Introduction
Osteoporosis (OP) and osteoarthritis (OA) rank among the most prevalent chronic musculoskeletal disorders affecting middle-aged and older women, and notably impair patient mobility and quality of life. The prevalence of both conditions rises with age, and these conditions often co-occur in postmenopausal women due to shared risk factors, including age, hormonal changes and chronic inflammation (1). OP is characterized by a reduction in bone mineral density (BMD) and altered bone microarchitecture, resulting in an increased risk of fracture and frailty (2). OA, a degenerative joint disease, is characterized by the deterioration of articular cartilage and aberrant subchondral bone remodeling, resulting in joint pain and functional impairment (3). The co-occurrence of these diseases typically exacerbates their combined impact on physical function (1). Despite the elevated prevalence and notable burden of these conditions, the underlying mechanisms driving the comorbidity of OP and OA remain incompletely elucidated, underscoring the necessity for further investigations into shared molecular pathways and risk factors.
An important factor underlying OP and OA in postmenopausal women is estrogen deficiency, which naturally occurs as a result of menopause. Estrogen plays an important role in maintaining homeostasis in bone and joint tissues. Estrogen has been shown to modulate bone remodeling, inhibit osteoclast-driven bone resorption and regulate cartilage and subchondral bone metabolism (4). The rapid decline in estrogen levels observed following menopause has been demonstrated to markedly accelerate bone loss, particularly in the initial postmenopausal years, indicating that estrogen deficiency is a primary driver of postmenopausal OP (5). Additionally, estrogen deficiency has been shown to increase patient susceptibility to OA by aggravating cartilage degradation, disrupting normal subchondral bone remodeling and promoting joint inflammation (6). In addition to its direct effects on bone and cartilage cells, estrogen deficiency has been shown to elicit systemic immune alterations such as low-grade chronic inflammation and modifications in immune cell function, further contributing to the pathogenesis of OP and OA (7). Notably, Shipov et al (8), employing ovariectomized animal models, which are widely accepted as representative models of postmenopausal estrogen deficiency, have demonstrated that estrogen deficiency results in both bone loss and joint degeneration, reinforcing the important role of hormonal changes in the coexistence of these conditions.
The clinical phenomenon of OP and OA comorbidity has garnered notable recognition in previous years; however, the molecular mechanisms underlying this relationship have yet to be fully elucidated (9,10). As highlighted in the research conducted by Liu et al (11), these prevalent skeletal disorders share overlapping genetic determinants and signaling cascades. Their work further identified pleiotropic genetic loci and shared genetic architecture underlying both osteoarthritis (OA) and osteoporosis (OP), which strongly underscores the inherent biological correlation between the two conditions. Additionally, both conditions are influenced by aging-related processes, such as cellular senescence, chronic inflammation and impaired autophagy, which may disrupt bone and cartilage homeostasis (12). Furthermore, additional comorbidities, including diabetes and cardiovascular disease, complicate the clinical management of patients with OP and OA (13,14), emphasizing the necessity for integrated, multidisciplinary therapeutic approaches. Despite these insights, notable research gaps persist regarding the elucidation of: i) Mechanisms by which estrogen deficiency regulates the molecular interactions between bone and joint tissues; and ii) how these modified interactions contribute to the onset and progression of coexisting OP and OA.
The present review systematically investigates the impact of estrogen deficiency on the pathogenesis of OP and OA comorbidity. The structure of the present review aims to extend beyond merely describing OP and OA in parallel and instead focus on how the loss of estrogen drives overlapping pathological processes. The present review also emphasizes the molecular foundations of OP and OA by discussing previous research advancements and their clinical implications. Furthermore, the present review examines the direct and indirect effects of estrogen deficiency on bone and joint cells while considering the roles of immune and inflammatory pathways. Additionally, the present review examines the impacts of genetic and environmental factors on the pathogenesis of OP and OA and explores novel biomarkers and therapeutic strategies targeting the common mechanisms of these conditions that have been associated with estrogen deficiency. By incorporating findings from basic research, animal studies and clinical investigations, the present review endeavors to provide a comprehensive framework for elucidating the complex interplay between estrogen deficiency, OP and OA, ultimately aiming to inform more effective clinical interventions for affected women.
Molecular mechanisms underlying estrogen deficiency and OP
Estrogen-mediated regulation of bone remodeling
Estrogen is important for preserving the balance of bone remodeling, as it impedes osteoclastogenesis and boosts osteoblast activity. Under physiological conditions, estrogen has been shown to suppress bone resorption through the activity of various pathways and cytokines by disrupting the differentiation of osteoclasts and reducing their lifespan, effectively restraining the initiation of bone remodeling and mitigating bone loss (15). Additionally, estrogen enhances the production of osteoprotegerin (OPG), which functions as a decoy receptor that binds to receptor activator of nuclear factor κ-B ligand (RANKL), thereby preventing RANKL from activating osteoclasts. In postmenopausal women, reduced estrogen levels have been shown to result in an increased RANKL/OPG ratio, promoting osteoclast differentiation and bone resorption (16). This accelerates bone loss and compromises skeletal microarchitecture, therefore increasing the risk of fractures (17). Furthermore, estrogen deficiency has been associated with the upregulation of pro-inflammatory cytokines, for example interleukin (IL)-1, IL-6 and tumor necrosis factor-α (TNF-α), which further supports osteoclastogenesis and exacerbates bone resorption (16).
In addition to exhibiting antiresorptive effects, estrogen also supports osteoblast survival and enhances bone formation by activating the Wnt signaling pathway, which is important for the differentiation and function of osteoblasts (18). Clinical and experimental evidence has suggested that interventions aimed at restoring estrogenic activity, such as hormone replacement therapy (HRT) or treatments with selective estrogen receptor (ER) modulators (SERMs), may lower elevated bone remodeling markers and partially reverse bone loss, although the optimal therapeutic regimen for these interventions remains under investigation (19). Therefore, these molecular mechanisms-through which estrogen suppresses osteoclastogenesis, promotes osteoblast survival and activates anabolic signaling-collectively underscore the critical role of estrogen as an active coordinator of skeletal health. The disruption of these mechanisms due to estrogen deficiency has been identified as a notable contributor to OP pathogenesis (Table I).
 |
Table I.
Shared molecular mechanisms linking estrogen deficiency to OP-OA comorbidity.
|
Table I.
Shared molecular mechanisms linking estrogen deficiency to OP-OA comorbidity.
| Mechanistic module |
Key alterations in OP |
Key alterations in OA |
Convergent role in OP-OA comorbidity |
(Refs.) |
| Bone-cartilage metabolism imbalance |
Increased RANKL/OPG ratio resulting in enhanced osteoclastogenesis and bone resorption; increased osteoblast and osteocyte apoptosis under estrogen-depleted stress |
Increased activity of matrix-degrading enzymes, such as MMP-13, with reduced type II collagen and aggrecan synthesis, leading to extracellular matrix breakdown |
Couples systemic bone loss with cartilage matrix degradation, fostering progressive osteochondral deterioration |
(37–41) |
| Chronic inflammation |
Elevated levels of pro-inflammatory cytokines, including IL-1β, IL-6 and TNF-α, promote osteoclast formation and resorption-dominant remodeling |
Synovitis and cytokine-driven catabolism accelerate cartilage damage and pain sensitization |
A shared systemic and local inflammatory state amplifies both bone resorption and cartilage degradation |
(55,56,65–67) |
| Immune dysregulation |
Osteoimmune imbalance, including B-cell expansion and Th17/Treg disequilibrium, supports osteoclast activation and impaired bone formation |
Inflammatory immune microenvironment sustains synovitis and cartilage degeneration |
Immune dysregulation bridges systemic bone and local joint pathology via persistent cytokine production and crosstalk |
(72–74) |
| Oxidative stress |
Reduced antioxidant defenses and increased ROS, leading to osteocyte apoptosis and impaired bone remodeling |
Elevated ROS drives chondrocyte apoptosis and senescence and promotes the expression of catabolic enzymes that accelerate cartilage degeneration |
Redox stress is a shared tissue-destabilizing mechanism compromising both bone and cartilage homeostasis |
(77–80) |
| Bone marrow microenvironment remodeling |
Increased marrow adiposity, cytokine burden, senescence, immune perturbation and ROS accumulation impair osteogenesis and favor bone loss |
Bone marrow-derived inflammatory and oxidative mediators impair chondrocyte function and cartilage repair; concomitant subchondral bone deterioration increases cartilage vulnerability and accelerates joint degeneration. |
Adipogenic drift, chronic inflammation, immune perturbation and ROS accumulation interfere with bone marrow microenvironmental homeostasis and underpin the concurrent progression of OP and OA |
(29–32) |
| Bone-joint unit interaction |
Deteriorated bone microarchitecture and subchondral changes alter load transmission and reduce mechanical support |
Altered biomechanics increase cartilage vulnerability and accelerate degeneration |
Results in a biomechanical-biological cycle, in which subchondral bone loss and cartilage damage reinforce each other |
(58–62) |
Estrogen deficiency and osteocyte apoptosis
Estrogen deficiency plays a central role in promoting apoptosis in osteoblasts and osteocytes, which adversely affects bone formation and contributes to OP pathogenesis. Numerous studies have demonstrated that estrogen deficiency, particularly after menopause, directly elevates the apoptotic rate of osteocytes and osteoblasts. This disruption in bone homeostasis ultimately weakens the bone matrix (20,21). Mechanistically, estrogen deficiency has been shown to alter the osteocyte microenvironment, resulting in heightened osteocyte apoptosis and modifications to the lacunar-canalicular network. These changes are temporally associated with accelerated bone loss and increased cortical microporosity (22,23). Additionally, estrogen deficiency has been shown to attenuate the mechanosensitivity of osteocytes and their paracrine regulation of osteoclasts, further aggravating bone resorption (22).
At the molecular level, estrogen has been shown to protect bone cells through a number of signaling pathways, such as the phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) and mitogen-activated protein kinase (MAPK) pathways. For example, the PI3K/Akt pathway helps to promote osteoblast survival and proliferation; however, the activity of this pathway has been shown to be notably diminished in osteoblasts derived from estrogen-deficient models. This reduction has been associated with increased apoptosis and a decreased capacity for bone formation (24). Similarly, the MAPK pathway, in particular the extracellular signal-regulated kinase and p38 cascades, has been shown to promote osteoblast proliferation and inhibit apoptosis. Dysregulation of this pathway under estrogen-deficient conditions has been shown to amplify bone cell loss (24,25). Furthermore, oxidative stress, which is amplified in the absence of estrogen, has been found to promote osteocyte apoptosis and disrupt bone remodeling. This has indicated that antioxidant therapies may play a role in alleviating bone loss in the context of estrogen deficiency (26,27). Furthermore, these findings highlight the important role of estrogen in maintaining bone cell viability and highlight the importance of the PI3K/Akt and MAPK pathways in mediating the anti-apoptotic effects of estrogen on osteoblasts and osteocytes (Table I). The disruption of these pathways induced by estrogen deficiency not only heightens bone cell apoptosis but also impedes bone formation, thereby accelerating the progression of OP.
Alterations in the bone marrow microenvironment
Estrogen deficiency leads to notable alterations to the bone marrow microenvironment, thus serving an important role in the development of OP and its relationship with OA. One of the key changes to the bone microenvironment observed in estrogen-deficient models is the increased accumulation of bone marrow adipocytes; this is driven by the shared lineage of adipocytes and osteoblasts, which originate from mesenchymal stromal cells. In postmenopausal OP, the expansion of marrow adipose tissue (MAT) caused by estrogen deficiency has been shown to impair bone remodeling via the release of adipokines and cytokines from MAT-derived adipocytes, which inhibit osteoblast function and contribute to bone loss (17). As such, MAT not only functions as a passive space filler upon estrogen deficiency-induced expansion, but has also been recognized as an active endocrine organ that modulates the bone microenvironment and exacerbates skeletal fragility (28).
Furthermore, estrogen deficiency triggers a pro-inflammatory state in the bone marrow, which is typified by elevated levels of inflammatory cytokines, such as TNF-α and IL-6, enhancing osteoclastogenesis and bone resorption (29). This inflammatory environment is sustained by immune cell dysregulation, particularly regarding B cells and monocytes, which upregulate RANKL expression and promote excessive osteoclast activation under estrogen-deficient conditions (30). Furthermore, cellular senescence becomes more pronounced in estrogen-deficient bone marrow. This process affects multiple resident cell populations, including immune cells, mesenchymal stem cells and osteocytes, and is accompanied by the senescence-associated secretory phenotype, which involves the release of pro-inflammatory cytokines and other factors, leading to heightened inflammation and the reduced regenerative capacity of mesenchymal stem cells (31). The accumulation of reactive oxygen species (ROS) induced by estrogen deficiency, combined with a shift in macrophage polarization toward the pro-inflammatory M1 phenotype, has been shown to contribute to the deterioration of the bone microenvironment by impairing osteogenesis and promoting bone loss (32). These interconnected processes, comprising adipogenic drift, chronic inflammation and oxidative stress, interfere with bone marrow microenvironmental homeostasis; therefore, these processes underpin both OP and the progression of OA in the context of estrogen deficiency (Table I). Elucidating these molecular and cellular modifications can reveal potential therapeutic targets for restoring marrow homeostasis and alleviating skeletal disease burdens.
