Sepsis is defined as life-threatening organ
dysfunction resulting from a dysregulated host response to
infection (1). Its pathogenesis is
complex and rapidly progressive, and it is associated with carrying
a high risk of mortality. Clinically, sepsis manifests as
hemostatic imbalance and fibrinolytic system dysfunction, with the
increased generation of thrombin and fibrin within the vasculature,
promoting microthrombus formation in small and medium-sized vessels
that can ultimately progress to systemic hemorrhage, multiple organ
failure and death (2,3). Although numerous randomized
controlled trials have investigated the efficacy of anticoagulant
therapy in sepsis, its effectiveness and the optimal timing of
intervention remain controversial (4,5).
Therefore, elucidating the pathophysiological mechanisms of
sepsis-induced coagulopathy (SIC) is essential.
Neutrophil extracellular traps (NETs) are
three-dimensional mesh-like scaffolds composed of a DNA backbone
embedded with various antimicrobial proteins, playing a dual role
in pathogen infection. Initially, NETs protect the host by
confining pathogen dissemination and exerting direct antimicrobial
effects (6,7). Conversely, excessive NET generation
may hyperactivate the coagulation system, inhibit anticoagulant
pathways, damage vascular endothelial cells (VECs) (8) and contribute to SIC through
interactions with platelets (9–11).
In severe cases, this may progress to disseminated intravascular
coagulation (DIC), leading to widespread hemorrhage and organ
dysfunction (12). The present
review discuses: i) The epidemiology and pathogenesis of SIC; ii)
NET production and the mechanistic pathways by which NETs regulate
SIC; and iii) the target therapy of NETs in SIC, aiming to clarify
the central role of NETs in SIC and identify potential therapeutic
targets.
In 2017, epidemiological data indicated 48.9 million
new sepsis cases globally, with 11 million related deaths,
accounting for 19.7% of total global mortality. Between 1990 and
2017, the incidence of sepsis decreased by 37.0% and related
mortality rates decreased by 52.8% (13). However, the COVID-19 pandemic
notably increased the global burden in 2021, with an estimated 166
million new cases (95% uncertainty interval 135–201 million) and
21.4 million related deaths (20.3–22.5 million), representing 31.5%
of total mortality rates. Adults aged ≥70 years exhibited the
highest mortality rates, with sepsis-related deaths in this age
group increasing from 8.34 million in 2019 to 15.5 million in 2021
(13). Research has demonstrated a
higher incidence of sepsis and mortality among males overall
(14). Notably, in the specific
context of postoperative sepsis, females exhibit higher incidence
and mortality rates (15–17). The progression of sepsis can induce
multiple organ dysfunction, increasing patient mortality,
prolonging periods of hospitalization, hindering clinical recovery
and imposing heavy economic burdens on society and families.
Coagulation dysfunction is a notable risk factors
for adverse outcomes in sepsis (18). Globally, 24.0–60.0% of patients
with sepsis develop SIC (19).
This condition exhibits a high incidence and diverse
manifestations, ranging from mild thrombocytopenia and
hyperfibrinolytic states to overt DIC if left untreated, with
doubling mortality rates (20). In
the initial phase of septic shock, circulating inflammatory
mediators damage VECs, activate the coagulation system and increase
capillary permeability, resulting in fluid and protein leakage. The
sympathoadrenal axis is activated, with catecholamines inducing
systemic vasoconstriction to ensure blood supply to vital organs.
At this stage, tissues may still receive adequate perfusion, and
hemodynamic alterations remain insignificant (21,22).
As the condition is aggravated however, the marked release of
inflammatory mediators causes damages to VECs and even death,
notably increasing capillary permeability with substantial fluid
and protein leakage, resulting in severe tissue edema. Marked
catecholamine release causes intense capillary constriction,
leading to blood stagnation within the microcirculation and
promoting extensive microthrombus formation. In the stage of
microcirculatory failure, persistent hypoxia and inflammation cause
extensive VEC death, and the microcirculation loses its
autoregulatory capacity. Circulating blood volume becomes notably
depleted, and tissue perfusion becomes markedly insufficient.
Vascular responsiveness to catecholamines decreases, and blood
pressure further declines. Blood flow becomes extensively stagnant
within the microcirculation, with the coagulation system markedly
activated while fibrinolysis is suppressed, leading to DIC and
ultimately resulting in severe tissue hypoxia and organ failure
(23–25).
The pathogenesis of SIC involves multiple regulatory
pathways. i) Tissue factor (TF) activation; following
lipopolysaccharide (LPS) binding to Toll-like receptor (TLR)-4 on
VEC/monocytes, the rapid recruitment of myeloid differentiation
primary response 88/TIR-domain-containing adapter-inducing
interferon β subsequently activates the NF-κB/interferon regulatory
factor 3 pathway. This leads to increased TF mRNA expression within
30 min. The newly expressed TF, a transmembrane glycoprotein, binds
to coagulation factor VIIa (FVIIa) to form the TF-FVIIa complex,
which subsequently activates coagulation factor X (FX) and factor
IX (FIX), converting them to FXa and FIXa, respectively. FXa, with
the assistance of FVa, converts prothrombin (FII) to thrombin
(FIIa), which then converts fibrinogen to fibrin, ultimately
forming a stable blood clot. Additionally, the TF-FVIIa complex
inhibits fibrinolysis by activating thrombin-activatable
fibrinolysis inhibitor (TAFI), thereby further promoting
coagulation (26–28). ii) Platelet-endothelial-glycocalyx
axis; inflammatory storms reduce endothelial glycocalyx thickness
from 0.5 µm to <0.1 µm, exposing collagen-von Willebrand factor
(vWF), and the platelet glycoprotein Ib-IX–V adhesion triggers
immediate release of ADP, thromboxane A2 and platelet factor 4
(PF4), forming protein aggregates and generating primary thrombi
within 30 sec (29,30). iii) Natural anticoagulant
consumption; TNF-α and IL-6 inhibit the hepatic synthesis of
antithrombin (AT) and protein C, while activated neutrophil
elastase (NE) degrades TF pathway inhibitor (TFPI), reducing plasma
AT activity (31–34). iv)
Inflammation-complement-coagulation cross-talk; TNF-α and IL-6
upregulate hepatic FVII and FVIII synthesis via JAK/STAT, and
complement C3a and C5a directly activate platelets and recruit
leukocytes, while membrane attack complex perforates the
endothelium, exposing procoagulant phospholipids and creating a
positive feedback loop among inflammation-complement-coagulation
(35–37). v) Mitochondria-reactive oxygen
species (RO)-Ca2+ axis; ROS disrupts mitochondrial
electron transport chains, reducing ATP by >50%, with
Na+/Ca2+ exchanger imbalance causing
cytoplasmic Ca2+ overload, tripling the efficiency of
Ca2+-dependent thrombin complex assembly, and lactate
accumulation with pH <7.2 further amplifies platelet deformation
and aggregation (38,39). vi) NETs; the ROS-PAD4-histone
citrullination pathway promotes NET release, directly providing TF
and anionic scaffolds that accelerate thrombus formation (40–42)
(Fig. 1).
