided drug discovery approaches. Int J Mol Sci.
20:1202018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: The next generation. Cell. 144:646–764. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gabay M, Li Y and Felsher DW: MYC
activation is a hallmark of cancer initiation and maintenance. Cold
Spring Harb Perspect Med. 4:a0142412014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sayyadi M, Safaroghli-Azar A,
Pourbagheri-Sigaroodi A, Abolghasemi H, Anoushirvani AA and Bashash
D: c-Myc inhibition using 10058-F4 increased the sensitivity of
acute promyelocytic leukemia cells to arsenic trioxide via blunting
PI3K/NF-κB axis. Arch Med Res. 51:636–644. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kumari N, Das K, Sharma S, Dahal S, Desai
SS, Roy U, Sharma A, Manjunath M, Gopalakrishnan V, Retheesh ST, et
al: Evaluation of potential role of R-loop and G-quadruplex DNA in
the fragility of c-MYC during chromosomal translocation associated
with Burkitt's lymphoma. J Biol Chem. 299:1054312023. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giacomini A, Taranto S, Gazzaroli G,
Faletti J, Capoferri D, Marcheselli R, Sciumè M, Presta M, Sacco A
and Roccaro AM: The FGF/FGFR/c-Myc axis as a promising therapeutic
target in multiple myeloma. J Exp Clin Cancer Res. 43:2942024.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu C, Jiang K, Ding Y, Yang A, Cai R, Bai
P, Xiong M, Fu C, Quan M, Xiong Z, et al: Kindlin-2 enhances c-Myc
translation through association with DDX3X to promote pancreatic
ductal adenocarcinoma progression. Theranostics. 13:4333–4355.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hua Q, Jin M, Mi B, Xu F, Li T, Zhao L,
Liu J and Huang G: LINC01123, a c-Myc-activated long non-coding
RNA, promotes proliferation and aerobic glycolysis of non-small
cell lung cancer through miR-199a-5p/c-Myc axis. J Hematol Oncol.
12:912019. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ireland AS, Micinski AM, Kastner DW, Guo
B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero
D, et al: MYC drives temporal evolution of small cell lung cancer
subtypes by reprogramming neuroendocrine fate. Cancer Cell.
38:60–78.e12. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bakiri L, Hasenfuss SC, Guío-Carrión A,
Thomsen MK, Hasselblatt P and Wagner EF: Liver cancer development
driven by the AP-1/c-Jun~Fra-2 dimer through c-Myc. Proc Natl Acad
Sci USA. 121:e24041881212024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Faskhoudi MA, Molaei P, Sadrkhanloo M,
Orouei S, Hashemi M, Bokaie S, Rashidi M, Entezari M, Zarrabi A,
Hushmandi K, et al: Molecular landscape of c-Myc signaling in
prostate cancer: A roadmap to clinical translation. Pathol Res
Pract. 233:1538512022. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gao FY, Li XT, Xu K, Wang RT and Guan XX:
c-MYC mediates the crosstalk between breast cancer cells and tumor
microenvironment. Cell Commun Signal. 21:282023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Evan G, Harrington E, Fanidi A, Land H,
Amati B and Bennett M: Integrated control of cell proliferation and
cell death by the c-myc oncogene. Philos Trans R Soc Lond B Biol
Sci. 345:269–275. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Boulos JC, Omer EA, Rigano D, Formisano C,
Chatterjee M, Leich E, Klauck SM, Shan LT and Efferth T:
Cynaropicrin disrupts tubulin and c-Myc-related signaling and
induces parthanatos-type cell death in multiple myeloma. Acta
Pharmacol Sin. 44:2265–2281. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang HY, Goldman AR, Zhang X, Speicher DW
and Dang CV: Measuring MYC-mediated metabolism in tumorigenesis.
Methods Mol Biol. 2318:231–239. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sim DY, Lee HJ, Ahn CH, Park J, Park SY,
Kil BJ, Shim BS, Kim B and Kim SH: Negative regulation of CPSF6
suppresses the warburg effect and angiogenesis leading to tumor
progression via c-Myc signaling network: Potential therapeutic
target for liver cancer therapy. Int J Biol Sci. 20:3442–3460.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yoshida GJ: Emerging roles of Myc in stem
cell biology and novel tumor therapies. J Exp Clin Cancer Res.
37:1732018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang J, Yang Y, Shao F, Meng Y, Guo D, He
J and Lu Z: Acetate reprogrammes tumour metabolism and promotes
PD-L1 expression and immune evasion by upregulating c-Myc. Nat
Metab. 6:914–932. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Amati B, Brooks MW, Levy N, Littlewood TD,
Evan GI and Land H: Oncogenic activity of the c-Myc protein
requires dimerization with Max. Cell. 72:233–245. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Panova S, Cliff MJ, Macek P, Blackledge M,
Jensen MR, Nissink JWM, Embrey KJ, Davies R and Waltho JP: Mapping
hidden residual structure within the Myc bHLH-LZ domain using
chemical denaturant titration. Structure. 27:1537–1546.e4. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee M, Seok J, Saha SK, Cho S, Jeong Y,
Gil M, Kim A, Shin HY, Bae H, Do JT, et al: Alterations and
co-occurrence of C-MYC, N-MYC, and L-MYC expression are related to
clinical outcomes in various cancers. Int J Stem Cells. 16:215–233.