Implications of estrogen deficiency-regulated bone remodeling for OP and OA comorbidity
The mechanism by which estrogen deficiency induces OP is not an isolated phenomenon; notably, estrogen deficiency creates a systemic and local environment that is harmful to joint health. The bone-derived alterations characteristic of OP have been increasingly recognized as important components of a broader bone-joint unit dysfunction that contributes to OA progression (33,34). Pro-inflammatory cytokines that drive bone resorption, such as IL-1β, IL-6 and TNF-α, can disseminate into the synovial fluid and subchondral bone, thereby triggering synovitis and accelerating cartilage degeneration (6,16). Estrogen deficiency also induces a higher RANKL/OPG ratio in the bone microenvironment, which potentiates osteoclast overactivation and subchondral bone deterioration, a key pathological change that facilitates OA progression. Similarly, oxidative stress, MAT expansion and alterations to the bone marrow microenvironment caused by aging cells have been shown to potentially impair chondrocyte function and affect cartilage repair (32,35). Furthermore, bone loss and microarchitectural deterioration, particularly in the subchondral compartment, can modify load transmission across the joint, thus increasing cartilage vulnerability and accelerating degenerative changes (36). Therefore, molecular dysregulation in the bone induced by estrogen deficiency directly influences the development of joint degeneration, contributing to OA. This establishes a fundamental association between alterations to the bone microenvironment and the concurrent onset of joint degeneration.
Molecular mechanisms underlying estrogen deficiency and OA
Imbalance of cartilage matrix metabolism
Estrogen plays an important role in maintaining the structural and functional integrity of articular cartilage. Notably, estrogen regulates chondrocyte activity, particularly regarding the production of type II collagen and aggrecan, which are important components of the cartilaginous extracellular matrix. As evidenced in studies on postmenopausal women and ovariectomized animal models, estrogen deficiency disrupts the balance of these components, resulting in impaired synthesis of the cartilaginous extracellular matrix and an increase in its degradation (37,38). Notably, estrogen deficiency has been shown to stimulate the upregulation of matrix metalloproteinases (MMPs), particularly MMP-13, the primary and specific collagenase responsible for targeted degradation of type II collagen in articular cartilage (39). Research has indicated that estrogen deficiency results in reduced proteoglycan levels, decreased type II collagen production and elevated MMP expression, culminating in cartilage degradation and OA-like alterations (40,41). Furthermore, interventions aimed at restoring estrogenic activity have been shown to partially reverse these degenerative changes. Therapeutic strategies that target downstream effectors of estrogen, such as galectin-3 or MMPs secreted by osteoclasts, have also yielded promising results, highlighting the importance of estrogen in preserving cartilage matrix homeostasis (40). Additional studies have revealed that therapeutic agents capable of enhancing chondrocyte-mediated matrix synthesis, such as actein, or combined metabolic interventions can ameliorate cartilage loss and restore extracellular matrix integrity in models of estrogen deficiency-induced OA (41,42). These findings underline the important role of estrogen in maintaining an equilibrium between anabolic and catabolic processes in cartilage.
Estrogen deficiency shifts this equilibrium towards matrix degradation by upregulating MMP expression and suppressing the synthesis of key matrix proteins (Table I). Therefore, therapeutic strategies targeting the molecular pathways influenced by estrogen loss, particularly those related to MMP regulation and matrix-protein synthesis, offer promise for preventing or alleviating cartilage degeneration in the context of OP and OA comorbidity.
Chondrocyte apoptosis and senescence
Estrogen deficiency has been recognized as an important factor in the induction of chondrocyte apoptosis, which undermines the ability of cartilage to repair and maintain itself. Estrogen deficiency, as observed in studies on postmenopausal women and ovariectomized animal models, creates an environment that fosters cartilage degeneration by amplifying oxidative stress and causing mitochondrial dysfunction (43). The research conducted by Song et al (44) indicated that estrogen replacement therapy (ERT), particularly using 17β-estradiol, can mitigate chondrocyte injury induced by acidic conditions by suppressing apoptosis and restoring mitochondrial function, underscoring the protective role of estrogen in maintaining chondrocyte viability. Mechanistically, estrogen deficiency is associated with heightened oxidative stress, which activates pro-apoptotic signaling pathways and disrupts mitochondrial function, ultimately leading to programmed cell death. Additionally, estrogen deficiency disrupts the Wnt/β-catenin signaling pathway in cartilage, decreasing chondrocyte proliferation and increasing apoptosis; these outcomes can be partially reversed by estrogen supplementation (45). Oxidative stress plays an important role, as excessive ROS levels not only induce apoptosis but also accelerate cellular aging processes, generating a feedback loop that drives cartilage deterioration (46,47) (Table I). Collectively, these molecular events emphasize how estrogen deficiency compromises chondrocyte survival and function, establishing a mechanistic link between postmenopausal OP and increased susceptibility to OA. Additionally, these findings highlight potential therapeutic targets for alleviating joint degeneration in individuals with estrogen deficiency.
Synovial inflammatory response
Estrogen deficiency reportedly exacerbates synovial inflammation, which is an important process in the pathogenesis of OP and OA comorbidity. Postmenopausal estrogen deficiency has been shown to alter the joint immune microenvironment by increasing the levels of pro-inflammatory cytokines, including IL-1β and TNF-α (48). These cytokines help to mediate the inflammatory cascade, facilitating the recruitment and stimulation of immune cells within the synovium (49). This inflammatory response not only accelerates joint damage but also promotes cartilage degradation. A recent study has indicated that follicle-stimulating hormone, which is upregulated in response to estrogen deficiency, can directly stimulate synovial macrophages to secrete inflammatory cytokines via activation of the nuclear factor κB (NF-κB) signaling pathway. This mechanism further aggravates inflammation in the joint and accelerates OA progression (50).
Clinical and histopathological examinations of synovial tissues from patients with OA and rheumatoid arthritis have revealed robust associations between sex-hormone receptor expression and both lymphocyte infiltration and subchondral inflammation, underscoring the impact of sex hormones on synovial pathology (51,52). A recent research indicated that the age-related decline in estrogen levels is associated with reduced levels of structural proteins in the cartilage and elevated inflammatory markers in the synovial fluid, which are particularly evident in female patients, suggesting a direct link between hormonal status and the joint microenvironment (53). The resulting inflammatory microenvironment not only promotes synovial hyperplasia and pannus formation but also accelerates the catabolic processes that lead to cartilage breakdown and joint degeneration (Table I). Interventions targeting the estrogen signaling pathway, such as SERMs or therapeutic agents that modify the retinoid X receptor α-ERα interaction, have demonstrated notable promise in reducing synovial inflammation and catabolic responses in preclinical models (54). Together, these findings emphasize the central role of estrogen deficiency in enhancing synovial inflammation by upregulating pro-inflammatory cytokines. This enhancement drives OA progression and contributes to OA-OP comorbidity.
Implications of estrogen deficiency-induced joint degeneration for the co-occurrence of OP and OA
The aforementioned mechanisms illustrate that estrogen deficiency promotes OA progression via disruption of cartilage matrix homeostasis, increased chondrocyte apoptosis and senescence, and the amplification of synovial inflammation. Notably, these joint-centered changes do not remain limited to the cartilage and synovium, instead contributing to a broader dysfunction of the osteochondral unit that may simultaneously exacerbate OP. Inflammatory mediators released from the synovium and deteriorating cartilage potentially accelerate subchondral bone loss by increasing the activation of osteoclasts and inhibiting osteogenic repair (55,56). Additionally, oxidative stress that triggers chondrocyte apoptosis has also been shown to promote osteocyte apoptosis and osteoclast activity (35,57). Consequently, the pathological processes initiated within the joint in response to estrogen deficiency, including inflammation, oxidative stress and cell death, do not solely impact the cartilage but actively contribute to the degradation of bone tissue (33). These interactions support a bidirectional model wherein cartilage degeneration and synovial inflammation increase subchondral bone turnover, whereas alterations in bone microarchitecture exacerbate cartilage vulnerability via biomechanical and biochemical feedback loops.
Overall, these findings reinforce the notion that estrogen deficiency serves as a common upstream trigger that orchestrates pathological processes across the bone-cartilage-synovium axis, thus providing a basis for integrated diagnostic methods and combined therapeutic strategies targeting OP-OA comorbidity.
Role of estrogen deficiency in OP and OA comorbidity
Bone-joint unit interactions
The complex relationship between bone and joint structures is important for maintaining the integrity of the musculoskeletal system, and by extension its functionality. Estrogen deficiency disrupts this delicate balance, thus contributing to the pathogenesis of OP-OA comorbidity. The bone-joint unit functions synergistically, as the health of the subchondral bone directly affects the biomechanics and homeostatic regulation of the overlying articular cartilage (58). Estrogen deficiency has been shown to lead to notable deterioration of the bone microarchitecture, resulting in decreased bone mass and weakened mechanical support (59). This altered bone environment may potentially modify joint loading patterns and increase the risk of cartilage damage, thereby accelerating OA progression (60). Notably, the connection between these comorbidities has been reinforced by evidence from studies that have concurrently evaluated both types of tissue. For example, in ovariectomized female rats and mice, which are well-established models for simulating postmenopausal estrogen deficiency, bone loss was characterized by decreased BMD and microstructural deterioration, and joint degeneration was typified by cartilage thinning and bone spur formation; these were observed simultaneously within the same animals. This provided direct experimental evidence for estrogen deficiency as a shared pathogenic driver underlying both OP and OA (61,62). Similarly, findings from clinical cohorts corroborate the concept of estrogen as a shared driver of OP and OA.
Postmenopausal populations often exhibit concomitant reductions in BMD alongside symptomatic or radiographic OA features, suggesting shared endocrine and inflammatory drivers affecting bone and joint tissues (1). Recent research has highlighted that chronic inflammation disrupts the homeostatic interactions between bone and joint cells. In particular, pyroptosis fosters an inflammatory environment that exacerbates both bone and joint degeneration, thereby disrupting cellular homeostasis within the skeletal environment (63) and further destabilizing the bone-joint complex (Fig. 1). Additionally, molecular mediators, such as irisin and S-equol, have emerged as important regulators of bone-muscle-adipose signaling, influencing inflammatory and metabolic pathways that affect bone and joint health (39,64). Estrogen deficiency potentially disrupts these molecular mediators, thereby damaging the structural and functional integration of the bone-joint unit. The relationship between bone and joint health, which is influenced by hormonal status and inflammation, underlines the necessity for a comprehensive approach to understanding and managing degenerative musculoskeletal diseases.