Neutrophils, as critical components of the host
immune system, participate in immune regulation against various
microorganisms primarily through three mechanisms: Degranulation,
phagocytosis and NET release. NETs are complex reticular structures
composed of a DNA backbone, embedded with multiple proteins
including citrullinated histone H3 (H3), NE, myeloperoxidase (MPO)
and cathepsin G (43). In
eukaryotes, DNA is tightly wound around octamers composed of two
molecules each of H3, H2B, H2A and H4, forming nucleosome core
particles. Histone citrullination reduces chromatin affinity for
DNA, allowing released DNA to interact with bacteria, thereby
mediating the antimicrobial activity of NETs (40,44).
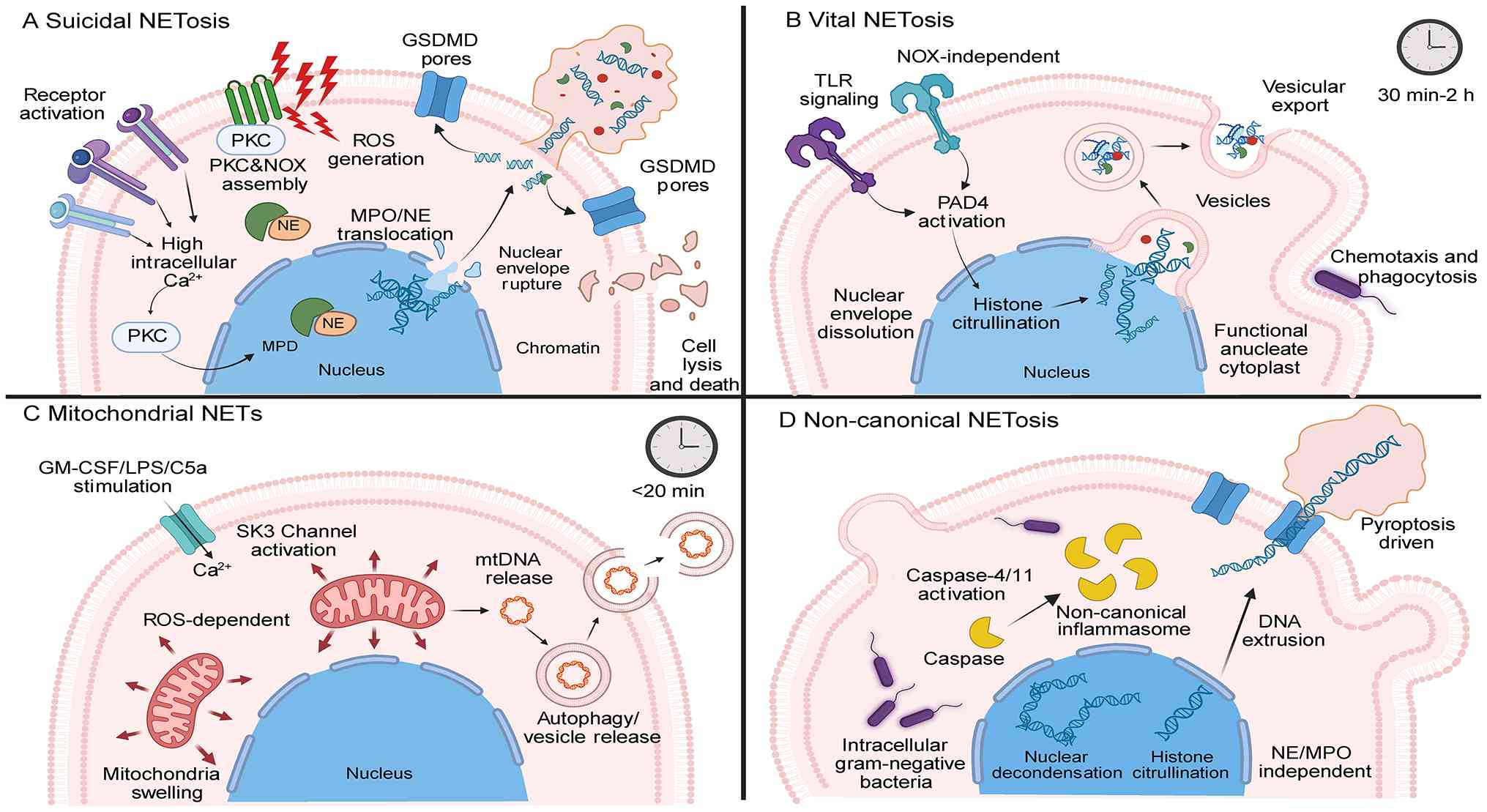
Suicidal NETosis is a NADPH oxidase (NOX)-dependent
form of cell death distinct from apoptosis and necrosis (45) (Fig.
2). This type of NETosis is initiated by receptor activation
(Toll-like receptors, Fc receptors or complement receptors),
leading to elevated intracellular Ca2+. Increased
Ca2+ stimulates protein kinase C and NOX activity,
thereby generating ROS (46).
Under the influence of ROS, activated MPO participates in chromatin
decondensation and nuclear membrane rupture (47). The resulting proteins and DNA
constitute the major components of NETs, further activating NE and
promoting its nuclear translocation. Subsequently, nuclear membrane
rupture occurs, and decondensed chromatin is released into the
cytoplasm, it is subsequently expelled extracellularly through
gasdermin D (GSDMD) pores or GSDMD-mediated membrane tearing,
ultimately resulting in neutrophil death (48,49).
Vital NETosis is TLR-mediated, with peptidylarginine
deiminase 4 (PAD4) and calpains synergistically promoting nuclear
envelope dissolution (50). Unlike
suicidal NETosis, vital NETosis is characterized by neutrophil
survival following NET release. In this pathway, neutrophils
respond to extracellular stimuli, triggering a series of reactions
that initiate NET formation (51).
In contrast to suicidal NETosis, this process does not require NOX
complex assembly. Following the activation of PAD4, histone
citrullination occurs, leading to chromatin decondensation.
DNA-protein complexes are packaged into vesicles and expelled
extracellularly within 30 min to 2 h, forming NETs (52,53).