2023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang C, Zhang J, Yin J, Gan Y, Xu S, Gu Y
and Huang W: Alternative approaches to target Myc for cancer
treatment. Signal Transduct Target Ther. 6:1172021. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lama D, Vosselman T, Sahin C, Liaño-Pons
J, Cerrato CP, Nilsson L, Teilum K, Lane DP, Landreh M and Arsenian
Henriksson M: A druggable conformational switch in the c-MYC
transactivation domain. Nat Commun. 15:18652024. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang Q, West-Osterfield K, Spears E, Li
Z, Panaccione A and Hann SR: MB0 and MBI are independent and
distinct transactivation domains in MYC that are essential for
transformation. Genes (Basel). 8:1342017. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kalkat M, Resetca D, Lourenco C, Chan PK,
Wei Y, Shiah YJ, Vitkin N, Tong Y, Sunnerhagen M, Done SJ, et al:
MYC protein interactome profiling reveals functionally distinct
regions that cooperate to drive tumorigenesis. Mol Cell.
72:836–848.e7. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Thomas LR, Foshage AM, Weissmiller AM,
Popay TM, Grieb BC, Qualls SJ, Ng V, Carboneau B, Lorey S, Eischen
CM and Tansey WP: Interaction of MYC with host cell factor-1 is
mediated by the evolutionarily conserved Myc box IV motif.
Oncogene. 35:3613–3618. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Cowling VH, Chandriani S, Whitfield ML and
Cole MD: A conserved Myc protein domain, MBIV, regulates DNA
binding, apoptosis, transformation, and G2 arrest. Mol Cell Biol.
26:4226–4239. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zinzalla G: Biophysical and structural
methods to study the bHLHZip region of human c-MYC. Methods Mol
Biol. 2318:21–43. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ghasemi N and Azizi H: Exploring Myc
puzzle: Insights into cancer, stem cell biology, and PPI networks.
Gene. 916:1484472024. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rahl PB and Young RA: MYC and
transcription elongation. Cold Spring Harb Perspect Med.
4:a0209902014. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Cargnello M and Topisirovic I: c-Myc
steers translation in lymphoma. J Exp Med. 216:1471–1473. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Amati B, Frank SR, Donjerkovic D and
Taubert S: Function of the c-Myc oncoprotein in chromatin
remodeling and transcription. Biochim Biophys Acta. 1471:M135–M145.
2001.PubMed/NCBI
|
|
47
|
Wang Y, Yang G, Zhang X, Bai R, Yuan D,
Gao D, He Q, Yuan Y, Zhang X, Kou J, et al: Antitumor effect of
anti-c-Myc aptamer-based PROTAC for degradation of the c-Myc
protein. Adv Sci (Weinh). 11:e23096392024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
See YX, Chen K and Fullwood MJ: MYC
overexpression leads to increased chromatin interactions at
super-enhancers and MYC binding sites. Genome Res. 32:629–642.
2022. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fatma H, Maurya SK and Siddique HR:
Epigenetic modifications of c-MYC: Role in cancer cell
reprogramming, progression and chemoresistance. Semin Cancer Biol.
83:166–176. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Dejure FR and Eilers M: MYC and tumor
metabolism: Chicken and egg. EMBO J. 36:3409–3420. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Stratton MR, Campbell PJ and Futreal PA:
The cancer genome. Nature. 458:719–724. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Martincorena I and Campbell PJ: Somatic
mutation in cancer and normal cells. Science. 349:1483–1489. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Stine ZE, Walton ZE, Altman BJ, Hsieh AL
and Dang CV: MYC, metabolism, and cancer. Cancer Discov.
5:1024–1039. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Baluapuri A, Wolf E and Eilers M: Target
gene-independent functions of MYC oncoproteins. Nat Rev Mol Cell
Biol. 21:255–267. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Zhao D, Pan C, Sun J, Gilbert C,
Drews-Elger K, Azzam DJ, Picon-Ruiz M, Kim M, Ullmer W, El-Ashry D,
et al: VEGF drives cancer-initiating stem cells through
VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene.
34:3107–3119. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Dadiani M, Seger D, Kreizman T, Badikhi D,
Margalit R, Eilam R and Degani H: Estrogen regulation of vascular
endothelial growth factor in breast cancer in vitro and in vivo:
The role of estrogen receptor alpha and c-Myc. Endocr Relat Cancer.