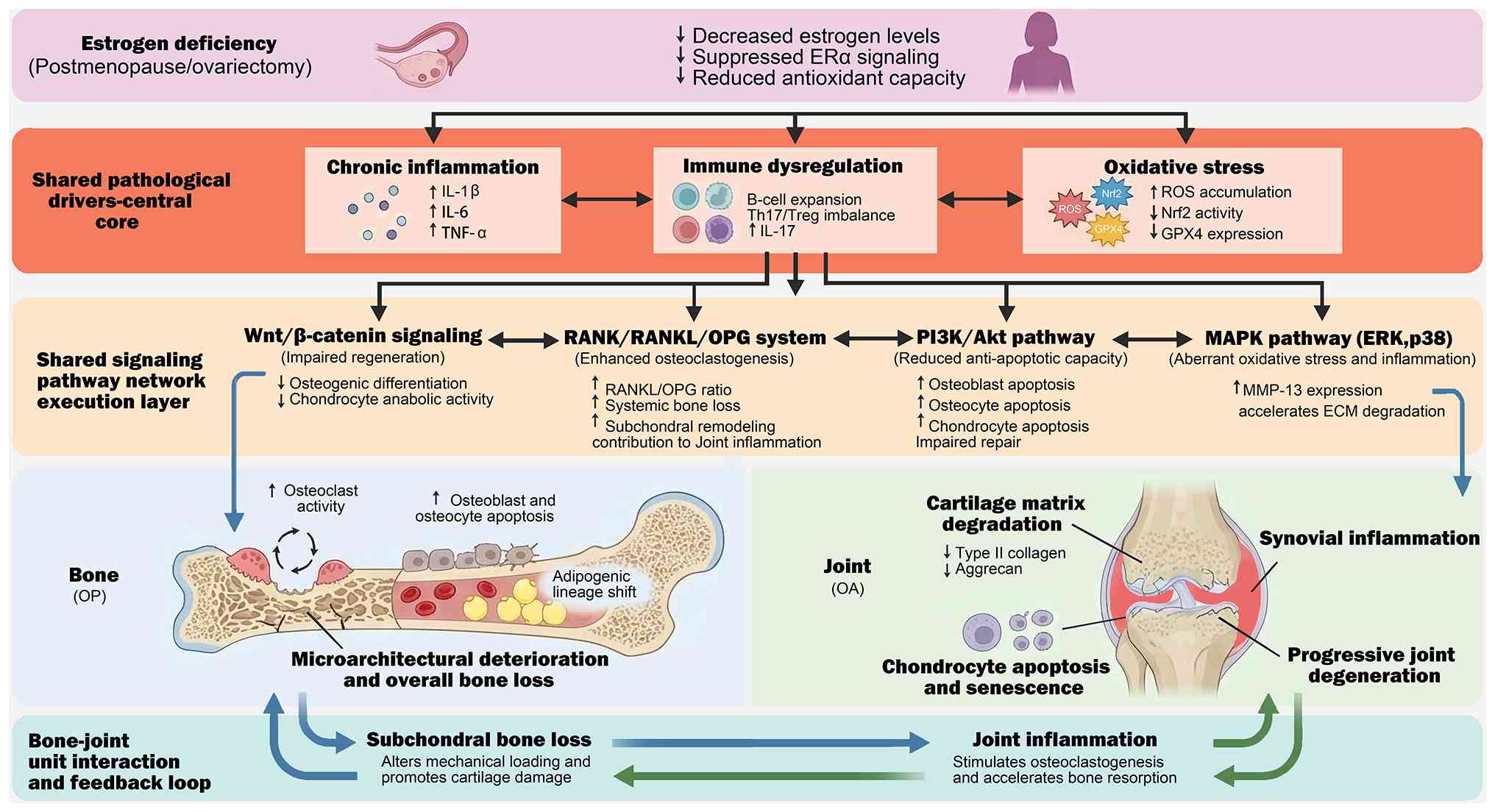 |
Figure 1.
Estrogen deficiency drives OP-OA comorbidity via shared inflammatory/immune/oxidative hubs and convergent signaling networks. Schematic overview illustrating how estrogen deficiency reduces estrogen receptor signaling and antioxidant capacity, thereby establishing a shared pathological core characterized by chronic inflammation (e.g., increased IL-1β/IL-6/TNF-α), immune dysregulation (including B-cell expansion and Th17/Treg imbalance with elevated IL-17) and oxidative stress (ROS accumulation with impaired Nrf2 activity and reduced GPX4). These upstream drivers converge on a shared signaling execution layer: i) Wnt/β-catenin attenuation impairs osteogenic differentiation and cartilage repair; ii) an increased RANK/RANKL/OPG ratio enhances osteoclastogenesis and promotes systemic bone loss and subchondral remodeling; iii) PI3K/Akt suppression and aberrant MAPK (ERK/p38) activation promote apoptosis, impaired repair, stress responses, and matrix catabolism (including upregulation of MMP-13), accelerating ECM degradation. Downstream, these mechanisms drive parallel tissue outcomes in the bone compartment (increased osteoclast activity, osteoblast/osteocyte apoptosis, marrow adipogenic shift, microarchitectural deterioration and overall bone loss) and the joint compartment (cartilage matrix degradation with loss of type II collagen/aggrecan, chondrocyte apoptosis/senescence, synovial inflammation and progressive joint degeneration). Finally, the figure highlights the bone-joint unit vicious cycle, in which subchondral bone loss alters mechanical loading to promote cartilage damage, while joint inflammation and oxidative stress feed back to stimulate osteoclastogenesis and accelerate bone resorption, collectively reinforcing OP-OA co-progression. OP, osteoporosis; OA, osteoarthritis; ERα, estrogen receptor-α; IL, interleukin; TNF-α, tumor necrosis factor-α; Th17, T helper 17; Treg, regulatory T cell; ROS, reactive oxygen species; Nrf2, nuclear factor erythroid 2-related factor 2; GPX4, glutathione peroxidase 4; RANKL, receptor activator of NF-κB ligand; OPG, osteoprotegerin; PI3K, phosphoinositide 3-kinase; Akt, protein kinase B; MAPK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; MMP-13, matrix metalloproteinase-13; ECM, extracellular matrix.
|
Activation of shared inflammatory pathways
Estrogen deficiency plays an important role in the development of OP and OA by upregulating pro-inflammatory cytokines, which activate overlapping inflammatory signaling pathways, including NF-κB and Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathways, thereby triggering chronic inflammation in bone and joint tissues. The NF-κB pathway is important for regulating immune and inflammatory responses; activation of this pathway has been associated with increased osteoclastogenesis and bone resorption, leading to bone loss in OP (55). Concurrently, NF-κB signaling enhances the expression of MMPs and other catabolic enzymes in chondrocytes, thus accelerating cartilage degradation in OA (65). The JAK/STAT pathway, another key inflammatory cascade, is also triggered by a number of cytokines that are elevated in estrogen-deficient states, further intensifying inflammatory responses and enhancing tissue destruction in both bone and cartilage (66). Notably, these pathways are often activated simultaneously under similar pathological conditions. For example, in postmenopausal women and ovariectomy models, an increase in IL-6 levels has been shown to activate the JAK/STAT signaling pathway, promoting osteoclastogenesis while also driving synovial inflammation and cartilage catabolism; this illustrates a shared inflammatory mechanism between OP and OA pathogenesis (66,67).
Chronic inflammation triggers pathways that secrete pro-inflammatory cytokines, such as IL-1β and TNF-α, which exert tissue-specific effects: In bone, they exacerbate bone loss by amplifying osteoclast activity; in cartilage, they promote degradation through enhanced catabolic activity and compromised repair (67) (Fig. 1). Furthermore, emerging evidence has suggested that interventions targeting these inflammatory pathways, whether through pharmacological agents or specific bioactive compounds such as flavonoids or gamabufotalin, can mitigate bone loss and cartilage breakdown (68–70). This underlines the notable role of inflammation in the co-occurrence of OP and OA. In summary, estrogen deficiency creates a chronic inflammatory environment by activating the NF-κB and JAK/STAT signaling pathways, as well as other related signaling pathways, thereby mediating the deleterious effects of inflammation on bone and joint tissues and establishing a mechanistic link between OP and OA (Table I).
Immune regulatory imbalance
Estrogen plays a notable role in modulating the differentiation and function of immune cells. Therefore, estrogen deficiency elicits notable immune dysregulation, which contributes to OP-OA comorbidity. Under normal physiological conditions, estrogen helps to maintain immune homeostasis in the bone marrow by regulating the activity of T cells, B cells and macrophages (71). In postmenopausal women with OP, estrogen deficiency results in the abnormal activation and proliferation of immune cells, particularly B cells (72). A single-cell transcriptomic study conducted on bone marrow cells isolated from ovariectomized mice has revealed a marked increase in B-cell proliferation and a concurrent reduction in neutrophil proportions compared with control mice. This transition has been attributed to the widespread downregulation of activator protein 1 subunits, which are regulated by ER-mediated signaling, particularly ERα (73). Notably, the pharmacological inhibition of B-cell development using anti-IL-7 antibodies has been shown to markedly reduce bone loss, accentuating the important role of immune cell imbalance in estrogen deficiency-induced OP (73).
This immune dysregulation extends beyond the bone tissue. The altered systemic immune profile, specifically the imbalance between helper T cells, resulting from estrogen deficiency has also been shown to affect joint health. In cases of estrogen deficiency, the equilibrium between pro-inflammatory T helper 17 (Th17) cells and anti-inflammatory regulatory T (Treg) cells is disrupted, resulting in increased IL-17 production (74). IL-17 is a potent cytokine that enhances osteoclast activity, which in turn promotes bone resorption. This cytokine also contributes to synovial inflammation and cartilage matrix degradation, thereby impacting bone and joint health simultaneously (67,74). As such, disruption of the Th17/Treg cell balance, which is influenced by systemic estrogen levels and gut microbiota, further exacerbates immune-mediated bone and joint pathology (74) (Fig. 1). Furthermore, integrative bioinformatic analyses of postmenopausal patients with OP have demonstrated that differentially expressed genes in immune cells, particularly monocytes and T-cell subsets, associate with immune-response mechanisms and their underlying signaling pathways that are instrumental in bone metabolism and inflammatory responses (75,76). These findings underscore the pathological role of immune dysregulation caused by estrogen deficiency, which is characterized by the aberrant activation of T cells, B cells and macrophages (Table I). This dysregulation not only intensifies bone resorption and joint inflammation but also serves as an important mechanistic link in the comorbidity of OP and OA.
Oxidative stress intensification: Common pathological mediators
Estrogen deficiency diminishes the activity of antioxidant enzymes, leading to the accumulation of ROS in bone tissue and the joint microenvironment, which in turn induces oxidative stress responses. Research conducted by Mohamad et al (77) demonstrated that menopause-related hormonal changes disrupt redox homeostasis in the body, resulting in elevated oxidative stress levels. Estrogen exerts antioxidant effects and helps to protect osteocytes from lipid-peroxidation damage by upregulating antioxidant enzymes, such as glutathione peroxidase 4 (GPX4) (78). Estrogen deficiency reduces GPX4 expression, resulting in the accumulation of phospholipid peroxides in osteoblasts. This accumulation has been shown to inhibit osteogenic differentiation and, by extension, impair bone formation (78). Additionally, estrogen has been shown to enhance the antioxidant capacity of bone-derived cells, including bone marrow mesenchymal stem cells (BMSCs), by activating the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway, promoting the expression of antioxidant genes. However, estrogen deficiency has been shown to inhibit Nrf2 activity, diminishing the ability of cells to respond to oxidative stress. This impairment leads to a shortened cellular lifespan and the reduced osteogenic differentiation capability of BMSCs, which ultimately affects bone formation (79) (Fig. 1).
In bone tissue, oxidative stress has been shown to activate apoptotic pathways, such as the Bcl-2-associated X protein/caspase 3 signaling pathway, resulting in a reduction in the number of osteocytes, decreased bone-matrix generation capacity and the destruction of trabecular structure, ultimately leading to bone loss and OP (57). Additionally, oxidative stress stimulates osteoclast differentiation by activating specific signaling pathways, such as the MAPK and NF-κB signaling pathways, while impeding osteoblast differentiation and mineralization. These effects culminate in increased bone resorption and reduced bone formation (80). Recent findings have suggested that fibroblast growth factor 7 protects osteoblasts from oxidative stress, reducing apoptosis and supporting osteogenic differentiation, thus exhibiting protective effects that prevent OP pathogenesis (81).
Oxidative stress acts as a common mediator that directly damages bone and cartilage tissue, accelerating joint degeneration. Research has indicated that excessive ROS accumulation potentially induces apoptosis and cellular senescence in osteoblasts and chondrocytes, leading to the dysfunction of these cells and disrupting the homeostasis of bone and cartilage (35). In articular cartilage, ROS can enhance the expression of catabolic enzymes, such as MMPs, thereby facilitating the degradation of the cartilaginous extracellular matrix and accelerating OA progression (39). Furthermore, oxidative stress can activate inflammatory pathways, such as the NF-κB and hypoxia-inducible factor 1-α signaling pathways, further aggravating the inflammatory response and contributing to synovitis and cartilage degeneration (82,83). In the joints, oxidative stress-induced apoptosis and matrix degradation generate a feedback loop that perpetuates degenerative changes in joint structure (82,84). Additionally, oxidative stress has been shown to potentially damage bone marrow microvessels and synovial cells, thereby exacerbating the pathological damage associated with OP and OA (85,86) (Fig. 1). Notably, studies utilizing ovariectomized models have demonstrated that antioxidant treatments can alleviate bone loss and cartilage damage, providing functional evidence that oxidative stress represents a common therapeutic target in these comorbidities (26,39). Summarily, oxidative stress has been shown to induce apoptosis in bone cells and chondrocytes, promote matrix degradation and regulate inflammatory responses and bone-remodeling signaling pathways, therefore constituting a common pathological basis for OP and OA (Table I).