Neutrophils remain viable following NET release, and the formed
anucleate cytoplasts retain chemotactic and phagocytic
capabilities, enabling rapid pathogen restriction during early
infection (54,55).
This type of NETosis is induced upon the detection
of Gram-negative bacteria in the cytoplasm. It depends on
non-canonical inflammasomes and exhibits key features of NETosis,
including nuclear decondensation, DNA extrusion, DNA-MPO
co-localization and histone citrullination (62,63).
Non-canonical NET formation requires pyroptosis involvement,
executed through caspase-4/11 and GSDMD, independent of NE and
MPO.
VECs represent primary targets in sepsis, and VEC
injury is a characteristic feature of SIC (64) (Fig.
3). During sepsis development, excessive neutrophil activation
leads to NET release, promoting the transition of VECs to
pro-inflammatory and procoagulant phenotypes. VECs amplify the
inflammation-coagulation cross-talk through the secretion of IL-6,
TNF-α, VEGF and other factors, providing an endothelial foundation
for early microthrombus formation in SIC (8). Additionally, NETs disrupt the
glycocalyx barrier on VEC surfaces, increasing vascular
permeability, or inducing ferroptosis, thereby causing endothelial
barrier collapse and resulting in microcirculatory flow
disturbances (8,65). Folco et al (66) reported that NETs promote the time-
and concentration- dependent expression of vascular cell adhesion
molecule-1 and intercellular adhesion molecule-1 on human VECs,
facilitating inflammatory cell recruitment on damaged endothelial
surfaces and further promoting thrombus formation. NETs can also
activate the cGAS-STING pathway in VECs and act on TLR2 on
endothelial surfaces, thereby inducing endothelial injury and TF
expression to promote coagulation (7). The activation of the pore-forming
protein GSDMD is the core driver of NET release. Recent research
has indicated that activation markers of the GSDMD-NET axis,
MPO-DNA, are notably associated with the elevated expression of
endothelial surface glycocalyx injury markers (syndecan-1, MMP-9)
in patients with SIC (67). It has
been found that direct VEC-neutrophil contact serves as the
initiation platform for NETosis in sepsis, with integrin Mac-1
identified as the molecular switch. Blocking Mac-1 function notably
inhibits the LPS induced increase in NETs and thrombus formation
(68). Currently, research on the
regulation of VEC function in SIC by NETs regulation of VECs
remains limited, representing an area requiring further
investigation.
In a previous study, in a model of sepsis induced by
cecal ligation and puncture, platelet-specific STK10 deletion
reduced P-selectin expression by ~50%, decreased peripheral
platelet-neutrophil aggregates to 40% of the controls, reduced the
pulmonary NET area by 60%, almost eliminated microvascular thrombi
and increased 24-h mouse survival from 35 to 70%, confirming that
STK10-regulated platelet-NET interaction is a notable pathway
driving thromboinflammation in sepsis (69). Similarly, in another study,
platelet-specific NLRP6 knockout activated intracellular NF-κB
signaling, promoting platelet-neutrophil interactions and NET
formation, exacerbating pulmonary and hepatic microvascular
thrombosis (70). Although heparin
and low-molecular-weight heparin are commonly used pharmacological
thrombo-prophylactic agents in Critical Care Medicine settings
(e.g., intensive care units), heparin-induced thrombocytopenia
(HIT) is a marked adverse reaction characterized by
thrombocytopenia, paradoxical hypercoagulability and severe
thrombotic complications. Without timely intervention, up to 50% of
patients progress to overt thrombosis (71). A study in 2019 revealed that NETs
are the mechanistic link between HIT and thrombosis. HIT immune
complexes (IgG-heparin-PF4) activate neutrophils through FcγRIIA
receptors, triggering NETosis and subsequent thrombus formation.
Notably, this NET-mediated procoagulant cascade is independent of
platelet activation and aggregation (72). Subsequent mechanistic research
demonstrated that targeting neutrophil-derived ROS completely
blocked NETosis and thrombosis in an ex vivo human whole
blood HIT model. Furthermore, the pharmacological or genetic
inhibition of NOX2 activity eliminated thrombosis in a murine HIT
model without improving thrombocytopenia. These findings indicate
that NOX2-mediated NET formation is an intervenable prothrombotic
target in HIT, operating independently of platelet count recovery
(73). Therefore, HIT therapeutic
strategies have shifted from simply changing anticoagulant
medications to precisely blocking NETosis, providing novel
molecular leverage points for precision diagnosis and treatment.
NETs and platelets exhibit bidirectional regulatory mechanisms in
immunothrombosis; platelets trigger NETosis through
P-selectin/PSGL-1, TLR and HMGB1 signaling axes, whereas the
DNA-histone scaffold of NETs reciprocally activates platelets,
provides anchoring sites for coagulation factors and amplifies the
inflammation-thrombosis positive feedback. The future development
of specific inhibitors targeting key nodes of NETs-platelet
interactions (such as PAD4, GSDMD and P-selectin) is required in
order to achieve multi-target synergistic intervention, breaking
the vicious cycle of inflammation-thrombosis and providing novel
strategies for the precision treatment of thrombotic diseases
(74).
NETs provide scaffolds for platelets, erythrocytes
and procoagulant factors, thereby obstructing vessels (75). NET-derived proteases and histones
exert cytotoxic effects on VECs through TF, while coagulation
factors such as prothrombin, fibrinogen and factor X deposit in
complexes formed between platelets and NETs, promoting thrombus
formation (76). In the extrinsic
pathway, extracellular histones, a key structural component of
NETs, increase TF expression at both VEC surfaces and increase mRNA
levels, while suppressing thrombomodulin expression. Similarly,
NET-associated proteases (such as NE and proteinase 3) can also
upregulate TF expression (77).
Mature IL-1α in NETs (activated by proteinase G) stimulates VECs,
further increasing TF expression and reinforcing the prothrombotic
effects of NETs in sepsis (66).
In the intrinsic coagulation pathway, NETs promote thrombin
generation through the activation of coagulation factors XII and
XI, and stimulate fibrin generation through binding to coagulation
factor XII, thereby promoting coagulation (78). Histone 4 and DNA can increase the
expression of hepatocyte nuclear factor 4α mRNA and TF mRNA,
thereby inducing the transcription of procoagulant factors
(79). The DNA backbone of NETs
activates coagulation factor XII independently of platelets, and
together with coagulation factor XI, they promote the coagulation
cascade and thrombin formation (80). Furthermore, NETs stabilize thrombus
formation by linking vWF and TFPI, thereby stabilizing thrombus
formation (81).