16:819–834. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zhu P, He F, Hou Y, Tu G, Li Q, Jin T,
Zeng H, Qin Y, Wan X, Qiao Y, et al: A novel hypoxic long noncoding
RNA KB-1980E6.3 maintains breast cancer stem cell stemness via
interacting with IGF2BP1 to facilitate c-Myc mRNA stability.
Oncogene. 40:1609–1627. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Klauber-DeMore N, Schulte BA and Wang GY:
Targeting MYC for triple-negative breast cancer treatment.
Oncoscience. 5:120–121. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Yang L, Li J, Li Y, Zhou Y, Wang Z, Zhang
D, Liu J and Zhang X: Diclofenac impairs the proliferation and
glucose metabolism of triple-negative breast cancer cells by
targeting the c-Myc pathway. Exp Ther Med. 21:5842021. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Yao B, Xing M, Zeng X, Zhang M, Zheng Q,
Wang Z, Peng B, Qu S, Li L, Jin Y, et al: KMT2D-mediated H3K4me1
recruits YBX1 to facilitate triple-negative breast cancer
progression through epigenetic activation of c-Myc. Clin Transl
Med. 14:e17532024. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Yuan XN, Shao YC, Guan XQ, Liu Q, Chu MF,
Yang ZL, Li H, Zhao S, Tian YH, Zhang JW and Wei L: METTL3
orchestrates glycolysis by stabilizing the c-Myc/WDR5 complex in
triple-negative breast cancer. Biochim Biophys Acta Mol Cell Res.
1871:1197162024. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Li J, Pan J, Liu Y, Luo X, Yang C, Xiao W,
Li Q, Yang L and Zhang X: 3-Bromopyruvic acid regulates glucose
metabolism by targeting the c-Myc/TXNIP axis and induces
mitochondria-mediated apoptosis in TNBC cells. Exp Ther Med.
24:5202022. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Pan JM, Li JC, Yang C, Xiao WF, Li QS, Luo
XH and Zhang XD: 3-Bromopyruvate inhibits the growth and glucose
metabolism of TNBC xenografts in nude mice by targeting c-Myc.
Anticancer Agents Med Chem. 23:1421–1428. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hsu WJ, Chen CH, Chang YC, Cheng CH, TsaI
YH and Lin CW: PRMT1 confers resistance to olaparib via modulating
MYC signaling in triple-negative breast cancer. J Pers Med.
11:10092021. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Öhlund D, Handly-Santana A, Biffi G,
Elyada E, Almeida AS, Ponz-Sarvise M, Corbo V, Oni TE, Hearn SA,
Lee EJ, et al: Distinct populations of inflammatory fibroblasts and
myofibroblasts in pancreatic cancer. J Exp Med. 214:579–596. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Han H, Jain AD, Truica MI,
Izquierdo-Ferrer J, Anker JF, Lysy B, Sagar V, Luan Y, Chalmers ZR,
Unno K, et al: Small-molecule MYC inhibitors suppress tumor growth
and enhance immunotherapy. Cancer Cell. 36:483–497.e15. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Michaels AD, Newhook TE, Adair SJ, Morioka
S, Goudreau BJ, Nagdas S, Mullen MG, Persily JB, Bullock TNJ,
Slingluff CL Jr, et al: CD47 blockade as an adjuvant immunotherapy
for resectable pancreatic cancer. Clin Cancer Res. 24:1415–1425.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Pan Y, Lu F, Fei Q, Yu X, Xiong P, Yu X,
Dang Y, Hou Z, Lin W, Lin X, et al: Single-cell RNA sequencing
reveals compartmental remodeling of tumor-infiltrating immune cells
induced by anti-CD47 targeting in pancreatic cancer. J Hematol
Oncol. 12:1242019. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Dang CV: MYC, metabolism, cell growth, and
tumorigenesis. Cold Spring Harb Perspect Med. 3:a0142172013.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ganguly K, Bhatia R, Rauth S, Kisling A,
Atri P, Thompson C, Vengoji R, Ram Krishn S, Shinde D, Thomas V, et
al: Mucin 5AC serves as the nexus for β-catenin/c-Myc interplay to
promote glutamine dependency during pancreatic cancer
chemoresistance. Gastroenterology. 162:253–268.e13. 2022.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Fang Y, Shen ZY, Zhan YZ, Feng XC, Chen
KL, Li YS, Deng HJ, Pan SM, Wu DH and Ding Y: CD36 inhibits
β-catenin/c-myc-mediated glycolysis through ubiquitination of GPC4
to repress colorectal tumorigenesis. Nat Commun. 10:39812019.
View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Fang Y, Zhou W, Rong Y, Kuang T, Xu X, Wu
W, Wang D and Lou W: Exosomal miRNA-106b from cancer-associated
fibroblast promotes gemcitabine resistance in pancreatic cancer.