Signaling pathways in the comorbidity of estrogen deficiency-related OP and OA
Wnt/β-catenin signaling pathway
The Wnt/β-catenin signaling pathway helps to regulate cell proliferation, differentiation and migration; as such, it is instrumental in mediating embryonic development and tissue homeostasis (87). Estrogen plays a notable role in maintaining bone homeostasis and cartilage integrity by regulating the Wnt/β-catenin signaling pathway, which is important for osteogenesis and cartilage repair (88,89). Typically, estrogen stimulates the Wnt/β-catenin pathway, thereby promoting osteoblast differentiation, bone matrix production and chondrocyte function, all of which promote bone formation and cartilage repair (90,91). This effect is facilitated by the ER-mediated transcriptional regulation of key Wnt pathway genes, leading to increased β-catenin signaling, which in turn results in greater bone mass and improved cartilage quality. Conversely, under conditions of estrogen deficiency, such as postmenopausal OP or ovariectomized animal models, Wnt/β-catenin pathway activity has been shown to be markedly diminished (92). This reduction in signaling has been shown to result in decreased osteogenic differentiation, impaired bone formation and accelerated cartilage degeneration (Table II). Animal studies using ovariectomized models have consistently demonstrated a marked decline in BMD and bone biomechanical strength, alongside histological evidence of cartilage deterioration; these phenotypic changes represent downstream pathological changes driven by reduced Wnt/β-catenin signaling activity (92,93). These findings underscore the importance of targeting the Wnt/β-catenin pathway in formulating treatments for postmenopausal women with OP and OA.
|
Table II.
Key signaling pathways dysregulated by estrogen deficiency in OP-OA comorbidity.
|
Table II.
Key signaling pathways dysregulated by estrogen deficiency in OP-OA comorbidity.
| Signaling pathway |
Estrogen deficiency-related alteration |
Major downstream consequences in OP |
Major downstream consequences in OA |
Translational notes on biomarkers and therapeutic implications |
(Refs.) |
| Wnt/β-catenin |
Reduced Wnt/β-catenin activity |
Reduced osteogenic differentiation and impaired bone formation |
Impaired cartilage repair and regeneration and exacerbated cartilage degeneration |
Wnt-related biomarkers, including sclerostin and DKK-1, may aid phenotyping; pathway modulation is a potential therapeutic direction |
(87–93,115) |
| RANK/RANKL/OPG |
Increased RANKL and decreased OPG expression, elevating the RANKL/OPG ratio |
Enhanced osteoclastogenesis and systemic bone loss |
Contributes to subchondral bone remodeling and osteochondral inflammation linked to OA severity |
Supports anti-RANKL strategies in high-resorption phenotypes; monitor remodeling-related markers where available |
(94–99) |
| PI3K/Akt |
Attenuated PI3K/Akt pro-survival signaling |
Increased osteoblast and osteocyte apoptosis and impaired repair capacity |
Increased chondrocyte apoptosis and reduced anabolic and repair responses |
Candidate axis for cytoprotective interventions aimed at restoring survival and repair signaling |
(100–104) |
| MAPK/ERK/p38 |
Aberrant MAPK activation, particularly of ERK and p38, driven by inflammation and oxidative stress |
Promotes stress responses and may facilitate resorption-dominant remodeling in inflammatory contexts |
Fosters matrix degradation and inflammatory responses, accelerating cartilage breakdown |
Potentially actionable by dampening excessive MAPK activation while restoring anabolic balance |
(102–108) |
| NF-κB |
Cytokine-driven activation of central inflammatory signaling cascade |
Promotes osteoclastogenesis and bone resorption; suppresses anabolic remodeling |
Induces catabolic enzymes and sustains synovial inflammation, accelerating cartilage degradation |
Rationale for anti-inflammatory strategies targeting shared inflammatory circuits; consider integration with disease phenotype |
(55,65,70) |
| JAK/STAT |
Activated by multiple upregulated cytokines, amplifying chronic inflammation |
Sustains elevated osteo-immune signaling, contributing to bone loss |
Enhances inflammatory signaling in the synovium and cartilage and promotes tissue destruction |
Supports cytokine-centered risk stratification and pathway-informed anti-inflammatory approaches |
(66) |
Receptor activator of NF-κB (RANK)/RANKL/OPG signaling pathway
The RANK/RANKL/OPG signaling pathway is important for regulating bone remodeling and joint inflammation, playing a notable role in the pathogenesis of OP and OA. Notably, the balance between RANKL and OPG is important for maintaining bone homeostasis. An increase in the RANKL/OPG ratio interferes with this homeostasis, resulting in increased bone resorption, a phenomenon evident in OP and subchondral bone remodeling in OA (94,95). Postmenopausal estrogen deficiency markedly contributes to this imbalance by raising RANKL levels and lowering OPG levels. These alterations have been shown to enhance osteoclast activity, resulting in systemic bone loss and localized joint degeneration (96,97). Furthermore, the RANK/RANKL/OPG pathway is notably involved in inflammatory processes within the joint, where elevated RANKL levels not only promote osteoclastogenesis but also associate with disease severity in OA (98). Therapeutic strategies targeting this axis, including RANKL-neutralizing antibodies such as denosumab, have proven effective in mitigating bone resorption and are currently being explored for broader applications in metabolic bone diseases and joint disorders (99). The RANK/RANKL/OPG pathway is important in linking estrogen deficiency to the pathophysiology of OP and OA (Table II), emphasizing its importance as a mechanistic and therapeutic target for managing these conditions.
PI3K/Akt and MAPK pathways
The PI3K/Akt and MAPK signaling pathways are important for regulating cell proliferation, apoptosis and inflammatory responses, indicating that these pathways are important for maintaining bone and joint homeostasis. Under physiological conditions, these pathways coordinate the functions of osteoblasts, osteoclasts and chondrocytes, preserving the integrity of bone and cartilage. However, estrogen deficiency, which is commonly observed in postmenopausal women, has been shown to disrupt the delicate balance of these signaling cascades, aggravating bone and joint damage (100,101). Research has revealed that estrogen potentially influences the PI3K/Akt and MAPK pathways, enhancing osteogenic differentiation, promoting angiogenesis and reducing apoptosis in BMSCs and chondrocytes (102,103). A decline in estrogen levels attenuates the protective effects of these pathways, resulting in heightened oxidative stress, increased inflammatory cytokine release and elevated osteoclast activity, which have been shown to contribute to the progression of OP and OA (104). The drug Ononin has been shown to exert chondroprotective effects in an IL-1β-induced chondrocyte inflammation model by inhibiting the expression of ECM-degrading enzymes such as MMP-13 and reducing type II collagen degradation, achieved through the downregulation of the MAPK signaling pathways (105).
The PI3K/Akt pathway is important for cell survival and the prevention of apoptosis, whereas the MAPK pathway is involved in responses to stress and inflammation (106). In the context of estrogen deficiency, both pathways become dysregulated: PI3K/Akt signaling diminishes, resulting in increased cell death and compromised tissue repair, whereas abnormal MAPK pathway activation fosters matrix degradation and inflammatory responses (107,108). Therapeutic interventions, including isoflavones and traditional Chinese medicines, have reportedly demonstrated protective effects by reactivating the PI3K/Akt pathway and inhibiting the overactivation of the MAPK pathway. This dual action helps to ameliorate inflammation, promote matrix synthesis and inhibit osteoclastogenesis (103,107,109). Collectively, these findings highlight the notable roles of the PI3K/Akt and MAPK signaling pathways in the pathogenesis of bone and joint comorbidities associated with estrogen deficiency, positioning these pathways as promising targets for clinical intervention in OP and OA (Table II).
Clinical manifestations and diagnostic challenges of estrogen deficiency-related comorbid OP and OA
Clinical characteristics of comorbid patients
Patients with comorbid OP and OA present a particularly complex clinical profile, marked by the simultaneous presence of reduced BMD and joint-related symptoms, such as pain and functional impairment. The co-occurrence of these degenerative diseases frequently results in overlapping and intertwined symptomatology, rendering clinical assessment and management of these conditions challenging (9,110). For example, individuals with postmenopausal OP may remain asymptomatic until a fracture occurs, whereas patients with OA localized in the knee typically experience chronic pain and progressive disability. When these conditions coexist, patients often report both generalized skeletal fragility and localized joint pain, leading to increased functional limitations and a reduced quality of life for patients (1). Interpreting imaging findings in patients with comorbid conditions can be particularly challenging. Patients with comorbid OP and OA may concurrently exhibit radiographic indicators characteristic of OA, such as joint-space shrinkage, subchondral sclerosis and osteophyte formation, alongside the reduced BMD evident in OP (111). This coexistence of imaging findings complicates the differential diagnosis of these conditions, as determining whether pain and dysfunction primarily originate from bone loss or joint degeneration becomes challenging (Fig. 2). Shared risk factors, for example age, hormonal shifts, particularly estrogen deficiency, and chronic low-grade inflammation, further obscure the clinical distinctions between these conditions. Recent insights into the mechanisms underlying degenerative bone and joint diseases, particularly the role of epigenetic modifications such as DNA methylation, have suggested that the pathogenesis of OP and OA may share molecular pathways that underscore their interrelated clinical features (112). Consequently, patients with OP and OA exhibit overlapping symptoms, complex imaging findings and shared risk factors, all of which increase diagnostic complexity and necessitate a nuanced, multidisciplinary approach to management.
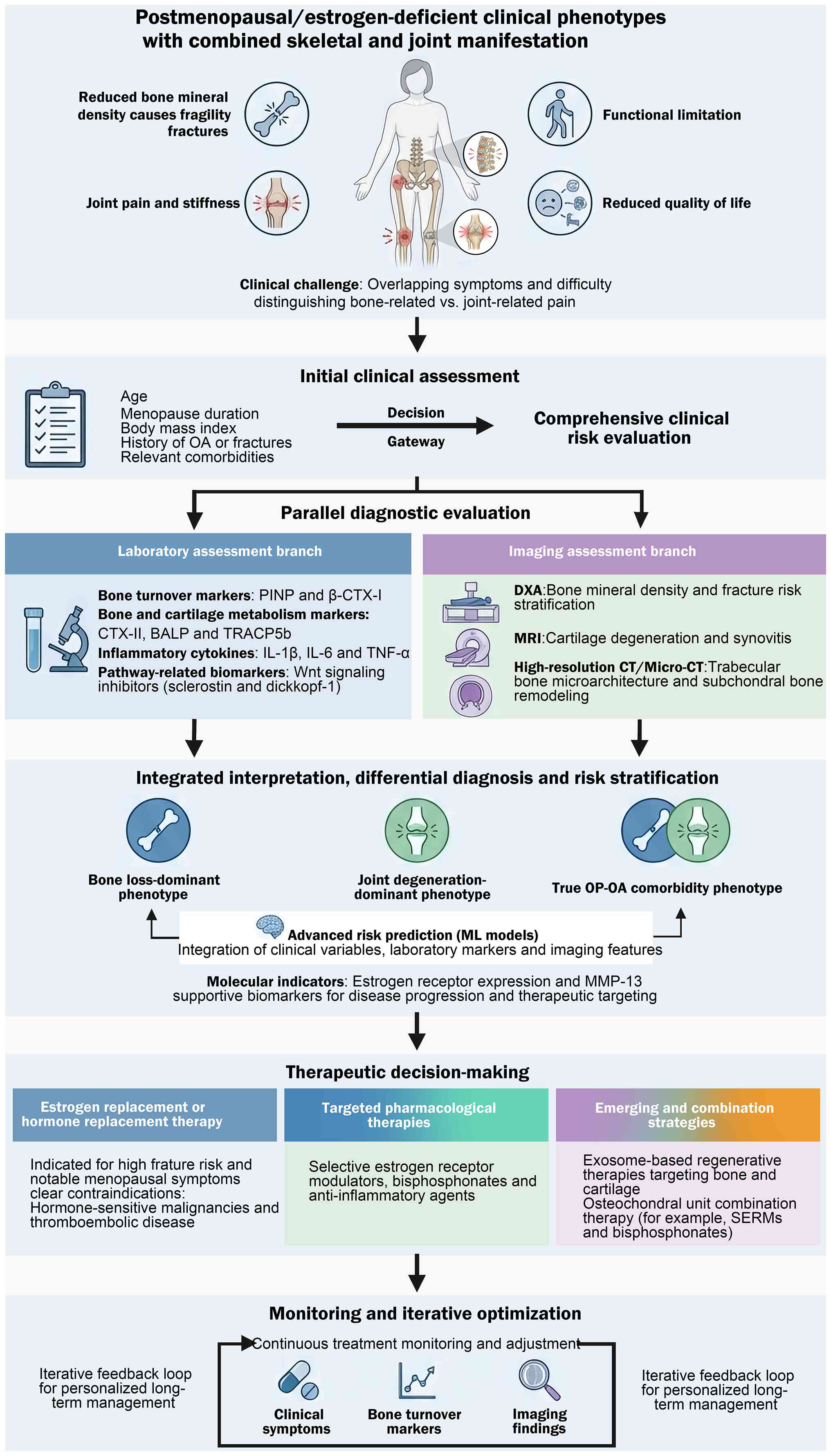 |
Figure 2.