The anticoagulant system comprises antithrombin, the
protein C system and TFPI. Research has demonstrated that during
sepsis, NETs can impair the anticoagulant system through their
protein components by degrading antithrombin, inhibiting activated
protein C generation and cleaving TFPI (82). It has been demonstrated that PAD4,
a notable protein in NET synthesis, can cause the citrullination of
arginine residues exposed on the TFPI surface, thereby impairing
its anticoagulant function and reducing coagulation inhibition
(83).
Laboratory findings in patients with sepsis have
demonstrated that fibrinogen degradation products, D-dimer and the
plasmin activation marker plasmin-α2 antiplasmin complex are only
mildly elevated, indicating that SIC manifests as marked
fibrinolysis suppression (84).
Varjú et al (85)
demonstrated that NETs and their components can directly
participate in clot formation and alter fibrin structure within
clots, enhancing mechanical resistance and reducing sensitivity to
tissue-type plasminogen activator (tPA)-induced fibrinolysis,
exerting antifibrinolytic effects. Mangold et al (86) further demonstrated that treatment
with deoxyribonuclease (DNase) increased tPA-mediated
fibrinolysis.
The precise regulation of NET formation and
clearance represents a promising strategy for mitigating their
deleterious effects in SIC. Potential anti-NET agents encompass
anti-inflammatory/immunomodulatory agents (such as aspirin and
cyclosporine A), anticoagulants (such as thrombomodulin, activated
protein C) and nucleases (such as DNase) (87–90).
These agents exert effects by inhibiting NET formation, degrading
the DNA backbone of NETs or neutralizing NET-associated proteins
(Fig. 4).
Key molecular targets include the Raf-MEK-ERK
signaling cascade, the NOX2-ROS axis and chromatin decondensation
enzymes critical in NETosis, such as NE, PAD4, MPO and histone
deacetylases. Preclinical studies have demonstrated that DNase
reduces sepsis-induced organ injury and bacterial dissemination,
synergizing with standard antimicrobial therapeutics (91,92).
α-1 antitrypsin inhibits NET formation by targeting NE, reducing
MPO activity by 50% in lung, liver, kidney and heart tissues,
thereby attenuating inflammation, coagulation and multi-organ
injury in mice with sepsis (93).
Pharmacological inhibition of PAD4 (the master regulator of NET
formation) similarly reduces NET release, ameliorates organ
dysfunction and improves the survival rates of mice with sepsis,
highlighting the therapeutic potential of PAD4-targeted strategies
(94,95). Recombinant thrombomodulin inhibits
LPS-induced NET formation in animal models, attenuates systemic
cytokine storms (apart from IL-1β) and improves survival rates
(96). GSDMD also represents an
emerging target; disulfiram or GSDMD gene knockout eliminates NET
formation, alleviates multi-organ dysfunction and reduces mortality
in sepsis models (97).
Circulating nucleosomes are established early
biomarkers reflecting cellular damage and NETosis in sepsis.
Nucleosome levels are higher in patients with sepsis than in
healthy volunteers and associate with disease severity (102). Nucleosomes are the basic
structural units of eukaryotic chromatin, composed of histones and
DNA (103). In sepsis, neutrophil
activation triggers the release of granule proteins and chromatin,
leading to apoptosis, necrosis or pyroptosis (104,105), increasing extracellular
nucleosome levels through the following pathways: i) The increased
release of cell-free DNA (cfDNA), leading to biphasic nucleosome
release (106,107); ii) suppressed or reduced DNase
activity, resulting in inadequate nucleosome clearance (108,109); and iii) histone release inducing
cytotoxicity, causing nucleosome cascade amplification (110). Therefore, neutralizing
extracellular histones can reduce cytokine release, improve tissue
perfusion and decrease mortality in experimental sepsis (111). In a previous study, following the
administration of human PF4 to septic mice, PF4 was found to
inhibit procoagulant and endothelial toxicity by physically
cross-linking cfDNA/NETs, reducing generation of short-fragment,
single-stranded DNA (112).
Collectively, these findings establish a solid foundation for
developing NET-targeted therapies for SIC and organ failure.
SIC is a notable pathophysiological process driven
by complex interactions between dysregulated host immune responses
and hemostatic system activation. As discussed in the present
review, NETs serve as pivotal mediators bridging innate immunity
and thrombo-inflammation in sepsis. Four distinct mechanisms of NET
release, suicidal NETosis, vital NETosis, mitochondrial NETs and
non-canonical NETosis, provide diverse pathways through which
neutrophils contribute to SIC pathogenesis. NETs exacerbate SIC
through multiple mechanisms, inducing VEC injury and barrier
dysfunction, amplifying platelet-neutrophil interactions,
activating both intrinsic and extrinsic coagulation cascades,
impairing natural anticoagulant systems and promoting fibrinolytic
resistance. These processes collectively drive microvascular
thrombosis, tissue hypoperfusion and ultimately multiple organ
failure. The identification of key molecular targets, particularly
PAD4, GSDMD, NOX2 and components of the platelet-NET axis, has
opened avenues for therapeutic intervention. Emerging strategies
targeting NET formation, accelerating NET clearance or disrupting
pathological platelet-neutrophil cross-talk exhibit notable
potential in preclinical models. However, clinical translation
requires further investigation to establish optimal timing, dosing
and patient selection criteria. Future research is required to
focus on developing specific biomarkers for NET burden, refining
targeted therapies with favorable risk-benefit profiles, and
conducting well-designed clinical trials to validate these
approaches. Elucidating the complex regulatory networks governing
NET-mediated coagulopathy will not only deepen the understanding of
sepsis pathophysiology but may also pave the way for precision
medicine strategies to improve outcomes in this life-threatening
condition.
Not applicable.
The present study was supported by the Tianjin Beichen Hospital
(Beichen District Health System Technology Project, grant no.
SHGY-2023005) and Tianjin Health Science and Technology Project
grant no. TJWJ2025QN113. The present study was also supported by
Tianjin Education Commission Research Program Project, grant no.
2025ZXZD011.
Not applicable.
HS and ZC conceived and organized the manuscript. JH
and BZ wrote the manuscript. JH and MJ designed the study and YZ
performed the literature search and the screening. JH, MJ, ZC, BZ,
YZ and HS contributed to editorial changes in the manuscript. All
authors read and approved the final version of the manuscript. Data
authentication not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Iba T, Helms J and Levy JH: Sepsis-induced
coagulopathy (SIC) in the management of sepsis. Ann Intensive Care.
14:1482024. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Girardis M, David S, Ferrer R, Helms J,
Juffermans NP, Martin-Loeches I, Povoa P, Russell L, Shankar-Hari
M, Iba T, et al: Understanding, assessing and treating immune,
endothelial and haemostasis dysfunctions in bacterial sepsis.