Exp Cell Res. 383:1115432019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Lin J, Wang X, Zhai S, Shi M, Peng C, Deng
X, Fu D, Wang J and Shen B: Hypoxia-induced exosomal circPDK1
promotes pancreatic cancer glycolysis via c-myc activation by
modulating miR-628-3p/BPTF axis and degrading BIN1. J Hematol
Oncol. 15:1282022. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Cui G, Huang Y, Feng W, Yao Y, Zhou H, Li
X, Gong H, Liu J, Luo Y, Sun Y, et al: Colon cancer-associated
transcript-1 enhances glucose metabolism and colon cancer cell
activity in a high-glucose environment in vitro and in vivo. J
Gastrointest Oncol. 11:1164–1185. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Selem NA, Youness RA and Gad MZ: What is
beyond LncRNAs in breast cancer: A special focus on colon
cancer-associated transcript-1 (CCAT-1). Noncoding RNA Res.
6:174–186. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhang J and Gao Y: CCAT-1 promotes
proliferation and inhibits apoptosis of cervical cancer cells via
the Wnt signaling pathway. Oncotarget. 8:68059–68070. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Li S, Ma YM, Zheng PS and Zhang P: GDF15
promotes the proliferation of cervical cancer cells by
phosphorylating AKT1 and Erk1/2 through the receptor ErbB2. J Exp
Clin Cancer Res. 37:802018. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wan F, Miao X, Quraishi I, Kennedy V,
Creek KE and Pirisi L: Gene expression changes during HPV-mediated
carcinogenesis: A comparison between an in vitro cell model and
cervical cancer. Int J Cancer. 123:32–40. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Li L, Zhang R, Yang H, Zhang D, Liu J, Li
J and Guo B: GDF15 knockdown suppresses cervical cancer cell
migration in vitro through the TGF-β/Smad2/3/Snail1 pathway. FEBS
Open Bio. 10:2750–2760. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Jiang Y, Han Y, Sun C, Han C, Han N, Zhi W
and Qiao Q: Rab23 is overexpressed in human bladder cancer and
promotes cancer cell proliferation and invasion. Tumour Biol.
37:8131–8138. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Jiang G and Huang C, Liao X, Li J, Wu XR,
Zeng F and Huang C: The RING domain in the anti-apoptotic protein
XIAP stabilizes c-Myc protein and preserves anchorage-independent
growth of bladder cancer cells. J Biol Chem. 294:5935–5944. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Wang J, Zhao X, Shi J, Pan Y, Chen Q, Leng
P and Wang Y: miR-451 suppresses bladder cancer cell migration and
invasion via directly targeting c-Myc. Oncol Rep. 36:2049–2058.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Lucas CM, Harris RJ, Giannoudis A and
Clark RE: c-Myc inhibition decreases CIP2A and reduces BCR-ABL1
tyrosine kinase activity in chronic myeloid leukemia.
Haematologica. 100:e179–e182. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Sanchez-Martin M and Ferrando A: The
NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia.
Blood. 129:1124–1133. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Chen JL, Ping YH, Tseng MJ, Chang YI, Lee
HC, Hsieh RH and Yeh TS: Notch1-promoted TRPA1 expression in
erythroleukemic cells suppresses erythroid but enhances
megakaryocyte differentiation. Sci Rep. 7:428832017. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Matsushita K, Kitamura K, Rahmutulla B,
Tanaka N, Ishige T, Satoh M, Hoshino T, Miyagi S, Mori T, Itoga S,
et al: Haploinsufficiency of the c-myc transcriptional repressor
FIR, as a dominant negative-alternative splicing model, promoted
p53-dependent T-cell acute lymphoblastic leukemia progression by
activating Notch1. Oncotarget. 6:5102–5117. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Qin T, Wang J, Wang J, Du Q, Wang L, Liu
H, Liu W, Li X, Jiang Y, Xu Q, et al: Nuclear to cytoplasmic
transport is a druggable dependency in HDAC7-driven small cell lung
cancer. Adv Sci (Weinh). 12:e24134452025. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Nishida Y, Ishizawa J, Ayoub E, Montoya
RH, Ostermann LB, Muftuoglu M, Ruvolo VR, Patsilevas T, Scruggs DA,
Khazaei S, et al: Enhanced TP53 reactivation disrupts MYC
transcriptional program and overcomes venetoclax resistance in
acute myeloid leukemias. Sci Adv. 9:eadh14362023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
García-Gutiérrez L, Bretones G, Molina E,
Arechaga I, Symonds C, Acosta JC, Blanco R, Fernández A, Alonso L,
Sicinski P, et al: Myc stimulates cell cycle progression through
the activation of Cdk1 and phosphorylation of p27. Sci Rep.