Integrated clinical workflow for evaluating and managing estrogen deficiency-related OP and OA comorbidity. Proposed diagnostic-therapeutic algorithm for postmenopausal/estrogen-deficient patients presenting with combined skeletal and joint manifestations (reduced BMD, fragility fracture risk, joint pain and stiffness, functional limitation, and impaired quality of life), where overlapping symptoms complicate discrimination between bone-derived and joint-derived pain. Following an initial clinical assessment and comprehensive risk evaluation, a parallel diagnostic evaluation is recommended. The laboratory branch incorporates bone turnover markers (e.g., PINP, β-CTX–I), bone-cartilage metabolism markers (e.g., CTX–II, BALP, TRACP5b), inflammatory cytokines (e.g., IL-1β, IL-6, TNF-α), and pathway-related biomarkers including Wnt inhibitors (sclerostin, DKK-1). The imaging branch integrates DXA for BMD and fracture-risk stratification, MRI for early detection of cartilage degeneration and synovitis, and high-resolution CT or micro-CT to characterize trabecular microarchitecture and subchondral remodeling. An integrated interpretation step supports differential diagnosis and risk stratification into bone loss-dominant, joint degeneration-dominant, and true OP-OA comorbidity phenotypes, potentially enhanced by machine-learning risk models that combine clinical variables (e.g., age, BMI, menopause duration, OA history, comorbidities) with laboratory and imaging features. Molecular indicators (e.g., estrogen receptor expression and MMP-13) may provide supportive biomarkers for disease progression and therapeutic targeting. Therapeutic decision-making includes i) ERT/HRT for appropriately selected high-fracture-risk patients with significant menopausal symptoms, while acknowledging major safety considerations and the need for individualized selection and monitoring; ii) targeted pharmacologic therapies such as SERMs, bisphosphonates, and anti-inflammatory agents; and iii) emerging/combination strategies, including exosome-based regenerative approaches and their combinations with established agents within the osteochondral niche. Finally, the workflow emphasizes iterative monitoring and optimization through longitudinal tracking of symptoms, bone turnover markers, and imaging outcomes to support personalized long-term management. OP, osteoporosis; OA, osteoarthritis; BMD, bone mineral density; DXA, dual-energy X-ray absorptiometry; MRI, magnetic resonance imaging; PINP, procollagen type I N-terminal propeptide; β-CTX–I, β-isomerized C-terminal telopeptide of type I collagen; CTX–II, C-terminal telopeptide of type II collagen; BALP, bone alkaline phosphatase; TRACP5b, tartrate-resistant acid phosphatase 5b; MMP-13, matrix metalloproteinase-13; ERT, estrogen replacement therapy; HRT, hormone replacement therapy; SERMs, selective estrogen receptor modulators.
|
Laboratory and imaging evaluation
Laboratory evaluation of bone metabolism and inflammation is important for differentiating and managing comorbid OP and OA. Bone turnover markers (BTMs), including serum procollagen type I N-propeptide (PINP) and β-isomerized C-terminal telopeptide of type I collagen (β-CTX–I), function as established biomarkers for OP pathogenesis. These markers assist not only risk stratification but also the monitoring of treatment response and adherence, as the notable alterations in PINP and β-CTX–I levels following anti-osteoporotic therapies have been shown to associate with reductions in fracture risk (113). Markers of bone and cartilage degradation, such as C-terminal telopeptide of type II collagen (CTX–II) and enzymes, such as bone alkaline phosphatase and tartrate-resistant acid phosphatase 5b, are important for evaluating bone turnover and joint metabolism, especially in patients with concurrent chronic kidney disease or other comorbidities that may confound biomarker interpretation (114). In OA, inflammatory cytokines and pathway-related molecules, including Wnt signaling inhibitors such as sclerostin and dickkopf-1, have been increasingly recognized as potential biomarkers of disease activity and progression (115,116) (Fig. 2). However, limitations persist, for example variability in assay standardization, confounding effects from impaired renal function secondary to chronic kidney disease and the insufficient integration of BTMs into clinical algorithms (113). These limitations underscore the need for further therapeutic validation and harmonization prior to routine clinical use. The interpretation of laboratory detection of these markers alongside clinical and imaging findings improves diagnostic accuracy and allows for tailored management strategies for patients with comorbid OP and OA.
Imaging modalities play an important role in assessing bone and joint pathology, particularly in patients with comorbid OP and OA. Each imaging technique exhibits specific applications and limitations. Dual-energy X-ray absorptiometry (DXA) is considered the benchmark for measuring BMD, which is important for diagnosing OP and assessing fracture risk. However, DXA offers limited insights into bone microarchitecture and cannot directly visualize cartilage or early joint structural modifications, restricting its effectiveness in the comprehensive evaluation of OA (117). By contrast, magnetic resonance imaging (MRI) excels at detecting early osteoarticular changes, such as cartilage degeneration, synovitis and adverse local tissue reactions, even in asymptomatic individuals. This capability supports its use in monitoring disease progression and identifying complications that may not be visible through clinical evaluation or plain radiographs (118). Ultra-high-resolution computed tomography (CT) enhances the visualization of trabecular bone microstructure and yields quantitative metrics that associate with bone quality and osteoarthritic disease status, surpassing conventional CT in accuracy (119) (Fig. 2). However, imaging modalities encounter several challenges, including variability in image interpretation, sensitivity to technical factors, such as spatial resolution, and the limited reproducibility of radiomic features, a specific type of quantitative imaging biomarkers, across various scanners and protocols (120). Additionally, recent evidence has indicated that the added value of imaging data in predicting clinical outcomes may be modest when compared with comprehensive clinical models. This emphasizes the necessity for integrated diagnostic algorithms that combine laboratory results, imaging data and clinical information to ensure optimal patient care (121).
Differential diagnosis and risk stratification
To ensure the accuracy of differential diagnosis and risk stratification in patients with comorbid OP and OA, especially in the context of estrogen deficiency, comprehensive assessments of bone and joint damage are needed in order to guide personalized treatment strategies. The pathophysiological mechanisms underlying these conditions possess both overlapping and distinct features, such as the relationship between BMD loss and cartilage degradation, necessitating integrated diagnostic approaches. Advanced imaging techniques, such as micro-CT and MRI microscopy, have proven valuable for quantitatively assessing trabecular bone microarchitecture and joint-space modifications (122). These technologies facilitate the early detection of bone loss and joint degeneration, improving preclinical models and clinical practice. Furthermore, DXA remains central to diagnosing OP and is effective at identifying and managing at-risk individuals, as evidenced by the association between DXA utilization and OP treatment rates in postmenopausal women (123). Incorporating clinical variables, such as age, body mass index, menopause duration, OA history and comorbidities, into machine learning models may enhance the accuracy of OP risk predictions compared with traditional methods, highlighting the potential of these models for personalized risk assessment (124,125).
At the molecular level, the expression levels of ERs and MMPs, such as MMP-13, in bone and cartilage tissues represent additional biomarkers of disease progression and therapeutic response in OP and OA. Furthermore, estrogen deficiency has been associated with an increase in catabolic enzymes and a decline in the integrity of bone and cartilage tissues (126,127). These molecular insights can guide the selection of targeted therapies, including SERMs or osteoclast activity modulators, which have exhibited promise in managing OP and OA (128,129) (Fig. 2). Ultimately, developing individualized diagnostic and treatment plans that incorporate imaging, clinical risk factors and molecular markers is important for optimizing outcomes in patients with estrogen-deficiency-related bone and joint comorbidities.
Estrogen-related intervention strategies and their limitations
Efficacy and risks of ERT
ERT and HRT represent well-established approaches for treating menopausal symptoms and preventing OP in postmenopausal women. Numerous randomized controlled trials and meta-analyses have demonstrated that ERT and HRT markedly improve BMD in postmenopausal female patients with OP, mitigating the risks of vertebral and non-vertebral fractures in this population (130,131). In addition to enhancing BMD, HRT has been shown to improve the quality of life of postmenopausal women by addressing vasomotor symptoms, such as hot flashes and night sweats, as well as urogenital atrophy (131,132). Furthermore, certain studies have indicated a beneficial effect of HRT on intervertebral disc height, which may help to alleviate the risk of vertebral fractures and enhance spinal health (133,134).
However, ERT also presents a number of risks. Well-documented adverse effects include an increased likelihood of breast and endometrial cancers, venous thromboembolism and stroke, particularly with long-term use or in women with predisposing factors (135). The risk profile can vary based on the formulation, route of administration and inclusion of progestogens, the latter of which is important for protecting the endometrium in women with an intact uterus (136). Large-scale studies have also revealed that HRT does not markedly impact cardiovascular outcomes, such as heart-failure risk, yet concerns regarding other cardiovascular events persist (135,137). Notably, the safety and efficacy of ERT and HRT may differ among various subgroups. Therefore, individualized therapy, in which the benefits of symptom relief and bone protection are carefully weighed against the potential for serious adverse events, is of notable importance (138). Clinical guidelines emphasize the importance of patient selection, recommending HRT primarily for women with notable menopausal symptoms or a high risk of fractures whereas advising against its use in patients with a history of hormone-sensitive malignancies or thromboembolic disease (139) (Fig. 2). Overall, ERT can provide notable clinical benefits by improving bone density and delaying intervertebral disc degeneration, but its use must be judicious, with careful consideration of individual risk factors and ongoing monitoring to optimize patient outcomes and minimize harm.
Novel targeted drugs and combination therapy
The management of comorbid OP and OA has increasingly shifted toward the development and application of novel targeted therapies. These therapies include SERMs, bisphosphonates and anti-inflammatory agents, which hold promise for improving outcomes in patients affected by both conditions through targeting shared molecular pathways and addressing the underlying pathophysiological mechanisms of bone and cartilage degeneration (140). SERMs modulate ERs to enhance BMD and may offer protection to cartilage (141). By contrast, bisphosphonates have proven effective in mitigating fracture risk in patients with OP by inhibiting osteoclast-mediated bone resorption (142). Furthermore, anti-inflammatory agents remain under investigation for their potential to alleviate chronic inflammation that contributes to the progression of OA and bone loss (143,144).
Beyond these traditional pharmacological strategies, advancements in regenerative medicine have introduced exosome-based therapies as an innovative intervention method. Exosomes, which carry transcription factors, proteins and targeting ligands, uniquely target cells to influence tissue repair, alleviate inflammation and maintain homeostasis (145). Preclinical models have demonstrated the potential of engineered exosomes to regenerate bone, osteochondral tissue and cartilage for the treatment of OA, OP and osteonecrosis (146,147). Combination treatments using exosomes and other established therapies, including SERMs and bisphosphonates, are currently being explored as a strategy for improving therapeutic efficacy synergistically, especially within the osteochondral niche (148–150) (Fig. 2). Despite the promising potential of these innovative combination therapies, robust evidence from well-designed clinical studies is important for validating their safety, efficacy and long-term benefits in managing comorbid OP and OA. Future research should focus on optimizing these combination strategies and elucidating their therapeutic mechanisms to enhance patient outcomes.
Conclusions
Estrogen deficiency has been increasingly recognized as an important factor underlying the comorbidity of OP and OA, corroborating the notion that these two prevalent age-related disorders represent interconnected manifestations of a shared bone-joint unit dysfunction rather than isolated conditions. By converging on common biological pathways, including chronic low-grade inflammation, immune imbalance, oxidative stress and impaired tissue remodeling, estrogen deficiency facilitates the coordinated deterioration of bone integrity and joint structure, thereby accelerating systemic bone loss and local osteochondral degeneration. From a clinical perspective, this mechanistic convergence underscores the necessity for integrated risk stratification and management strategies that concurrently address skeletal fragility and joint degeneration. Future research should prioritize refined phenotyping, biomarker-guided risk assessments and pathway-based therapeutic strategies, reinforced by longitudinal cohorts and multi-omics approaches, to enable the development of precision treatments for patients OP-OA comorbidity.
Acknowledgements
We are grateful to Dr Mingming Liu (Department of Orthopedics, Lianyungang Clinical College of Xuzhou Medical University) for their valuable assistance with figure preparation.
Funding
The present study was financially supported by the Basic Research Program of the Xuzhou Science and Technology Bureau (grant no. KC22037), the Medical Science and Technology Innovation Project of Xuzhou Municipal Health Commission (grant no. XWKYHT20240048) and the Henan Provincial Medical Science and Technology Research Plan (grant no. LHGJ20250751).
Availability of data and materials
Not applicable.