Intensive Care Med. 50:1580–1592. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xia T, Yu J, Du M, Chen X, Wang C and Li
R: Vascular endothelial cell injury: Causes, molecular mechanisms,
and treatments. MedComm (2020). 6:e700572025. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schmoch T, Mohnle P, Weigand MA, Briegel
J, Bauer M, Bloos F, Briegel J, Radke DI, Bauer M, Bloos F, et al:
The prevalence of sepsis-induced coagulopathy in patients with
sepsis-a secondary analysis of two German multicenter randomized
controlled trials. Ann Intensive Care. 13:32023. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Matsuoka T, Yamakawa K, Umemura Y, Homma
K, Iba T and Sasaki J: The transition of the criteria for
disseminated intravascular coagulation and the targeted patients in
randomized controlled trials over the decades: A scoping review.
Thromb J. 22:1122024. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Middleton EA, He XY, Denorme F, Campbell
RA, Ng D, Salvatore SP, Mostyka M, Baxter-Stoltzfus A, Borczuk AC,
Loda M, et al: Neutrophil extracellular traps contribute to
immunothrombosis in COVID-19 acute respiratory distress syndrome.
Blood. 136:1169–1179. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhu S, Yu Y, Qu M, Qiu Z, Zhang H, Miao C
and Guo K: Neutrophil extracellular traps contribute to
immunothrombosis formation via the STING pathway in
sepsis-associated lung injury. Cell Death Discov. 9:3152023.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang H, Wang Y, Qu M, Li W, Wu D, Cata JP
and Miao C: Neutrophil, neutrophil extracellular traps and
endothelial cell dysfunction in sepsis. Clin Transl Med.
13:e11702023. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Iba T and Levy JH: Inflammation and
thrombosis: Roles of neutrophils, platelets and endothelial cells
and their interactions in thrombus formation during sepsis. J
Thromb Haemost. 16:231–241. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dehghani T and Panitch A: Endothelial
cells, neutrophils and platelets: Getting to the bottom of an
inflammatory triangle. Open Biol. 10:2001612020. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang Y and Liu Y: Neutrophil-induced liver
injury and interactions between neutrophils and liver sinusoidal
endothelial cells. Inflammation. 44:1246–1262. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Castanheira FVS and Kubes P: Neutrophils
and NETs in modulating acute and chronic inflammation. Blood.
133:2178–2185. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rudd KE, Johnson SC, Agesa KM, Shackelford
KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer
S, et al: Global, regional, and national sepsis incidence and
mortality, 1990–2017: Analysis for the global burden of disease
study. Lancet. 395:200–211. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thompson KJ, Finfer SR, Woodward M, Leong
RNF and Liu B: Sex differences in sepsis hospitalisations and
outcomes in older women and men: A prospective cohort study. J
Infect. 84:770–776. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Angele MK, Pratschke S, Hubbard WJ and
Chaudry IH: Gender differences in sepsis: Cardiovascular and
immunological aspects. Virulence. 5:12–19. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dockman RL, Carpenter JM, Diaz AN, Benbow
RA and Filipov NM: Sex differences in behavior, response to LPS,
and glucose homeostasis in middle-aged mice. Behav Brain Res.
418:1136282022. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sarkar P, Mazgaeen L, Saini S, Gautam N,
Paden A, Paton H, Boevers P, Duvall J, Parlet C, Bermick J and
Gurung P: Neutrophils are key modulators of sex differences in
LPS-induced shock in mice. Blood Vessel Thromb Hemost.
2:1001042025. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Iba T, Helms J, Connors JM and Levy JH:
The pathophysiology, diagnosis, and management of sepsis-associated
disseminated intravascular coagulation. J Intensive Care.
11:242023. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Tan R, Ge C, Wang J, Yang Z, Guo H, Yan Y
and Du Q: Interpretable machine learning model for early morbidity
risk prediction in patients with sepsis-induced coagulopathy: A
multi-center study. Front Immunol. 16:15522652025. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Williams B, Zou L, Pittet JF and Chao W:
Sepsis-induced coagulopathy: A comprehensive narrative review of
pathophysiology, clinical presentation, diagnosis, and management
strategies. Anesth Analg. 138:696–711. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yajnik V and Maarouf R: Sepsis and the
microcirculation: The impact on outcomes. Curr Opin Anaesthesiol.
35:230–235. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rovas A, Seidel LM, Vink H, Pohlkotter T,
Pavenstadt H, Ertmer C, Hessler M and Kümpers P: Association of
sublingual microcirculation parameters and endothelial glycocalyx
dimensions in resuscitated sepsis. Crit Care. 23:2602019.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bateman RM, Sharpe MD, Jagger JE and Ellis
CG: Sepsis impairs microvascular autoregulation and delays
capillary response within hypoxic capillaries. Crit Care.
19:3892015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
De Backer D, Ricottilli F and
Ospina-Tascon GA: Septic shock: A microcirculation disease. Curr
Opin Anaesthesiol. 34:85–91. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Colbert JF and Schmidt EP: Endothelial and
microcirculatory function and dysfunction in sepsis. Clin Chest
Med. 37:263–275. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li X, Sim MMS and Wood JP: Recent insights
into the regulation of coagulation and thrombosis. Arterioscler
Thromb Vasc Biol. 40:e119–e125. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang X, Cheng X, Tang Y, Qiu X, Wang Y,
Kang H, Wu J, Wang Z, Liu Y, Chen F, et al: Bacterial endotoxin
activates the coagulation cascade through gasdermin D-dependent
phosphatidylserine exposure. Immunity. 51:983–996. e62019.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Deng M, Tang Y, Li W, Wang X, Zhang R,
Zhang X, Zhao X, Liu J, Tang C, Liu Z, et al: The endotoxin
delivery protein HMGB1 mediates caspase-11-dependent lethality in
sepsis. Immunity. 49:740–753. e72018. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Rodrigues M, Kosaric N, Bonham CA and
Gurtner GC: Wound healing: A cellular perspective. Physiol Rev.
99:665–706. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bloom SI, Islam MT, Lesniewski LA and
Donato AJ: Mechanisms and consequences of endothelial cell
senescence. Nat Rev Cardiol. 20:38–51. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schlommer C, Brandtner A and Bachler M:
Antithrombin and its role in host defense and inflammation. Int J
Mol Sci. 22:42832021. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wilhelm AR, Parsons NA, Samelson-Jones BJ,
Davidson RJ, Esmon CT, Camire RM and George LA: Activated protein C
has a regulatory role in factor VIII function. Blood.