9:186932019. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Niu Z, Liu H, Zhou M, Wang H, Liu Y, Li X,
Xiong W, Ma J, Li X and Li G: Knockdown of c-Myc inhibits cell
proliferation by negatively regulating the Cdk/Rb/E2F pathway in
nasopharyngeal carcinoma cells. Acta Biochim Biophys Sin
(Shanghai). 47:183–191. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mao A, Zhao Q, Zhou X, Sun C, Si J, Zhou
R, Gan L and Zhang H: MicroRNA-449a enhances radiosensitivity by
downregulation of c-Myc in prostate cancer cells. Sci Rep.
6:273462016. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Seth A, Gupta S and Davis RJ: Cell cycle
regulation of the c-Myc transcriptional activation domain. Mol Cell
Biol. 13:4125–4136. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Hydbring P, Castell A and Larsson LG: MYC
modulation around the CDK2/p27/SKP2 axis. Genes (Basel). 8:1742017.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Zhang Y, Li Z, Hao Q, Tan W, Sun J, Li J,
Chen CW, Li Z, Meng Y, Zhou Y, et al: The Cdk2-c-Myc-miR-571 axis
regulates DNA replication and genomic stability by targeting
geminin. Cancer Res. 79:4896–4910. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Henriksson S, Calderón-Montaño JM, Solvie
D, Warpman Berglund U and Helleday T: Overexpressed c-Myc
sensitizes cells to TH1579, a mitotic arrest and oxidative DNA
damage inducer. Biomolecules. 12:17772022. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Fan Y, Jia X, Xie T, Zhu L and He F:
Radiosensitizing effects of c-myc gene knockdown-induced G2/M phase
arrest by intrinsic stimuli via the mitochondrial signaling
pathway. Oncol Rep. 44:2669–2677. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Boxer LM and Dang CV: Translocations
involving c-myc and c-myc function. Oncogene. 20:5595–5610. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Bretones G, Delgado MD and León J: Myc and
cell cycle control. Biochim Biophys Acta. 1849:506–516. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Chen J, Li W, Cui K, Ji K, Xu S and Xu Y:
Artemisitene suppresses tumorigenesis by inducing DNA damage
through deregulating c-Myc-topoisomerase pathway. Oncogene.
37:5079–5087. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Cui F, Fan R, Chen Q, He Y, Song M, Shang
Z, Zhang S, Zhu W, Cao J, Guan H and Zhou PK: The involvement of
c-Myc in the DNA double-strand break repair via regulating
radiation-induced phosphorylation of ATM and DNA-PKcs activity. Mol
Cell Biochem. 406:43–51. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Fan L, Xu S, Zhang F, Cui X, Fazli L,
Gleave M, Clark DJ, Yang A, Hussain A, Rassool F and Qi J: Histone
demethylase JMJD1A promotes expression of DNA repair factors and
radio-resistance of prostate cancer cells. Cell Death Dis.
11:2142020. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Gravina GL, Festuccia C, Popov VM, Di
Rocco A, Colapietro A, Sanità P, Monache SD, Musio D, De Felice F,
Di Cesare E, et al: c-Myc sustains transformed phenotype and
promotes radioresistance of embryonal rhabdomyosarcoma cell lines.
Radiat Res. 185:411–422. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Mac Partlin M, Homer E, Robinson H,
McCormick CJ, Crouch DH, Durant ST, Matheson EC, Hall AG, Gillespie
DA and Brown R: Interactions of the DNA mismatch repair proteins
MLH1 and MSH2 with c-MYC and MAX. Oncogene. 22:819–825. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Olave MC and Graham RP: Mismatch repair
deficiency: The what, how and why it is important. Genes
Chromosomes Cancer. 61:314–321. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Li Z, Pearlman AH and Hsieh P: DNA
mismatch repair and the DNA damage response. DNA Repair (Amst).
38:94–101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Fishel R: Mismatch repair. J Biol Chem.
290:26395–26403. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Ijsselsteijn R, Jansen JG and de Wind N:
DNA mismatch repair-dependent DNA damage responses and cancer. DNA
Repair (Amst). 93:1029232020. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Bucci B, D'Agnano I, Amendola D, Citti A,
Raza GH, Miceli R, De Paula U, Marchese R, Albini S, Felsani A, et
al: Myc down-regulation sensitizes melanoma cells to radiotherapy
by inhibiting MLH1 and MSH2 mismatch repair proteins. Clin Cancer
Res. 11:2756–2767. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Carey JPW, Karakas C, Bui T, Chen X,
Vijayaraghavan S, Zhao Y, Wang J, Mikule K, Litton JK, Hunt KK and
Keyomarsi K: Synthetic lethality of PARP inhibitors in combination
with MYC blockade is independent of BRCA status in triple-negative
breast cancer. Cancer Res. 78:742–757. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Kuzyk A and Mai S: c-MYC-induced genomic
instability. Cold Spring Harb Perspect Med. 4:a0143732014.