Authors' contributions
JC was responsible for conceptualization, acquiring the funding and writing the original draft of the manuscript. XW contributed towards writing the original draft of the manuscript. LZ was responsible for investigation and methodology. SJ also contributed toward the methodology of the present review. WZ reviewed and edited the manuscript. WZ was responsible for supervision and reviewing and editing the manuscript. Data authentication is not applicable. All authors read and approved the final version of the manuscript.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Glossary
Abbreviations
Abbreviations:
|
BMSCs
|
bone marrow mesenchymal stem cells
|
|
BTMs
|
bone turnover markers
|
|
BMD
|
bone mineral density
|
|
DXA
|
dual-energy X-ray absorptiometry
|
|
ER
|
estrogen receptor
|
|
ERT
|
estrogen replacement therapy
|
|
GPX4
|
glutathione peroxidase 4
|
|
HRT
|
hormone replacement therapy
|
|
IL
|
interleukin
|
|
JAK
|
Janus kinase
|
|
STAT
|
signal transducer and activator of transcription
|
|
MMPs
|
matrix metalloproteinases
|
|
MAPK
|
mitogen-activated protein kinase
|
|
MAT
|
marrow adipose tissue
|
|
MRI
|
magnetic resonance imaging
|
|
NF-κB
|
nuclear factor κB
|
|
OP
|
osteoporosis
|
|
OA
|
osteoarthritis
|
|
OPG
|
osteoprotegerin
|
|
PINP
|
procollagen type I N-propeptide
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
Akt
|
protein kinase B
|
|
RANKL
|
receptor activator of nuclear factor κ-B ligand
|
|
ROS
|
reactive oxygen species
|
|
SERMs
|
selective estrogen receptor modulators
|
|
TNF-α
|
tumor necrosis factor-α
|
|
β-CTX–I
|
β-isomerized C-terminal telopeptide of type I collagen
|
References
|
1
|
Lu Z, Feng W, Wang Y, Zhang X, Wei X, Chen M, Wang S, Cheng T, Cui X and Xie Y: Prevalence, clinical characteristics, and biological markers of postmenopausal osteoporosis and knee osteoarthritis in Beijing: Study protocol for a cross-sectional and prospective study. Front Med (Lausanne). 12:15825332025. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shevroja E, Cafarelli FP, Guglielmi G and Hans D: DXA parameters, trabecular bone score (TBS) and bone mineral density (BMD), in fracture risk prediction in endocrine-mediated secondary osteoporosis. Endocrine. 74:20–28. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kim GM, Kim J, Lee JY, Park MC and Lee SY: IgSF11 deficiency alleviates osteoarthritis in mice by suppressing early subchondral bone changes. Exp Mol Med. 55:2576–2585. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Palacios S, González SP, Sánchez-Prieto M and Fasero M: Clinical challenges and considerations in pharmacotherapy of osteoporosis due to menopause. Expert Opin Pharmacother. 25:1359–1372. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gosset A, Pouillès JM and Trémollieres F: Menopausal hormone therapy for the management of osteoporosis. Best Pract Res Clin Endocrinol Metab. 35:1015512021. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Karsdal MA, Bay-Jensen AC, Henriksen K and Christiansen C: The pathogenesis of osteoarthritis involves bone, cartilage and synovial inflammation: May estrogen be a magic bullet? Menopause Int. 18:139–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fischer V and Haffner-Luntzer M: Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. Semin Cell Dev Biol. 123:14–21. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Shipov A, Zaslansky P, Riesemeier H, Segev G, Atkins A, Kalish-Achrai N, Weiner S and Shahar R: The influence of estrogen deficiency on the structural and mechanical properties of rat cortical bone. PeerJ. 9:e102132021. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huang K and Cai H: The interplay between osteoarthritis and osteoporosis: Mechanisms, implications, and treatment considerations-a narrative review. Exp Gerontol. 197:1126142024. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cheng J, Liu M, Li Q, Zhao L and Zhang Q: Macrophage polarization: A bridge connecting osteoarthritis and osteoporosis. Biochem Biophys Res Commun. 801:1533222026. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu Z, Chen X, Ruan Z, Wang C, Yuan D, Xiao W, Li Y and Zhao S: Genetic analysis of comorbidities between osteoarthritis, sarcopenia, and osteoporosis. Exp Gerontol. 206:1127882025. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Rayson A, Boudiffa M, Naveed M, Griffin J, Dall'Ara E and Bellantuono I: Geroprotectors and skeletal health: Beyond the headlines. Front Cell Dev Biol. 10:6820452022. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li B, Yang Z, Li Y, Zhang J, Li C and Lv N: Exploration beyond osteoarthritis: The association and mechanism of its related comorbidities. Front Endocrinol (Lausanne). 15:13526712024. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Azeez TA: Osteoporosis and cardiovascular disease: A review. Mol Biol Rep. 50:1753–1763. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deng L and Guo Y: Estrogen effects on orthodontic tooth movement and orthodontically-induced root resorption. Arch Oral Biol. 118:1048402020. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hsu SH, Chen LR and Chen KH: Primary osteoporosis induced by androgen and estrogen deficiency: The molecular and cellular perspective on pathophysiological mechanisms and treatments. Int J Mol Sci. 25:121392024. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li J, Chen X, Lu L and Yu X: The relationship between bone marrow adipose tissue and bone metabolism in postmenopausal osteoporosis. Cytokine Growth Factor Rev. 52:88–98. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yu AXD, Xu ML, Yao P, Kwan KKL, Liu YX, Duan R, Dong TTX, Ko RKM and Tsim KWK: Corylin, a flavonoid derived from Psoralea Fructus, induces osteoblastic differentiation via estrogen and Wnt/β-catenin signaling pathways. FASEB J. 34:4311–4328. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Davies HO: A local audit evaluating bone health in patients with functional hypothalamic amenorrhoea secondary to an eating disorder and a review of the application of hormone therapy in this clinical setting. Post Reprod Health. 30:182–189. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
He B, Zhu Y, Cui H, Sun B, Su T and Wen P: Comparison of necroptosis with apoptosis for OVX-induced osteoporosis. Front Mol Biosci. 8:7906132021. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xu SY, Shi P and Zhou RM: Post-menopausal oestrogen deficiency induces osteoblast apoptosis via regulating HOTAIR/miRNA-138 signalling and suppressing TIMP1 expression. J Cell Mol Med. 25:4572–4582. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McNamara LM: Osteocytes and estrogen deficiency. Curr Osteoporos Rep. 19:592–603. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Allison H, O'Sullivan LM and McNamara LM: Temporal changes in cortical microporosity during estrogen deficiency associated with perilacunar resorption and osteocyte apoptosis: A pilot study. Bone Rep. 16:1015902022. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang Y, Wang X, Wang K, Qin W and Li N: Extract of Curculigo capitulata ameliorates postmenopausal osteoporosis by promoting osteoblast proliferation and differentiation. Cells. 13:20282024. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lu PY, Huang M, Shao MH, Hu JX, Ding CY, Feng YJ, Zhang M, Lin HP and Tian HS: Effect and mechanism of recombinant human fibroblast growth factor 18 on osteoporosis in OVX mice. Climacteric. 27:305–313. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Marcucci G, Domazetovic V, Nediani C, Ruzzolini J, Favre C and Brandi ML: Oxidative stress and natural antioxidants in osteoporosis: Novel preventive and therapeutic approaches. Antioxidants (Basel). 12:3732023. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yang K, Cao F, Xue Y, Tao L and Zhu Y: Three classes of antioxidant defense systems and the development of postmenopausal osteoporosis. Front Physiol. 13:8402932022. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Li J, Lu L, Liu L, Wang C, Xie Y, Li H, Tian L and Yu X: The unique role of bone marrow adipose tissue in ovariectomy-induced bone loss in mice. Endocrine. 83:77–91. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li M, Sun H, Liu L, Ning Y, Cao Y, Lu B, Zhao Y, Kuang M and Wang D: Pro-inflammatory immune microenvironment and Thrombospondin-1-positive monocytes as drivers of osteoclastogenesis in postmenopausal osteoporosis. J Bone Miner Res. 40:1061–1076. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meng X, Lin Z, Cao S, Janowska I, Sonomoto K, Andreev D, Katharina K, Wen J, Knaup KX, Wiesener MS, et al: Estrogen-mediated downregulation of HIF-1α signaling in B lymphocytes influences postmenopausal bone loss. Bone Res. 10:152022. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu W, Fu J, Gu Y, Wei Y, Ma P and Wu J: JAK2/STAT3 regulates estrogen-related senescence of bone marrow stem cells. J Endocrinol. 245:141–153. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ye Y, Zhong H, Huang S, Lai W, Huang Y, Sun C, Zhang Y and Zheng S: Reactive oxygen species scavenging hydrogel regulates stem cell behavior and promotes bone healing in osteoporosis. Tissue Eng Regen Med. 20:981–992. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yang Z, Tan Q, Zhao Z, Niu G, Li S, Li W, Song C and Leng H: Distinct pathological changes of osteochondral units in early OVX-OA involving TGF-β signaling. Front Endocrinol (Lausanne). 13:10741762022. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ziemian SN, Ayobami OO, Rooney AM, Kelly NH, Holyoak DT, Ross FP and van der Meulen MCH: Low bone mass resulting from impaired estrogen signaling in bone increases severity of load-induced osteoarthritis in female mice. Bone. 152:1160712021. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Riegger J, Schoppa A, Ruths L, Haffner-Luntzer M and Ignatius A: Oxidative stress as a key modulator of cell fate decision in osteoarthritis and osteoporosis: A narrative review. Cell Mol Biol Lett. 28:762023. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wada H, Aso K, Izumi M and Ikeuchi M: The effect of postmenopausal osteoporosis on subchondral bone pathology in a rat model of knee osteoarthritis. Sci Rep. 13:29262023. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Maneix L, Servent A, Porée B, Ollitrault D, Branly T, Bigot N, Boujrad N, Flouriot G, Demoor M, Boumediene K, et al: Up-regulation of type II collagen gene by 17β-estradiol in articular chondrocytes involves Sp1/3, Sox-9, and estrogen receptor α. J Mol Med (Berl). 92:1179–1200. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Naqvi SM, O'Sullivan LM, Allison H, Casey VJ, Schiavi-Tritz J and McNamara LM: Altered extracellular matrix and mechanotransduction gene expression in rat bone tissue following long-term estrogen deficiency. JBMR Plus. 8:ziae0982024. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hu YC, Huang TC, Huang LW, Cheng HL, Hsieh BS and Chang KL: S-Equol ameliorates menopausal osteoarthritis in rats through reducing oxidative stress and cartilage degradation. Nutrients. 16:23642024. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen L, Hong H, Yang Z, Zhong Y, Sun S, Li Z, Song C, Li W and Leng H: Osteoclast-secreted galectin-3 promotes articular cartilage degeneration in OVX rats via LRP1/beta-catenin axis. Osteoarthritis Cartilage. 34:536–549. 2026. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Li H, Gou Y, Tian F, Zhang Y, Lian Q, Hu Y and Zhang L: Combination of metformin and exercise alleviates osteoarthritis in ovariectomized mice fed a high-fat diet. Bone. 157:1163232022. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou L, Li Y, Ma J, Zhang Q, Tang S, Zou K, Zeng Q, Huang H, Jin H, Zhang Q and Feng J: Role and mechanism of Actein on condylar bone metabolism in APOE deletion-induced osteoporotic mice. Bone. 190:1173042025. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Fernández-Moreno M, Rego-Pérez I and Blanco FJ: Is osteoarthritis a mitochondrial disease? What is the evidence. Curr Opin Rheumatol. 34:46–53. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Song SJ, Tao JJ, Li SF, Qian XW, Niu RW, Wang C, Zhang YH, Chen Y, Wang K, Zhu F, et al: 17β-estradiol attenuates rat articular chondrocyte injury by targeting ASIC1a-mediated apoptosis. Mol Cell Endocrinol. 505:1107422020. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gou Y, Li H, Sun X, Chen D and Tian F: Parathyroid hormone (1–34) retards the lumbar facet joint degeneration and activates Wnt/β-catenin signaling pathway in ovariectomized rats. J Orthop Surg Res. 19:3522024. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Portal-Núñez S, Esbrit P, Alcaraz MJ and Largo R: Oxidative stress, autophagy, epigenetic changes and regulation by miRNAs as potential therapeutic targets in osteoarthritis. Biochem Pharmacol. 108:1–10. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang DK, Zheng HL, Zhou WS, Duan ZW, Jiang SD, Li B, Zheng XF and Jiang LS: Mitochondrial dysfunction in oxidative stress-mediated intervertebral disc degeneration. Orthop Surg. 14:1569–1582. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Albrahim T, Alangry R, Alotaibi R, Almandil L and Alburikan S: Effects of regular exercise and intermittent fasting on neurotransmitters, inflammation, oxidative stress, and brain-derived neurotrophic factor in cortex of ovariectomized rats. Nutrients. 