137:2532–2543. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li X, Song X, Mahmood DFD, Sim MMS,
Bidarian SJ and Wood JP: Activated protein C, protein S, and tissue
factor pathway inhibitor cooperate to inhibit thrombin activation.
Thromb Res. 230:84–93. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo H, Liu D, Gelbard H, Cheng T, Insalaco
R, Fernandez JA, Griffin JH and Zlokovic BV: Activated protein C
prevents neuronal apoptosis via protease activated receptors 1 and
3. Neuron. 111:41162023. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Nooijer AH, Kotsaki A, Kranidioti E,
Kox M, Pickkers P, Toonen EJM, Giamarellos-Bourboulis EJ and Netea
MG: Complement activation in severely ill patients with sepsis: No
relationship with inflammation and disease severity. Crit Care.
27:632023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Noris M and Galbusera M: The complement
alternative pathway and hemostasis. Immunol Rev. 313:139–161. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rawish E, Sauter M, Sauter R, Nording H
and Langer HF: Complement, inflammation and thrombosis. Br J
Pharmacol. 178:2892–2904. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Andrieux P, Chevillard C, Cunha-Neto E and
Nunes JPS: Mitochondria as a cellular hub in infection and
inflammation. Int J Mol Sci. 22:113382021. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Flora GD, Nayak MK, Ghatge M, Kumskova M,
Patel RB and Chauhan AK: Mitochondrial pyruvate dehydrogenase
kinases contribute to platelet function and thrombosis in mice by
regulating aerobic glycolysis. Blood Adv. 7:2347–2359. 2023.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hidalgo A, Libby P, Soehnlein O, Aramburu
IV, Papayannopoulos V and Silvestre-Roig C: Neutrophil
extracellular traps: from physiology to pathology. Cardiovasc Res.
118:2737–2753. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Breda LCD, Breda CNS, de Almeida JRF,
Paulo LNM, Jannuzzi GP, Menezes IG, Albuquerque RC, Câmara NOS,
Ferreira KS and de Almeida SR: Fonsecaeapedrosoi conidia and hyphae
activate neutrophils distinctly: Requirement of TLR-2 and TLR-4 in
neutrophil effector functions. Front Immunol. 11:5400642020.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shao Y, Guo Z, Yang Y, Liu L, Huang J,
Chen Y, Li L and Sun B: Neutrophil extracellular traps contribute
to myofibroblast differentiation and scar hyperplasia through the
Toll-like receptor 9/nuclear factor Kappa-B/interleukin-6 pathway.
Burns Trauma. 10:tkac0442022. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wigerblad G and Kaplan MJ: Neutrophil
extracellular traps in systemic autoimmune and autoinflammatory
diseases. Nat Rev Immunol. 23:274–288. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Y, Du C, Zhang Y and Zhu L:
Composition and function of neutrophil extracellular traps.
Biomolecules. 14:4162024. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Jiang X, Li Q, Huang R, Qian Y, Jiang Y,
Liu T, Wang Y, Hu K, Huang J, Huang W, et al: Giardia duodenalis
triggered neutrophil extracellular traps in goats. Immunobiology.
230:1528942025. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Huang J, Hong W, Wan M and Zheng L:
Molecular mechanisms and therapeutic target of NETosis in diseases.
MedComm (2020). 3:e1622022. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Mou Z, Chen Y, Hu J, Hu Y, Zou L, Chen X,
Liu S, Yin Q, Gong J, Li S, et al: Icaritin inhibits the
progression of urothelial cancer by suppressing PADI2-mediated
neutrophil infiltration and neutrophil extracellular trap
formation. Acta Pharm Sin B. 14:3916–3930. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Burgener SS and Schroder K: Neutrophil
extracellular traps in host defense. Cold Spring Harb Perspect
Biol. 12:a0370282020. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zou S, Han X, Luo S, Tan Q, Huang H, Yao
Z, Hou W, Jie H and Wang J: Bay-117082 treats sepsis by inhibiting
neutrophil extracellular traps (NETs) formation through
down-regulating NLRP3/N-GSDMD. Int Immunopharmacol. 141:1128052024.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Gosswein S, Lindemann A, Mahajan A,
Maueroder C, Martini E, Patankar J, Schett G, Becker C, Wirtz S,
Naumann-Bartsch N, et al: Citrullination licenses calpain to
decondense nuclei in neutrophil extracellular trap formation. Front
Immunol. 10:24812019. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Thirugnanasambandham I, Radhakrishnan A,
Kuppusamy G, Singh SK and Dua K: Peptidylarginine deiminase-4:
Medico-formulative strategy towards management of rheumatoid
arthritis. Biochem Pharmacol. 200:1150402022. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Singhal A and Kumar S: Neutrophil and
remnant clearance in immunity and inflammation. Immunology.
165:22–43. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Akk A, Springer LE and Pham CT: Neutrophil
extracellular traps enhance early inflammatory response in sendai
virus-induced asthma phenotype. Front Immunol. 7:3252016.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Liu X, Arfman T, Wichapong K,
Reutelingsperger CPM, Voorberg J and Nicolaes GAF: PAD4 takes
charge during neutrophil activation: Impact of PAD4 mediated NET
formation on immune-mediated disease. J Thromb Haemost.
19:1607–1617. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lv G, Wang H, Wang J, Lian S and Wu R:
Effect of BLV infection on the immune function of polymorphonuclear
neutrophil in dairy cows. Front Vet Sci. 8:7376082021. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yousefi S, Mihalache C, Kozlowski E,
Schmid I and Simon HU: Viable neutrophils release mitochondrial DNA
to form neutrophil extracellular traps. Cell Death Differ.
16:1438–1444. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Denning NL, Aziz M, Gurien SD and Wang P:
DAMPs and NETs in sepsis. Front Immunol. 10:25362019. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xia L, Yan X and Zhang H: Mitochondrial
DNA-activated cGAS-STING pathway in cancer: Mechanisms and
therapeutic implications. Biochim Biophys Acta Rev Cancer.
1880:1892492025. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu L, Mao Y, Xu B, Zhang X, Fang C, Ma Y,
Men K, Qi X, Yi T, Wei Y and Wei X: Induction of neutrophil
extracellular traps during tissue injury: Involvement of STING and
toll-like receptor 9 pathways. Cell Prolif. 53:e127752020.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Zheng Y, Zhu Y, Liu X, Zheng H, Yang Y, Lu
Y, Zhou H, Zheng J and Dong Z: The screening of albumin as a key
serum component in preventing release of neutrophil extracellular
traps by selectively inhibiting mitochondrial ROS generation. Can J
Physiol Pharmacol. 99:427–438. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zeng FL, Zhang Y, Wang ZH, Zhang H, Meng
XT, Wu YQ, Qian ZZ, Ding YH, Li J, Ma TT and Huang C: Neutrophil
extracellular traps promote acetaminophen-induced acute liver
injury in mice via AIM2. Acta Pharmacol Sin. 45:1660–1672. 2024.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chen KW, Monteleone M, Boucher D,
Sollberger G, Ramnath D, Condon ND, von Pein JB, Broz P, Sweet MJ
and Schroder K: Noncanonical inflammasome signaling elicits
gasdermin D-dependent neutrophil extracellular traps. Sci Immunol.