View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Das SK, Karmakar S, Venkatachalapathy H,
Jha RK, Batchelor E and Levens D: Excessive MYC-topoisome activity
triggers acute DNA damage, MYC degradation, and replacement by a
p53-topoisome. Mol Cell. 84:4059–4078.e10. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Faubert B, Solmonson A and DeBerardinis
RJ: Metabolic reprogramming and cancer progression. Science.
368:eaaw54732020. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Li M, Yu J, Ju L, Wang Y, Jin W, Zhang R,
Xiang W, Ji M, Du W, Wang G, et al: USP43 stabilizes c-Myc to
promote glycolysis and metastasis in bladder cancer. Cell Death
Dis. 15:442024. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Guo X, Zhu Y and Hong X, Zhang M, Qiu X,
Wang Z, Qi Z and Hong X: miR-181d and c-myc-mediated inhibition of
CRY2 and FBXL3 reprograms metabolism in colorectal cancer. Cell
Death Dis. 8:e29582017. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Nieminen AI, Partanen JI, Hau A and
Klefstrom J: c-Myc primed mitochondria determine cellular
sensitivity to TRAIL-induced apoptosis. EMBO J. 26:1055–1067. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Shen S, Yao T, Xu Y, Zhang D, Fan S and Ma
J: CircECE1 activates energy metabolism in osteosarcoma by
stabilizing c-Myc. Mol Cancer. 19:1512020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Hsieh AL, Walton ZE, Altman BJ, Stine ZE
and Dang CV: MYC and metabolism on the path to cancer. Semin Cell
Dev Biol. 43:11–21. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Xiao L, Hu ZY, Dong X, Tan Z, Li W, Tang
M, Chen L, Yang L, Tao Y, Jiang Y, et al: Targeting Epstein-Barr
virus oncoprotein LMP1-mediated glycolysis sensitizes
nasopharyngeal carcinoma to radiation therapy. Oncogene.
33:4568–4578. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Wise DR, DeBerardinis RJ, Mancuso A, Sayed
N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon
SB and Thompson CB: Myc regulates a transcriptional program that
stimulates mitochondrial glutaminolysis and leads to glutamine
addiction. Proc Natl Acad Sci USA. 105:18782–18777. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Gao P, Tchernyshyov I, Chang TC, Lee YS,
Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT and
Dang CV: c-Myc suppression of miR-23a/b enhances mitochondrial
glutaminase expression and glutamine metabolism. Nature.
458:762–765. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
121
|
Peng Y, Fu S, Hu W, Qiu Y, Zhang L, Tan R
and Sun LQ: Glutamine synthetase facilitates cancer cells to
recover from irradiation-induced G2/M arrest. Cancer Biol Ther.
21:43–51. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B,
Zhou Q, He J, Qiu Y, Wen M, et al: Glutamine synthetase promotes
radiation resistance via facilitating nucleotide metabolism and
subsequent DNA damage repair. Cell Rep. 28:1136–1143.e4. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Rashmi R, Jayachandran K, Zhang J, Menon
V, Muhammad N, Zahner M, Ruiz F, Zhang S, Cho K, Wang Y, et al:
Glutaminase inhibitors induce thiol-mediated oxidative stress and
radiosensitization in treatment-resistant cervical cancers. Mol
Cancer Ther. 19:2465–2475. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Zimmerli D, Brambillasca CS, Talens F,
Bhin J, Linstra R, Romanens L, Bhattacharya A, Joosten SEP, Da
Silva AM, Padrao N, et al: MYC promotes immune-suppression in
triple-negative breast cancer via inhibition of interferon
signaling. Nat Commun. 13:65792022. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Yao X, Xue Y, Ma Q, Bai Y, Jia P, Zhang Y,
Lai B, He S, Ma Q, Zhang J, et al: 221S-1a inhibits endothelial
proliferation in pathological angiogenesis through ERK/c-Myc
signaling. Eur J Pharmacol. 952:1758052023. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Yao J, Huang M, Shen Q, Ding M, Yu S, Guo
Y, Lin Y, Zheng Y, Chen W, Yan W, et al: c-Myc-PD-L1 axis sustained
gemcitabine-resistance in pancreatic cancer. Front Pharmacol.
13:8515122022. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Casey SC, Tong L, Li Y, Do R, Walz S,
Fitzgerald KN, Gouw AM, Baylot V, Gütgemann I, Eilers M and Felsher
DW: MYC regulates the antitumor immune response through CD47 and
PD-L1. Science. 352:227–231. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Liang MQ, Yu FQ and Chen C: C-Myc
regulates PD-L1 expression in esophageal squamous cell carcinoma.
Am J Transl Res. 12:379–388. 2020.PubMed/NCBI
|
|
129
|
Oh ET, Kim CW, Kim HG, Lee JS and Park HJ:
Brusatol-mediated inhibition of c-Myc increases HIF-1α degradation
and causes cell death in colorectal cancer under hypoxia.