15:42702023. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Ghosh R and Bishayi B: Endogenous blocking of TLR2 along with TNF-α and IL-1β ameliorates the severity of the S. aureus arthritis via modulating STAT3/SOCS3 expressions in tissue resident macrophages. Microb Pathog. 187:1065182024. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen Y, Xu N, Zhang WW, Wang Y, Su T, Zhou YM and Xu J: FSH enhances the inflammatory response of macrophages in the knee joint possibly through the NFκB pathway. FEBS Open Bio. 15:622–633. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Kültür T and Zengin M: Patients with rheumatoid arthritis and osteoarthritis in terms of sex hormone receptors and histopathological comparison of features. Arch Rheumatol. 36:192–200. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Capellino S, Riepl B, Rauch L, Angele P, Cutolo M and Straub RH: Quantitative determination of steroid hormone receptor positive cells in the synovium of patients with rheumatoid arthritis and osteoarthritis: Is there a link to inflammation? Ann Rheum Dis. 66:53–58. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kaur B, Rana D, Konar M, Sharma R, Chouhan DK, Saini UC, Prakash M, Arora A, Dhillon MS, Kaur J, et al: Comparative proteomic analysis of osteoarthritis and rheumatoid arthritis: Identifying potential biomarkers. J Orthop Res. 43:1396–1412. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Li H, Li X, Yang B, Su J, Cai S, Huang J, Hu T, Chen L, Xu Y and Li Y: The retinoid X receptor α modulator K-80003 suppresses inflammatory and catabolic responses in a rat model of osteoarthritis. Sci Rep. 11:169562021. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Abu-Amer Y: NF-κB signaling and bone resorption. Osteoporos Int. 24:2377–2386. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hügle T and Geurts J: What drives osteoarthritis?-synovial versus subchondral bone pathology. Rheumatology (Oxford). 56:1461–1471. 2017.PubMed/NCBI
|
|
57
|
Zhao K, Han D, He SR, Wu LY, Liu WY and Zhong ZM: N-acetyl-L-cysteine attenuates oxidative stress-induced bone marrow endothelial cells apoptosis by inhibiting BAX/caspase 3 pathway. Biochem Biophys Res Commun. 656:115–121. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Zhang S, Li T, Feng Y, Zhang K, Zou J, Weng X, Yuan Y and Zhang L: Exercise improves subchondral bone microenvironment through regulating bone-cartilage crosstalk. Front Endocrinol (Lausanne). 14:11593932023. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Liu Y, Tan H, Huang C, Li L and Wu S: Olive oil effectively mitigates ovariectomy-induced marrow adiposity assessed by MR spectroscopy in estrogen-deficient rabbits. Acta Radiol. 63:245–252. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Rapagna S, Roberts BC, Solomon LB, Reynolds KJ, Thewlis D and Perilli E: Relationships between tibial articular cartilage, in vivo external joint moments and static alignment in end-stage knee osteoarthritis: A micro-CT study. J Orthop Res. 40:1125–1134. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bei MJ, Tian FM, Xiao YP, Cao XH, Liu N, Zheng ZY, Dai MW, Wang WY, Song HP and Zhang L: Raloxifene retards cartilage degradation and improves subchondral bone micro-architecture in ovariectomized rats with patella baja-induced-patellofemoral joint osteoarthritis. Osteoarthritis Cartilage. 28:344–355. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Miyatake K, Muneta T, Ojima M, Yamada J, Matsukura Y, Abula K, Sekiya I and Tsuji K: Coordinate and synergistic effects of extensive treadmill exercise and ovariectomy on articular cartilage degeneration. BMC Musculoskelet Disord. 17:2382016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Li Z, Cheng W, Gao K, Liang S, Ke L, Wang M, Fan J, Li D, Zhang P, Xu Z and Li N: Pyroptosis: A spoiler of peaceful coexistence between cells in degenerative bone and joint diseases. J Adv Res. 71:227–262. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Wang YT, Zheng SY, Jiang SD, Luo Y, Wu YX, Naranmandakh S, Li YS, Liu SG and Xiao WF: Irisin in degenerative musculoskeletal diseases: Functions in system and potential in therapy. Pharmacol Res. 210:1074802024. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Chen WP, Tang JL, Bao JP, Hu PF, Shi ZL and Wu LD: Anti-arthritic effects of chlorogenic acid in interleukin-1β-induced rabbit chondrocytes and a rabbit osteoarthritis model. Int Immunopharmacol. 11:23–28. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Godoi MA, Camilli AC, Gonzales KGA, Costa VB, Papathanasiou E, Leite FRM and Guimarães-Stabili MR: JAK/STAT as a potential therapeutic target for osteolytic diseases. Int J Mol Sci. 24:102902023. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Tantowi NACA, Mohamed S, Lau SF and Hussin P: Comparison of diclofenac with apigenin-glycosides rich Clinacanthus nutans extract for amending inflammation and catabolic protease regulations in osteoporotic-osteoarthritis rat model. Daru. 28:443–453. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Yu X, Wu Q, Ren Z, Chen B, Wang D, Yuan T, Ding H, Wang Y, Yuan G, Wang Y, et al: Kaempferol attenuates wear particle-induced inflammatory osteolysis via JNK and p38-MAPK signaling pathways. J Ethnopharmacol. 318:1170192024. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Damani JJ, De Souza MJ, VanEvery HL, Strock NCA and Rogers CJ: The role of prunes in modulating inflammatory pathways to improve bone health in postmenopausal women. Adv Nutr. 13:1476–1492. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Sun K, Zhu J, Deng Y, Xu X, Kong F, Sun X, Huan L, Ren C, Sun J and Shi J: Gamabufotalin inhibits osteoclastgenesis and counteracts estrogen-deficient bone loss in mice by suppressing RANKL-induced NF-κB and ERK/MAPK pathways. Front Pharmacol. 12:6299682021. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Marie JC and Bonnelye E: Effects of estrogens on osteoimmunology: A role in bone metastasis. Front Immunol. 13:8991042022. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Breuil V, Ticchioni M, Testa J, Roux CH, Ferrari P, Breittmayer JP, Albert-Sabonnadière C, Durant J, De Perreti F, Bernard A, et al: Immune changes in post-menopausal osteoporosis: The immunos study. Osteoporos Int. 21:805–814. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Zhang Z, Tao Z, Zhang Z, Zhang W, Zhang X, Xu X, Gao R, Tao X and Zhou X: Single-cell transcriptomic analysis reveals AP-1 downregulation remodels bone marrow environment and contributes to osteopenia in ovariectomized mice. J Orthop Translat. 52:1–13. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Qi P, Xie R, Liu H, Zhang Z, Cheng Y, Ma J, Wan K and Xie X: Mechanisms of gut homeostasis regulating Th17/Treg cell balance in PMOP. Front Immunol. 15:14973112024. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Deng YX, He WG, Cai HJ, Jiang JH, Yang YY, Dan YR, Luo HH, Du Y, Chen L and He BC: Analysis and validation of hub genes in blood monocytes of postmenopausal osteoporosis patients. Front Endocrinol (Lausanne). 12:8152452022. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Zhou X, Chen Y, Zhang Z, Miao J, Chen G and Qian Z: Identification of differentially expressed genes, signaling pathways and immune infiltration in postmenopausal osteoporosis by integrated bioinformatics analysis. Heliyon. 10:e237942023. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Mohamad NV, Ima-Nirwana S and Chin KY: Are Oxidative stress and inflammation mediators of bone loss due to estrogen deficiency? A review of current evidence. Endocr Metab Immune Disord Drug Targets. 20:1478–1487. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Zhang QY, Gong HB, Jiang MY, Jin F, Wang G, Yan CY, Luo X, Sun WY, Ouyang SH, Wu YP, et al: Regulation of enzymatic lipid peroxidation in osteoblasts protects against postmenopausal osteoporosis. Nat Commun. 16:7582025. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Yang R, Li J, Zhang J, Xue Q, Qin R, Wang R, Goltzman D and Miao D: 17β-estradiol plays the anti-osteoporosis role via a novel ESR1-Keap1-Nrf2 axis-mediated stress response activation and Tmem119 upregulation. Free Radic Biol Med. 195:231–244. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zhang C, Li H, Li J, Hu J, Yang K and Tao L: Oxidative stress: A common pathological state in a high-risk population for osteoporosis. Biomed Pharmacother. 163:1148342023. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li LC, Liu L, Xu F, Wang YZ and Mao KL: Unraveling the role of FGF7 in osteoarthritis and osteoporosis: Therapeutic implications and challenges. FASEB J. 39:e704792025. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zahan OM, Serban O, Gherman C and Fodor D: The evaluation of oxidative stress in osteoarthritis. Med Pharm Rep. 93:12–22. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Chen X, Feng G, Shao L, Lin X, He X, Yang Y, Zhao X, Yan J, Ma L, Zhou Y, et al: Maintaining high levels of HIF-1α protects osteoarthritis cartilage by activating autophagy. Tissue Cell. 96:1030272025. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Ansari MY, Ahmad N and Haqqi TM: Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed Pharmacother. 129:1104522020. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Kim D, Piao J, Park JS, Lee D, Hwang DY and Hong HS: Substance P-mediated vascular protection ameliorates bone loss. Oxid Med Cell Longev. 2023:99033362023. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Huang J, Wu L, Zhao Y and Zhao H: Programmed cell death of chondrocytes, synovial cells, osteoclasts, and subchondral bone cells in osteoarthritis. J Inflamm Res. 18:12323–12360. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Nusse R and Clevers H: Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell. 169:985–999. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Yang C, Yang P, Liu P, Wang H, Ke E, Li K and Yan H: Targeting Filamin A alleviates ovariectomy-induced bone loss in mice via the WNT/β-catenin signaling pathway. Cell Signal. 90:1101912022. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yuasa T, Kondo N, Yasuhara R, Shimono K, Mackem S, Pacifici M, Iwamoto M and Enomoto-Iwamoto M: Transient activation of Wnt/{beta}-catenin signaling induces abnormal growth plate closure and articular cartilage thickening in postnatal mice. Am J Pathol. 175:1993–2003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Yan X, Fan DY, Pi YL, Zhang YN, Fu PJ and Zhang HF: ERα/β/DMP1 axis promotes trans-differentiation of chondrocytes to bone cells through GSK-3β/β-catenin pathway. J Anat. 240:1152–1161. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Yin X, Wang X, Hu X, Chen Y, Zeng K and Zhang H: ERβ induces the differentiation of cultured osteoblasts by both Wnt/β-catenin signaling pathway and estrogen signaling pathways. Exp Cell Res. 335:107–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Park OJ, Kwon Y, Kim J, Park C, Yun CH and Han SH: Muramyl dipeptide alleviates estrogen deficiency-induced osteoporosis through canonical Wnt signaling. J Pathol. 260:137–147. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Ye X and Liu X: Wnt16 signaling in bone homeostasis and osteoarthristis. Front Endocrinol (Lausanne). 13:10957112022. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Tat SK, Pelletier JP, Vergés J, Lajeunesse D, Montell E, Fahmi H, Lavigne M and Martel-Pelletier J: Chondroitin and glucosamine sulfate in combination decrease the pro-resorptive properties of human osteoarthritis subchondral bone osteoblasts: A basic science study. Arthritis Res Ther. 9:R1172007. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