3:eaar66762018. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhou L, Li Y, You J, Wu C, Zuo L, Chen Y,
Kang L, Zhou Z, Huang R and Wu S: Salmonella spvC gene suppresses
macrophage/neutrophil antibacterial defense mediated by gasdermin
D. Inflamm Res. 73:19–33. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Iba T, Levy JH, Raj A and Warkentin TE:
Advance in the management of sepsis-induced coagulopathy and
disseminated intravascular coagulation. J Clin Med. 8:7282019.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Fei Y and Huang X, Ning F, Qian T, Cui J,
Wang X and Huang X: NETs induce ferroptosis of endothelial cells in
LPS-ALI through SDC-1/HS and downstream pathways. Biomed
Pharmacother. 175:1166212024. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Folco EJ, Mawson TL, Vromman A,
Bernardes-Souza B, Franck G, Persson O, Nakamura M, Newton G,
Luscinskas FW and Libby P: Neutrophil extracellular traps induce
endothelial cell activation and tissue factor production through
interleukin-1alpha and cathepsin G. Arterioscler Thromb Vasc Biol.
38:1901–1912. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Shan X, Yu S, Tang Z, Yang P and Dong L:
GSDMD-NETs in patients with sepsis-induced coagulopathy and their
interaction with glycocalyx damage. Front Immunol. 16:16241282025.
View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fang J, Ding H, Huang J, Liu W, Hong T,
Yang J, Wu Z, Li Z, Zhang S, Liu P, et al: Mac-1 blockade impedes
adhesion-dependent neutrophil extracellular trap formation and
ameliorates lung injury in LPS-induced sepsis. Front Immunol.
16:15489132025. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Li Y, Zhu H, Liu Y, Li X, Zu X, Wang C, Xu
X, Sun Y, Dai Y, Zhang J, et al: STK10 regulates platelet function
in arterial thrombosis and thromboinflammation. Blood. 147:73–86.
2026. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Jiang H, Chen S, Gui X, Li Y, Sun Y, Zhu
H, Dai Y, Zhang J, Li X, Ju W, et al: Platelet NLRP6 protects
against microvascular thrombosis in sepsis. Blood. 146:382–395.
2025. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Hvas AM, Favaloro EJ and Hellfritzsch M:
Heparin-induced thrombocytopenia: Pathophysiology, diagnosis and
treatment. Expert Rev Hematol. 14:335–346. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Perdomo J, Leung HHL, Ahmadi Z, Yan F,
Chong JJH, Passam FH and Chong BH: Neutrophil activation and
NETosis are the major drivers of thrombosis in heparin-induced
thrombocytopenia. Nat Commun. 10:13222019. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Leung HHL, Perdomo J, Ahmadi Z, Yan F,
McKenzie SE and Chong BH: Inhibition of NADPH oxidase blocks
NETosis and reduces thrombosis in heparin-induced thrombocytopenia.
Blood Adv. 5:5439–5451. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li J, Geng Y, Luo Y, Sun X, Guo Y and Dong
Z: Pathological roles of NETs-platelet synergy in thrombotic
diseases: From molecular mechanisms to therapeutic targeting. Int
Immunopharmacol. 159:1149342025. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Thalin C, Hisada Y, Lundstrom S, Mackman N
and Wallen H: Neutrophil extracellular traps: villains and targets
in arterial, venous, and cancer-associated thrombosis. Arterioscler
Thromb Vasc Biol. 39:1724–1738. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou P, Li T, Jin J, Liu Y, Li B, Sun Q,
Tian J, Zhao H, Liu Z, Ma S, et al: Interactions between neutrophil
extracellular traps and activated platelets enhance procoagulant
activity in acute stroke patients with ICA occlusion. EBioMedicine.
53:1026712020. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Pignataro G, Gemma S, Petrucci M, Barone
F, Piccioni A, Franceschi F and Candelli M: Unraveling NETs in
sepsis: From cellular mechanisms to clinical relevance. Int J Mol
Sci. 26:74642025. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhao Z, Pan Z, Zhang S, Ma G, Zhang W,
Song J, Wang Y, Kong L and Du G: Neutrophil extracellular traps: A
novel target for the treatment of stroke. Pharmacol Ther.
241:1083282023. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Reyes-Garcia AML, Aroca A, Arroyo AB,
Garcia-Barbera N, Vicente V, Gonzalez-Conejero R and Martínez C:
Neutrophil extracellular trap components increase the expression of
coagulation factors. Biomed Rep. 10:195–201. 2019.PubMed/NCBI
|
|
80
|
Rao AN, Kazzaz NM and Knight JS: Do
neutrophil extracellular traps contribute to the heightened risk of
thrombosis in inflammatory diseases? World J Cardiol. 7:829–842.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Demyanets S, Stojkovic S, Mauracher LM,
Kopp CW, Wojta J, Thaler J, Panzer S and Gremmel T: Surrogate
markers of neutrophil extracellular trap formation are associated
with ischemic outcomes and platelet activation after peripheral
angioplasty and stenting. J Clin Med. 9:3042020. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Li RHL and Tablin F: A comparative review
of neutrophil extracellular traps in sepsis. Front Vet Sci.
5:2912018. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Thomassen M, Bouwens BRC, Wichapong K,
Suylen DP, Bouwman FG, Hackeng TM and Koenen RR: Protein arginine
deiminase 4 inactivates tissue factor pathway inhibitor-alpha by
enzymatic modification of functional arginine residues. J Thromb
Haemost. 21:1214–1226. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Asakura H: Classifying types of
disseminated intravascular coagulation: Clinical and animal models.