Theranostics. 7:3415–3431. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Weili Z, Zhikun L, Jianmin W and Qingbao
T: Knockdown of USP28 enhances the radiosensitivity of esophageal
cancer cells via the c-Myc/hypoxia-inducible factor-1 alpha
pathway. J Cell Biochem. 120:201–212. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Steinbichler TB, Dudás J, Skvortsov S,
Ganswindt U, Riechelmann H and Skvortsova II: Therapy resistance
mediated by cancer stem cells. Semin Cancer Biol. 53:156–167. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Okita K, Ichisaka T and Yamanaka S:
Generation of germline-competent induced pluripotent stem cells.
Nature. 448:313–317. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Aguirre M, Escobar M, Forero Amézquita S,
Cubillos D, Rincón C, Vanegas P, Tarazona MP, Atuesta Escobar S,
Blanco JC and Celis LG: Application of the yamanaka transcription
factors Oct4, Sox2, Klf4, and c-Myc from the laboratory to the
clinic. Genes (Basel). 14:16972023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Chen H, Jin K, Song J, Zuo Q, Yang H,
Zhang Y and Li B: Functional characterization of the Sox2, c-Myc,
and Oct4 promoters. J Cell Biochem. 120:332–342. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Rennoll S and Yochum G: Regulation of MYC
gene expression by aberrant Wnt/β-catenin signaling in colorectal
cancer. World J Biol Chem. 6:290–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Suo S, Zhao D, Li F, Zhang Y,
Rodriguez-Rodriguez S, Nguyen LXT, Ghoda L, Carlesso N, Marcucci G,
Zhang B and Jin J: Homoharringtonine inhibits the NOTCH/MYC pathway
and exhibits antitumor effects in T-cell acute lymphoblastic
leukemia. Blood. 144:1343–1347. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Wang H, Zhang S, Zhang Y, Jia J, Wang J,
Liu X, Zhang J, Song X, Ribback S, Cigliano A, et al: TAZ is
indispensable for c-MYC-induced hepatocarcinogenesis. J Hepatol.
76:123–134. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Zhong Q, Liu ZH, Lin ZR, Hu ZD, Yuan L,
Liu YM, Zhou AJ, Xu LH, Hu LJ, Wang ZF, et al: The RARS-MAD1L1
fusion gene induces cancer stem cell-like properties and
therapeutic resistance in nasopharyngeal carcinoma. Clin Cancer
Res. 24:659–673. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Das B, Pal B, Bhuyan R, Li H, Sarma A,
Gayan S, Talukdar J, Sandhya S, Bhuyan S, Gogoi G, et al: MYC
regulates the HIF2α stemness pathway via nanog and Sox2 to maintain
self-renewal in cancer stem cells versus non-stem cancer cells.
Cancer Res. 79:4015–4025. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Yang A, Qin S, Schulte BA, Ethier SP, Tew
KD and Wang GY: MYC inhibition depletes cancer stem-like cells in
triple-negative breast cancer. Cancer Res. 77:6641–6650. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Lin CH, Lin C, Tanaka H, Fero ML and
Eisenman RN: Gene regulation and epigenetic remodeling in murine
embryonic stem cells by c-Myc. PLoS One. 4:e78392009. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Beaulieu ME, Jauset T, Massó-Vallés D,
Martínez-Martín S, Rahl P, Maltais L, Zacarias-Fluck MF,
Casacuberta-Serra S, Serrano Del Pozo E, Fiore C, et al: Intrinsic
cell-penetrating activity propels Omomyc from proof of concept to
viable anti-MYC therapy. Sci Transl Med. 11:eaar50122019.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Struntz NB, Chen A, Deutzmann A, Wilson
RM, Stefan E, Evans HL, Ramirez MA, Liang T, Caballero F, Wildschut
MHE, et al: Stabilization of the Max homodimer with a small
molecule attenuates Myc-driven transcription. Cell Chem Biol.
26:711–723.e14. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Shao R, Liu C, Xue R, Deng X, Liu L, Song
C, Xie J, Tang H and Liu W: Tumor-derived exosomal ENO2 modulates
polarization of tumor-associated macrophages through reprogramming
glycolysis to promote progression of diffuse large B-cell lymphoma.
Int J Biol Sci. 20:848–863. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Morelli E, Biamonte L, Federico C, Amodio
N, Di Martino MT, Gallo Cantafio ME, Manzoni M, Scionti F, Samur
MK, Gullà A, et al: Therapeutic vulnerability of multiple myeloma
to MIR17PTi, a first-in-class inhibitor of pri-miR-17-92. Blood.