De Leon-Oliva D, Barrena-Blázquez S, Jiménez-Álvarez L, Fraile-Martinez O, García-Montero C, López-González L, Torres-Carranza D, García-Puente LM, Carranza ST, Álvarez-Mon MÁ, et al: The RANK-RANKL-OPG system: A multifaceted regulator of homeostasis, immunity, and cancer. Medicina (Kaunas). 59:17522023. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Ni X, Wu B, Li S, Zhu W, Xu Z, Zhang G, Cui H, Bai Q and Wang J: Equol exerts a protective effect on postmenopausal osteoporosis by upregulating OPG/RANKL pathway. Phytomedicine. 108:1545092023. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Karim K, Giribabu N and Salleh N: Marantodes pumilum Var Alata (Kacip Fatimah) ameliorates derangement in RANK/RANKL/OPG pathway and reduces inflammation and oxidative stress in the bone of estrogen-deficient female rats with type-2 diabetes. Phytomedicine. 91:1536772021. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Mendez-Frausto G, Uresti-Rivera EE, Godina-Gonzalez S, Portales-Perez DP, Gonzalez-Amaro R and Garcia-Hernandez MH: Expression of mBD4, mBD3 and CRAMP during type II collagen-induced arthritis/CIA and their association with inflammation and bone-remodeling markers. Exp Mol Pathol. 123:1046892021. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Lu J, Hu D, Zhang Y, Ma C, Shen L and Shuai B: Current comprehensive understanding of denosumab (the RANKL neutralizing antibody) in the treatment of bone metastasis of malignant tumors, including pharmacological mechanism and clinical trials. Front Oncol. 13:11338282023. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Chen RB, Yang YD, Sun K, Liu S, Guo W, Zhang JX and Li Y: Potential mechanism of Ziyin Tongluo Formula in the treatment of postmenopausal osteoporosis: Based on network pharmacology and ovariectomized rat model. Chin Med. 16:882021. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Liu C, Zhang J, Ye Z, Luo J, Peng B and Wang Z: Research on the role and mechanism of the PI3K/Akt/mTOR signalling pathway in osteoporosis. Front Endocrinol (Lausanne). 16:15417142025. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Zhang X, Liu L, Liu D, Li Y, He J and Shen L: 17β-Estradiol promotes angiogenesis of bone marrow mesenchymal stem cells by upregulating the PI3K-Akt signaling pathway. Comput Struct Biotechnol J. 20:3864–3873. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Huang LW, Huang TC, Hu YC, Hsieh BS, Cheng HL, Chiu PR and Chang KL: S-Equol protects chondrocytes against sodium nitroprusside-caused matrix loss and apoptosis through activating PI3K/Akt pathway. Int J Mol Sci. 22:70542021. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Iantomasi T, Romagnoli C, Palmini G, Donati S, Falsetti I, Miglietta F, Aurilia C, Marini F, Giusti F and Brandi ML: Oxidative stress and inflammation in osteoporosis: Molecular mechanisms involved and the relationship with microRNAs. Int J Mol Sci. 24:37722023. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Xu F, Zhao LJ, Liao T, Li ZC, Wang LL, Lin PY, Jiang R and Wei QJ: Ononin ameliorates inflammation and cartilage degradation in rat chondrocytes with IL-1β-induced osteoarthritis by downregulating the MAPK and NF-κB pathways. BMC Complement Med Ther. 22:252022. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Shorning BY, Dass MS, Smalley MJ and Pearson HB: The PI3K-AKT-mTOR pathway and prostate cancer: At the crossroads of AR, MAPK, and WNT signaling. Int J Mol Sci. 21:45072020. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Xiao J, Zhang G, Mai J, He Q, Chen W, Li J, Ma Y, Pan Z, Yang J, Li S, et al: Bioinformatics analysis combined with experimental validation to explore the mechanism of XianLing GuBao capsule against osteoarthritis. J Ethnopharmacol. 294:1152922022. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Housmans BAC, van den Akker GGH, Neefjes M, Timur UT, Cremers A, Peffers MJ, Caron MMJ, van Rhijn LW, Emans PJ, Boymans TAEJ, et al: Direct comparison of non-osteoarthritic and osteoarthritic synovial fluid-induced intracellular chondrocyte signaling and phenotype changes. Osteoarthritis Cartilage. 31:60–71. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Gu ZX, Wang ZH, Zhang XQ, Zhou GH, Liang GH, Zeng LF and Liu J: Research advances in the study of traditional Chinese medicine formula granules on signaling pathway-mediated disease mechanisms. Front Pharmacol. 16:16092112025. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Dell'Isola A, Pihl K, Turkiewicz A, Hughes V, Zhang W, Bierma-Zeinstra S, Prieto-Alhambra D and Englund M: Risk of comorbidities following physician-diagnosed knee or hip osteoarthritis: A register-based cohort study. Arthritis Care Res (Hoboken). 74:1689–1695. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Park SY, Kim MJ, Cho JH, Nam HS, Ho JPY and Lee YS: Osteoarthritis progression pattern based on patient specific characteristics using machine learning. NPJ Digit Med. 8:4642025. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Zhang L, Cai R, Wang C, Liu J, Kuang Z and Wang H: Prediction of multiple degenerative diseases based on DNA methylation in a co-physiology mechanisms perspective. Int J Mol Sci. 25:95142024. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Bhattoa HP, Vasikaran S, Trifonidi I, Kapoula G, Lombardi G, Jørgensen NR, Pikner R, Miura M, Chapurlat R, Hiligsmann M, et al: Update on the role of bone turnover markers in the diagnosis and management of osteoporosis: A consensus paper from the European Society for clinical and economic aspects of osteoporosis, osteoarthritis and musculoskeletal diseases (ESCEO), international osteoporosis foundation (IOF), and international federation of clinical chemistry and laboratory medicine (IFCC). Osteoporos Int. 36:579–608. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Nagy E, Nagy-Finna C, Popoviciu H and Kovács B: Soluble biomarkers of osteoporosis and osteoarthritis, from pathway mapping to clinical trials: An update. Clin Interv Aging. 15:501–518. 2020. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Huang X, Zhong L, van Helvoort E, Lafeber F, Mastbergen S, Hendriks J, Post JN and Karperien M: The expressions of dickkopf-related protein 1 and frizzled-related protein are negatively correlated to local inflammation and osteoarthritis severity. Cartilage. 12:496–504. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Schubert M, Miyatake K, Kumagai K, Imai S, Yamaguchi Y and Inaba Y: Sclerostin inhibits interleukin-1β-induced late stage chondrogenic differentiation through downregulation of Wnt/β-catenin signaling pathway. PLoS One. 15:e02396512020. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Shi G, Subramanian S, Cao Q, Demehri S, Siewerdsen JH and Zbijewski W: Application of a novel ultra-high resolution multi-detector CT in quantitative imaging of trabecular microstructure. Proc SPIE Int Soc Opt Eng. 11317:113171E2020.PubMed/NCBI
|
|
118
|
Koff MF, Gao MA, Neri JP, Chiu YF, Lin BQ, Burge AJ, Su E, Padgett DE and Potter HG: Adverse local tissue reactions are common in asymptomatic individuals after hip resurfacing arthroplasty: Interim report from a prospective longitudinal study. Clin Orthop Relat Res. 479:2633–2650. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Shi G, Quevedo Gonzalez FJ, Breighner RE, Moseley KF, Witham T, Carrino JA, Siewerdsen JH and Zbijewski W: Effects of nonstationary system blur on radiomic texture features of trabecular bone in normal and ultra-high resolution CT. Med Phys. 52:e179432025. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Cardín-Pereda A, García-Sánchez D, Terán-Villagrá N, Alfonso-Fernández A, Fakkas M, Pérez-Del Barrio A, Marín-Díez E, Fernández-Lobo V, Sanz-Bellón P, Montes-Figueroa E, et al: Diagnostic reliability of plain radiography in osteonecrosis of the femoral head: General radiological features revised. Curr Med Imaging. 20:e2908232204842024. View Article : Google Scholar
|
|
121
|
Nair A, Alagha MA, Cobb J and Jones G: Assessing the value of imaging data in machine learning models to predict patient-reported outcome measures in knee osteoarthritis patients. Bioengineering (Basel). 11:8242024. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Batiste DL, Kirkley A, Laverty S, Thain LMF, Spouge AR, Gati JS, Foster PJ and Holdsworth DW: High-resolution MRI and micro-CT in an ex vivo rabbit anterior cruciate ligament transection model of osteoarthritis. Osteoarthritis Cartilage. 12:614–626. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Rossi LMM, Copes RM, Dal Osto LC, Flores C, Comim FV and Premaor MO: Factors related with osteoporosis treatment in postmenopausal women. Medicine (Baltimore). 97:e115242018. View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Yoo TK, Kim SK, Kim DW, Choi JY, Lee WH, Oh E and Park EC: Osteoporosis risk prediction for bone mineral density assessment of postmenopausal women using machine learning. Yonsei Med J. 54:1321–1330. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Tu JB, Liao WJ, Liu WC and Gao XH: Using machine learning techniques to predict the risk of osteoporosis based on nationwide chronic disease data. Sci Rep. 14:52452024. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Achari Y, Lu T and Hart DA: Polymorphisms in the promoter regions for human MMP-1 and MMP-13 lead to differential responses to the alpha and beta isoforms of estrogen receptor and their ligand in vitro. Biochim Biophys Acta. 1782:391–400. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
127
|
Sun GH, Liao Y, Liao Y, Ni ZY, Li N, Zhong PR, Li XH and Zhou J: Electroacupuncture intervention improves cartilage degeneration and subchondral bone osteoporosis of knee-joint possibly by adjusting OPG/RANK/RANKL signaling in ovariectomized rats. Zhen Ci Yan Jiu. 43:781–787. 2018.(In Chinese). PubMed/NCBI
|
|
128
|
Ho WC, Chang CC, Wu WT, Lee RP, Yao TK, Peng CH and Yeh KT: Effect of osteoporosis treatments on osteoarthritis progression in postmenopausal women: A review of the literature. Curr Rheumatol Rep. 26:188–195. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Xiao YP, Tian FM, Dai MW, Wang WY, Shao LT and Zhang L: Are estrogen-related drugs new alternatives for the management of osteoarthritis? Arthritis Res Ther. 18:1512016. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Schroeder RJ, Staszkiewicz J, O'Quin C, Carroll B, Doan N, Patel S, Ahmadzadeh S, Kallurkar A, Viswanath O, Varrassi G, et al: Oral therapeutics post menopausal osteoporosis. Cureus. 15:e428702023.PubMed/NCBI
|
|
131
|
Tang Y, Ma R, Zhang L, Sun X and Wang Y: Effectiveness and safety of hormone replacement therapy in the treatment of menopausal syndrome: A meta-analysis. Am J Transl Res. 17:1–15. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Ali A, Iftikhar A, Tabassum M, Imran R, Shaid MU, Hashmi MR, Saad M, Humayun M, Imtiaz S and Baig E: Efficacy and safety of intravaginal estrogen in the treatment of atrophic vaginitis: A systematic review and meta-analysis. J Menopausal Med. 30:88–103. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
133
|
Stevenson TEJ, Brincat MP, Pollacco J and Stevenson JC: Effect of hormone replacement therapy on intervertebral disc height. Climacteric. 26:110–113. 2023. View Article : Google Scholar : PubMed/NCBI
|
|
134
|
Muscat Baron Y, Brincat MP, Galea R and Calleja N: Low intervertebral disc height in postmenopausal women with osteoporotic vertebral fractures compared to hormone-treated and untreated postmenopausal women and premenopausal women without fractures. Climacteric. 10:314–319. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Cameron CR, Cohen S, Sewell K and Lee M: The art of hormone replacement therapy (HRT) in menopause management. J Pharm Pract. 37:736–740. 2024. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Ruan X and Mueck AO: Primary choice of estrogen and progestogen as components for HRT: A clinical pharmacological view. Climacteric. 25:443–452. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Chen Z, Wu C and Huang Z: Association between estrogen replacement therapy and heart failure in postmenopausal women: A systematic review and meta-analysis. Prev Med. 181:1079092024. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Flores VA, Pal L and Manson JE: Hormone therapy in menopause: Concepts, controversies, and approach to treatment. Endocr Rev. 42:720–752. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Panay N, Anderson RA, Bennie A, Cedars M, Davies M, Ee C, Gravholt CH, Kalantaridou S, Kallen A, Kim KQ, et al: Evidence-based guideline: Premature ovarian insufficiency. Hum Reprod Open. 2024:hoae0652024. View Article : Google Scholar : PubMed/NCBI
|
|
140
|
Schuiling KD, Robinia K and Nye R: Osteoporosis update. J Midwifery Womens Health. 56:615–627. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Mitlak BH and Cohen FJ: Selective estrogen receptor modulators: A look ahead. Drugs. 57:653–663. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Reid IR and Billington EO: Drug therapy for osteoporosis in older adults. Lancet. 399:1080–1092. 2022. View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Fu S, Wang Y, Zhou J, Liu Y, Wan J, Xu L, Wang J, Zhang Z and Wang S: Engineered EVs from 3D-cultured MSCs for synergistic modulation of inflammatory microenvironment and cartilage regeneration in osteoarthritis. ACS Appl Mater Interfaces. 17:42658–42673. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Zhang X, Xu D, Zhang R, Wang H and Yang G: Interaction between diabetes and osteoporosis: Imbalance between inflammation and bone remodeling. Osteoporos Int. 36:2401–2409. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
145
|
Brezgin S, Danilik O, Yudaeva A, Kachanov A, Kostyusheva A, Karandashov I, Ponomareva N, Zamyatnin AA, Parodi A, Chulanov V and Kostyushev D: Basic guide for approaching drug delivery with extracellular vesicles. Int J Mol Sci. 25:104012024. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Wang X and Thomsen P: Mesenchymal stem cell-derived small extracellular vesicles and bone regeneration. Basic Clin Pharmacol Toxicol. 128:18–36. 2021. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Roszkowski S: Therapeutic potential of mesenchymal stem cell-derived exosomes for regenerative medicine applications. Clin Exp Med. 24:462024. View Article : Google Scholar : PubMed/NCBI
|
|
148
|
Bei HP, Hung PM, Yeung HL, Wang S and Zhao X: Bone-a-petite: Engineering exosomes towards bone, osteochondral, and cartilage repair. Small. 17:e21017412021. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Chen G, Li H, Chen R and Chen G: Exosomes as emerging therapeutic strategies in primary osteoporosis: A narrative review. Drug Des Devel Ther. 19:9099–9115. 2025. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Lu CH, Chen YA, Ke CC and Liu RS: Mesenchymal stem cell-derived extracellular vesicle: A promising alternative therapy for osteoporosis. Int J Mol Sci. 22:127502021. View Article : Google Scholar : PubMed/NCBI
|