J Intensive Care. 2:202014. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Varju I, Longstaff C, Szabo L, Farkas AZ,
Varga-Szabo VJ, Tanka-Salamon A, Machovich R and Kolev K: DNA,
histones and neutrophil extracellular traps exert anti-fibrinolytic
effects in a plasma environment. Thromb Haemost. 113:1289–1298.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Mangold A, Alias S, Scherz T, Hofbauer M,
Jakowitsch J, Panzenbock A, Simon D, Laimer D, Bangert C,
Kammerlander A, et al: Coronary neutrophil extracellular trap
burden and deoxyribonuclease activity in ST-elevation acute
coronary syndrome are predictors of ST-segment resolution and
infarct size. Circ Res. 116:1182–1192. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei
R, Lin ZF, Wang XY, Wang CQ, Lu M, et al: Increased neutrophil
extracellular traps promote metastasis potential of hepatocellular
carcinoma via provoking tumorous inflammatory response. J Hematol
Oncol. 13:32020. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Xu C, Ye Z, Jiang W, Wang S and Zhang H:
Cyclosporine A alleviates colitis by inhibiting the formation of
neutrophil extracellular traps via the regulating pentose phosphate
pathway. Mol Med. 29:1692023. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Li Y, Li H, Wang Y, Guo J and Zhang D:
Potential biomarkers for early diagnosis, evaluation, and prognosis
of sepsis-induced coagulopathy. Clin Appl Thromb Hemost.
29:107602962311950892023. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Zhao J, Zhen N, Zhou Q, Lou J, Cui W,
Zhang G and Tian B: NETs promote inflammatory injury by activating
cGAS-STING pathway in acute lung injury. Int J Mol Sci.
24:51252023. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Mai SH, Khan M, Dwivedi DJ, Ross CA, Zhou
J, Gould TJ, Gross PL, Weitz JI, Fox-Robichaud AE and Liaw PC;
Canadian Critical Care Translational Biology Group, : Delayed but
not early treatment with dnase reduces organ damage and improves
outcome in a murine model of sepsis. Shock. 44:166–172. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zalghout S and Martinod K: Therapeutic
potential of DNases in immunothrombosis: Promising succor or
uncertain future? J Thromb Haemost. 23:760–778. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Cai M, Deng J, Wu S, Cao Y, Chen H, Tang
H, Zou C, Zhu H and Qi L: Alpha-1 antitrypsin targeted neutrophil
elastase protects against sepsis-induced inflammation and
coagulation in mice via inhibiting neutrophil extracellular trap
formation. Life Sci. 353:1229232024. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Liu X, Li T, Chen H, Yuan L and Ao H: Role
and intervention of PAD4 in NETs in acute respiratory distress
syndrome. Respir Res. 25:632024. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
He W, Xi Q, Cui H, Zhang P, Huang R, Wang
T and Wang D: Liang-ge decoction ameliorates coagulation
dysfunction in cecal ligation and puncture-induced sepsis model
rats through inhibiting PAD4-dependent neutrophil extracellular
trap formation. Evid Based Complement Alternat Med.
2023:50429532023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Kato Y, Nishida O, Kuriyama N, Nakamura T,
Kawaji T, Onouchi T, Hasegawa D and Shimomura Y: Effects of
thrombomodulin in reducing lethality and suppressing neutrophil
extracellular trap formation in the lungs and liver in a
lipopolysaccharide-induced murine septic shock model. Int J Mol
Sci. 22:49332021. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Silva CMS, Wanderley CWS, Veras FP, Sonego
F, Nascimento DC, Goncalves AV, Martins TV, Cólon DF, Borges VF,
Brauer VS, et al: Gasdermin D inhibition prevents multiple organ
dysfunction during sepsis by blocking NET formation. Blood.
138:2702–2713. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mansour A, Bachelot-Loza C, Nesseler N,
Gaussem P and Gouin-Thibault I: P2Y(12) inhibition beyond
thrombosis: effects on inflammation. Int J Mol Sci. 21:13912020.
View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Vardon-Bounes F, Ruiz S, Gratacap MP,
Garcia C, Payrastre B and Minville V: Platelets are critical key
players in sepsis. Int J Mol Sci. 20:34942019. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
A feedback loop between platelets and NETs
amplifies inflammation in severe sepsis. Nat Cardiovasc Res.
1:698–699. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Chen S, Wu A, Shen X, Kong J and Huang Y:
Disrupting the dangerous alliance: Dual anti-inflammatory and
anticoagulant strategy targets platelet-neutrophil crosstalk in
sepsis. J Control Release. 379:814–831. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Su F, Moreau A, Savi M, Salvagno M, Annoni
F, Zhao L, Xie K, Vincent JL and Taccone FS: Circulating
nucleosomes as a novel biomarker for sepsis: A scoping review.
Biomedicines. 12:13852024. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Cutter AR and Hayes JJ: A brief review of
nucleosome structure. FEBS Lett. 589((20 Pt A)): 2914–2922. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Chen L, Zhao Y, Lai D, Zhang P, Yang Y, Li
Y, Fei K, Jiang G and Fan J: Neutrophil extracellular traps promote
macrophage pyroptosis in sepsis. Cell Death Dis. 9:5972018.
View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Magna M and Pisetsky DS: The role of HMGB1
in the pathogenesis of inflammatory and autoimmune diseases. Mol
Med. 20:138–146. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Nija RJ, Sanju S, Sidharthan N and Mony U:
Extracellular trap by blood cells: Clinical implications. Tissue
Eng Regen Med. 17:141–153. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Loureiro A, Pais C and Sampaio P:
Relevance of macrophage extracellular traps in C. albicans killing.
Front Immunol. 10:27672019. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Koyama R, Arai T, Kijima M, Sato S, Miura
S, Yuasa M, Kitamura D and Mizuta R: DNase gamma, DNase I and
caspase-activated DNase cooperate to degrade dead cells. Genes
Cells. 21:1150–1163. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Janovicova L, Conka J, Laukova L and Celec
P: Variability of endogenous deoxyribonuclease activity and its
pathophysiological consequences. Mol Cell Probes. 65:1018442022.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Li Y, Wan D, Luo X, Song T, Wang Y, Yu Q,
Jiang L, Liao R, Zhao W and Su B: Circulating histones in sepsis:
Potential outcome predictors and therapeutic targets. Front
Immunol. 12:6501842021. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Garcia B, Su F, Dewachter L, Wang Y, Li N,
Remmelink M, Eycken MV, Khaldi A, Favory R, Herpain A, et al:
Neutralization of extracellular histones by sodium-Beta-O-methyl
cellobioside sulfate in septic shock. Crit Care. 27:4582023.
View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ngo AT, Skidmore A, Oberg J, Yarovoi I,
Sarkar A, Levine N, Bochenek V, Zhao G, Rauova L, Kowalska MA, et
al: Platelet factor 4 limits neutrophil extracellular trap- and
cell-free DNA-induced thrombogenicity and endothelial injury. JCI
Insight. 8:e1710542023. View Article : Google Scholar : PubMed/NCBI
|