132:1050–1063. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Oprea M and Ionita M: Antisense
oligonucleotides-based approaches for the treatment of multiple
myeloma. Int J Biol Macromol. 291:1391862025. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Mazur PK, Herner A, Mello SS, Wirth M,
Hausmann S, Sánchez-Rivera FJ, Lofgren SM, Kuschma T, Hahn SA,
Vangala D, et al: Author correction: combined inhibition of BET
family proteins and histone deacetylases as a potential
epigenetics-based therapy for pancreatic ductal adenocarcinoma. Nat
Med. 30:20902024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Glover-Cutter K, Larochelle S, Erickson B,
Zhang C, Shokat K, Fisher RP and Bentley DL: TFIIH-associated Cdk7
kinase functions in phosphorylation of C-terminal domain Ser7
residues, promoter-proximal pausing, and termination by RNA
polymerase II. Mol Cell Biol. 29:5455–5464. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Coudé MM, Braun T, Berrou J, Dupont M,
Bertrand S, Masse A, Raffoux E, Itzykson R, Delord M, Riveiro ME,
et al: BET inhibitor OTX015 targets BRD2 and BRD4 and decreases
c-MYC in acute leukemia cells. Oncotarget. 6:17698–17712. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Huang WL, Abudureheman T, Xia J, Chu L,
Zhou H, Zheng WW, Zhou N, Shi RY, Li MH, Zhu JM, et al: CDK9
inhibitor induces the apoptosis of B-cell acute lymphocytic
leukemia by inhibiting c-Myc-mediated glycolytic metabolism. Front
Cell Dev Biol. 9:6412712021. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Cho Y, Park MJ, Kim K, Kim SW, Kim W, Oh S
and Lee JH: Reactive oxygen species-induced activation of
Yes-associated protein-1 through the c-Myc pathway is a therapeutic
target in hepatocellular carcinoma. World J Gastroenterol.
26:6599–6613. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Lahlali T, Plissonnier ML, Romero-López C,
Michelet M, Ducarouge B, Berzal-Herranz A, Zoulim F, Mehlen P and
Parent R: Netrin-1 protects hepatocytes against cell death through
sustained translation during the unfolded protein response. Cell
Mol Gastroenterol Hepatol. 2:281–301.e9. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Monsen RC, DeLeeuw LW, Dean WL, Gray RD,
Chakravarthy S, Hopkins JB, Chaires JB and Trent JO: Long promoter
sequences form higher-order G-quadruplexes: An integrative
structural biology study of c-Myc, k-Ras and c-Kit promoter
sequences. Nucleic Acids Res. 50:4127–4147. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Huppert JL and Balasubramanian S:
G-quadruplexes in promoters throughout the human genome. Nucleic
Acids Res. 35:406–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Yang D: G-quadruplex DNA and RNA. Methods
Mol Biol. 2035:1–24. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
156
|
Robinson J, Raguseo F, Nuccio SP, Liano D
and Di Antonio M: DNA G-quadruplex structures: More than simple
roadblocks to transcription? Nucleic Acids Res. 49:8419–8431. 2021.
View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Dhanasekaran R, Park J, Yevtodiyenko A,
Bellovin DI, Adam SJ, Kd AR, Gabay M, Fernando H, Arzeno J, Arjunan
V, et al: MYC ASO impedes tumorigenesis and elicits oncogene
addiction in autochthonous transgenic mouse models of HCC and RCC.
Mol Ther Nucleic Acids. 21:850–859. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
158
|
Webb MS, Tortora N, Cremese M, Kozlowska
H, Blaquiere M, Devine DV and Kornbrust DJ: Toxicity and
toxicokinetics of a phosphorothioate oligonucleotide against the
c-myc oncogene in cynomolgus monkeys. Antisense Nucleic Acid Drug
Dev. 11:155–163. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
159
|
Massó-Vallés D and Soucek L: Blocking Myc
to treat cancer: Reflecting on two decades of omomyc. Cells.
9:8832020. View Article : Google Scholar : PubMed/NCBI
|
|
160
|
Garralda E, Beaulieu ME, Moreno V,
Casacuberta-Serra S, Martínez-Martín S, Foradada L, Alonso G,
Massó-Vallés D, López-Estévez S, Jauset T, et al: MYC targeting by
OMO-103 in solid tumors: A phase 1 trial. Nat Med. 30:762–771.
2024. View Article : Google Scholar : PubMed/NCBI
|
|
161
|
Senapedis W, Gallagher KM, Figueroa E,
Farelli JD, Lyng R, Hodgson JG, O'Donnell CW, Newman JV, Pacaro M,
Siecinski SK, et al: Targeted transcriptional downregulation of MYC
using epigenomic controllers demonstrates antitumor activity in
hepatocellular carcinoma models. Nat Commun. 15:78752024.
View Article : Google Scholar : PubMed/NCBI
|
|
162
|
Xu Y, Yu Q, Wang P, Wu Z, Zhang L, Wu S,
Li M, Wu B, Li H, Zhuang H, et al: A selective small-molecule c-Myc
degrader potently regresses lethal c-Myc overexpressing tumors. Adv
Sci (Weinh). 9:e21043442022. View Article : Google Scholar : PubMed/NCBI
|